 Stefanie K. Wculek
Stefanie K. Wculek Sofía C. Khouili
Sofía C. Khouili Elena Priego
Elena Priego Ignacio Heras-Murillo
Ignacio Heras-Murillo David Sancho
David Sancho- Immunobiology Laboratory, Centro Nacional de Investigaciones Cardiovasculares (CNIC), Madrid, Spain
Dendritic cells (DCs) control innate and adaptive immunity by patrolling tissues to gather antigens and danger signals derived from microbes and tissue. Subsequently, DCs integrate those environmental cues, orchestrate immunity or tolerance, and regulate tissue homeostasis. Recent advances in the field of immunometabolism highlight the notion that immune cells markedly alter cellular metabolic pathways during differentiation or upon activation, which has important implications on their functionality. Previous studies showed that active oxidative phosphorylation in mitochondria is associated with immature or tolerogenic DCs, while increased glycolysis upon pathogen sensing can promote immunogenic DC functions. However, new results in the last years suggest that regulation of DC metabolism in steady state, after immunogenic activation and during tolerance in different pathophysiological settings, may be more complex. Moreover, ontogenically distinct DC subsets show different functional specializations to control T cell responses. It is, thus, relevant how metabolism influences DC differentiation and plasticity, and what potential metabolic differences exist among DC subsets. Better understanding of the emerging connection between metabolic adaptions and functional DC specification will likely allow the development of therapeutic strategies to manipulate immune responses.
Metabolic Control of Dendritic Cell Development
Natural dendritic cells (DCs) present in steady state comprise type 1 conventional DCs (cDC1s), type 2 cDCs (cDC2s), double negative (CD8/CD103– CD11b–) DCs (DN-DCs), and plasmacytoid DCs (pDCs; Table 1). Natural DCs derive from myeloid progenitors in the bone marrow and require FMS-like tyrosine kinase 3 ligand (FLT3L) to differentiate via the common DC progenitor (CDP) and DC precursors (pre-DCs). In addition, other cells that are functionally similar to DCs, such as Langerhans cells (LCs), can derive from embryonic precursors. Moreover, during inflammatory settings, DCs can develop from blood monocytes (moDCs; Table 1).
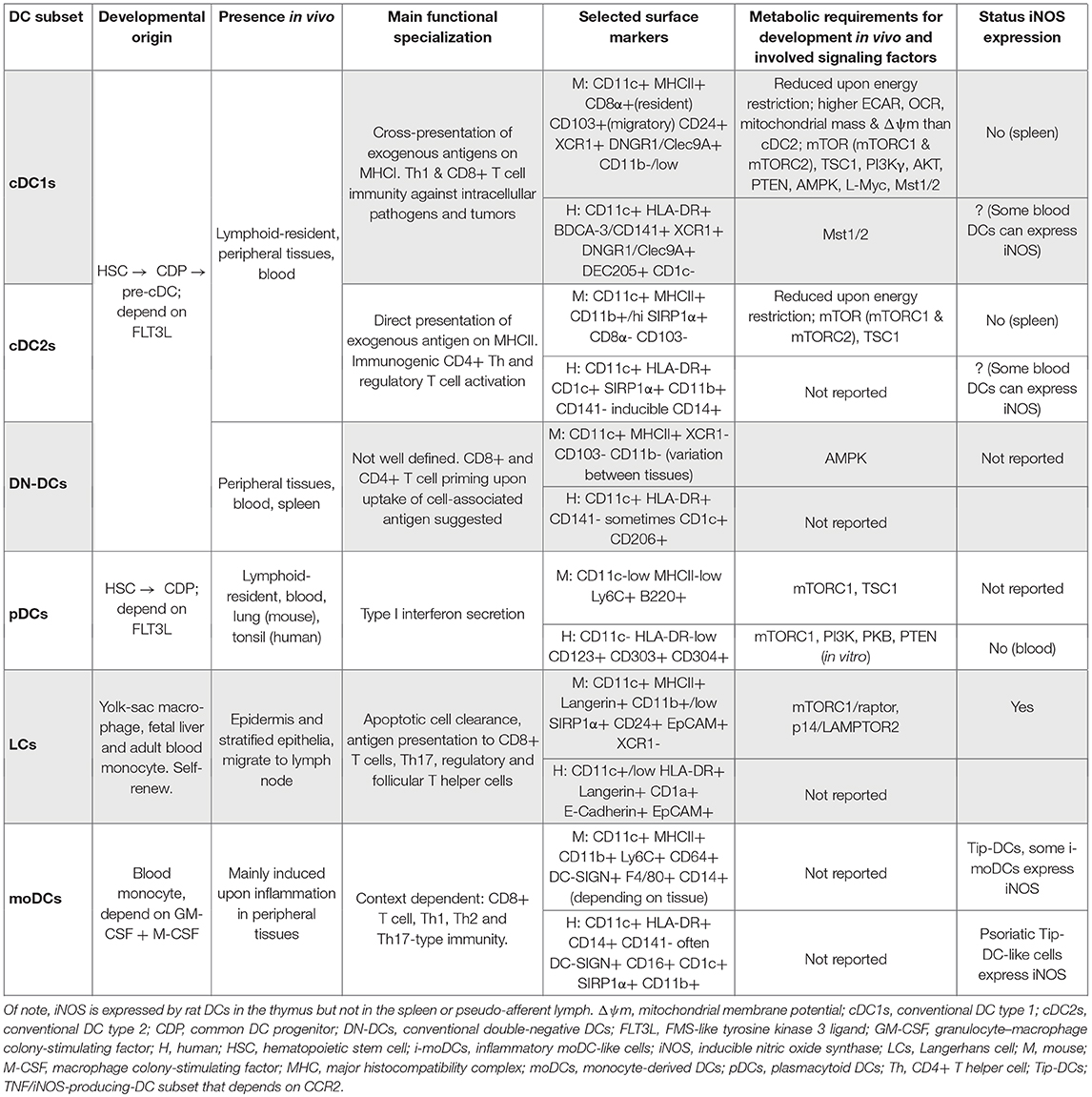
Table 1. Dendritic cell subsets in vivo.
Energy Metabolism During Dendritic Cell Generation
Differentiation of Dendritic Cells From Monocytes With GM-CSF
The importance of energy metabolism was first established in the development of human moDCs in vitro. Granulocyte–macrophage colony-stimulating factor (GM-CSF) and interleukin (IL)-4-induced differentiation and survival of DCs from human monocytes rely on the mammalian target of rapamycin (mTOR) complex 1 (mTORC1) activation via phosphoinositide 3-kinase (PI3K; Figure 1) and are abrogated by rapamycin, an mTOR/mTORC1 inhibitor [Table 2 and (1, 2)]. The mTORC1 downstream target peroxisomal proliferator-activated receptor γ (PPARγ) is upregulated early in moDC differentiation, affecting cell maturation and function largely through control of lipid metabolism (3–6). Indeed, inhibition of cytosolic fatty acid synthesis (FAS) via blocking acetyl-CoA carboxylase (ACC) 1 reduces moDC differentiation (7). Moreover, PPARγ co-activator-1α (PGC1α) and mitochondrial transcription factor A (TFAM), fundamental inducers of mitochondrial biogenesis and also indirect mTORC1 targets (8, 9), are also elevated during moDC differentiation (10). In line, differentiated moDCs show a higher oxygen consumption rate (OCR), contain more mitochondria, and produce more adenosine triphosphate (ATP) compared to monocytes (10, 11). Importantly, blocking the electron transport chain (ETC) with the complex I (CI) inhibitor rotenone (Figure 2) partially prevents moDC differentiation, despite causing a notable increase in glycolysis/lactate production (10, 11). Hence, moDC differentiation depends on oxidative phosphorylation (OXPHOS) and a balanced fatty acid metabolism.
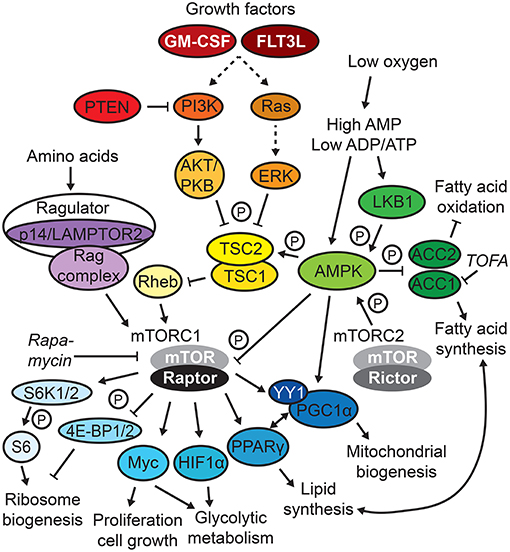
Figure 1. mTOR/AMPK signaling. Selected signaling circuits of the complex mammalian target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) signaling network are depicted. Frequently used metabolic inhibitors are displayed in italics, and P indicates phosphorylation.
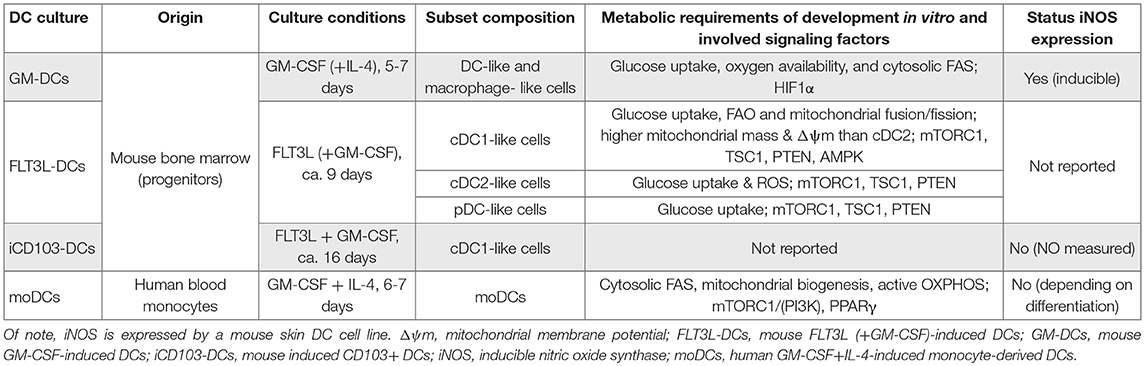
Table 2. Culture systems of dendritic cells.
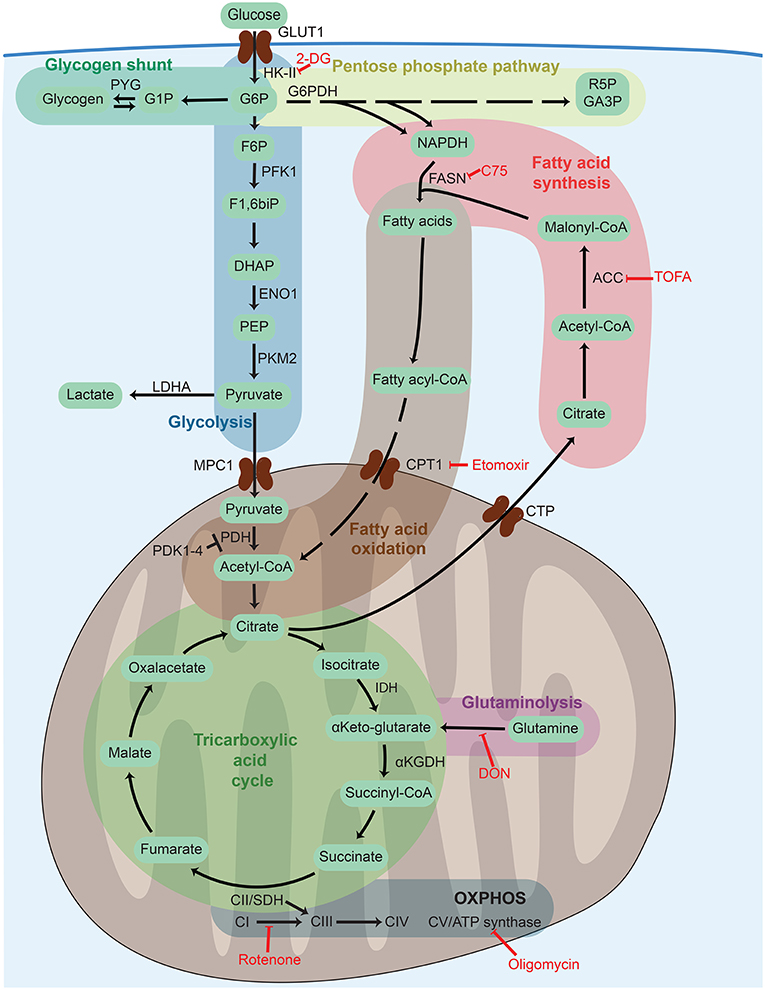
Figure 2. Cellular metabolism networks. Glucose is imported from the extracellular environment and can generate glycogen stores, be used in the pentose phosphate pathway to generate reducing power, or be oxidized during glycolysis to obtain adenosine triphosphate (ATP). Pyruvate generated from glycolysis can either be partially oxidized to lactate to quickly regenerate the consumed nicotinamide adenine dinucleotide (NADH) or translocate into the mitochondria to be completely oxidized thought the tricarboxylic acid (TCA) cycle. The TCA cycle can also be fueled by fatty acids via fatty acid oxidation or glutamine via glutaminolysis. The electrons released by glycolysis and the TCA cycle enter into the electron transport chain composed of complex I–V (CI–CV) where ATP is generated by oxidative phosphorylation (OXPHOS). Frequently used metabolic inhibitors are indicated in red. 2-DG, 2-deoxy-D-glucose; ACC, acetyl-CoA carboxylase; αKGDH, α-ketoglutarate dehydrogenase; CoA, coenzyme A; CPT1, carnitine palmitoyltransferase 1, CTP, citrate transport protein; DHAP, dihydroxyacetone phosphate; DON, 6-Diazo-5-oxo-L-norleucine; ENO1, enolase 1; F1,6biP, fructose 1,6 biphosphate; F5P, fructose 5 phosphate; F6P, fructose 6 phosphate; FASN, fatty acid synthase; G1P, glucose 1 phosphate, G6P, glucose 6 phosphate; G6PDH, glucose 6 phosphate dehydrogenase; GA3P, glyceraldehyde 3 phosphate, GLUT1, glucose transporter 1; HK-II, hexokinase 2; IDH, isocitrate dehydrogenase; LDHA, lactate dehydrogenase A; MPC1, mitochondrial pyruvate carrier 1; NADPH, nicotinamide adenine dinucleotide phosphate; PDH, pyruvate dehydrogenase PDK1-4, pyruvate dehydrogenase kinase 1-4, PEP, phosphoenolpyruvate; PFK1, phosphofructokinase-1; PKM2, pyruvate kinase isozyme M2; PYG, glycogen phosphorylase; R5P, ribose 5-phosphate; SDH, succinate dehydrogenase; TOFA, 5-(Tetradecyloxy)-2-furoic acid.
Likewise, the DC-like cells differentiated from mouse bone marrow cultured with GM-CSF in vitro, a culture system composed of a mixed population of DCs and macrophages [Table 2, GM-DCs and (12)], also show glucose uptake together with high mitochondrial membrane potential (ΔΨm) and oxygen consumption (13). Indeed, GM-DC differentiation under hypoxic conditions yields fewer total cells, and hypoxia-inducible factor (HIF)-1α deficiency further reduces the frequency of CD11c+ GM-DCs, linked to decreased ATP (14). As HIF1α is a key metabolic regulator and many of its target genes drive glycolysis (see the section Sustained Glycolysis: The Role of HIF1α), these observations point toward the importance of an active glucose metabolism involving oxidative and glycolytic pathways in GM-DCs. However, CD11c-Cre HIF1αflox/− mice display unaltered DC homeostasis in the steady state (15). Moreover, impairment of cytosolic FAS by blocking ACC1 with the inhibitor 5-(tetradecyloxy)-2-furoic acid (TOFA) in GM-DC cultures or by the administration of the fatty acid synthase (FASN) inhibitor methylene-2-octyl-5-oxotetrahydrofuran-3-carboxylic acid (C75; Figure 2) in vivo reduces the generation of DCs (7), further suggesting that balanced FA metabolism contributes to DC development. However, it is noteworthy that the inhibitor C75 can also cause mitochondrial dysfunction (16).
Natural Dendritic Cell Differentiation
Generally, the presence of CDPs, pre-DCs, cDCs, and pDCs is reduced in energy-restricted mice, while myeloid progenitors, blood monocytes, and spleen macrophages are increased. FLT3L administration is unable to rescue the effect (17), highlighting the intrinsic importance of uncompromised energy metabolism for in vivo DC differentiation compared to monocytes. In concert, natural mouse DC progenitors in the bone marrow (Table 2; FLT3L-DC cultures) are dependent on nutrient transporters and glucose uptake for proliferation upon FLT3L stimulation in vitro (18). Those FLT3L-stimulated bone marrow cultures allow for the separate evaluation of mouse CDP-derived DC subsets [Table 2; FLT3L-DCs and (19)]. Notably, the inhibition of fatty acid oxidation (FAO) with etomoxir (Figure 2), promoting mitochondrial fusion with M1 or blocking fission with Mdivi-1, does not affect pDCs but strongly skews cDC differentiation toward cDC2s, while reactive oxygen species (ROS) inhibition favors cDC1s (18). Of note, apart from inhibition of carnitine palmitoyltransferase 1 (Cpt1a), a crucial enzyme for long-chain FAO, etomoxir displays off-target effects and can independently block mitochondrial respiration or enhance the ΔΨm in T cells (20). Indeed, cDC1s generally display higher mitochondrial mass and ΔΨm than cDC2s in vitro and in vivo (18, 21, 22). The non-canonical Hippo pathway kinases mammalian sterile twenty-like (Mst) 1 and 2 are crucial for mitochondrial homeostasis, energy metabolism, and immunogenic function of cDC1s, but less for cDC2s, and are activated by FLT3L in cDC1s (21). In line, in vivo FLT3L administration to CD11c-Cre Mst1/2flox/flox mice yields reduced splenic cDC1 numbers compared to controls. Unexpectedly, CD11c-Cre Mst1/2flox/flox mice exhibit elevated frequencies of splenic cDC1s, unaltered pDCs, and reduced cDC2s in the steady state (21); hence, the precise role of (non-canonical) Hippo signaling in DC development needs further investigation.
Overall, these data highlight differential energy requirements for DC subset generation, where moDCs and spleen cDC1s appear more dependent on functional mitochondrial metabolism and OXPHOS than cDC2s or pDCs (Tables 1, 2).
Nutrient-Sensing Pathways Affecting Dendritic Cell Development
Adaption to extra- and intracellular nutrient sensing via the mTOR network composed of mTORC1 and 2 complexes (Figure 1) is central for the development of DCs (23). This notion is supported by the fact that the DC differentiation-inducing factors GM-CSF and FLT3L directly induce mTOR activation (2, 24, 25).
Monocyte-Derived Dendritic Cells and Embryo-Derived Langerhans Cells
The generation and survival of the non-CDP-derived human moDCs and self-maintaining LCs depend on mTORC1 (Tables 1, 2). As mentioned in the previous section, mTOR is constitutively active in cultured human moDCs, and the mTOR inhibitor rapamycin, which affects mTORC1 stronger than mTORC2, abrogates their differentiation, inducing apoptosis, in line with GM-CSF/IL-4 activating mTOR to sustain survival (1, 2). Mice deficient in the mTORC1 component Raptor in CD11c-expressing cells, but not the mTORC2 component Rictor (Figure 1), progressively lose epidermal LCs over time (26). In concert, LCs deficient in the Ragulator complex component p14 [a.k.a. lysosomal adaptor and mitogen-activated protein kinase and mTOR activator/regulator 2 (LAMPTOR2)], which display abrogated extracellular signaling-regulated kinase (ERK) and mTOR signaling, are increasingly mature and unable to self-renew due to reduced responsiveness to tumor growth factor (TGF)-β1 (27, 28), which is crucial for LC differentiation and maintenance (29).
Dendritic Cells Generated From Common Dendritic Cell Progenitors
Despite the Ras/PI3K/AKT/mTOR signaling axis (Figure 1) being activated by FLT3L (24, 25), the precise role of mTOR signaling is more ambiguous in FLT3L-dependent, CDP-derived DC subsets (Tables 1, 2). There are conflicting observations depending on how mTOR signaling is targeted. A line of evidence suggests that active mTOR signaling promotes generation of proper natural DC numbers and subset distribution. In vitro, generation of pDCs, cDC1s, and cDC2s in FLT3L-DCs is reduced by rapamycin and enhanced by loss of phosphatase and tensin homolog (PTEN), a negative regulator of PI3K/AKT/mTOR signaling (24) (Figure 1). Similarly, rapamycin administration to mice in the steady state decreases CDPs and pre-DCs in the bone marrow as well as total CD11c+ DCs, pDCs, and cDC2s in the spleen (25, 30). cDC1s and, to a lesser extent, cDC2s are profoundly reduced in the spleens and lungs of CD11c-Cre mTORflox/flox mice, CD11c-Cre Raptorflox/flox, Rictorflox/flox double-knockout mice and mice lacking functional PI3Kγ or AKT, upstream activators of mTOR (25, 31). In accordance, cDC1s are strongly expanded in lymphoid and peripheral organs in mice deficient for PTEN (CD11c-Cre PTENflox/flox mice), a phenotype reversed by rapamycin administration (24). While pDC development is largely unaffected in PI3Kγ-deficient mice (25), human pDC differentiation in vitro is blocked by rapamycin, PI3K, and AKT/PKB inhibitors and facilitated by PTEN inhibition or enforced AKT activation (32).
In contrast, other reports suggest an inhibitory function of mTOR signaling for natural DC development. FLT3L-DCs show induction of AMP-activated protein kinase (AMPK) signaling, which antagonizes mTORC1 (Figure 1) (18, 33). AMPKα1 deficiency does not affect pDC or overall cDC differentiation but results in relative loss of cDC1s and DN-DCs (18, 33). Moreover, mTOR inhibition by rapamycin increases spleen cDC1 and cDC2 subsets and several DC subsets in peripheral organs upon FLT3L-mediated DC expansion in vivo (25). Loss of mTORC1 in DCs in CD11c-Cre Raptorflox/flox mice also expands CD11c+ DCs in the bone marrow, cDC1s in the spleen, and cDC2s in the small intestine (26, 34). Similarly, tuberous sclerosis 1 (TSC1) deficiency (Figure 1), using tamoxifen-inducible Rosa-Cre TSC1flox/flox mice, enhances mTOR activation and reduces pDCs, cDC1s, and cDC2s generated in FLT3L-DCs and in vivo, which is rescued by rapamycin (35). Conversely, CD11c-Cre TSC1flox/flox mice show no major alterations in DC development (24, 36). In humans, rapamycin treatment of kidney transplant patients does not affect cDC/pDC differentiation, while DCs appear more immunogenic (2).
In conclusion, a delicate balance of the complex system of nutrient sensing and mTOR (mTORC1) signaling is crucial to ensure appropriate development of DCs (23). Strikingly, loss of both mTOR complexes results in opposite effects on in vivo DC development compared with loss of mTORC1 alone, probably indicating differential inhibition of mTOR downstream targets and collaboration of mTOR complexes. Indeed, DC loss upon TSC1 deficiency is accompanied by increased DC apoptosis and enhanced metabolic activity due to TSC1-dependent inhibition of Myc, an effector downstream of mTOR (Figure 1), and reversed upon Myc loss (35). Of note, Myc itself regulates glucose and glutamine catabolism in activated T cells (37). Moreover, apart from controlling mTORC1 activity, AMPK is an important regulator of fatty acid metabolism limiting ACC1/2 activity (Figure 1), which is crucial for T cell activation (38). AMPK loss generally favors cytosolic FAS over mitochondrial FAO, which likely accounts for the decrease in differentiation in AMPKα1-deficient cDC1s, as this process was shown to be sensitive to FAO block (18) and, hence, could be independent from mTOR signaling.
Moreover, the context dependence of balanced mTOR signaling in DCs may be strongly influenced by FLT3L. First, rapamycin and Mst1/2 deficiency have different or even opposing effects on DC generation in the steady state compared with FLT3L-mediated DC expansion in vivo (21, 25, 30). Second, while FLT3L-mediated differentiation of DC subsets from mouse bone marrow in vitro clearly relies on appropriate mTOR activity (18, 24, 33), GM-CSF-induced DC development in vitro was not affected by mTOR deregulation. FLT3L and GM-CSF have both been shown to activate mTOR (2, 24, 25); however, this activation might serve different purposes. Third, deregulated mTOR signaling appears to have stronger effects on the generation of cDC1s than other natural DC subsets, in line with spleen cDC1s being more metabolically active and their reliance on functional mitochondrial respiration (18, 21). The notion that cDC1s appear to rely more on FLT3L than other subsets, especially in peripheral tissues (39), might provide a potential explanation. Indeed, cDC1s in the spleen have higher basal phosphorylation levels of S6 protein, a readout for mTORC1 activity (Figure 1), than other DC subsets and upregulate mTOR activation to a greater extent upon FLT3L administration in vivo. Moreover, the increase of cDC1s upon PTEN deficiency is specific to the FLT3L-responsive CX3CR1-negative subset (24).
Last, caution has to be taken when interpreting the effect of manipulating mTOR signaling in DCs. For instance, deletion of the positive mTORC1 regulator p14/LAMPTOR2 in CD11c-expressing cells increases pre-DCs in the bone marrow and amplifies DC subsets in spleen and lymph nodes (LNs) due to accumulation of FLT3 receptor on the DC surface, leading to activation of mTOR (40). Also, while the requirement of mTOR and its signaling components was assessed, the specific mechanisms or the direct role of this nutrient sensor in regulating metabolic pathways such as glycolysis, OXPHOS, or fatty acid metabolism during DC development largely remain to be defined and could account for some of the observed controversies.
Metabolic Rearrangements Upon Immunogenic Dendritic Cell Stimulation
Increasing efforts have been made over the past years to better understand metabolic changes that occur in DCs upon stimulation and how those affect DC functionalities. Resting DCs show a catabolic metabolism and continuously break down nutrients for energy generation and cell maintenance. This metabolic state manifests active OXPHOS, driven by the tricarboxylic acid (TCA) cycle fueled via FAO and glutaminolysis, and is largely regulated by AMPK (13, 41–45), as discussed in the section Metabolic Control of Dendritic Cell Development. Apart from glucose, steady-state DCs use intracellular glycogen to support basal glycolytic demands, which provides metabolic substrates for mitochondrial respiration (46). Upon immunogenic activation, DCs often adopt an anabolic metabolism for the generation of substrates for biosynthesis and cell growth. Activated DCs switch to glycolysis and lactic fermentation that provide energy and additionally reroute glycolytic intermediates into the pentose phosphate pathway (PPP). Moreover, production of nitric oxide (NO), which inhibits the ETC, is induced by some activated DC subsets (Tables 1, 2). The TCA cycle is rewired, leading to accumulation of TCA intermediates that can serve as immunomodulatory signals and support FAS and production of ROS and NO upon DC activation (41–45) (Figure 3). Of note, most of the current knowledge on DC metabolism was obtained using DC-like cells differentiated with GM-CSF from mouse bone marrow in vitro (Table 2; GM-DCs), which also contain a significant proportion of macrophage-like cells (12). This DC culture model provides important insights on the basis of metabolic adaptions of DCs after activation but does not allow investigation of different DC subsets, which appear more and more relevant in light of the differential metabolic requirements for their development.
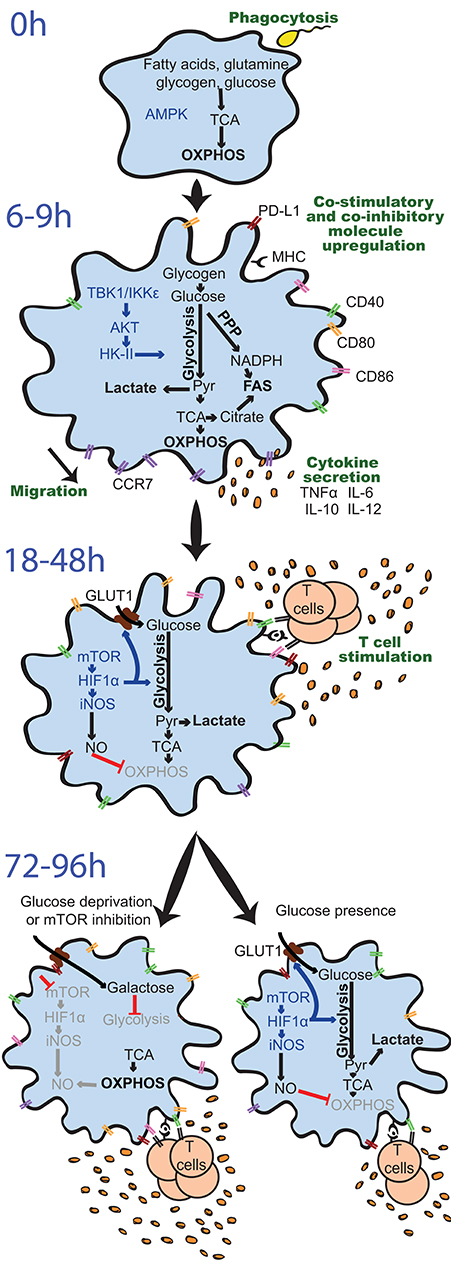
Figure 3. Differential regulation and effects of glycolysis induction in GM-DCs upon stimulation over time. Resting GM-DCs (top) display a basal metabolism with active AMPK (AMP-activated protein kinase) and fatty acids, glutamine, glycogen, and glucose being fully oxidized to generate energy by oxidative phosphorylation (OXPHOS). Upon early stimulation after 6–9 h, GM-DCs are activated and exhibit transiently enhanced OXPHOS/mitochondrial membrane potential and an increased glycolytic metabolism mainly using glucose from intracellular glycogen stores. The induction of glycolysis is predominantly driven by a TBK1-IKKε/AKT/HK-II axis and largely devoted to fatty acid synthesis (FAS). Moreover, enhanced early glycolytic activity of GM-DCs is vital for their migration and upregulation of co-stimulatory/inhibitory molecules as well as cytokines. At later time points about 18–48 h after robust stimulation, a mTOR/HIF1α/iNOS axis is activated in GM-DCs, leading to enforced glycolysis via upregulation of glucose importers such as GLUT1 and inhibition of OXPHOS via nitric oxide (NO). This fostered glycolytic activity appears crucial for the interaction of GM-DCs with T cells. Nevertheless, the sustained inhibition of OXPHOS by NO and reliance on glycolysis for energy generation can reduce the ability of GM-DCs to stimulate T cells in the long term. Glucose deprivation or mTOR inhibition can preserve metabolic flexibility and functional OXPHOS in GM-DCs, sustaining their activity at least during 72–96 h and extending their life span. AKT, protein kinase B; CCR7, C-C chemokine receptor type 7; CD, cluster of differentiation; GLUT1, glucose transporter 1; GM-DC, GM-CSF, mouse GM-CSF-induced DCs; HIF1α, hypoxia-inducible factor 1-alpha; HK-II, hexokinase II; IKKε, IkB kinase; IL, interleukin; iNOS, inducible nitric oxide synthase; MHC, major histocompatibility complex; mTOR, mammalian target of rapamycin; NADPH, nicotinamide adenine dinucleotide phosphate; PD-L1, programmed death-ligand 1, Pyr, pyruvate; PPP, pentose phosphate pathway; TBK1, TANK-binding kinase 1; TCA, Tricarboxcylic acid cycle; TNFα, tumor necrosis factor α.
Increased Glycolytic Activity Determines Inflammatory Dendritic Cell Functions—A Consensus Among Activated DC Subsets?
An early elevation of glycolysis is a metabolic hallmark of activated DCs and occurs in different mouse DC cultures, human moDCs in vitro, and mouse/human DC subsets in vivo/ex vivo (Figures 3, 4) shortly after pattern recognition receptor (PRR) stimulation with a wide range of pure pathogen-associated molecular patterns (PAMPs) or complex stimuli, such as lipopolysaccharides (LPSs) (13, 47–51), CpG oligodeoxynucleotides (13, 49), poly(I:C) (15, 49), R848/Resiquimod (49, 52), protamine–RNA complexes (pRNA) (53), zymosan (50), Pam3CSK4/Pam2CSK4 (49), Aspergillus fumigatus (54), Chlamydia (55), heat-killed Propionibacterium acnes (13), and influenza A virus or rhinovirus infection (52). Interestingly, stimulants such as LPS and zymosan strongly induce upregulation of costimulatory molecules and cytokines, whereas weak activators such as house dust mite (HDM) or zymosan lacking TLR ligands (ZymD) provoke a milder GM-DC maturation profile (56). Importantly, the potency of stimulants inducing GM-DC activation is directly correlated with enhanced degree and maintenance of glycolysis induction (56).
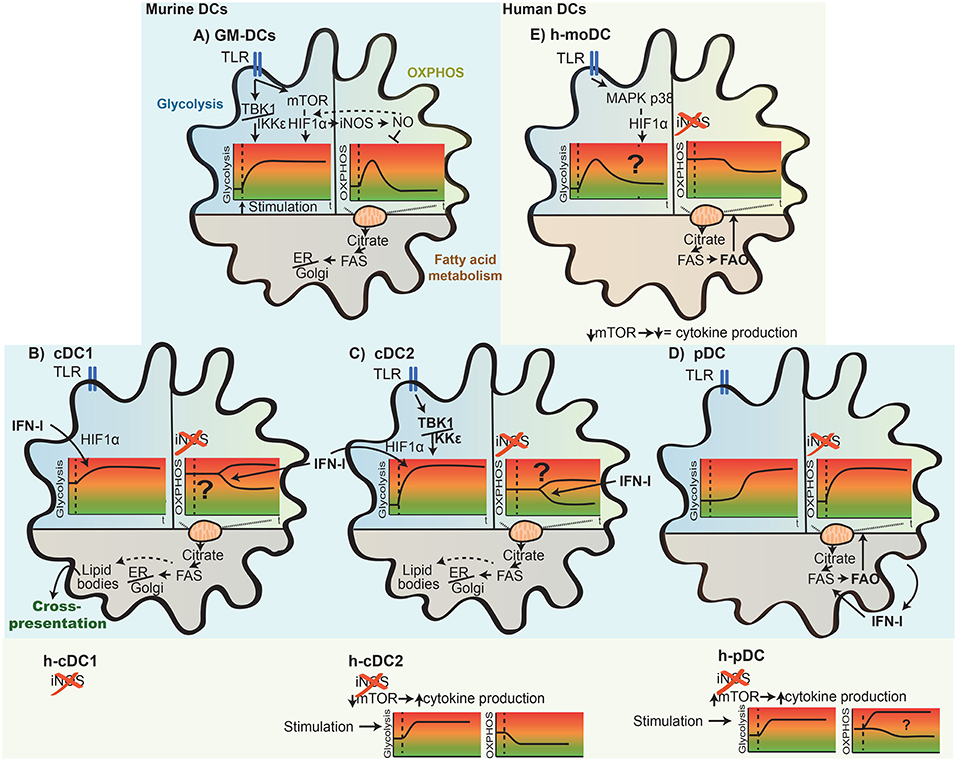
Figure 4. Differential metabolic rearrangement in mouse and human DC subsets upon activation. Depicted here are key adaptions of the main metabolic pathways [glycolysis, OXPHOS (oxidative phosphorylation), and fatty acid metabolism] of DCs upon TLR stimulation. The glycolytic and OXPHOS state of the cells over time (t) is indicated as a schematic representation. In GM-DCs (A), TLR stimulation leads first to induction of glycolysis, and later, mitochondrial OXPHOS is reduced (see also Figure 3). Whereas, this increase in glycolysis is consistently observed after stimulation, differences in the basal glycolytic state, promptness of the glycolytic induction, increased rate, and signaling factors driving these changes in distinct DC subsets are illustrated for naturally occurring mouse and human (h-) cDC1s (B), cDC2s (C), pDCs (D), and human in vitro-generated moDCs (h-moDCs; E). The impact of TLR stimulation on OXPHOS metabolism among DC subsets likely differs due to the lack of iNOS expression in naturally occurring DCs and h-moDCs. In addition, OXPHOS rearrangements of activated cDCs are context dependent and appear to be down-modulated in splenic cDCs in an IFN-I-dependent manner but remain high in cultured FLT3L-cDCs (B,C). An increase in fatty acid synthesis is generally ascribed to most DC subsets upon stimulation; however, differences in fatty acid use emerge, such as fuel of fatty acid oxidation to drive OXPHOS in h-moDCs and pDCs (D,E) or for organelle biosynthesis in GM-DCs, cDC1s, and cDC2s (A–C). In line, fatty acids can accumulate within DCs and form lipid bodies that associate with enhanced cross-presentation potential of cDC1s. Lastly, the thus far reported role of TBK1/IKKε and mTOR/HIF1α regulating cDC, pDC, and h-moDC metabolism and function upon activation is displayed.
Requirement of Glycolysis for Functions of Activated Dendritic Cells
Interrupting the glucose-to-pyruvate pathway significantly impairs DC maturation, upregulation of co-stimulatory molecules, cytokine secretion, and T cell stimulatory capacity in the long term (Figure 3). For example, pharmacological blockade of glycolysis using 2-deoxyglucose (2-DG), genetic deficiency of glycolytic enzymes such as α-enolase (ENO1), or overexpression of lactate dehydrogenase A (LDHA) or pyruvate dehydrogenase kinase 1 (PDK1) (Figure 2) prevents GM-DC maturation and immunogenicity upon stimulation with LPS or Chlamydia (13, 47, 49, 57) and can skew GM-DCs toward inducing Th17 and regulatory T cells (Treg) rather than Th1 and Th2 responses (49). In line, natural mouse cDC1s and cDC2s isolated from the spleen decrease expression of co-stimulatory molecules, IL-12 production, and activation of CD4+ and CD8+ T cells when activated by LPS in the presence of 2-DG (49). pRNA-stimulated human blood cDC2s require glycolytic activity for activation, evidenced by TNFα production, CD86, and programmed death ligand 1 (PD-L1) expression (53). Treatment of primary human pDCs with 2-DG upon influenza A virus stimulation also reduces co-stimulatory molecule and type I interferon (IFN-I) expression (52), while another study rather suggests induction of glutamine-fueled OXPHOS upon pRNA stimulation of human blood pDCs (53). However, the effects of inhibition of glycolysis by 2-DG in DCs have to be taken with caution, as 2-DG itself deregulates cytokine expression of human moDCs in vitro by activation of the endoplasmic reticulum (ER) stress response via the sensor inositol-requiring protein 1α (IRE1α) (50). In addition, 2-DG can impair the TCA cycle, OXPHOS, and ATP levels, as recently described in macrophages (58).
Other DC functions such as phagocytosis do not seem to be affected by inhibition of glycolysis during stimulation of human moDCs (50). However, reduced endocytic/phagocytic activity in aging mouse spleen cDC1s and DN-DCs [termed merocytic DCs (mcDCs)] and a resulting decline in antigen cross-presentation are linked to mitochondrial dysfunction with decreased basal OCR and ΔΨm as well as enhanced proton leakage and ROS. Importantly, inhibition of ATP synthase by oligomycin or the uncoupling agent carbonyl cyanide 4-(trifluromethoxy)phenyl-hydrazone (FCCP) corroborates the diminished phagocytosis of cDC1s and DN-DCs/mcDCs (22). Moreover, antigen uptake seems to decrease in GM-DCs in hypoxia, when glycolytic activity is increased by HIF1α stabilization, which is also observed in human moDCs after stimulation (47, 50).
In contrast, glucose and enhanced glycolytic activity are required for the ability of DCs to migrate (Figure 3). Independently of stimulation, glucose-deprived GM-DCs show reduced mobility, increased rounded morphology losing dendrites, and impaired oligomerization of CCR7, the chemokine receptor driving DC migration toward LNs. Subsequently, glucose limitation or 2-DG presence prevents migration of GM-DCs as well as splenic CD11c+ cDCs both in vitro and in vivo (49, 56). In line, HIF1α-deficient GM-DCs, which largely fail to induce glycolysis (see the section Sustained Glycolysis: The Role of HIF1α), display reduced CCR7 levels, and GM-DCs differentiated in hypoxic conditions exhibit elevated migratory potential in vitro and in vivo that is dependent on HIF1α (14).
Overall, early induction of glycolysis emerges as a general feature of immunogenic activation of most cultured DCs and primary DC subsets and appears necessary for several aspects of their maturation such as upregulation of co-stimulatory surface molecules and cytokine production, despite having no major effects on phagocytosis or antigen uptake. However, DC activation leads to cytoskeletal changes that support increased migratory capacity to migrate toward LNs and T cell zones, which is also affected by early induced glycolysis. Ultimately, in light of those findings, glycolytic increase in DCs upon stimulation is vital for adequate induction of adaptive T cell responses (59) and, hence, regulates immune homeostasis (Figure 3).
Mechanisms That Control Glycolytic Reprogramming in Activated Dendritic Cells
Fuels for Glycolytic Induction Upon Dendritic Cell Stimulation
Extracellular glucose consumption by DCs is required for some aspects of induction of glycolysis, functionality, and survival in activated DCs (13, 56). However, glucose uptake and its effects on DC activation emerge to be time and DC subset dependent. Expression of glycolytic enzymes is not increased in GM-DCs at 4 or 8 h after LPS, HDM, curdlan, or zymosan stimulation (56, 60), when cells already display an enhanced glycolytic activity (56), but is only detectable 18–24 h after stimulation. Moreover, switching GM-DCs from a glucose-containing to a galactose-containing medium, which only supports a low glycolytic rate, 8 h after LPS stimulation actually enhances co-stimulatory molecule expression, IL-12 production, and their potential to activate CD8+ T cells, which is ascribed to deregulation of the mTORC1/HIF1α network (60) (Figure 3). Indeed, increased glycolysis may be preferentially supported by glycogenolysis of intracellular glycogen reserves during the first 6 h post-stimulation of DCs, rather than extracellular glucose (46). GM-DCs activated with LPS or IL-4 during differentiation accumulate intracellular glycogen, which correlates with their enhanced T cell stimulation potential (61). At later stages after GM-DC stimulation, extracellular glucose uptake is enhanced via the upregulation of glucose transporters such as the glucose transporter 1 (GLUT1) (13, 46, 56), and GLUT1 inhibition 24 h after LPS stimulation reduces CD40 and CD86 expression (46). Of note, expression levels of GLUT1 might be a suboptimal readout for its induction or activity. In fact, GLUT1 is translocated from intracellular vesicles to the cell membrane for glucose uptake upon LPS stimulation in macrophages, which does not entirely correlate with mRNA expression levels (62). Moreover, a significant amount of glucose imported from the extracellular environment by activated DCs still appears to be metabolized to glycogen first before entering glycolysis (glycogen shunt; Figure 2) (46). Additionally, upon 6 h pRNA stimulation of primary human blood cDC2s, glycolytic metabolism appears to rely on BCL2 interacting protein 3 (BNIP3)-dependent mitophagy, despite reported 2-DG-sensitive glucose uptake and ENO2 upregulation (53).
Early Glycolytic Induction: The TBK1/IKKε/AKT/HK-II Axis
Glycolytic reprogramming upon activation of DCs appears to be largely driven by TANK-binding kinase-1 (TBK1)/IκB kinase-ε (IKKε)/AKT/hexokinase (HK)-II activation in the short term and regulated by AMPK loss and induction of mTOR and/or HIF1α in the long term (Figure 3). TBK1 and IKKε, both non-canonical IκB kinase homologs downstream of TLRs, are activated in GM-DCs within minutes after LPS stimulation, leading to PI3K-independent AKT phosphorylation and association of the rate-limiting glycolytic enzyme HK-II with mitochondria. These events promote glycolytic flux and support early induction of glycolysis in LPS-stimulated GM-DCs as well as in primary mouse spleen cDC2s ex vivo (49) (Figure 4). Indeed, early induction of TBK1, AKT, and mTORC1 occurs upon stimulation with potent and weak stimuli, correlating with early increase in glycolytic activity (56). LPS-stimulated human moDCs in vitro also enhance HK-II expression and activity in concert with enhanced glycolysis and cytokine production in the long term; however, HK-II induction and glycolysis in this setting appear to rely on HIF1α activity mediated by p38/mitogen-activated protein kinase (MAPK; Figure 4). Nevertheless, this p38/MAPK/HIF1α axis does not seem to be involved in enhanced glycolysis by human moDCs after TLR2/6-mediated activation but relies on TBK1 (51). Notably, HK-II itself can act as a PRR and cause inflammasome activation (63).
Glycolytic Reprogramming: AMPK vs. the PI3K/AKT/mTOR Pathway
Loss of AMPK and induction of the PI3K/AKT/mTOR pathway (Figure 1) at longer time points after LPS stimulation of GM-DCs (18–24 h) ultimately lead to upregulation of glycolytic enzymes such as LDHA, pyruvate kinase 2 (PKM2), or phosphofructokinase (PFK), as well as glucose transporters like GLUT1 (13, 56), which depend on glucose availability (60) (Figure 3). Mechanistically, inactivation of AMPK occurs upon LPS stimulation, alleviating mTORC1 inhibition (13, 60). In line, activation of AKT, mTORC1, and mTORC2 declines 18 h after weak stimulation of GM-DCs hand in hand with loss of increased glycolysis activity (56). Enforced AMPK activation or inhibition/loss can prevent or foster GM-DC maturation, respectively (13, 33), associating active AMPK with diminishing proinflammatory DC functions (59). Human pRNA-activated cDC2s downregulate AMPKα1 levels, which appears to be dependent on mitophagy in this system (53). A reduction in glycolysis and activation of GM-DCs upon early inhibition of glycogenolysis also associate with a rapid drop in intracellular ATP and AMPK activation (46). Inhibition of mTOR/mTORC1 blunts glucose consumption, lactate production, upregulation of glycolytic enzymes/glucose transporters, and increased extracellular acidification rate (ECAR) in GM-DCs 20 h or longer after LPS stimulation (60, 64). Hence, mTOR activation appears to control DC activation, especially maintaining it for prolonged periods of time (43) (Figure 3). Indeed, ectopic AKT/PKB activation, which sustains mTOR activation, enhances co-stimulatory molecule expression and cytokine secretion in human pDCs (32). Also, mTOR signaling is essential for induction of IFN-I responses of (primary) mouse and human pDCs (65). In concert, rapamycin treatment of anti-CD40-stimulated GM-DCs in vitro or IL-4-treated spleen CD11c+ DCs in vivo downregulates co-stimulatory molecules/cytokines and promotes activation of Tregs, but not allogeneic CD4+ T cells (30, 66).
Nevertheless, sustained mTOR signaling may also be detrimental for proinflammatory DC functions (23, 41, 59, 67). For example, knockdown or pharmacological inhibition of mTOR enhances life span, prolongs the expression of co-stimulatory molecules, cytokine production, and promotes T cell stimulatory activity of LPS-stimulated GM-DCs (64, 68, 69) (Figure 3). Indeed, mTOR promotes NO production by activated GM-DCs, which limits their mitochondrial energy metabolism, while mTOR inhibition restores the metabolic flexibility of those cells in the long term (68) (Figures 3, 4). However, loss of the negative mTORC1 regulator TSC1 in mouse DCs causes impaired cytokine production and antigen presentation upon TLR4 stimulation (36). mTORC1 inhibition in human CD1c+ cDC2s enhances proinflammatory cytokine production upon stimulation with various agents but has the opposite effect on LPS-stimulated human moDCs. Those contrasting effects are ascribed to differential activation of NFκB upon mTORC1 blockade, which increases in LPS-stimulated CD1c+ cDC2s but remains unchanged in moDCs (2). A spatiotemporal model to integrate the ambiguous roles of mTOR regulating DC functions has been proposed (23).
Sustained Glycolysis: The Role of HIF1α
HIF1α stabilization is also involved in enhanced glycolytic activity of GM-DCs and human moDCs upon stimulation in vitro and of natural mouse cDCs in vivo (15, 47, 51) (Figures 3, 4). Many glycolytic genes are HIF1α targets and are downregulated in DCs upon HIF1α loss, such as GLUT1 and LDHA (47, 50, 54, 60). Moreover, only potent GM-DC stimulation that leads to long-term induction of glycolysis causes HIF1α stabilization and induction of its target genes, while weak activation fails to do so (56). In line, GM-DCs in the steady state express higher MHCII and co-stimulatory molecule levels in hypoxic conditions (14). LPS stimulation of GM-DCs in hypoxia compared with normoxia further elevates HIF1α activation, glucose consumption, glycolytic enzyme expression, and lactate and ATP production, enhancing GM-DC activation (47). Similar effects are also observed upon in vitro Aspergillus fumigatus stimulation of human moDCs in hypoxia in vitro (54). Inhibition or loss of HIF1α in GM-DCs or human moDCs prevents the increase in glycolytic rate and upregulation of glycolytic genes upon LPS or Aspergillus fumigatus stimulation and reduces co-stimulatory molecule expression, proinflammatory cytokine production (including IL-12), and CD4+ T cell stimulatory capacity in the long term (47, 51, 54). However, LPS-stimulated HIF1α-deficient GM-DCs show enhanced IL-12 expression and CD8+ T cell activation (60). Hence, further efforts will be necessary to clarify the exact role of HIF1α on DC functions. Nevertheless, spleen CD11c+ MHCII+ cDCs of mice lacking HIF1α in CD11c-expressing cells also fail to induce higher glycolysis and display reduced immunogenicity 14–18 h after poly(I:C) stimulation. However, some of those effects might be ascribed to elevated death of HIF1α-deficient spleen cDCs (15). Importantly, HIF1α can be induced or stabilized by many other mechanisms apart from mTORC1 or hypoxia, such as glucose withdrawal (60), which might differentially influence the effects on immunogenic DC activation. Moreover, HIF1α can be activated by intracellular pyruvate or lactate produced by glycolysis (70, 71). Indeed, the timing of HIF1α stabilization occurring in human moDCs 4 h after LPS or zymosan stimulation trails the immediate increase in glycolysis (50). Notably, weakly stimulated GM-DCs do not accumulate HIF1α while still inducing early glycolysis, in contrast to strongly activated GM-DCs that stabilize HIF1α and maintain high glycolytic activity at later stages (56). Taken together, HIF1α is implicated in the maintenance rather than in the early induction of glycolysis after DC stimulation (50) (Figures 3, 4) and appears to partially depend on glucose availability (60).
Extracellular Cues Influencing Glycolytic Metabolism of Activated Dendritic Cells
Signals in the microenvironment can strongly influence DC function via modulating their glucose metabolism. For example, the anti-inflammatory cytokine IL-10 inhibits the LPS-mediated increase in glycolysis and GM-DC maturation likely via maintaining active AMPK (13), and IL-10-deficient GM-DCs display higher levels of the glycolytic enzyme ENO1 (57). Similarly, IL-10 loss in macrophages causes enhanced glycolytic reprogramming upon LPS stimulation, which is ascribed to mTORC1 inhibition by autocrine IL-10 via signal transducer and activator of transcription 3 (STAT3) and DNA damage inducible transcript 4 (DDIT4). Notably, they also accumulate dysfunctional mitochondria due to reduced autophagy independent of NO (62).
Metabolic reprogramming of mouse spleen cDCs may rely on type I IFNs in concert with PRR signaling, as IFNα/β receptor (IFNAR)-deficient cDCs fail to elevate glycolytic activity after poly(I:C) stimulation in vivo while maintaining active OXPHOS (15), and mouse pDCs from FLT3L-DC cultures increase their OCR and ECAR upon 24 h exposure to IFNα (72) (Figure 4). However, IFNα treatment or IFNAR inhibition in primary human blood pDCs ex vivo does not affect induction of glycolysis after stimulation with influenza A virus (52).
Last, exogenous metabolites such as fatty acids or lactate are sensed by DCs, leading to an adaption of their metabolism and functions [reviewed in Pearce and Everts (41)], such as lactate-mediated effects on HIF1α (70, 71, 73). For example, the short-chain fatty acid butyrate can prevent maturation and glycolytic reprogramming of human moDCs upon LPS stimulation, driving them to induce Tregs (74).
Fatty Acid Synthesis and ER Stress During Dendritic Cell Activation
Generation of TCA cycle intermediates regulates function and de novo FAS upon DC stimulation. Indeed, while glycolysis-derived ATP appears to be dispensable for early GM-DC activation, incorporation of pyruvate into the mitochondrial TCA cycle is essential, as knockdown of the mitochondrial pyruvate carrier MPC-1 (Figure 2) limits GM-DC maturation and cytokine production (49). Accumulation of TCA intermediates such as citrate, succinate, and fumarate in stimulated DCs contributes to the regulation of inflammatory responses as well as cytokine production (45, 75). Additionally, citrate escaping the mitochondria serves as an important substrate for protein acetylation, nicotinamide adenine dinucleotide phosphate (NADPH) production, and, importantly, cytosolic FAS in activated DCs (49, 75) (Figures 2, 3). In addition, knockdown of the PPP enzyme glucose-6-phosphate dehydrogenase (G6PDH) reduces LPS-induced maturation of GM-DCs (49). The PPP produces ribose 5-phosphate (R5P), a precursor for biosynthesis of nucleotides, and NADPH, which is needed for production of ROS and NO as well as for cytosolic FAS (Figure 2).
De novo FAS and accumulation of phospholipids increase upon GM-DC stimulation with LPS (49) and after activation of in vitro bone marrow-derived cDC1-like cells (iCD103; Table 2) with LPS, CpG, and Mycobacterium bovis Bacille Calmette Guerin (BCG) (76) (Figure 4). Indeed, accumulation of intracellular fat in LPS- or IL-4-stimulated GM-DCs correlates with enhanced T cell activation capacity (61). FAS also leads to increased lipid storage in lipid bodies (LBs) in GM-DCs (49), organelles composed of a core of neutral lipids such as cholesteryl esters or triglycerides (TAG) surrounded by a single layer of phospholipids (77). Notably, intracellular LB formation associates with induction of cross-presentation potential in GM-DCs, FLT3L-DCs, and mouse spleen cDCs that is at least partially dependent on inflammasome activation or IFNγ-induced protein immunity-related GTPase family member m3 (Irgm3) (78, 79). Accordingly, the specialized cross-presenting CD8+ cDC1 subset (Table 1) in the spleen harbors more LBs than CD8– cDCs (79). Human and mouse liver DCs with high lipid content are more potent activators of NK, CD4+, and CD8+ T cells, which is reduced by inhibition of FAS (80). In line, FAS blockade in GM-DCs by knockdown of the mitochondria–cytosol citrate shuttle citrate transport protein (CTP) or by the FASN or ACC inhibitors C75 and TOFA (Figure 2) prevents LPS-induced activation and proinflammatory functions of GM-DCs (49). However, non-activated GM-DCs or human moDCs differentiated in the presence of TOFA show high levels of ER stress, ERK and AKT signaling, and PPARγ expression, linked to enhanced DC immunogenicity and T cell priming (7). In the iCD103 culture system that rather represents cDC1-like DCs (Table 2), deficiency in ACC1 or 2 or their inhibition by TOFA does not affect co-stimulatory surface marker expression and their inflammatory cytokine profile upon CpG or Mycobacterium bovis BCG stimulation. T cell priming capacity or in vivo mycobacterial control of iCD103 DCs also remains unaffected by interference with FAS (76). Of note, FAS impairment in iCD103s also results in enhanced uptake of extracellular fatty acids, which might represent a compensatory mechanism for fatty acid generation. Nevertheless, the actual role and subsequent usage of fatty acids produced by DCs appear to be dependent on the context and DC subsets (Figure 4). For example, de novo synthesized fatty acids provide building blocks for expansion of the Golgi apparatus and the ER in LPS-stimulated GM-DCs and are ultimately required for activated DCs to produce and secrete large amounts of cytokines, which can lead to ER stress and the unfolded protein response (41, 49). Liver DCs containing high amounts of lipids have an increased ER stress, and its blockade reduces their ability to induce immune responses (80). Indeed, ER stress can enhance IL-23 production in zymosan-stimulated human moDCs via IRE1α and X-box binding protein 1 (XBP1) (50). In contrast, in mouse pDCs sorted from FLT3L-DC cultures, an increase in ECAR late after CpG or IFNα stimulation associates with enhanced FAS, which, in this setting, serves as a source of fatty acids for FAO to maintain high OXPHOS levels (72) (Figure 4).
Overall, regulation of ER stress and lipid metabolism in activated DCs can notably influence their function to release cytokines and to present antigen (41, 81), and further efforts will be needed to understand the precise functions in different settings. In that regard, the importance of de novo FAS and lipid accumulation in tolerogenic or dysfunctional DCs in cancer is discussed in the section Lipid Accumulation and Dendritic Cell Dysfunction in Cancer.
Mitochondrial Energy Generation Regulating Dendritic Cell Activation—Specific to Dendritic Cell Subsets and the Context
Mouse GM-CSF Dendritic Cell Cultures
Development of natural DCs largely relies on FAO to fuel OXPHOS (see the section Metabolic Control of Dendritic Cell Development). However, in cultured GM-DCs, mitochondrial energy metabolism is dramatically reduced upon immunogenic stimulation in the long term (13) (Figures 3, 4). Indeed, the FAO inhibitor etomoxir, the glutaminolysis inhibitor 6-diazo-5-oxo-L-norleucine (DON), or glutamine deprivation has no effect on GM-DC maturation upon LPS stimulation (46, 49). Furthermore, GM-DCs display irresponsiveness to ETC inhibitors and exhibit decreased OCR and ΔΨm 18 or 24 h post-LPS stimulation, which is independent of PI3K/AKT signaling (13, 48). The production of NO via the enzyme inducible NO synthase (iNOS) is central to the collapse of OXPHOS of activated GM-DCs in the long term and their functions (48) and was recently reviewed (82). In brief, NO is induced in GM-DCs within 6 h after LPS stimulation, and their enhanced glycolytic rate becomes NO dependent about 9 h after stimulation, when OXPHOS declines (49). Stabilized HIF1α enhances NO generation by increasing the expression of iNOS, which, in turn, leads to the inhibition of prolyl hydroxilases (PHDs) that label HIF1α for degradation. This positive loop causes NO accumulation, which leads to nitrosilation of some ETC complexes and inhibits their functionality (48, 60, 82) (Figures 3, 4). A small proportion of mouse moDCs induced by Listeria monocytogenes infection also display a comparable NO-mediated inhibition of OCR late after stimulation that is compensated by enhanced glycolysis (48). Based on those and other studies in tolerogenic DCs (see the section DC Metabolism in Tolerance), anabolic metabolism, and glycolysis are generally associated with immunogenicity of DCs, while catabolic metabolism and active mitochondrial respiration, regulated via AMPK/PGC1α, are related to tolerogenicity of DCs (41, 43, 83).
However, several pieces of evidence point toward a potential role of mitochondrial energy metabolism and functional OXPHOS in immunogenic, activated DCs. Indeed, ΔΨm and OCR are actually increased in GM-DCs in the short term up to 6 h after LPS stimulation before iNOS becomes expressed (Figure 4), which is prevented by 2-DG (48, 49), and weak stimuli like HDM or ZymD do not reduce mitochondrial respiration 18 h post-activation (56). Moreover, decreased mitochondrial abundance is usually not associated with NO-mediated OXPHOS inhibition upon GM-DC activation (13), and 24 h LPS-activated GM-DCs or mouse moDCs fully restore their mitochondrial respiratory profile when NO production is diminished (48). Also, ENO1 loss causes a profound dysregulation of mitochondrial morphology in short-term (2 h) Chlamydia-stimulated GM-DCs associated with a drop of intracellular pyruvate levels and enhanced cell death (57). Additionally, antiviral responses of DCs promoted by cytoplasmic RNA sensor RIG-I-like receptor (RLR) signaling depend on the mitochondrial localization of the antiviral signaling protein (MAVS), which requires active ΔΨm (41).
These observations suggest that mitochondrial energy generation contributes to DC activation in certain settings. Indeed, deficiency or inhibition of iNOS in LPS-activated GM-DCs maintains active OXPHOS and even enhances aspects of DC activation, such as CD8+ T cell stimulation and CD86 and MHC molecule expression in the long term (48) (Figure 3). The presence of the mTORC1 inhibitor rapamycin attenuates NO production and ameliorates the decrease in mitochondrial-dependent OCR in activated GM-DCs (60, 68). The maintenance of functional OXPHOS permits the cells to use FAO and glutaminolysis for energy generation (68). Also, the culture of LPS-activated GM-DCs in galactose enhances OCR, while ECAR levels plummet (60). Indeed, in the long term, rapamycin-treated or galactose-cultured activated GM-DCs display a prolonged life span together with extended co-stimulatory molecule and IL-12 expression that leads to more potent activation of CD8+ T cells, which is at least partially dependent on suppression of HIF1α/iNOS signaling (60, 64, 68) (Figure 3).
Natural Mouse and Human Dendritic Cell Subsets
Crucially, contrary to cultured GM-DCs, most DC subsets present in lymphoid organs do not express detectable levels of iNOS, foremost naturally occurring cDC1s and cDC2s, as well as cultured human moDCs (82) (Tables 1, 2 and Figure 4). In line, mitochondrial energy metabolism and OXPHOS remain intact in in vitro LPS- or zymosan-stimulated human moDCs (50). Also, splenic mouse cDC1 and cDC2 increase their ECAR shortly after in vivo LPS stimulation (49); however, notably, they do not display any differences in the ECAR/OCR ratio 24 h after ex vivo LPS stimulation (48) (Figure 4). Uptake of dead cell material and cross-presentation potential of unstimulated natural mouse spleen cDC1s and DN-DCs/mcDCs (Table 1) are diminished upon abrogated mitochondrial function caused by aging or ETC inhibition (22). Conversely, 14 h in vivo poly(I:C) stimulation reduces ΔΨm and OCR of total spleen cDCs, which is prevented by IFNAR deletion (15) (Figure 4), suggesting an additional context-mediated mechanism. In the same study, maintenance of mitochondrial energy metabolism and reduction in ECAR upon poly(I:C) stimulation by HIF1α loss in spleen cDCs reduce their T cell activation potential. However, this effect is ascribed to unbalancing cellular metabolism leading to enhanced ROS production, lower ATP levels, and increased cell death (15). Nevertheless, in 6 h-stimulated human blood cDC2s, mitochondrial morphology and dynamics are altered, the OCR is strongly reduced, and BNIP3-dependent mitophagy is triggered, which appears necessary for glycolytic activity and activation (53).
Notably, pDCs appear to show distinctive rewiring of their mitochondrial energy metabolism in different settings (Figure 4). While human pDCs mildly decrease their OCR after 24 h ex vivo influenza or rhinovirus infection (52), they elevate OXPHOS 6 h post-pRNA stimulation, which appears to be mediated by autophagy-induced glutaminolysis (53). Importantly, the induction of mitochondrial energy metabolism in human pDCs is required for the production of IFNα, CD80, and PD-L1 expression (53). Mouse pDCs sorted from FLT3L-DC cultures enhance mitochondrial pyruvate import and FAO that fuel elevated OXPHOS 24 h post-CpC stimulation. This effect is due to IFN-I induction, with IFNα itself promoting FAO via PPARα (72), in contrast to mouse spleen cDCs where IFNAR deficiency maintains high OCR (15) (Figure 4).
Hence, no general conclusion can be reached as to the importance and function of mitochondrial energy metabolism, OCR, and OXPHOS in activated DCs, and it appears context and DC subset dependent (Figure 4). Metabolic flexibility of activated DCs to switch their carbon source for ATP generation from glucose to galactose, glutamine or fatty acids would be of benefit in DC function and indeed, prevention of OXPHOS collapse and metabolic plasticity enhance DC survival and activation upon glucose deprivation and mTOR or iNOS inhibition (48, 60, 68). In the future, it will be interesting to determine the influence of the microenvironment in which DCs are activated. Not only nutrient or oxygen availability but also other environmental factors can strongly influence DC metabolism, such as extracellular lactate, fatty acids (41), the TCA intermediates citrate, succinate, and fumarate (45, 75), as well as IL-10 (13, 57) or IFN-I (15, 52, 72), as discussed in the section Extracellular Cues Influencing Glycolytic Metabolism of Activated Dendritic Cells. Moreover, NO produced by neighboring cells can cause HIF1α stabilization and trigger a cellular loop in DCs, leading to a glycolytic switch (60, 68).
Development and maintenance of different DC subsets display differential metabolic requirements (discussed in the section Metabolic Control of Dendritic Cell Development), which will likely reflect on their metabolic reprogramming upon activation. Considering that different DC subsets specialize on distinct functions (Table 1), their metabolic requirements to exert those tasks might differ, as suggested in a recent study (53). Moreover, a fine regulation of OXPHOS activity, such as reported in the case of supercomplex assembly in macrophages (84, 85), may also have a functional effect on DCs.
Dendritic Cell Metabolism in Tolerance
DCs contribute to the maintenance of immunological tolerance in order to prevent hyperactivation of the immune system and subsequent autoimmune diseases. Generally, such tolerogenic DCs arise in the steady state during uptake of (self-) antigen in the absence of danger signals, upon sensing of anti-inflammatory cytokines/factors, and during various pathological states, including cancer, due to tolerizing signals (86, 87). Tolerogenic or semimature DCs can be identified by upregulation of regulatory surface molecules or receptors such as PD-L1 and tolerogenic cytokines IL-10, IL-27, and TGFβ, leading to induction of Treg activation at the expense of effector T cells (83, 86). Much of the functionality of tolerogenic DCs is intertwined with metabolic activity, such as lipid accumulation or catabolism of amino acids [tryptophan (Trp) and arginine (Arg)].
Metabolic State(s) and Their Regulatory Cellular Pathways in Tolerogenic Dendritic Cells
Metabolic Adaptions of Tolerized Dendritic Cells
Our understanding of energy metabolism of tolerogenic DCs is largely based on observations in human moDC cultures treated with vitamin D3 or D2 (VitD3 or VitD2), dexamethasone (DEX), and/or resveratrol (83, 88–91). Resveratrol is a plant-derived polyphenol that induces regulatory properties in mouse and human DCs, preventing their maturation and immunogenic activation (92, 93). Glucocorticoid receptor engagement by DEX modulates many aspects of DC maturation, including antigen presentation and cytokine production, leading to a tolerant phenotype (83, 94, 95). VitD3 skews DC functionality toward an inhibitory phenotype inducing Tregs and enhancing expression of inhibitory receptors (96, 97).
Tolerogenic human moDCs, generated either by treatment with DEX+VitD3 for 48 h or 1,25-dihydroxyvitamin D3 [1,25(OH)2-VitD3, the active form of VitD3] for 24 h, exhibit enhanced catabolism and metabolic plasticity, increased expression of genes involved in OXPHOS, glycolysis/glucose metabolism, and FAO in concert with higher mitochondrial respiration (OCR) and glycolytic activity (ECAR) than untreated moDCs (88, 89). Intriguingly, LPS stimulation of DEX+VitD3-tolerized moDCs slightly decreases their OXPHOS capacity; however, their glycolytic capacity drops to levels of immunogenic LPS-stimulated DCs, which are, in this study, lower than those of untreated moDCs (88). Functionally, while MHCII expression of LPS-stimulated immunogenic moDCs is sensitive to glycolysis inhibition, LPS-stimulated DEX+VitD3-tolerogenic moDCs remain unaffected. DEX+VitD3-tolerogenic moDCs increase their MHCII levels upon inhibition of FAO instead. In line, FAO inhibition rescues the ability of DEX+VitD3-tolerogenic moDCs to induce expression of activation markers on CD4+ T cells upon LPS stimulation (88). Moreover, in the context of melanoma, a Wnt5a/β-catenin and PPARγ pathway induces FAO and a tolerogenic indoleamine 2,3-dioxygenase (IDO)-producing and Treg-activating phenotype in DCs (98). In contrast, in moDCs tolerized by DEX+VitD2, immunogenic stimulation induces even higher glycolysis/cellular LDH activity than in activated moDCs (91). Nevertheless, the maintenance of tolerogenic features of both 1,25(OH)2-VitD3-treated and (re-stimulated) DEX+VitD2-treated moDCs relies on glycolysis, and their tolerogenic phenotype is abrogated by 2-DG treatment (89, 91). Notably, levels of FAO are unaltered in 1,25(OH)2-VitD3-treated vs. control moDCs, and FAO inhibition by etomoxir does not affect their tolerogenic hallmarks (89). Accumulation of pyruvate during glycolysis may partially cause the concomitant increase in OXPHOS in those tolerogenic moDCs in concert with elevated OXPHOS-related gene expression (89). Those results indicate a metabolic plasticity and responsiveness of tolerogenic moDCs, which display a very active metabolism, despite showing differential dependencies on glycolysis vs. FAO/OXPHOS. Those controversies may be due to the different experimental settings, presence or absence of immunogenic stimulation, and the fact that 1,25(OH)2-VitD3 has stronger effects on OXPHOS, lipid, and glucose metabolism of tolerogenic moDCs than DEX (99). Nevertheless, tolerogenic DCs appear to rely less on glycolysis than LPS-activated immunogenic DCs for their functionality and, as they largely upregulate functional OXPHOS, might be able to adapt their metabolism depending on the context. However, those conclusions are solely based on cultured human moDCs, and the metabolism of other tolerized DC subsets in complex in vivo settings largely remains to be investigated.
AMPK and mTOR Pathways Influence Tolerogenicity of Dendritic Cells
The tolerogenic status of DCs is also influenced by a balance of the nutrient-sensing pathways AMPK and mTOR, which appear to be equally context dependent as for immunogenic stimulation of DCs. Inflammatory activation of DCs involves enhanced glycolytic activity and anabolic metabolism compared to immature DCs that largely appear to be controlled by mTOR signaling (see the section Mechanisms That Control Glycolytic Reprogramming in Activated Dendritic Cells), and, despite controversial findings (64, 68), mTOR inhibition by rapamycin can cause DC tolerization (30, 66). Accordingly, DEX or resveratrol treatment of macrophages can block iNOS expression and NO generation (100, 101), whose upregulation associates with LPS-activated GM-DCs (82), while VitD3 had varying effects (102). Indeed, an axis involving AMPK, PGC1α, and PPARγ is suggested to control tolerogenicity of DCs, largely by preventing biosynthetic metabolic adaptions or pathways driving immunogenic DC activation such as mTOR (41, 43, 59, 83). This concept is founded on the observations that tolerogenic DCs show enhanced mitochondrial respiration and that AMPK activation favors catabolic metabolism, FAO, and OXPHOS, largely via PPARγ and the mitochondrial biogenesis inducer PGC1α (41, 43, 59, 83). Indeed, DEX+VitD3- and 1,25(OH)2-VitD3-treated human moDCs upregulate AMPK activity and signaling (88, 89), human cDC2s (53) and GM-DCs reduce AMPK activation upon pRNA or LPS exposure, and the inhibitory effect of IL-10 on LPS-mediated maturation of mouse GM-DCs appears to be AMPK dependent (13). Further, AMPKα1-deficient LPS-stimulated GM-DCs show augmented proinflammatory features such as enhanced co-stimulatory molecule expression and CD40 signaling, increased IL-6 and TNFα, but decreased IL-10 production and skewing of CD4+ T cell activation toward a Th1 and Th17 phenotype (33). The AMPK inducer 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) is equally potent in blocking glucose consumption by LPS-stimulated GM-DCs as 2-DG (13) and AMPK activation after uptake of dead cells induces autophagy, tolerogenic properties, and reduced anti-tumor immune responses (103). Intriguingly, several studies also implicate VitD3, resveratrol, and DEX in enhancing AMPK activation in various other settings and cell types (104–110). Moreover, resveratrol treatment promotes OXPHOS and mitochondrial biosynthesis in mice and humans via mechanisms similar to AMPK, such as activating the histone deacetylase Sirtuin 1 and augmenting PGC1α expression (90, 111). Loss of the PGC1α targets PPARγ or nuclear factor erythroid 2-related factor 2 (NRF2) enhances DC maturity and proimmunogenic functionality (41).
However, the precise role of balanced mTOR/AMPK signaling in tolerogenic DCs remains controversial. Indeed, the PI3K/AKT/mTOR axis is reported to be vital for tolerogenic features of moDCs, independent from AMPK (89, 91). Human restimulated DEX+VitD2-tolerized moDCs strongly upregulate mTOR phosphorylation and signaling compared to non-tolerized controls (91). PI3K or mTOR inhibition (by LY294002 or rapamycin, respectively) enhances MHC and co-stimulatory molecule expression and reduces co-inhibitory molecules as well as the IL-10/IL-12p70 expression ratio by 1,25(OH)2-VitD3-treated and DEX+VitD2-treated moDCs without or after immunogenic activation. Induction of CD4+ and CD8+ T cell proliferation and IFNγ production is also enhanced by mTOR inhibition in both tolerogenic human moDC cultures (89, 91). Importantly, in this setting, AMPK activation by AICAR is ineffective in altering the tolerogenic phenotype of 1,25(OH)2-VitD3-treated moDCs (89). Moreover, the context dependence of cellular metabolism associated with active mTOR signaling is highlighted by a recent study of allergic airway inflammation in mice harboring mTOR-deficient CD11c-expressing cells (31). There, HDM exposure induces the generation of lung CD11c+ MHCII+ CD11b+ DCs that depend on macrophage CSF (M-CSF) and, hence, likely represent moDCs (Table 1). Upon loss of mTOR, those induced CD11b+ DCs show enhanced expression of CD80 and CD86 co-stimulatory molecules and skew the HDM-mediated Th2-polarized allergy toward a neutrophilic Th17-mediated lung inflammation. Moreover, mTOR-deficient CD11b+ DCs accumulate fatty acid metabolites, and FAO inhibition by etomoxir diminishes their activated phenotype (31). Those observations suggest anti-inflammatory/tolerizing effects of mTOR associated with inhibition of FAO that, in turn, appears functionally important for an activated state and Th17 polarization capacity of lung CD11b+ inflammatory DCs in allergic airway inflammation.
In summary, research on primary DC subsets in settings of immune tolerance, additional to tolerized DC cultures, will be needed to advance our knowledge on tolerogenic DC metabolism.
Lipid Accumulation and Dendritic Cell Dysfunction in Cancer
The role of lipid metabolism for immunogenic and tolerogenic DC function is ambiguous. Although lipid accumulation in DCs seems to support immunogenic immune responses and cross-presentation (78, 79) (see the section Fatty Acid Synthesis and ER Stress During Dendritic Cell Activation), it also associates with DC dysfunction in tumor settings. Tumor-associated DCs accumulate high amounts of cytosolic lipids in both mice and humans. Lipid-laden DCs isolated from tumor-bearing mice exhibit defective T cell stimulation ability due to altered antigen processing and presentation (112). The aberrant lipid accumulation in DCs is fostered by yet-unknown factors secreted by tumor cells and mediated by macrophage scavenger receptor 1 (Msr1) on DCs (112), a receptor that binds primarily modified lipoproteins (113). Inhibition of Msr1 or blockade of FAS with TOFA restores lipid content and DC immunogenicity, indicating that enhanced lipid uptake, FAS, or a combination impairs DC-mediated antitumor immunity. Interestingly, this effect is observed in cDC1s and cDC2s but not in pDCs (112), which might be a reflection of the different functions and/or metabolic pathway usage among DC subsets in vivo (Table 1 and Figure 4). Indeed, CD103+ cDC1s from draining LNs (dLNs) of tumor-bearing mice accumulate more LBs compared to the CD103– DC counterparts, which substantially reduces their ability to cross-present antigens (114). Cross-presentation plays a central role in the generation of efficacious anticancer CD8+ cytotoxic T cell responses (115), and these data provide a metabolic explanation for the impaired ability of tumor-infiltrating DCs to induce potent antitumor adaptive responses.
The differential effect of lipid accumulation in DCs seen in tumor settings may be due to accumulation and/or signaling by modified lipid species. For instance, tumor-derived factors act on DCs activating liver X receptor (LXR)-α signaling, whose natural ligands are oxidized cholesterol (oxysterols), and reduce the expression of CCR7, inhibiting their migration to the dLNs (116). Consistently, LXR-α/LXR-β-deficient GM-DCs show impaired migration in response to the CCR7 ligands CCL19 and CCL21, and this response is partially dependent on the LXR target CD38, a molecule that is linked to leukocyte trafficking (117). Oxidized lipids contained in tumor-associated DCs also affect cross-presentation (118). Accumulation of oxidized polyunsaturated fatty acids, cholesterol esters, and TAG impairs cross-presentation without affecting the presentation of endogenous antigens. Notably, the accumulation of non-oxidized lipids does not alter cross-presentation, supporting the idea that it is not the mere storage of lipids but the accumulation of modified lipids that alters DC function (114, 118). Consistent with these observations, tumor-derived factors trigger lipid peroxidation in tumor-associated DCs, which activates the ER stress response mediated by IRE-1α and its target XBP1. XBP1 activation, in turn, induces a lipid biosynthetic program that results in the accumulation of LBs and blunted antigen presentation, leading to a reduced ability to control tumor growth (119). Regarding the mechanisms by which LBs and modified lipids could impair cross-presentation, oxidatively truncated TAG accumulate on the surface of LBs and bind the heat shock-induced chaperone heat shock protein 70 (HSP70). As a result of this interaction, peptide–MHCI complexes do not traffic to the cell surface and rather accumulate in lysosomal/late endosomal compartments (114), although the mechanism by which HSP70 controls antigen cross-presentation remains to be elucidated.
Taken together, these data illustrate mechanisms by which capabilities of DCs are suppressed in tumors through modification of their lipid metabolism, either by secreted factors or indirectly by an altered tumor microenvironment. Tumor-associated DCs exert their functions in a tissue where glucose is scarce due to the high glycolytic rates of tumor cells (120), and the inability to adopt a glycolytic metabolism can impair DC effector functions (see the section Metabolic Rearrangements Upon Immunogenic Dendritic Cell Stimulation). Alternatively, tumor-derived factors can enforce FAO and OXPHOS in DCs and promote accumulation of lipids, which can, in turn, inhibit secretion of proinflammatory cytokines and antigen cross-presentation, respectively (98, 112, 119). Nonetheless, it remains unanswered why and how tumor-associated DCs accumulate high amounts of lipids. Some reports indicate that lipid accumulation is due to activation of a lipogenic program (119), while others suggest increased lipid uptake (112). Moreover, tumor cells also secrete other factors to the local milieu that act on tumor-infiltrating DCs and support the acquisition of a tolerogenic phenotype such as adenosine (121) or lactate (70, 71, 73). Thus, the metabolic reprogramming of tumor-associated DCs can contribute to tumor progression.
Amino Acid Metabolism and Tolerizing Dendritic Cell Functions
Catabolism of the essential amino acid Trp is critical in balancing inflammation and tolerance. Trp is metabolized by the enzyme IDO1, generating kynurenine (Kyn) in a process that consumes oxygen (122). This enzyme is highly expressed by tumor cells and exploited as a mechanism for immune evasion (123). IDO1-mediated Trp catabolism promotes local immunosuppression by two means: (1) Trp starvation limits T cell proliferation by impairing the T cell cycle machinery (124, 125), and (2) Kyn products induce T cell apoptosis (126), inhibit T cell cytotoxicity via downregulation of T cell receptor (TCR) CD3 ζ-chain (127), and induce differentiation of Tregs (127, 128). Notably, a subset of tumor-associated pDCs that accumulate in tumor-draining LNs (tdLNs) express IDO and mediate antigen-specific T cell anergy, contributing to tumor progression (129, 130). Cytokines such as IFNγ and TGFβ (131–134) and immunosuppressive drugs such as DEX (131) induce IDO in pDCs. Remarkably, cytotoxic T-lymphocyte-associated protein (CTLA)-4-expressing Tregs bind B7 family receptors on pDCs also triggering IDO1 expression (132, 133). This bidirectional conditioning also happens upon glucocorticoid-inducible TNF receptor-related protein (GITR) ligand (GITRL) engagement by GITR, expressed by Tregs and pDCs, respectively, inducing IDO1 expression via activation of the IKB–IKKα non-canonical NFkB pathway in pDCs in an IFNα-dependent manner (131). This crosstalk would establish a positive feedback loop to favor long-term immunosuppression. DEX induces this tolerogenic pathway by concomitant upregulation of GITR on CD4+ T cells and GITRL on pDCs (131). DEX treatment is a frequently used treatment to tolerize human moDCs in vitro (see the section Metabolic Adaptions of Tolerized Dendritic Cells), which often display high FAO and OXPHOS rates (88, 89). Therefore, one could hypothesize that FAO and IDO1 activities collaborate in establishing a tolerogenic program in DCs. Indeed, an oxidative metabolic profile adopted by tolerogenic DCs supports IDO1 function, providing a direct link between FAO and tolerogenic DC responses in vivo (98).
Arg is another amino acid that has a central immunomodulatory role. In immune cells, Arg is metabolized by iNOS under inflammatory conditions to generate L-citruline and NO (135), the latter being associated with activated GM-DCs (82). Alternatively, Arg can be metabolized by arginases 1 and 2 (Arg1 and 2) to produce ornithine, a precursor for polyamines that can support tumor cell proliferation (135, 136). Notably, tumor-infiltrating DCs act as Arg sinks, contributing to local Arg depletion and indirectly inhibiting T cell antitumor responses (137). Additionally, Arg1-dependent production of the polyamine spermidine by DCs induces both IDO1 enzymatic and signaling activities, allowing the establishment of a tolerogenic phenotype in response to TGFβ (138). Interestingly, myeloid-derived suppressor cells also release polyamines that condition DCs to express IDO1 and, therefore, amplify the immune suppression exerted through joint modulation of amino acid catabolism in cancer (138).
Enhanced Trp and Arg catabolism causes amino acid depletion in the local microenvironment, which is sensed by T cells via the Ser/Thr kinase general control non-derepressible 2 kinase (GCN2) and results in limited protein synthesis and proliferative arrest (139–141). Intriguingly, GCN2 activation in response to amino acid scarcity improves antigen presentation by human moDCs in vitro in response to yellow fever vaccine YF-17D by enhancing autophagy (142). Indeed, human CD8+ T cell responses after YF-17D vaccination correlate with increased expression of GCN2 and autophagy-related genes, and mice deficient for GCN2 or autophagy related-proteins 5 or 7 in the CD11c compartment show impaired antiviral T cell responses (142). Hence, active Trp and Arg amino acid metabolism by DCs influences the microenvironment and T cell responses and is involved in immune suppression.
Concluding Remarks
DCs are functionally defined by their ability to prime immunity and tolerance, but how their cellular metabolism (Figure 2) is affected by sensing of environmental cues and how this metabolic rewiring affects, in turn, DC function is an emerging fascinating field. The diversity of DCs (Tables 1, 2) and the fact that a great body of literature has been generated using DC-like cells from mouse bone marrow cultures with GM-CSF (12) limit our ability to predict what are the regulation and consequences of metabolic rearrangements in natural DCs in vivo.
Moreover, the use of inhibitors or genetic deletion of metabolic regulators to interrogate modulation of metabolic pathways is debated. Metabolic inhibitors have the advantage of immediate action on otherwise unaltered DCs and universal application on primary mouse and human DCs ex vivo. However, their applicability for DC-specific in vivo studies is limited, and they can have off-target effects, such as reported for C75, etomoxir, and 2-DG (16, 20, 58). On the other hand, genetic deletion of metabolic regulators in DCs using Cre-expressing mouse lines or other genetic approaches such as shRNA or CRISPR/Cas9 largely circumvents side effects and allows investigation of DCs with metabolic impairment in vivo. Nevertheless, genetic deficiency of important metabolic regulators can cause a deregulation of DC development (see the section Metabolic Control of Dendritic Cell Development) that challenges investigation of their functions, and unrelated compensating mechanisms that are difficult to control. While there is probably no consensus on the ideal strategy, studying consequences of manipulation of DC metabolism in vivo, rather than in vitro, may be of high relevance, as the microenvironment is crucial for cellular metabolism. Additionally, future studies of DC metabolism employing combined approaches of pharmacological inhibition and genetic deficiency will be most convincing.
Nevertheless, some patterns are starting to emerge showing that moDC and cDC1 generation is more dependent on functional mitochondrial metabolism and OXPHOS than cDC2s or pDCs (Tables 1, 2). Early induction of glycolysis characterizes and is required for immunogenic activation of cultured DCs and primary DC subsets, while long-term glycolytic reprogramming is finely regulated and may have suboptimal consequences (Figure 3). Indeed, important differences of metabolic/glycolytic adaptions of DCs early or late after stimulation are emerging, such as the different signaling pathways regulating early (49) and, likely, rather late glycolytic reprogramming (50, 56) or the time-dependent substrate use for glycolysis (46). Moreover, while weak and potent stimulants induce early glycolytic activity in GM-DCs, only strong activation achieves maintenance of increased glycolysis for 18 h or longer (56), further supporting the action of different mechanisms. Notably, metabolic flexibility for energy generation of long-term activated GM-DCs (3 days or more) seems to benefit their immunogenic functions (60, 64, 68) (Figure 3).
In contrast, tolerogenic DCs appear to generally rely more on OXPHOS than glycolysis, based on cultured human moDCs tolerized with specific stimuli. However, we only understand fragments of the cellular energy metabolism of tolerogenic DCs and the signaling pathways controlling their induction and maintenance of their functions.
Importantly, different DC subsets (Table 1) emerge to display pronounced variations in their adaption of mitochondrial energy metabolism upon immunogenic activation (Figure 4), reaching from strong induction of OXPHOS in pDCs, context-dependent alterations in cDCs, to a long-term reduction in cultured GM-DCs or human moDCs. Additionally, while enhanced glycolysis and FAS appear as general features of activated DCs, the further application of fatty acids as building blocks for the ER/Golgi or substrate for FAO also largely varies among DC subsets (Figure 4). Further efforts in primary DC subsets in different settings will likely contribute to a better understanding of context dependence and regulation of immunogenic and tolerogenic DC subset metabolism, as highlighted for lung inflammatory DCs (31).
Overall, integration of nutrient sensing and adequate adaption of mTOR/AMPK signaling (Figure 1) are crucial for metabolic adjustments by DCs. However, the complexity of metabolic reprogramming of DCs (upon stimulation) is highlighted by the fact that the signaling mechanisms involved in inducing glycolytic activity show context dependency and even contradictory effects with regard to regulating DC function. This controversy might be explained by differential routes of activation and additional functions and nutrient-dependent regulations of those important cellular signaling networks in DCs, apart from controlling glycolytic metabolism, that remain to be defined. For example, mTOR signaling is often linked with immunogenic DC activation due to increasing glycolytic and anabolic metabolism (41, 43, 59). However, tolerized moDCs also exhibit increased glycolysis compared to control moDCs in the steady state or after additional stimulation (88, 89), which was, indeed, also dependent on mTOR and reduced by rapamycin (91). Those observations indicate that the general association of a metabolic state, anabolic glycolysis vs. catabolic FAO/mitochondrial respiration (Figure 2), and concomitantly pathways controlling metabolic adaption to nutrients, mTOR vs. AMPK activation, cannot be generally ascribed to immunogenic vs. tolerogenic DCs.
Indeed, the influence of the particular immunogenic or tolerogenic context, ontogenic constraints of distinct DC subsets, and additional (environmental) factors on the balance of nutrient-sensing pathways and metabolic adaptions of DCs will have to be carefully assessed in the future.
Author Contributions
SKW prepared tables and figures and conceptualized and wrote the manuscript. SCK conceptualized and wrote part of the manuscript. EP and IH-M helped conceptualize the manuscript and prepared the figures. DS contributed to funding acquisition and supervised, conceptualized, and wrote the manuscript. All authors declare no conflict of interest, contributed to manuscript revision, and read and approved the final version.
Funding
The DS laboratory is funded by the CNIC and grant SAF2016-79040-R from Ministerio de Ciencia, Innovacióne Universidades (MCIU), Agencia Estatal de Investigación, and Fondo Europeo de Desarrollo Regional (FEDER); B2017/BMD-3733 Immunothercan-CM from Comunidad de Madrid; RD16/0015/0018-REEM from FIS-Instituto de Salud Carlos III, MICINN, and FEDER; Acteria Foundation; Constantes y Vitales prize (Atresmedia); La Marató de TV3 Foundation (201723); the European Commission (635122-PROCROP H2020); and the European Research Council (ERC-2016-Consolidator Grant 725091). SKW is supported by a European Molecular Biology Organization Long-term Fellowship (grant ALTF 438-2016) and a CNIC-International Postdoctoral Program Fellowship (grant 17230-2016). SCK is a recipient of a FPU fellowship (FPU16/03142) from the Spanish Ministry of Education, Culture and Sports. EP is supported by a predoctoral grant from the Spanish Ministry of Economy and Competitiveness (BES-2017-079717). IH-M receives the support of a fellowship from la Caixa Foundation (ID 100010434, fellowship code: LCF/BQ/IN17/11620074) and from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 713673. The CNIC is supported by the MCIU and the Pro-CNIC Foundation and is a Severo Ochoa Center of Excellence (SEV-2015-0505).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors DS.
Acknowledgments
We thank all members of the DS laboratory at CNIC for scientific discussions and are especially grateful to Gillian Dunphy for comments and corrections of the manuscript.
References
1. Woltman AM, van der Kooij SW, Coffer PJ, Offringa R, Daha MR, van Kooten C. Rapamycin specifically interferes with GM-CSF signaling in human dendritic cells, leading to apoptosis via increased p27KIP1 expression. Blood. (2003) 101:1439–45. doi: 10.1182/blood-2002-06-1688
2. Haidinger M, Poglitsch M, Geyeregger R, Kasturi S, Zeyda M, Zlabinger GJ, et al. A versatile role of mammalian target of rapamycin in human dendritic cell function and differentiation. J Immunol. (2010) 185:3919–31. doi: 10.4049/jimmunol.1000296
3. Szatmari I, Gogolak P, Im JS, Dezso B, Rajnavolgyi E, Nagy L. Activation of PPARγ specifies a dendritic cell subtype capable of enhanced induction of iNKT cell expansion. Immunity. (2004) 21:95–106. doi: 10.1016/j.immuni.2004.06.003
4. Szatmari I, Töröcsik D, Agostini M, Nagy T, Gurnell M, Barta E, et al. PPARgamma regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood. (2007) 110:3271–80. doi: 10.1182/blood-2007-06-096222
5. Nencioni A, Grunebach F, Zobywlaski A, Denzlinger C, Brugger W, Brossart P. Dendritic cell immunogenicity is regulated by peroxisome proliferator-activated receptor. J Immunol. (2002) 169:1228–35. doi: 10.4049/jimmunol.169.3.1228
6. Gogolak P, Rethi B, Szatmari I, Lanyi A, Dezso B, Nagy L, et al. Differentiation of CD1a- and CD1a+ monocyte-derived dendritic cells is biased by lipid environment and PPARγ. Blood. (2007) 109:643–52. doi: 10.1182/blood-2006-04-016840
7. Rehman A, Hemmert KC, Ochi A, Jamal M, Henning JR, Barilla R, et al. Role of fatty-acid synthesis in dendritic cell generation and function. J Immunol. (2013) 190:4640–9. doi: 10.4049/jimmunol.1202312
8. Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1α transcriptional complex. Nature. (2007) 450:736–40. doi: 10.1038/nature06322
9. Morita M, Gravel SP, Hulea L, Larsson O, Pollak M, St-Pierre J, et al. MTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle. (2015) 14:473–80. doi: 10.4161/15384101.2014.991572
10. Zaccagnino P, Saltarella M, Maiorano S, Gaballo A, Santoro G, Nico B, et al. An active mitochondrial biogenesis occurs during dendritic cell differentiation. Int J Biochem Cell Biol. (2012) 44:1962–9. doi: 10.1016/j.biocel.2012.07.024
11. Del Prete A, Zaccagnino P, Di Paola M, Saltarella M, Oliveros Celis C, Nico B, et al. Role of mitochondria and reactive oxygen species in dendritic cell differentiation and functions. Free Radic Biol Med. (2008) 44:1443–51. doi: 10.1016/j.freeradbiomed.2007.12.037
12. Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c+MHCII+ macrophages and dendritic cells. Immunity. (2015) 42:1197–211. doi: 10.1016/j.immuni.2015.05.018
13. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. (2010) 115:4742–9. doi: 10.1182/blood-2009-10-249540
14. Köhler T, Reizis B, Johnson RS, Weighardt H, Förster I. Influence of hypoxia-inducible factor 1α on dendritic cell differentiation and migration. Eur J Immunol. (2012) 42:1226–36. doi: 10.1002/eji.201142053
15. Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol. (2014) 12:59. doi: 10.1371/journal.pbio.1001759
16. Chen C, Han X, Zou X, Li Y, Yang L, Cao K, et al. 4-Methylene-2-octyl-5-oxotetrahydrofuran-3-carboxylic acid (C75), an inhibitor of fatty-acid synthase, suppresses the mitochondrial fatty acid synthesis pathway and impairs mitochondrial function. J Biol Chem. (2014) 289:17184–94. doi: 10.1074/jbc.M114.550806
17. Duriancik DM, Gardner EM. Energy restriction impairs dendritic cell development in C57BL/6J mice. Mech Ageing Dev. (2016) 154:9–19. doi: 10.1016/j.mad.2016.02.002
18. Kratchmarov R, Viragova S, Kim MJ, Rothman NJ, Liu K, Reizis B, et al. Metabolic control of cell fate bifurcations in a hematopoietic progenitor population. Immunol Cell Biol. (2018) 8:1–9. doi: 10.1111/imcb.12040
19. Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, et al. Cutting edge: generation of splenic CD8+ and CD8- dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J Immunol. (2005) 174:6592–7. doi: 10.4049/jimmunol.174.11.6592
20. Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, et al. Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab. (2018) 28:504–15.e7. doi: 10.1016/j.cmet.2018.06.002
21. Du X, Wen J, Wang Y, Karmaus PWF, Khatamian A, Tan H, et al. Hippo/Mst signalling couples metabolic state and immune function of CD8α+ dendritic cells. Nature. (2018) 558:141–5. doi: 10.1038/s41586-018-0177-0
22. Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, et al. Loss of phagocytic and antigen cross-presenting capacity in aging dendritic cells is associated with mitochondrial dysfunction. J Immunol. (2015) 195:2624–32. doi: 10.4049/jimmunol.1501006
23. Sukhbaatar N, Hengstschläger M, Weichhart T. mTOR-mediated regulation of dendritic cell differentiation and function. Trends Immunol. (2016) 37:778–89. doi: 10.1016/j.it.2016.08.009
24. Sathaliyawala T, O'Gorman WE, Greter M, Bogunovic M, Konjufca V, Hou ZE, et al. Mammalian target of rapamycin controls dendritic cell development downstream of Flt3 ligand signaling. Immunity. (2010) 33:597–606. doi: 10.1016/j.immuni.2010.09.012
25. Nobs SP, Schneider C, Dietrich MG, Brocker T, Rolink A, Hirsch E, et al. PI3-kinase-γ has a distinct and essential role in lung-specific dendritic cell development. Immunity. (2015) 43:674–89. doi: 10.1016/j.immuni.2015.09.006
26. Kellersch B, Brocker T. Langerhans cell homeostasis in mice is dependent on mTORC1 but not mTORC2 function. Blood. (2013) 121:298–307. doi: 10.1182/blood-2012-06-439786
27. Sparber F, Scheffler JM, Amberg N, Tripp CH, Heib V, Hermann M, et al. The late endosomal adaptor molecule p14 (LAMTOR2) represents a novel regulator of Langerhans cell homeostasis. Blood. (2014) 123:217–27. doi: 10.1182/blood-2013-08-518555
28. Sparber F, Tripp CH, Komenda K, Scheffler JM, Clausen BE, Huber LA, et al. The late endosomal adaptor molecule p14 (LAMTOR2) regulates TGFβ1-mediated homeostasis of langerhans cells. J Invest Dermatol. (2015) 135:119–29. doi: 10.1038/jid.2014.324
29. Collin M, Milne P. Langerhans cell origin and regulation. Curr Opin Hematol. (2016) 23:28–35. doi: 10.1097/MOH.0000000000000202
30. Hackstein H, Taner T, Zahorchak AF, Morelli AE, Logar AJ, Gessner A, et al. Rapamycin inhibits IL-4-induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood. (2003) 101:4457–63. doi: 10.1182/blood-2002-11-3370
31. Sinclair C, Bommakanti G, Gardinassi L, Loebbermann J, Johnson MJ, Hakimpour P, et al. MTOR regulates metabolic adaptation of APCs in the lung and controls the outcome of allergic inflammation. Science. (2017) 357:1014–21. doi: 10.1126/science.aaj2155
32. van de Laar L, van den Bosch A, Boonstra A, Binda RS, Buitenhuis M, Janssen HLA, et al. PI3K-PKB hyperactivation augments human plasmacytoid dendritic cell development and function. Blood. (2012) 120:4982–91. doi: 10.1182/blood-2012-02-413229
33. Carroll KC, Viollet B, Suttles J. AMPKα1 deficiency amplifies proinflammatory myeloid APC activity and CD40 signaling. J Leukoc Biol. (2013) 94:1113–21. doi: 10.1189/jlb.0313157
34. Ohtani M, Hoshii T, Fujii H, Koyasu S, Hirao A, Matsuda S. Cutting edge: mTORC1 in intestinal CD11c+CD11b+ dendritic cells regulates intestinal homeostasis by promoting IL-10 production. J Immunol. (2012) 188:4736–40. doi: 10.4049/jimmunol.1200069
35. Wang Y, Huang G, Zeng H, Yang K, Lamb RF, Chi H. Tuberous sclerosis 1 (Tsc1)-dependent metabolic checkpoint controls development of dendritic cells. Proc Natl Acad Sci. (2013) 110:E4894–903. doi: 10.1073/pnas.1308905110
36. Pan H, O'Brien TF, Wright G, Yang J, Shin J, Wright KL, et al. Critical role of the tumor suppressor tuberous sclerosis complex 1 in dendritic cell activation of CD4 T cells by promoting MHC class II expression via IRF4 and CIITA. J Immunol. (2013) 191:699–707. doi: 10.4049/jimmunol.1201443
37. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
38. Byersdorfer CA. The role of fatty acid oxidation in the metabolic reprograming of activated T-cells. Front Immunol. (2014) 5:641. doi: 10.3389/fimmu.2014.00641
39. Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. (2013) 38:970–83. doi: 10.1016/j.immuni.2013.04.011
40. Scheffler JM, Sparber F, Tripp CH, Herrmann C, Humenberger A, Blitz J, et al. LAMTOR2 regulates dendritic cell homeostasis through FLT3-dependent mTOR signalling. Nat Commun. (2014) 5:5138. doi: 10.1038/ncomms6138
41. Pearce EJ, Everts B. Dendritic cell metabolism. Nat Rev Immunol. (2015) 15:18–29. doi: 10.1038/nri3771
42. Kelly B, O'Neill LAJ. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. (2015) 25:771–84. doi: 10.1038/cr.2015.68
43. O'Neill LAJ, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. (2016) 213:15–23. doi: 10.1084/jem.20151570
44. O'Neill LAJ, Kishton RJ. Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
45. Ryan DG, O'Neill LAJ. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. (2017) 591:2992–3006. doi: 10.1002/1873-3468.12744
46. Thwe PM, Pelgrom L, Cooper R, Beauchamp S, Reisz JA, D'Alessandro A, et al. Cell-intrinsic glycogen metabolism supports early glycolytic reprogramming required for dendritic cell immune responses. Cell Metab. (2017) 26:558–67.e5. doi: 10.1016/j.cmet.2017.08.012
47. Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, et al. Hypoxia and hypoxia-inducible factor-1 modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol. (2008) 180:4697–705. doi: 10.4049/jimmunol.180.7.4697
48. Everts B, Amiel E, van der Windt GJW, Freitas TC, Chott R, Yarasheski KE, et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood. (2012) 120:1422–31. doi: 10.1182/blood-2012-03-419747
49. Everts B, Amiel E, Huang SCC, Smith AM, Chang CH, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol. (2014) 15:323–32. doi: 10.1038/ni.2833
50. Márquez S, Fernández JJ, Terán-Cabanillas E, Herrero C, Alonso S, Azogil A, et al. Endoplasmic reticulum stress sensor IRE1α enhances IL-23 expression by human dendritic cells. Front Immunol. (2017) 8:639. doi: 10.3389/fimmu.2017.00639
51. Perrin-Cocon L, Aublin-Gex A, Diaz O, Ramière C, Peri F, André P, et al. Toll-like receptor 4–induced glycolytic burst in human monocyte-derived dendritic cells results from p38-dependent stabilization of HIF-1α and increased hexokinase II expression. J Immunol. (2018) 2018:ji1701522. doi: 10.4049/jimmunol.1701522
52. Bajwa G, DeBerardinis RJ, Shao B, Hall B, Farrar JD, Gill MA. Cutting edge: critical role of glycolysis in human plasmacytoid dendritic cell antiviral responses. J Immunol. (2016) 2016:1501557. doi: 10.4049/jimmunol.1501557
53. Basit F, Mathan T, Sancho D, de Vries IJM. Human dendritic cell subsets undergo distinct metabolic reprogramming for immune response. Front Immunol. (2018) 9:2489. doi: 10.3389/fimmu.2018.02489
54. Fliesser M, Morton CO, Bonin M, Ebel F, Hünniger K, Kurzai O, et al. Hypoxia-inducible factor 1α modulates metabolic activity and cytokine release in anti-Aspergillus fumigatus immune responses initiated by human dendritic cells. Int J Med Microbiol. (2015) 305:865–73. doi: 10.1016/j.ijmm.2015.08.036
55. McKeithen DN, Omosun YO, Ryans K, Mu J, Xie Z, Simoneaux T, et al. The emerging role of ASC in dendritic cell metabolism during Chlamydia infection. PLoS ONE. (2017) 12:e0188643. doi: 10.1371/journal.pone.0188643
56. Guak H, Al Habyan S, Ma EH, Aldossary H, Al-Masri M, Won SY, et al. Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat Commun. (2018) 9:2463. doi: 10.1038/s41467-018-04804-6
57. Ryans K, Omosun Y, McKeithen DN, Simoneaux T, Mills CC, Bowen N, et al. The immunoregulatory role of alpha enolase in dendritic cell function during Chlamydia infection. BMC Immunol. (2017) 18:27. doi: 10.1186/s12865-017-0212-1
58. Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab. (2018) 28:463–75.e4. doi: 10.1016/j.cmet.2018.08.012
59. de Lima Thomaz L, Peron G, Oliveira J, da Rosa LC, Thomé R, Verinaud L. The impact of metabolic reprogramming on dendritic cell function. Int Immunopharmacol. (2018) 63:84–93. doi: 10.1016/j.intimp.2018.07.031
60. Lawless SJ, Kedia-Mehta N, Walls JF, McGarrigle R, Convery O, Sinclair LV, et al. Glucose represses dendritic cell-induced T cell responses. Nat Commun. (2017) 8:1–14. doi: 10.1038/ncomms15620
61. Maroof A, English NR, Bedford PA, Gabrilovich DI, Knight SC. Developing dendritic cells become “lacy” cells packed with fat and glycogen. Immunology. (2005) 115:473–83. doi: 10.1111/j.1365-2567.2005.02181.x
62. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. (2017) 356:513–9. doi: 10.1126/science.aal3535
63. Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell. (2016) 166:624–36. doi: 10.1016/j.cell.2016.05.076
64. Amiel E, Everts B, Freitas TC, King IL, Curtis JD, Pearce EL, et al. Inhibition of mechanistic target of rapamycin promotes dendritic cell activation and enhances therapeutic autologous vaccination in mice. J Immunol. (2012) 189:2151–8. doi: 10.4049/jimmunol.1103741
65. Cao W, Manicassamy S, Tang H, Kasturi SP, Pirani A, Murthy N, et al. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat Immunol. (2008) 9:1157–64. doi: 10.1038/ni.1645
66. Turnquist HR, Raimondi G, Zahorchak AF, Fischer RT, Wang Z, Thomson AW. Rapamycin-conditioned dendritic cells are poor stimulators of allogeneic CD4+ T cells, but enrich for antigen-specific Foxp3+ T regulatory cells and promote organ transplant tolerance. J Immunol. (2007) 178:7018–31. doi: 10.4049/jimmunol.178.11.7018
67. Boor PPC, Metselaar HJ, Mancham S, Van Der Laan LJW, Kwekkeboom J. Rapamycin has suppressive and stimulatory effects on human plasmacytoid dendritic cell functions. Clin Exp Immunol. (2013) 174:389–401. doi: 10.1111/cei.12191
68. Amiel E, Everts B, Fritz D, Beauchamp S, Ge B, Pearce EL, et al. Mechanistic target of rapamycin inhibition extends cellular lifespan in dendritic cells by preserving mitochondrial function. J Immunol. (2014) 193:2821–30. doi: 10.4049/jimmunol.1302498
69. Ohtani M, Nagai S, Kondo S, Mizuno S, Nakamura K, Tanabe M, et al. Mammalian target of rapamycin and glycogen synthase kinase 3 differentially regulate lipopolysaccharide-induced interleukin-12 production in dendritic cells. Blood. (2008) 112:635–43. doi: 10.1182/blood-2008-02-137430
70. Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem. (2002) 277:23111–5. doi: 10.1074/jbc.M202487200
71. Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
72. Wu D, Sanin DE, Everts B, Chen Q, Qiu J, Buck MD, et al. Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity. (2016) 44:1325–36. doi: 10.1016/j.immuni.2016.06.006
73. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood. (2006) 107:2013–21. doi: 10.1182/blood-2005-05-1795
74. Kaisar MMM, Pelgrom LR, van der Ham AJ, Yazdanbakhsh M, Everts B. Butyrate conditions human dendritic cells to prime type 1 regulatory T cells via both histone deacetylase inhibition and G protein-coupled receptor 109A signaling. Front Immunol. (2017) 8:1429. doi: 10.3389/fimmu.2017.01429
75. Williams NC, O'Neill LAJ. A role for the Krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation. Front Immunol. (2018) 9:141. doi: 10.3389/fimmu.2018.00141
76. Stüve P, Minarrieta L, Erdmann H, Arnold-Schrauf C, Swallow M, Guderian M, et al. De novo fatty acid synthesis during mycobacterial infection is a prerequisite for the function of highly proliferative T cells, but not for dendritic cells or macrophages. Front Immunol. (2018) 9:495. doi: 10.3389/fimmu.2018.00495
77. den Brok MH, Raaijmakers TK, Collado-Camps E, Adema GJ. Lipid droplets as immune modulators in myeloid cells. Trends Immunol. (2018) 39:380–92. doi: 10.1016/j.it.2018.01.012
78. den Brok MH, Büll C, Wassink M, de Graaf AM, Wagenaars JA, Minderman M, et al. Saponin-based adjuvants induce cross-presentation in dendritic cells by intracellular lipid body formation. Nat Commun. (2016) 7:13324. doi: 10.1038/ncomms13324
79. Bougnères L, Helft J, Tiwari S, Vargas P, Chang BHJ, Chan L, et al. A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC Class I in dendritic cells. Immunity. (2009) 31:232–44. doi: 10.1016/j.immuni.2009.06.022
80. Ibrahim J, Nguyen AH, Rehman A, Ochi A, Jamal M, Graffeo CS, et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology. (2012) 143:1061–72. doi: 10.1053/j.gastro.2012.06.003
81. Jennelle LT, Dandekar AP, Magoro T, Hahn YS. Immunometabolic signaling pathways contribute to macrophage and dendritic cell function. Crit Rev Immunol. (2016) 36:379–94. doi: 10.1615/CritRevImmunol.2017018803
82. Thwe PM, Amiel E. The role of nitric oxide in metabolic regulation of dendritic cell immune function. Cancer Lett. (2018) 412:236–42. doi: 10.1016/j.canlet.2017.10.032
83. Sim WJ, Ahl PJ, Connolly JE. Metabolism is central to tolerogenic dendritic cell function. Med Inflamm. (2016) 2016:1–10. doi: 10.1155/2016/2636701
84. Garaude J, Acín-Pérez R, Martínez-Cano S, Enamorado M, Ugolini M, Nistal-Villán E, et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat Immunol. (2016) 17:1037–45. doi: 10.1038/ni.3509
85. Sancho D, Enamorado M, Garaude J. Innate immune function of mitochondrial metabolism. Front Immunol. (2017) 8:527. doi: 10.3389/fimmu.2017.00527
86. Takenaka MC, Quintana FJ. Tolerogenic dendritic cells. Semin Immunopathol. (2017) 39:113–20. doi: 10.1007/s00281-016-0587-8
87. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity. (2015) 43:435–49. doi: 10.1016/j.immuni.2015.09.001
88. Malinarich F, Duan K, Hamid RA, Bijin A, Lin WX, Poidinger M, et al. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J Immunol. (2015) 194:5174–86. doi: 10.4049/jimmunol.1303316
89. Ferreira GB, Vanherwegen AS, Eelen G, Gutiérrez ACF, VanLommel L, Marchal K, et al. Vitamin D3 induces tolerance in human dendritic cells by activation of intracellular metabolic pathways. Cell Rep. (2015) 10:711–25. doi: 10.1016/j.celrep.2015.01.013
90. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell. (2006) 127:1109–22. doi: 10.1016/j.cell.2006.11.013
91. Dánová K, Klapetková A, Kayserová J, Šedivá A, Špíšek R, Jelínková LP. NF-κB, p38 MAPK, ERK1/2, mTOR, STAT3 and increased glycolysis regulate stability of paricalcitol/dexamethasone-generated tolerogenic dendritic cells in the inflammatory environment. Oncotarget. (2015) 6:14123–38. doi: 10.18632/oncotarget.4234
92. Švajger U, Obermajer N, Jeras M. Dendritic cells treated with resveratrol during differentiation from monocytes gain substantial tolerogenic properties upon activation. Immunology. (2010) 129:525–35. doi: 10.1111/j.1365-2567.2009.03205.x
93. Kim GY, Cho H, Ahn SC, Oh YH, Lee CM, Park YM. Resveratrol inhibits phenotypic and functional maturation of murine bone marrow-derived dendritic cells. Int Immunopharmacol. (2004) 4:245–53. doi: 10.1016/j.intimp.2003.12.009
94. Piemonti L, Monti P, Allavena P, Sironi M, Soldini L, Leone BE, et al. Glucocorticoids affect human dendritic cell differentiation and maturation. J Immunol. (1999) 162:6473–81.
95. Xia CQ, Peng R, Beato F, Clare-Salzler MJ. Dexamethasone induces IL-10-producing monocyte-derived dendritic cells with durable immaturity. Scand J Immunol. (2005) 62:45–54. doi: 10.1111/j.1365-3083.2005.01640.x
96. Bscheider M, Butcher EC. Vitamin D immunoregulation through dendritic cells. Immunology. (2016) 148:227–36. doi: 10.1111/imm.12610
97. Adorini L, Penna G. Induction of tolerogenic dendritic cells by vitamin D receptor agonists. Handb Exp Pharmacol. (2009) 188:251–73. doi: 10.1007/978-3-540-71029-5_12
98. Zhao F, Xiao C, Evans KS, Theivanthiran T, DeVito N, Holtzhausen A, et al. Paracrine Wnt5a-β-catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity. (2018) 48:147–60.e7. doi: 10.1016/j.immuni.2017.12.004
99. Ferreira GB, Kleijwegt FS, Waelkens E, Lage K, Nikolic T, Hansen DA, et al. Differential protein pathways in 1,25-dihydroxyvitamin D 3 and dexamethasone modulated tolerogenic human dendritic cells. J Proteome Res. (2012) 11:941–71. doi: 10.1021/pr200724e
100. Korhonen R, Lahti A, Hämäläinen M, Kankaanranta H, Moilanen E. Dexamethasone inhibits inducible nitric-oxide synthase expression and nitric oxide production by destabilizing mRNA in lipopolysaccharide-treated macrophages. Mol Pharmacol. (2002) 62:698–704. doi: 10.1124/mol.62.3.698
101. Man-Ying Chan M, Mattiacci JA, Hwang HS, Shah A, Fong D. Synergy between ethanol and grape polyphenols, quercetin, and resveratrol, in the inhibition of the inducible nitric oxide synthase pathway. Biochem Pharmacol. (2000) 60:1539–48. doi: 10.1016/S0006-2952(00)00471-8
102. Di Rosa M, Malaguarnera M, Nicoletti F, Malaguarnera L. Vitamin D3: a helpful immuno-modulator. Immunology. (2011) 134:123–39. doi: 10.1111/j.1365-2567.2011.03482.x
103. Baghdadi M, Yoneda A, Yamashina T, Nagao H, Komohara Y, Nagai S, et al. TIM-4 glycoprotein-mediated degradation of dying tumor cells by autophagy leads to reduced antigen presentation and increased immune tolerance. Immunity. (2013) 39:1070–81. doi: 10.1016/j.immuni.2013.09.014
104. Li W, Wang Q-L, Liu X, Dong S-H, Li H-X, Li C-Y, et al. Combined use of vitamin D3 and metformin exhibits synergistic chemopreventive effects on colorectal neoplasia in rats and mice. Cancer Prev Res. (2015) 8:139–48. doi: 10.1158/1940-6207.CAPR-14-0128
105. Chang E, Kim Y. Vitamin D insufficiency exacerbates adipose tissue macrophage infiltration and decreases AMPK/SIRT1 activity in obese rats. Nutrients. (2017) 9:1–15. doi: 10.3390/nu9040338
106. Bakhshalizadeh S, Amidi F, Shirazi R, Shabani Nashtaei M. Vitamin D3 regulates steroidogenesis in granulosa cells through AMP-activated protein kinase (AMPK) activation in a mouse model of polycystic ovary syndrome. Cell Biochem Funct. (2018) 36:183–93. doi: 10.1002/cbf.3330
107. Manna P, Achari AE, Jain SK. Vitamin D supplementation inhibits oxidative stress and upregulate SIRT1/AMPK/GLUT4 cascade in high glucose-treated 3T3L1 adipocytes and in adipose tissue of high fat diet-fed diabetic mice. Arch Biochem Biophys. (2017) 615:22–34. doi: 10.1016/j.abb.2017.01.002
108. Lan F, Weikel KA, Cacicedo JM, Ido Y. Resveratrol-induced AMP-activated protein kinase activation is cell-type dependent: lessons from basic research for clinical application. Nutrients. (2017) 9:751. doi: 10.3390/nu9070751
109. Kim J, Yang G, Kim Y, Kim J, Ha J. AMPK activators: mechanisms of action and physiological activities. Exp Mol Med. (2016) 48:e224–12. doi: 10.1038/emm.2016.16
110. Viana AYI, Sakoda H, Anai M, Fujishiro M, Ono H, Kushiyama A, et al. Role of hepatic AMPK activation in glucose metabolism and dexamethasone-induced regulation of AMPK expression. Diabetes Res Clin Pract. (2006) 73:135–42. doi: 10.1016/j.diabres.2005.12.011
111. Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and health span. Nat Rev Mol Cell Biol. (2012) 13:225–38. doi: 10.1038/nrm3293
112. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. (2010) 16:880–6. doi: 10.1038/nm.2172
113. Platt N, Gordon S. Is the class A macrophage scavenger receptor (SR-A) multifunctional?—The mouse's tale. J Clin Invest. (2001) 108:649–54. doi: 10.1172/JCI13903
114. Veglia F, Tyurin VA, Mohammadyani D, Blasi M, Duperret EK, Donthireddy L, et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun. (2017) 8:2122. doi: 10.1038/s41467-017-02186-9
115. Sánchez-Paulete AR, Teijeira A, Cueto FJ, Garasa S, Pérez-Gracia JL, Sánchez-Arráez A, et al. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann Oncol. (2017) 28:xii44–xii55. doi: 10.1093/annonc/mdx237
116. Villablanca EJ, Raccosta L, Zhou D, Fontana R, Maggioni D, Negro A, et al. Tumor-mediated liver X receptor-alpha activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat Med. (2010) 16:98–105. doi: 10.1038/nm.2074
117. Beceiro S, Pap A, Czimmerer Z, Sallam T, Guillén JA, Gallardo G, et al. Liver X receptor nuclear receptors are transcriptional regulators of dendritic cell chemotaxis. Mol Cell Biol. (2018) 38:17. doi: 10.1128/MCB.00534-17
118. Ramakrishnan R, Tyurin VA, Veglia F, Condamine T, Amoscato A, Mohammadyani D, et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J Immunol. (2014) 192:2920–31. doi: 10.4049/jimmunol.1302801
119. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. (2015) 161:1527–38. doi: 10.1016/j.cell.2015.05.025
120. Ho P-C, Liu P-S. Metabolic communication in tumors: a new layer of immunoregulation for immune evasion. J ImmunoTher Cancer. (2016) 4:4. doi: 10.1186/s40425-016-0109-1
121. Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood. (2008) 112:1822–31. doi: 10.1182/blood-2008-02-136325
122. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. (2004) 4:762–74. doi: 10.1038/nri1457
123. Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. (2003) 9:1269–74. doi: 10.1038/nm934
124. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. (1999) 189:1363–72. doi: 10.1084/jem.189.9.1363
125. Hwu P, Du MX, Lapointe R, Do M, Taylor MW, Young HA. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J Immunol. (2000) 164:3596–9. doi: 10.4049/jimmunol.164.7.3596
126. Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. (2002) 9:1069–77. doi: 10.1038/sj.cdd.4401073
127. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol. (2006) 176:6752–61. doi: 10.4049/jimmunol.176.11.6752
128. Curti A, Pandolfi S, Valzasina B, Aluigi M, Isidori A, Ferri E, et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood. (2007) 109:2871–7. doi: 10.1182/blood-2006-07-036863
129. Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, Extermann M, et al. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int J Cancer. (2002) 101:151–5. doi: 10.1002/ijc.10645
130. Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. (2004) 114:280–90. doi: 10.1172/JCI21583
131. Grohmann U, Volpi C, Fallarino F, Bozza S, Bianchi R, Vacca C, et al. Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat Med. (2007) 13:579–86. doi: 10.1038/nm1563
132. Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. (2002) 3:1097–101. doi: 10.1038/ni846
133. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. (2003) 4:1206–12. doi: 10.1038/ni1003
134. Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. (2011) 12:870–8. doi: 10.1038/ni.2077
135. Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front Immunol. (2017) 8:93. doi: 10.3389/fimmu.2017.00093
136. Casero RA, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov. (2007) 6:373–90. doi: 10.1038/nrd2243
137. Norian LA, Rodriguez PC, O'Mara LA, Zabaleta J, Ochoa AC, Cella M, et al. Tumor-infiltrating regulatory dendritic cells inhibit CD8+ T cell function via L-arginine metabolism. Cancer Res. (2009) 69:3086–94. doi: 10.1158/0008-5472.CAN-08-2826
138. Mondanelli G, Bianchi R, Pallotta MT, Orabona C, Albini E, Iacono A, et al. A relay pathway between arginine and tryptophan metabolism confers immunosuppressive properties on dendritic cells. Immunity. (2017) 46:233–44. doi: 10.1016/j.immuni.2017.01.005
139. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. (2005) 5:641–54. doi: 10.1038/nri1668
140. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. (2005) 22:633–42. doi: 10.1016/j.immuni.2005.03.013
141. Rodriguez PC, Quiceno DG. Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. (2007) 109:1568–73. doi: 10.1182/blood-2006-06-031856
Keywords: dendritic cell, metabolism, mitochondria, glycolysis, mammalian target of rapamycin, hypoxia-inducible factor, AMP-activated protein kinase, DC subsets
Citation: Wculek SK, Khouili SC, Priego E, Heras-Murillo I and Sancho D (2019) Metabolic Control of Dendritic Cell Functions: Digesting Information. Front. Immunol. 10:775. doi: 10.3389/fimmu.2019.00775
Received: 07 November 2018; Accepted: 25 March 2019;
Published: 25 April 2019.
Edited by:
Bart Everts, Leiden University Medical Center, NetherlandsReviewed by:
Michael D. Buck, Francis Crick Institute, United KingdomEyal Amiel, University of Vermont, United States
Copyright © 2019 Wculek, Khouili, Priego, Heras-Murillo and Sancho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefanie K. Wculek, c3RlZmFuaWUud2N1bGVrQGNuaWMuZXM=
David Sancho, ZHNhbmNob0BjbmljLmVz