 Chris John Weston
Chris John Weston Henning Wolfgang Zimmermann
Henning Wolfgang Zimmermann David H. Adams
David H. Adams
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 24 April 2019
Sec. Molecular Innate Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00893
This article is part of the Research Topic The Role of Myeloid-Derived Cells in the Progression of Liver Disease View all 16 articles
Control of homeostasis and rapid response to tissue damage in the liver is orchestrated by crosstalk between resident and infiltrating inflammatory cells. A crucial role for myeloid cells during hepatic injury and repair has emerged where resident Kupffer cells, circulating monocytes, macrophages, dendritic cells and neutrophils control local tissue inflammation and regenerative function to maintain tissue architecture. Studies in humans and rodents have revealed a heterogeneous population of myeloid cells that respond to the local environment by either promoting regeneration or driving the inflammatory processes that can lead to hepatitis, fibrogenesis, and the development of cirrhosis and malignancy. Such plasticity of myeloid cell responses presents unique challenges for therapeutic intervention strategies and a greater understanding of the underlying mechanisms is needed. Here we review the role of myeloid cells in the establishment and progression of liver disease and highlight key pathways that have become the focus for current and future therapeutic strategies.
Myeloid cells arise from the common myeloid precursor and give rise to monocytes, dendritic cells and macrophages, and granulocytes. Myeloid cell functions include the recognition, ingestion and degradation of cellular debris, foreign material or pathogens, subsequent control of inflammatory responses, and maintenance of tissue architecture. There is increasing evidence implicating granulocytes in liver homeostasis and disease but this review will focus mainly on monocytes and macrophages. Macrophages are a diverse, heterogeneous population derived from short-lived, but plastic, precursor monocyte populations. Monocytes are rapidly recruited to sites of injury and their functions are imprinted in the bone marrow, whereas macrophages tend to be long-lived and tissue-resident, where their functions are dictated by environmental cues (1). A highly coordinated pathway of monocyte recruitment and subsequent imprinting of macrophage “identities,” the mechanisms of which are only now beginning to be understood, controls local tissue inflammatory and regenerative functions and is critical in maintaining tissue architecture (2–4). Extensive rodent and human studies have demonstrated key roles for monocytes and macrophages in the establishment, progression and regression of liver disease including a critical role in directing tissue regeneration (5–7). This fine balance of pro- and anti-inflammatory mediators is crucial to determining the path of disease progression, and understanding how myeloid cells contribute to injury and repair will enable the rational design of novel therapies. In this review we summarize the identities and roles of myeloid cell populations in the liver, and describe approaches that are being developed to reduce inflammation through targeting this innate immune cell population.
The liver contains a population of self-renewing resident macrophages, termed Kupffer cells (KC), derived from yolk sac-derived progenitor cells (8, 9) or hematopoietic stem cells (10). In mice KC phenotype is thought to be controlled by a specific set of transcription factors including ID3 and ZEB2 through progenitor cell development and maintenance of the expression of LXRα permitting replenishment of the KC niche by progenitors from the circulation (2, 11–13) (Figure 1, Table 1). They are non-migratory, being retained in the sinusoids where they maintain a tolerogenic environment despite the presence of low levels of food particles and bacterial antigens delivered from the gut via the portal vein (4, 14, 15). This is achieved through highly effective phagocytic and scavenging mechanisms triggered by toll-like receptor (TLR) signaling and scavenger receptors such as CD36, scavenger receptor-A and galectin-3 (16, 17). Their expression of high levels of pattern recognition receptors (PRRs) allows macrophages to respond to a wide range of danger-associated molecular patterns (DAMPs) released during tissue injury, such as high mobility group protein B1 (HMGB1), ATP, uric acid, DNA fragments and cholesterol crystals (5) and pathogen-associated molecular patterns (PAMPs, such as lipopolysaccharide and flagellin) released from microbes. Activation of PRRs leads to the formation of the inflammasome (18) via multi-protein complexes including the NOD-, LRR- and pyrin domain-containing 3 (NLRP3) (19). Formation of the inflammasome promotes the release of potent signaling molecules including IL-1β, PGE2, HMGB1, TNF-α, and IL-17, driving inflammation and fibrosis (20–24). KC from both mouse and human liver also secrete the anti-inflammatory cytokine IL-10 (25–27), express low levels of MHC class II and co-stimulatory molecules combined with high levels of the T-cell inhibitory molecule PDL-1 (27). This makes them unable to fully activate T cell effector function but rather to promote the development of regulatory T-cells (Treg). This is further enhanced through their secretion of PGE2 (28) and upregulation of the indolamine 2,3-dioxygenase pathway which promotes immune cell tolerance (29).
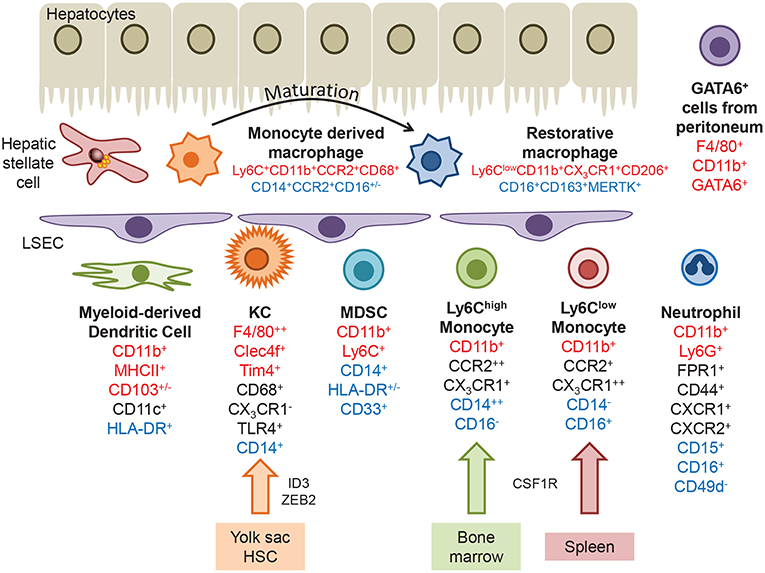
Figure 1. Myeloid populations present within the liver. Kupffer cells are derived from the yolk sac or hematopoietic stem cells, under the control of transcription factors such as ID3 and ZEB2. Infiltrating monocytes originating in the bone marrow or spleen express the chemokine receptors CCR2 and CX3CR1, and can differentiate into monocyte-derived dendritic cells. Following injury monocytes undergo transendothelial migration across LSEC and differentiate into monocyte derived macrophages, which can mature into a more restorative phenotype or replenish the KC pool. A GATA6+ macrophage population that migrates from the peritoneum during hepatic injury has been identified in mice. Markers that identify hepatic macrophages in mice, humans or are common to both are highlighted in red, blue and black text, respectively.
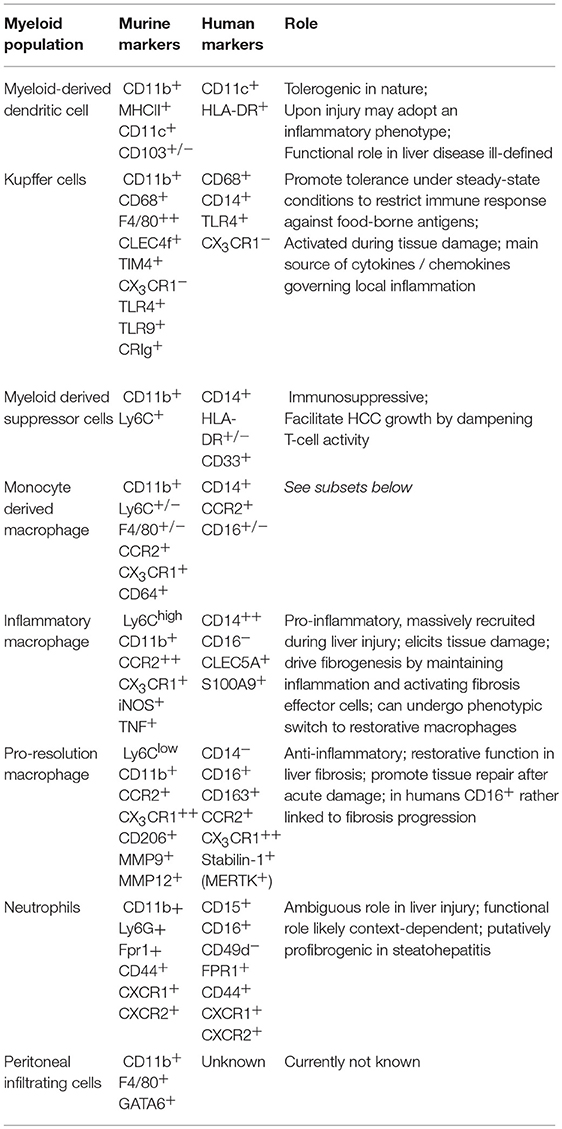
Table 1. Myeloid cell populations in humans and mice.
Murine KC have been well characterized under homeostatic conditions and in experimental models of hepatic injury, where they express CD11b+F4/80++CD68+CD11c+/−CLEC4F+TIM4+ in addition to TLR4, TLR9, and CRIg, but are negative for the chemokine receptor CX3CR1. Recent advances in proteomic analysis has revealed circadian regulation of not only KC numbers in uninjured mouse liver, but also components of the immune response pathway which peak during the daytime, including Tlr4, Myd88, Irak4, and Tak1 (30). Human KC are less well described but can be identified through expression of CD68+CD14+TLR4+ and lack of CX3CR1.
Circulating monocytes are actively recruited to the liver, guided by adhesion molecules and chemokine gradients generated at the sinusoidal endothelial interface (see below). In mice bone marrow derived myeloid cells expressing high levels of Ly6C and CCR2 rapidly infiltrate tissue and are associated with the expression of pattern recognition receptors (PRR) and inflammatory cytokines (CD11b+CCR2+CX3CR1+CD43−). In contrast Ly6Clow monocytes from the spleen express a broad range of scavenger receptors and exhibit a patrolling behavior that may enable the engulfment of apoptotic cells (CD11b+CCR2−CX3CR1++CD43+) (27, 31–36). In humans there is no discriminatory expression of Ly6C and monocytes are classified according to the expression of CD14 and CD16 giving rise to classical (CD14++CD16−), intermediate (CD14+CD16+), and non-classical (CD14−CD16+) populations. Gene expression profiling of these subsets has determined that CD14++CD16− monocytes resemble murine Ly6Chigh infiltrative cells and CD14−CD16+ monocytes more closely align with the patrolling Ly6Clow population (31). Potent immunomodulatory myeloid derived suppressor cells (MDSC) are also present in both murine and human liver tissue. MDSCs are a heterogeneous population of cells which express markers shared with other immune cell populations (CD11b+Ly6C+ in mice, CD14+HLA-DR+/−CD33+ in humans), therefore identification is usually confirmed by means of a T-cell suppression assay (37). MDSC suppress immune responses through production of arginase 1 (Arg1), inducible nitric oxide synthase (iNOS) and generation of reactive oxygen species (ROS), or secretion of IL-10 (38).
Damage to tissue results in an upregulation of adhesion molecules on liver sinusoidal endothelium (LSEC) and the secretion of chemokines, cytokines and other bioactive molecules that promote immune cell recruitment [reviewed in (39)]. Circulating CCR2+ monocytes are recruited in response to local CCL2, released primarily by hepatic stellate cells (HSC) (40, 41), or through the CCR8/CCL1 and CXCR3/CXCL10 axes (42–44). In humans the migration of CD14−CD16+ monocytes is promoted through activation of CX3CR1 by endothelial CX3CL1, a transmembrane chemokine that is expressed at high levels during inflammation (45). Intermediate CD14+CD16+ monocyte populations are enriched in the diseased liver (46), partly due to their increased propensity when compared with other monocytes to migrate across LSEC. These cells exhibit high phagocytic activity and secrete pro-inflammatory and fibrogenic mediators (47). Bidirectional migration of monocytes affects the local balance of inflammatory and anti-inflammatory cells. Pro-inflammatory CD14−CD16+ subsets undergo reverse migration from tissue back into the circulation via across LSEC from where they may contribute to systemic inflammatory responses, whereas anti-inflammatory cells remain in the tissue where they suppress T-cells and promote endotoxin tolerance (48). A phenotypic switch in macrophage phenotype is observed during acute liver injury in humans where MAC387 (S100A9) can be used to identify circulation-derived macrophages in contrast to CD68+ resident populations (49). Infiltrating monocytes also undergo local intrahepatic differentiation into anti-inflammatory MDSC following injury via contact-dependent mechanisms such as communication with hepatic stellate cells (HSC), or interaction with soluble mediators such as catalase (50, 51).
In mouse models of sterile injury CCR2+Ly6Chigh monocytes form rings to demarcate the extent of injury (14), and subsequently mature into Ly6Clow monocytes that promote the resolution of injury and fibrosis (52). These “pro-restorative” macrophages exhibited a phenotype distinct from the classical M1 (pro-inflammatory) or M2 (pro-resolution) dichotomy with increased expression of genes that promote tissue restoration including matrix metalloproteinases (MMPs), growth factors, and phagocytosis-related genes. Murine monocytes can also take unconventional routes into liver tissue. In a model of sterile liver injury GATA6-positive macrophages (CD11b+F4/80+Gata6+) originating in the peritoneal compartment were observed within the hepatic compartment at a very early stage of tissue damage (53). These cells migrate directly across the mesothelium, dependent on adenosine triphosphate and the adhesion molecule CD44. The contribution of these cells to disease pathogenesis is currently unknown.
Thus, local polarization of myeloid cell populations, and recruitment of macrophages from other sites, has important implications in disease pathogenesis where the balance of pro-/anti- inflammatory mediators and fibrogenic responses dictates the course of the disease.
Acute liver failure (ALF) is associated with high mortality and toxic liver injury in response to overdose of drugs such as acetaminophen is a more common cause of ALF than immune-mediated injury arising from acute viral hepatitis (54, 55). Much of what we know of macrophage function during early disease is derived from experimental models of acute liver injury in rodents such as carbon tetrachloride toxicity (hepatocyte necrosis) (56), bacterial infection (57), concanavalin A (T cell mediated hepatocyte destruction) (58), ischemia-reperfusion (I-R) injury (59), sterile injury (14), and viral infection (60). These models show that extensive hepatocyte damage mediated by heat/toxin/immune-mediated killing releases DAMPs such as HMGB1 and nuclear DNA which are sensed by KC leading to the release of cytokines and chemokines, creating an environment that drives the recruitment of inflammatory macrophage subsets (5). In the absence of persistent injury, tissue repair is initiated by the maturation of pro-inflammatory populations to a more restorative phenotype, associated with anti-inflammatory and pro-angiogenic responses (52) (Figure 2).
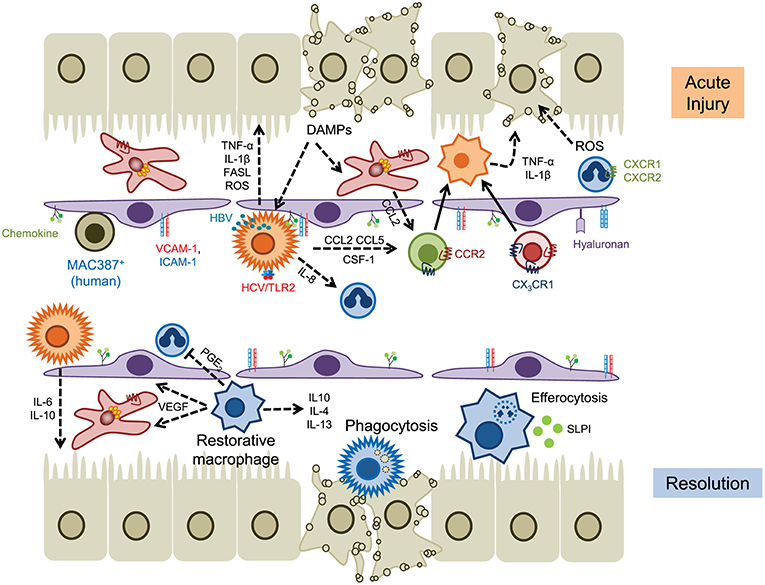
Figure 2. The role of myeloid cells in acute liver injury. Hepatocyte cell death releases DAMPs that activate KC and hepatic stellate cells, leading to the release of chemokines such as CCL2 and IL-8 (CXCL8) that drives the recruitment of myeloid cells into local areas of inflammation (top). Neutrophils are recruited via CD44 and hyaluronan and generate ROS that promotes hepatocyte death, whereas infiltrating monocytes (and KCs) secrete proinflammatory cytokines such as TNFα and IL-1β. Hepatic viruses can also stimulate KC through internalization (HBV) or binding to TLR2 (HCV). Homeostasis is restored through the action of restorative macrophages (matured by phagocytosis) that secrete anti-inflammatory cytokines and promote angiogenesis, and secretion of IL-6 and IL-10 by KC (lower panel). Infiltrating neutrophils are removed through efferocytosis mediated by MERTK+ macrophages and SLPI. Solid lines indicate cell migration, dashed lines represent the secretion of soluble mediators.
In acetaminophen (APAP) induced liver injury, perhaps the best described rodent model of ALF, KC respond to tissue damage through the rapid release of cytokines and chemokines including IL-1β, TNF-α, CCL2, and CCL5 (61). Initially the numbers of KC are reduced (<24 h) and early injury is associated with high numbers of infiltrating Ly6Chigh monocytes which produce proinflammatory cytokines such as TNF-α and IL-1β and chemokines such as CCL2 and CCL5 (62–65). Evidence that these early entrants drive tissue injury comes from data showing that (i) infiltration of these cells during acetaminophen-induced injury can be reduced through blockade of CCR2-mediated recruitment (mNOX-E36, a CCL2 inhibitor, or cenicriviroc, a CCR2/CCR5 dual inhibitor) and (ii) that adoptive transfer of bone marrow monocytes exacerbated tissue damage (66). Initiation of repair and control of inflammation is mediated following a phenotypic switch in hepatic macrophages toward a pro-resolution, hepatoprotective subset expressing IL-10, IL-4 and IL-13 (67–69). This maturation event is dependent on colony stimulating factor 1 (CSF1) and secretory leukocyte protease inhibitor (SLPI) in areas of hepatic necrosis (70–72). The emergence of CCR2lowCX3CR1high cells is also associated with the expression of vascular endothelial growth factor A (VEGF-A) which promotes repair of the vascular architecture, and increased phagocytic capacity to remove dead and dying cells (72–74). In murine models this reparative pathway can be disrupted via modulation of CCR2 signaling or depletion of macrophages through treatment with liposomal clodronate, indicating that both tissue resident and infiltrating myeloid cell populations orchestrate repair (62, 63, 75).
Similar findings have been described in patients with ALF. Clusters of CCR2+ macrophages are seen in patients with APAP-induced liver failure (66) and increased serum CCL2 levels are associated with an unfavorable prognosis (49). A pro-resolution population of MerTK+HLA-DRhigh cells has been identified in circulatory and tissue compartments of patients with ALF (72, 76). Analysis of these macrophages determined that they secreted anti-inflammatory mediators and exhibited reduced responses to bacterial challenge, consistent with an anti-inflammatory immune tolerant function. This is supported by the fact that APAP-treated Mer knockout animals exhibited persistent liver injury and inflammation associated with a defect in efferocytosis (72).
The pathways involved in other acute injury settings also result in activation of KC following hepatocyte damage mediated by T-cells (concanavalin A), oxidative stress (I-R), heat (sterile injury), or virus induced apoptosis (hepatitis viruses). During viral infection of humans KC increase in number and drive the infiltration of other immune cell populations through the production of inflammatory cytokines such as IL-1β, IL-18, and TNF-α (77–80). KC expression of IL-6, IFN-γ, reactive oxygen species, FAS ligand, granzyme B and TRAIL has been shown to inhibit hepatitis C (HCV) replication, and induces apoptosis of infected hepatocytes (81, 82). Triggering of KC responses arises as a result of engulfment of hepatitis B viral particles (leading to production of IL-18 and NK cell stimulation) (83) or via TLR2 signaling and formation of the inflammasome, with concomitant secretion of IL-18 and IL-1β, in the case of HCV (84, 85). Conversely in the setting of chronic hepatitis B viral infection the immune response is impaired through release of IL-10 (86), reduced IL-12 expression (87) or T-cell exhaustion (88) mediated by TLR2 signaling on KCs, via upregulation of galectin-9 expression driving further immune cell exhaustion following engagement with Tim-3 (89), or through increased expression of the inhibitory ligand PDL1 (90). An excess of hepatitis B virus antigen can also dampen TLR responses which contribute to viral evasion of innate and adaptive immune responses (91). This is thought to occur through suppression of proinflammatory cytokines and expression of tolerogenic mediators (IL-10 in particular) reminiscent of the tolerogenic effects of LPS, although the signaling pathways mediating this effect may be distinct.
A prolonged cycle of iterative bursts of tissue damage and inflammation underlies chronic liver disease leading to fibrogenesis and ultimately in some cases cirrhosis. A proportion of patients will develop hepatocellular carcinoma on the background of continuing inflammation and fibrogenesis (92). The incidence of non-alcoholic fatty liver disease (NAFLD) and alcohol related liver disease (ARLD) has increased rapidly in recent years and following advances in the treatment of chronic viral hepatitis, attention is now switching to treating these increasingly common chronic conditions (93) (Figure 3).
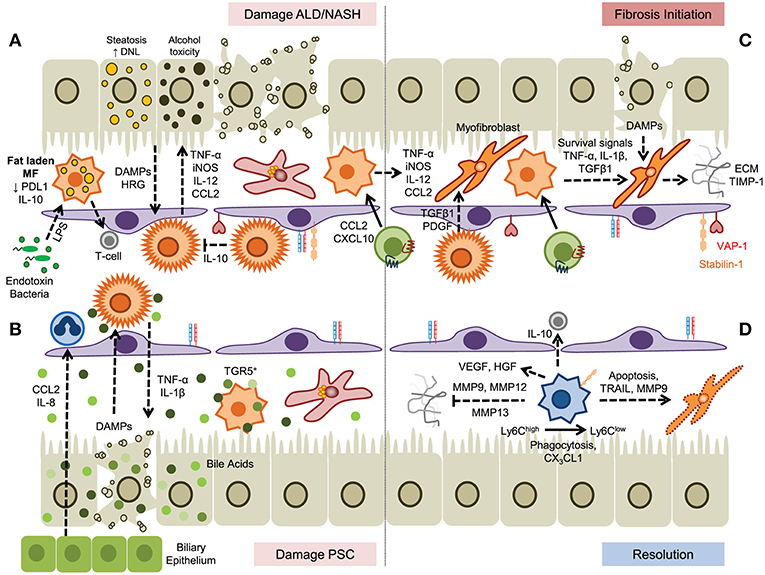
Figure 3. A dual role for myeloid cells in the establishment and resolution of chronic liver disease. (A) Hepatocyte damage driven by steatosis or alcohol toxicity activates KC which secrete proinflammatory cytokines that drive disease progression and promotes infiltration of myeloid cells. In steatotic livers fat laden macrophages exhibit impaired endotoxin responses but may prime T-cell mediated immunity. (B) Cholangiocyte-derived chemokines promote recruitment of hepatic neutrophils and subsequent damage to hepatocytes promotes further inflammation. Bile acids promote KC inflammasome formation; however this can be suppressed through binding of bile salts to TGR5 expressed by monocyte-derived macrophages. (C) Secretion of soluble factors by KC and monocyte-derived macrophages promotes fibrosis through the activation and differentiation of hepatic stellate cells, promoting survival of myofibroblasts and the generation of extracellular matrix proteins. (D) Resolution of fibrosis is mediated by Ly6Clow macrophages, generated from Ly6Chigh precursors, by degradation of ECM by matrix metalloproteinases, induced apoptosis of hepatic stellate cells and myofibroblasts, and secretion of anti-inflammatory cytokines.
NAFLD is a spectrum of disease ranging from simple steatosis (fatty liver) to non-alcoholic steatohepatitis (NASH), fibrosis and cirrhosis (with or without malignancy). The underlying pathology is driven by dysregulation of lipid metabolism and accumulation of lipid in hepatocytes. It is a systemic disease where dysregulated inflammation in adipose, and liver tissue and changes in the gut microbiome all drive the production of inflammatory mediators such as cytokines and chemokines (94). In patients with NAFLD enlarged and aggregated KC populations are seen in the liver and their presence correlates with the severity of the disease (95).
This is consistent with observations in diet-induced murine models of NAFLD where KC activation leads to triglyceride accumulation and production of proinflammatory cytokines such as TNF-α (96, 97). Murine hepatic macrophages can also receive activation signals from lipid-stimulated hepatocyte-derived extracellular vesicles via tumor necrosis factor-related apoptosis-inducing ligand receptor 2 (TRAIL-R2, also known as DR5) and receptor-interacting protein kinase 1 (98), and obese mice also show reduced expression of the glucocorticoid-induced leucine zipper (GILZ) in macrophages associated with a proinflammatory phenotype (99). In this context the development of steatohepatitis arises from chronic inflammation associated with an influx of Ly6C+ monocytes that enhances the proinflammatory environment through activation of liver resident cell populations (100, 101). These infiltrating monocyte subsets are recruited via chemokine receptor pairs such as CCR2-CCL2 (40, 102) and CXCR3-CXCL10 (44), or atypical adhesion molecules including vascular adhesion protein-1 and scavenger receptors (103).
Intestinal dysbiosis and hepatocyte apoptosis contribute to the inflammatory response via DAMP- and PAMP-mediated pathways respectively (104, 105) associated with increased expression and activation of receptors such as TLR4 and TLR9 in both humans and murine models of NASH (106). Changes in the microbiome can have complex effects on the liver altering metabolic response through the production of metabolites that enter the liver via the portal vein as well as through bacterial products such as LPS, and in the presence of a leaky gut intact bacteria are taken up by KC (107). KC also regulate anti-inflammatory responses through secretion of IL-10. In addition to its more general anti-inflammatory properties KC derived IL-10 also induces the apoptosis of proinflammatory KC allowing KC to self-regulate toward a more tolerogenic environment (108). The induction of a pro-resolution M2 KC phenotype is dependent on activation of RORα and KLF4, and provision of an activator of RORα (JC1-40) improved the symptoms of NASH in a high fat diet murine model suggesting that KC polarization is a viable therapeutic strategy (109). Immune checkpoint proteins such as Tim-3 have been detected on a range of macrophage subsets in murine models of NASH. The presence of TIM-3 limits steatohepatitis by controlling ROS induced activation of NOX2 and the NLRP3 inflammasome and secretion of IL-1β and IL-18 (110). Thus, therapeutic strategies could look at promoting the recruitment or differentiation of TIM-3 macrophages to shift the local environment toward resolution and suppression of inflammation.
Similar mechanisms of disease progression have been described for ARLD, where metabolism of alcohol in the liver drives hepatocyte cell death. In rodent models of ARLD such as the Lieber-DeCarli diet hepatic macrophages become activated to produce TNF-α, IL-6, CCL2 and ROS (111, 112) and depletion of macrophage populations with either GdCl3 or liposomal clodronate attenuated alcohol-induced liver inflammation (111, 113). Expression of myeloid NADPH oxidase, specifically the catalytic subunit gp91phox, contributes to the pathogenesis of murine ARLD by driving a switch between pro-inflammatory and pro-resolution macrophage populations. Thus, gp91phox-deficient animals show an increased ratio of Ly6Chigh/Ly6Clow intrahepatic macrophages and a diminished capacity for efferocytosis (114). Clustering of myeloid cells close to portal tracts is observed in ARLD patients (115) associated with increased levels of cytokines (IL-6, IL-8, IL-18), chemokines and macrophage activation markers that correlate with outcome and severity of disease (116–119). Gut permeability is increased in patients with ARLD leading to high levels of endotoxin in the liver resulting in a greater sensitivity of circulating monocytes from these patients to LPS (120, 121); a phenomenon also reported for resident KC isolated from alcohol-fed mice where increased sensitivity to endotoxin promoted expression of TNF-α and CCL2 (122, 123).
The role of macrophages in cholestatic diseases such as primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) is not well described. Accumulation of perisinusoidal hepatic macrophages in human tissue is reported in PSC but not in PBC (124) and increased infiltration of CD68+/CCR2+ cells was observed at later stages of disease in PSC including both CD206+ (anti-inflammatory) and iNOS+ (pro-inflammatory) macrophages (125). In diseases associated with cholestasis dysregulated bile acid production and excretion by cholangiocytes directly affects macrophage function and differentiation although the effects are complex. Although in mice hydrophobic bile acids have been reported to promote the formation of macrophage inflammasomes and IL-1β secretion (126, 127) other studies report activation of anti-inflammatory pathways in human macrophages by taurolithocholic acid through a PKA-mediated increase in IL-10 (128). Mice lacking the bile acid transporter Mdr2 (Abcb4) develop hepatobiliary inflammation and fibrosis with some, but not all, features of PSC including an accumulation of peribiliary, proinflammatory macrophages recruited in response to cholangiocyte secretion of IL-8 and CCL2. Pharmacological treatment of mice with the CCR2/CCR5 antagonist cenicriviroc attenuated macrophage infiltration and liver injury consistent with an effector role for macrophages (125) and other rodent models have shown that the G-protein-coupled bile acid receptor, Gpbar1 (TGR5) is expressed by macrophages to sense and respond to bile acids (129, 130). Activation of murine TGR5 leads to PKA-induced ubiquitination of NLRP3, acting as a brake on inflammasome activation (131) and dampening cytokine responses (129). In a murine model of colitis treatment with the TGR5 agonist BAR501 reduced the trafficking of Ly6Chigh monocytes into the intestinal mucosa, reduced the expression of inflammatory genes (Tnfa, Ifng, Il1b, Il6, and Ccl2), and induced a regulatory T-cell environment through the production of IL-10 and TGF-β (132). The therapeutic potential of other TGR5 agonists such as 6α-ethyl-23(S)-methyl-cholic acid (6-EMCA, INT-777) are currently being explored in cholestatic liver disease (126, 133).
Neutrophils are derived from bone marrow and are released into the peripheral circulation where they play an important role in host defense and tissue healing (134), characterized by a high phagocytic capacity, the production of antimicrobial molecules and ability to shape immune responses (134, 135). The identities of neutrophil subsets and their functions are not clearly defined, with much of our knowledge arising from murine models. As a result their important roles in liver homeostasis and disease are only beginning to be understood (136, 137).
Neutrophil recruitment from the circulation into the liver is independent of selectins (138) and in many conditions is also independent of α2 integrin and ICAM-1 (139). Instead, neutrophils use CD44 to bind hyaluronan (HA) on LSEC and respond to chemokine ligands of CXCR2. A signaling network of TLR2, S100A9 and CXCL2 was shown to be necessary for neutrophil recruitment in a chronic model of liver injury in the mouse (140), while activation of TLR4 on LSEC was sufficient to induce the deposition of serum-associated hyaluronan-associated protein within the hepatic sinusoids which promoted CD44-dependent neutrophil migration in a murine model of endotoxemia (141). In sterile rodent injury models such as local thermal injury, HA-CD44 driven recruitment is less important and neutrophils use αMβ2 (Mac-1) binding to ICAM-1 (21). This pathway plays little role in septic injury because IL-10 leads to a loss of cell surface αMβ2 (142). Invading neutrophils in septic injury tend to arrest soon after infiltrating the tissue, whereas in sterile injury these cells migrate toward the focus of damage and adopt a swarming behavior which restricts neutrophil motility to within the boundary of the injury. This behavior is amplified by leukotriene B4 (LTB4) produced by the first invading neutrophils (21, 143). In sterile injury neutrophil recruitment can be promoted by ATP release from necrotic hepatocytes leading to activation of the inflammasome, and presentation of ligands for CXCR2 on the surface of the hepatic sinusoids (21). Alternatively N-formyl peptides released from dying and dead hepatocytes are detected by the formylated peptide receptor-1 on neutrophils which guide them toward the site of injury (21, 144, 145). This enables neutrophils to prioritize their responses to chemoattractant gradients that arise directly from damaged tissue over competing signals from chemokines or LTB4 and remain within the boundaries of the necrotized tissue (146–149). Live cell imaging in mice identified a non-muscle myosin II protein that was essential for neutrophil trafficking, demonstrating that myosin heavy chain 9 (Myh9) was localized in branching lamellipodia and in the uropod where it may enable fast neutrophil migration (150).
During acute liver injury, neutrophils use the receptor for advanced glycation end products (RAGE) to respond to HMGB1 released by necrotic hepatocytes (151). However, this pathway also contributes to sepsis through diminished bacterial killing by neutrophils and reduced NADPH oxidase activation (152). Neutrophils form extracellular traps by a process known as NETosis to enhance antibacterial defenses [reviewed in (153)]. Defects in NET formation have been linked to impaired efferocytosis and contribute to liver injury and sepsis in models of liver disease (154). In murine models of chronic liver disease neutrophils drive hepatocellular damage but are also associated with mechanisms of tissue repair. Myeloperoxidase secreted by neutrophils drives oxidative damage and contributes to the development of NASH in mice (155) and increased levels of myeloperoxidase activity have been detected in patients with NASH (156). The development of obesity-related inflammation in patients with NASH also correlated with an increase in the ratio of neutrophil elastase to its inhibitor α1-antitrypsin, although the ratio reduced as the disease progressed to fibrosis (157). Conversely murine neutrophils can alleviate fibrosis through secretion of MMP8 and MMP9 (158), and depletion of neutrophils improved liver function in a diet-induced model of NASH (159). Following resolution of tissue damage in sterile injury neutrophils migrate out of the tissue and back into the vasculature and, following passage through the lungs where they upregulate CXCR4, return to the bone marrow where they undergo apoptosis (160). It is not currently known if this process contributes to the pathogenesis of other hepatic diseases.
In contrast to hepatic macrophages liver dendritic cells are scarce and mostly scattered in the portal region where they capture antigens delivered by via the portal vein (161). Dendritic cells can also translocate from blood to lymph via the hepatic sinusoids to concentrate in regional perihepatic lymph nodes (162). Hepatic dendritic cells comprise plasmacytoid DCs (pDCs) and classical (myeloid) DCs (cDCs) which express high levels of MHC-Class II molecules (e.g., HLA-DR) but are negative for other hematopoietic lineage markers (163). In general hepatic DCs are tolerogenic and inherently anti-inflammatory, but they can gain pro-inflammatory properties in the setting of chronic liver injury (164, 165). Plasmacytoid DCs identified as lin−CD11cintMHC-IIintPDCA-1+Siglec-H+ in some respects resemble B-cells and represent the most abundant subset in the murine liver under steady-state conditions (163, 166). Human pDCs are characterized by BDCA-2 and CD123 expression, but occur less frequently than in mice (167). This cell population responds to TLR7/8 ligands and mediate antiviral immunity by secreting type I interferons such as IFN-α but are less potent T-cell inductors. Classical DCs comprise two subtypes: cross-presenting lin−CD11c+CD11b−CD103+CX3CR1− DCs (mainly interacting with CD8+ T-cells via MHC-I) and conventional lin−CD11c+CD11b+CD103−CX3CR1+ DCs (presenting MHC-II bound antigens to CD4+ T-cells) which correspond to human CD141 (BDCA-3) and CD1c (BDCA-1) DCs respectively (61, 163). In human liver CD1c+ DCs prevail in contrast to mice (168). Another recent nomenclature differentiates hepatic DCs based on lipid content with high-lipid liver DCs inducing robust T-cell activation and cytokine secretion whereas low-lipid DC promote immune tolerance in both mice and humans (169).
Several factors contribute to the tolerogenic nature of hepatic DCs. When compared to splenic DCs, hepatic DCs were shown to be relatively immature (less CD40, CD80, CD86, CD83) with a reduced capacity to cross-present antigen to T-cells. Human DCs secrete high levels of IL-10, but less IL12p70 upon LPS-exposure (165) thereby contributing to endotoxin tolerance in the healthy liver (170). According to some reports hepatic DCs also predominantly induce regulatory and IL-4 secreting T-cells (171). During homeostasis low levels of circulating LPS trigger the expression of indoleamine-2,3-dioxygenase in human pDCs which catalyzes the production of immunoregulatory metabolites (172, 173). Following CpG stimulation murine pDC also fail to release abundant class I interferons owing to high NOD2 expression (174). Interestingly, circulating DCs that cross the hepatic sinusoids to reach the afferent lymphatics are educated by the hepatic microenvironment to adopt a regulatory phenotype, emphasizing the inherent tolerogenic phenotype of the hepatic niche (175).
Compared to other myeloid cells such as macrophages and monocytes the role of DCs in the initiation and progression of liver diseases is poorly defined. After switching from a regulatory to a proinflammatory state hepatic DCs can exacerbate acute liver injury in certain murine models (176, 177) whereas in human fatty liver disease there is emerging evidence they are protective by removing cellular debris and restricting DAMP driven activation of innate effector CD8+ T-cells (178). CD103+ DCs might be central to this response as Batf3 deficient mice that lack CD103 displayed a more aggressive course in an experimental model of NASH (179). The failure to clear HCV has been associated with a reduced capacity of pDCs to secrete antiviral IFN-α and their ability to stimulate inhibitory T-cell receptors such as PD-1, TIM-3, and CTLA-4 (180–182). Thus, impaired DC activation in HCV infection might favor T-cell unresponsiveness leading to viral immune escape and persistence. DCs are not the only APCs within the liver. The liver's unique metabolic functions and constant exposure to gut antigens and gut-derived microbial products has resulted in a complex system for regulating immune responses in which DCs, endothelial cells and stromal cells may all contribute to presenting antigens and maintaining immune homeostasis (175).
Some studies have suggested that DCs may play a role in driving fibrogenesis beyond their ability to activate immune responses. However, the data are not compelling and most evidence points to DCs being largely dispensable for the progression of fibrosis. Although the expansion of CD11b+ DCs has been observed during hepatic fibrogenesis in mice (183, 184), DCs are thought to promote resolution rather than progression of fibrosis. For example depletion of DCs during the regression phase of murine liver fibrosis significantly impairs tissue repair whereas in vivo expansion or adoptive transfer of purified DCs enhanced fibrosis reversal. This pro-resolution effect was mediated by MMP9 activity and clearance of activated hepatic stellate cells (185). Moreover, due to their anti-angiogenic properties DCs can counteract the profibrotic effect of VEGF mainly by expressing the VEGF receptor 1 (sFLT1) thus reducing the bioavailability of VEGF (186).
Liver fibrogenesis was previously regarded as a unidirectional process with little chance of resolution once scar tissue has formed. However, evidence now shows that even advanced fibrosis and in some circumstances cirrhosis are at least partially reversible if the cause of liver injury can be eliminated (187). This concept has been demonstrated in both experimental models of chronic liver injury (188–190) and in human liver disease (191). For example in humans successful treatment of chronic viral hepatitis can lead to a marked improvement in liver architecture indicating that the liver has the potential for regeneration and remodeling of scar tissue. A landmark study by Marcellin et al. demonstrated that following 5 years of treatment of chronic Hepatitis B infection with tenofovir disoproxil fumarate, cirrhosis could be reverted in 74% of cases (192).
Kupffer cells and infiltrating monocyte-derived macrophages are crucially involved in this process of tissue remodeling (Figure 3) and it is clear that hepatic macrophages can play context-dependent fibrogenic and fibrolytic roles due to their heterogeneity and plasticity. For example hepatocyte-derived HRG, a non-inflammasome activating factor contributing to KC stimulation, favors a profibrotic phenotype of murine hepatic macrophages. This was demonstrated in HRG-deficient knockout mice where liver fibrosis was significantly attenuated in diet and toxin-induced models of liver injury (193). Similarly in humans interleukin-34 and macrophage colony-stimulating factor (M-CSF) promote a profibrotic phenotype in hepatic macrophages in the setting of chronic viral hepatitis (194). KC neutralize circulating endotoxins during homeostasis and release anti-inflammatory mediators such as IL-10 during low-level lipopolysaccharide (LPS) exposure (26). However, dysbiosis and translocation of gut bacteria and bacterial products due to intestinal barrier dysfunction result in the excessive presence of PAMPs within the hepatic microvasculature that reach the liver via the portal vein (195) which, in murine models, drives inflammasome activation of profibrotic hepatic stellate cells (196, 197). In a recent publication the cell-specific innate immune receptor triggering receptor expressed on myeloid cells-1 (TREM-1) was reported to promote hepatic inflammation and fibrosis in mice and humans (198), and inhibition of TREM-1 in mice ameliorated inflammation and macrophage and neutrophil activation in a mouse model of ARLD (199).
Chemokines released by KCs shapes the subsequent phase of hepatic inflammation. The CXC chemokines CXCL1, CXCL2, CXCL8 attract neutrophils whereas CCL2 is the major chemokine that governs influx of bone-marrow-derived monocytes (5). Hepatic stellate cells are another important source of CCL2 and there is a bidirectional relationship between pro-fibrotic effector cells and hepatic macrophages (197) as shown by the ability of HSC to respond to CCL2 and CCL5 produced by hepatic macrophages. Bone-marrow chimeric mice were used to show that activation of CCR2 and CCR5 in HSC drives fibrogenesis through stimulation of HSC migration and collagen production, whereas CCR1 acts solely on monocytes/macrophages (200–202). In humans macrophages exposed to HCV serum synthesize CCL5 and activate hepatic stellate cell confirming the murine data (203). CCL3 deficient mice display reduced HSC proliferation and migration and attenuated fibrogenesis (204). Secretion of CCL3 was shown to be dependent on the expression of the scavenger receptor Stabilin-1 by a specific macrophage population, and genetic deletion of Stab1 led to diminished anti-fibrotic responses in diet and toxin-induced murine models of liver disease (205).
The release of CCL2 during early hepatic injury in mice augments the intrahepatic pool of macrophages by selectively attracting bone-marrow derived inflammatory CCR2+CX3CR1lowLy6Chigh monocytes but not CCR2−CX3CR1highLy6Clow counterparts (56). The expansion of hepatic macrophages is maintained during iterative episodes of liver injury that drive fibrogenesis (56) and CCR2 directed inhibition of monocyte recruitment during liver injury in murine models reduces liver scarring (56, 200, 206, 207). Targeting CCR2 with either the small molecule inhibitor cenicriviroc (208–210) or the L-enantiomeric RNA oligonucleotide mNOX-E36 (211) achieved similar results. In line with this data from the phase 2b Centaur trial revealed that treatment with cenicriviroc reduces fibrosis in patients with NASH after 1 year of treatment (212). Despite these advances, the profibrotic role of monocytic CCR2 is not fully understood following recent studies demonstrating that CCR2 expressed by monocytes/macrophages is dispensable for liver fibrogenesis (200). This was confirmed following further studies in mice which revealed that CCR1 (which binds CCL3 and CCL4), CCR8 (which binds mainly CCL1) and CCR9 which binds CCL25 are also involved in recruiting monocytes to the site of hepatic injury during fibrogenesis (42, 201, 213).
Both monocyte-derived macrophages and Kupffer cells promote fibrogenesis by secreting TGF-β and galectin-3, which drive transdifferentiation of HSCs into matrix secreting myofibroblasts (56, 214–216). Hepatic macrophages are also implicated in the survival and activation of HSC through secretion of IL-1β and TNF-α [in a NF-κB activation-dependent fashion (183)] or via IL-4 and IL-13 secretion in Th2-dominated rodent injury models such as parasitic infections (217). Oncostatin M (OSM) might also function as a potent regulator of hepatic macrophage/HSC interaction by enhancing the expression of profibrotic and mitogenic genes such as TGF-β and PDGF in bone-marrow derived infiltrating macrophages, with macrophage-depleted livers being largely protected from OSM-induced fibrosis (218). Interestingly, the profibrotic effect of KC-secreted TGF-β is retained following inhibition of CCL2-dependent monocyte in experimental steatohepatitis (102) which might impede the effectiveness of CCL2/CCR2 based therapies to treat liver fibrogenesis.
The first reports of hepatic macrophages driving fibrosis resolution were published alongside data describing the profibrogenic nature of Kupffer cells and monocyte-derived macrophages in liver injury. This apparent paradox was clarified when Duffield and colleagues reported a dual role for macrophages during different phases of chronic liver injury in mice. They used a CCl4 model to show that mice in which hepatic macrophages were selectively depleted exhibited less matrix deposition at advanced stages of fibrogenesis but more fibrosis when macrophages were depleted during the resolution phase (219). These data suggested the existence of distinct macrophage populations within the liver that fulfill opposing functions according to the disease stage. A subsequent study by the same group confirmed this by reporting the accumulation of macrophages around scar fibers during the resolution phase that were capable of degrading ECM through expression of matrix metalloproteinase protein 13 (MMP13) (220). These scar-associated macrophages are also equipped with MMP9, MMP12 and TRAIL, and contribute to the disruption of scar tissue and induction of fibroblast apoptosis (187). However, these studies did not identify which hepatic macrophage population gave rise to this profibrolytic subset. One of the first lines of evidence that monocyte-derived macrophages might be responsible stems from a paper showing that CCR2 deficiency is protective during fibrogenesis but hinders scar removal during the regression phase following cessation of CCl4 challenge in rodents. The putative mechanism is a balance between levels of tissue inhibitor of metalloproteinase-1 (TIMP1) and MMP1 and MMP13 mRNA, in the liver (207). This study also determined that profibrotic and pro-resolution macrophages share the same precursor cells; a hypothesis supported by a seminal paper in 2012 showing that in mice Ly6Chigh inflammatory macrophages undergo a phenotypic switch to an anti-inflammatory and anti-fibrotic “restorative” CD11bhighF4/80intLy6Clow subtype (52). The accumulation of Ly6Clow macrophages producing matrilytic MMPs peaked at the maximum point of fibrosis resolution whereas the early phase of liver parenchyma damage is dominated by freshly recruited inflammatory CCR2+Ly6Chigh macrophages. Phagocytosis of cell debris drives macrophage transdifferentiation toward a restorative Ly6Clow phenotype (52). The concept of a context dependent hepatic macrophage plasticity was demonstrated using sterile liver inflammation models in which CCR2highCX3CR1low macrophages accumulate early after focal tissue injury in a ring-like structure and then give rise to a reparative CCR2lowCX3CR1high phenotype which facilitate wound repair (14). There is also evidence that in situ reprogramming of infiltrating macrophages from a profibrotic to an antifibrotic subset is controlled by the CX3CR1/CX3CL1 axis, which promotes macrophage survival and imprints an anti-inflammatory state. Consequently, CX3CR1 knockout mice display enhanced tissue damage and fibrosis after bile duct ligation and CCl4 exposure (221, 222). Though circulating Gr1low (Ly6Clow) CX3CR1+ monocytes show patrolling behavior in blood stream (34) there is no data so far to support the idea that these cells are directly recruited to the inflamed liver and thereby perpetuate fibrosis resolution. Never the less, this cannot be excluded since extravasation of CX3CR1+ monocytes into affected organs has been demonstrated in models of myocardial infarction and lung injury (223, 224).
Translating findings from rodent models into patients is not straightforward. Most importantly—as outlined above—human liver macrophage subsets lack well-defined surface marker patterns that allow for distinction of resident Kupffer cells from infiltrating monocytes. For example CD68+ which is deemed to be a macrophage marker in mice can also be detected on circulating monocytes in human, and although gene profiles show overlap between murine Ly6Chigh and “classical” human CD14++CD16− monocytes and murine Ly6Clow and “non-classical” human CD14+CD16++ monocytes (31) there are clear functional differences. In addition, it is difficult to integrate the “intermediate” CD14++CD16+ subset into the murine nomenclature (225). In general, CD16+ monocytes are enriched in the liver in comparison to peripheral blood even under steady state conditions (46), with increased numbers being observed in patients with cirrhosis (47). CD14+CD16− cells can acquire CD16 expression under the influence of soluble factors present in the diseased liver such as IL-10 and TGF-β, and CD16+ monocytes display higher phagocytic capacity and can secrete both pro- and anti-inflammatory cytokines upon LPS stimulation thus resembling both Ly6Clow and Ly6Chigh monocytes/macrophages in mice. Of note, CD16+ but not CD16− monocytes can directly activate human HSC (47). Rodent models also lack the highly-crosslinked scar tissue observed for patients with advanced fibrosis, and as a result macrophage-mediated resolution of fibrosis is accelerated in murine models compared to humans. Therefore, further research is warranted to better define disease-specific characteristics of hepatic monocyte and macrophage subsets in human diseases. Despite these differences there are some striking parallels between mice and humans. For example, the CCL2/CCR2 axis plays a similar role in fibrosis and macrophage infiltration to the inflamed liver in mice and humans, and TREM-1 is emerging as an additional shared marker.
Myeloid cell subsets are linked to virtually all steps in the natural course of tumor formation and spreading in the liver. Extensive research has shown a prominent role for tumor associated macrophages (TAM) and MDSC in the development of hepatocellular carcinoma (HCC), an archetypical inflammatory tumor in which chronic inflammation drives liver cancer pathogenesis, invasion, and metastasis (226). In the context of liver carcinogenesis monocytes/macrophages exert dualistic functions in a stage-dependent manner with CCR2+ CCL2-responsive monocytes promoting tumor surveillance through the elimination of senescent premalignant hepatocytes in healthy livers (Figure 4), whereas in established tumors monocytes/macrophages are reprogrammed to silence NK cells resulting in tumor growth (227, 228). Tumor associated macrophages derived from infiltrating monocytes are a dominant cellular component of human tumor stroma with increased density of TAMs in peritumoral margins being closely linked to poor prognosis (229, 230). Consistent with this the absence of macrophages correlated with improved patient survival (231). In both humans and murine models TAMs drive tumorigenesis by sustaining inflammatory pathways mediated through secretion of cytokines such as TNF-α and IL-6 that suppress hepatocyte apoptosis and induce proliferation in response to tissue damage (232–234). The surface receptor TREM-1 expressed on myeloid cells governs the secretion of proinflammatory mediators and engagement of TREM-1 in hepatic macrophages has been shown to trigger carcinogenesis (235). Trem1-deficient mice treated with diethylnitrosamine (DEN) were protected from malignancy due to attenuation of cytokine secretion (e.g., IL-6, IL-1β, TNF, CCL2) and ablation of inflammatory signaling pathways (p38, ERK1/2, JNK, MAPK, and NF-κB) in KC (235).
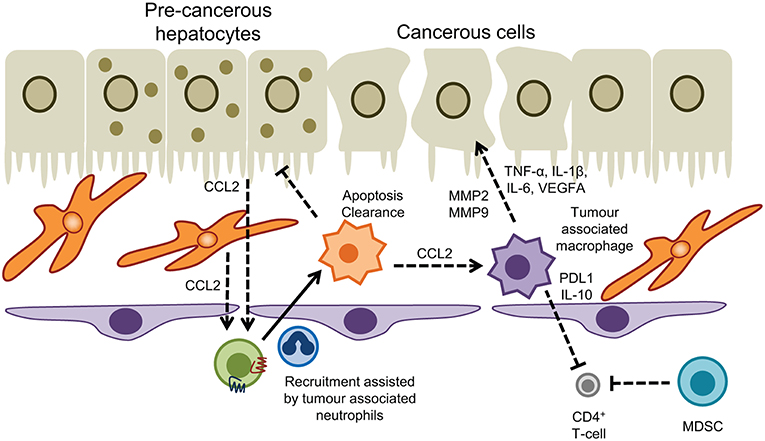
Figure 4. Myeloid cells in hepatic malignancy. Tumor associated macrophages promote cancer cell proliferation and neoangiogenesis, and act in concert with myeloid-derived suppressor cells to dampen T-cell immunosurveillance. Conversely recruitment of monocytes into the tumor microenvironment driven by CCL2 produced by hepatocytes and hepatic stellate cells, and interactions with tumor associated neutrophils promotes apoptosis and clearance of pre-cancerous hepatocytes to prevent HCC.
The inflammatory environment present in chronic liver injury facilitates the recruitment and retention of monocyte-derived TAMs which promote tumorigenesis in a MMP2/MMP9 dependent fashion. This was demonstrated by comparing the seeding of injected HCC cells in healthy livers with CCl4 preconditioned livers where an alternatively-activated macrophage population (M2-like) were enriched in the tumor environment (236). Infiltrating TAMs are frequently reported to resemble alternatively-activated macrophages (237–239) although the dichotomous approach of M1/M2 polarization does not fully reflect the entire spectrum and heterogeneity of tissue macrophages. Nevertheless, reversal of M2-like polarization in experimental HCC has yielded promising results in containing tumor progression (240) with TIM-3 and Wnt ligands identified as critical drivers of alternative activation of TAMs and HCC growth (239, 241). In patients, total immune cell infiltration into HCC correlated with M1-like macrophage populations and a more favorable prognosis (242).
The CCL2/CCR2 axis is a promising novel target in HCC therapy. Antagonism of CCR2 by the compound RDC018 not only reduced TAM infiltration but also restored anti-tumor immune response and ameliorated HCC outcome in murine models of HCC (243). Tumor associated neutrophils (TAN) provide an important source of CCL2 in HCC and can act synergistically with TAM to support liver tumor progression (244). In humans neutrophil extracellular traps can also promote inflammation and development of HCC on the background of NASH, driven by the presence of free fatty acids (245); however our understanding of the precise role played by neutrophils in liver cancer remains elusive. One striking feature of TAM is the induction of an immune suppressive microenvironment that disrupts anti-tumor immunity. For example, release of regulatory cytokines such as TGF-β and IL-10 by TAM impair Th1 and cytotoxic T-cells but promote regulatory T cells and Th2 activity all of which facilitate tumor growth. TAMs also express high levels of PDL1, galectin-9, and indoleamine-pyrrole 2,3-dioxygenase (IDO) that foster T-cell exhaustion and prevent effective anti-tumor immune response (241). In HCC the expression of PDL1 by TAMs correlated with increased tumor burden and the intensity of the protein was associated with high mortality and reduced survival (246). MDSC share many mechanisms with TAM to protect from HCC-targeted T-cell activity, and the net effect of MDSCs in HCC nodules and peritumoral stroma is progression of the tumor (241). Furthermore, MDSC reduce the tissue availability of arginine and cysteine, which are essential for T-cell proliferation and impede NK cell cytotoxicity and development via NKp30 receptor (241).
There is a major unmet need for effective therapies to prevent or reverse liver fibrosis particularly in the context of a major increase in fatty liver disease and the continuing high prevalence of alcoholic cirrhosis (247). Macrophages have the dual potential to serve as therapeutic targets and as treatment vehicles for inflammation-induced liver fibrosis and carcinogenesis (248). In principle, macrophages can be targeted at different stages of disease and different subsets of monocyte macrophages can be targeted. Such strategies include (i) attenuation of Kupffer cell activation by anti-inflammatory compounds; (ii) inhibition of macrophage precursor cell (i.e., monocyte) recruitment to the injured liver; (iii) manipulation of macrophage polarization and differentiation to facilitate transition toward a restorative reparative phenotype (iv) infusion of beneficial pro-restorative macrophages (248) (Figure 5). Interference with chemokine pathways to restrict influx of inflammatory monocytes is one of the most advanced approaches. As stated earlier, the CCR2/CCR5 antagonist Cenicriviroc has entered phase 2b clinical trials with promising results reported after 12 months treatment of NASH-related fibrosis (212). The current options in targeting macrophages in the context of liver disease have recently been comprehensively summarized (248). Adoptive cell therapy using hematopoietic stem cells or macrophages is an approach that is attracting increasing interest. The first studies reporting efficacy of bone marrow cell transfer in murine models of liver fibrosis were published almost 15 years ago when injection of bone marrow cells was shown to cause MMP9-dependent reduction in ECM deposition in response to CCl4 (249). In a study by Thomas et al. in 2011 bone-marrow derived macrophages (BMM) were prepared in vitro by stimulation with CSF-1 and subsequently injected into the portal vein of mice with long-term CCl4 induced fibrosis. The infused cells did not conform to the M1/M2 paradigm but expressed IL-10, TWEAK, and MMP13, which are known to suppress inflammation and to promote cell regeneration and fibrolysis. Treatment significantly reduced liver scarring by promoting myofibroblasts apoptosis, MMP-induced degradation of ECM and by stimulating liver regeneration. In contradistinction, non-purified whole bone marrow cells increased liver fiber content (250). Similar results were obtained in another study showing that IL-10 producing CD11b+Gr1+ myeloid cells account for the tissue remodeling effect of bone marrow transplantation in liver fibrosis (251). BMM also ameliorate oxidative stress and reduce production of the potent profibrotic cytokine IL-13 (252). Interestingly, macrophages derived from pluripotent embryonic stem cells exhibit comparable antifibrotic effects to BMM though these cells tend to resemble resident Kupffer cells rather than infiltrating macrophages (253). A contributing factor to the success of bone marrow derived macrophage transplantation in liver fibrosis could be activation of the sphingosine-1-phosphate receptor (S1PR) that critically controls BMM motility (254). Mice treated with FTY720 which triggers S1PR internalization retained infused c-kit+/sca1+/lin− hematopoietic stem cells in the liver due to a failure of the cells to egress into the draining lymph. This was associated with reduced scarring in methionine-choline-deficient diet fed and CCl4 treated mice (255). Further studies are needed to dissect whether S1P/S1PR antagonism also augments the antifibrotic effects of transplanted BMM. Despite the promising experimental findings human cell therapy trials in advanced clinical cirrhosis have so far proven disappointing. The REALISTIC trial tested the efficacy of G-CSF mobilized and autologous infusions of CD133+ stem cell therapy in cirrhosis but failed to show any improvement in liver function with more complications in the treatment group (256). This is perhaps unsurprising given that resolution will only occur if the right cells are infused into the right microenvironment at the right disease stage. This requires the design of more sophisticated precision medicine trials. Such studies are underway (257, 258) but we are only at the start of understanding macrophage therapy for liver diseases.
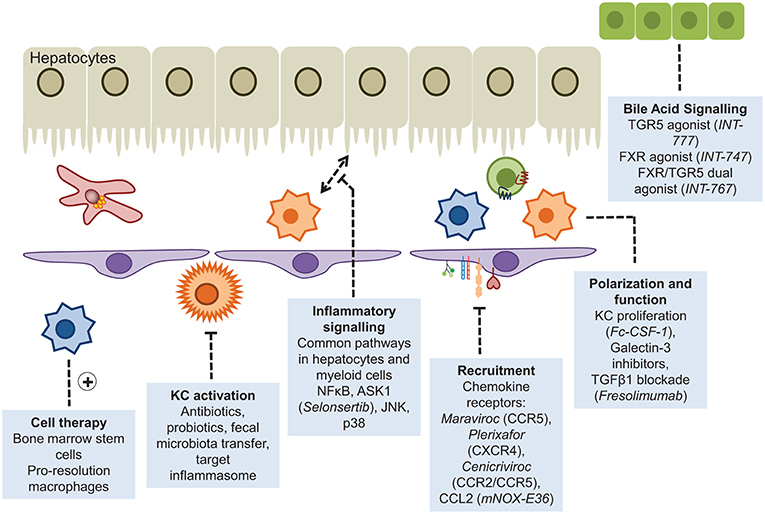
Figure 5. Myeloid cells as therapeutic targets. Approaches that have been adopted to enhance or diminish the role of myeloid cells in liver disease include disruption of the recruitment cascade or inflammatory signaling pathways, and augmented pro-resolution responses through cellular infusions of stem cells or the provision of agonists driving macrophage polarization.
CW, HZ wrote parts of the manuscript. DA edited and finalized the manuscript.
CW is funded by the BBSRC (BB/N018869/1) and HZ received funding from the German Research Council (Deutsche Forschungsgemeinschaft). This paper presents independent research supported by the NIHR Birmingham Biomedical Research Centre at the University Hospitals Birmingham NHS Foundation Trust and the University of Birmingham. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Jung S. Macrophages and monocytes in 2017: Macrophages and monocytes: of tortoises and hares. Nat Rev Immunol. (2018) 18:85–6. doi: 10.1038/nri.2017.158
2. Scott CL, T'Jonck W, Martens L, Todorov H, Sichien D, Soen B, et al. The transcription factor ZEB2 is required to maintain the tissue-specific identities of macrophages. Immunity. (2018) 49:312–25. doi: 10.1016/j.immuni.2018.07.004
3. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. (2014) 159:1312–26. doi: 10.1016/j.cell.2014.11.018
4. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. (2014) 40:274–88. doi: 10.1016/j.immuni.2014.01.006
5. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. (2017) 17:306–21. doi: 10.1038/nri.2017.11
6. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. (2014) 60:1090–6. doi: 10.1016/j.jhep.2013.12.025
7. Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, Pellicoro A, et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med. (2012) 18:572–9. doi: 10.1038/nm.2667
8. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518:547–51. doi: 10.1038/nature13989
9. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity. (2015) 42:665–78. doi: 10.1016/j.immuni.2015.03.011
10. Sheng J, Ruedl C, Karjalainen K. Fetal HSCs versus EMP2s. Immunity. (2015) 43:1025. doi: 10.1016/j.immuni.2015.11.023
11. Mass E, Ballesteros I, Farlik M, Halbritter F, Gunther P, Crozet L, et al. Specification of tissue-resident macrophages during organogenesis. Science. (2016) 353:aaf4238. doi: 10.1126/science.aaf4238
12. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun. (2016) 7:10321. doi: 10.1038/ncomms10321
13. Beattie L, Sawtell A, Mann J, Frame TCM, Teal B, de Labastida Rivera F, et al. Bone marrow-derived and resident liver macrophages display unique transcriptomic signatures but similar biological functions. J Hepatol. (2016) 65:758–68. doi: 10.1016/j.jhep.2016.05.037
14. Dal-Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CH, Petri B, et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med. (2015) 212:447–56. doi: 10.1084/jem.20141539
15. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. (2014) 41:21–35. doi: 10.1016/j.immuni.2014.06.013
16. Bieghs V, Wouters K, van Gorp PJ, Gijbels MJ, de Winther MP, Binder CJ, et al. Role of scavenger receptor A and CD36 in diet-induced nonalcoholic steatohepatitis in hyperlipidemic mice. Gastroenterology. (2010) 138:2477–86, 2486.e1–3. doi: 10.1053/j.gastro.2010.02.051
17. Iacobini C, Menini S, Ricci C, Scipioni A, Sansoni V, Cordone S, et al. Accelerated lipid-induced atherogenesis in galectin-3-deficient mice: role of lipoxidation via receptor-mediated mechanisms. Arterioscler Thromb Vasc Biol. (2009) 29:831–6. doi: 10.1161/ATVBAHA.109.186791
18. Zannetti C, Roblot G, Charrier E, Ainouze M, Tout I, Briat F, et al. Characterization of the inflammasome in human kupffer cells in response to synthetic agonists and pathogens. J Immunol. (2016) 197:356–67. doi: 10.4049/jimmunol.1502301
19. Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. (2012) 143:1158–72. doi: 10.1053/j.gastro.2012.09.008
20. Toki Y, Takenouchi T, Harada H, Tanuma S, Kitani H, Kojima S, et al. Extracellular ATP induces P2X7 receptor activation in mouse Kupffer cells, leading to release of IL-1beta, HMGB1, and PGE2, decreased MHC class I expression and necrotic cell death. Biochem Biophys Res Commun. (2015) 458:771–6. doi: 10.1016/j.bbrc.2015.02.011
21. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. (2010) 330:362–6. doi: 10.1126/science.1195491
22. Amaral SS, Oliveira AG, Marques PE, Quintao JL, Pires DA, Resende RR, et al. Altered responsiveness to extracellular ATP enhances acetaminophen hepatotoxicity. Cell Commun Signal. (2013) 11:10. doi: 10.1186/1478-811X-11-10
23. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. (2012) 482:179–85. doi: 10.1038/nature10809
24. Wree A, McGeough MD, Inzaugarat ME, Eguchi A, Schuster S, Johnson CD, et al. NLRP3 inflammasome driven liver injury and fibrosis: roles of IL-17 and TNF in mice. Hepatology. (2018) 67:736–49. doi: 10.1002/hep.29523
25. Knolle PA, Uhrig A, Protzer U, Trippler M, Duchmann R, Meyer zum Buschenfelde KH, et al. Interleukin-10 expression is autoregulated at the transcriptional level in human and murine Kupffer cells. Hepatology. (1998) 27:93–9. doi: 10.1002/hep.510270116
26. Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer zum Buschenfelde KH, Gerken G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J Hepatol. (1995) 22:226–9. doi: 10.1016/0168-8278(95)80433-1
27. Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology. (2015) 62:279–91. doi: 10.1002/hep.27793
28. You Q, Cheng L, Kedl RM, Ju C. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology. (2008) 48:978–90. doi: 10.1002/hep.22395
29. Yan ML, Wang YD, Tian YF, Lai ZD, Yan LN. Inhibition of allogeneic T-cell response by Kupffer cells expressing indoleamine 2,3-dioxygenase. World J Gastroenterol. (2010) 16:636–40. doi: 10.3748/wjg.v16.i5.636
30. Wang Y, Song L, Liu M, Ge R, Zhou Q, Liu W, et al. A proteomics landscape of circadian clock in mouse liver. Nat Commun. (2018) 9:1553. doi: 10.1038/s41467-018-03898-2
31. Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. (2010) 115:e10–9. doi: 10.1182/blood-2009-07-235028
32. Tacke F, Randolph GJ. Migratory fate and differentiation of blood monocyte subsets. Immunobiology. (2006) 211:609–18. doi: 10.1016/j.imbio.2006.05.025
33. David BA, Rezende RM, Antunes MM, Santos MM, Freitas Lopes MA, Diniz AB, et al. Combination of mass cytometry and imaging analysis reveals origin, location, and functional repopulation of liver myeloid cells in mice. Gastroenterology. (2016) 151:1176–91. doi: 10.1053/j.gastro.2016.08.024
34. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. (2007) 317:666–70. doi: 10.1126/science.1142883
35. Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell. (2013) 153:362–75. doi: 10.1016/j.cell.2013.03.010
36. Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Ann Rev Immunol. (2015) 33:643–75. doi: 10.1146/annurev-immunol-032414-112220
37. Poschke I, Kiessling R. On the armament and appearances of human myeloid-derived suppressor cells. Clin Immunol. (2012) 144:250–68. doi: 10.1016/j.clim.2012.06.003
38. Hammerich L, Tacke F. Emerging roles of myeloid derived suppressor cells in hepatic inflammation and fibrosis. World J Gastrointest Pathophysiol. (2015) 6:43–50. doi: 10.4291/wjgp.v6.i3.43
39. Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells—gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol. (2018) 15:555–67. doi: 10.1038/s41575-018-0020-y
40. Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G1310–21. doi: 10.1152/ajpgi.00365.2011
41. Galastri S, Zamara E, Milani S, Novo E, Provenzano A, Delogu W, et al. Lack of CC chemokine ligand 2 differentially affects inflammation and fibrosis according to the genetic background in a murine model of steatohepatitis. Clin Sci. (2012) 123:459–71. doi: 10.1042/CS20110515
42. Heymann F, Hammerich L, Storch D, Bartneck M, Huss S, Russeler V, et al. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C-C motif chemokine receptor 8 in mice. Hepatology. (2012) 55:898–909. doi: 10.1002/hep.24764
43. Tomita K, Freeman BL, Bronk SF, LeBrasseur NK, White TA, Hirsova P, et al. CXCL10-mediates macrophage, but not other innate immune cells-associated inflammation in murine nonalcoholic steatohepatitis. Sci Rep. (2016) 6:28786. doi: 10.1038/srep28786
44. Zhang X, Han J, Man K, Li X, Du J, Chu ES, et al. CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophages and autophagy. J Hepatol. (2016) 64:160–70. doi: 10.1016/j.jhep.2015.09.005
45. Aspinall AI, Curbishley SM, Lalor PF, Weston CJ, Blahova M, Liaskou E, et al. CX(3)CR1 and vascular adhesion protein-1-dependent recruitment of CD16(+) monocytes across human liver sinusoidal endothelium. Hepatology. (2010) 51:2030–9. doi: 10.1002/hep.23591
46. Liaskou E, Zimmermann HW, Li KK, Oo YH, Suresh S, Stamataki Z, et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology. (2013) 57:385–98. doi: 10.1002/hep.26016
47. Zimmermann HW, Seidler S, Nattermann J, Gassler N, Hellerbrand C, Zernecke A, et al. Functional contribution of elevated circulating and hepatic non-classical CD14CD16 monocytes to inflammation and human liver fibrosis. PLoS ONE. (2010) 5:e11049. doi: 10.1371/journal.pone.0011049
48. Zimmermann HW, Bruns T, Weston CJ, Curbishley SM, Liaskou E, Li KK, et al. Bidirectional transendothelial migration of monocytes across hepatic sinusoidal endothelium shapes monocyte differentiation and regulates the balance between immunity and tolerance in liver. Hepatology. (2016) 63:233–46. doi: 10.1002/hep.28285
49. Antoniades CG, Quaglia A, Taams LS, Mitry RR, Hussain M, Abeles R, et al. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology. (2012) 56:735–46. doi: 10.1002/hep.25657
50. Hochst B, Schildberg FA, Sauerborn P, Gabel YA, Gevensleben H, Goltz D, et al. Activated human hepatic stellate cells induce myeloid derived suppressor cells from peripheral blood monocytes in a CD44-dependent fashion. J Hepatol. (2013) 59:528–35. doi: 10.1016/j.jhep.2013.04.033
51. Resheq YJ, Li KK, Ward ST, Wilhelm A, Garg A, Curbishley SM, et al. Contact-dependent depletion of hydrogen peroxide by catalase is a novel mechanism of myeloid-derived suppressor cell induction operating in human hepatic stellate cells. J Immunol. (2015) 194:2578–86. doi: 10.4049/jimmunol.1401046
52. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci USA. (2012) 109:E3186–95. doi: 10.1073/pnas.1119964109
53. Wang J, Kubes P. A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell. (2016) 165:668–78. doi: 10.1016/j.cell.2016.03.009
54. Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol. (2015) 62(1 Suppl.):S112–20. doi: 10.1016/j.jhep.2014.12.016
55. Bernal W, Wendon J. Acute liver failure. N Engl J Med. (2013) 369:2525–34. doi: 10.1056/NEJMra1208937
56. Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. (2009) 50:261–74. doi: 10.1002/hep.22950
57. Hitchcock JR, Cook CN, Bobat S, Ross EA, Flores-Langarica A, Lowe KL, et al. Inflammation drives thrombosis after Salmonella infection via CLEC-2 on platelets. J Clin Invest. (2015) 125:4429–46. doi: 10.1172/JCI79070
58. Erhardt A, Biburger M, Papadopoulos T, Tiegs G. IL-10, regulatory T cells, and Kupffer cells mediate tolerance in concanavalin A-induced liver injury in mice. Hepatology. (2007) 45:475–85. doi: 10.1002/hep.21498
59. Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. Am J Transplant. (2011) 11:1563–9. doi: 10.1111/j.1600-6143.2011.03579.x
60. Movita D, van de Garde MD, Biesta P, Kreefft K, Haagmans B, Zuniga E, et al. Inflammatory monocytes recruited to the liver within 24 hours after virus-induced inflammation resemble Kupffer cells but are functionally distinct. J Virol. (2015) 89:4809–17. doi: 10.1128/JVI.03733-14
61. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol. (2016) 13:88–110. doi: 10.1038/nrgastro.2015.200
62. Holt MP, Cheng L, Ju C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J Leukoc Biol. (2008) 84:1410–21. doi: 10.1189/jlb.0308173
63. Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol. (2014) 193:344–53. doi: 10.4049/jimmunol.1400574
64. Michael SL, Pumford NR, Mayeux PR, Niesman MR, Hinson JA. Pretreatment of mice with macrophage inactivators decreases acetaminophen hepatotoxicity and the formation of reactive oxygen and nitrogen species. Hepatology. (1999) 30:186–95. doi: 10.1002/hep.510300104
65. Laskin DL, Gardner CR, Price VF, Jollow DJ. Modulation of macrophage functioning abrogates the acute hepatotoxicity of acetaminophen. Hepatology. (1995) 21:1045–50. doi: 10.1002/hep.1840210424
66. Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology. (2016) 64:1667–82. doi: 10.1002/hep.28682
67. Bourdi M, Masubuchi Y, Reilly TP, Amouzadeh HR, Martin JL, George JW, et al. Protection against acetaminophen-induced liver injury and lethality by interleukin 10: role of inducible nitric oxide synthase. Hepatology. (2002) 35:289–98. doi: 10.1053/jhep.2002.30956
68. Yee SB, Bourdi M, Masson MJ, Pohl LR. Hepatoprotective role of endogenous interleukin-13 in a murine model of acetaminophen-induced liver disease. Chem Res Toxicol. (2007) 20:734–44. doi: 10.1021/tx600349f
69. Bourdi M, Eiras DP, Holt MP, Webster MR, Reilly TP, Welch KD, et al. Role of IL-6 in an IL-10 and IL-4 double knockout mouse model uniquely susceptible to acetaminophen-induced liver injury. Chem Res Toxicol. (2007) 20:208–16. doi: 10.1021/tx060228l
70. Stutchfield BM, Antoine DJ, Mackinnon AC, Gow DJ, Bain CC, Hawley CA, et al. CSF1 restores innate immunity after liver injury in mice and serum levels indicate outcomes of patients with acute liver failure. Gastroenterology. (2015) 149:1896–909.e14. doi: 10.1053/j.gastro.2015.08.053
71. Antoniades CG, Khamri W, Abeles RD, Taams LS, Triantafyllou E, Possamai LA, et al. Secretory leukocyte protease inhibitor: a pivotal mediator of anti-inflammatory responses in acetaminophen-induced acute liver failure. Hepatology. (2014) 59:1564–76. doi: 10.1002/hep.26933
72. Triantafyllou E, Pop OT, Possamai LA, Wilhelm A, Liaskou E, Singanayagam A, et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut. (2018) 67:333–47. doi: 10.1136/gutjnl-2016-313615
73. Krenkel O, Mossanen JC, Tacke F. Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surg Nutr. (2014) 3:331–43. doi: 10.3978/j.issn.2304-3881.2014.11.01
74. Campana L, Starkey Lewis PJ, Pellicoro A, Aucott RL, Man J, O'Duibhir E, et al. The STAT3-IL-10-IL-6 pathway is a novel regulator of macrophage efferocytosis and phenotypic conversion in sterile liver injury. J Immunol. (2018) 200:1169–87. doi: 10.4049/jimmunol.1701247
75. Ju C, Reilly TP, Bourdi M, Radonovich MF, Brady JN, George JW, et al. Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem Res Toxicol. (2002) 15:1504–13. doi: 10.1021/tx0255976
76. Bernsmeier C, Pop OT, Singanayagam A, Triantafyllou E, Patel VC, Weston CJ, et al. Patients with acute-on-chronic liver failure have increased numbers of regulatory immune cells expressing the receptor tyrosine kinase MERTK. Gastroenterology. (2015) 148:603–15.e14. doi: 10.1053/j.gastro.2014.11.045
77. Khakoo SI, Soni PN, Savage K, Brown D, Dhillon AP, Poulter LW, et al. Lymphocyte and macrophage phenotypes in chronic hepatitis C infection. Correlation with disease activity. Am J Pathol. (1997) 150:963–70.
78. McGuinness PH, Painter D, Davies S, McCaughan GW. Increases in intrahepatic CD68 positive cells, MAC387 positive cells, and proinflammatory cytokines (particularly interleukin 18) in chronic hepatitis C infection. Gut. (2000) 46:260–9. doi: 10.1136/gut.46.2.260
79. Shrivastava S, Mukherjee A, Ray R, Ray RB. Hepatitis C virus induces interleukin-1beta (IL-1beta)/IL-18 in circulatory and resident liver macrophages. J Virol. (2013) 87:12284–90. doi: 10.1128/JVI.01962-13
80. Hosomura N, Kono H, Tsuchiya M, Ishii K, Ogiku M, Matsuda M, et al. HCV-related proteins activate Kupffer cells isolated from human liver tissues. Digest Dis Sci. (2011) 56:1057–64. doi: 10.1007/s10620-010-1395-y
81. Boltjes A, Movita D, Boonstra A, Woltman AM. The role of Kupffer cells in hepatitis B and hepatitis C virus infections. J Hepatol. (2014) 61:660–71. doi: 10.1016/j.jhep.2014.04.026
82. Broering R, Wu J, Meng Z, Hilgard P, Lu M, Trippler M, et al. Toll-like receptor-stimulated non-parenchymal liver cells can regulate hepatitis C virus replication. J Hepatol. (2008) 48:914–22. doi: 10.1016/j.jhep.2008.01.028
83. Boltjes A, van Montfoort N, Biesta PJ, Op den Brouw ML, Kwekkeboom J, van der Laan LJ, et al. Kupffer cells interact with hepatitis B surface antigen in vivo and in vitro, leading to proinflammatory cytokine production and natural killer cell function. J Infect Dis. (2015) 211:1268–78. doi: 10.1093/infdis/jiu599
84. Chang S, Dolganiuc A, Szabo G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J Leukoc Biol. (2007) 82:479–87. doi: 10.1189/jlb.0207128
85. Huang LR, Wohlleber D, Reisinger F, Jenne CN, Cheng RL, Abdullah Z, et al. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8(+) T cells and successful immunotherapy against chronic viral liver infection. Nat Immunol. (2013) 14:574–83. doi: 10.1038/ni.2573
86. Liu J, Yu Q, Wu W, Huang X, Broering R, Werner M, et al. TLR2 stimulation strengthens intrahepatic myeloid-derived cell-mediated T cell tolerance through inducing Kupffer cell expansion and IL-10 production. J Immunol. (2018) 200:2341–51. doi: 10.4049/jimmunol.1700540
87. Wang S, Chen Z, Hu C, Qian F, Cheng Y, Wu M, et al. Hepatitis B virus surface antigen selectively inhibits TLR2 ligand-induced IL-12 production in monocytes/macrophages by interfering with JNK activation. J Immunol. (2013) 190:5142–51. doi: 10.4049/jimmunol.1201625
88. Li M, Sun R, Xu L, Yin W, Chen Y, Zheng X, et al. Kupffer cells support hepatitis B virus-mediated CD8+ T cell exhaustion via hepatitis B core antigen-TLR2 interactions in mice. J Immunol. (2015) 195:3100–9. doi: 10.4049/jimmunol.1500839
89. Nebbia G, Peppa D, Schurich A, Khanna P, Singh HD, Cheng Y, et al. Upregulation of the Tim-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS ONE. (2012) 7:e47648. doi: 10.1371/journal.pone.0047648
90. Tian Y, Kuo CF, Akbari O, Ou JH. Maternal-derived hepatitis B virus e antigen alters macrophage function in offspring to drive viral persistence after vertical transmission. Immunity. (2016) 44:1204–14. doi: 10.1016/j.immuni.2016.04.008
91. Jiang M, Broering R, Trippler M, Poggenpohl L, Fiedler M, Gerken G, et al. Toll-like receptor-mediated immune responses are attenuated in the presence of high levels of hepatitis B virus surface antigen. J Viral Hepatitis. (2014) 21:860–72. doi: 10.1111/jvh.12216
92. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. (2014) 14:181–94. doi: 10.1038/nri3623
93. Ndugga N, Lightbourne TG, Javaherian K, Cabezas J, Verma N, Barritt ASt, et al. Disparities between research attention and burden in liver diseases: implications on uneven advances in pharmacological therapies in Europe and the USA. BMJ Open. (2017) 7:e013620. doi: 10.1136/bmjopen-2016-013620
94. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol. (2018) 15:349–64. doi: 10.1038/s41575-018-0009-6
95. Park JW, Jeong G, Kim SJ, Kim MK, Park SM. Predictors reflecting the pathological severity of non-alcoholic fatty liver disease: comprehensive study of clinical and immunohistochemical findings in younger Asian patients. J Gastroenterol Hepatol. (2007) 22:491–7. doi: 10.1111/j.1440-1746.2006.04758.x
96. Clementi AH, Gaudy AM, van Rooijen N, Pierce RH, Mooney RA. Loss of Kupffer cells in diet-induced obesity is associated with increased hepatic steatosis, STAT3 signaling, and further decreases in insulin signaling. Biochim Biophys Acta. (2009) 1792:1062–72. doi: 10.1016/j.bbadis.2009.08.007
97. Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes. (2010) 59:347–57. doi: 10.2337/db09-0016
98. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology. (2016) 150:956–67. doi: 10.1053/j.gastro.2015.12.037
99. Robert O, Boujedidi H, Bigorgne A, Ferrere G, Voican CS, Vettorazzi S, et al. Decreased expression of the glucocorticoid receptor-GILZ pathway in Kupffer cells promotes liver inflammation in obese mice. J Hepatol. (2016) 64:916–24. doi: 10.1016/j.jhep.2015.11.023
100. Morinaga H, Mayoral R, Heinrichsdorff J, Osborn O, Franck N, Hah N, et al. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes. (2015) 64:1120–30. doi: 10.2337/db14-1238
101. Miyachi Y, Tsuchiya K, Komiya C, Shiba K, Shimazu N, Yamaguchi S, et al. Roles for cell-cell adhesion and contact in obesity-induced hepatic myeloid cell accumulation and glucose intolerance. Cell Rep. (2017) 18:2766–79. doi: 10.1016/j.celrep.2017.02.039
102. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. (2012) 61:416–26. doi: 10.1136/gutjnl-2011-300304
103. Weston CJ, Shepherd EL, Claridge LC, Rantakari P, Curbishley SM, Tomlinson JW, et al. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J Clin Invest. (2015) 125:501–20. doi: 10.1172/JCI73722
104. Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. (2007) 47:571–9. doi: 10.1016/j.jhep.2007.04.019
105. Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest. (2016) 126:859–64. doi: 10.1172/JCI83885
106. Mridha AR, Haczeyni F, Yeh MM, Haigh WG, Ioannou GN, Barn V, et al. TLR9 is up-regulated in human and murine NASH: pivotal role in inflammatory recruitment and cell survival. Clin Sci. (2017) 131:2145–59. doi: 10.1042/CS20160838
107. Balmer ML, Slack E, de Gottardi A, Lawson MA, Hapfelmeier S, Miele L, et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med. (2014) 6:237ra66. doi: 10.1126/scitranslmed.3008618
108. Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. (2014) 59:130–42. doi: 10.1002/hep.26607
109. Han YH, Kim HJ, Na H, Nam MW, Kim JY, Kim JS, et al. RORalpha induces KLF4-mediated M2 polarization in the liver macrophages that protect against nonalcoholic steatohepatitis. Cell Rep. (2017) 20:124–35. doi: 10.1016/j.celrep.2017.06.017
110. Du X, Wu Z, Xu Y, Liu Y, Liu W, Wang T, et al. Increased Tim-3 expression alleviates liver injury by regulating macrophage activation in MCD-induced NASH mice. Cell Mol Immunol. (2018). doi: 10.1038/s41423-018-0032-0. [Epub ahead of print].
111. Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. (2012) 122:3476–89. doi: 10.1172/JCI60777
112. Enomoto N, Ikejima K, Bradford BU, Rivera CA, Kono H, Goto M, et al. Role of Kupffer cells and gut-derived endotoxins in alcoholic liver injury. J Gastroenterol Hepatol. (2000) 15(Suppl):D20–5. doi: 10.1046/j.1440-1746.2000.02179.x
113. Koop DR, Klopfenstein B, Iimuro Y, Thurman RG. Gadolinium chloride blocks alcohol-dependent liver toxicity in rats treated chronically with intragastric alcohol despite the induction of CYP2E1. Mol Pharmacol. (1997) 51:944–50. doi: 10.1124/mol.51.6.944
114. Wang M, Frasch SC, Li G, Feng D, Gao B, Xu L, et al. Role of gp91(phox) in hepatic macrophage programming and alcoholic liver disease. Hepatol Commun. (2017) 1:765–79. doi: 10.1002/hep4.1078
115. Ju C, Mandrekar P. Macrophages and alcohol-related liver inflammation. Alcohol Res. (2015) 37:251–62.
116. Afford SC, Fisher NC, Neil DA, Fear J, Brun P, Hubscher SG, et al. Distinct patterns of chemokine expression are associated with leukocyte recruitment in alcoholic hepatitis and alcoholic cirrhosis. J Pathol. (1998) 186:82–9. doi: 10.1002/(SICI)1096-9896(199809)186:1<82::AID-PATH151>3.0.CO;2-D
117. Luna-Casado L, Diez-Ruiz A, Gutierrez-Gea F, Santos-Perez JL, Rico-Irles J, Wachter H, et al. Increased peripheral mononuclear cells expression of adhesion molecules in alcoholic cirrhosis: its relation to immune activation. J Hepatol. (1997) 27:477–83. doi: 10.1016/S0168-8278(97)80351-0
118. Fisher NC, Neil DA, Williams A, Adams DH. Serum concentrations and peripheral secretion of the beta chemokines monocyte chemoattractant protein 1 and macrophage inflammatory protein 1alpha in alcoholic liver disease. Gut. (1999) 45:416–20. doi: 10.1136/gut.45.3.416
119. McClain CJ, Barve S, Deaciuc I, Kugelmas M, Hill D. Cytokines in alcoholic liver disease. Semin Liver Dis. (1999) 19:205–19. doi: 10.1055/s-2007-1007110
120. Gobejishvili L, Barve S, Joshi-Barve S, Uriarte S, Song Z, McClain C. Chronic ethanol-mediated decrease in cAMP primes macrophages to enhanced LPS-inducible NF-kappaB activity and TNF expression: relevance to alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. (2006) 291:G681–8. doi: 10.1152/ajpgi.00098.2006
121. Zhang Z, Bagby GJ, Stoltz D, Oliver P, Schwarzenberger PO, Kolls JK. Prolonged ethanol treatment enhances lipopolysaccharide/phorbol myristate acetate-induced tumor necrosis factor-alpha production in human monocytic cells. Alcohol Clin Exp Res. (2001) 25:444–9. doi: 10.1111/j.1530-0277.2001.tb02233.x
122. Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology. (2011) 54:2185–97. doi: 10.1002/hep.24599
123. Nagy LE. The role of innate immunity in alcoholic liver disease. Alcohol Res. (2015) 37:237–50.
124. Cameron RG, Blendis LM, Neuman MG. Accumulation of macrophages in primary sclerosing cholangitis. Clin Biochem. (2001) 34:195–201. doi: 10.1016/S0009-9120(01)00215-6
125. Guicciardi ME, Trussoni CE, Krishnan A, Bronk SF, Lorenzo Pisarello MJ, O'Hara SP, et al. Macrophages contribute to the pathogenesis of sclerosing cholangitis in mice. J Hepatol. (2018) 69:676–86. doi: 10.1016/j.jhep.2018.05.018
126. Calmus Y, Poupon R. Shaping macrophages function and innate immunity by bile acids: mechanisms and implication in cholestatic liver diseases. Clin Res Hepatol Gastroenterol. (2014) 38:550–6. doi: 10.1016/j.clinre.2014.07.007
127. Gong Z, Zhou J, Zhao S, Tian C, Wang P, Xu C, et al. Chenodeoxycholic acid activates NLRP3 inflammasome and contributes to cholestatic liver fibrosis. Oncotarget. (2016) 7:83951–63. doi: 10.18632/oncotarget.13796
128. Haselow K, Bode JG, Wammers M, Ehlting C, Keitel V, Kleinebrecht L, et al. Bile acids PKA-dependently induce a switch of the IL-10/IL-12 ratio and reduce proinflammatory capability of human macrophages. J Leukoc Biol. (2013) 94:1253–64. doi: 10.1189/jlb.0812396
129. Keitel V, Donner M, Winandy S, Kubitz R, Haussinger D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun. (2008) 372:78–84. doi: 10.1016/j.bbrc.2008.04.171
130. Pean N, Doignon I, Garcin I, Besnard A, Julien B, Liu B, et al. The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Hepatology. (2013) 58:1451–60. doi: 10.1002/hep.26463
131. Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity. (2016) 45:802–16. doi: 10.1016/j.immuni.2016.09.008
132. Biagioli M, Carino A, Cipriani S, Francisci D, Marchiano S, Scarpelli P, et al. The bile acid receptor GPBAR1 regulates the M1/M2 phenotype of intestinal macrophages and activation of GPBAR1 rescues mice from murine colitis. J Immunol. (2017) 199:718–33. doi: 10.4049/jimmunol.1700183
133. Hegade VS, Speight RA, Etherington RE, Jones DE. Novel bile acid therapeutics for the treatment of chronic liver diseases. Therap Adv Gastroenterol. (2016) 9:376–91. doi: 10.1177/1756283X16630712
134. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. (2006) 6:173–82. doi: 10.1038/nri1785
135. Segal AW. How neutrophils kill microbes. Ann Rev Immunol. (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
136. Deniset JF, Kubes P. Neutrophil heterogeneity: bona fide subsets or polarization states? J Leukoc Biol. (2018) 103:829–38. doi: 10.1002/JLB.3RI0917-361R
137. Kubes P. The enigmatic neutrophil: what we do not know. Cell Tiss Res. (2018) 371:399–406. doi: 10.1007/s00441-018-2790-5
138. Wong J, Johnston B, Lee SS, Bullard DC, Smith CW, Beaudet AL, et al. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J Clin Invest. (1997) 99:2782–90. doi: 10.1172/JCI119468
139. McDonald B, McAvoy EF, Lam F, Gill V, de la Motte C, Savani RC, et al. Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J Exp Med. (2008) 205:915–27. doi: 10.1084/jem.20071765
140. Moles A, Murphy L, Wilson CL, Chakraborty JB, Fox C, Park EJ, et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J Hepatol. (2014) 60:782–91. doi: 10.1016/j.jhep.2013.12.005
141. McDonald B, Jenne CN, Zhuo L, Kimata K, Kubes P. Kupffer cells and activation of endothelial TLR4 coordinate neutrophil adhesion within liver sinusoids during endotoxemia. Am J Physiol Gastrointest Liver Physiol. (2013) 305:G797–806. doi: 10.1152/ajpgi.00058.2013
142. Menezes GB, Lee WY, Zhou H, Waterhouse CC, Cara DC, Kubes P. Selective down-regulation of neutrophil Mac-1 in endotoxemic hepatic microcirculation via IL-10. J Immunol. (2009) 183:7557–68. doi: 10.4049/jimmunol.0901786
143. Lammermann T, Afonso PV, Angermann BR, Wang JM, Kastenmuller W, Parent CA, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. (2013) 498:371–5. doi: 10.1038/nature12175
144. Ng LG, Qin JS, Roediger B, Wang Y, Jain R, Cavanagh LL, et al. Visualizing the neutrophil response to sterile tissue injury in mouse dermis reveals a three-phase cascade of events. J Invest Dermatol. (2011) 131:2058–68. doi: 10.1038/jid.2011.179
145. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) 464:104–7. doi: 10.1038/nature08780
146. Volmering S, Block H, Boras M, Lowell CA, Zarbock A. The neutrophil Btk signalosome regulates integrin activation during sterile inflammation. Immunity. (2016) 44:73–87. doi: 10.1016/j.immuni.2015.11.011
147. Heit B, Robbins SM, Downey CM, Guan Z, Colarusso P, Miller BJ, et al. PTEN functions to ‘prioritize’ chemotactic cues and prevent ‘distraction’ in migrating neutrophils. Nat Immunol. (2008) 9:743–52. doi: 10.1038/ni.1623
148. Foxman EF, Campbell JJ, Butcher EC. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J Cell Biol. (1997) 139:1349–60. doi: 10.1083/jcb.139.5.1349
149. Heit B, Tavener S, Raharjo E, Kubes P. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol. (2002) 159:91–102. doi: 10.1083/jcb.200202114
150. Zehrer A, Pick R, Salvermoser M, Boda A, Miller M, Stark K, et al. A fundamental role of Myh9 for neutrophil migration in innate immunity. J Immunol. (2018) 201:1748–64. doi: 10.4049/jimmunol.1701400
151. Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest. (2015) 125:539–50. doi: 10.1172/JCI76887
152. Tadie JM, Bae HB, Banerjee S, Zmijewski JW, Abraham E. Differential activation of RAGE by HMGB1 modulates neutrophil-associated NADPH oxidase activity and bacterial killing. Am J Physiol Cell Physiol. (2012) 302:C249–56. doi: 10.1152/ajpcell.00302.2011
153. Honda M, Kubes P. Neutrophils and neutrophil extracellular traps in the liver and gastrointestinal system. Nat Rev Gastroenterol Hepatol. (2018) 15:206–21. doi: 10.1038/nrgastro.2017.183
154. Bukong TN, Cho Y, Iracheta-Vellve A, Saha B, Lowe P, Adejumo A, et al. Abnormal neutrophil traps and impaired efferocytosis contribute to liver injury and sepsis severity after binge alcohol use. J Hepatol. (2018). 69:1145–54. doi: 10.1016/j.jhep.2018.07.005
155. Rensen SS, Bieghs V, Xanthoulea S, Arfianti E, Bakker JA, Shiri-Sverdlov R, et al. Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PLoS ONE. (2012) 7:e52411. doi: 10.1371/journal.pone.0052411
156. Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol. (2009) 175:1473–82. doi: 10.2353/ajpath.2009.080999
157. Zang S, Ma X, Zhuang Z, Liu J, Bian D, Xun Y, et al. Increased ratio of neutrophil elastase to alpha1-antitrypsin is closely associated with liver inflammation in patients with nonalcoholic steatohepatitis. Clin Exp Pharmacol Physiol. (2016) 43:13–21. doi: 10.1111/1440-1681.12499
158. Saijou E, Enomoto Y, Matsuda M, Yuet-Yin Kok C, Akira S, Tanaka M, et al. Neutrophils alleviate fibrosis in the CCl4-induced mouse chronic liver injury model. Hepatol Commun. (2018) 2:703–17. doi: 10.1002/hep4.1178
159. Ou R, Liu J, Lv M, Wang J, Wang J, Zhu L, et al. Neutrophil depletion improves diet-induced non-alcoholic fatty liver disease in mice. Endocrine. (2017) 57:72–82. doi: 10.1007/s12020-017-1323-4
160. Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Meininger C, Kubes P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science. (2017) 358:111–6. doi: 10.1126/science.aam9690
161. Thomson AW, O'Connell PJ, Steptoe RJ, Lu L. Immunobiology of liver dendritic cells. Immunol Cell Biol. (2002) 80:65–73. doi: 10.1046/j.0818-9641.2001.01058.x
162. Kudo S, Matsuno K, Ezaki T, Ogawa M. A novel migration pathway for rat dendritic cells from the blood: hepatic sinusoids-lymph translocation. J Exp Med. (1997) 185:777–84. doi: 10.1084/jem.185.4.777
163. Rahman AH, Aloman C. Dendritic cells and liver fibrosis. Biochim Biophys Acta. (2013) 1832:998–1004. doi: 10.1016/j.bbadis.2013.01.005
164. Lukacs-Kornek V, Schuppan D. Dendritic cells in liver injury and fibrosis: shortcomings and promises. J Hepatol. (2013) 59:1124–6. doi: 10.1016/j.jhep.2013.05.033
165. Goddard S, Youster J, Morgan E, Adams DH. Interleukin-10 secretion differentiates dendritic cells from human liver and skin. Am J Pathol. (2004) 164:511–9. doi: 10.1016/S0002-9440(10)63141-0
166. Zhang J, Raper A, Sugita N, Hingorani R, Salio M, Palmowski MJ, et al. Characterization of Siglec-H as a novel endocytic receptor expressed on murine plasmacytoid dendritic cell precursors. Blood. (2006) 107:3600–8. doi: 10.1182/blood-2005-09-3842
167. Lai WK, Curbishley SM, Goddard S, Alabraba E, Shaw J, Youster J, et al. Hepatitis C is associated with perturbation of intrahepatic myeloid and plasmacytoid dendritic cell function. J Hepatol. (2007) 47:338–47. doi: 10.1016/j.jhep.2007.03.024
168. Collin M, McGovern N, Haniffa M. Human dendritic cell subsets. Immunology. (2013) 140:22–30. doi: 10.1111/imm.12117
169. Ibrahim J, Nguyen AH, Rehman A, Ochi A, Jamal M, Graffeo CS, et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology. (2012) 143:1061–72. doi: 10.1053/j.gastro.2012.06.003
170. Strauss O, Dunbar PR, Bartlett A, Phillips A. The immunophenotype of antigen presenting cells of the mononuclear phagocyte system in normal human liver–a systematic review. J Hepatol. (2015) 62:458–68. doi: 10.1016/j.jhep.2014.10.006
171. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Ann Rev Immunol. (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
172. Jurgens B, Hainz U, Fuchs D, Felzmann T, Heitger A. Interferon-gamma-triggered indoleamine 2,3-dioxygenase competence in human monocyte-derived dendritic cells induces regulatory activity in allogeneic T cells. Blood. (2009) 114:3235–43. doi: 10.1182/blood-2008-12-195073
173. Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. (2002) 196:459–68. doi: 10.1084/jem.20020121
174. Castellaneta A, Sumpter TL, Chen L, Tokita D, Thomson AW. NOD2 ligation subverts IFN-alpha production by liver plasmacytoid dendritic cells and inhibits their T cell allostimulatory activity via B7-H1 up-regulation. J Immunol. (2009) 183:6922–32. doi: 10.4049/jimmunol.0900582
175. Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol. (2010) 10:753–66. doi: 10.1038/nri2858
176. Wang J, Cao X, Zhao J, Zhao H, Wei J, Li Q, et al. Critical roles of conventional dendritic cells in promoting T cell-dependent hepatitis through regulating natural killer T cells. Clin Exp Immunol. (2017) 188:127–37. doi: 10.1111/cei.12907
177. Connolly MK, Ayo D, Malhotra A, Hackman M, Bedrosian AS, Ibrahim J, et al. Dendritic cell depletion exacerbates acetaminophen hepatotoxicity. Hepatology. (2011) 54:959–68. doi: 10.1002/hep.24429
178. Henning JR, Graffeo CS, Rehman A, Fallon NC, Zambirinis CP, Ochi A, et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology. (2013) 58:589–602. doi: 10.1002/hep.26267
179. Heier EC, Meier A, Julich-Haertel H, Djudjaj S, Rau M, Tschernig T, et al. Murine CD103(+) dendritic cells protect against steatosis progression towards steatohepatitis. J Hepatol. (2017) 66:1241–50. doi: 10.1016/j.jhep.2017.01.008
180. Velazquez VM, Hon H, Ibegbu C, Knechtle SJ, Kirk AD, Grakoui A. Hepatic enrichment and activation of myeloid dendritic cells during chronic hepatitis C virus infection. Hepatology. (2012) 56:2071–81. doi: 10.1002/hep.25904
181. Cervantes-Barragan L, Lewis KL, Firner S, Thiel V, Hugues S, Reith W, et al. Plasmacytoid dendritic cells control T-cell response to chronic viral infection. Proc Natl Acad Sci USA. (2012) 109:3012–7. doi: 10.1073/pnas.1117359109
182. Lee J, Suh WI, Shin EC. T-cell dysfunction and inhibitory receptors in hepatitis C virus infection. Immune Netw. (2010) 10:120–5. doi: 10.4110/in.2010.10.4.120
183. Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. (2013) 58:1461–73. doi: 10.1002/hep.26429
184. Connolly MK, Bedrosian AS, Mallen-St Clair J, Mitchell AP, Ibrahim J, Stroud A, et al. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-alpha. J Clin Invest. (2009) 119:3213–25. doi: 10.1172/JCI37581
185. Jiao J, Sastre D, Fiel MI, Lee UE, Ghiassi-Nejad Z, Ginhoux F, et al. Dendritic cell regulation of carbon tetrachloride-induced murine liver fibrosis regression. Hepatology. (2012) 55:244–55. doi: 10.1002/hep.24621
186. Blois SM, Piccioni F, Freitag N, Tirado-Gonzalez I, Moschansky P, Lloyd R, et al. Dendritic cells regulate angiogenesis associated with liver fibrogenesis. Angiogenesis. (2014) 17:119–28. doi: 10.1007/s10456-013-9382-5
187. Ramachandran P, Iredale JP, Fallowfield JA. Resolution of liver fibrosis: basic mechanisms and clinical relevance. Semin Liver Dis. (2015) 35:119–31. doi: 10.1055/s-0035-1550057
188. Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Nakatani T, et al. Tissue inhibitor of metalloproteinases-1 attenuates spontaneous liver fibrosis resolution in the transgenic mouse. Hepatology. (2002) 36:850–60. doi: 10.1053/jhep.2002.35625
189. Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. (1998) 102:538–49. doi: 10.1172/JCI1018
190. Issa R, Williams E, Trim N, Kendall T, Arthur MJ, Reichen J, et al. Apoptosis of hepatic stellate cells: involvement in resolution of biliary fibrosis and regulation by soluble growth factors. Gut. (2001) 48:548–57. doi: 10.1136/gut.48.4.548
191. Ellis EL, Mann DA. Clinical evidence for the regression of liver fibrosis. J Hepatol. (2012) 56:1171–80. doi: 10.1016/j.jhep.2011.09.024
192. Marcellin P, Gane E, Buti M, Afdhal N, Sievert W, Jacobson IM, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet. (2013) 381:468–75. doi: 10.1016/S0140-6736(12)61425-1
193. Bartneck M, Fech V, Ehling J, Govaere O, Warzecha KT, Hittatiya K, et al. Histidine-rich glycoprotein promotes macrophage activation and inflammation in chronic liver disease. Hepatology. (2016) 63:1310–24. doi: 10.1002/hep.28418
194. Preisser L, Miot C, Le Guillou-Guillemette H, Beaumont E, Foucher ED, Garo E, et al. IL-34 and macrophage colony-stimulating factor are overexpressed in hepatitis C virus fibrosis and induce profibrotic macrophages that promote collagen synthesis by hepatic stellate cells. Hepatology. (2014) 60:1879–90. doi: 10.1002/hep.27328
195. Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol. (2014) 60:197–209. doi: 10.1016/j.jhep.2013.07.044
196. Aoyama T, Paik YH, Seki E. Toll-like receptor signaling and liver fibrosis. Gastroenterol Res Pract. (2010) 2010:8. doi: 10.1155/2010/192543
197. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. (2007) 13:1324–32. doi: 10.1038/nm1663
198. Nguyen-Lefebvre AT, Ajith A, Portik-Dobos V, Horuzsko DD, Arbab AS, Dzutsev A, et al. The innate immune receptor TREM-1 promotes liver injury and fibrosis. J Clin Invest. (2018) 128:4870–83. doi: 10.1172/JCI98156
199. Tornai D, Furi I, Shen ZT, Sigalov AB, Coban S, Szabo G. Inhibition of triggering receptor expressed on myeloid cells 1 ameliorates inflammation and macrophage and neutrophil activation in alcoholic liver disease in mice. Hepatol Commun. (2019) 3:99–115. doi: 10.1002/hep4.1269
200. Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology. (2009) 50:185–97. doi: 10.1002/hep.22952
201. Seki E, De Minicis S, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, et al. CCR1 and CCR5 promote hepatic fibrosis in mice. J Clin Invest. (2009) 119:1858–70. doi: 10.1172/JCI37444
202. Berres ML, Koenen RR, Rueland A, Zaldivar MM, Heinrichs D, Sahin H, et al. Antagonism of the chemokine Ccl5 ameliorates experimental liver fibrosis in mice. J Clin Invest. (2010) 120:4129–40. doi: 10.1172/JCI41732
203. Sasaki R, Devhare PB, Steele R, Ray R, Ray RB. Hepatitis C virus-induced CCL5 secretion from macrophages activates hepatic stellate cells. Hepatology. (2017) 66:746–57. doi: 10.1002/hep.29170
204. Heinrichs D, Berres ML, Nellen A, Fischer P, Scholten D, Trautwein C, et al. The chemokine CCL3 promotes experimental liver fibrosis in mice. PLoS ONE. (2013) 8:e66106. doi: 10.1371/journal.pone.0066106
205. Rantakari P, Patten DA, Valtonen J, Karikoski M, Gerke H, Dawes H, et al. Stabilin-1 expression defines a subset of macrophages that mediate tissue homeostasis and prevent fibrosis in chronic liver injury. Proc Natl Acad Sci USA. (2016) 113:9298–303. doi: 10.1073/pnas.1604780113
206. Imamura M, Ogawa T, Sasaguri Y, Chayama K, Ueno H. Suppression of macrophage infiltration inhibits activation of hepatic stellate cells and liver fibrogenesis in rats. Gastroenterology. (2005) 128:138–46. doi: 10.1053/j.gastro.2004.10.005
207. Mitchell C, Couton D, Couty JP, Anson M, Crain AM, Bizet V, et al. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol. (2009) 174:1766–75. doi: 10.2353/ajpath.2009.080632
208. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. (2018) 67:1270–83. doi: 10.1002/hep.29544
209. Lefebvre E, Moyle G, Reshef R, Richman LP, Thompson M, Hong F, et al. Antifibrotic effects of the dual CCR2/CCR5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PLoS ONE. (2016) 11:e0158156. doi: 10.1371/journal.pone.0158156
210. Kruger AJ, Fuchs BC, Masia R, Holmes JA, Salloum S, Sojoodi M, et al. Prolonged cenicriviroc therapy reduces hepatic fibrosis despite steatohepatitis in a diet-induced mouse model of nonalcoholic steatohepatitis. Hepatol Commun. (2018) 2:529–45. doi: 10.1002/hep4.1160
211. Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N, et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology. (2014) 59:1060–72. doi: 10.1002/hep.26783
212. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. (2018) 67:1754–67. doi: 10.1002/hep.29477
213. Chu PS, Nakamoto N, Ebinuma H, Usui S, Saeki K, Matsumoto A, et al. C-C motif chemokine receptor 9 positive macrophages activate hepatic stellate cells and promote liver fibrosis in mice. Hepatology. (2013) 58:337–50. doi: 10.1002/hep.26351
214. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. (2005) 115:209–18. doi: 10.1172/JCI24282
215. Li H, Zheng HW, Chen H, Xing ZZ, You H, Cong M, et al. Hepatitis B virus particles preferably induce Kupffer cells to produce TGF-beta1 over pro-inflammatory cytokines. Digest Liver Dis. (2012) 44:328–33. doi: 10.1016/j.dld.2011.11.005
216. Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, Iredale JP, et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci USA. (2006) 103:5060–5. doi: 10.1073/pnas.0511167103
217. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. (2010) 30:245–57. doi: 10.1055/s-0030-1255354
218. Matsuda M, Tsurusaki S, Miyata N, Saijou E, Okochi H, Miyajima A, et al. Oncostatin M causes liver fibrosis by regulating cooperation between hepatic stellate cells and macrophages in mice. Hepatology. (2018) 67:296–312. doi: 10.1002/hep.29421
219. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. (2005) 115:56–65. doi: 10.1172/JCI200522675
220. Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. (2007) 178:5288–95. doi: 10.4049/jimmunol.178.8.5288
221. Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology. (2010) 52:1390–400. doi: 10.1002/hep.23795
222. Karlmark KR, Zimmermann HW, Roderburg C, Gassler N, Wasmuth HE, Luedde T, et al. The fractalkine receptor CX(3)CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology. (2010) 52:1769–82. doi: 10.1002/hep.23894
223. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. (2007) 204:3037–47. doi: 10.1084/jem.20070885
224. Landsman L, Varol C, Jung S. Distinct differentiation potential of blood monocyte subsets in the lung. J Immunol. (2007) 178:2000–7. doi: 10.4049/jimmunol.178.4.2000
225. Ziegler-Heitbrock L, Hofer TP. Toward a refined definition of monocyte subsets. Front Immunol. (2013) 4:23. doi: 10.3389/fimmu.2013.00023
226. Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. (2002) 31:339–46. doi: 10.1038/ng0802-339
227. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. (2011) 479:547–51. doi: 10.1038/nature10599
228. Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell. (2016) 30:533–47. doi: 10.1016/j.ccell.2016.09.003
229. Ding T, Xu J, Wang F, Shi M, Zhang Y, Li SP, et al. High tumor-infiltrating macrophage density predicts poor prognosis in patients with primary hepatocellular carcinoma after resection. Hum Pathol. (2009) 40:381–9. doi: 10.1016/j.humpath.2008.08.011
230. Zhu XD, Zhang JB, Zhuang PY, Zhu HG, Zhang W, Xiong YQ, et al. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J Clin Oncol. (2008) 26:2707–16. doi: 10.1200/JCO.2007.15.6521
231. Foerster F, Hess M, Gerhold-Ay A, Marquardt JU, Becker D, Galle PR, et al. The immune contexture of hepatocellular carcinoma predicts clinical outcome. Sci Rep. (2018) 8:5351. doi: 10.1038/s41598-018-21937-2
232. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. (2004) 431:461–6. doi: 10.1038/nature02924
233. Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. (2005) 121:977–90. doi: 10.1016/j.cell.2005.04.014
234. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. (2014) 147:1393–404. doi: 10.1053/j.gastro.2014.08.039
235. Wu J, Li J, Salcedo R, Mivechi NF, Trinchieri G, Horuzsko A. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. (2012) 72:3977–86. doi: 10.1158/0008-5472.CAN-12-0938
236. Delire B, Henriet P, Lemoine P, Leclercq IA, Starkel P. Chronic liver injury promotes hepatocarcinoma cell seeding and growth, associated with infiltration by macrophages. Cancer Sci. (2018) 109:2141–52. doi: 10.1111/cas.13628
237. Yeung OW, Lo CM, Ling CC, Qi X, Geng W, Li CX, et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J Hepatol. (2015) 62:607–16. doi: 10.1016/j.jhep.2014.10.029
238. Takai H, Ashihara M, Ishiguro T, Terashima H, Watanabe T, Kato A, et al. Involvement of glypican-3 in the recruitment of M2-polarized tumor-associated macrophages in hepatocellular carcinoma. Cancer Biol Ther. (2009) 8:2329–38. doi: 10.4161/cbt.8.24.9985
239. Yang Y, Ye YC, Chen Y, Zhao JL, Gao CC, Han H, et al. Crosstalk between hepatic tumor cells and macrophages via Wnt/beta-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. (2018) 9:793. doi: 10.1038/s41419-018-0818-0
240. Ye Y, Xu Y, Lai Y, He W, Li Y, Wang R, et al. Long non-coding RNA cox-2 prevents immune evasion and metastasis of hepatocellular carcinoma by altering M1/M2 macrophage polarization. J Cell Biochem. (2018) 119:2951–63. doi: 10.1002/jcb.26509
241. Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, et al. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut. (2015) 64:1593–604. doi: 10.1136/gutjnl-2014-307671
242. Rohr-Udilova N, Klinglmuller F, Schulte-Hermann R, Stift J, Herac M, Salzmann M, et al. Deviations of the immune cell landscape between healthy liver and hepatocellular carcinoma. Sci Rep. (2018) 8:6220. doi: 10.1038/s41598-018-24437-5
243. Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. (2017) 66:157–67. doi: 10.1136/gutjnl-2015-310514
244. Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology. (2016) 150:1646–58e17. doi: 10.1053/j.gastro.2016.02.040
245. van der Windt DJ, Sud V, Zhang H, Varley PR, Goswami J, Yazdani HO, et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology. (2018). 68:1347–60. doi: 10.1002/hep.29914
246. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. (2009) 206:1327–37. doi: 10.1084/jem.20082173
247. Lens S, Alvarado-Tapias E, Marino Z, Londono MC, E LL, Martinez J, et al. Effects of all-oral anti-viral therapy on HVPG and systemic hemodynamics in patients with hepatitis C virus-associated cirrhosis. Gastroenterology. (2017) 153:1273–83e1. doi: 10.1053/j.gastro.2017.07.016
248. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. (2017) 66:1300–12. doi: 10.1016/j.jhep.2017.02.026
249. Sakaida I, Terai S, Yamamoto N, Aoyama K, Ishikawa T, Nishina H, et al. Transplantation of bone marrow cells reduces CCl4-induced liver fibrosis in mice. Hepatology. (2004) 40:1304–11. doi: 10.1002/hep.20452
250. Thomas JA, Pope C, Wojtacha D, Robson AJ, Gordon-Walker TT, Hartland S, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology. (2011) 53:2003–15. doi: 10.1002/hep.24315
251. Suh YG, Kim JK, Byun JS, Yi HS, Lee YS, Eun HS, et al. CD11b(+) Gr1(+) bone marrow cells ameliorate liver fibrosis by producing interleukin-10 in mice. Hepatology. (2012) 56:1902–12. doi: 10.1002/hep.25817
252. de Souza VCA, Pereira TA, Teixeira VW, Carvalho H, de Castro M, D'Assuncao C G, et al. Bone marrow-derived monocyte infusion improves hepatic fibrosis by decreasing osteopontin, TGF-beta1, IL-13 and oxidative stress. World J Gastroenterol. (2017) 23:5146–57. doi: 10.3748/wjg.v23.i28.5146
253. Haideri SS, McKinnon AC, Taylor AH, Kirkwood P, Starkey Lewis PJ, O'Duibhir E, et al. Injection of embryonic stem cell derived macrophages ameliorates fibrosis in a murine model of liver injury. NPJ Regen Med. (2017) 2:14. doi: 10.1038/s41536-017-0017-0
254. Yang L, Han Z, Tian L, Mai P, Zhang Y, Wang L, et al. Sphingosine 1-phosphate receptor 2 and 3 mediate bone marrow-derived monocyte/macrophage motility in cholestatic liver injury in mice. Sci. Rep. (2015) 5:13423. doi: 10.1038/srep13423
255. King A, Houlihan DD, Kavanagh D, Haldar D, Luu N, Owen A, et al. Sphingosine-1-phosphate prevents egress of hematopoietic stem cells from liver to reduce fibrosis. Gastroenterology. (2017) 153:233–48e16. doi: 10.1053/j.gastro.2017.03.022
256. Newsome PN, Fox R, King AL, Barton D, Than NN, Moore J, et al. Granulocyte colony-stimulating factor and autologous CD133-positive stem-cell therapy in liver cirrhosis (REALISTIC): an open-label, randomised, controlled phase 2 trial. Lancet Gastroenterol Hepatol. (2018) 3:25–36. doi: 10.1016/S2468-1253(17)30326-6
257. Thomas JA, Ramachandran P, Forbes SJ. Studies of macrophage therapy for cirrhosis - From mice to men. J Hepatol. (2018) 68:1090–1. doi: 10.1016/j.jhep.2017.11.043
258. Fraser AR, Pass C, Burgoyne P, Atkinson A, Bailey L, Laurie A, et al. Development, functional characterization and validation of methodology for GMP-compliant manufacture of phagocytic macrophages: A novel cellular therapeutic for liver cirrhosis. Cytotherapy. (2017) 19:1113–24. doi: 10.1016/j.jcyt.2017.05.009
Keywords: hepatitis (general), hepatocellular carcinoma, cirrhosis, fibrosis, myeloid derived suppressor cell (MDSC), neutrophil (PMN), macrophage, circulating monocytes
Citation: Weston CJ, Zimmermann HW and Adams DH (2019) The Role of Myeloid-Derived Cells in the Progression of Liver Disease. Front. Immunol. 10:893. doi: 10.3389/fimmu.2019.00893
Received: 13 September 2018; Accepted: 08 April 2019;
Published: 24 April 2019.
Edited by:
Rajiv Jalan, University College London, United KingdomReviewed by:
Cordula M. Stover, University of Leicester, United KingdomCopyright © 2019 Weston, Zimmermann and Adams. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chris John Weston, Yy5qLndlc3RvbkBiaGFtLmFjLnVr
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.