 Zvi Fishelson
Zvi Fishelson Michael Kirschfink
Michael Kirschfink- 1Department of Cell and Developmental Biology, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel
- 2Institute of Immunology, University of Heidelberg, Heidelberg, Germany
The interactions of cancer cells with components of the complement system are highly complex, leading to an outcome that is either favorable or detrimental to cancer cells. Currently, we perceive only the “tip of the iceberg” of these interactions. In this review, we focus on the complement terminal C5b-9 complex, known also as the complement membrane attack complex (MAC) and discuss the complexity of its interaction with cancer cells, starting with a discussion of its proposed mode of action in mediating cell death, and continuing with a portrayal of the strategies of evasion exhibited by cancer cells, and closing with a proposal of treatment approaches targeted at evasion strategies. Upon intense complement activation and membrane insertion of sufficient C5b-9 complexes, the afflicted cells undergo regulated necrotic cell death with characteristic damage to intracellular organelles, including mitochondria, and perforation of the plasma membrane. Several pro-lytic factors have been proposed, including elevated intracellular calcium ion concentrations and activated JNK, Bid, RIPK1, RIPK3, and MLKL; however, further research is required to fully characterize the effective cell death signals activated by the C5b-9 complexes. Cancer cells over-express a multitude of protective measures which either block complement activation, thus reducing the number of membrane-inserted C5b-9 complexes, or facilitate the elimination of C5b-9 from the cell surface. Concomitantly, cancer cells activate several protective pathways that counteract the death signals. Blockage of complement activation is mediated by the complement membrane regulatory proteins CD46, CD55, and CD59 and by soluble complement regulators, by proteases that cleave complement proteins and by protein kinases, like CK2, which phosphorylate complement proteins. C5b-9 elimination and inhibition of cell death signals are mediated by caveolin and dynamin, by Hsp70 and Hsp90, by the mitochondrial stress protein mortalin, and by the protein kinases PKC and ERK. It is conceivable that various cancers and cancers at different stages of development will utilize distinct patterns of these and other MAC resistance strategies. In order to enhance the impact of antibody-based therapy on cancer, novel precise reagents that block the most effective protective strategies will have to be designed and applied as adjuvants to the therapeutic antibodies.
Preface
The complement system may affect cancer in several forms, ranging from promotion of cancer growth and metastasis, on the one hand, to antibody-based cancer eradication, on the other. Upon encounter of the cancer cells with the complement system, activation may proceed via the classical, alternative, and/or lectin pathways (1) (Figure 1). This initiation step leads to formation of a C3 convertase (C4b2a or C3bBb) that deposits C3b molecules on the cells, followed by formation of a C5 convertase (C4b2a3b or C3bBb3b) that cleaves C5 and initiates formation of the C5b-9 complexes, termed the membrane attack complexes (MAC). Here, we will focus on the anti-cancer cytotoxic activity of complement, with an emphasis on the mode of action of the MAC. Reviews on the cancer-promoting activities of complement (2–4) and on complement activation by clinical anti-cancer antibodies (5–7) have been published recently; therefore, these topics will not be covered in this review. Another topic recently reviewed is the insights into the fine structural details of the complement MAC (8–11). MAC expresses a plethora of non-lytic and sublytic activities that have been reviewed elsewhere (12–15) and are thus excluded from this review. Here we will describe the current status of research on the cytotoxic effects of MAC, emphasizing the findings, dogmas, and open questions in our quest to better understand the fine mechanistic details of MAC-induced cancer cell death. Next, we will present the currently recognized counter-mechanisms utilized by cancer cells to resist complement-dependent cytotoxicity (CDC). Finally, we will discuss several potential therapeutic approaches for the intervention and potentiation of antibody-based anti-cancer immunotherapy that have been proposed and tested.
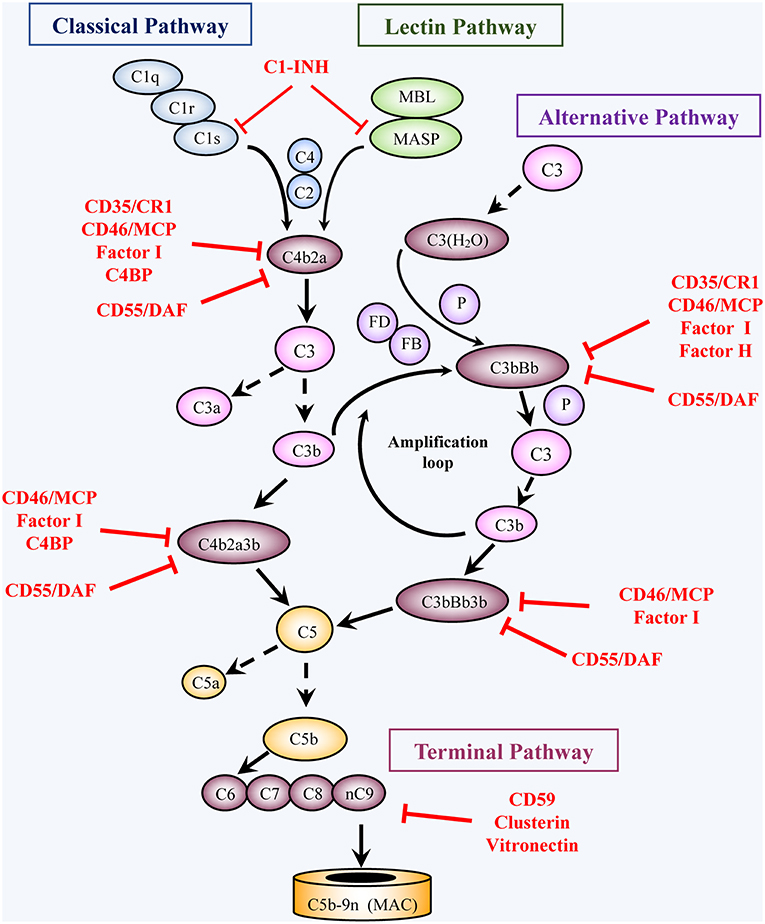
Figure 1. Activation and regulation of the complement pathways. Activation: Complement activation proceeds through four converging pathways shown in this simplified scheme, i.e., the classical (CP), lectin (LP), alternative (AP), and terminal (TP) pathways. Activation of the CP and LP can be potentiated by components of the AP (Amplification loop). Binding of C1 (a complex of C1q, 2C1r, 2C1s) via C1q to antigen-bound antibodies initiates the CP, whereas binding of MBL or ficolin (in complex with MBL-associated serine proteases, MASP) to carbohydrates (e.g., microbial) initiates activation of the LP. The AP is initiated by C3 spontaneously hydrolyzed at a low rate into C3(H2O) or following another C3-tickover event. The three pathways generate a C3 converting enzyme, a C3 convertase (that cleaves C3 into C3a and C3b), by activation of C4 and C2 (CP and LP: C4b2a), or of factors B and D (AP: C3bBb). AP activation is facilitated by properdin (P). The resulting C3b not only opsonizes target cells but also joins the C3 convertases and turns them into C5 convertases, which convert C5 into C5a and C5b. Subsequent TP activation by assembly of C5b with C6, C7, C8 and multiple C9 molecules, generates the membrane attack complex, (C5b-9, MAC). By binding to specific receptors, C3a and C5a exert multiple cell stimulatory activities, ranging from allergy and anaphylaxis to promotion of acquired immunity by stimulation of lymphocytes and antigen presenting cells. Regulation: Complement activation is tightly regulated by multiple soluble and membrane proteins. Soluble inhibitors include: C1 inhibitor (C1-INH), C4 binding protein (C4BP), factor H (FH), factor I (FI), Clusterin and Vitronectin. The membrane regulatory proteins are: Decay Accelerating Factor (DAF, CD55), Membrane Cofactor Protein (MCP, CD46), Complement Receptor 1 (CR1, CD35), and CD59. As shown in the figure, C1-INH interferes with activation of C1r, C1s, and MASP. C4BP, FH, CD55, and CD35 restrict formation and stability of the CP and AP C3/C5 convertases or promote FI-mediated inactivation of C4b (CD35/CR1, CD46/MCP, C4BP) or C3b (CD35/CR1, CD46/MCP, FH). Clusterin and vitronectin prevent the association of the forming C5b-9 complexes with the membrane, whereas CD59 limits cell damage by preventing MAC complex formation.
Mechanisms Underlying Complement-mediated Cancer Cell Damage
Perspective: The Early Studies on Osmotic Cell Death
Studies on cancer cell killing by complement have been conducted long before the identification of the complement terminal pathway responsible for mediating cell damage and death. As early as 1950s, Kalfayan et al. (16), Ellem (17), and Green et al. (18) investigated the action of antibody and complement on rabbit Brown-Pearce carcinoma cells, rat Ehrlich and mouse Krebs ascites tumor cells, respectively. They observed cell swelling and increased plasma membrane leakiness. They proposed that complement impairs cell membrane integrity, increases cell permeability to anions, cations, and water, and causes osmotic cell swelling up to the point that the membrane collapses, culminating in osmotic cell lysis (19). The leakage from the cells was proposed to occur through functional, stretching, and possibly reversible “holes” in the swelling cells, which could be blocked, to some extent, by increasing the osmotic pressure of the extracellular medium (20). The concept of complement-induced osmotic lysis of target cells is still popular today but, as discussed later, it must be viewed with a grain of salt. Kim et al. (21) subjected Ehrlich ascites tumor cells to CDC and demonstrated that osmotic protection effectively prevented cell swelling but did not rescue the cells from death. They hypothesized that the cells died following activation of metabolic events that were detrimental to cell survival or through activation of a “suicidal” mechanism of programmed cell death. In conclusion, osmotic burst of inflated complement-damaged cells may occur, but these bursts are most likely a consequence of metabolic collapse of the cell rather than the cause of cell death.
The Complement Cell Death Mediator: A Concerted Action of Toxic Moieties
Membrane pores caused by complement were first visualized by electron microscopy on red blood cell membranes as large ring structures (22). Similar lesions were viewed on E. coli cell walls (23). Over the years, ample information on the fine ultrastructure of the MAC that can activate cell death has been gathered (24) and has been recently further examined (8–11, 25–27). For a complete updated view of the MAC structure, the reader is referred to those publications. The observed ring structure apparently corresponds to the structure of polymerized C9 molecules attached to their polymerization accelerator, the C5b-8 complex (28). However, even today we have only a partial view of the fine details of the cytotoxic mechanisms activated by MAC, eventually leading to the point of no return and cell death. Besides the paucity of investigations on the subject, several reasons account for that. First, the early dogmas were based on investigations with complement-targeted artificial membranes and red blood cells, which are clearly different, largely passive targets, compared with nucleated cells (29–34). Second, very large variation exists in refractoriness to the MAC, even among closely related cancer cell lines and even within a supposedly homogenous population of cultured cancer cell lines. Third, in target cells MAC activates concomitantly several signaling pathways and biochemical events, some cytotoxic and others pro-survival, and it is the particular balance among them that dictates cell fate, survival, or death. Finally, activation of the terminal complement pathway may result in generating, in the target cell membrane, a cocktail of membrane-inserted protein complexes: C5b-8, C5b-91, C5b-92, C5b-93, and so on, up to C5b-9 with 12-18 polymerized C9 molecules (28, 35). Each of these complexes may induce in the target cell slightly different signals that have not yet been discretely characterized. Detailed analysis of the effect of the terminal complement complex size on the lysis of rat Ehrlich ascites tumor cells by human complement indicated that complexes containing more C9 per C5b-8 are cytolytically more potent. Nevertheless, the kinetics of cell death appeared similar in cells bearing C5b-9 complexes that have either 1 or 4 C9 molecules per C5b-8 (36). Moreover, some human cancer cells, such as U938, HL60, and B-CLL cells, could be lysed by C5b-8 alone, in the absence of C9, when a sufficient number of complexes were deposited on them (37, 38). Hemolysis of sheep red blood cells could be efficiently activated by C5b-9 complexes generated with thrombin-cleaved C9, which cannot undergo classic ring-like polymerization, but forms apparently, string-like oligomeric structures that may lead to leakage of membranes (39, 40). Hence, it is improbable that MAC, with its various intermediary complexes, activates a unified mechanism of cell death in all cell types. An additional level of complexity has been introduced by reports of apoptotic cell death induced by MAC (41), but this has not been observed so far with cancer cells undergoing CDC.
Calcium Ions Influx: Dose-Dependent Dichotomy
At non-toxic or sublytic doses, MAC has been shown to trigger numerous signals in many types of cells, normal and malignant. This topic has been extensively discussed recently and will not be covered here (12–15). Initially, measurements with pigeon erythrocyte sealed “ghosts” revealed an increase in intracellular calcium ions, which begins within seconds after binding of MAC and supposedly precedes the cell death process activated by lytic doses of MAC (42). This transient rise of intracellular free Ca2+ in target cells was thought to be required for cell death. However, later it became apparent that the rise in the level of intracellular calcium ions is essential for cell survival and recovery (43). Reduction of the extracellular Ca2+ concentration by chelation delays the onset of cell death, as measured by LDH release, but the cells eventually die like control cells (44, 45). Similarly, increasing the concentration of Ca2+ around the cells accelerates the rate of cell death without affecting the final percentage of dead cells (36). An intriguing question is: can CDC be blocked by intracellular chelation of the calcium ions? Intracellular Ca2+ chelation with BAPTA-AM was shown to efficiently block mitochondrial distress in human lung epithelial cells responding to a non-lytic dose of MAC, cells that do not undergo cell death (46). Furthermore, calcium ionophores that pump Ca2+ into the cell induce in K562, human erythroleukemia cells, a state of resistance to CDC (47). Can BAPTA-AM block CDC when cells are exposed to lytic MAC doses? BAPTA-AM reduced the release of LDH from rat hepatocytes subjected to lytic antibody and complement by ~40% without affecting the rate of cell death (48). Clearly, MAC activates a surge of [Ca2+]i in target cells but its exact impact on the process of cell death still awaits clarification. Furthermore, based on earlier findings, the involvement of calcium-independent processes in the critical events determining cell death cannot be ruled out.
Beyond Calcium Ions: The Cell Death Propagators in a Regulated Necrotic Process
The molecular checkpoints that tilt the balance within MAC-bearing cells between a protective state and cell collapse have not yet been identified. It is well-accepted that exposure of nucleated cells to multiple (“lytic”) MAC hits (34) is needed to overcome the cells' innate resistance (described below) and to kill the cells by necrotic-type cell death. Intensive research on apoptosis, and more recently on necroptosis induced by numerous effector molecules, has clearly revealed that compound regulated molecular processes accompany and/or lead to cell death (49–52). Those findings have prompted adopting a similar research approach in the analysis of the mechanism underlying CDC. Recently, MAC was shown to activate RIPK1, RIPK3, and MLKL, known transducers of necroptotic cell death activated by several exogenous ligands of TNF receptor, Fas, TLR, and other membrane receptors (53). Necroptotic cell death, also termed regulated necrosis, is characterized by increased membrane permeabilization and mitochondrial damage (49, 50, 54, 55), much like CDC. Inhibitors of RIPK1, RIPK3, and MLKL reduce the extent of CDC, whereas overexpression of these proteins enhances cell sensitivity to CDC (53). Two additional intracellular proteins that may play a role in the multi-factorial cell death process activated by lytic MAC are the c-Jun kinase JNK (56) and the BH3-only protein Bid (57). Apparently, in some cells, the RIP kinases, MLKL, JNK, and Bid, act as components in one or more lined cascades of intracellular molecular interactions activated by sublytic and lytic MAC concentrations (53, 57). At lytic MAC concentrations, this cascade may promote a regulated necrotic cell death (Figure 2). Blocking any of these five proteins markedly lowers the extent of CDC but does not block it completely. Therefore, it appears that this cascade acts in concert with other death-promoting processes, calcium-dependent or -independent, which still await characterization. Of note, activated MLKL was shown in necroptotic cells to oligomerize at the plasma membrane, increase membrane permeabilization, and induce a Ca2+ influx (55, 58–60). Co-localization of MAC with MLKL at the plasma membrane (53) suggests that they may collaborate in mediating cell death. In general, cancer cells that express sufficient levels of the RIPKs, MLKL, and Bid might be sensitive to this necroptotic-like pathway once activated by MAC. In contrast, cancers that suppress the expression or function of any or all of these proteins are expected to be protected from this cytotoxic pathway even if triggered by MAC.
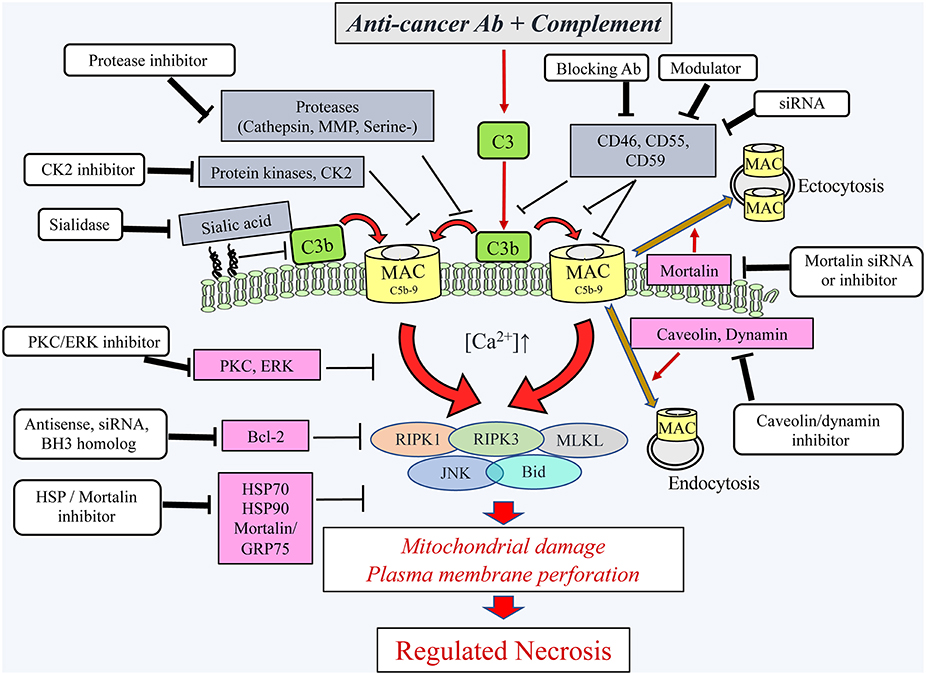
Figure 2. Schematic presentation of the cytotoxic pathways, induced in cancer cells by the complement C5b-9, the counteractive cellular resistance mechanisms, and postulated approaches to overcome this cancer evasion. Following the binding of antibodies to cancer cells, the complement system is activated and deposits C4b and C3b molecules that serve as initiators of C3/C5 convertase activation. The C5 convertases initiate the activation of the terminal complement pathway and the formation of the C5b-9 complexes (24). Upon insertion of the C5b-9 complexes into the plasma membrane of cancer cells, they induce calcium ion influx and activate pro- and anti-lytic signals. This scheme depicts the proteins proposed to be involved in the ensuing cancer cell death (encircled) and the proteins protecting the cancer cells from the lytic processes. Extracellular (gray boxes) and intracellular (purple boxes) protective proteins are indicated. Several reagents (white boxes) that will block the protective proteins are indicated and proposed for adjuvant therapy to therapeutic antibodies. Ab, antibody; Bcl-2, B-cell lymphoma/leukemia-2; BH3, Bcl-2 homolog domain-3; Bid, BH3 interacting domain death agonist; CK2, casein kinase 2; ERK, extracellular signal-regulated kinase; HSP90, heat shock protein 90; HSP70, heat shock protein 70; JNK, c-jun N-terminal kinase; MAC, complement membrane attack complex; MLKL, mixed lineage kinase domain-like protein; MMP, matrix metalloproteinase; PKC, protein kinase C; RIPK1, receptor-interacting protein kinase 1; RIPK3, receptor-interacting protein kinase 3; serine-, serine protease; siRNA, small interfering RNA.
Lytic MAC: Mitotoxicity and Metabolic Depletion
Mitochondria play a pivotal active role in activating the intrinsic pathway of apoptotic cell death, mostly after mitochondrial outer membrane permeabilization (61). Mitochondrial swelling and damage were observed in cells undergoing necrotic cell death induced by complement (62, 63). The cellular ATP level drops rapidly in cells attacked by MAC in copy numbers that are above the lytic threshold, apparently after mitochondrial dysfunction, accompanied by leakage of cytosolic ATP from the cells through pores in the plasma membrane (64–66). In theory, mitochondrial damage and cellular metabolic depletion beyond a point of “no return” may induce cell collapse and death. However, to date, there is no strong evidence supporting an active role for mitochondria in CDC. Perhaps, they are mere innocent bystanders damaged by the necrosis executioners? Reactive oxygen species are generated throughout the necrotic process (15). Still, whether they take part in the MAC-induced cell death process is also an open question. The involvement of mitochondria and mitochondrial ROS in necroptosis triggered by various necroptosis inducers was extensively investigated in several types of target cells (67). Ample earlier findings have supported a pivotal role for mitochondria in necroptosis, but more recently, several investigations casted doubt on that notion (67). Thus, for example, mitochondrially deficient cells were shown to be responsive to TNF/zVAD treatment and to undergo necroptosis (68). It will be of interest to examine the relative sensitivity of these cells to CDC. At present, we can conclude that whether mitochondria are dispensable or essential for MAC-induced necrotic cell death remains to be further investigated. Similar to necroptosis (67), it is likely that different cell types may mediate CDC by an array of distinct mitochondria-dependent and -independent strategies.
Normal Cells and Even More so, Cancer Cells Can Resist Mac-induced Cell Death
The fate of a target cell attacked by MAC is dictated by two mutually exclusive processes: (a) the rate and extent of the formation of the C5b-9 complexes and their insertion into the target cell membrane, and (b) the capacity of the target cell to block C5b-9 complex formation and to resist cell damage inflicted by C5b-9. Cancer cells can resist CDC by using a plethora of extracellular and intracellular mechanisms (Figure 2). The number of membrane-inserted C5b-9 complexes may be restricted by inhibiting the complement activation cascade earlier at the C3/C5 activation stage, by blocking complex assembly and/or membrane insertion, and by facilitating complex removal from the cell surface. All these protective strategies have been identified in cancer cells and are described below. It is generally accepted that cancer cells are more resistant to CDC than are normal cells due to their elevated expression of protective mechanisms. Apparently, during the tumorigenic process, the complement system reacts against the transformed cells by eliminating or modifying the complement-sensitive malignant cells, thus enriching the cancer cell population for complement-resistant cells. This process resembles the selection of antibiotic-resistant bacteria (69). This hypothesis remains to be supported by in vivo evidence; however, in vitro studies show that sensitive cancer cells may be transformed into cells expressing increased complement resistance, transiently, after a brief treatment with a sublytic dose of MAC (70) and more stably, following several cycles of exposures to cytolytic MAC (71). A MAC-resistant phenotype may be acquired upon the elevation or reduction in the expression level of microRNAs such as miR-200, miR-217 (72), and others that are currently under investigation.
Basal Physiological Cell Resistance to CDC
As shown already in 1974 (73, 74), nucleated cells can resist MAC-induced damage. Several inhibitors of protein synthesis were shown to increase the cell's susceptibility to CDC. Since the elevated sensitivity to CDC was acquired hours after the complete shutdown of protein synthesis (75), it is likely that the treated cells became sensitive only after catabolism of long-lived protective proteins. These earlier findings were followed by the development of the concept of multi-hit characteristics of nucleated cell death by MAC, implicating the cooperation of multiple MACs in cell death (34). Another interesting earlier finding was that damaged tumor and mast cells could be rescued by exogenous application of cAMP (76, 77). Consequently, activation of cAMP by sublytic MAC supports cell recovery from MAC damage (78). These findings were confirmed in leukemia cells treated with dibutyryl cAMP or with activators of intracellular cAMP (3-isobutyl 1-methyl xanthine and forskolin), which were shown to reduce cell death (79). In contrast, H-89, an inhibitor of the cAMP-dependent kinase PKA, enhanced carcinoma cell sensitivity to CDC. Apparently, phosphorylation events mediated by several protein kinases dictate the basal capacity of cells to resist MAC damage (79, 80). Protein phosphorylation events involving PKC, MEK, and ERK support the survival of cancer cells undergoing a complement attack (81–83). Protein phosphorylation may upregulate the expression of the complement membrane regulators on cancer cells (84–86) and facilitate MAC elimination from K562 cells (87, 88). The transcription factor NF-κB also plays a role in cell protection from CDC (89). One of its postulated functions is upregulation of a protein phosphatase that inactivates JNK, thus reducing cell death signaling. However, further investigation is required to fully identify the pro-survival phosphoproteins and phosphatases and their precise mode of action.
Proteins of the heat shock protein family (HSPs), well-known general house keepers, damage/ repair proteins and targets in cancer therapy (90–92), most probably also contribute to the basal resistance of cancer cells to CDC. Thus, far, a role for Hsc70/Hsp70 (93) and Hsp90 (94, 95) in cell protection from CDC has been shown. Pharmaceutical inhibitors of Hsp70 and Hsp90 sensitize cancer cells to CDC. Hsc70 relocates within minutes from the cytoplasm to the cell surface after exposing K562 cells to sublytic complement (93). Upon inhibition of Hsp90, Ramos cells become more sensitive to the action of Rituximab and complement (95). Additional thorough experimentation is required to fully comprehend how Hsc70/Hsp70, Hsp90, and other HSPs regulate CDC. The fact that Hsp90 can directly interact with C9 and that Hsp90 inhibitor enhances MAC deposition (95) suggest that Hsp90 down-regulates MAC deposition by blocking its assembly and/or facilitating its rate of removal from the cell surface. Hsp90 can potentially reduce CDC by suppressing mitochondria-initiated calcium-mediated stress responses (96).
Anticomplementary Response on the Cancer Cell Surface
Like all normal cells, cancer cells are protected from autologous complement attack by several specific cell-surface complement inhibitors: CD55 (decay accelerating factor, DAF), CD46 (membrane cofactor protein, MCP), CD59, and CD35 (complement receptor type 1, CR1) (1, 97–99) (Figure 1). In addition, certain proteolytic enzymes, protein kinases, and sialic acid residues (described below) confer on the cells elevated resistance to CDC (100).
Membrane Complement Regulatory Proteins
Immunohistochemical analysis revealed the expression of CD59, CD55, and CD46 on uveal melanoma (101), thyroid carcinoma (102), lung and kidney cancer (103, 104), colon adenocarcinoma (105), and prostate cancer (106). This was supported by analysis of human tumor cell lines derived from human malignant gastrointestinal tumors (107), melanoma (108), breast cancer (109, 110), renal tumor (110), Burkitt lymphoma (111), neuroblastoma (112), and ovarian (113), and prostate carcinoma (79). In primary uterine cervix tissue, the expression of CD46, but not of CD55, was found to increase during transition from normal to premalignant to malignant cells (114). CR1/CD35 was identified in malignant endometrial tissue (115) and on leukemic blasts (116). Increased membrane regulator expression relative to the corresponding normal tissue has been reported in many tumors (103, 105, 114, 115, 117–122). Colorectal and gastric carcinomas and osteosarcoma have increased the expression of CD55 (123), whereas gastric carcinoma exhibits high levels of both CD55 and CD59 (124). Overexpression of CD59 was also identified by expression profiling for pancreatic cancer (125). Upregulation of membrane regulator expression on tumor cells is often correlated with increased complement resistance (86, 111). In ovarian cancer, resistance to complement correlated with high levels of CD55 expression (113). In melanoma cell lines with variable CD59 expression, resistance to death by anti-ganglioside antibody and homologous complement positively correlated with the expression level of CD59 (126).
Several clinical studies support a postulated function of membrane complement regulatory proteins (and thus, the extent of complement resistance) in cancer progression. Poorer prognosis in colorectal carcinoma correlates with the expression level of CD59 (127). Local tumor progression and tissue dedifferentiation of prostate cancer also correlate with CD59 expression (128). Analysis of 120 breast cancer patients revealed a worse prognosis associated with CD59 overexpression (129). In contrast, another report concluded that loss of CD59 correlated with poor survival in 520 breast cancer patients (130). In colorectal cancer patients, a 7-year survival was significantly reduced when the tumors expressed high levels of CD55 (131). CD55 overexpression was also reported as an independent risk factor for recurrence of breast cancer in patients receiving postoperative adjuvant therapy containing trastuzumab (132).
The expression level of the membrane complement regulators may also be shaped by cytokines, growth factors, or hormones, which are released into the tumor microenvironment (83, 133, 134). For example, TNFα and IL-1ß enhanced the expression of CD55 and CD59 in colon adenocarcinoma cells (135). TNFα, IL-1α, and INFγ enhanced CD55 expression in lung cancer cells (136). In hepatoma cells, TNFα, combined with IL-1ß and IL-6, enhanced CD55 and CD59 expression but decreased CD46 expression (134). Transcription abnormalities (137) and the microRNA level of expression (72) may also affect the expression level of the membrane complement regulators. Evidently, the factors and molecular mechanisms that determine the expression level of each of the membrane regulator proteins in vivo in each cancer type (and in normal cells) remain to be further investigated.
Exposure to chemotherapeutic drugs may also modify the level of the regulators′ expression. 5-azacytidine was shown to elevate the levels of CD55 and CD59 in Burkitt lymphoma cell lines (111) but only of CD59 in melanoma cells. In contrast, levamisole reduces CD59 levels in colon adenocarcinoma cell lines (138) and after pretreatment of breast carcinoma cells with tamoxifen, trastuzumab-induced CDC was enhanced due to CD55 down-regulation (132). Conversion of cancer cells from being drug-sensitive to drug-resistant is also associated with modification of their complement sensitivity. Doxorubicin-resistant human colon carcinoma cells are more sensitive to CDC than are doxorubicin-sensitive cells (139). KB-V1, a multidrug-resistant variant of KB-3-1, the human oral carcinoma cell line, exhibits a higher susceptibility to CDC than do its parental multidrug-sensitive cells (140). The increased complement sensitivity was associated with a reduced expression of CD55. Inversely, drug resistance was associated with CDC resistance in the HL60 myeloid leukemia cell lines (141). In ovarian carcinoma cells, drug resistance was associated with complement resistance and with membrane complement regulator overexpression (142). Hence, the impact of any drug on the expression of membrane regulators and on CDC resistance needs to be determined for each drug and cancer type.
Released or secreted membrane complement regulators in the cancer microenvironment may also support cell resistance to CDC. Soluble forms of membrane regulators have been identified in several body fluids, even under normal conditions. They are either produced by alternative splicing or released from the cell surface through enzymatic cleavage. Thus, sera of cancer patients contain active, soluble forms of CD46 (143). Elevated CD55 concentrations in stool specimens have been proposed to have diagnostic value for patients with colorectal cancer (144). A constitutive release of soluble CD59, which retains its activity as well as its GPI-anchor from human melanoma cells, was reported (145). In primary tumor sections, CD55 and/or CD59 were found in the stroma of breast, colorectal, lung, renal, and cervical carcinomas (103, 123, 146). In vitro, endothelial cells, HeLa cells (147) as well as osteosarcoma and colorectal cells (123, 148) release CD55 in a soluble form or deposit it into their extracellular matrix. K562 erythroleukemia cells (83) and breast, ovarian, and prostate carcinoma cell lines (79) secrete soluble CD59. Elevated plasma levels of soluble CR1 were found in leukemia patients (149).
The observed correlations between elevated expression or secretion of one or more of the membrane complement regulatory proteins on cancer cells and (a) enhanced resistance to CDC or (b) poor cancer prognosis, suggests that the membrane complement regulatory proteins have an effect on prognosis through their impact on complement resistance. Thus, by suppressing C3 deposition on the cancer cells, CD46 and CD55 can lower, on one hand, the extent of MAC generation and CDC, and on the other hand, reduce immune protection through complement-dependent cellular cytotoxicity. CD59 can down-regulate MAC generation and CDC. However, a direct correlation between cancer patients' prognosis and the complement resistance level of their cancer, still remains to be established. We cannot rule out non-complement-mediated effects of the membrane complement regulatory proteins of cancer cells on the patients' immune response. Thus, the membrane complement regulatory proteins on cancer cells, through intracellular signaling, or cooperation with other cell surface receptors may potentially modulate cell resistance to immune effector cells such as natural killer cells and cytotoxic T lymphocytes (150, 151).
Membrane Surface Proteases, Protein Kinases, and Sialic Acid
Cancer cells become increasingly protected from CDC by expression on their cell surface of proteases that proteolytically degrade the deposited complement proteins (152). Thus, degradation of bound C3b by a C3-cleaving serine (153) or cysteine protease (154), respectively, was demonstrated on human and murine melanoma cells. C3-cleaving serine protease activity was also identified on the surface of U937 cells (155). Membrane serine proteases on K562 erythroleukemia cells also appear to contribute to their complement resistance (156). Matrix metalloproteinases (MMP) membrane type-1 (MT1) can cleave bound C3b off breast cancer cells and protect in vitro breast carcinoma and melanoma cells from CDC (157). Transfection of B16F1 melanoma cells with MT1-MMP enhanced their capacity to form lung metastases in normal but not in C3-deficient C57BL/6 mice (157). The effect of those proteases on proteins of the terminal complement pathway has not been tested. However, it is conceivable that these and other membrane proteases have a similar degradative impact on C5-C9. This still awaits determination.
Ecto-protein kinases (ecto-PK) are extracellular protein kinases that can phosphorylate both cell-surface and external proteins. Serine/threonine and tyrosine ecto-PKs were found on the surfaces of K562, U937, and HL-60 cells (158), and an ecto-casein kinase 2 (CK2)-like activity was associated with breast and ovarian carcinoma cells (159). C9 phosphorylation by ecto-CK2 was shown to be protective in K562 cells against CDC, possibly by inhibiting MAC formation or by leading to the production of an inactive or unstable MAC (160). Further investigation of this strategy of CDC evasion is warranted.
Brief treatment with sialidase, which removes sialic acid from the cell surface, has been shown to confer on several cell types increased sensitivity to CDC. Thus, removal of sialic acid from red blood cells (161, 162), murine sarcoma cells (163), and human bladder carcinoma cells (164) sensitized them to lysis by complement. Human prostate, breast, and ovarian carcinoma cells also utilize surface sialylation for protection from complement (79). High sialic acid expression correlates with lower complement activation, probably because of inactivation of C3b by factors H and I, which is more efficient on surfaces rich in sialic acid (162). The sialic acid inhibitory activity on CDC of mouse erythrolukemia MEL cells is apparently abrogated by O-acetylation at its 9-hydroxyl group (165). α2-6 hypersialylation apparently lowers the response of CLL cells to Rituximab therapy through its action on complement (166). Thus, by limiting the extent of C3 deposition, sialic acid may also control the assembly of C5b-9 complexes on the cancer cells.
Soluble Complement Regulators in the Cancer Microenvironment
Soluble complement inhibitors such as C1 Inhibitor, factor H, and factor I are predominantly synthesized by hepatocytes and macrophages but can also be released from other tissues, although in considerably smaller amounts. In the cancer microenvironment, these secreted inhibitors may contribute to protection of cancer cells from complement attack by blocking complement activation at the C1 and C3 activation steps (99). In support of this, a growing number of reports indicate that cancer cells of various origins secrete one or more complement inhibitor. Synthesis of C1 Inhibitor has been described in astroglioma and neuroblastoma (112), breast cancer cell lines, and in a primary ovarian carcinoma cell line (156). Factor H is expressed both in lung adenocarcinoma and cutaneous squamous cell carcinoma (167, 168) and high levels of factor H and factor H-like protein-1 were shown to be secreted by ovarian tumor cells (83, 169). Additionally, factor H was found to be elevated in bronchoalveolar lavage fluids and the sputum of patients with lung cancer (170). Chronic lymphocytic leukemia (CLL) cells that bind factor H to their surface resisted Rituximab-mediated CDC (171). Factor H was coexpressed with factor I in glioma and rhabdomyosarcoma cells in its plasma form and in a truncated form (172). Tumor-associated factor I is postulated to promote the progression of cutaneous squamous cell carcinoma (173) and positively correlates with poor survival and recurrence of breast cancer (174).
Active Removal of the Membrane-Inserted MAC
An additional important defensive tactic used by cancer cells to resist CDC is rapid elimination of MAC from the cell surface. This was first shown with U937 histiocytic leukemia cells, Ehrlich ascites tumor cells (175, 176), and neutrophils (177). Neutrophils remove MAC both by endocytosis and exocytosis (178). Elimination of MAC by exo-vesiculation has been described in glomerular epithelial cells, platelets, and oligodendrocytes (179–181). The intracellular signals involved in MAC elimination include Gi proteins (182), PKC and ERK (88, 183). The process of MAC removal through outward and inward vesiculation was imaged in MAC-bearing K562 erythroleukemia cells (184). Membrane vesicles shed from MAC-bearing neutrophils contain MAC and have elevated levels of cholesterol and diacylglycerol, suggesting selective membrane protein and lipid sorting during the ectocytosis process (185). In support, the elimination of MAC by endocytosis is inhibited in K562 cells after cholesterol depletion (186). MAC endocytosis in K562 cells largely depends on caveolae and dynamin-dependent intracellular release of MAC-loaded endosomes (186). The process of MAC removal by exo-vesiculation was also partially characterized in K562 cells and was found to require the expression of the mitochondrial stress protein mortalin/GRP75 (87). Mortalin is over-expressed in many cancer types and is an essential survival stress protein (187). It was shown to be significantly protective from CDC (188). Its exact mode of action remains to be elucidated; however, evidently, mortalin inhibitors efficiently sensitize K562 cells and colorectal carcinoma HCT116 cells to CDC (189).
Intervention Strategies to Overcome Cancer Resistance to CDC
As previously described, cancer cells escape CDC through amplification of an array of resistance strategies that block the formation of MAC, facilitate MAC elimination from the cell surface, or inhibit the cytotoxic consequences of MAC insertion into the plasma membrane (Figure 2). In order to overcome that resistance, more potent antibodies and polymeric antibodies have been engineered (5–7). Attachment of complement-activating proteins such as CVF, C3b, C7, or C9 directly to therapeutic antibodies represents an alternative means to strengthen complement attack and thereby to overcome complement resistance of cancer cells (190–193). Here, we will restrict our description to intervention strategies that may be or have been developed to augment the CDC of cancer cells by weakening their anti-MAC resistance mechanisms. These include the following: (1) blocking or silencing the membrane complement regulatory proteins, (2) inhibiting the extracellular enzymes that interfere with complement activation, and (3) inhibiting the intracellular pathways that support cell resistance and recovery (Figure 2). An additional, yet unexplored approach, which is based on the earlier findings, is targeting a sialidase to the cancer microenvironment or blocking the sialylation of surface glycoconjugates in cancer cells, which is expected to sensitize them to CDC.
Antibody-Mediated Neutralization of Complement Regulator Expression
Specific inhibition of complement regulators' activity is best achieved with monoclonal antibodies that enhance the susceptibility of cancer cells to CDC (86, 194, 195). Thus, blocking antibodies markedly enhance the anti-tumor activity of Rituximab in vitro and in vivo (196). Neutralization of CD55 in Burkitt lymphoma cells (111), leukemia cells (196–199), melanoma cells (200), and breast cancer cells (86) increased their sensitivity to complement. Similarly, inhibition of CD59 with a monoclonal antibody led to efficient sensitization to CDC of neuroblastoma cells (112), leukemic cells (83, 199), breast (86), ovarian (113), renal (201), and prostate carcinoma cells (106). Mini-antibodies targeting both CD55 and CD59 were shown to enhance Rituximab-dependent CDC in vitro and to increase the survival of Rituximab-treated SCID mice in a xenograft model of human CD20+ B-cell lymphoma (196). Bispecific antibodies targeting both CD20 and CD55 or CD20 and CD59 were also shown to potentiate the CDC of CD20-positive lymphoma cells in vitro and to prevent the growth of human lymphoma cells in SCID mice (202). Neutralization of the soluble complement regulators may also be applied to cancer immunotherapy. Thus, anti-factor H antibody increased antibody-dependent CDC of colorectal cancer treated with anti-CEA monoclonal antibody (203). Inhibition of factor H activity with a recombinant protein reflecting the factor H short-consensus repeat 18–20 improved the CDC of CLL cells in the presence of Rituximab and the blockage of CD55 and CD59 further enhanced CDC (171).
Silencing of Complement Regulators' Expression by RNAi
Another specific approach is to knock down the expression of the membrane complement regulators' expression by siRNAs. RNA interference (RNAi), mediated by small interfering RNA (siRNA), is the most efficient strategy for specific silencing of therapeutically relevant genes (204). In the last years numerous strategies have been developed for a better delivery of siRNAs in vitro and in vivo (205). We have shown that silencing of single or multiple complement regulators by anti-sense oligonucleotides or siRNAs results in a significant increase of opsonization and CDC of tumor cell lines of various histological origin (194, 206). Silencing of CD55 and CD59 in breast cancer cells with specific shRNA enhanced CDC (207). Using chemically stabilized anti-complement regulators, siRNAs and AtuPLEX, we observed a significant knockdown of regulator expression on HER2-positive carcinoma cells. Subsequently, treatment with a combination of two anti-HER2 antibodies, trastuzumab and pertuzumab, and normal human serum, augmented C3 binding and CDC could be recorded (208). Similar results were observed with lymphoma cells in which silencing of complement regulators enhanced antibody-dependent CDC (209). For specific delivery of liposomes or lipoplexes loaded with siRNA molecules into cancer cells, transferrin may be attached to them to facilitate their binding to cancer cells through transferrin receptor (TfR/CD71) and their active entry into the cells (210). By using this approach, delivery of siRNA molecules specific to CD46, CD55, and CD59 to transferrin receptor-positive carcinoma cells was achieved and promoted the knockdown of the complement regulators and enhanced CDC (211).
Neutralization of Extracellular Protective Enzymes
Considering the aforementioned anti-complement effects of certain proteases and protein kinases, it is likely that tailor-made protease and/or kinase inhibitors will promote antibody-based immunotherapy. In vitro and a few in vivo results support this hypothesis. Treatment of K562 cells with serine protease inhibitors markedly enhanced their sensitivity to CDC (156). CK2 inhibitors also augmented Raji cell killing by Rituximab and complement (160). Single-chain variable fragment (ScFv) directed to cathepsin L was used to inhibit the tumorigenic and metastatic phenotype of human melanoma cells in nude mice (212). In addition, injection of an anti-cathepsin L ScFv lentiviral vector into tumors already induced in nude mice inhibited tumor growth and associated angiogenesis (213). Whether or not the complement system is involved in the latter anti-tumor effects of the anti-cathepsin L treatment remains unresolved.
Inhibition of Intracellular Protective Pathways
As described above, the list of intracellular molecular pathways supporting cancer cell resistance to CDC is increasing. Currently, we can hypothesize that a coordinated inhibition of any of the following active molecules in the following cancer cells: cAMP, PKC, MEK/ERK, Hsp70, Hsc70, Hsp90, and mortalin, combined with complement-activating antibody, will amplify cancer cell death and increase the sensitivity of cancer to immunotherapy. For each of these molecules, this claim has been clearly supported in vitro by data and now awaits in vivo testing. Inhibition of PKC and MEK1, the ERK kinase, lowers the rate of MAC elimination from the cells and sensitizes them to CDC (81, 82, 88, 183). Inhibitors of MEK-ERK are in clinical use now in cancer therapy (214–216), and testing their impact on the therapeutic efficacy of anti-cancer antibodies is highly warranted. Heat shock proteins are over-expressed in cancer and play a significant role in resistance to various types of therapy (217). The list of inhibitors of heat shock proteins that have been developed for clinical use is growing and a few have entered clinical trials (90–92, 218, 219). The use of these heat shock protein inhibitors as adjuvants to antibody-based therapy may yield a superior clinical outcome. Mortalin belongs to the family of heat shock proteins and is also over-expressed in cancer (187). The mortalin expression level in colorectal adenocarcinoma cells correlates with poor patient survival (220). Mortalin inhibitors like MKT-077 could be considered as complementary treatment to anticancer antibody therapy. In support, pretreatment with MKT-077 sensitized K562 cells to CDC (87, 189). Unfortunately, thus far, testing of MKT-077 in patients has been stalled due to toxicity effects (221) and alternative inhibitors are being sought. Mortalin silencing with specific siRNA reduced MAC elimination and increased the sensitivity of K562 cells to CDC (189). Therefore, it is reasonable to predict that combining reagents that knockdown or inhibit mortalin with anti-cancer antibody therapy will be advantageous to cancer patients.
Concluding Remarks
Complement activation on and around cancer cells has been postulated to elicit several concomitant physiological and immunological responses that may act cooperatively to either mediate cancer cell death or promote cell survival, growth, and metastasis. In theory, these responses may also negate and annul each other. Multiple strategies to overcome complement resistance, as described here, open up new opportunities for improving antibody-based immunotherapy. Undoubtedly, applying any of the intervention treatments described above, together with a therapeutic antibody, will produce on and around the cancer cells/mass, besides C5b-9 complexes, additional complement activation products, such as cancer-bound iC3b, which promotes antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cellular cytotoxicity (CDCC) as well as C3a and C5a, which may suppress cellular anti-cancer immune response. Consequently, in the worst scenario, intervention strategies to augment complement activation may worsen the outcome of the anti-cancer antibody therapy. Hence, for each cancer type, therapeutic antibody, and intervention strategy, an optimal protocol will have to be developed that favors cancer destruction over cancer promotion.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the German Federal Ministry of Education and Research (BMBF BIODISC program), the Cooperation Program in Cancer Research of the Deutsches Krebsforschungszentrum (DKFZ) and Israeli's Ministry of Science (MK and ZF), and grants from the Israel Science Foundation and the Israel Cancer Association (ZF).
References
1. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
2. Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD. Complement in cancer: untangling an intricate relationship. Nat Rev Immunol. (2018) 18:5–18. doi: 10.1038/nri.2017.97
3. Sayegh ET, Bloch O, Parsa AT. Complement anaphylatoxins as immune regulators in cancer. Cancer Med. (2014) 3:747–58. doi: 10.1002/cam4.241
4. Kochanek DM, Ghouse SM, Karbowniczek MM, Markiewski MM. Complementing cancer metastasis. Front Immunol. (2018) 9:1629. doi: 10.3389/fimmu.2018.01629
5. Taylor RP, Lindorfer MA. Cytotoxic mechanisms of immunotherapy: harnessing complement in the action of anti-tumor monoclonal antibodies. Semin Immunol. (2016) 28:309–16. doi: 10.1016/j.smim.2016.03.003
6. Stern M, Herrmann R. Overview of monoclonal antibodies in cancer therapy: present and promise. Crit Rev Oncol Hematol. (2005) 54:11–29. doi: 10.1016/j.critrevonc.2004.10.011
7. Mamidi S, Hone S, Kirschfink M. The complement system in cancer: ambivalence between tumour destruction and promotion. Immunobiology. (2017) 222:45–54. doi: 10.1016/j.imbio.2015.11.008
8. Sharp TH, Koster AJ, Gros P. Heterogeneous MAC initiator and pore structures in a lipid bilayer by phase-plate cryo-electron tomography. Cell Rep. (2016) 15:1–8. doi: 10.1016/j.celrep.2016.03.002
9. Bayly-Jones C, Bubeck D, Dunstone MA. The mystery behind membrane insertion: a review of the complement membrane attack complex. Philos Trans R Soc Lond B Biol Sci. (2017) 372:20160221. doi: 10.1098/rstb.2016.0221
10. Morgan BP, Boyd C, Bubeck D. Molecular cell biology of complement membrane attack. Semin Cell Dev Biol. (2017) 72:124–32. doi: 10.1016/j.semcdb.2017.06.009
11. Dudkina NV, Spicer BA, Reboul CF, Conroy PJ, Lukoyanova N, Elmlund H, et al. Structure of the poly-C9 component of the complement membrane attack complex. Nat Commun. (2016) 7:10588. doi: 10.1038/ncomms10588
12. Tegla CA, Cudrici C, Patel S, Trippe R III, Rus V, Niculescu F, et al. Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunol Res. (2011) 51:45–60. doi: 10.1007/s12026-011-8239-5
13. Morgan BP. The membrane attack complex as an inflammatory trigger. Immunobiology. (2016) 221:747–51. doi: 10.1016/j.imbio.2015.04.006
14. Bohana-Kashtan O, Ziporen L, Donin N, Kraus S, Fishelson Z. Cell signals transduced by complement. Mol Immunol. (2004) 41:583–97. doi: 10.1016/j.molimm.2004.04.007
15. Takano T, Elimam H, Cybulsky AV. Complement-mediated cellular injury. Semin Nephrol. (2013) 33:586–601. doi: 10.1016/j.semnephrol.2013.08.009
16. Kalfayan B, Kidd JG. Structural changes produced in Brown-Pearce carcinoma cells by means of a specific antibody and complement. J Exp Med. (1953) 97:145–62. doi: 10.1084/jem.97.1.145
17. Ellem KA. Some aspects of the ascites tumor cell response to a heterologous antiserum. Cancer Res. (1958) 18:1179–85.
18. Green H, Fleischer RA, Barrow P, Goldberg B. The cytotoxic action of immune gamma globulin and complement on Krebs ascites tumor cells. II. Chemical studies. J Exp Med. (1959) 109:511–21. doi: 10.1084/jem.109.5.511
19. Green H, Goldberg B. The action of antibody and complement on mammalian cells. Ann N Y Acad Sci. (1960) 87:352–62. doi: 10.1111/j.1749-6632.1960.tb23205.x
20. Green H, Barrow P, Goldberg B. Effect of antibody and complement on permeability control in ascites tumor cells and erythrocytes. J Exp Med. (1959) 110:699–713. doi: 10.1084/jem.110.5.699
21. Kim SH, Carney DF, Papadimitriou JC, Shin ML. Effect of osmotic protection on nucleated cell killing by C5b-9: cell death is not affected by the prevention of cell swelling. Mol Immunol. (1989) 26:323–31. doi: 10.1016/0161-5890(89)90087-4
22. Rosse WF, Dourmashkin R, Humphrey JH. Immune lysis of normal human and paroxysmal nocturnal hemoglobinuria (PNH) red blood cells. 3. The membrane defects caused by complement lysis. J Exp Med. (1966) 123:969–84. doi: 10.1084/jem.123.6.969
23. Medhurst FA, Glynn AA, Dourmashkin RR. Lesions in Escherichia coli cell walls caused by the action of mouse complement. Immunology. (1971) 20:441–50.
24. Muller-Eberhard HJ. The membrane attack complex of complement. Annu Rev Immunol. (1986) 4:503–28. doi: 10.1146/annurev.iy.04.040186.002443
25. Heesterbeek DA, Bardoel BW, Parsons ES, Bennett I, Ruyken M, Doorduijn DJ, et al. Bacterial killing by complement requires membrane attack complex formation via surface-bound C5 convertases. EMBO J. (2019) 38:e99852. doi: 10.15252/embj.201899852
26. Spicer BA, Law RHP. Caradoc-Davies TT, Ekkel SM, Bayly-Jones C, Pang SS, et al., The first transmembrane region of complement component-9 acts as a brake on its self-assembly. Nat Commun. (2018) 9:3266. doi: 10.1038/s41467-018-05717-0
27. Menny A, Serna M, Boyd CM, Gardner S, Joseph AP, Morgan BP, et al. CryoEM reveals how the complement membrane attack complex ruptures lipid bilayers. Nat Commun. (2018) 9:5316. doi: 10.1038/s41467-018-07653-5
28. Tschopp J, Podack ER, Muller-Eberhard HJ. Ultrastructure of the membrane attack complex of complement: detection of the tetramolecular C9-polymerizing complex C5b-8. Proc Natl Acad Sci USA. (1982) 79:7474–8. doi: 10.1073/pnas.79.23.7474
29. Haxby JA, Gotze O, Muller-Eberhard HJ, Kinsky SC. Release of trapped marker from liposomes by the action of purified complement components. Proc Natl Acad Sci USA. (1969) 64:290–5. doi: 10.1073/pnas.64.1.290
30. Michaels DW, Abramovitz AS, Hammer CH, Mayer MM. Increased ion permeability of planar lipid bilayer membranes after treatment with the C5b-9 cytolytic attack mechanism of complement. Proc Natl Acad Sci USA. (1976) 73:2852–6. doi: 10.1073/pnas.73.8.2852
31. Benz R, Schmid A, Wiedmer T, Sims PJ. Single-channel analysis of the conductance fluctuations induced in lipid bilayer membranes by complement proteins C5b-9. J Membr Biol. (1986) 94:37–45. doi: 10.1007/BF01901011
32. Bhakdi S, Tranum-Jensen J. Membrane damage by complement. Biochim Biophys Acta. (1983) 737:343–72. doi: 10.1016/0304-4157(83)90006-0
33. Esser AF, Kolb WP, Podack ER, Muller-Eberhard HJ. Molecular reorganization of lipid bilayers by complement: a possible mechanism for membranolysis. Proc Natl Acad Sci USA. (1979) 76:1410–4. doi: 10.1073/pnas.76.3.1410
34. Koski CL, Ramm LE, Hammer CH, Mayer MM, Shin ML. Cytolysis of nucleated cells by complement: cell death displays multi-hit characteristics. Proc Natl Acad Sci USA. (1983) 80:3816–20. doi: 10.1073/pnas.80.12.3816
35. Bhakdi S, Tranum-Jensen J. C5b-9 assembly: average binding of one C9 molecule to C5b-8 without poly-C9 formation generates a stable transmembrane pore. J Immunol. (1986) 136:2999–3005.
36. Kim SH, Carney DF, Hammer CH, Shin ML. Nucleated cell killing by complement: effects of C5b-9 channel size and extracellular Ca2+ on the lytic process. J Immunol. (1987) 138:1530–6.
37. Morgan BP, Imagawa DK, Dankert JR, Ramm LE. Complement lysis of U937, a nucleated mammalian cell line in the absence of C9: effect of C9 on C5b-8 mediated cell lysis. J Immunol. (1986) 136:3402–6.
38. Taylor RP, Lindorfer MA, Cook EM, Beurskens FJ, Schuurman J, Parren P, et al. Hexamerization-enhanced CD20 antibody mediates complement-dependent cytotoxicity in serum genetically deficient in C9. Clin Immunol. (2017) 181:24–8. doi: 10.1016/j.clim.2017.05.016
39. Biesecker G, Gerard C, Hugli TE. An amphiphilic structure of the ninth component of human complement. Evidence from analysis of fragments produced by alpha-thrombin. J Biol Chem. (1982) 257:2584–90.
40. Dankert JR, Esser AF. Proteolytic modification of human complement protein C9: loss of poly(C9) and circular lesion formation without impairment of function. Proc Natl Acad Sci USA. (1985) 82:2128–32. doi: 10.1073/pnas.82.7.2128
41. Nauta AJ, Daha MR, Tijsma O, van de Water B, Tedesco F, Roos A. The membrane attack complex of complement induces caspase activation and apoptosis. Eur J Immunol. (2002) 32:783–92. doi: 10.1002/1521-4141(200203)32:3<783::AID-IMMU783>3.0.CO;2-Q
42. Campbell AK, Daw RA, Hallett MB, Luzio JP. Direct measurement of the increase in intracellular free calcium ion concentration in response to the action of complement. Biochem J. (1981) 194:551–60. doi: 10.1042/bj1940551
43. Morgan BP, Luzio JP, Campbell AK. Intracellular Ca2+ and cell injury: a paradoxical role of Ca2+ in complement membrane attack. Cell Calcium. (1986) 7:399–411. doi: 10.1016/0143-4160(86)90042-4
44. Papadimitriou JC, Phelps PC, Shin ML, Smith MW, Trump BF. Effects of Ca2+ deregulation on mitochondrial membrane potential and cell viability in nucleated cells following lytic complement attack. Cell Calcium. (1994) 15:217–27. doi: 10.1016/0143-4160(94)90061-2
45. Beum PV, Lindorfer MA, Peek EM, Stukenberg PT, de Weers M, Beurskens FJ, et al. Penetration of antibody-opsonized cells by the membrane attack complex of complement promotes Ca(2+) influx and induces streamers. Eur J Immunol. (2011) 41:2436–46. doi: 10.1002/eji.201041204
46. Triantafilou K, Hughes TR, Triantafilou M, Morgan BP. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci. (2013) 126:2903–13. doi: 10.1242/jcs.124388
47. Reiter Y, Ciobotariu A, Jones J, Morgan BP, Fishelson Z. Complement membrane attack complex, perforin, and bacterial exotoxins induce in K562 cells calcium-dependent cross-protection from lysis. J Immunol. (1995) 155:2203–10.
48. Nishikawa Y, Ukida M, Matsuo R, Omori N, Tsuji T. Ca2+ influx initiates death of hepatocytes injured by activation of complement. Liver. (1994) 14:200–5. doi: 10.1111/j.1600-0676.1994.tb00074.x
49. Galluzzi L, Kepp O, Chan FK, Kroemer G. Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol. (2017) 12:103–30. doi: 10.1146/annurev-pathol-052016-100247
50. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. (2017) 18:127–36. doi: 10.1038/nrm.2016.149
51. Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. (2002) 2:277–88. doi: 10.1038/nrc776
52. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. (2004) 116:205–19. doi: 10.1016/S0092-8674(04)00046-7
53. Lusthaus M, Mazkereth N, Donin N, Fishelson Z. Receptor-interacting protein Kinases 1 and 3, and mixed lineage kinase domain-like protein are activated by sublytic complement and participate in complement-dependent cytotoxicity. Front Immunol. (2018) 9:306. doi: 10.3389/fimmu.2018.00306
54. Vanden Berghe T, Hassannia B, Vandenabeele P. An outline of necrosome triggers. Cell Mol Life Sci. (2016) 73:2137–52. doi: 10.1007/s00018-016-2189-y
55. Petrie EJ, Czabotar PE, Murphy JM. The structural basis of necroptotic cell death signaling. Trends Biochem Sci. (2019) 44:53–63. doi: 10.1016/j.tibs.2018.11.002
56. Gancz D, Donin N, Fishelson Z. Involvement of the c-jun N-terminal kinases JNK1 and JNK2 in complement-mediated cell death. Mol Immunol. (2009) 47:310–7. doi: 10.1016/j.molimm.2009.09.016
57. Ziporen L, Donin N, Shmushkovich T, Gross A, Fishelson Z. Programmed necrotic cell death induced by complement involves a Bid-dependent pathway. J Immunol. (2009) 182:515–21. doi: 10.4049/jimmunol.182.1.515
58. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. (2014) 16:55–65. doi: 10.1038/ncb2883
59. Xia B, Fang S, Chen X, Hu H, Chen P, Wang H, et al. MLKL forms cation channels. Cell Res. (2016) 26:517–28. doi: 10.1038/cr.2016.26
60. Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. (2014) 7:971–81. doi: 10.1016/j.celrep.2014.04.026
61. Tait SW, Green DR. Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol. (2013) 5:a008706. doi: 10.1101/cshperspect.a008706
62. Papadimitriou JC, Drachenberg CB, Shin ML, Trump BF. Ultrastructural studies of complement mediated cell death: a biological reaction model to plasma membrane injury. Virchows Arch. (1994) 424:677–85. doi: 10.1007/BF00195784
63. Goldberg B, Green H. The cytotoxic action of immune gamma globulin and complement on Krebs ascites tumor cells. I. Ultrastructural studies. J Exp Med. (1959) 109:505–10. doi: 10.1084/jem.109.5.505
64. Sala-Newby GB, Taylor KM, Badminton MN, Rembold CM, Campbell AK. Imaging bioluminescent indicators shows Ca2+ and ATP permeability thresholds in live cells attacked by complement. Immunology. (1998) 93:601–9. doi: 10.1046/j.1365-2567.1998.00004.x
65. Papadimitriou JC, Ramm LE, Drachenberg CB, Trump BF, Shin ML. Quantitative analysis of adenine nucleotides during the prelytic phase of cell death mediated by C5b-9. J Immunol. (1991) 147:212–7.
66. Tirosh R, Degani H, Berke G. Prelytic reduction of high-energy phosphates induced by antibody and complement in nucleated cells. 31P-NMR study. Complement. (1984) 1:207–12. doi: 10.1159/000467839
67. Marshall KD, Baines CP. Necroptosis: is there a role for mitochondria? Front Physiol. (2014) 5:323. doi: 10.3389/fphys.2014.00323
68. Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. (2013) 5:878–85. doi: 10.1016/j.celrep.2013.10.034
69. Courvalin P. Why is antibiotic resistance a deadly emerging disease? Clin Microbiol Infect. (2016) 22:405–7. doi: 10.1016/j.cmi.2016.01.012
70. Reiter Y, Ciobotariu A, Fishelson Z. Sublytic complement attack protects tumor cells from lytic doses of antibody and complement. Eur J Immunol. (1992) 22:1207–13. doi: 10.1002/eji.1830220515
71. Fishelson Z, Reiter Y. Monoclonal antibody-C3b conjugates: killing of K562 cells and selection of a stable complement resistant variant. In: Groopman J, Evans C, and Golde D, editors. Mechanisms of Action and Therapeutic Application of Biologicals in Cancer and Immune Deficiency Disorders. New York, NY: Alan R. Liss (1989). p. 273–82.
72. Hillman Y, Mazkereth N, Farberov L, Shomron N, Fishelson Z. Regulation of complement-dependent cytotoxicity by MicroRNAs miR-200b, miR-200c, and miR-217. J Immunol. (2016) 196:5156–65. doi: 10.4049/jimmunol.1502701
73. Segerling M, Ohanian SH, Borsos T. Effect of metabolic inhibitors on killing of tumor cells by antibody and complement. J Natl Cancer Inst. (1974) 53:1411–3. doi: 10.1093/jnci/53.5.1411
74. Ferrone S, Pellegrino MA, Dierich MP, Reisfeld RA. Effect of inhibitors of macromolecular synthesis on HL-A antibody mediated lysis of cultured lymphoblasts. Tissue Antigens. (1974) 4:275–82. doi: 10.1111/j.1399-0039.1974.tb00253.x
75. Schlager SI, Boyle MD, Ohanian SH, Borsos T. Effect of inhibiting DNA, RNA, and protein synthesis of tumor cells on their susceptibility to killing by antibody and complement. Cancer Res. (1977) 37:1432–7.
76. Boyle MD, Ohanian SH, Borsos T. Studies on the terminal stages of antibody-complement-mediated killing of a tumor cell. II. Inhibition of transformation of T to dead cells by 3'5' cAMP. J Immunol. (1976) 116:1276–9.
77. Kaliner M, Austen KF. Adenosine 3'5'-monophosphate: inhibition of complement-mediated cell lysis. Science. (1974) 183:659–61. doi: 10.1126/science.183.4125.659
78. Patel AK, Campbell AK. The membrane attack complex of complement induces permeability changes via thresholds in individual cells. Immunology. (1987) 60:135–40.
79. Donin N, Jurianz K, Ziporen L, Schultz S, Kirschfink M, Fishelson Z. Complement resistance of human carcinoma cells depends on membrane regulatory proteins, protein kinases and sialic acid. Clin Exp Immunol. (2003) 131:254–63. doi: 10.1046/j.1365-2249.2003.02066.x
80. Fishelson Z, Kopf E, Paas Y, Ross L, Reiter Y. Protein phosphorylation as a mechanism of resistance against complement damage. In: Melchers FEA, editor. Progress in Immunology. Berlin: Springer-Verlag (1989). p. 205–08.
81. Kraus S, Fishelson Z. Cell desensitization by sublytic C5b-9 complexes and calcium ionophores depends on activation of protein kinase C. Eur J Immunol. (2000) 30:1272–80. doi: 10.1002/(SICI)1521-4141(200005)30:5<1272::AID-IMMU1272>3.0.CO;2-9
82. Kraus S, Seger R, Fishelson Z. Involvement of the ERK mitogen-activated protein kinase in cell resistance to complement-mediated lysis. Clin Exp Immunol. (2001) 123:366–74. doi: 10.1046/j.1365-2249.2001.01477.x
83. Jurianz K, Ziegler S, Donin N, Reiter Y, Fishelson Z. Kirschfink M, K562 erythroleukemic cells are equipped with multiple mechanisms of resistance to lysis by complement. Int J Cancer. (2001) 93:848–54. doi: 10.1002/ijc.1406
84. Holguin MH, Martin CB, Weis JH, Parker CJ. Enhanced expression of the complement regulatory protein, membrane inhibitor of reactive lysis (CD59), is regulated at the level of transcription. Blood. (1993) 82:968–77.
85. Marchbank KJ, Morgan BP, van den Berg CW. Regulation of CD59 expression on K562 cells: effects of phorbol myristate acetate, cross-linking antibody and non-lethal complement attack. Immunology. (1995) 85:146–52.
86. Jurianz K, Maslak S, Garcia-Schuler H, Fishelson Z, Kirschfink M. Neutralization of complement regulatory proteins augments lysis of breast carcinoma cells targeted with rhumAb anti-HER2. Immunopharmacology. (1999) 42:209–18. doi: 10.1016/S0162-3109(99)00006-5
87. Pilzer D, Fishelson Z. Mortalin/GRP75 promotes release of membrane vesicles from immune attacked cells and protection from complement-mediated lysis. Int Immunol. (2005) 17:1239–48. doi: 10.1093/intimm/dxh300
88. Pilzer D, Gasser O, Moskovich O, Schifferli JA, Fishelson Z. Emission of membrane vesicles: roles in complement resistance, immunity and cancer. Springer Semin Immunopathol. (2005) 27:375–87. doi: 10.1007/s00281-005-0004-1
89. Gancz D, Lusthaus M, Fishelson Z. A role for the NF-kappaB pathway in cell protection from complement-dependent cytotoxicity. J Immunol. (2012) 189:860–6. doi: 10.4049/jimmunol.1103451
90. Saini J, Sharma PK. Clinical, prognostic and therapeutic significance of heat shock proteins in cancer. Curr Drug Targets. (2018) 19:1478–90. doi: 10.2174/1389450118666170823121248
91. Chatterjee S, Burns TF. Targeting heat shock proteins in cancer: a promising therapeutic approach. Int J Mol Sci. (2017) 18:E1978. doi: 10.3390/ijms18091978
92. Wu J, Liu T, Rios Z, Mei Q, Lin X, Cao S. Heat shock proteins and cancer. Trends Pharmacol Sci. (2017) 38:226–56. doi: 10.1016/j.tips.2016.11.009
93. Fishelson Z, Hochman I, Greene LE, Eisenberg E. Contribution of heat shock proteins to cell protection from complement-mediated lysis. Int Immunol. (2001) 13:983–91. doi: 10.1093/intimm/13.8.983
94. Sreedhar AS, Nardai G, Csermely P. Enhancement of complement-induced cell lysis: a novel mechanism for the anticancer effects of Hsp90 inhibitors. Immunol Lett. (2004) 92:157–61. doi: 10.1016/j.imlet.2003.11.025
95. Rozenberg P, Ziporen L, Gancz D, Saar-Ray M, Fishelson Z. Cooperation between Hsp90 and mortalin/GRP75 in resistance to cell death induced by complement C5b-9. Cell Death Dis. (2018) 9:150. doi: 10.1038/s41419-017-0240-z
96. Park HK, Lee JE, Lim J, Kang BH. Mitochondrial Hsp90s suppress calcium-mediated stress signals propagating from mitochondria to the ER in cancer cells. Mol Cancer. (2014) 13:148. doi: 10.1186/1476-4598-13-148
97. Kim DD, Song WC. Membrane complement regulatory proteins. Clin Immunol. (2006) 118:127–36. doi: 10.1016/j.clim.2005.10.014
98. Liszewski MK, Atkinson JP. Complement regulator CD46: genetic variants and disease associations. Hum Genomics. (2015) 9:7. doi: 10.1186/s40246-015-0029-z
99. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. (2009) 9:729–40. doi: 10.1038/nri2620
100. Gancz D, Fishelson Z. Cancer resistance to complement-dependent cytotoxicity (CDC): problem-oriented research and development. Mol Immunol. (2009) 46:2794–800. doi: 10.1016/j.molimm.2009.05.009
101. Goslings WR, Blom DJ, de Waard-Siebinga I, van Beelen E, Claas FH, Jager MJ, et al. Membrane-bound regulators of complement activation in uveal melanomas. CD46, CD55, and CD59 in uveal melanomas. Invest Ophthalmol Vis Sci. (1996) 37:1884–91.
102. Yamakawa M, Yamada K, Tsuge T, Ohrui H, Ogata T, Dobashi M, et al. Protection of thyroid cancer cells by complement-regulatory factors. Cancer. (1994) 73:2808–17. doi: 10.1002/1097-0142(19940601)73:11<2808::AID-CNCR2820731125>3.0.CO;2-P
103. Niehans GA, Cherwitz DL, Staley NA, Knapp DJ, Dalmasso AP. Human carcinomas variably express the complement inhibitory proteins CD46 (membrane cofactor protein), CD55 (decay-accelerating factor), and CD59 (protectin). Am J Pathol. (1996) 149:129–42.
104. Terachi T, Stanescu G, Pontes JE, Medof ME, Caulfield MJ. Coexistence of autologous antibodies and decay-accelerating factor, an inhibitor of complement, on human renal tumor cells. Cancer Res. (1991) 51:2521–3.
105. Bjorge L, Vedeler CA, Ulvestad E, Matre R. Expression and function of CD59 on colonic adenocarcinoma cells. Eur J Immunol. (1994) 24:1597–603. doi: 10.1002/eji.1830240722
106. Jarvis GA, Li J, Hakulinen J, Brady KA, Nordling S, Dahiya R, et al. Expression and function of the complement membrane attack complex inhibitor protectin (CD59) in human prostate cancer. Int J Cancer. (1997) 71:1049–55. doi: 10.1002/(SICI)1097-0215(19970611)71:6<1049::AID-IJC22>3.0.CO;2-7
107. Juhl H, Helmig F, Baltzer K, Kalthoff H, Henne-Bruns D, Kremer B. Frequent expression of complement resistance factors CD46, CD55, and CD59 on gastrointestinal cancer cells limits the therapeutic potential of monoclonal antibody 17-1A. J Surg Oncol. (1997) 64:222–30. doi: 10.1002/(SICI)1096-9098(199703)64:3<222::AID-JSO9>3.0.CO;2-C
108. Brasoveanu LI, Altomonte M, Gloghini A, Fonsatti E, Coral S, Gasparollo A, et al. Expression of protectin (CD59) in human melanoma and its functional role in cell- and complement-mediated cytotoxicity. Int J Cancer. (1995) 61:548–56. doi: 10.1002/ijc.2910610420
109. Thorsteinsson L, O'Dowd GM, Harrington PM, Johnson PM. The complement regulatory proteins CD46 and CD59, but not CD55, are highly expressed by glandular epithelium of human breast and colorectal tumour tissues. APMIS. (1998) 106:869–78. doi: 10.1111/j.1699-0463.1998.tb00233.x
110. Gorter A, Blok VT, Haasnoot WH, Ensink NG, Daha MR, Fleuren GJ. Expression of CD46, CD55, and CD59 on renal tumor cell lines and their role in preventing complement-mediated tumor cell lysis. Lab Invest. (1996) 74:1039–49.
111. Kuraya M, Yefenof E, Klein G, Klein E. Expression of the complement regulatory proteins CD21, CD55 and CD59 on Burkitt lymphoma lines: their role in sensitivity to human serum-mediated lysis. Eur J Immunol. (1992) 22:1871–6. doi: 10.1002/eji.1830220729
112. Gasque P, Thomas A, Fontaine M, Morgan BP. Complement activation on human neuroblastoma cell lines in vitro: route of activation and expression of functional complement regulatory proteins. J Neuroimmunol. (1996) 66:29–40. doi: 10.1016/0165-5728(96)00015-X
113. Bjorge L, Hakulinen J, Wahlstrom T, Matre R, Meri S. Complement-regulatory proteins in ovarian malignancies. Int J Cancer. (1997) 70:14–25. doi: 10.1002/(SICI)1097-0215(19970106)70:1<14::AID-IJC3>3.0.CO;2-9
114. Simpson KL, Jones A, Norman S, Holmes CH. Expression of the complement regulatory proteins decay accelerating factor (DAF, CD55), membrane cofactor protein (MCP, CD46) and CD59 in the normal human uterine cervix and in premalignant and malignant cervical disease. Am J Pathol. (1997) 151:1455–67.
115. Murray KP, Mathure S, Kaul R, Khan S, Carson LF, Twiggs LB, et al. Expression of complement regulatory proteins-CD 35, CD 46, CD 55, and CD 59-in benign and malignant endometrial tissue. Gynecol Oncol. (2000) 76:176–82. doi: 10.1006/gyno.1999.5614
116. Guc D, Canpinar H, Kucukaksu C, Kansu E. Expression of complement regulatory proteins CR1, DAF, MCP and CD59 in haematological malignancies. Eur J Haematol. (2000) 64:3–9. doi: 10.1034/j.1600-0609.2000.80097.x
117. Hofman P, Hsi BL, Manie S, Fenichel P, Thyss A, Rossi B. High expression of the antigen recognized by the monoclonal antibody GB24 on human breast carcinomas: a preventive mechanism of malignant tumor cells against complement attack? Breast Cancer Res Treat. (1994) 32:213–9. doi: 10.1007/BF00665772
118. Varsano S, Rashkovsky L, Shapiro H, Ophir D, Mark-Bentankur T. Human lung cancer cell lines express cell membrane complement inhibitory proteins and are extremely resistant to complement-mediated lysis; a comparison with normal human respiratory epithelium in vitro, and an insight into mechanism(s) of resistance. Clin Exp Immunol. (1998) 113:173–82. doi: 10.1046/j.1365-2249.1998.00581.x
119. Blok VT, Daha MR, Tijsma OM, Weissglas MG, van den Broek LJ, Gorter A. A possible role of CD46 for the protection in vivo of human renal tumor cells from complement-mediated damage. Lab Invest. (2000) 80:335–44. doi: 10.1038/labinvest.3780038
120. Nowicki S, Nowicki B, Pham T, Hasan R, Nagamani M. Expression of decay accelerating factor in endometrial adenocarcinoma is inversely related to the stage of tumor. Am J Reprod Immunol. (2001) 46:144–8. doi: 10.1111/j.8755-8920.2001.460205.x
121. Nakagawa M, Mizuno M, Kawada M, Uesu T, Nasu J, Takeuchi K, et al. Polymorphic expression of decay-accelerating factor in human colorectal cancer. J Gastroenterol Hepatol. (2001) 16:184–9. doi: 10.1046/j.1440-1746.2001.02418.x
122. Li B, Lin H, Fan J, Lan J, Zhong Y, Yang Y, et al. CD59 is overexpressed in human lung cancer and regulates apoptosis of human lung cancer cells. Int J Oncol. (2013) 43:850–8. doi: 10.3892/ijo.2013.2007
123. Li L, Spendlove I, Morgan J, Durrant LG. CD55 is over-expressed in the tumour environment. Br J Cancer. (2001) 84:80–6. doi: 10.1054/bjoc.2000.1570
124. Kiso T, Mizuno M, Nasu J, Shimo K, Uesu T, Yamamoto K, et al. Enhanced expression of decay-accelerating factor and CD59/homologous restriction factor 20 in intestinal metaplasia, gastric adenomas and intestinal-type gastric carcinomas but not in diffuse-type carcinomas. Histopathology. (2002) 40:339–47. doi: 10.1046/j.1365-2559.2002.01350.x
125. Crnogorac-Jurcevic T, Efthimiou E, Nielsen T, Loader J, Terris B, Stamp G, et al. Expression profiling of microdissected pancreatic adenocarcinomas. Oncogene. (2002) 21:4587–94. doi: 10.1038/sj.onc.1205570
126. Brasoveanu LI, Altomonte M, Fonsatti E, Colizzi F, Coral S, Nicotra MR, et al. Levels of cell membrane CD59 regulate the extent of complement-mediated lysis of human melanoma cells. Lab Invest. (1996) 74:33–42.
127. Watson NF, Durrant LG, Madjd Z, Ellis IO, Scholefield JH, Spendlove I. Expression of the membrane complement regulatory protein CD59 (protectin) is associated with reduced survival in colorectal cancer patients. Cancer Immunol Immunother. (2006) 55:973–80. doi: 10.1007/s00262-005-0055-0
128. Xu C, Jung M, Burkhardt M, Stephan C, Schnorr D, Loening S, et al. Increased CD59 protein expression predicts a PSA relapse in patients after radical prostatectomy. Prostate. (2005) 62:224–32. doi: 10.1002/pros.20134
129. Ouyang Q, Zhang L, Jiang Y, Ni X, Chen S, Ye F, et al. The membrane complement regulatory protein CD59 promotes tumor growth and predicts poor prognosis in breast cancer. Int J Oncol. (2016) 48:2015–24. doi: 10.3892/ijo.2016.3408
130. Madjd Z, Pinder SE, Paish C, Ellis IO, Carmichael J, Durrant LG. Loss of CD59 expression in breast tumours correlates with poor survival. J Pathol. (2003) 200:633–9. doi: 10.1002/path.1357
131. Durrant LG, Chapman MA, Buckley DJ, Spendlove I, Robins RA, Armitage NC. Enhanced expression of the complement regulatory protein CD55 predicts a poor prognosis in colorectal cancer patients. Cancer Immunol Immunother. (2003) 52:638–42. doi: 10.1007/s00262-003-0402-y
132. Liu M, Yang YJ, Zheng H, Zhong XR, Wang Y, Wang Z, et al. Membrane-bound complement regulatory proteins are prognostic factors of operable breast cancer treated with adjuvant trastuzumab: a retrospective study. Oncol Rep. (2014) 32:2619–27. doi: 10.3892/or.2014.3496
133. Hyc A, Osiecka-Iwan A, Strzelczyk P, Moskalewski S. Effect of IL-1beta, TNF-alpha and IL-4 on complement regulatory protein mRNA expression in human articular chondrocytes. Int J Mol Med. (2003) 11:91–4. doi: 10.3892/ijmm.11.1.91
134. Spiller OB, Criado-Garcia O, Rodriguez De Cordoba S, Morgan BP. Cytokine-mediated up-regulation of CD55 and CD59 protects human hepatoma cells from complement attack. Clin Exp Immunol. (2000) 121:234–41. doi: 10.1046/j.1365-2249.2000.01305.x
135. Bjorge L, Jensen TS, Ulvestad E, Vedeler CA, Matre R. The influence of tumour necrosis factor-alpha, interleukin-1 beta and interferon-gamma on the expression and function of the complement regulatory protein CD59 on the human colonic adenocarcinoma cell line HT29. Scand J Immunol. (1995) 41:350–6. doi: 10.1111/j.1365-3083.1995.tb03578.x
136. Varsano S, Rashkovsky L, Shapiro H, Radnay J. Cytokines modulate expression of cell-membrane complement inhibitory proteins in human lung cancer cell lines. Am J Respir Cell Mol Biol. (1998) 19:522–9. doi: 10.1165/ajrcmb.19.3.3181
137. Hatanaka M, Seya T, Matsumoto M, Hara T, Nonaka M, Inoue N, et al. Mechanisms by which the surface expression of the glycosyl-phosphatidylinositol-anchored complement regulatory proteins decay-accelerating factor (CD55) and CD59 is lost in human leukaemia cell lines. Biochem J. (1996) 314 (Pt 3):969–76.
138. Bjorge L, Matre R. Down-regulation of CD59 (protectin) expression on human colorectal adenocarcinoma cell lines by levamisole. Scand J Immunol. (1995) 42:512–6. doi: 10.1111/j.1365-3083.1995.tb03688.x
139. Gambacorti-Passerini C, Rivoltini L, Supino R, Rodolfo M, Radrizzani M, Fossati G, et al. Susceptibility of chemoresistant murine and human tumor cells to lysis by interleukin 2-activated lymphocytes. Cancer Res. (1988) 48:2372–6.
140. Bomstein Y, Fishelson Z. Enhanced sensitivity of P-glycoprotein-positive multidrug resistant tumor cells to complement-mediated lysis. Eur J Immunol. (1997) 27:2204–11. doi: 10.1002/eji.1830270913
141. Weisburg JH, Curcio M, Caron PC, Raghu G, Mechetner EB, Roepe PD, et al. The multidrug resistance phenotype confers immunological resistance. J Exp Med. (1996) 183:2699–704. doi: 10.1084/jem.183.6.2699
142. Odening KE, Li W, Rutz R, Laufs S, Fruehauf S, Fishelson Z, et al. Enhanced complement resistance in drug-selected P-glycoprotein expressing multi-drug-resistant ovarian carcinoma cells. Clin Exp Immunol. (2009) 155:239–48. doi: 10.1111/j.1365-2249.2008.03817.x
143. Seya T, Hara T, Iwata K, Kuriyama S, Hasegawa T, Nagase Y, et al. Purification and functional properties of soluble forms of membrane cofactor protein (CD46) of complement: identification of forms increased in cancer patients' sera. Int Immunol. (1995) 7:727–36. doi: 10.1093/intimm/7.5.727
144. Iwagaki N, Mizuno M, Nasu J, Mizuno M, Okazaki H, Hori S, et al. Advances in the development of a reliable assay for the measurement of stool decay-accelerating factor in the detection of colorectal cancer. J Immunoassay Immunochem. (2002) 23:497–507. doi: 10.1081/IAS-120015480
145. Brasoveanu LI, Fonsatti E, Visintin A, Pavlovic M, Cattarossi I, Colizzi F, et al. Melanoma cells constitutively release an anchor-positive soluble form of protectin (sCD59) that retains functional activities in homologous complement-mediated cytotoxicity. J Clin Invest. (1997) 100:1248–55. doi: 10.1172/JCI119638
146. Gelderman KA, Blok VT, Fleuren GJ, Gorter A. The inhibitory effect of CD46, CD55, and CD59 on complement activation after immunotherapeutic treatment of cervical carcinoma cells with monoclonal antibodies or bispecific monoclonal antibodies. Lab Invest. (2002) 82:483–93. doi: 10.1038/labinvest.3780441
147. Hindmarsh EJ, Marks RM. Decay-accelerating factor is a component of subendothelial extracellular matrix in vitro, and is augmented by activation of endothelial protein kinase C. Eur J Immunol. (1998) 28:1052–62. doi: 10.1002/(SICI)1521-4141(199803)28:03<1052::AID-IMMU1052>3.0.CO;2-W
148. Nasu J, Mizuno M, Uesu T, Takeuchi K, Inaba T, Ohya S, et al. Cytokine-stimulated release of decay-accelerating factor (DAF;CD55) from HT-29 human intestinal epithelial cells. Clin Exp Immunol. (1998) 113:379–85. doi: 10.1046/j.1365-2249.1998.00660.x
149. Sadallah S, Lach E, Schwarz S, Gratwohl A, Spertini O, Schifferli JA. Soluble complement receptor 1 is increased in patients with leukemia and after administration of granulocyte colony-stimulating factor. J Leukoc Biol. (1999) 65:94–101. doi: 10.1002/jlb.65.1.94
150. Miyagawa S, Kubo T, Matsunami K, Kusama T, Beppu K, Nozaki H, et al. Delta-short consensus repeat 4-decay accelerating factor (DAF: CD55) inhibits complement-mediated cytolysis but not NK cell-mediated cytolysis. J Immunol. (2004) 173:3945–52. doi: 10.4049/jimmunol.173.6.3945
151. Kimberley FC, Sivasankar B, Paul Morgan B Alternative roles for CD59. Mol Immunol. (2007) 44:73–81. doi: 10.1016/j.molimm.2006.06.019
152. Macor P, Capolla S, Tedesco F. Complement as a biological tool to control tumor growth. Front Immunol. (2018) 9:2203. doi: 10.3389/fimmu.2018.02203
153. Ollert MW, Frade R, Fiandino A, Panneerselvam M, Petrella EC, Barel M, et al. C3-cleaving membrane proteinase. A new complement regulatory protein of human melanoma cells. J Immunol. (1990) 144:3862–7.
154. Jean D, Hermann J, Rodrigues-Lima F, Barel M, Balbo M, Frade R. Identification on melanoma cells of p39, a cysteine proteinase that cleaves C3, the third component of complement: amino-acid-sequence identities with procathepsin L. Biochem J. (1995) 312 (Pt 3):961–9.
155. Maison CM, Villiers CL, Colomb MG. Proteolysis of C3 on U937 cell plasma membranes. Purification of cathepsin G. J Immunol. (1991) 147:921–6.
156. Jurianz K, Ziegler S, Garcia-Schuler H, Kraus S, Bohana-Kashtan O, Fishelson Z, et al. Complement resistance of tumor cells: basal and induced mechanisms. Mol Immunol. (1999) 36:929–39. doi: 10.1016/S0161-5890(99)00115-7
157. Rozanov DV, Savinov AY, Golubkov VS, Tomlinson S, Strongin AY. Interference with the complement system by tumor cell membrane type-1 matrix metalloproteinase plays a significant role in promoting metastasis in mice. Cancer Res. (2006) 66:6258–63. doi: 10.1158/0008-5472.CAN-06-0539
158. Paas Y, Fishelson Z. Shedding of tyrosine and serine/threonine ecto-protein kinases from human leukemic cells. Arch Biochem Biophys. (1995) 316:780–8. doi: 10.1006/abbi.1995.1104
159. Paas Y, Bohana-Kashtan O, Fishelson Z. Phosphorylation of the complement component, C9, by an ecto-protein kinase of human leukemic cells. Immunopharmacology. (1999) 42:175–85. doi: 10.1016/S0162-3109(99)00027-2
160. Bohana-Kashtan O, Pinna LA, Fishelson Z. Extracellular phosphorylation of C9 by protein kinase CK2 regulates complement-mediated lysis. Eur J Immunol. (2005) 35:1939–48. doi: 10.1002/eji.200425716
161. Lauf PK. Immunological and physiological characteristics of the rapid immune hemolysis of neuraminidase-treated sheep red cells produced by fresh guinea pig serum. J Exp Med. (1975) 142:974–88. doi: 10.1084/jem.142.4.974
162. Pangburn MK, Muller-Eberhard HJ. Complement C3 convertase: cell surface restriction of beta1H control and generation of restriction on neuraminidase-treated cells. Proc Natl Acad Sci USA. (1978) 75:2416–20. doi: 10.1073/pnas.75.5.2416
163. Turianskyj FH, Gyenes L. The effect of neuraminidase on the sensitivity of tumor cells toward lysis by antibody and complement or by sensitized lymphocytes. Transplantation. (1976) 22:24–30. doi: 10.1097/00007890-197607000-00004
164. Jacobsen F. Increase of the in vitro complement-dependent cytotoxicity against autologous invasive human bladder tumor cells by neuraminidase treatment. Acta Pathol Microbiol Immunol Scand. (1982) 90:187–92. doi: 10.1111/j.1699-0463.1982.tb01437.x
165. Shi WX, Chammas R, Varki NM, Powell L, Varki A. Sialic acid 9-O-acetylation on murine erythroleukemia cells affects complement activation, binding to I-type lectins, and tissue homing. J Biol Chem. (1996) 271:31526–32. doi: 10.1074/jbc.271.49.31526
166. Bordron A, Bagacean C, Mohr A, Tempescul A, Bendaoud B, Deshayes S, et al. Resistance to complement activation, cell membrane hypersialylation and relapses in chronic lymphocytic leukemia patients treated with rituximab and chemotherapy. Oncotarget. (2018) 9:31590–605. doi: 10.18632/oncotarget.25657
167. Cui T, Chen Y, Knosel T, Yang L, Zoller K, Galler K, et al. Human complement factor H is a novel diagnostic marker for lung adenocarcinoma. Int J Oncol. (2011) 39:161–8. doi: 10.3892/ijo.2011.1010
168. Riihila PM, Nissinen LM, Ala-Aho R, Kallajoki M, Grenman R, Meri S, et al. Complement factor H: a biomarker for progression of cutaneous squamous cell carcinoma. J Invest Dermatol. (2014) 134:498–506. doi: 10.1038/jid.2013.346
169. Junnikkala S, Hakulinen J, Jarva H, Manuelian T, Bjorge L, Butzow R, et al. Secretion of soluble complement inhibitors factor H and factor H-like protein (FHL-1) by ovarian tumour cells. Br J Cancer. (2002) 87:1119–27. doi: 10.1038/sj.bjc.6600614
170. Hsu YF, Ajona D, Corrales L, Lopez-Picazo JM, Gurpide A, Montuenga LM, et al. Complement activation mediates cetuximab inhibition of non-small cell lung cancer tumor growth in vivo. Mol Cancer. (2010) 9:139. doi: 10.1186/1476-4598-9-139
171. Horl S, Banki Z, Huber G, Ejaz A, Windisch D, Muellauer B, et al. Reduction of complement factor H binding to CLL cells improves the induction of rituximab-mediated complement-dependent cytotoxicity. Leukemia. (2013) 27:2200–8. doi: 10.1038/leu.2013.169
172. Legoedec J, Gasque P, Jeanne JF, Fontaine M. Expression of the complement alternative pathway by human myoblasts in vitro: biosynthesis of C3, factor B, factor H and factor I. Eur J Immunol. (1995) 25:3460–6. doi: 10.1002/eji.1830251238
173. Riihila P, Nissinen L, Farshchian M, Kivisaari A, Ala-Aho R, Kallajoki M, et al. Complement factor I promotes progression of cutaneous squamous cell carcinoma. J Invest Dermatol. (2015) 135:579–88. doi: 10.1038/jid.2014.376
174. Okroj M, Holmquist E, Nilsson E, Anagnostaki L, Jirstrom K, Blom AM. Local expression of complement factor I in breast cancer cells correlates with poor survival and recurrence. Cancer Immunol Immunother. (2015) 64:467–78. doi: 10.1007/s00262-015-1658-8
175. Carney DF, Koski CL, Shin ML. Elimination of terminal complement intermediates from the plasma membrane of nucleated cells: the rate of disappearance differs for cells carrying C5b-7 or C5b-8 or a mixture of C5b-8 with a limited number of C5b-9. J Immunol. (1985) 134:1804–9.
176. Ramm LE, Whitlow MB, Koski CL, Shin ML, Mayer MM. Elimination of complement channels from the plasma membranes of U937, a nucleated mammalian cell line: temperature dependence of the elimination rate. J Immunol. (1983) 131:1411–5.
177. Campbell AK, Morgan BP. Monoclonal antibodies demonstrate protection of polymorphonuclear leukocytes against complement attack. Nature. (1985) 317:164–6. doi: 10.1038/317164a0
178. Morgan BP, Dankert JR, Esser AF. Recovery of human neutrophils from complement attack: removal of the membrane attack complex by endocytosis and exocytosis. J Immunol. (1987) 138:246–53.
179. Scolding NJ, Morgan BP, Houston WA, Linington C, Campbell AK, Compston DA. Vesicular removal by oligodendrocytes of membrane attack complexes formed by activated complement. Nature. (1989) 339:620–2. doi: 10.1038/339620a0
180. Kerjaschki D, Schulze M, Binder S, Kain R, Ojha PP, Susani M, et al. Transcellular transport and membrane insertion of the C5b-9 membrane attack complex of complement by glomerular epithelial cells in experimental membranous nephropathy. J Immunol. (1989) 143:546–52.
181. Sims PJ, Wiedmer T. Repolarization of the membrane potential of blood platelets after complement damage: evidence for a Ca++ -dependent exocytotic elimination of C5b-9 pores. Blood. (1986) 68:556–61.
182. Niculescu F, Rus H, Shin ML. Receptor-independent activation of guanine nucleotide-binding regulatory proteins by terminal complement complexes. J Biol Chem. (1994) 269:4417–23.
183. Carney DF, Lang TJ, Shin ML. Multiple signal messengers generated by terminal complement complexes and their role in terminal complement complex elimination. J Immunol. (1990) 145:623–9.
184. Moskovich O, Fishelson Z. Live cell imaging of outward and inward vesiculation induced by the complement c5b-9 complex. J Biol Chem. (2007) 282:29977–86. doi: 10.1074/jbc.M703742200
185. Stein JM, Luzio JP. Ectocytosis caused by sublytic autologous complement attack on human neutrophils. The sorting of endogenous plasma-membrane proteins and lipids into shed vesicles. Biochem J. (1991) 274 (Pt 2):381–6.
186. Moskovich O, Herzog LO, Ehrlich M, Fishelson Z. Caveolin-1 and dynamin-2 are essential for removal of the complement C5b-9 complex via endocytosis. J Biol Chem. (2012) 287:19904–15. doi: 10.1074/jbc.M111.333039
187. Wadhwa R, Takano S, Kaur K, Deocaris CC, Pereira-Smith OM, Reddel RR, et al. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int J Cancer. (2006) 118:2973–80. doi: 10.1002/ijc.21773
188. Saar Ray M, Moskovich O, Iosefson O, Fishelson Z. Mortalin/GRP75 binds to complement C9 and plays a role in resistance to complement-dependent cytotoxicity. J Biol Chem. (2014) 289:15014–22. doi: 10.1074/jbc.M114.552406
189. Pilzer D, Saar M, Koya K, Fishelson Z. Mortalin inhibitors sensitize K562 leukemia cells to complement-dependent cytotoxicity. Int J Cancer. (2010) 126:1428–35. doi: 10.1002/ijc.24888
190. Gelderman KA, Kuppen PJ, Bruin W, Fleuren GJ, Gorter A. Enhancement of the complement activating capacity of 17-1A mAb to overcome the effect of membrane-bound complement regulatory proteins on colorectal carcinoma. Eur J Immunol. (2002) 32:128–35. doi: 10.1002/1521-4141(200201)32:1<128::AID-IMMU128>3.0.CO;2-P
191. Reiter Y, Fishelson Z. Targeting of complement to tumor cells by heteroconjugates composed of antibodies and of the complement component C3b. J Immunol. (1989) 142:2771–7.
192. Juhl H, Petrella EC, Cheung NK, Bredehorst R, Vogel CW. Additive cytotoxicity of different monoclonal antibody-cobra venom factor conjugates for human neuroblastoma cells. Immunobiology. (1997) 197:444–59. doi: 10.1016/S0171-2985(97)80078-2
193. Raitses Gurevich M, Fishelson Z. Construction and characterization of recombinant human C9 or C7 linked to single chain Fv directed to CD25. Mol Immunol. (2013) 55:400–8. doi: 10.1016/j.molimm.2013.03.013
194. Geis N, Zell S, Rutz R, Li W, Giese T, Mamidi S, et al. Inhibition of membrane complement inhibitor expression (CD46, CD55, CD59) by siRNA sensitizes tumor cells to complement attack in vitro. Curr Cancer Drug Targets. (2010) 10:922–31. doi: 10.2174/156800910793357952
195. Macor P, Tedesco F. Complement as effector system in cancer immunotherapy. Immunol Lett. (2007) 111:6–13. doi: 10.1016/j.imlet.2007.04.014
196. Macor P, Tripodo C, Zorzet S, Piovan E, Bossi F, Marzari R, et al. In vivo targeting of human neutralizing antibodies against CD55 and CD59 to lymphoma cells increases the antitumor activity of rituximab. Cancer Res. (2007) 67:10556–63. doi: 10.1158/0008-5472.CAN-07-1811
197. Zhong RK, Kozii R, Ball ED. Homologous restriction of complement-mediated cell lysis can be markedly enhanced by blocking decay-accelerating factor. Br J Haematol. (1995) 91:269–74. doi: 10.1111/j.1365-2141.1995.tb05289.x
198. Ziller F, Macor P, Bulla R, Sblattero D, Marzari R, Tedesco F. Controlling complement resistance in cancer by using human monoclonal antibodies that neutralize complement-regulatory proteins CD55 and CD59. Eur J Immunol. (2005) 35:2175–83. doi: 10.1002/eji.200425920
199. Golay J, Lazzari M, Facchinetti V, Bernasconi S, Borleri G, Barbui T, et al. CD20 levels determine the in vitro susceptibility to rituximab and complement of B-cell chronic lymphocytic leukemia: further regulation by CD55 and CD59. Blood. (2001) 98:3383–9. doi: 10.1182/blood.V98.12.3383
200. Cheung NK, Walter EI, Smith-Mensah WH, Ratnoff WD, Tykocinski ML, Medof ME. Decay-accelerating factor protects human tumor cells from complement-mediated cytotoxicity in vitro. J Clin Invest. (1988) 81:1122–8. doi: 10.1172/JCI113426
201. Gorter A, Meri S. Immune evasion of tumor cells using membrane-bound complement regulatory proteins. Immunol Today. (1999) 20:576–82. doi: 10.1016/S0167-5699(99)01537-6
202. Macor P, Secco E, Mezzaroba N, Zorzet S, Durigutto P, Gaiotto T, et al. Bispecific antibodies targeting tumor-associated antigens and neutralizing complement regulators increase the efficacy of antibody-based immunotherapy in mice. Leukemia. (2015) 29:406–14. doi: 10.1038/leu.2014.185
203. Wilczek E, Rzepko R, Nowis D, Legat M, Golab J, Glab M, et al. The possible role of factor H in colon cancer resistance to complement attack. Int J Cancer. (2008) 122:2030–7. doi: 10.1002/ijc.23238
204. Aagaard L, Rossi JJ. RNAi therapeutics: principles, prospects and challenges. Adv Drug Deliv Rev. (2007) 59:75–86. doi: 10.1016/j.addr.2007.03.005
205. Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol. (2012) 19:60–71. doi: 10.1016/j.chembiol.2011.12.008
206. Zell S, Geis N, Rutz R, Schultz S, Giese T, Kirschfink M. Down-regulation of CD55 and CD46 expression by anti-sense phosphorothioate oligonucleotides (S-ODNs) sensitizes tumour cells to complement attack. Clin Exp Immunol. (2007) 150:576–84. doi: 10.1111/j.1365-2249.2007.03507.x
207. Wang Y, Yang YJ, Wang Z, Liao J, Liu M, Zhong XR, et al. CD55 and CD59 expression protects HER2-overexpressing breast cancer cells from trastuzumab-induced complement-dependent cytotoxicity. Oncol Lett. (2017) 14:2961–69. doi: 10.3892/ol.2017.6555
208. Mamidi S, Cinci M, Hasmann M, Fehring V, Kirschfink M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol Oncol. (2013) 7:580–94. doi: 10.1016/j.molonc.2013.02.011
209. Mamidi S, Hone S, Teufel C, Sellner L, Zenz T, Kirschfink M. Neutralization of membrane complement regulators improves complement-dependent effector functions of therapeutic anticancer antibodies targeting leukemic cells. Oncoimmunology. (2015) 4:e979688. doi: 10.4161/2162402X.2014.979688
210. Tros de Ilarduya C, Duzgunes N. Delivery of therapeutic nucleic acids via transferrin and transferrin receptors: lipoplexes and other carriers. Expert Opin Drug Deliv. (2013) 10:1583–91. doi: 10.1517/17425247.2013.837447
211. Cinci M, Mamidi S, Li W, Fehring V, Kirschfink M. Targeted delivery of siRNA using transferrin-coupled lipoplexes specifically sensitizes CD71 high expressing malignant cells to antibody-mediated complement attack. Target Oncol. (2015) 10:405–13. doi: 10.1007/s11523-014-0345-6
212. Rousselet N, Mills L, Jean D, Tellez C, Bar-Eli M, Frade R. Inhibition of tumorigenicity and metastasis of human melanoma cells by anti-cathepsin L single chain variable fragment. Cancer Res. (2004) 64:146–51. doi: 10.1158/0008-5472.CAN-03-1717
213. Frade R, Rousselet N, Jean D. Intratumoral gene delivery of anti-cathepsin L single-chain variable fragment by lentiviral vector inhibits tumor progression induced by human melanoma cells. Cancer Gene Ther. (2008) 15:591–604. doi: 10.1038/cgt.2008.51
214. Heigener DF, Gandara DR, Reck M. Targeting of MEK in lung cancer therapeutics. Lancet Respir Med. (2015) 3:319–27. doi: 10.1016/S2213-2600(15)00026-0
215. Sarkisian S, Davar D. MEK inhibitors for the treatment of NRAS mutant melanoma. Drug Des Devel Ther. (2018) 12:2553–65. doi: 10.2147/DDDT.S131721
216. Liu F, Yang X, Geng M, Huang M. Targeting ERK, an Achilles' Heel of the MAPK pathway, in cancer therapy. Acta Pharm Sin B. (2018) 8:552–62. doi: 10.1016/j.apsb.2018.01.008
217. Powers MV, Clarke PA, Workman P. Death by chaperone: HSP90, HSP70 or both? Cell Cycle. (2009) 8:518–26. doi: 10.4161/cc.8.4.7583
218. Taldone T, Gozman A, Maharaj R, Chiosis G. Targeting Hsp90: small-molecule inhibitors and their clinical development. Curr Opin Pharmacol. (2008) 8:370–4. doi: 10.1016/j.coph.2008.06.015
219. Donnelly A, Blagg BS. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr Med Chem. (2008) 15:2702–17. doi: 10.2174/092986708786242895
220. Dundas SR, Lawrie LC, Rooney PH, Murray GI. Mortalin is over-expressed by colorectal adenocarcinomas and correlates with poor survival. J Pathol. (2005) 205:74–81. doi: 10.1002/path.1672
Keywords: complement, C5b-9, complement-dependent cytotoxicity, regulated necrosis, cancer immune resistance
Citation: Fishelson Z and Kirschfink M (2019) Complement C5b-9 and Cancer: Mechanisms of Cell Damage, Cancer Counteractions, and Approaches for Intervention. Front. Immunol. 10:752. doi: 10.3389/fimmu.2019.00752
Received: 31 January 2019; Accepted: 20 March 2019;
Published: 10 April 2019.
Edited by:
Maciej M. Markiewski, Texas Tech University Health Sciences Center, United StatesReviewed by:
Michelle A. Dunstone, Monash University, AustraliaJamie Honeychurch, University of Manchester, United Kingdom
Copyright © 2019 Fishelson and Kirschfink. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zvi Fishelson, bGlmaXNoQHRhdWV4LnRhdS5hYy5pbA==