 Ruben Pio
Ruben Pio Daniel Ajona
Daniel Ajona Sergio Ortiz-Espinosa1
Sergio Ortiz-Espinosa1 Alberto Mantovani
Alberto Mantovani John D. Lambris
John D. Lambris
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 April 2019
Sec. Molecular Innate Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00774
This article is part of the Research Topic Therapeutic Modulation of the Complement System: Clinical Indications and Emerging Drug Leads View all 15 articles
Reactivation of cytotoxic CD8+ T-cell responses has set a new direction for cancer immunotherapy. Neutralizing antibodies targeting immune checkpoint programmed cell death protein 1 (PD-1) or its ligand (PD-L1) have been particularly successful for tumor types with limited therapeutic options such as melanoma and lung cancer. However, reactivation of T cells is only one step toward tumor elimination, and a substantial fraction of patients fails to respond to these therapies. In this context, combination therapies targeting more than one of the steps of the cancer-immune cycle may provide significant benefits. To find the best combinations, it is of upmost importance to understand the interplay between cancer cells and all the components of the immune response. This review focuses on the elements of the complement system that come into play in the cancer-immunity cycle. The complement system, an essential part of innate immunity, has emerged as a major regulator of cancer immunity. Complement effectors such as C1q, anaphylatoxins C3a and C5a, and their receptors C3aR and C5aR1, have been associated with tolerogenic cell death and inhibition of antitumor T-cell responses through the recruitment and/or activation of immunosuppressive cell subpopulations such as myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), or M2 tumor-associated macrophages (TAMs). Evidence is provided to support the idea that complement blocks many of the effector routes associated with the cancer-immunity cycle, providing the rationale for new therapeutic combinations aimed to enhance the antitumor efficacy of anti-PD-1/PD-L1 checkpoint inhibitors.
Profound advances in our understanding of the interactions between tumors and the immune system have allowed the development of therapeutic approaches that boost the body's natural defenses against cancer. These therapies are aimed to mount effective antitumor immune responses and include immunomodulators, vaccines, and adoptive transfer of immune cells (1). Some of the most clinically effective immunotherapies to date target the programmed cell death protein 1 (PD-1) immune checkpoint. PD-1 is expressed by T cells during priming or expansion and binds to one of its two ligands PD-L1 or PD-L2 (2). Tumor cells upregulate PD-L1 in response to cytokines such as interferon (IFN)-γ (3). Interaction of PD-L1 with PD-1 on T cells causes T-cell apoptosis, anergy, and exhaustion, protecting tumor cells from CD8+ T cell-mediated cytolysis (3). PD-L1 can also deliver intrinsic intracellular signals that enhance cancer cell survival, regulate stress responses, and confer resistance toward apoptotic stimuli (4, 5). PD-1 and other checkpoints are also expressed by NK cells which may contribute to the antitumor activity of some therapeutic strategies under development (6, 7).
Neutralizing monoclonal antibodies against PD-1 or PD-L1 have transformed the therapeutic landscape of a wide range of cancers (Table 1), being particularly successful for tumors with limited therapeutic options such as melanoma or lung cancer (3). Notably, these antibodies generate durable responses without causing serious side effects. However, a significant fraction of patients manifests innate or acquired resistance to these therapies. Immune escape mechanisms stem from different cell interactions within the tumor microenvironment, and emphasize the need of developing rational combination strategies to obtain more potent anticancer responses (8).
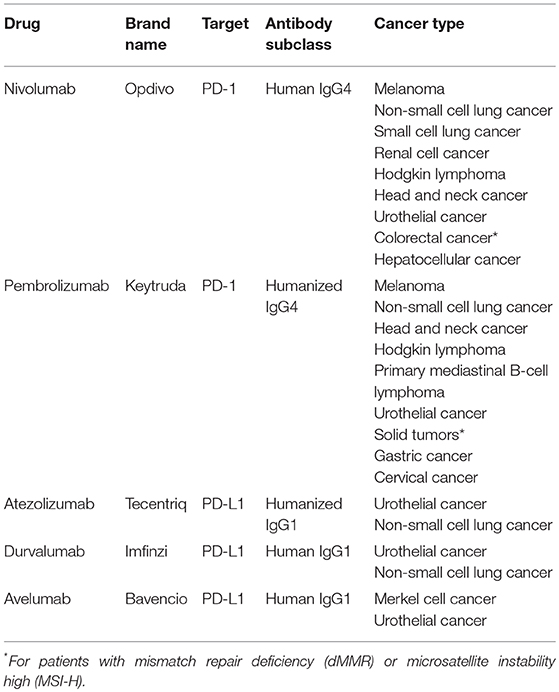
Table 1. FDA-approved immune-checkpoint inhibitors (monoclonal antibodies) for cancer treatment.
In 2013, Daniel Chen and Ira Mellman described a series of self-sustaining stepwise events, referred as the cancer-immunity cycle, by which the anti-cancer immune responses lead to an effective elimination of cancer cells (9). The existence of negative feedback mechanisms developed by tumors hinders this cycle of cancer immunity and may pose a barrier to the development of effective clinical responses. Anticancer immunotherapies should be aimed to reactivate all the steps of the cycle, which include immunogenic cell death, maturation of antigen-presenting cells, T-cell priming and activation, promotion of immune infiltration, blockade of immunosuppression, and enhancement of effector T-cell activity. In this context, combination therapies would provide synergistic effects for the maintenance of the cancer-immunity cycle.
In the last years, the complement system, an essential part of innate immunity, has surged as a master regulator of cancer immunity (10). We, and others, have actively contributed to this field, leading to the proposal that modulation of complement activation can improve the antitumor efficacy of inhibitors targeting the PD-1/PD-L1 pathway. In 2008, making a paradigm shift in tumor immunology, we demonstrated that complement activation, followed by C5a signaling, has a tumor-promoting role in cancer (11). In 2012, using a lung cancer model, we first demonstrated an association between the inhibition of C5a receptor 1 (C5aR1) and the expression of PD-L1 within the tumor microenvironment (12). These results suggested the possibility of blocking complement factors to increase the efficacy of other immune therapeutic strategies (12). Following this line, we demonstrated that inhibition of PD-1/PD-L1 synergizes with the inhibition of C5a/C5aR1 in various preclinical models of lung cancer (13). This rationale provided the basis for a clinical trial in which the anti-C5aR1 antibody IPH5401 is being evaluated in combination with the anti-PD-L1 antibody durvalumab in patients with solid tumors (STELLAR-001).
In this review we describe the participation of complement elements in the steps of the cancer-immunity cycle. We propose that a combinational therapy using anti-PD-1/PD-L1 antibodies together with modulators of the complement system may open new therapeutic opportunities for tumors resistant to PD-1/PD-L1 blockade.
The cancer-immunity cycle is defined as a series of functional stepwise events needed to obtain an efficient control of cancer growth by the immune system (9). The process is initiated by the release of neo-antigens generated as a result of genomic instability. Cancer-associated antigens are captured by dendritic cells which, upon migration to lymph nodes, prime and activate tumor-specific cytolytic CD8+ T cells. These effector cells migrate and infiltrate the tumor stroma, where potentially are able to recognize and eliminate cancer cells. T cell-mediated cytotoxic responses release new tumor antigens, fueling the cancer-immunity cycle. Interestingly, this model provides the rationale for targeting different steps of the cycle in order to maintain its functionality. An effective cancer immunotherapy should be designed based on the specific resistance mechanisms underlining the rate-limiting steps in each particular patient (14, 15). In the case of anti-PD-1/PD-L1 therapies, a variety of biological factors contribute to treatment resistance, including lack of cancer antigens recognizable by T cells, impaired cancer-antigen presentation, impaired activation of cancer-specific T cells, poor infiltration of T cells into tumors, and accumulation of immunosuppressive factors and cells in the tumor microenvironment (15). The evading strategies present in a given tumor would determine whether this tumor shows an inflamed or a noninflamed phenotype (16). Clinical evidence suggests that anti-PD-1/PD-L1 inhibitors are most effective in inflamed tumors characterized by high tumor PD-L1 expression, CD8+ T-cell infiltration or mutational burden (17–20). Jerby-Arnon et al. recently analyzed the relationship between malignant cell states and CD8+ T-cell infiltration and identified a T-cell exclusion program that predicts responses to PD-1/PD-L1 blockade (21). This program was enriched for genes involved in predictable processes, such as antigen processing and presentation, IFN-γ signaling, and immune modulation; but also identified genes associated with activation and modulation of the complement system (21). Elements of the complement cascade are also present in a signature of serum proteins that predicts survival in patients receiving PD-1 blocking antibodies, suggesting that complement activation may inhibit the efficacy of adaptive antitumor immunity (22).
The complement system, a central element of innate immunity, represents a first line of defense against unwanted non-self and host elements, and orchestrates many immunological and inflammatory processes that substantially contribute to body homeostasis (23, 24). Complement activities are mediated by more than 50 circulating, cell surface-bound and intracellular proteins. There are three main mechanisms of complement activation, known as classical, lectin, and alternative pathways. The classical pathway is commonly initiated by the binding of C1q to complement-fixing antibodies (mostly IgM and IgG types); although C1q can also recognize non-immunoglobulin ligands such as C-reactive protein (CRP), pentraxin 3 (PTX3), or apoptotic cells (25). The lectin pathway is activated by homologous proteins to C1q (mannose-binding lectin, collectins, and ficolins) that recognize repetitive carbohydrate patterns (26). Lastly, the alternative pathway is initiated by spontaneous cleavage of C3 on activating surfaces (27). Although the three complement pathways differ in their mechanisms of target recognition, in all cases, initiation of the complement cascade leads to the formation of C3 convertases and the activation of the central component C3. After this activation, C5 convertases are formed, C5 is cleaved, and the assembly of the pore-like membrane attack complex (MAC) is initiated. The enzymatic cleavage of complement elements leads to the release of proteolytic fragments such as C3a and C5a, and the deposition of other fragments such as C3b and iC3b. These molecules modulate a diverse set of processes (23), including the initiation and regulation of effector T-cell responses (28). Prevention of inappropriate activation by complement regulators takes place at three main levels: inhibition of protease activities in the activation cascade, decay and destruction of convertases, and control of MAC formation (29). Finally, recent experimental and clinical evidences suggest that intracellular complement components have important roles in cell physiology (30).
Complement has been traditionally regarded as playing a role in the elimination of tumor cells. Accordingly, an effective control of tumor growth may be achieved by complement-fixing antibodies (31). However, growing evidence, starting for the initial observation in a model of cervical cancer (11), strongly supports a tumor-promoting role of complement in several tumor types. This topic has been extensively reviewed elsewhere (10, 32–35). Briefly, complement establishes an immunosuppressive microenvironment, promotes angiogenesis, sustains cellular proliferation, and participates in tumor cell invasion and migration. In light of the various contributions of complement to cancer progression, it is not surprising that expression of complement effectors and receptors is associated with disease progression and poor prognosis (36–42). Among all the complement elements with potential pro-cancer activities, C1q, C3-derived fragments, and C5a are recognized as major modulators of tumor progression (43–45).
In this section we will discuss the potential implication of effectors and regulators of the complement system in the steps of the cancer-immunity cycle. In light of the breadth and complexity of the immune response, we will focus our review on the specific aspects of the regulation of CD8+ cytotoxic T cells, which are in large part the mediators of anti-PD-1/PD-L1 therapies. We will also examine evidence supporting the participation of complement in the regulation of the type 1 T helper (Th1) response, as it has a profound influence on the quality and extension of cytotoxic T-cell responses (46). Recent reviews have extensively addressed other complement-mediated immune functions not covered in the present review (10, 28, 47–50).
The cancer-immunity cycle is initiated by tumor-specific neo-antigens generated by somatic mutations (51). Dying cancer cells release these antigens to the tumor microenvironment, where are captured and processed by dendritic cells, the principal cell type responsible for instructing naïve T cells to undergo antigen-specific effector functions. Depending on the stimuli provided by dying cancer cells, their interaction with dendritic cells can have immunogenic or tolerogenic consequences (52). The generation of an immunogenic or a tolerogenic cell death is mainly regulated by damage-associated molecular patterns (DAMPs). DAMPs are endogenous co-stimulatory signals secreted or presented on the cell surface of dying cells that interact with pattern-recognition receptors (PRRs) alerting the host of danger. Complement is required for efficient sensing of DAMPs (53–55). The specific interactions of danger sensors with complement elements allow to differentiate between physiological and pathological danger, shaping the maturation of dendritic cells (23). This activity depends mainly on the classical complement pathway. Direct binding of complement C1q to apoptotic cells promotes a phagocytic-mediated uptake of dying cells, which sustains an anti-inflammatory innate immune response through the expression of cytokines such as transforming growth factor (TGF)-β (56). In fact, genetic deficiencies in C1q, as well as other elements of the classical complement pathway, can compromise the induction of self-tolerance and result in systemic autoimmune diseases (57, 58). Another complement element involved in the recognition of danger signals is factor H, a soluble complement inhibitor produced and secreted by cancer cells (59, 60). Upon opsonization of apoptotic cells, factor H induces an anti-inflammatory cytokine profile (61, 62) and a tolerogenic stage (63). CD46, a membrane-bound complement regulatory protein able to interact with C3 activation fragments and found at high levels in some cancer types (64, 65), has also been proposed as a negative regulator of immune recognition (66). Complement proteins are easily detectable in various types of cancer, consistent with complement activation by these tumors (32). Therefore, upregulation of complement components in the surface of dying cancer cells may be associated with a tolerogenic cell death, in contrast to the immunogenic cell death required for an effective anticancer immune response (67, 68).
Progress of the cancer-immunity cycle requires the presence of activation signals that allow dendritic cells to mature, migrate to the lymph nodes, and present the neo-antigens to naïve T cells. Efficient priming also relies on the contextual information provided by the microenvironment. Mature dendritic cells in the presence of suitable signals are able to induce T-cell effector functions; whereas in the absence of appropriate conditions, antigen presentation leads to T-cell anergy or generation of regulatory T cells (Tregs) that suppress effector responses.
Locally-produced complement elements determine the state of dendritic cell activation (69), and are critical in the regulation of T-cell responses (28). Production of C1q and C3 by dendritic cells induces their maturation and their capacity to stimulate Th1-cell responses (70, 71). C3 may also facilitate intracellular antigen processing and presentation (72). In agreement with these observations, optimal priming and expansion of CD4+ and CD8+ T cells in infection models is dampened by C3 deficiency (73, 74), and the complement fragment C3d amplifies antitumor T-cell responses (75). In the case of C3a and C5a, through activation of their respective receptors C3aR and C5aR1, these anaphylatoxins enhance the capacity of human monocyte-derived dendritic cells to stimulate T cells (76). In accordance, C3aR pathway inhibition in dendritic cells results in defective T-cell priming, associated with a reduced surface expression of major histocompatibility complex (MHC) and costimulatory molecules (77). Finally, downregulation of the expression of the complement regulator CD55 in antigen-presenting cells during T-cell activation increases the local production of C3a and C5a, providing costimulatory signals to induce T-cell proliferation and differentiation (78, 79).
Complement elements can also exert a direct influence on T cells. Activation of human CD4+ T cells by CD46 stimulates the effector potential of Th1 cells (80–82). As on dendritic cells, paracrine and autocrine interactions of C3a and C5a with their respective receptors C3aR and C5aR1 mediate Th1 cytokine production and T-cell induction (78, 83). It has also been suggested that Tregs express C3aR and C5aR1, and that signaling through these receptors inhibits Treg function (84, 85).
A central role in T-cell homeostasis has been recently assigned to intracellular elements of the complement system (30). Activation of lysosomal C3aR by intracellularly generated C3a contributes to the survival of resting CD4+ T cells. Upon activation, the intracellular stores of the C3 system translocate to the cell surface triggering the upregulation of IFN-γ and Th1-cell responses in conjunction with the extracellular engagement of C3b to CD46 (86). Intracellular C5a can also be generated from endogenous C5. Upon T-cell activation, C5a binds to intracellular C5aR1, inducing the activation of the NLRP3 inflammasome and, consequently, the initiation of a Th1 response (87).
All these activities underline the importance of complement effectors and regulators in the initiation of T-cell responses. In contrast, the complement system has also been associated with the prevention of T-cell priming and the induction of tolerance; probably as a regulatory mechanism to facilitate the timely resolution of the immune response. Thus, C1q can suppress macrophage-mediated inflammation and dendritic cell-mediated Th1-cell proliferation (88, 89). Binding of the C3 fragment iC3b to complement receptor type 3 (CR3) on antigen-presenting cells results in the production of TGF-β2 and interleukin (IL)-10, and the induction of antigen-specific tolerance (90). C3a and C3b also participate in the contraction phase of human Th1 responses (80, 82). CD46 promotes the switching of CD4+ T cells toward IL-10 producing cells with a regulatory phenotype (80, 91), and negatively regulates Th1 activity through the binding of endogenous C5a to surface-expressed C5aR2 (87). C5aR2 was first proposed as a negative regulator of C5aR1, but some specific functions have been ascribed to this C5a receptor (92). CD55 may also have a role in the suppression of adaptive immune responses. Mice lacking CD55 experience enhanced T-cell responses to active immunization, characterized by an increased production of INF-γ and IL-2, as well as downregulation of IL-10 (93).
All these studies point to the dual role played by complement in the activation of effector T-cell responses. On the one hand, complement elements are central in the primary phase of effector expansion. On the other hand, complement can mediate a suboptimal T-cell activation associated with the contraction phase or the establishment of tolerance. With this duality in mind, it is interesting to analyze the relative role played by complement in the context of well-established tumors. It has been suggested that C5a affects T-cell responses in a concentration-dependent manner (94). Tumor-bearing mice with low C5a-producing tumor cells exhibit a reduced tumor burden with increased IFN-γ-producing CD4+ and CD8+ T cells in the spleen and tumor-draining lymph nodes. In contrast, tumor-bearing mice with high C5a-producing cancer cells have an accelerated tumor progression with less CD4+ and CD8+ T cells in the tumor, tumor-draining lymph nodes, and the spleen (94). This effect was associated with the presence of more myeloid-derived suppressor cells (MDSCs) in the spleen. Interestingly, other studies have found elevated levels of C5a in cancer patients (12), which have been implicated in the recruitment of MDSCs to tumors (11). MDSCs are immunosuppressive immature myeloid cells able to disrupt major mechanisms of antitumor immune responses (95–97). In models of breast cancer, C5aR1 signaling in MDSCs induces the production of immunosuppressive cytokines, such as TGF-β, and reduces Th1 immune responses (98, 99). Treatment of mouse squamous cell carcinomas with paclitaxel and PMX-53, a C5aR1 inhibitor, results in peripheral priming and expansion of antigen-specific clones (100). Therefore, we can conclude that complement-mediated effects may have evolved at established tumors to interfere with the generation of antitumor T-cell responses.
A range of tumors escape from antitumor immune responses even after activation and expansion of T cells. In some cases, this can be attributed to the ability of the tumor endothelium to prevent T-cell trafficking. A complex network of endothelial adhesion molecules, Th1 cytokines, and surface receptors regulates T-cell homing and infiltration (101). Tumor cells, in concert with the endothelium, interfere with T-cell infiltration through a variety of molecular mechanisms, including the downregulation of endothelial adhesion molecules and the expression of T-cell inhibitory ligands (102).
Although complement elements can directly act on the endothelium (103–105), little is known about its contribution to the biology of tumor-associated endothelial cells. In the context of adoptive T-cell transfer, the capacity of tumor-reactive CD4+ and CD8+ T cells to infiltrate tumors requires the local production of C3, complement activation and release of C5a (106). Accordingly, the abrogation of CD55 expression enhances tumor T-cell infiltration (106). Radiotherapy has been found to upregulate the release of C3a and C5a within the irradiated tumors, leading to pronounced infiltration of CD8+ effector T cells (107). The presence of C3d on melanoma cells yields greater infiltration by CD4+ and CD8+ lymphocytes (75). However, in apparent contradiction to these findings, blocking of C3aR or C5aR1 in most cancer models has been associated with an increased infiltration of T cells both in the primary tumor (11, 13, 42, 99, 100, 108, 109) and the metastatic niche (98, 109). The mechanisms underlying this outcome are not fully understood but may be related to the downregulation of immunosuppressive cell populations, such as MDSCs, able to impair T-cell trafficking (11, 12). Complement inhibition also reduces the levels of VEGF (110), which may normalize the tumor vasculature, increasing the infiltration of lymphocytes into tumors (111).
An immunosuppressive microenvironment hampers the killing capacity of cytotoxic CD8+ T-cells. The pro-tumorigenic effect of complement activity is mediated, in an important way, by promoting immunosuppressing responses within the tumor microenvironment. The critical contribution of complement to regulating immunosuppressive cell populations, such as TAMs, MDSCs, or Tregs, has been recently reviewed (47). C5a acts as a potent chemoattractant for polymorphonuclear MDSCs and stimulates the production of immunosuppressive reactive oxygen and nitrogen species by tumor infiltrating monocytic MDSCs (11). Accordingly, pharmacological blockade of C5aR1 decreases the frequency of MDSCs and impairs tumor growth (11–13, 112). C5aR1 inhibition also downregulates the expression of immunosuppression-related genes within the tumor milieu (12). In addition, C5a contributes to conditioning the premetastatic niche through TGF-β and IL-10-mediated accumulation of Tregs, proliferation of resident alveolar macrophages, and decrease in number and maturation of dendritic cells (98). As a consequence, effector CD4+ T-cell responses skew toward a Th2 phenotype, limiting Th1 responses (98, 113).
C5a also affects the biology of macrophages. C5a skews macrophage polarization toward an M2 phenotype via C5aR1 signaling upon Leishmania infection (114). After ex vivo challenge of human whole blood with heat-killed Pseudomonas aeruginosa, C5a induces PD-L1 expression on monocytes, and the production of IL-10 and TGF-β (115). Elevation of PD-L1 expression has also been reported after C1q-mediated polarization of macrophages (89). M2 tumor-associated macrophages (TAMs) are an essential component of the tumor microenvironment that contribute to tumor progression by blocking CD8+ T-cell responses (116). Recruitment of tumor-promoting TAMs with a M2-like phenotype is also observed in mouse sarcomas induced in a PTX3-deficient context and characterized by an increase in C5a and CCL2 (44). C5a also promotes hepatic metastases of colon cancer associated with an increase of monocyte chemoattractant protein-1 (MCP1), anti-inflammatory modulators such as arginase-1, IL-10, or TGF-β, and M2-like macrophages (117, 118). Similarly, in a model of squamous carcinogenesis, C5a regulates the protumorogenic properties of C5aR1-expressing mast cells and macrophages, leading to hampered antitumor CD8+ T-cell responses (100). A combined treatment with cytotoxic chemotherapy and the blockade of C5aR1 synergistically inhibits the recruitment of effector memory CD8+ T cells by both the modification of macrophage- and IFNγ-dependent mechanisms (100). Interestingly, this study suggests that C5a is not generated in the tumors through C3 activation, although further studies are needed to rule out this possibility (119).
Complement C3 activation fragments can also precondition the tumor microenvironment toward immunosuppression. The C3 degradation product iC3b promotes the development of MDSCs in vitro (120). Inhibition of complement C3 abrogates the suppressor phenotype of polymorphonuclear MDSCs in the ovarian tumor microenvironment (121). Deletion of C3 in tumor cells also inhibits M2 polarization (122). Signaling mediated by C3a contributes to melanoma tumorigenesis by inhibiting neutrophil and CD4+ T-cell responses (108). Interestingly, some studies have suggested a direct effect of complement effectors in the functionality of T cells. C3 inhibits IL-10-mediated cytotoxic properties of tumor-infiltrating CD8+ T lymphocytes in an autocrine manner, enhancing melanoma and breast cancer growth (123). Alterations in CD4+ T cells by C3/C5-dependent pathways may also have a major role in lung cancer progression (109).
Finally, complement can also slow down the feeding of the cancer-immunity cycle by dying cancer cells. Ribosomal protein S19 (RPS19), upon release from dying tumor cells, interacts with C5aR1 expressed on MDSCs, promoting its recruitment to tumors, the generation of Tregs, the production of immunosuppressive cytokines (including TGF-β), and the reduction of CD8+ T-cell tumor infiltration (99).
Overall, tumor-associated complement activation deeply influences the tumor microenvironment, leading to an immunosuppressive state and the attenuation of tumor-specific cytotoxic T-cell responses.
As reviewed in the previous section, a growing body of evidence supports the notion that complement activities support cancer growth and metastasis in the context of established tumors (124). Many mechanisms related to immune escape and resistance to checkpoint inhibitors can be modulated by elements of the complement system (summarized in Figure 1). The non-immunology-related effects of complement on cancer cell biology, including cancer cell proliferation, survival and invasion capacity (42, 43, 117, 125–137), further reinforces the impact of complement activation in cancer progression.
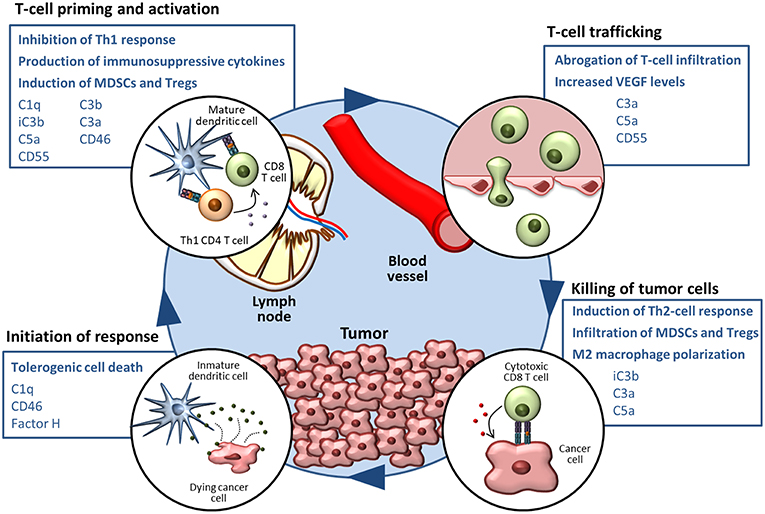
Figure 1. The complement system in the cancer-immunity cycle. The cancer-immunity cycle is summarized in four steps. Complement-mediated mechanisms associated with the inhibition of the cancer-immunity cycle, together with complement components that participate in these processes, are shown in blue boxes.
Based on the regulatory functions of complement in the cancer-immunity cycle, we sought to evaluate whether complement inhibition may represent an effective target for combined immunotherapies in preclinical syngeneic models of cancer. Clinical successes and limitations of anti-PD-1/PD-L1 monotherapy prompted us to use this target as the primary building block for the combination. The C5a/C5aR1 axis was selected as the complement-related target based on the abundant evidence supporting the role of this pathway in the establishment of an immunosuppressive microenvironment (Table 2) (45). Using different lung cancer models, we observed a remarkable synergistic control of lung tumor burden and metastatic progression in animals simultaneously treated with an aptamer against C5a (AON-D21) and an anti-PD-1 monoclonal antibody (13). This effect is accompanied by a negative association between the frequency of CD8+ T cells and the presence of MDSCs within tumors, and by a reduction of CD8+ T-cell exhaustion markers (13). The synergistic benefit of this combination was later confirmed in models of melanoma and colon cancer (138). Interestingly, PD-1/PD-L1 antibodies induce the production of C5a (138), establishing a regulatory loop between both pathways.
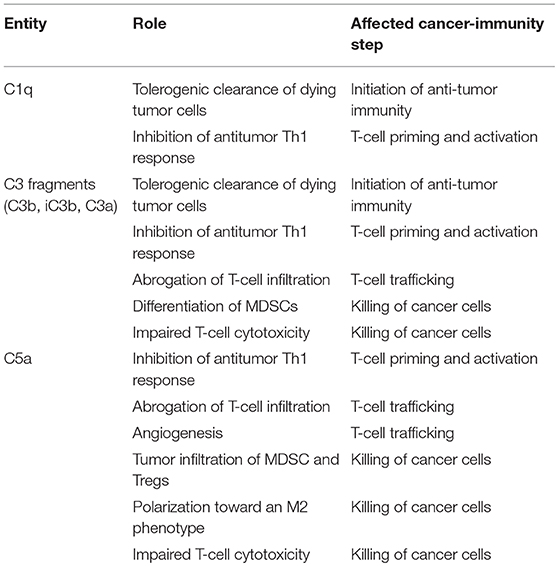
Table 2. Contribution of some elements of the complement system to the inhibition of the cancer-immunity cycle.
Other complement elements, such as C1q, C3, or C3a, may be also targeted to re-educate the tumor microenvironment and sensitize it to the subsequent administration of immune checkpoint blockers (Table 2). A multifaceted repertoire of therapeutic inhibitors targeting these complement elements has been developed, and are currently in preclinical or clinical development (139, 140). Figure 2 shows examples of compounds that may be used to target complement in the context of cancer immunotherapy. Complement C3, the centerpiece of complement activation, represents a particularly attractive target for therapeutic complement inhibition (141). Nonresponsive patients to PD-1/PD-L1 blockade frequently have noninflamed tumors with a defect in the early stages of the cancer-immunity cycle (15). Opsonization of dying cells with C3 fragments induces the production of anti-inflammatory cytokines and reduces the costimulatory molecules needed for the maturation of dendritic cells, resulting in T-cell tolerance (90). Therefore, C3 blockade may have a beneficial impact in the early stages of the cancer-immunity cycle, converting a noninflamed tumor into an inflamed tumor susceptible to PD-1/PD-L1 blockade. Moreover, both C3a and C5a production would be impaired (at least in the case of inhibitors, such as compstatin, that blocks C3 activation by all pathways). Interestingly, a simultaneous blockade of C3aR and C5aR1 has been reported to enhance the efficacy of anti-PD-1 therapy against melanoma cells (123). Deletion of C3 in tumor cells that had high C3 expression enhanced efficacy of anti–PD-L1 treatment (122). Additionally, complement C3 inhibition may have antitumor potential in the context of other immune combinations. For example, C3 inhibition by complement depletion or the use of the inhibitor compstatin enhances the antitumor efficacy of oncolytic virus (142, 143) and induces natural killer (NK)-mediated antitumoral responses (144). Inhibition of complement activation upstream of all complement effectors also appears to be a rational approach. In this sense, combinatorial therapies involving inhibitors of C1q (e.g., C1-INH), which presents both complement-dependent and -independent tumor promoting activities, merit further investigation. Finally, it has to be noted that most complement inhibitors target the extracellular complement system, preserving its intracellular activity. This may be of upmost importance, since intracellular C3aR and C5aR1 signaling pathways seem to be required for T-cell survival (28).
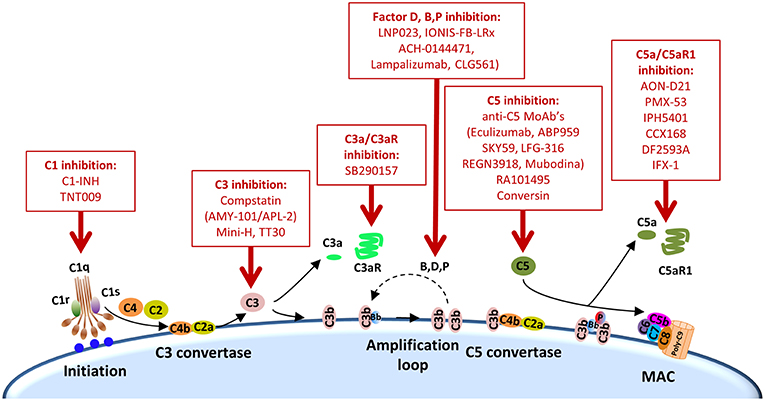
Figure 2. Points of complement inhibition. Steps of the classical complement activation pathway and some inhibitors available for targeting these steps are shown. These points of therapeutic intervention may render synergistic antitumor activities in combination with anti-PD-1/PD-L1 therapies.
Finally, it is interesting to point that the preclinical findings showing the feasibility and value of blocking C5a/C5aR1 to increase tumor-killing efficacy of checkpoint inhibitors have been the basis for the design of a phase I/II study (STELLAR-001). In this trial, the safety and efficacy of durvalumab (an anti-PD-L1 monoclonal antibody) is being tested in combination with IPH5401 (an anti-C5aR1 monoclonal antibody) in patients with selected solid tumors, including non-small cell lung cancer and hepatocellular carcinoma (NCT03665129). We are looking forward to the outcome of this trial, as well as to the clinical evaluation of novel combinations involving complement inhibitors.
This is an exciting time for the complement field, in which new biological concepts have brought new therapeutic opportunities. Based on the extensive literature associating complement activation and cancer progression, we propose here that substantial clinical benefits can be achieved by multi-modal anticancer immunotherapies targeting both complement-mediated mechanisms (to reverse immunosuppression), and PD-1/PD-L1 immune checkpoints (to re-activate T-cell functionality). Our preclinical studies supporting the idea that C5a/C5aR1 inhibition creates a “window of opportunity” for the administration of anti-PD-1/PD-L1 checkpoint inhibitors pave the way for the evaluation of other complement-based combinations. The challenge being that many potential combinations can be evaluated. Insights into how complement switches from tumor suppressing to tumor promoting activities at the onset of disease, as well as how to manage this dichotomy should be important research areas in order to establish the best therapeutic strategies. The differences between mice and humans in complement-mediated T-cell responses should also be considered (28, 87). To overcome this limitation, faithful mouse models that recapitulate the complexity of the human immune context in the tumor microenvironment are urgently needed (145).
RP and JL designed the concept. All authors wrote the manuscript. SO-E prepared the figures. All authors read and approved the final version of the manuscript.
Authors' research was supported by FIMA, CIBERONC, Fundación Científica de la Asociación Española Contra el Cáncer, Fundación Ramón Areces, Juan Serrano and Fondo de Investigación Sanitaria-Fondo Europeo de Desarrollo Regional Una manera de hacer Europa (PI17/00411) to RP and DA; U.S. National Institutes of Health (AI068730 and AI030040) to JL; AIRC IG, AIRC 5x1000, and Ministero della Salute to AM. SO-E was supported from fellowships from Amigos de la Universidad de Navarra and Formación de Personal Universitario (FPU 17/00032).
JL is the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors (including third-generation compstatin analogs such as AMY-101) and inventor of patents or patent applications that describe the use of complement inhibitors for therapeutic purposes, some of which are developed by Amyndas Pharmaceuticals. JL is also the inventor of the compstatin technology licensed to Apellis Pharmaceuticals (i.e., 4(1MeW)7W/POT-4/APL-1 and PEGylated derivatives). AM is inventor of patents related to PTX3 and gets royalties from related immunoassays. RP and DA are inventors of patents related to the use of complement C4 fragments as diagnostic and prognostic cancer biomarkers.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Lohmueller J, Finn OJ. Current modalities in cancer immunotherapy: Immunomodulatory antibodies, CARs and vaccines. Pharmacol Ther. (2017) 178:31–47. doi: 10.1016/j.pharmthera.2017.03.008
2. Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res. (2013) 19:4917–24. doi: 10.1158/1078-0432.CCR-12-1972
3. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. (2016) 8:328rv4. doi: 10.1126/scitranslmed.aad7118
4. Gato-Cañas M, Zuazo M, Arasanz H, Ibañez-Vea M, Lorenzo L, Fernandez-Hinojal G, et al. PDL1 signals through conserved sequence motifs to overcome interferon-mediated cytotoxicity. Cell Rep. (2017) 20:1818–29. doi: 10.1016/j.celrep.2017.07.075
5. Escors D, Gato-Cañas M, Zuazo M, Arasanz H, García-Granda MJ, Vera R, et al. The intracellular signalosome of PD-L1 in cancer cells. Signal Transduct Target Ther. (2018) 3:26. doi: 10.1038/s41392-018-0022-9
6. Molgora M, Bonavita E, Ponzetta A, Riva F, Barbagallo M, Jaillon S, et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature. (2017) 551:110–4. doi: 10.1038/nature24293
7. Moretta L, Pietra G, Vacca P, Pende D, Moretta F, Bertaina A, et al. Human NK cells: from surface receptors to clinical applications. Immunol Lett. (2016) 178:15–9. doi: 10.1016/j.imlet.2016.05.007
8. Melero I, Berman DM, Aznar MA, Korman AJ, Pérez Gracia JL, Haanen J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer. (2015) 15:457–72. doi: 10.1038/nrc3973
9. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
10. Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD. Complement in cancer: untangling an intricate relationship. Nat Rev Immunol. (2018) 18:5–18. doi: 10.1038/nri.2017.97
11. Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. (2008) 9:1225–35. doi: 10.1038/ni.1655
12. Corrales L, Ajona D, Rafail S, Lasarte JJJ, Riezu-Boj JIJI, Lambris JDJD, et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. (2012) 189:4674–83. doi: 10.4049/jimmunol.1201654
13. Ajona D, Ortiz-Espinosa S, Moreno H, Lozano T, Pajares MJ, Agorreta J, et al. A combined PD-1/C5a blockade synergistically protects against lung cancer growth and metastasis. Cancer Discov. (2017) 7:694–703. doi: 10.1158/2159-8290.CD-16-1184
14. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. (2017) 541:321–30. doi: 10.1038/nature21349
15. Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol. (2016) 27:1492–504. doi: 10.1093/annonc/mdw217
16. Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res. (2016) 22:1865–74. doi: 10.1158/1078-0432.CCR-15-1507
17. Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. (2014) 515:563–67. doi: 10.1038/nature14011
18. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) 515:568–71. doi: 10.1038/nature13954
19. Buder-Bakhaya K, Hassel JC. Biomarkers for clinical benefit of immune checkpoint inhibitor treatment-A review from the melanoma perspective and beyond. Front Immunol. (2018) 9:1474. doi: 10.3389/fimmu.2018.01474
20. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. doi: 10.1126/science.aaa1348
21. Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. (2018) 175:984–97. doi: 10.1016/j.cell.2018.09.006
22. Weber JS, Sznol M, Sullivan RJ, Blackmon S, Boland G, Kluger HM, et al. A serum protein signature associated with outcome after anti–PD-1 therapy in metastatic melanoma. Cancer Immunol Res. (2018) 6:79–86. doi: 10.1158/2326-6066.CIR-17-0412
23. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
24. Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol. (2017) 18:1288–98. doi: 10.1038/ni.3858
25. Kojouharova M, Reid K, Gadjeva M. New insights into the molecular mechanisms of classical complement activation. Mol Immunol. (2010) 47:2154–60. doi: 10.1016/j.molimm.2010.05.011
26. Garred P, Genster N, Pilely K, Bayarri-Olmos R, Rosbjerg A, Ma YJ, et al. A journey through the lectin pathway of complement-MBL and beyond. Immunol Rev. (2016) 274:74–97. doi: 10.1111/imr.12468
27. Rodríguez de Córdoba S, Harris CL, Morgan BP, Llorca O. Lessons from functional and structural analyses of disease-associated genetic variants in the complement alternative pathway. Biochim Biophys Acta. (2011) 1812:12–22. doi: 10.1016/j.bbadis.2010.09.002
28. West EE, Kolev M, Kemper C. Complement and the regulation of T cell responses. Annu Rev Immunol. (2018) 36:309–38. doi: 10.1146/annurev-immunol-042617-053245
29. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. (2009) 9:729–40. doi: 10.1038/nri2620
30. Arbore G, Kemper C, Kolev M. Intracellular complement - the complosome - in immune cell regulation. Mol Immunol. (2017) 89:2–9. doi: 10.1016/j.molimm.2017.05.012
31. Macor P, Capolla S, Tedesco F. Complement as a biological tool to control tumor growth. Front Immunol. (2018) 9:2203. doi: 10.3389/fimmu.2018.02203
32. Afshar-Kharghan V. The role of the complement system in cancer. J Clin Invest. (2017) 127:780–9. doi: 10.1172/JCI90962
33. Mamidi S, Höne S, Kirschfink M. The complement system in cancer: ambivalence between tumour destruction and promotion. Immunobiology. (2017) 222:45–54. doi: 10.1016/j.imbio.2015.11.008
34. Pio R, Corrales L, Lambris JD. The role of complement in tumor growth. Adv Exp Med Biol. (2014) 772:229–62. doi: 10.1007/978-1-4614-5915-6_11
35. Kochanek DM, Ghouse SM, Karbowniczek MM, Markiewski MM. Complementing cancer metastasis. Front Immunol. (2018) 9:1629. doi: 10.3389/fimmu.2018.01629
36. Ajona D, Pajares MJ, Chiara MD, Rodrigo JP, Jantus-Lewintre E, Camps C, et al. Complement activation product C4d in oral and oropharyngeal squamous cell carcinoma. Oral Dis. (2015) 21:899–904. doi: 10.1111/odi.12363
37. Ajona D, Pajares MJ, Corrales L, Perez-Gracia JL, Agorreta J, Lozano MD, et al. Investigation of complement activation product C4d as a diagnostic and prognostic biomarker for lung cancer. J Natl Cancer Inst. (2013) 105:1385–93. doi: 10.1093/jnci/djt205
38. Imamura T, Yamamoto-Ibusuki M, Sueta A, Kubo T, Irie A, Kikuchi K, et al. Influence of the C5a–C5a receptor system on breast cancer progression and patient prognosis. Breast Cancer. (2016) 23:876–85. doi: 10.1007/s12282-015-0654-3
39. Gu J, Ding JY, Lu CL, Lin ZW, Chu YW, Zhao GY, et al. Overexpression of CD88 predicts poor prognosis in non-small cell lung cancer. Lung Cancer. (2013) 81:259–65. doi: 10.1016/j.lungcan.2013.04.020
40. Wada Y, Maeda Y, Kubo T, Kikuchi K, Eto M, Imamura T. C5a receptor expression is associated with poor prognosis in urothelial cell carcinoma patients treated with radical cystectomy or nephroureterectomy. Oncol Lett. (2016) 12:3995–4000. doi: 10.3892/ol.2016.5137
41. Xi W, Liu L, Wang J, Xia Y, Bai Q, Long Q, et al. High level of anaphylatoxin C5a predicts poor clinical outcome in patients with clear cell renal cell carcinoma. Sci Rep. (2016) 6:29177. doi: 10.1038/srep29177
42. Cho MS, Vasquez HG, Rupaimoole R, Pradeep S, Wu S, Zand B, et al. Autocrine effects of tumor-derived complement. Cell Rep. (2014) 6:1085–95. doi: 10.1016/j.celrep.2014.02.014
43. Bulla R, Tripodo C, Rami D, Ling GS, Agostinis C, Guarnotta C, et al. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat Commun. (2016) 7:10346. doi: 10.1038/ncomms10346
44. Bonavita E, Gentile S, Rubino M, Maina V, Papait R, Kunderfranco P, et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell. (2015) 160:700–14. doi: 10.1016/j.cell.2015.01.004
45. Ajona D, Ortiz-Espinosa S, Pio R. Complement anaphylatoxins C3a and C5a: emerging roles in cancer progression and treatment. Semin Cell Dev Biol. (2019) 85:153–63. doi: 10.1016/j.semcdb.2017.11.023
46. Kennedy R, Celis E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol Rev. (2008) 222:129–44. doi: 10.1111/j.1600-065X.2008.00616.x
47. Kolev M, Markiewski MM. Targeting complement-mediated immunoregulation for cancer immunotherapy. Semin Immunol. (2018) 37:85–97. doi: 10.1016/j.smim.2018.02.003
48. Janelle V, Lamarre A. Role of the complement system in NK cell-mediated antitumor T-cell responses. Oncoimmunology. (2014) 3:e27897. doi: 10.4161/onci.27897
49. Berraondo P, Minute L, Ajona D, Corrales L, Melero I, Pio R. Innate immune mediators in cancer: between defense and resistance. Immunol Rev. (2016) 274:290–306. doi: 10.1111/imr.12464
50. Marin AV, Cárdenas PP, Jiménez-Reinoso A, Muñoz-Ruiz M, Regueiro JR. Lymphocyte integration of complement cues. Semin Cell Dev Biol. (2018) 85:132–42. doi: 10.1016/j.semcdb.2018.02.005
51. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. (2014) 515:577–81. doi: 10.1038/nature13988
52. Garg AD, Romano E, Rufo N, Agostinis P. Immunogenic versus tolerogenic phagocytosis during anticancer therapy: mechanisms and clinical translation. Cell Death Differ. (2016) 23:938–51. doi: 10.1038/cdd.2016.5
53. Köhl J. Self, non-self, and danger: a complementary view. In: Lambris JD, editor. Current Topics in Complement. Boston, MA: Springer US (2006). p. 71–94.
54. Ogden CA, DeCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med. (2001) 194:781–95. doi: 10.1084/jem.194.6.781
55. Botto M, Dell'Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. (1998) 19:56–9. doi: 10.1038/ng0598-56
56. Gershov D, Kim S, Brot N, Elkon KB. C-reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. J Exp Med. (2000) 192:1353–64. doi: 10.1084/jem.192.9.1353
57. Carneiro-Sampaio M, Liphaus BL, Jesus AA, Silva CAA, Oliveira JB, Kiss MH. Understanding systemic lupus erythematosus physiopathology in the light of primary immunodeficiencies. J Clin Immunol. (2008) 28:34–41. doi: 10.1007/s10875-008-9187-2
58. Ling GS, Crawford G, Buang N, Bartok I, Tian K, Thielens NM, et al. C1q restrains autoimmunity and viral infection by regulating CD8+ T cell metabolism. Science. (2018) 360:558–63. doi: 10.1126/science.aao4555
59. Ajona D, Castaño Z, Garayoa M, Zudaire E, Pajares MJ, Martinez A, et al. Expression of complement factor H by lung cancer cells: effects on the activation of the alternative pathway of complement. Cancer Res. (2004) 64:6310–8. doi: 10.1158/0008-5472.CAN-03-2328
60. Junnikkala S, Hakulinen J, Jarva H, Manuelian T, Bjørge L, Bützow R, et al. Secretion of soluble complement inhibitors factor H and factor H-like protein (FHL-1) by ovarian tumour cells. Br J Cancer. (2002) 87:1119–27. doi: 10.1038/sj.bjc.6600614
61. Martin M, Leffler J, Smolag KI, Mytych J, Björk A, Chaves LD, et al. Factor H uptake regulates intracellular C3 activation during apoptosis and decreases the inflammatory potential of nucleosomes. Cell Death Differ. (2016) 23:903–11. doi: 10.1038/cdd.2015.164
62. Mihlan M, Stippa S, Józsi M, Zipfel PF. Monomeric CRP contributes to complement control in fluid phase and on cellular surfaces and increases phagocytosis by recruiting factor H. Cell Death Differ. (2009) 16:1630–40. doi: 10.1038/cdd.2009.103
63. Olivar R, Luque A, Cárdenas-Brito S, Naranjo-Gómez M, Blom AM, Borràs FE, et al. The complement inhibitor factor H generates an anti-inflammatory and tolerogenic state in monocyte-derived dendritic cells. J Immunol. (2016) 196:4274–90. doi: 10.4049/jimmunol.1500455
64. Su Y, Liu Y, Behrens CR, Bidlingmaier S, Lee N-K, Aggarwal R, et al. Targeting CD46 for both adenocarcinoma and neuroendocrine prostate cancer. JCI Insight. (2018) 3:121497. doi: 10.1172/jci.insight.121497
65. Madjd Z, Durrant LG, Pinder SE, Ellis IO, Ronan J, Lewis S, et al. Do poor-prognosis breast tumours express membrane cofactor proteins (CD46)? Cancer Immunol Immunother. (2005) 54:149–56. doi: 10.1007/s00262-004-0590-0
66. Elward K, Griffiths M, Mizuno M, Harris CL, Neal JW, Morgan BP, et al. CD46 plays a key role in tailoring innate immune recognition of apoptotic and necrotic cells. J Biol Chem. (2005) 280:36342–54. doi: 10.1074/jbc.M506579200
67. Krysko O, Løve Aaes T, Bachert C, Vandenabeele P, Krysko DV. Many faces of DAMPs in cancer therapy. Cell Death Dis. (2013) 4:e631. doi: 10.1038/cddis.2013.156
68. Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta. (2010) 1805:53–71. doi: 10.1016/j.bbcan.2009.08.003
69. Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol. (2017) 188:183–94. doi: 10.1111/cei.12952
70. Baruah P, Dumitriu IE, Malik TH, Cook HT, Dyson J, Scott D, et al. C1q enhances IFN-gamma production by antigen-specific T cells via the CD40 costimulatory pathway on dendritic cells. Blood. (2009) 113:3485–93. doi: 10.1182/blood-2008-06-164392
71. Peng Q, Li K, Patel H, Sacks SH, Zhou W. Dendritic cell synthesis of C3 is required for full T cell activation and development of a Th1 phenotype. J Immunol. (2006) 176:3330–41. doi: 10.4049/jimmunol.176.6.3330
72. Baudino L, Sardini A, Ruseva MM, Fossati-Jimack L, Cook HT, Scott D, et al. C3 opsonization regulates endocytic handling of apoptotic cells resulting in enhanced T-cell responses to cargo-derived antigens. Proc Natl Acad Sci USA. (2014) 111:1503–8. doi: 10.1073/pnas.1316877111
73. Kopf M, Abel B, Gallimore A, Carroll M, Bachmann MF. Complement component C3 promotes T-cell priming and lung migration to control acute influenza virus infection. Nat Med. (2002) 8:373–8. doi: 10.1038/nm0402-373
74. Suresh M, Molina H, Salvato MS, Mastellos D, Lambris JD, Sandor M. Complement component 3 is required for optimal expansion of CD8 T cells during a systemic viral infection. J Immunol. (2003) 170:788–94. doi: 10.4049/jimmunol.170.2.788
75. Platt JL, Silva I, Balin SJ, Lefferts AR, Farkash E, Ross TM, et al. C3d regulates immune checkpoint blockade and enhances antitumor immunity. JCI Insight. (2017) 2:e90201. doi: 10.1172/jci.insight.90201
76. Li K, Fazekasova H, Wang N, Peng Q, Sacks SH, Lombardi G, et al. Functional modulation of human monocytes derived DCs by anaphylatoxins C3a and C5a. Immunobiology. (2012) 217:65–73. doi: 10.1016/j.imbio.2011.07.033
77. Peng Q, Li K, Anderson K, Farrar CA, Lu B, Smith RAG, et al. Local production and activation of complement up-regulates the allostimulatory function of dendritic cells through C3a-C3aR interaction. Blood. (2008) 111:2452–61. doi: 10.1182/blood-2007-06-095018
78. Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. (2008) 28:425–35. doi: 10.1016/j.immuni.2008.02.001
79. Heeger PS, Lalli PN, Lin F, Valujskikh A, Liu J, Muqim N, et al. Decay-accelerating factor modulates induction of T cell immunity. J Exp Med. (2005) 201:1523–30. doi: 10.1084/jem.20041967
80. Cardone J, Le Friec G, Vantourout P, Roberts A, Fuchs A, Jackson I, et al. Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat Immunol. (2010) 11:862–71. doi: 10.1038/ni.1917
81. Le Friec G, Sheppard D, Whiteman P, Karsten CM, Shamoun SA-T, Laing A, et al. The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat Immunol. (2012) 13:1213–21. doi: 10.1038/ni.2454
82. Ghannam A, Fauquert J-L, Thomas C, Kemper C, Drouet C. Human complement C3 deficiency: Th1 induction requires T cell-derived complement C3a and CD46 activation. Mol Immunol. (2014) 58:98–107. doi: 10.1016/j.molimm.2013.11.010
83. Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. (2008) 112:1759–66. doi: 10.1182/blood-2008-04-151068
84. Kwan W, van der Touw W, Paz-Artal E, Li MO, Heeger PS. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med. (2013) 210:257–68. doi: 10.1084/jem.20121525
85. van der Touw W, Cravedi P, Kwan W -H., Paz-Artal E, Merad M, Heeger PS. Receptors for C3a and C5a modulate stability of alloantigen-reactive induced regulatory T cells. J Immunol. (2013) 190:5921–5. doi: 10.4049/jimmunol.1300847
86. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. (2013) 39:1143–57. doi: 10.1016/j.immuni.2013.10.018
87. Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science. (2016) 352:aad1210. doi: 10.1126/science.aad1210
88. Benoit ME, Clarke EV, Morgado P, Fraser DA, Tenner AJ. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J Immunol. (2012) 188:5682–93. doi: 10.4049/jimmunol.1103760
89. Clarke E V, Weist BM, Walsh CM, Tenner AJ. Complement protein C1q bound to apoptotic cells suppresses human macrophage and dendritic cell-mediated Th17 and Th1 T cell subset proliferation. J Leukoc Biol. (2015) 97:147–60. doi: 10.1189/jlb.3A0614-278R
90. Sohn J-H, Bora PS, Suk H-J, Molina H, Kaplan HJ, Bora NS. Tolerance is dependent on complement C3 fragment iC3b binding to antigen-presenting cells. Nat Med. (2003) 9:206–12. doi: 10.1038/nm814
91. Kemper C, Chan AC, Green JM, Brett KA, Murphy KM, Atkinson JP. Activation of human CD4+ cells with CD3 and CD46 induces a T-regulatory cell 1 phenotype. Nature. (2003) 421:388–92. doi: 10.1038/nature01315
92. Croker DE, Halai R, Kaeslin G, Wende E, Fehlhaber B, Klos A, et al. C5a2 can modulate ERK1/2 signaling in macrophages via heteromer formation with C5a1 and β-arrestin recruitment. Immunol Cell Biol. (2014) 92:631–9. doi: 10.1038/icb.2014.32
93. Liu J, Miwa T, Hilliard B, Chen Y, Lambris JD, Wells AD, et al. The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med. (2005) 201:567–77. doi: 10.1084/jem.20040863
94. Gunn L, Ding C, Liu M, Ma Y, Qi C, Cai Y, et al. Opposing roles for complement component C5a in tumor progression and the tumor microenvironment. J Immunol. (2012) 189:2985–94. doi: 10.4049/jimmunol.1200846
95. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
96. Ibáñez-Vea M, Zuazo M, Gato M, Arasanz H, Fernández-Hinojal G, Escors D, et al. Myeloid-derived suppressor cells in the tumor microenvironment: current knowledge and future perspectives. Arch Immunol Ther Exp. (2018) 66:113–23. doi: 10.1007/s00005-017-0492-4
97. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. (2018) 120:16–25. doi: 10.1038/s41416-018-0333-1
98. Vadrevu SK, Chintala NK, Sharma SK, Sharma P, Cleveland C, Riediger L, et al. Complement C5a receptor facilitates cancer metastasis by altering T cell responses in the metastatic niche. Cancer Res. (2014) 74:3454–65. doi: 10.1158/0008-5472.CAN-14-0157
99. Markiewski MM, Vadrevu SK, Sharma SK, Chintala NK, Ghouse S, Cho J-H, et al. The ribosomal protein S19 suppresses antitumor immune responses via the complement C5a receptor 1. J Immunol. (2017) 198:2989–99. doi: 10.4049/jimmunol.1602057
100. Medler TR, Murugan D, Horton W, Kumar S, Cotechini T, Forsyth AM, et al. Complement C5a fosters squamous carcinogenesis and limits T cell response to chemotherapy. Cancer Cell. (2018) 34:561–78.e6. doi: 10.1016/j.ccell.2018.09.003
101. Marelli-Berg FM, Cannella L, Dazzi F, Mirenda V. The highway code of T cell trafficking. J Pathol. (2008) 214:179–89. doi: 10.1002/path.2269
102. Motz GT, Santoro SP, Wang L-P, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. (2014) 20:607–15. doi: 10.1038/nm.3541
103. Collard CD, Agah A, Reenstra W, Buras J, Stahl GL. Endothelial nuclear factor-kappaB translocation and vascular cell adhesion molecule-1 induction by complement: Inhibition with anti-human C5 therapy or cGMP analogues. Arterioscler Thromb Vasc Biol. (1999) 19:2623–9.
104. Schraufstatter IU, Trieu K, Sikora L, Sriramarao P, DiScipio R. Complement C3a and C5a induce different signal transduction cascades in endothelial cells. J Immunol. (2002) 169:2102–10. doi: 10.4049/jimmunol.169.4.2102
105. Bossi F, Tripodo C, Rizzi L, Bulla R, Agostinis C, Guarnotta C, et al. C1q as a unique player in angiogenesis with therapeutic implication in wound healing. Proc Natl Acad Sci USA. (2014) 111:4209–14. doi: 10.1073/pnas.1311968111
106. Facciabene A, De Sanctis F, Pierini S, Reis ES, Balint K, Facciponte J, et al. Local endothelial complement activation reverses endothelial quiescence, enabling T-cell homing, and tumor control during T-cell immunotherapy. Oncoimmunology. (2017) 6:e1326442. doi: 10.1080/2162402X.2017.1326442
107. Surace L, Lysenko V, Fontana AOO, Cecconi V, Janssen H, Bicvic A, et al. Complement is a central mediator of radiotherapy-induced tumor-specific immunity and clinical response. Immunity. (2015) 42:767–77. doi: 10.1016/j.immuni.2015.03.009
108. Nabizadeh JA, Manthey HD, Steyn FJ, Chen W, Widiapradja A, Md Akhir FN, et al. The complement C3a receptor contributes to melanoma tumorigenesis by inhibiting neutrophil and CD4+ T cell responses. J Immunol. (2016) 196:4783–92. doi: 10.4049/jimmunol.1600210
109. Kwak JW, Laskowski J, Li HY, McSharry MV, Sippel TR, Bullock BL, et al. Complement activation via a C3a receptor pathway alters CD4+ T lymphocytes and mediates lung cancer progression. Cancer Res. (2018) 78:143–56. doi: 10.1158/0008-5472.CAN-17-0240
110. Nunez-Cruz S, Gimotty PA, Guerra MW, Connolly DC, Wu Y-QQ, DeAngelis RA, et al. Genetic and pharmacologic inhibition of complement impairs endothelial cell function and ablates ovarian cancer neovascularization. Neoplasia. (2012) 14:994–1004. doi: 10.1593/neo.121262
111. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. (2010) 70:6171–80. doi: 10.1158/0008-5472.CAN-10-0153
112. Han X, Zha H, Yang F, Guo B, Zhu B. Tumor-derived tissue factor aberrantly activates complement and facilitates lung tumor progression via recruitment of myeloid-derived suppressor cells. Int J Mol Sci. (2017) 18:22. doi: 10.3390/ijms18010022
113. Sharma SK, Chintala NK, Vadrevu SK, Patel J, Karbowniczek M, Markiewski MM. Pulmonary alveolar macrophages contribute to the premetastatic niche by suppressing antitumor T cell responses in the lungs. J Immunol. (2015) 194:5529–38. doi: 10.4049/jimmunol.1403215
114. Hawlisch H, Belkaid Y, Baelder R, Hildeman D, Gerard C, Köhl J. C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity. (2005) 22:415–26. doi: 10.1016/j.immuni.2005.02.006
115. An L-L, Gorman JV, Stephens G, Swerdlow B, Warrener P, Bonnell J, et al. Complement C5a induces PD-L1 expression and acts in synergy with LPS through Erk1/2 and JNK signaling pathways. Sci Rep. (2016) 6:33346. doi: 10.1038/srep33346
116. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
117. Piao C, Cai L, Qiu S, Jia L, Song W, Du J. Complement 5a enhances hepatic metastases of colon cancer via monocyte chemoattractant protein-1-mediated inflammatory cell infiltration. J Biol Chem. (2015) 290:10667–76. doi: 10.1074/jbc.M114.612622
118. Piao C, Zhang W-M, Li T-T, Zhang C-C, Qiu S, Liu Y, et al. Complement 5a stimulates macrophage polarization and contributes to tumor metastases of colon cancer. Exp Cell Res. (2018) 366:127–38. doi: 10.1016/j.yexcr.2018.03.009
119. Mastellos DC, Reis ES, Lambris JD. Complement C5a-mediated TAM-ing of antitumor immunity drives squamous carcinogenesis. Cancer Cell. (2018) 34:531–3. doi: 10.1016/j.ccell.2018.09.005
120. Hsieh C-C, Chou H-S, Yang H-R, Lin F, Bhatt S, Qin J, et al. The role of complement component 3 (C3) in differentiation of myeloid-derived suppressor cells. Blood. (2013) 121:1760–8. doi: 10.1182/blood-2012-06-440214
121. Singel KL, Emmons TR, Khan ANH, Mayor PC, Shen S, Wong JT, et al. Mature neutrophils suppress T cell immunity in ovarian cancer microenvironment. JCI insight. (2019) 4:e122311. doi: 10.1172/jci.insight.122311
122. Zha H, Wang X, Zhu Y, Chen D, Han X, Yang F, et al. Intracellular activation of complement C3 leads to PD-L1 antibody treatment resistance by modulating tumor-associated macrophages. Cancer Immunol Res. (2019) 7:193–207. doi: 10.1158/2326-6066.CIR-18-0272
123. Wang Y, Sun S-N, Liu Q, Yu Y-Y, Guo J, Wang K, et al. Autocrine complement inhibits IL10-dependent T-cell mediated antitumor immunity to promote tumor progression. Cancer Discov. (2016) 6:1022–35. doi: 10.1158/2159-8290.CD-15-1412
124. Pio R, Ajona D, Lambris JD. Complement inhibition in cancer therapy. Semin Immunol. (2013) 25:54–64. doi: 10.1016/j.smim.2013.04.001
125. Riihilä P, Nissinen L, Farshchian M, Kallajoki M, Kivisaari A, Meri S, et al. Complement component C3 and complement factor B promote growth of cutaneous squamous cell carcinoma. Am J Pathol. (2017) 187:1186–97. doi: 10.1016/j.ajpath.2017.01.006
126. Abdelbaset-Ismail A, Borkowska-Rzeszotek S, Kubis E, Bujko K, Brzezniakiewicz-Janus K, Bolkun L, et al. Activation of the complement cascade enhances motility of leukemic cells by downregulating expression of HO-1. Leukemia. (2017) 31:446–58. doi: 10.1038/leu.2016.198
127. Ajona D, Zandueta C, Corrales L, Moreno H, Pajares MJ, Ortiz-Espinosa S, et al. Blockade of the complement C5a/C5aR1 axis impairs lung cancer bone metastasis by CXCL16-mediated effects. Am J Respir Crit Care Med. (2018) 197:1164–76. doi: 10.1164/rccm.201703-0660OC
128. Kaida T, Nitta H, Kitano Y, Yamamura K, Arima K, Izumi D, et al. C5a receptor (CD88) promotes motility and invasiveness of gastric cancer by activating RhoA. Oncotarget. (2016) 7:84798–809. doi: 10.18632/oncotarget.12656
129. Hu W-H, Hu Z, Shen X, Dong L-Y, Zhou W-Z, Yu X-X. C5a receptor enhances hepatocellular carcinoma cell invasiveness via activating ERK1/2-mediated epithelial-mesenchymal transition. Exp Mol Pathol. (2016) 100:101–8. doi: 10.1016/j.yexmp.2015.10.001
130. Maeda Y, Kawano Y, Wada Y, Yatsuda J, Motoshima T, Murakami Y, et al. C5aR is frequently expressed in metastatic renal cell carcinoma and plays a crucial role in cell invasion via the ERK and PI3 kinase pathways. Oncol Rep. (2015) 33:1844–50. doi: 10.3892/or.2015.3800
131. Cai K, Wan Y, Wang Z, Wang Y, Zhao X, Bao X. C5a promotes the proliferation of human nasopharyngeal carcinoma cells through PCAF-mediated STAT3 acetylation. Oncol Rep. (2014) 32:2260–6. doi: 10.3892/or.2014.3420
132. Lu Y, Hu X. C5a stimulates the proliferation of breast cancer cells via Akt-dependent RGC-32 gene activation. Oncol Rep. (2014) 32:2817–23. doi: 10.3892/or.2014.3489
133. Nitta H, Murakami Y, Wada Y, Eto M, Baba H, Imamura T. Cancer cells release anaphylatoxin C5a from C5 by serine protease to enhance invasiveness. Oncol Rep. (2014) 32:1715–9. doi: 10.3892/or.2014.3341
134. Nitta H, Wada Y, Kawano Y, Murakami Y, Irie A, Taniguchi K, et al. Enhancement of human cancer cell motility and invasiveness by anaphylatoxin C5a via aberrantly expressed C5a receptor (CD88). Clin Cancer Res. (2013) 19:2004–13. doi: 10.1158/1078-0432.CCR-12-1204
135. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10+ GPR77+ cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. (2018) 172:841–56. doi: 10.1016/j.cell.2018.01.009
136. Zhao C, Li Y, Qiu W, He F, Zhang W, Zhao D, et al. C5a induces A549 cell proliferation of non-small cell lung cancer via GDF15 gene activation mediated by GCN5-dependent KLF5 acetylation. Oncogene. (2018) 37:4821–37. doi: 10.1038/s41388-018-0298-9
137. Chen J, Li G-Q, Zhang L, Tang M, Cao X, Xu G-L, et al. Complement C5a/C5aR pathway potentiates the pathogenesis of gastric cancer by down-regulating p21 expression. Cancer Lett. (2018) 412:30–6. doi: 10.1016/j.canlet.2017.10.003
138. Zha H, Han X, Zhu Y, Yang F, Li Y, Li Q, et al. Blocking C5aR signaling promotes the anti-tumor efficacy of PD-1/PD-L1 blockade. Oncoimmunology. (2017) 6:e1349587. doi: 10.1080/2162402X.2017.1349587
139. Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: therapeutic interventions. J Immunol. (2013) 190:3839–47. doi: 10.4049/jimmunol.1203200
140. Hawksworth OA, Li XX, Coulthard LG, Wolvetang EJ, Woodruff TM. New concepts on the therapeutic control of complement anaphylatoxin receptors. Mol Immunol. (2017) 89:36–43. doi: 10.1016/j.molimm.2017.05.015
141. Ricklin D, Lambris JD. Therapeutic control of complement activation at the level of the central component C3. Immunobiology. (2016) 221:740–6. doi: 10.1016/j.imbio.2015.06.012
142. Evgin L, Ilkow CS, Bourgeois-Daigneault M-C, de Souza CT, Stubbert L, Huh MS, et al. Complement inhibition enables tumor delivery of LCMV glycoprotein pseudotyped viruses in the presence of antiviral antibodies. Mol Ther Oncolytics. (2016) 3:16027. doi: 10.1038/mto.2016.27
143. Evgin L, Acuna SA, Tanese de Souza C, Marguerie M, Lemay CG, Ilkow CS, et al. Complement inhibition prevents oncolytic vaccinia virus neutralization in immune humans and cynomolgus macaques. Mol Ther. (2015) 23:1066–76. doi: 10.1038/mt.2015.49
144. Janelle V, Langlois M-P, Tarrab E, Lapierre P, Poliquin L, Lamarre A. Transient complement inhibition promotes a tumor-specific immune response through the implication of natural killer cells. Cancer Immunol Res. (2014) 2:200–6. doi: 10.1158/2326-6066.CIR-13-0173
Keywords: cancer immunity, immunotherapy, complement system, C3a, C5a, C1q, PD-1, PD-L1
Citation: Pio R, Ajona D, Ortiz-Espinosa S, Mantovani A and Lambris JD (2019) Complementing the Cancer-Immunity Cycle. Front. Immunol. 10:774. doi: 10.3389/fimmu.2019.00774
Received: 01 February 2019; Accepted: 25 March 2019;
Published: 12 April 2019.
Edited by:
Carlo Pucillo, University of Udine, ItalyReviewed by:
Martin V. Kolev, GlaxoSmithKline, United KingdomCopyright © 2019 Pio, Ajona, Ortiz-Espinosa, Mantovani and Lambris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruben Pio, cnBpb0B1bmF2LmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.