 Annelot Breedveld1,2
Annelot Breedveld1,2 Marjolein van Egmond1,2,3*
Marjolein van Egmond1,2,3*- 1Department of Molecular Cell Biology and Immunology, Amsterdam UMC, Amsterdam, Netherlands
- 2Amsterdam Infection and Immunity Institute, Amsterdam UMC, Amsterdam, Netherlands
- 3Department of Surgery, Amsterdam UMC, Amsterdam, Netherlands
Immunoglobulin A (IgA) is the most abundant antibody class present at mucosal surfaces. The production of IgA exceeds the production of all other antibodies combined, supporting its prominent role in host-pathogen defense. IgA closely interacts with the intestinal microbiota to enhance its diversity, and IgA has a passive protective role via immune exclusion. Additionally, inhibitory ITAMi signaling via the IgA Fc receptor (FcαRI; CD89) by monomeric IgA may play a role in maintaining homeostatic conditions. By contrast, IgA immune complexes (e.g., opsonized pathogens) potently activate immune cells via cross-linking FcαRI, thereby inducing pro-inflammatory responses resulting in elimination of pathogens. The importance of IgA in removal of pathogens is emphasized by the fact that several pathogens developed mechanisms to break down IgA or evade FcαRI-mediated activation of immune cells. Augmented or aberrant presence of IgA immune complexes can result in excessive neutrophil activation, potentially leading to severe tissue damage in multiple inflammatory, or autoimmune diseases. Influencing IgA or FcαRI-mediated functions therefore provides several therapeutic possibilities. On the one hand (passive) IgA vaccination strategies can be developed for protection against infections. Furthermore, IgA monoclonal antibodies that are directed against tumor antigens may be effective as cancer treatment. On the other hand, induction of ITAMi signaling via FcαRI may reduce allergy or inflammation, whereas blocking FcαRI with monoclonal antibodies, or peptides may resolve IgA-induced tissue damage. In this review both (patho)physiological roles as well as therapeutic possibilities of the IgA-FcαRI axis are addressed.
Introduction
Immunoglobulins have important functions in immunity (1, 2). In mucosal areas like the gastrointestinal, genitourinary, and respiratory tracts, IgA is the predominant antibody present (3). It plays a key role at these surfaces, which are continuously exposed to antigens, food, and (commensal) microorganisms. Keeping a tight balance by tolerating commensals and harmless (food) antigens on the one hand and providing protection against harmful pathogens on the other hand is a challenging role of mucosal immunity. Mucosal IgA protects the host by diversifying the microbiota, neutralizing toxins and viruses, blocking colonization and penetration of pathogenic bacteria, clearing unwanted particles, and promoting sampling of antigens (4). Mucosal IgA is generally considered as a neutralizing, non-activating antibody. In serum, IgA is the second most abundant antibody after IgG. In contrast to mucosal IgA, the roles of serum IgA are relatively unexplored (5).
IgA deficiency is a relatively common disease with limited effects on human health (6), which supported the notion of IgA as a non-inflammatory antibody. By contrast, increased serum IgA levels or IgA autoantibodies have been reported in multiple (inflammatory) diseases including IgA nephropathy (IgAN), IgA vasculitis, dermatitis herpetiformis, celiac disease, inflammatory bowel disease (IBD), Sjögren's syndrome, ankylosing spondylitis, alcoholic liver cirrhosis, and Acquired immunodeficiency syndrome (7–9), although the role of IgA in pathology is still ill-understood.
IgA can interact with several receptors that are present on a variety of (immune) cells. The polymeric immunoglobulin receptor (pIgR) is present on the basolateral side of epithelial cells and mediates transport of dimeric IgA (dIgA) to the mucosal lumen, where it is released as secretory IgA (SIgA) (10). Retrograde transport of SIgA immune complexes back into the lamina propria can occur via the transferrin receptor (Tfr or CD71) on epithelial cells or through microfold cells (M cells), possible via interaction with Dectin-1 (11, 12). It has been reported that SIgA immune complexes in the lamina propria can be taken up by sub-epithelial dendritic cells (DCs) through interaction with Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN) (13). B cells were shown to express the inhibitory IgA receptor Fc receptor-like 4 (FcRL4), which was suggested to contribute to the regulation of mucosal IgA responses (14). In the liver, hepatocytes express the asialoglycoprotein receptor (ASGPR) that is involved in clearance of IgA from the circulation (15). Mesangial cells in the glomeruli of the kidneys express CD71 and the recently identified β-1,4-galactosyltransferase as potential receptors to clear IgA (16). Furthermore, an Fc alpha/mu receptor is expressed in gut, spleen and lymph nodes, although its functions remain to be elucidated (17). Finally, the IgA Fc receptor FcαRI (CD89) is expressed by myeloid cells (18). It was demonstrated that interaction of monomeric serum IgA with FcαRI induces inhibitory signals (19, 20). As such, it is thought that IgA has an anti-inflammatory role during homeostatic conditions. By contrast, IgA immune complexes bind avidly to FcαRI, resulting in cross-linking and induction of pro-inflammatory responses, which may play an important role in resolving (mucosal) infections. Additionally, it was suggested that the presence of excessive IgA immune complexes can lead to uncontrolled and disproportionate immune cell activation, which leads to severe tissue damage in autoimmune diseases, such as IgA blistering diseases and rheumatoid arthritis (RA) (18). The number of diverse IgA receptors, their differential expression on cells and the distinct IgA-induced effector functions support the complex roles of this antibody in maintaining homeostatic conditions, which go beyond the dogma of IgA as a non-inflammatory regulator of mucosal immunity. This is further supported by the fact that several bacteria have evolved to escape IgA-mediated functions by for instance producing IgA1 proteases or expressing molecules that hamper interaction with IgA receptors (21–23).
This review addresses the different functions of IgA, its interaction with FcαRI,—as the best characterized IgA receptor -, in health and disease and the possibilities to target either molecule for therapeutic strategies.
Structure of IgA
IgA is the most predominant antibody produced by humans with a synthesis rate of 66 mg/kg each day. IgA can be subdivided into IgA1 and IgA2 (Figure 1A). The IgA1 hinge region contains multiple O-linked glycans and two N-linked glycosylation sites per heavy chain. The truncated hinge of IgA2 lacks O-glycans and each heavy chain contains two additional N-glycans (25).
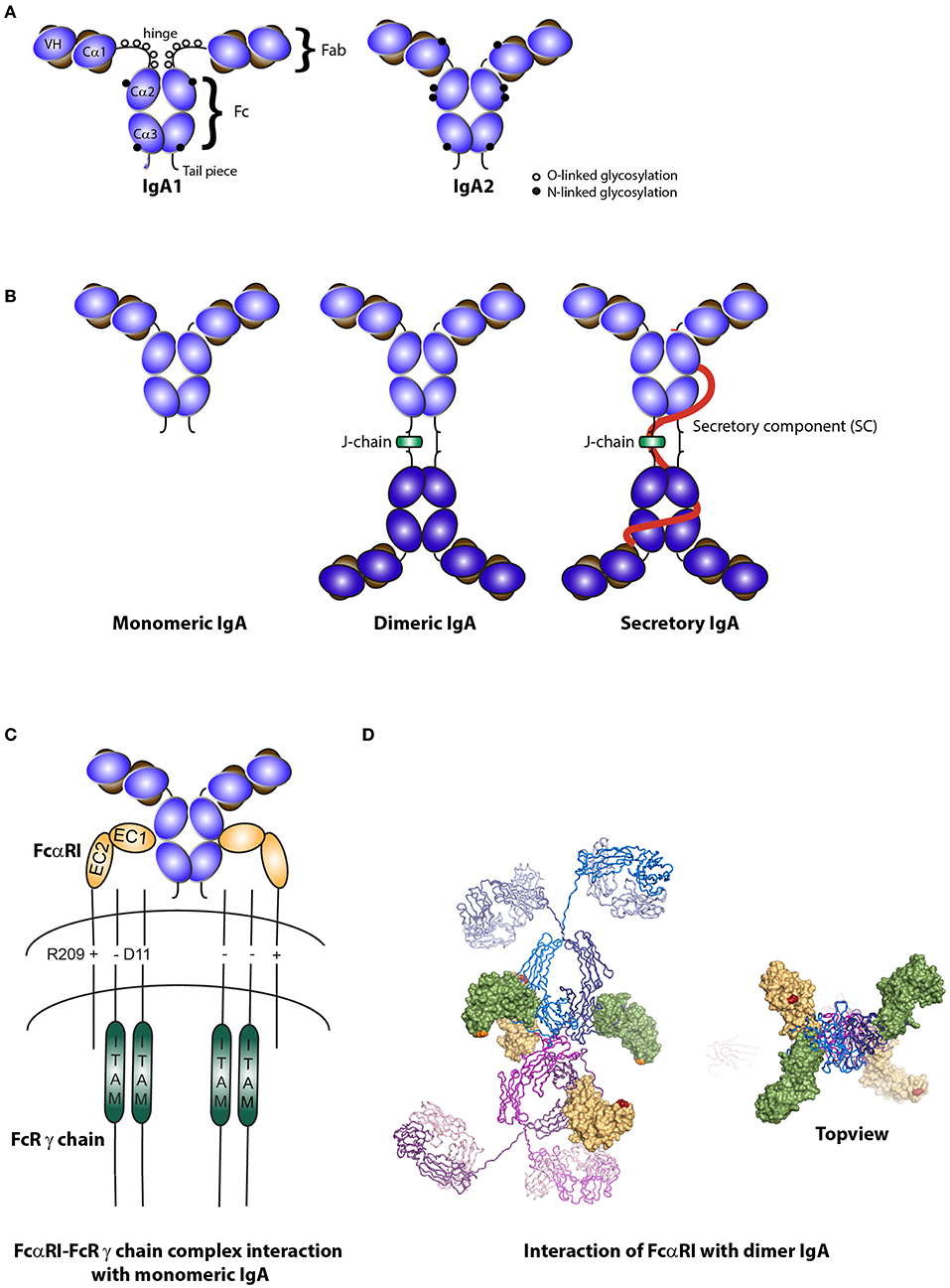
Figure 1. Structure of IgA isotypes and the IgA Fc receptor (FcαRI). (A) IgA1 vs. IgA2. IgA consists of two heavy chains (blue), each composed of three constant regions and one variable region, and two light chains (brown) that consist of one constant and one variable region. IgA1 contains a hinge region with O-linked glycosylation. (B) Monomeric, dimeric IgA, and secretory IgA. Dimeric IgA consist of two IgA molecules that are linked with a J-chain (green). Secretory IgA contains an additional molecule, the secretory component (SC; red). (C) Structure of FcαRI. FcαRI consist of a transmembrane domain, a short cytoplasmic tail and two extracellular domains (EC1 & EC2). It is associated with the signaling FcR γ chain via an electrostatic interaction. The IgA heavy chain junction of Cα2 and Cα3 binds to the EC1 domain of FcαRI in a 2:1 stoichiometry. (D) A model of dIgA1 bound to four FcαRI molecules. The FcαRI:IgA1-Fc complex (PDB 1OW0) was superimposed onto the solution structure of dIgA1 published by Bonner et al. (24) (PDB 2QTJ). FcαRI molecules bound to the top or bottom IgA1 antibodies are colored green or yellow, respectively. The C-terminal residue of each receptor is shown in orange or red to illustrate the membrane-proximal region. Kindly provided by Andrew B. Herr, PhD (Cincinnati Children's Hospital).
IgA1 comprises 80–85% of total human serum IgA (1–3 mg/ml) and is prevalent on many mucosal surfaces including the nasal, bronchial, gastric, and small intestinal mucosa. IgA2 is predominantly present in the colon (26). This division of IgA1 and IgA2 between the small and large intestine may reflect the luminal distribution of food proteins and gram-negative bacteria, which is supported by findings that bacterial overgrowth in the small intestine shifts production toward IgA2 (27). Whereas, IgA1 is susceptible to proteolytic cleavage due to a longer hinge region, IgA2 is resistant to bacterial proteases that are present in the lumen of mucosal areas (6, 21).
Serum IgA is predominantly monomeric in humans and primates (while dimeric in other animals) and is produced by local plasma cells in the bone marrow, spleen, and lymph nodes. IgA at mucosal surfaces is produced by local plasma cells as dimeric molecules, although small amounts of monomers, trimers and tetramers or polymers can also be present (1, 3). Dimeric IgA is composed of two monomers, which are linked with a J-chain (Figure 1B). It associates with the pIgR present at the basolateral side of epithelial cells in mucosal surfaces, after which dIgA is transported across the epithelium and released into the lumen. For every molecule of dIgA that is transported across the epithelium, one pIgR is needed (10, 28, 29). At the luminal side, pIgR is cleaved and a part, referred to as secretory component (SC), remains attached thereby forming SIgA (Figure 1B). Secretory component is a hydrophilic and highly (N- and O-linked) glycosylated negatively charged molecule, which protects SIgA from degradation when present in luminal secretions (30). Glycan removal led to reduced binding capacity of SIgA to gram-positive bacteria, indicating the essential role of carbohydrate structures in efficient IgA coating of (mainly commensal) bacteria (31).
Structure and Expression of FcαRI
FcαRI is a member of the Fc receptor immunoglobulin superfamily (32). It is expressed on cells of the myeloid lineage, including neutrophils, eosinophils, monocytes, macrophages, Kupffer cells, and certain DC subsets (18, 33, 34). Additionally, FcαRI expression on human platelets was described (35). Unlike for other Fc receptors, the FcαRI gene is located on chromosome 19 (19q13.4) within the leukocyte receptor cluster (LRC). This region also encodes natural killer cell inhibitory/activatory receptors and leukocyte Ig-like receptors. The FcαRI amino acid sequence has therefore more similarity with these receptors than with other Fc receptors (19, 36). The EC1 and EC2 exons each encode an extracellular Ig-like domain. These domains are folded in an angle of ~90°. The TM/C exon encodes both the transmembrane domain and a short cytoplasmic tail (Figure 1C) (36).
Surface expression of two isoforms of FcαRI has been described on human phagocytes. The a.1 isoform is a 32 kDa single pass transmembrane receptor, which can be extensively glycosylated (six N-linked sites and seven O-linked sites) (37). The molecular weight of the a.1 isoform varies between 55 and 75 kDa on neutrophils and monocytes, while the molecule is heavier (70–100 kDa) on eosinophils due to more extensive glycosylation (21, 37). The a.2 isoform misses 66 nucleotides in the EC2 exon resulting in loss of one O-linked glycosylation site and has a weight of 28 kDa (without glycosylation) (38). The a.2 isoform is solely expressed on alveolar macrophages (38).
The expression of FcαRI was estimated to represent 66,000 and 57,000 molecules on neutrophils and monocytes, respectively (33). Expression level can be modulated by several mediators. On neutrophils, N-formylmethionyl-leucyl-phenylalanine (fMLP), interleukin (IL)-8, tumor necrosis factor-alpha (TNF-α), lipopolysaccharide (LPS), and granulocyte-macrophage colony–stimulating factor (GM-CSF) enhanced the expression of FcαRI (39–41). GM-CSF has also an important role in mobilizing neutrophils from the bone marrow (42) and was shown to induce high affinity binding of IgA by human neutrophils (43). Neutrophil FcαRI upregulation was rapid and either induced by de novo synthesis or via transport from an intracellular pool to the cell surface (44). On monocytes and monocyte-like cell lines FcαRI expression was enhanced by calcitriol, LPS, TNF-α, GM-CSF, and IL-1β, while downregulation was observed in response to transforming growth factor-beta (TGF-β) or interferon-gamma (IFN-γ) (45, 46). Both monomeric and, to a greater extent, polymeric IgA were able to downregulate FcαRI, possibly due to receptor aggregation, resulting in internalization (47–49).
IgA and FcαRI
Binding of IgA to FcαRI
FcαRI is a low affinity Fc receptor for monomeric IgA and dIgA (Ka = 106 M−1), while IgA immune complexes bind with high avidity and cross-link FcαRI (50). Monomeric IgA binds to the EC1 domain of FcαRI via its Cα2 and Cα3 domains in a 2:1 stoichiometry (i.e., one IgA molecule binds two FcαRI molecules) (Figure 1C) (51, 52). Presence of residues Pro440-Phe443 and Leu257-Leu258 in these domains is essential for IgA binding to FcαRI (53).
Dimeric IgA contains four FcαRI binding sites and can therefore theoretically bind four FcαRI, although this is presumably not possible due to steric hindrance (Figure 1D) (24). It remains to be elucidated how dIgA exactly interacts with the FcαRI. Binding of SIgA to FcαRI is hampered because of steric hindrance by SC. In order for SIgA to activate cells, co-stimulation of FcαRI, and the lectin Mac-1 (CD11b/CD18) was necessary (54).
Little is known about the difference between IgA1 and IgA2 binding to FcαRI (if any) or the influence of glycosylation on binding capacity. It was however shown that a specific mutation (Asn58 to Glu58) resulted in an altered glycosylation pattern of FcαRI, which increased the binding capacity of IgA nearly 2-fold (55). Removal of sialic acids led to a nearly 4-fold increase of IgA binding. This demonstrates the importance of glycosylation at position 58 of FcαRI in binding affinity for IgA (55). N-glycans located at the external surface of the IgA heavy chain were important for interaction with FcαRI as well (56). Furthermore, it was demonstrated that alterations in IgA1 glycosylation and impaired sialylation of FcαRI were linked to increased binding of IgA1 to FcαRI on neutrophils of patients with IgA nephropathy, which may influence pro-inflammatory functions (47). In transfectants, eosinophils, and monocytes FcαRI binding capacity for IgA immune complexes was enhanced by incubation with several cytokines like GM-CSF, IL-4, and IL-5, without affecting the expression level of the FcαRI on the cell surface (43, 57).
Competitive binding for FcαRI has been described for pentraxins, including the acute phase C reactive protein and serum amyloid P component, resulting in cell activation (58). These proteins are characterized by a pentameric ring-like structure containing five subunits, which recognize a similar site on FcαRI as IgA. However, mutations in FcαRI outside the IgA binding site did not affect IgA binding, but enhanced pentraxin binding 2-fold, suggesting that pentraxins bind to a broader region on FcαRI than IgA (58).
Importantly, Staphylococcus aureus and group A and B streptococci developed evasion strategies for IgA-mediated elimination by FcαRI-expressing immune cells by producing several decoy proteins that obstruct binding of IgA to FcαRI (22, 23). Anti-IgA proteins from group B streptococci include Sir22, Arp4 and an unrelated β protein, whereas Staphylococcus aureus produces staphylococcal superantigen-like protein seven. These proteins bind to specific Fc residues (amino acids Pro440-Phe443; i.e. PLAF) in the Cα2 and Cα3 domains of IgA (22, 23, 59). The fact that pathogens evolved and developed mechanisms to evade IgA-mediated elimination by FcαRI-expressing immune cells (60), challenges the dogma of IgA as a non-inflammatory and possibly redundant antibody.
Cell Activation After FcαRI Cross-Linking
Cross-linking of FcαRI by IgA immune complexes (or IgA-opsonized pathogens) induces a variety of processes, including phagocytosis, antibody-dependent cellular cytotoxicity, superoxide generation, release of inflammatory mediators, and cytokines as well as antigen presentation (61–65). Due to hampered binding of SIgA to FcαRI, SIgA does not induce efficient uptake of pathogens by neutrophils or Kupffer cells. However, respiratory burst in neutrophils can be triggered by SIgA, although less efficiently compared to serum IgA (54). Phagocytosis of IgA-coated bacteria or yeast particles by neutrophils was enhanced after priming with GM-CSF or IL-8 (43, 66). Uptake of IgA-coated particles induces increased reactive oxygen species production, subsequently resulting in the release of neutrophil extracellular traps (NETs), which are web-like structures consisting of DNA and proteins (e.g., elastase and myeloperoxidase) able to catch pathogens (61). FcαRI cross-linking furthermore induces release of the neutrophil chemoattractant leukotriene B4 (LTB4) by neutrophils, thereby generating a positive migration feedback loop (64). LTB4 was also shown to mediate migration of monocyte derived DCs (67). As such, in case of mucosal infection neutrophils can function as potent phagocytes, which eliminate invading IgA-coated pathogens, and attract DCs that present antigens to T cells. It was recently shown that in vitro generated human mucosal CD103+ DCs (cultured with retinoic acid) express FcαRI. After stimulation with IgA-coated Staphylococcus aureus in the presence of Pam3CSK4 CD103+ DCs produced pro-inflammatory cytokines including TNF-α, IL-1β, IL-6, and IL-23, which was dependent on FcαRI (68). Furthermore, IgA-stimulated CD103+ DCs enhanced IL-17 production by allogeneic T cells and IL-22 production by intestinal type three innate lymphoid cells, respectively. This suggests that FcαRI crosslinking by IgA induces a pro-inflammatory T helper 17 response accompanied with IL-22 induced activation of epithelial cells, which might contribute to tissue repair (68).
Signaling via FcαRI
In order to initiate optimal effector functions after FcαRI cross-linking, FcαRI needs association with the FcR γ-chain, which contains an immunoreceptor tyrosine-based activation motif (ITAM) in its intracellular domain (69–72). After cross-linking of FcαRI, the src family kinase Fyn aggregates with the receptor complex and phosphorylates the tyrosines in the ITAM in order to act as docking sites for signaling molecules (69, 73, 74). Recruited Syk associates with the phosphorylated ITAM and plays an essential role in the signaling cascade. It directly activates phosphoinositide 3-kinase and phospholipase Cγ, which are involved in two separate signaling routes. These pathways are interconnected and can therefore modulate each other (69, 72, 75). Eventually the signaling pathways will activate the cell, resulting in pro-inflammatory functions (18, 66). Alternatively, it was demonstrated that inhibitory signals can also be transduced via the FcαRI-FcR γ-chain complex. Monomeric targeting of FcαRI (not leading to cross-linking) results in an inhibitory signal and is dependent on Lyn instead of Fyn kinase (74). Phosphorylation of Syk, LAT, and ERK is hampered by “inhibisomes” that contain signaling molecules and both inhibitory and activating receptors (20, 76). Inhibisomes impair activating Fc receptor functioning via a process referred to as inhibitory ITAM (ITAMi) receptor signaling that initiates anti-inflammatory responses to dampen pro-inflammatory responses induced by other Fc receptors (77). Thus, Lyn and Fyn are essential molecules controlling ITAM inhibition and activation, respectively (74). Signaling via FcαRI was recently described in more detail in Aleyd et al. (18).
IgA and FcαRI mediated functions at different locations
In the Lumen of Mucosal Sites
The functions of SIgA in the intestinal lumen have been well-characterized (Figure 2A). SIgA and commensal microbes have important roles in maintaining balance between tolerance toward non-harmful commensals and food compounds vs. immunity against pathogens (78, 79). When SIgA is released from the epithelial layer into the lumen, it remains attached to the outer mucus layer (80). This close localization to bacteria enables SIgA to disturb bacterial motility and to surround pathogens that have a hydrophilic shell. SIgA coating blocks entrance of pathogens into the intestinal epithelium (a process called immune exclusion). This coating leads to agglutination, because antibody-mediated crosslinking of surface antigens will lead to formation of bacterial clumps. Peristaltic bowel movements ensure removal of bacterial aggregates. Bacterial products like enzymes and toxins can be neutralized by SIgA and adherence to host cells, including epithelial cells, is prevented (78). Thus, together with the mucus layer, SIgA forms a barrier against pathogens, and commensals by preventing colonization and penetration of the mucosal epithelium, thereby avoiding infection and antigen leakage into the systemic circulation (81).
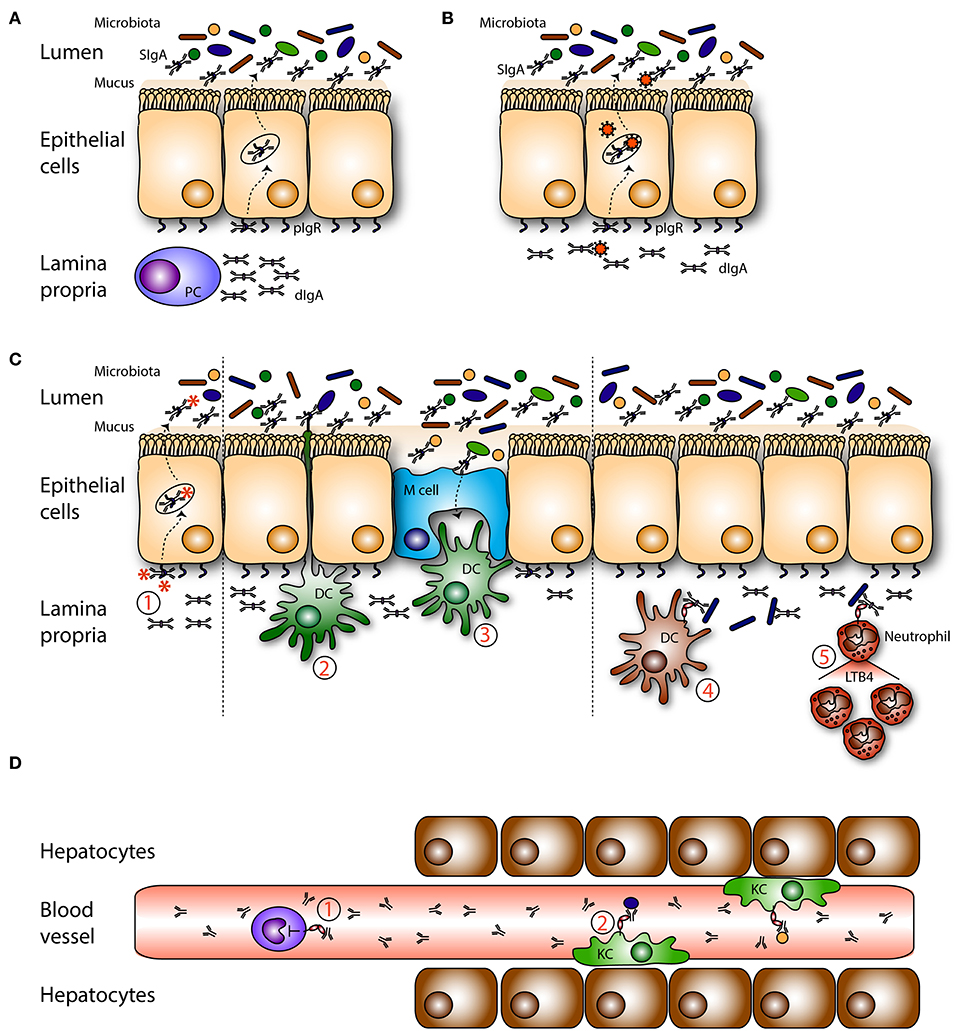
Figure 2. Roles of mucosal IgA in homeostasis. (A) Dimeric IgA (dIgA) is produced by local plasma cells (PC) in the lamina propria. Dimeric IgA is transported to the intestinal lumen by binding to the polymeric immunoglobulin receptor (pIgR) present on epithelial cells. In the lumen it is released as secretory IgA (SIgA) where it can coat (commensal) bacteria. (B) On route dIgA can bind, neutralize and eliminate viruses. (C) (1) Infiltrated antigens and pathogens are opsonized by dIgA and transported back into the lumen. Sub epithelial dendritic cells (DCs) can (2) sample antigens or (3) take up SIgA-coated pathogens that enter via microfold cells (M cells). Pathogens in the lamina propria are coated with dIgA after which this immune complex is taken up by FcαRI- expressing (4) DCs, and (5) neutrophils. In response neutrophils secrete leukotriene B4 (LTB4), hereby attracting more neutrophils, which will clear the infection. (D) Serum IgA is (1) capable of inhibiting (unwanted) pro inflammatory responses in the circulation via ITAMi signaling in monocytes. (2) IgA-opsonized bacteria which have leaked into the circulation are taken up by FcαRI-expressing Kupffer cells (KC) in the liver.
Recently the interplay between luminal microbiota and SIgA was investigated in more detail (4, 82) (Figure 2A). It is still unclear how SIgA and the microbiota exactly relate to each other, but it was described that SIgA shapes and diversifies the gut microflora on the one hand, while the microbiota on the other hand has an important role in regulating IgA levels (82–84). During weaning up to 70% of intestinal commensals are coated with SIgA in mice and the majority of human fecal bacteria in healthy donors are opsonized with IgA, emphasizing the importance of this association in maintaining homeostasis (31, 85). A wide variety of commensals can be opsonized with polyreactive IgA, although it remains unclear how these IgA antibodies are generated, what the relevant targets are and how binding affects the fitness and physiology of commensals (86). In contrast to commensals, pathogens elicit highly specific T-cell dependent IgA responses (86, 87). Genetic background was shown to influence the repertoire diversity and abundance of innate IgA as germ free (GF) naïve BALB/c mice already had substantial innate IgA levels, independent of microbiota colonization, while this was not the case for C57BL/6 mice (83).
Furthermore, highly glycosylated IgA was able to bind to several gram negative bacteria, which modulated their gene expression, leading to enhanced processing of fibers and production of butyrate as well as a diversified microbiota composition (82). Moreover, it was shown that the disbalanced microbiota of colitogenic mice became more balanced and diversified after oral administration of the monoclonal IgA W27, which is a high-affinity polyreactive antibody (84). Importantly, mice lacking IgA, J-chain or pIgR were unable to efficiently protect themselves against mucosal infections. These mice have an altered gut microbiota as well as increased mucosal permeability, which renders them more susceptible to develop colitis (80, 88–94). Of note, lack of J-chain or pIgR also led to absence of SIgM which may have contributed to altered microbiota and compromised immunity (88, 91, 94). A diversified microbiota can regulate the amount of SIgA present in luminal secretions by enhancing the level of pIgR on epithelial cells (1, 31, 95), illustrating the complex interrelationship between IgA (and IgM) and the microbiota in maintaining homeostatic conditions.
In Epithelial Layers
It was described that SIgA can be translocated back into the lamina propria (retrograde transport). SIgA and SIgA immune complexes can be transported across specialized epithelial cells, referred to as M cells, which are located in the Peyer's patches of the intestine. Dectin-1 that is expressed by M cells may facilitate transepithelial transport (11, 96). Additionally, a role for TfR1 on epithelial cells in retrotransport of SIgA-coated gliadin was reported in studies investigating celiac disease (12). Retrotransported SIgA immune complexes can be taken up by DCs that are present in the subepithelial dome. In vitro models support an essential role for DC-SIGN in recognizing SIgA immune complexes (13). The retrograde transport mechanism and recognition of SIgA by DCs is thought to be essential in monitoring the antigenic status of the intestinal lumen.
On route through epithelial cells, dIgA has the ability to intercept and disarm viruses and redirect them into the lumen, after which they are removed with the feces (Figure 2B). Neutralization of several viruses by IgA including sendai, influenza, rota, measles, and human immunodeficiency virus (HIV), as well as bacterial LPS was demonstrated in vitro (97–104). IgA has enhanced neutralizing capacity compared to other Ig isotypes due to the N-linked glycosylation of position N459D in the heavy chain Cα3 (105). This effect is independent of the antigen binding site as glycosylated non-specific IgA neutralized the virus as well.
IgA deficient mice, J-chain deficient mice (lacking dIgA, SIgA, and SIgM) and pIgR deficient mice (lacking SIgA and SIgM) have defects in clearing viruses, are more susceptible to reinfection and lack protective immunity (106–108), which emphasizes the important role of IgA in anti-viral immune responses. By contrast, in a recent study pIgR knock-out mice showed a reduced acute norovirus infection rate, suggesting that pIgR and natural polymeric immunoglobulins may promote viral infections. It was suggested that the less diversified intestinal microbiota present in pIgR deficient mice was responsible for reduced infection. pIgR deficient mice had enhanced levels of the anti-viral cytokine IFN-γ, which might explain the reduced infection rates (109).
In the Lamina Propria
Food particles and commensal microbiota are abundantly present in the lumen and can continuously reach the lamina propria through diffusion (via epithelial tight junctions) or transcytosis (Figure 2C). These non-harmful components need to be tolerated and they need to be cleared from the mucosa to avoid the formation of immune complexes, which may trigger undesired immune responses. In the lamina propria dIgA can eliminate both non-harmful and harmful components from the tissue by transporting immune complexes back into the lumen through association with the pIgR (97, 101). Additionally, FcαRI+ residing immune cells may help to clear IgA-opsonized pathogens. Under homeostatic conditions, only few FcαRI+ cells are found in mucosal areas (own unpublished data). However, as dIgA-opsonized complexes have the ability to cross-link FcαRI, leading to release of LTB4, neutrophils can be rapidly recruited (64) (Figure 2C). Furthermore, in vitro generated FcαRI+ CD103+ DCs were shown to produce pro-inflammatory cytokines after stimulation with IgA-coated Staphylococcus aureus in the presence of Pam3CSK4, suggesting that a protective adaptive immune response is initiated during mucosal infections (68).
Systemic Protection
It was demonstrated that a variety of commensal microbes in the gut influenced serum IgA levels, which protected mice against lethal sepsis when the intestinal barrier was damaged (110). Serum IgA was shown to target the same epitopes and used the same V-gene segments as plasma cells in the gut (111), suggesting that systemic and mucosal plasma cells originated from the same B cell clone. Diminished serum IgA titers were found in GF mice compared to specific pathogen free mice, supporting the essential role for microbiota diversity in IgA production (110, 112). Mice lacking pIgR and SIgA have epithelial barrier disruption, enhanced numbers of IgA-secreting plasma cells and systemic immune activation indicated by increased levels of serum IgG and IgA (113, 114). Higher levels of albumin were found in the feces, suggesting leakage of serum proteins across the epithelium (113). At the same time intestinal proteins and microorganisms can leak into the tissue and enter the bloodstream. After entering the portal circulation they will encounter Kupffer cells in the liver, which are specialized FcαRI+ macrophages that can eliminate IgA-coated bacteria from the bloodstream (Figure 2D) (34). Kupffer cells, macrophages, and monocytes produced increased levels of pro-inflammatory cytokines (e.g., TNF-α, IL-1β, and IL-6) after cross-linking of FcαRI in the presence of pathogen recognition receptor ligands (such as Pam3CSK4, LPS, flagellin) (reflecting IgA-opsonized pathogens) (115).
In contrast to the activating properties of complexed serum IgA, monomeric serum IgA was capable of downregulating cell responses and promoted powerful anti-inflammatory effects (Figure 2D). This is referred to as ITAMi signaling (see above) (19). Monomeric targeting of the FcαRI with serum IgA may protect against enhanced receptor activation, which is beneficial in inflammatory diseases characterized by the presence of IgG immune complexes or enhanced FcεRI signaling.
IgA-associated Diseases
In babies circulating IgA levels are physiologically low because IgA cannot be transported across the placenta. Colostrum and breast milk are important sources of SIgA antibodies to provide local protection against infections. As soon as exposure to microbiota takes place, the SIgA immune system is rapidly maturing. Nonetheless, adult serum IgA levels are not reached before puberty as the systemic IgA compartment develops very slowly. The daily production rate of IgA exceeds that of all other combined antibody classes (66 mg/kg/day), suggesting that the role of IgA in immune defense must be considerable for the body to spend high energy levels (30). Abnormal IgA levels in serum and external secretions (either higher or lower) have been described in numerous pathologies. Very low or absent IgA is referred to as IgA deficiency (116). High levels of (aberrantly glycosylated) IgA are present in multiple diseases including IgA nephropathy, dermatitis herpetiformis, IgA vasculitis, and rheumatoid arthritis.
IgA Deficiency
Selective IgA deficiency is the most common human immunodeficiency and is characterized by a serum IgA concentration of <7 mg/dL. Both serum IgA and mucosal IgA are very low or absent in selective IgA deficient patients (6, 117). Defects in B cell isotype switching, terminal differentiation of IgA+ plasma cells into secretory cells or long-term survival of IgA-secreting plasma cells can result in IgA deficiency (118, 119). Unbalanced cytokine production (including IL-4, IL-6, IL-7, IL-10, and IL-21) has been reported with a key role for TGF-β, as it induces isotype switching and differentiation of antigen-stimulated B cells into IgA-secreting plasma cells (120, 121). The initial defect may lie in the stem cell compartment as IgA deficiency can be transferred by bone marrow transplantation (122).
A significant heterogeneity exists among IgA deficient patients. Many patients are asymptomatic and only few show minor clinical symptoms. This may in part be due to better hygiene, vaccination, and antibiotic use in modern Western society. Furthermore, it was shown that increased production of IgM and/or IgG can partially compensate for the lack of IgA (Figure 3A) (123, 124). IgA deficient patients can develop severe pathology when IgG and/or IgM does not compensate for IgA loss (117, 125). Conflicting reports on the influence of IgA deficiency on fecal microbiota in patients have been reported. One study showed a significantly less diverse fecal microbiota of IgA deficient patients compared to healthy controls, and specific taxa were lost in IgA deficient patients (124). Another study reported a mild loss in microbial diversity in IgA deficient patients (117). SIgM could partially rescue the microbiota composition in IgA deficient patients (117). However, SIgM targeted a broader range of microbes and showed less specificity for microbes compared to SIgA (124). Patients can have increased C reactive protein levels, which is indicative for enhanced inflammation, along with an increased risk of death 10–15 years after initial diagnosis (126, 127). In a recent study no risk association was found between IgA deficiency and hospital infections (127). IgA deficiency was also not more common among hospitalized individuals compared to healthy blood donors (123). However, patients have moderate enhanced susceptibility to gastrointestinal, urinary, and recurrent respiratory infections, allergies, celiac disease, and autoimmune diseases (6, 128, 129). Forty-five percent of IgA deficient patients have the 8.1 haplotype (HLA-A1, B8, DR3, DQ2 haplotype) compared to 16% in the general population (123, 130). The 8.1 haplotype is also commonly found in patients with autoimmune diseases (including rheumatoid arthritis and systemic lupus erythematosus), which are strongly associated with IgA deficiency (123). These findings suggest that patients share predisposing genes, which may explain the increased prevalence of selective IgA deficiency in certain autoimmune disorders.
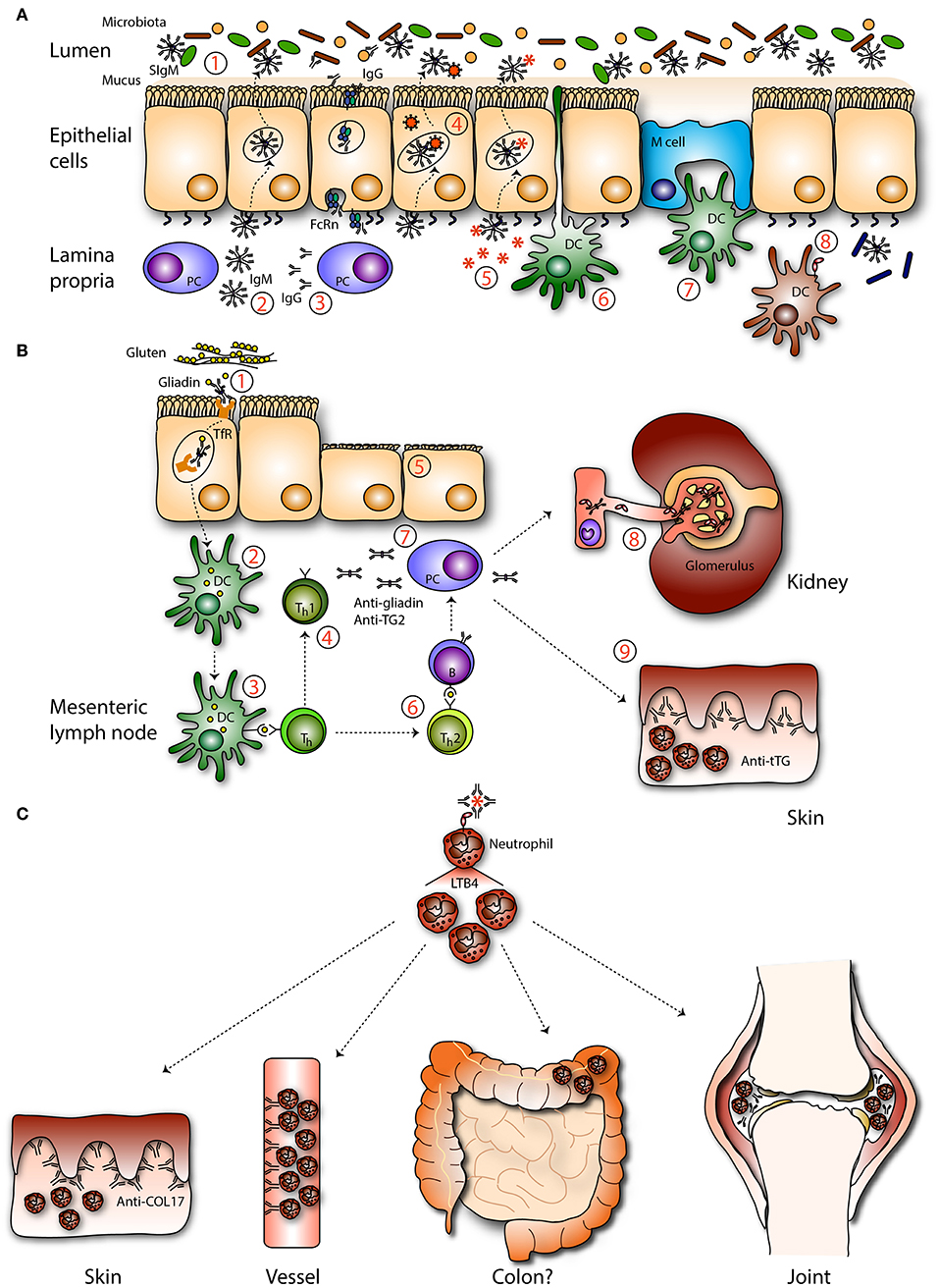
Figure 3. Mucosal IgA in pathogenesis. (A) IgA deficiency. In absence of IgA the intestinal microbiota is (1) less diverse and unbalanced. (2) IgM, and (3) IgG can be transported across the epithelium by associating with the pIgR or the neonatal Fc receptor (FcRn), respectively, to compensate IgA loss. Theoretically, IgM may be able to (4) neutralize viruses and (5) exclude infiltrated antigens. Sub epithelial DCs are not described to (6) sample IgM coated bacteria. (7) IgM-coated bacteria are not described to enter M cells. (8) Invaded IgM-coated pathogens are not recognized by FcαRI-expressing immune cells, resulting in less efficient pathogen elimination. (B) Celiac disease and gluten-associated diseases. (1) IgA-gliadin complexes bind to the transferrin receptor (TfR) and are retrotransported across the epithelium. (2) In the lamina propria deamidated gliadin is taken up by DCs and after processing (3) presented to T helper (Th) cells. (4) Th1 cell activation leads to the release of pro-inflammatory mediators (5) causing tissue damage. (6) Activated Th2 cells stimulate autoantibody production directed against gliadin and tissue transglutaminase by B cells. (7) Plasma cells produce autoantibodies which (8) can be detected in the circulation and form deposits with soluble FcαRI in the kidney resulting in damage (IgA nephropathy). (9) IgA anti-tissue transglutaminase forms complexes in the dermis, resulting in neutrophil activation and concomitant tissue damage (dermatitis herpetiformis). (C) IgA-FcαRI induced pathology. IgA immune complexes activate neutrophils by cross-linking FcαRI, resulting in release of LTB4 and enhanced neutrophil influx, which induces tissue damage as seen in the skin (LABD), vessels (IgA vasculitis), joints (RA), and potentially in colon (IBD).
Eighty-four percent of IgA deficient patients were positive for a wide variety of allergens in skin prick tests (131). The link between IgA deficiency and allergies may be explained by higher levels of circulating antigens due to increased permeability at mucosal surfaces. Alternatively FcαRI may not be able to induce ITAMi signaling in the absence of monomeric serum IgA, possibly resulting in overactivation of immune cells that can lead to development of allergies and autoimmune diseases (132).
Anti-IgA antibodies, usually of the IgG subclass, are commonly found in IgA deficient patients (40%), although their etiology remains unknown. Transfusing IgA-containing blood products to treat IgA deficiency is therefore complicated, as IgA-anti-IgA immune complexes can induce severe reactions, especially when anti-IgA antibodies of the IgE subclass are present (133, 134).
Allergic Diseases
Allergic diseases are inflammatory conditions of which the majority are characterized by high specific IgE levels and activated mast cells (135). Alterations in microbiota diversity reduced immunological tolerance, potentially resulting in food allergies, allergic rhinitis, and asthma (136). It was suggested that the current hygienic life style in the western world altered normal microbiota colonization in infants thereby contributing to the enhanced prevalence of allergies (“hygiene hypothesis”) (137). A less diverse composition of the microbiota was associated with lower IgA levels, suggesting that IgA can be involved in the development of allergic diseases as well. Studies investigating the link between IgA levels and allergy and asthma have however been conflicting. In children, serum IgA levels correlated negatively with asthma severity (138). Moreover, it was shown that lower levels of intestinal bacteria were coated with IgA in infants with asthma (139). In adults, both house dust mite sensitization and airway hyper-responsiveness correlated negatively with serum IgA (140). Furthermore, asthma symptoms like shortness of breath and sputum production inversely correlated with, respectively mucosal SIgA and serum IgA levels (141). These results suggest a potential link between low IgA levels and the risk and severity of allergic asthma, which supports a protective role for IgA in allergy. However, data has also been reported showing enhanced specific IgA levels in patients with allergic rhinitis and atopic asthma (142, 143). Moreover, allergen specific IgA in combination with eosinophilia were common characteristics of asthma and allergic rhinitis (142), possibly because IgA can induce eosinophil survival, thereby contributing to disease severity (144). These results demonstrate a dual role of IgA in allergies, which is currently ill-understood.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a systemic and chronic autoimmune disorder characterized by infiltration of inflammatory immune cells in the joints, resulting in swelling and pain. Presence of autoantibodies including rheumatoid factor (RF) and anti-citrullinated protein antibodies are commonly present, which can be of the IgM, IgG or IgA isotype. High IgA anti-citrullinated protein antibodies and IgA RF titers correlate with worse disease prognosis and severity, and have been used as predictive value for disease progression (145–147). This suggests that IgA contributes to disease pathology. It was demonstrated that blocking the interaction between IgA RF and macrophage FcαRI resulted in reduced levels of TNF-α (148). Additionally, neutrophils stimulated with plasma of RA patients, containing IgA RF, induced NET release, which was inhibited by blocking FcαRI (149). Thus, IgA-immune complexes in patients can induce pro-inflammatory functions of neutrophils and macrophages, which are prominently present in inflamed joints (150), and as such contribute to inflammation in RA.
IgA Nephropathy
IgA nephropathy (IgAN) is the most common form of glomerulonephritis and progresses to end-stage kidney failure in 50% of patients. IgA levels are increased in both serum and urine of IgAN patients. A link with RA was suggested, as patients can also have enhanced IgA RF in their serum. This condition is referred to as rheumatoid nephropathy (151). It was hypothesized that galactose-deficient O-glycans in IgA1, produced by plasma cells in the gut, can trigger the production of anti-glycan IgG/IgA antibodies, after which formed immune complexes deposit in the glomeruli where renal injury is initiated (152, 153). The enzymes responsible for production of O-linked glycans are glycosyltransferases, which expression are amongst others regulated by bacterial products, suggesting that the microbiome regulates the specific glycosylation pattern of IgA1 during the initial phase of IgAN (154, 155). In mice, galactose-deficient IgA1 was not cleared from the murine circulation and deposited in the kidney (15). FcαRI+ Kupffer cells were unable to clear circulating IgA of IgAN patients in vivo (156). In transgenic mice expressing the human FcαRI on monocytes and macrophages, soluble FcαRI-IgA complexes that were deposited in the mesangium, induced glomerular and interstitial macrophage infiltration, hematuria, mesangial matrix expansion, and mild proteinuria (157, 158). Moreover, in FcαRI transgenic and human IgA knock-in mice soluble FcαRI-IgA complexes induced kidney inflammation by interacting with TfR1 on mesangial cells, which induced the release of pro-inflammatory mediators. Additionally, the expression of transglutaminase 2 was induced, which subsequently enhanced the expression of TfR1, thereby inducing a pathogenic amplification loop (157). Of note, shedding of FcαRI from macrophages of transgenic mice may have aggravated disease, which will likely not occur in patients as human macrophages express lower FcαRI levels. Nonetheless, IgA-soluble FcαRI complexes have been found in patients with IgAN, and serum IgA immune complexes bound more avidly to TfR1 in vitro compared to those from healthy controls (158, 159). It was demonstrated that IgAN and Henoch-Schönlein purpura nephritis patients had reduced levels of soluble FcαRI and transglutaminase 2 in their urine (160), which makes it plausible that soluble FcαRI immune complexes deposit in the human kidney.
IgA Vasculitis
IgA vasculitis, also known as Henoch-Schönlein purpura, is the most common form of vasculitis. The condition is characterized by IgA1 immune deposits and neutrophil infiltrates affecting the small vessels. As a result, red blood cells can leak into the skin leading to typical cutaneous hemorrhages, which leads to diagnosing the disease (161, 162). The pathology of IgA vasculitis remains unclear. However, it is suggested to represent a systemic equivalent of IgAN, as IgA vasculitis can be accompanied with nephropathy, resembling IgAN (163). Unlike IgAN, which is characterized by deposits of galactose-deficient IgA1, it is unknown which type of IgA accumulates in IgA vasculitis (152, 164).
Increased levels of soluble FcαRI-IgA complexes were found in sera of adult and pediatric vasculitis patients with or without co-existing nephritis, which was associated with decreased FcαRI expression on monocytes (165, 166). Furthermore, it was proposed that IgA1 anti-endothelial cell antibodies might play a role. Serum IgA from IgA vasculitis patients was shown to bind in vitro to human but not bovine glomerular endothelial cells (167). IgA anti-endothelial cell antibodies induced the production of IL-8 by endothelial cells, thereby contributing to an inflammatory environment and neutrophil recruitment (168, 169). In addition to enhanced levels of serum TNF-α, which promoted anti-endothelial cell antibody binding to endothelial cells, IL-8 production increased inflammation in IgA vasculitis patients (167, 169, 170). It was hypothesized that neutrophils become activated by IgA-FcαRI mediated cross-linking, resulting in inflammatory processes like reactive oxygen species production and NET formation. Moreover, IgA-activated neutrophils release LTB4, inducing neutrophil migration, thereby enhancing a positive feedback loop, which may result in pathogenesis observed in IgA vasculitis (162).
Inflammatory Bowel Disease
Inflammatory bowel disease is characterized by chronic inflammation in the intestinal tract and is subdivided into ulcerative colitis and Crohn's disease (171). The barrier function in these patients is disrupted allowing bacteria to invade the subepithelial lamina propria (171). Downregulation of pIgR on intestinal epithelial cells and a disturbed microbiota were observed in IBD patients (172). It was proposed that intracellular signaling pathways and protein trafficking was altered due to damage caused by the inflammatory environment resulting in pIgR regulation defects (10). Mice lacking pIgR have enhanced numbers of IgA-secreting plasma cells, possibly as compensation. Increased production, combined with diminished transcytosis of dIgA into the lumen may potentially result in accumulation of dIgA in the lamina propria. Invading bacteria can become opsonized with dIgA, potentially resulting in activation of neutrophils by cross-linking of FcαRI, leading to tissue damage. Neutrophils that had taken up IgA were observed in the mucosa of ulcerative colitis patients (64). Cross-talk between Fc receptors and TLRs was induced by antibody-coated bacteria, resulting in release of pro-inflammatory mediators (e.g., LTB4) by DCs, which may also contribute to inflammation in IBD. LTB4 induces recruitment of neutrophils and monocytes. Additionally, simultaneous triggering of FcαRI and TLR4 on neutrophils resulted in enhanced release of pro-inflammatory TNF-α (173). Furthermore, due to diminished SIgA levels in the lumen, neutralization, and immune exclusion of microbes is decreased, which may worsen disease in patients (10). Patients have increased levels of specific IgA against microbiota in their serum (174). Highly IgA-coated bacteria obtained from IBD patients induced colitis in GF mice (175). The extent to which IgA responses against microbiota differ in homeostatic conditions vs. IBD, remain unclear (86).
Surprisingly, an increased amount of fecal bacteria of IBD patients was opsonized with IgA compared to those obtained from healthy control feces (85, 175, 176). It was suggested that leakage of serum IgA or dIgA due to a disrupted epithelial layer contributed to enhanced fecal bacteria coating (176, 177). The exact role of IgA and FcαRI in inflammatory bowel disease however remains to be elucidated.
Celiac Disease and Dermatitis Herpetiformis
Celiac disease is a multifactorial autoimmune disease characterized by a damaged small intestinal mucosa and nutrient malabsorption following gluten ingestion. Individuals carrying the DQ2 and DQ8 HLA haplotypes have an increased risk to develop celiac disease (178). Gliadin, a glycoprotein of gluten, can complex with specific IgA, after which it can be retrotransported across the epithelium via the TfR (Figure 3B). Celiac disease patients have increased expression of TfR on their epithelial cells, allowing increased retrotransport (12). Intracellular degradation of these peptides is disturbed in patients and after entrance in the lamina propria gliadin peptides are deamidated by tissue transglutaminase after which they are presented to HLA-DQ2 or HLA-DQ8 expressing CD4+ T cells. This results in the release of pro-inflammatory mediators, concomitant tissue damage and production of autoantibodies (179). IgA anti-tissue transglutaminase antibodies play a key role in disease pathogenesis (180). Presence of these antibodies serve to diagnose the disease and are furthermore linked to other IgA-related diseases like dermatitis herpetiformis and IgA nephropathy (Figure 3B). In dermatitis herpetiformis aberrant IgA antibodies are directed against structural proteins maintaining cell-cell adhesion in the epidermis leading to skin tissue damage (181). It is hypothesized that celiac disease patients can develop dermatitis herpetiformis as high avidity IgA anti-tissue transglutaminase antibodies form immune complexes and deposit in the dermis of these patients, although it is ill understood why some patients develop dermatitis herpetiformis, and others do not. Furthermore, small bowel inflammation in celiac disease is mostly the result of mononuclear cell infiltration, whereas neutrophils accumulate in the skin of dermatitis herpetiformis patients (181). Patient neutrophils have increased ability to bind IgA without FcαRI expression alteration, supporting that FcαRI is primed (e.g., by cytokines or differential glycosylation) (182).
Gluten exposure can also lead to glomerular IgA deposition and IgAN (183). Transgenic human IgA and FcαRI knock-in mice developed severe pathology when gluten were present in their diet, including intestinal injury, increased IgA1–soluble FcαRI complexes, mesangial IgA1 deposition, and elevated serum IgA1 anti-gliadin antibodies. IgAN patients also have increased IgA1 anti-gliadin antibodies in their serum, suggesting that entrance of gluten can potentially result in loss of oral tolerance thereby contributing to IgAN (184). Consumption of a gluten-free diet can resolve manifestations of celiac disease, dermatitis herpetiformis, and IgAN.
Linear IgA Bullous Disease
Linear IgA Bullous Disease (LABD) patients suffer from extensive skin damage and blister formation caused by the presence of IgA autoantibodies against collagen XVII, and concomitant neutrophil accumulation (Figure 3C). Collagen XVII plays a critical role in maintaining adhesion between dermis and epidermis in the skin (185). Neutrophil activation is likely the direct result of FcαRI triggering by IgA autoantibodies (186). Tissue damage was induced in the presence of neutrophils and serum of LABD patients (containing IgA anti-collagen XVII autoantibodies) in an ex vivo skin model, as presence of activated neutrophils led to separation of the dermis from the epidermis (reflecting blister formation) (186). Eosinophil influx has been observed in the skin of LABD patients as well, suggesting that these cells might also contribute to disease pathology through FcαRI mediated respiratory burst activity (185).
IgA and FcαRI as therapy
IgA plays an important role in dampening mucosal infections, but can also have detrimental effects in inflammatory or autoimmune diseases. Lack of IgA may increase susceptibility to infection, whereas overabundant IgA complexes or autoantibodies can be harmful as enhanced activation of the FcαRI may lead to various pathologies. Several therapeutic strategies have been proposed that influence the IgA-FcαRI axis (Figure 4). For instance, in order to benefit from increased IgA levels during infections it was proposed to increase pathogen-specific IgA levels through passive or active vaccination thereby resulting in efficient clearance of pathogens. Furthermore, IgA might be used as a therapeutic antibody to mediate efficient tumor cell killing. Additionally, two different strategies of targeting FcαRI have been proposed. First, the induction of ITAMi signaling via FcαRI may dampen inflammation that is caused by other pro-inflammatory receptors (e.g., FcεRI). Second, blocking FcαRI may reduce activation of immune cells in IgA-mediated inflammation.
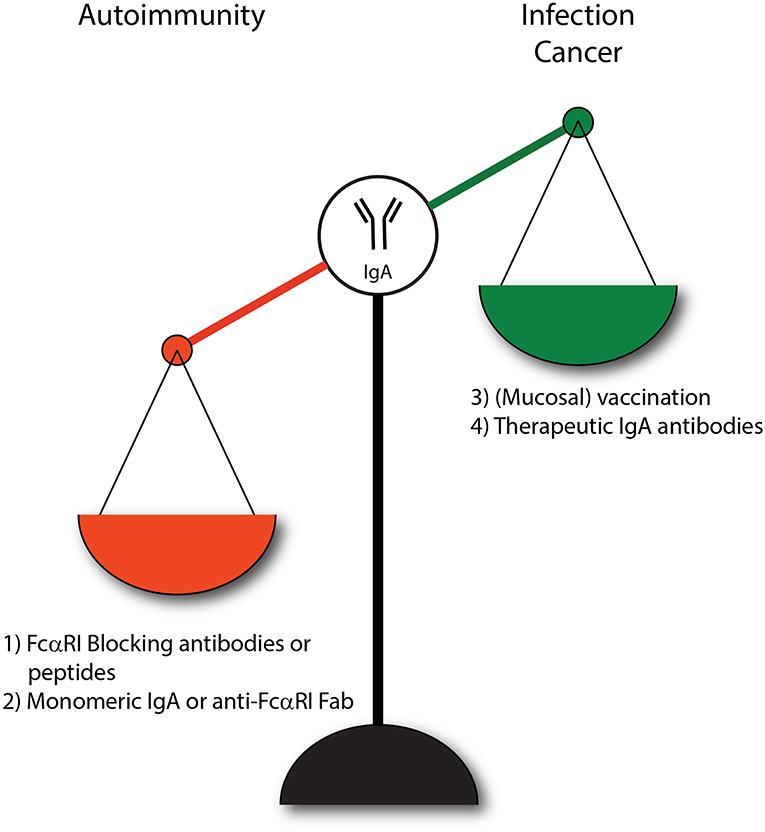
Figure 4. IgA and FcαRI as therapeutic targets. (1) Enhanced IgA-FcαRI activation in IgA-associated autoimmune diseases is unwanted. Blocking IgA-FcαRI interactions by monoclonal antibodies or peptides may reduce tissue damage in these diseases. (2) Treatment with monomeric IgA or anti-FcαRI Fabs may induce ITAMi signaling thereby inhibiting IgG-induced phagocytosis and IgE-mediated allergic diseases. (3) To combat infections, administration of IgA (passive vaccination) or induction of IgA levels via active vaccination may result in enhanced protective immunity. (4) IgA monoclonal antibody therapy may result in efficient killing of tumor cells by activating FcαRI-expressing immune cells.
IgA in Vaccination Strategies
Administration of specific IgA (passive immunization) or enhancing IgA production through active immunization might be effective strategies to combat viral and bacterial infections (104). Intranasal passive administration of either specific monomeric or polymeric IgA against a mycobacterium tuberculosis antigen led to short, but effective protection against infection in mice (187). The short protection duration was probably due to degradation of IgA by bacterial proteases present in the respiratory tract fluid. Furthermore, mice expressing human FcαRI on blood neutrophils and monocytes had a lower mycobacterium tuberculosis infection rate compared to control mice after inoculation of human IgA mAb, supporting an additive beneficial role for FcαRI in clearing the infection (188). Wild type (WT) mice which orally received Salmonella typhimurium that had been complexed with human plasma-derived IgA and IgM had reduced intestinal infection rates compared to mice exposed to Salmonella typhimurium only. Enhanced bacterial clearance was observed when SC was coupled to plasma-derived IgA and IgM (189). Polymeric IgA against influenza virus reached the nasal mucosa after intravenous administration and protected WT mice against infection, and was 10 times more effective than IgG in reducing viral shedding (190, 191). Passive immunization with recombinant dIgA showed better prevention against intra-rectal simian HIV transmission in rhesus macaques. Dimeric IgA could either directly neutralize simian HIV or the virus was trapped by large immune complexes preventing entrance in the epithelial barrier (103). It was demonstrated that IgA isolated from serum of HIV survivors and vaccinated HIV patients neutralized HIV through occupying the CD4 binding site of the virus (192, 193). By contrast, in another study, HIV specific IgA levels in plasma interfered with IgG effector functions in vitro, thereby increasing HIV infection risk (194). Recently, it was reported that oral and nasal administrated HIV antigens bound to SIgA were retro transported through M cells and reached sub-epithelial DCs, thereby inducing both mucosal and systemic humoral and cellular immune responses in CX3CR1/GFP transgenic mice (195). Vaccination with SIgA-HIV antigen protected these mice from infection after challenge with a recombinant vaccinia virus expressing the HIV antigen (195, 196). This demonstrates the potential of SIgA to serve as a vaccine carrier for HIV via mucosal administration.
Vaccination with live attenuated polio induced intestinal IgA production and induced long-lived memory immune responses in elderly who still had detectable serum and salivary IgA levels (197–199), which suggested that priming with live virus is needed to obtain IgA responses after booster vaccination with inactivated polio virus. Furthermore, WT mice that were immunized and re-challenged with reovirus had enhanced serum and intestinal IgA levels and were protected from infection (108, 200). IgA−/− mice were unable to clear reovirus infection (108). Mice immunized with hemagglutinin (surface protein on influenza virus) produced anti-influenza specific SIgA, which protected mice from infection after intranasal administration (201). Mice deficient of IgA, J-chain or pIgR showed compromised immune protection (106–108), supporting that IgA enhances protective immunity against viral and bacterial infections.
FcαRI as Therapeutic Target in Cancer Antibody Therapy
Antibodies targeting specific tumor-associated antigens like epidermal growth factor receptor (EGFR) and human epidermal growth factor two (HER2) are increasingly used to treat solid tumors (respectively colorectal cancer and breast cancer) (202). These therapeutic antibodies are often of the IgG isotype and have long half-life, can activate complement, and recruit natural killer cells as well as macrophages as cytotoxic effector cells (202, 203). Alternatively, IgA or FcαRI bispecific antibodies (BsAb) may represent promising novel drugs to treat cancer by enhancing activation of FcαRI-expressing immune cells (204). For instance, BsAb targeting both tumor antigens and FcαRI efficiently recruited neutrophils in vitro, which was not observed after targeting Fcγ receptors (205–207). Moreover, (immature) neutrophils were able to kill tumor cells more efficiently due to FcαRI-induced antibody dependent cellular cytotoxicity (62, 203, 208). Similarly, IgA anti-tumor mAbs mediated tumor killing more efficient compared to IgG mAbs. In vitro studies demonstrated the superior ability of FcαRI to induce neutrophil-mediated tumor cell killing for multiple tumor antigens, including EGFR, HER2, EpCAM, HLA-II, CD20, CD30, and carcinoembryonic antigen (203, 205, 209).
Unfortunately, in vivo tumor targeting using IgA anti-tumor mAbs has been difficult as mice do not express a homolog of the human FcαRI, and human IgA has a short half-life in mice (158). The generation and utilization of FcαRI transgenic mice (71, 158) can contribute to study IgA as therapeutic antibody in vivo. Treatment with IgA anti-HER2 or IgA anti-EGFR anti-tumor mAbs resulted in significantly enhanced anti-tumor cytotoxicity in FcαRI transgenic mice compared to WT littermates, which was mediated by FcαRI-expressing macrophages (210, 211). Furthermore, IgA anti-CD20 mAbs inhibited B cell lymphoma cell proliferation in vitro and recruited FcαRI-expressing immune cells in vivo in FcαRI transgenic mice (212, 213).
Thus, IgA antibodies have been shown to recruit neutrophils and macrophages as effector cells, but the short half-life of IgA in serum is a major drawback for use of IgA as therapeutic antibody. Compared to IgG, the glycosylation sites of therapeutic IgA mAbs that are produced in non-human systems (like rodent cells) have higher immunogenic potential (high glycosylation) and therapeutic IgA will be likely cleared efficiently from the human (or murine) circulation. Increasing the sialylation of glycans on IgA mAbs enhanced their serum half-life due to decreased clearance by the ASGPR in the liver (210). Inducing sialylation of IgA mAbs was done by using fully human systems thereby augmenting their in vivo therapeutic potential (211, 214–216). Increased sialylation of the N-glycans in IgA anti-HER2 mAbs resulted in significantly reduced tumor growth in FcαRI-transgenic SCID mice bearing BT-474 tumors (214). Addition of an albumin-binding domain to IgA1 (IgA1-albumin) enhanced interaction of the antibody with the FcRn, which extended the half-life of IgA1 in vivo. Although ex vivo studies showed that IgA1-albumin induced lower maximal tumor cell lysis, enhanced tumor cell killing was observed in vivo (211). Another modified IgA molecule against EGFR lacking glycosylation and free cysteine's, and with a stabilized heavy and light chain linkage, showed increased efficacy by interacting with FcαRI-expressing myeloid cells in vivo. Some therapeutic activity was observed in non-FcαRI transgenic mice demonstrating contribution of Fab-mediated effector functions as well (216). Although IgA antibodies are not used in clinical studies yet, engineering IgA with increased half-life represents a promising strategy for targeting tumors in patients.
Induction of ITAMi Signaling
Enhancing ITAMi signaling by monomeric targeting of the FcαRI may be a promising strategy to inhibit IgG-induced phagocytosis and IgE-mediated allergic diseases (20). Monovalent targeting of FcαRI with the anti-FcαRI mAb A77 inhibited degranulation of RBL-2H3 transfected cells and reduced airway inflammation in FcαRI transgenic mice after crosslinking FcεRI with IgE immune complexes in an allergic asthma model (20, 217). Furthermore, naturally occurring serum IgA dampened immune responses by inducing ITAMi signaling via FcαRI (19). FcαRI transgenic mice (expression on monocytes/macrophages), which developed RA after injection with IgG anti-collagen had reduced manifestations or even complete resolution of arthritis after treatment with monomeric IgA. IgG-induced ITAM signaling was blocked efficiently, implicating a potential role of monomeric IgA in treatment of autoimmune diseases with IgG autoantibodies (218). Moreover, prior targeting of FcαRI by Fab A77 suppressed inflammation in transgenic mice with FcαRI-expressing monocytes and macrophages that suffered from IgG immune complex glomerulonephritis and obstructive nephropathy (219). Renal inflammation induced by pristane was characterized by enhanced serum IgG levels, pro-inflammatory cytokines and immune infiltration in FcαRI transgenic mice with receptor expression on monocytes and macrophages. Blocking FcαRI with Fab MIP8a inhibited cytokine production, leukocyte recruitment and inflammation (77). Furthermore, renal inflammation induced by CpG (TLR9 agonist) in these FcαRI transgenic mice was downregulated by monomeric occupancy of FcαRI (220). Thus, monomeric targeting of FcαRI is suggested to induce anti-inflammatory properties, which could be useful in treatment of inflammatory diseases with involvement of myeloid cells.
FcαRI Blocking
Blocking FcαRI might be effective in IgA-mediated inflammation. In vitro it was shown that neutrophils stimulated with IgA immune complexes obtained from RA patients released neutrophil extracellular traps. FcαRI blocking on neutrophils with the anti-FcαRI mAb MIP8a reduced NET formation and might alleviate neutrophil induced tissue damage in RA patients (149). IgA autoantibodies in serum of LABD patients induced neutrophil-mediated tissue damage in an ex vivo human skin model. FcαRI blocking with MIP8a prevented IgA induced tissue damage (186). Similarly, peptides targeting the interaction sites of IgA and FcαRI showed effective blocking of IgA binding to FcαRI, and reduced IgA-induced neutrophil migration in vitro. These peptides were able to penetrate into human skin, supporting that they might function as novel therapy in skin autoimmune diseases (221). As such, blocking FcαRI might serve as therapeutic strategy for IgA-associated inflammatory diseases.
Conclusion
IgA is important in maintaining balance of mucosal immunity. SIgA regulates immune exclusion by neutralizing pathogens and more recently the role of SIgA in diversifying the intestinal microbiota has become clear. The mechanisms involved in IgA-mediated regulation of microbiota diversity and reciprocal regulation of IgA levels by microbes are incompletely understood. Moreover, it remains unclear why certain SIgA coated antigens associate with the epithelial layer while others are eliminated via immune exclusion. In addition to the traditional role of IgA as non-inflammatory regulator of homeostasis, several pro-inflammatory functions have been described, which need to be clarified in more detail.
After binding to FcαRI, IgA plays important roles in pathogen elimination. It can also have detrimental effects on human health when aberrant IgA is present. Perpetual IgA-FcαRI interaction results in enhanced activation of immune cells with concomitant tissue damage as seen in autoimmune diseases like LABD (64). It is yet undefined why neutrophil influx is observed in some IgA-FcαRI mediated diseases, including LABD and dermatitis herpetiformis, but not others. For instance, Crohn's disease is generally less characterized by neutrophil influx in the intestinal mucosa, while in patients with ulcerative colitis massive neutrophil infiltration is present (171). Similarly, and maybe even more confusing, is the fact that celiac disease patients develop IgA anti-tissue transglutaminase antibodies resulting in infiltration of mainly mononuclear cells in the intestinal tract, while in dermatitis herpetiformis (skin manifestation of celiac disease) binding of IgA autoantibodies to epidermal transglutaminase results in neutrophil recruitment (181).
Targeting the IgA-FcαRI axis with blocking monoclonal antibodies or peptides may alleviate inflammation and concomitant tissue damage. Moreover, inhibitory ITAMi signaling induced by monomeric targeting of FcαRI has been suggested to represent a promising strategy in allergies or IgG immune complex-mediated diseases. By contrast, in infectious diseases and cancer enhancing pro-inflammatory effects of IgA-FcαRI interaction might be very beneficial.
Lack of an FcαRI equivalent in mice has hampered our understanding of the functions and therapeutic applications of IgA and FcαRI. Future research will be facilitated by the use of several human FcαRI transgenic and human IgA knock-in models (71, 158, 222). This will help to increase our knowledge on the complex roles of IgA and FcαRI in (patho)physiology as well as the therapeutic possibilities for targeting these multifaceted molecules.
Author Contributions
AB wrote the paper and designed the figures. MvE designed and illustrated the figures, supervised, and revised the paper.
Funding
MvE is supported by the Netherlands Organization for Scientific Research (VICI 91814650).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ASGPR, Asialoglycoprotein receptor; BsAb, Bispecific antibodies; DC-SIGN, Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin; DCs, Dendritic cells; dIgA, Dimeric IgA; FcαRI, Fc alpha receptor; FcRL4, Fc receptor-like 4; fMLP, N-formylmethionyl-leucyl-phenylalanine; GF, Germ free; GM-CSF, Granulocyte-macrophage colony–stiulating factor, ; IBD, Inflammatory bowel disease; IFN-γ, Interferon-gamma; IgA, Immunoglobulin A; IgAN, IgA nephropathy; IL, Interleukin; ITAM, Immunoreceptor tyrosine-based activation motif; ITAMi, Inhibitory immunoreceptor tyrosine-based activation motif; kDa, Kilodalton; LABD, Linear IgA Bullous disease; LPS, Lipopolysaccharide; LTB4, Leukotriene B4; M cells, Microfold cells; pIgR, Polymeric immunoglobulin receptor; RF, Rheumatoid factor; SC, Secretory component; SIgA, Secretory IgA; SIgM, Secretory IgM; Tfr, Transferrin receptor; TGF-β, Transforming growth factor-beta; TLR, Toll like receptor; TNF-α, Tumor necrosis factor-alpha.
References
1. Woof JM, Kerr MA. The function of immunoglobulin A in immunity. J Pathol. (2006) 208:270–82. doi: 10.1002/path.1877
2. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. (2014) 5:520. doi: 10.3389/fimmu.2014.00520
3. Wines BD, Hogarth PM. IgA receptors in health and disease. Tissue Antigens. (2006) 68:103–14. doi: 10.1111/j.1399-0039.2006.00613.x
4. Macpherson AJ, Yilmaz B, Limenitakis JP, Ganal-Vonarburg SC. IgA function in relation to the intestinal microbiota. Annu Rev Immunol. (2018) 36:359–81. doi: 10.1146/annurev-immunol-042617-053238
5. Leong KW, Ding JL. The unexplored roles of human serum IgA. DNA Cell Biol. (2014) 33:823–9. doi: 10.1089/dna.2014.2639
7. Delacroix DL, Elkom KB, Geubel AP, Hodgson HF, Dive C, Vaerman JP. Changes in size, subclass, and metabolic properties of serum immunoglobulin A in liver diseases and in other diseases with high serum immunoglobulin A. J Clin Invest. (1983) 71:358–67. doi: 10.1172/JCI110777
8. Lauret E, Rodrigo L. Celiac disease and autoimmune-associated conditions. Biomed Res Int. (2013) 2013:127589. doi: 10.1155/2013/127589
9. Robert T, Berthelot L, Cambier A, Rondeau E, Monteiro RC. Molecular insights into the pathogenesis of IgA nephropathy. Trends Mol Med. (2015) 21:762–75. doi: 10.1016/j.molmed.2015.10.003
10. Johansen FE, Kaetzel CS. Regulation of the polymeric immunoglobulin receptor and IgA transport: new advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol. (2011) 4:598–602. doi: 10.1038/mi.2011.37
11. Rochereau N, Drocourt D, Perouzel E, Pavot V, Redelinghuys P, Brown GD, et al. Dectin-1 is essential for reverse transcytosis of glycosylated SIgA-antigen complexes by intestinal M cells. PLoS Biol. (2013) 11:e1001658. doi: 10.1371/journal.pbio.1001658
12. Matysiak-Budnik T, Moura IC, Arcos-Fajardo M, Lebreton C, Menard S, Candalh C, et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J Exp Med. (2008) 205:143–54. doi: 10.1084/jem.20071204
13. Baumann J, Park CG, Mantis NJ. Recognition of secretory IgA by DC-SIGN: implications for immune surveillance in the intestine. Immunol Lett. (2010) 131:59–66. doi: 10.1016/j.imlet.2010.03.005
14. Wilson TJ, Fuchs A, Colonna M. Cutting edge: human FcRL4 and FcRL5 are receptors for IgA and IgG. J Immunol. (2012) 188:4741–5. doi: 10.4049/jimmunol.1102651
15. Rifai A, Fadden K, Morrison SL, Chintalacharuvu KR. The N-glycans determine the differential blood clearance and hepatic uptake of human immunoglobulin (Ig)A1 and IgA2 isotypes. J Exp Med. (2000) 191:2171–82. doi: 10.1084/jem.191.12.2171
16. Molyneux K, Wimbury D, Pawluczyk I, Muto M, Bhachu J, Mertens PR, et al. beta1,4-galactosyltransferase 1 is a novel receptor for IgA in human mesangial cells. Kidney Int. (2017) 92:1458–68. doi: 10.1016/j.kint.2017.05.002
17. Shibuya A, Sakamoto N, Shimizu Y, Shibuya K, Osawa M, Hiroyama T, et al. Fc alpha/mu receptor mediates endocytosis of IgM-coated microbes. Nat Immunol. (2000) 1:441–6. doi: 10.1038/80886
18. Aleyd E, Heineke MH, van Egmond M. The era of the immunoglobulin A Fc receptor FcalphaRI; its function and potential as target in disease. Immunol Rev. (2015) 268:123–38. doi: 10.1111/imr.12337
19. Mkaddem SB, Christou I, Rossato E, Berthelot L, Lehuen A, Monteiro RC. IgA, IgA receptors, and their anti-inflammatory properties. Curr Top Microbiol Immunol. (2014) 382:221–35. doi: 10.1007/978-3-319-07911-0_10
20. Pasquier B, Launay P, Kanamaru Y, Moura IC, Pfirsch S, Ruffie C, et al. Identification of FcalphaRI as an inhibitory receptor that controls inflammation: dual role of FcRgamma ITAM. Immunity. (2005) 22:31–42. doi: 10.1016/j.immuni.2004.11.017
21. van Egmond M, Damen CA, van Spriel AB, Vidarsson G, van Garderen E, van de Winkel JG. IgA and the IgA Fc receptor. Trends Immunol. (2001) 22:205–11. doi: 10.1016/S1471-4906(01)01873-7
22. Pleass RJ, Areschoug T, Lindahl G, Woof JM. Streptococcal IgA-binding proteins bind in the Calpha 2-Calpha 3 interdomain region and inhibit binding of IgA to human CD89. J Biol Chem. (2001) 276:8197–204. doi: 10.1074/jbc.M009396200
23. Ramsland PA, Willoughby N, Trist HM, Farrugia W, Hogarth PM, Fraser JD, et al. Structural basis for evasion of IgA immunity by Staphylococcus aureus revealed in the complex of SSL7 with Fc of human IgA1. Proc Natl Acad Sci USA. (2007) 104:15051–6. doi: 10.1073/pnas.0706028104
24. Bonner A, Furtado PB, Almogren A, Kerr MA, Perkins SJ. Implications of the near-planar solution structure of human myeloma dimeric IgA1 for mucosal immunity and IgA nephropathy. J Immunol. (2008) 180:1008–18. doi: 10.4049/jimmunol.180.2.1008
25. Woof JM, Russell MW. Structure and function relationships in IgA. Mucosal Immunol. (2011) 4:590–7. doi: 10.1038/mi.2011.39
26. Brandtzaeg P, Johansen FE. Mucosal B cells: phenotypic characteristics, transcriptional regulation, and homing properties. Immunol Rev. (2005) 206:32–63. doi: 10.1111/j.0105-2896.2005.00283.x
27. Kett K, Baklien K, Bakken A, Kral JG, Fausa O, Brandtzaeg P. Intestinal B-cell isotype response in relation to local bacterial load: evidence for immunoglobulin A subclass adaptation. Gastroenterology. (1995) 109:819–25. doi: 10.1016/0016-5085(95)90389-5
28. Kaetzel CS. Cooperativity among secretory IgA, the polymeric immunoglobulin receptor, and the gut microbiota promotes host-microbial mutualism. Immunol Lett. (2014) 162(2 Pt. A):10–21. doi: 10.1016/j.imlet.2014.05.008
29. Bonner A, Almogren A, Furtado PB, Kerr MA, Perkins SJ. Location of secretory component on the Fc edge of dimeric IgA1 reveals insight into the role of secretory IgA1 in mucosal immunity. Mucosal Immunol. (2009) 2:74–84. doi: 10.1038/mi.2008.68
30. Brandtzaeg P, Prydz H. Direct evidence for an integrated function of J chain and secretory component in epithelial transport of immunoglobulins. Nature. (1984) 311:71–3. doi: 10.1038/311071a0
31. Mathias A, Corthesy B. Recognition of gram-positive intestinal bacteria by hybridoma- and colostrum-derived secretory immunoglobulin A is mediated by carbohydrates. J Biol Chem. (2011) 286:17239–47. doi: 10.1074/jbc.M110.209015
32. Maliszewski CR, March CJ, Schoenborn MA, Gimpel S, Shen L. Expression cloning of a human Fc receptor for IgA. J Exp Med. (1990) 172:1665–72. doi: 10.1084/jem.172.6.1665
33. Monteiro RC, Kubagawa H, Cooper MD. Cellular distribution, regulation, and biochemical nature of an Fc alpha receptor in humans. J Exp Med. (1990) 171:597–613. doi: 10.1084/jem.171.3.597
34. van Egmond M, van Garderen E, van Spriel AB, Damen CA, van Amersfoort ES, van Zandbergen G, et al. FcalphaRI-positive liver Kupffer cells: reappraisal of the function of immunoglobulin A in immunity. Nat Med. (2000) 6:680–5. doi: 10.1038/76261
35. Qian K, Xie F, Gibson AW, Edberg JC, Kimberly RP, Wu J. Functional expression of IgA receptor FcalphaRI on human platelets. J Leukoc Biol. (2008) 84:1492–500. doi: 10.1189/jlb.0508327
36. de Wit TP, Morton HC, Capel PJ, van de Winkel JG. Structure of the gene for the human myeloid IgA Fc receptor (CD89). J Immunol. (1995) 155:1203–9.
37. Morton HC, Schiel AE, Janssen SW, van de Winkel JG. Alternatively spliced forms of the human myeloid Fc alpha receptor (CD89) in neutrophils. Immunogenetics. (1996) 43:246–7. doi: 10.1007/s002510050057
38. Patry C, Sibille Y, Lehuen A, Monteiro RC. Identification of Fc alpha receptor (CD89) isoforms generated by alternative splicing that are differentially expressed between blood monocytes and alveolar macrophages. J Immunol. (1996) 156:4442–8.
39. Hostoffer RW, Krukovets I, Berger M. Increased Fc alpha R expression and IgA-mediated function on neutrophils induced by chemoattractants. J Immunol. (1993) 150:4532–40.
40. Nikolova EB, Russell MW. Dual function of human IgA antibodies: inhibition of phagocytosis in circulating neutrophils and enhancement of responses in IL-8-stimulated cells. J Leukoc Biol. (1995) 57:875–82. doi: 10.1002/jlb.57.6.875
41. Wehrli M, Cortinas-Elizondo F, Hlushchuk R, Daudel F, Villiger PM, Miescher S, et al. Human IgA Fc receptor FcalphaRI (CD89) triggers different forms of neutrophil death depending on the inflammatory microenvironment. J Immunol. (2014) 193:5649–59. doi: 10.4049/jimmunol.1400028
42. Khajah M, Millen B, Cara DC, Waterhouse C, McCafferty DM. Granulocyte-macrophage colony-stimulating factor (GM-CSF): a chemoattractive agent for murine leukocytes in vivo. J Leukoc Biol. (2011) 89:945–53. doi: 10.1189/jlb.0809546
43. Weisbart RH, Kacena A, Schuh A, Golde DW. GM-CSF induces human neutrophil IgA-mediated phagocytosis by an IgA Fc receptor activation mechanism. Nature. (1988) 332:647–8. doi: 10.1038/332647a0
44. Yin N, Peng M, Xing Y, Zhang W. Intracellular pools of FcalphaR (CD89) in human neutrophils are localized in tertiary granules and secretory vesicles, and two FcalphaR isoforms are found in tertiary granules. J Leukoc Biol. (2007) 82:551–8. doi: 10.1189/jlb.0207112
45. Maliszewski CR, Shen L, Fanger MW. The expression of receptors for IgA on human monocytes and calcitriol-treated HL-60 cells. J Immunol. (1985) 135:3878–81.
46. Shen L, Collins JE, Schoenborn MA, Maliszewski CR. Lipopolysaccharide and cytokine augmentation of human monocyte IgA receptor expression and function. J Immunol. (1994) 152:4080–6.
47. Grossetete B, Launay P, Lehuen A, Jungers P, Bach JF, Monteiro RC. Down-regulation of Fc alpha receptors on blood cells of IgA nephropathy patients: evidence for a negative regulatory role of serum IgA. Kidney Int. (1998) 53:1321–35. doi: 10.1046/j.1523-1755.1998.00885.x
48. Geissmann F, Launay P, Pasquier B, Lepelletier Y, Leborgne M, Lehuen A, et al. A subset of human dendritic cells expresses IgA Fc receptor (CD89), which mediates internalization and activation upon cross-linking by IgA complexes. J Immunol. (2001) 166:346–52. doi: 10.4049/jimmunol.166.1.346
49. Pasquier B, Lepelletier Y, Baude C, Hermine O, Monteiro RC. Differential expression and function of IgA receptors (CD89 and CD71) during maturation of dendritic cells. J Leukoc Biol. (2004) 76:1134–41. doi: 10.1189/jlb.0204101
50. Wines BD, Sardjono CT, Trist HH, Lay CS, Hogarth PM. The interaction of Fc alpha RI with IgA and its implications for ligand binding by immunoreceptors of the leukocyte receptor cluster. J Immunol. (2001) 166:1781–9. doi: 10.4049/jimmunol.166.3.1781
51. Posgai MT, Tonddast-Navaei S, Jayasinghe M, Ibrahim GM, Stan G, Herr AB. FcalphaRI binding at the IgA1 CH2-CH3 interface induces long-range conformational changes that are transmitted to the hinge region. Proc Natl Acad Sci USA. (2018) 115:E8882–91. doi: 10.1073/pnas.1807478115
52. Herr AB, Ballister ER, Bjorkman PJ. Insights into IgA-mediated immune responses from the crystal structures of human FcalphaRI and its complex with IgA1-Fc. Nature. (2003) 423:614–20. doi: 10.1038/nature01685
53. Pleass RJ, Dehal PK, Lewis MJ, Woof JM. Limited role of charge matching in the interaction of human immunoglobulin A with the immunoglobulin A Fc receptor (Fc alpha RI) CD89. Immunology. (2003) 109:331–5. doi: 10.1046/j.1365-2567.2003.01677.x
54. Van Spriel AB, Leusen JH, Vile H, Van De Winkel JG. Mac-1 (CD11b/CD18) as accessory molecule for Fc alpha R (CD89) binding of IgA. J Immunol. (2002) 169:3831–6. doi: 10.4049/jimmunol.169.7.3831
55. Xue J, Zhao Q, Zhu L, Zhang W. Deglycosylation of FcalphaR at N58 increases its binding to IgA. Glycobiology. (2010) 20:905–15. doi: 10.1093/glycob/cwq048
56. Gomes MM, Wall SB, Takahashi K, Novak J, Renfrow MB, Herr AB. Analysis of IgA1 N-glycosylation and its contribution to FcalphaRI binding. Biochemistry. (2008) 47:11285–99. doi: 10.1021/bi801185b
57. Bracke M, Dubois GR, Bolt K, Bruijnzeel PL, Vaerman JP, Lammers JW, et al. Differential effects of the T helper cell type 2-derived cytokines IL-4 and IL-5 on ligand binding to IgG and IgA receptors expressed by human eosinophils. J Immunol. (1997) 159:1459–65.
58. Lu J, Marjon KD, Mold C, Marnell L, Du Clos TW, Sun P. Pentraxins and IgA share a binding hot-spot on FcalphaRI. Protein Sci. (2014) 23:378–86. doi: 10.1002/pro.2419
59. Langley R, Wines B, Willoughby N, Basu I, Proft T, Fraser JD. The staphylococcal superantigen-like protein 7 binds IgA and complement C5 and inhibits IgA-Fc alpha RI binding and serum killing of bacteria. J Immunol. (2005) 174:2926–33. doi: 10.4049/jimmunol.174.5.2926
60. Woof JM. Immunoglobulins and their receptors, and subversion of their protective roles by bacterial pathogens. Biochem Soc Trans. (2016) 44:1651–8. doi: 10.1042/BST20160246
61. Aleyd E, van Hout MWM, Ganzevles SH, Hoeben KA, Everts V, Bakema JE, et al. IgA enhances NETosis and release of neutrophil extracellular traps by polymorphonuclear cells via Fcα receptor I. 2016. p. 2374–83.
62. Otten MA, Rudolph E, Dechant M, Tuk CW, Reijmers RM, Beelen RH, et al. Immature neutrophils mediate tumor cell killing via IgA but not IgG Fc receptors. J Immunol. (2005) 174:5472–80. doi: 10.4049/jimmunol.174.9.5472
63. Gorter A, Hiemstra PS, Leijh PC, van der Sluys ME, van den Barselaar MT, van Es LA, et al. IgA- and secretory IgA-opsonized S. aureus induce a respiratory burst and phagocytosis by polymorphonuclear leucocytes. Immunology. (1987) 61:303–9.
64. van der Steen L, Tuk CW, Bakema JE, Kooij G, Reijerkerk A, Vidarsson G, et al. Immunoglobulin A: Fc(alpha)RI interactions induce neutrophil migration through release of leukotriene B4. Gastroenterology. (2009) 137:2018–29 e1–3.
65. Shen L, van Egmond M, Siemasko K, Gao H, Wade T, Lang ML, et al. Presentation of ovalbumin internalized via the immunoglobulin-A Fc receptor is enhanced through Fc receptor gamma-chain signaling. Blood. (2001) 97:205–13. doi: 10.1182/blood.V97.1.205
66. Bakema JE, van Egmond M. The human immunoglobulin A Fc receptor FcalphaRI: a multifaceted regulator of mucosal immunity. Mucosal Immunol. (2011) 4:612–24. doi: 10.1038/mi.2011.36
67. Shin EH, Lee HY, Bae YS. Leukotriene B4 stimulates human monocyte-derived dendritic cell chemotaxis. Biochem Biophys Res Commun. (2006) 348:606–11. doi: 10.1016/j.bbrc.2006.07.084
68. Hansen IS, Krabbendam L, Bernink JH, Loayza-Puch F, Hoepel W, van Burgsteden JA, et al. FcalphaRI co-stimulation converts human intestinal CD103(+) dendritic cells into pro-inflammatory cells through glycolytic reprogramming. Nat Commun. (2018) 9:863. doi: 10.1038/s41467-018-03318-5
69. Lang ML, Chen YW, Shen L, Gao H, Lang GA, Wade TK, et al. IgA Fc receptor (FcalphaR) cross-linking recruits tyrosine kinases, phosphoinositide kinases and serine/threonine kinases to glycolipid rafts. Biochem J. (2002) 364(Pt 2):517–25. doi: 10.1042/bj20011696
70. Morton HC, van den Herik-Oudijk IE, Vossebeld P, Snijders A, Verhoeven AJ, Capel PJ, et al. Functional association between the human myeloid immunoglobulin A Fc receptor (CD89) and FcR gamma chain. Molecular basis for CD89/FcR gamma chain association. J Biol Chem. (1995) 270:29781–7. doi: 10.1074/jbc.270.50.29781
71. van Egmond M, van Vuuren AJ, Morton HC, van Spriel AB, Shen L, Hofhuis FM, et al. Human immunoglobulin A receptor (FcalphaRI, CD89) function in transgenic mice requires both FcR gamma chain and CR3 (CD11b/CD18). Blood. (1999) 93:4387–94.
72. Pfefferkorn LC, Yeaman GR. Association of IgA-Fc receptors (Fc alpha R) with Fc epsilon RI gamma 2 subunits in U937 cells. Aggregation induces the tyrosine phosphorylation of gamma 2. J Immunol. (1994) 153:3228–36.
73. Gulle H, Samstag A, Eibl MM, Wolf HM. Physical and functional association of Fc alpha R with protein tyrosine kinase Lyn. Blood. (1998) 91:383–91.
74. Mkaddem SB, Murua A, Flament H, Titeca-Beauport D, Bounaix C, Danelli L, et al. Lyn and Fyn function as molecular switches that control immunoreceptors to direct homeostasis or inflammation. Nat Commun. (2017) 8:246. doi: 10.1038/s41467-017-00294-0
75. Park RK, Izadi KD, Deo YM, Durden DL. Role of Src in the modulation of multiple adaptor proteins in FcalphaRI oxidant signaling. Blood. (1999) 94:2112–20.
76. Pfirsch-Maisonnas S, Aloulou M, Xu T, Claver J, Kanamaru Y, Tiwari M, et al. Inhibitory ITAM signaling traps activating receptors with the phosphatase SHP-1 to form polarized “inhibisome” clusters. Sci Signal. (2011) 4:ra24. doi: 10.1126/scisignal.2001309
77. Liu C, Kanamaru Y, Watanabe T, Tada N, Horikoshi S, Suzuki Y, et al. Targeted IgA Fc receptor I (FcalphaRI) therapy in the early intervention and treatment of pristane-induced lupus nephritis in mice. Clin Exp Immunol. (2015) 181:407–16. doi: 10.1111/cei.12647
78. Mantis NJ, Rol N, Corthesy B. Secretory IgA's complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. (2011) 4:603–11. doi: 10.1038/mi.2011.41
79. Martin-Gallausiaux C, Beguet-Crespel F, Marinelli L, Jamet A, Ledue F, Blottiere HM, et al. Butyrate produced by gut commensal bacteria activates TGF-beta1 expression through the transcription factor SP1 in human intestinal epithelial cells. Sci Rep. (2018) 8:9742. doi: 10.1038/s41598-018-28048-y
80. Rogier EW, Frantz AL, Bruno ME, Kaetzel CS. Secretory IgA is concentrated in the outer layer of colonic mucus along with gut bacteria. Pathogens. (2014) 3:390–403. doi: 10.3390/pathogens3020390
81. Corthesy B. Multi-faceted functions of secretory IgA at mucosal surfaces. Front Immunol. (2013) 4:185. doi: 10.3389/fimmu.2013.00185
82. Nakajima A, Vogelzang A, Maruya M, Miyajima M, Murata M, Son A, et al. IgA regulates the composition and metabolic function of gut microbiota by promoting symbiosis between bacteria. J Exp Med. (2018) 215:2019–34. doi: 10.1084/jem.20180427
83. Fransen F, Zagato E, Mazzini E, Fosso B, Manzari C, El Aidy S, et al. BALB/c and C57BL/6 mice differ in polyreactive IgA abundance, which impacts the generation of antigen-specific IgA and microbiota diversity. Immunity. (2015) 43:527–40. doi: 10.1016/j.immuni.2015.08.011
84. Okai S, Usui F, Yokota S, Hori IY, Hasegawa M, Nakamura T, et al. High-affinity monoclonal IgA regulates gut microbiota and prevents colitis in mice. Nat Microbiol. (2016) 1:16103. doi: 10.1038/nmicrobiol.2016.103
85. van der Waaij LA, Limburg PC, Mesander G, van der Waaij D. In vivo IgA coating of anaerobic bacteria in human faeces. Gut. (1996) 38:348–54. doi: 10.1136/gut.38.3.348
86. Bunker JJ, Erickson SA, Flynn TM, Henry C, Koval JC, Meisel M, et al. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science. (2017) 358:eaan6619. doi: 10.1126/science.aan6619
87. Bunker JJ, Bendelac A. IgA responses to microbiota. Immunity. (2018) 49:211–24. doi: 10.1016/j.immuni.2018.08.011
88. Reikvam DH, Derrien M, Islam R, Erofeev A, Grcic V, Sandvik A, et al. Epithelial-microbial crosstalk in polymeric Ig receptor deficient mice. Eur J Immunol. (2012) 42:2959–70. doi: 10.1002/eji.201242543
89. Frantz AL, Bruno ME, Rogier EW, Tuna H, Cohen DA, Bondada S, et al. Multifactorial patterns of gene expression in colonic epithelial cells predict disease phenotypes in experimental colitis. Inflamm Bowel Dis. (2012) 18:2138–48. doi: 10.1002/ibd.22923
90. Blutt SE, Miller AD, Salmon SL, Metzger DW, Conner ME. IgA is important for clearance and critical for protection from rotavirus infection. Mucosal Immunol. (2012) 5:712–9. doi: 10.1038/mi.2012.51
91. Davids BJ, Palm JE, Housley MP, Smith JR, Andersen YS, Martin MG, et al. Polymeric immunoglobulin receptor in intestinal immune defense against the lumen-dwelling protozoan parasite Giardia. J Immunol. (2006) 177:6281–90. doi: 10.4049/jimmunol.177.9.6281
92. Sun K, Johansen FE, Eckmann L, Metzger DW. An important role for polymeric Ig receptor-mediated transport of IgA in protection against Streptococcus pneumoniae nasopharyngeal carriage. J Immunol. (2004) 173:4576–81. doi: 10.4049/jimmunol.173.7.4576
93. Schwartz-Cornil I, Benureau Y, Greenberg H, Hendrickson BA, Cohen J. Heterologous protection induced by the inner capsid proteins of rotavirus requires transcytosis of mucosal immunoglobulins. J Virol. (2002) 76:8110–7. doi: 10.1128/JVI.76.16.8110-8117.2002
94. Lycke N, Erlandsson L, Ekman L, Schon K, Leanderson T. Lack of J chain inhibits the transport of gut IgA and abrogates the development of intestinal antitoxic protection. J Immunol. (1999) 163:913–9.
95. Kvale D, Brandtzaeg P. Butyrate differentially affects constitutive and cytokine-induced expression of HLA molecules, secretory component (SC), and ICAM-1 in a colonic epithelial cell line (HT-29, clone m3). Adv Exp Med Biol. (1995) 371A:183–8. doi: 10.1007/978-1-4615-1941-6_37
96. Kadaoui KA, Corthesy B. Secretory IgA mediates bacterial translocation to dendritic cells in mouse Peyer's patches with restriction to mucosal compartment. J Immunol. (2007) 179:7751–7. doi: 10.4049/jimmunol.179.11.7751
97. Yan H, Lamm ME, Bjorling E, Huang YT. Multiple functions of immunoglobulin A in mucosal defense against viruses: an in vitro measles virus model. J Virol. (2002) 76:10972–9. doi: 10.1128/JVI.76.21.10972-10979.2002
98. Mazanec MB, Kaetzel CS, Lamm ME, Fletcher D, Nedrud JG. Intracellular neutralization of virus by immunoglobulin A antibodies. Proc Natl Acad Sci USA. (1992) 89:6901–5. doi: 10.1073/pnas.89.15.6901
99. Fernandez MI, Pedron T, Tournebize R, Olivo-Marin JC, Sansonetti PJ, Phalipon A. Anti-inflammatory role for intracellular dimeric immunoglobulin a by neutralization of lipopolysaccharide in epithelial cells. Immunity. (2003) 18:739–49. doi: 10.1016/S1074-7613(03)00122-5
100. Mantis NJ, Palaia J, Hessell AJ, Mehta S, Zhu Z, Corthesy B, et al. Inhibition of HIV-1 infectivity and epithelial cell transfer by human monoclonal IgG and IgA antibodies carrying the b12 V region. J Immunol. (2007) 179:3144–52. doi: 10.4049/jimmunol.179.5.3144
101. Wright A, Lamm ME, Huang YT. Excretion of human immunodeficiency virus type 1 through polarized epithelium by immunoglobulin A. J Virol. (2008) 82:11526–35. doi: 10.1128/JVI.01111-08
102. Hur EM, Patel SN, Shimizu S, Rao DS, Gnanapragasam PN, An DS, et al. Inhibitory effect of HIV-specific neutralizing IgA on mucosal transmission of HIV in humanized mice. Blood. (2012) 120:4571–82. doi: 10.1182/blood-2012-04-422303
103. Kulkarni V, Ruprecht RM. Mucosal IgA responses: damaged in established HIV infection-yet, effective weapon against HIV transmission. Front Immunol. (2017) 8:1581. doi: 10.3389/fimmu.2017.01581
104. Blutt SE, Conner ME. The gastrointestinal frontier: IgA and viruses. Front Immunol. (2013) 4:402. doi: 10.3389/fimmu.2013.00402
105. Maurer MA, Meyer L, Bianchi M, Turner HL, Le NPL, Steck M, et al. Glycosylation of human IgA directly inhibits influenza A and other sialic-acid-binding viruses. Cell Rep. (2018) 23:90–9. doi: 10.1016/j.celrep.2018.03.027
106. McNeal MM, Stone SC, Basu M, Bean JA, Clements JD, Hendrickson BA, et al. Protection against rotavirus shedding after intranasal immunization of mice with a chimeric VP6 protein does not require intestinal IgA. Virology. (2006) 346:338–47. doi: 10.1016/j.virol.2005.11.016
107. Asahi Y, Yoshikawa T, Watanabe I, Iwasaki T, Hasegawa H, Sato Y, et al. Protection against influenza virus infection in polymeric Ig receptor knockout mice immunized intranasally with adjuvant-combined vaccines. J Immunol. (2002) 168:2930–8. doi: 10.4049/jimmunol.168.6.2930
108. Silvey KJ, Hutchings AB, Vajdy M, Petzke MM, Neutra MR. Role of immunoglobulin A in protection against reovirus entry into Murine Peyer's patches. J Virol. (2001) 75:10870–9. doi: 10.1128/JVI.75.22.10870-10879.2001
109. Turula H, Bragazzi Cunha J, Mainou BA, Ramakrishnan SK, Wilke CA, Gonzalez-Hernandez MB, et al. Natural secretory immunoglobulins promote enteric viral infections. J Virol. (2018). doi: 10.1128/JVI.00826-18
110. Wilmore JR, Gaudette BT, Gomez Atria D, Hashemi T, Jones DD, Gardner CA, et al. Commensal microbes induce serum IgA responses that protect against polymicrobial sepsis. Cell Host Microbe. (2018) 23:302–11 e3. doi: 10.1016/j.chom.2018.01.005
111. Iversen R, Snir O, Stensland M, Kroll JE, Steinsbo O, Korponay-Szabo IR, et al. Strong clonal relatedness between serum and gut IgA despite different plasma cell origins. Cell Rep. (2017) 20:2357–67. doi: 10.1016/j.celrep.2017.08.036
112. Kozakova H, Schwarzer M, Tuckova L, Srutkova D, Czarnowska E, Rosiak I, et al. Colonization of germ-free mice with a mixture of three lactobacillus strains enhances the integrity of gut mucosa and ameliorates allergic sensitization. Cell Mol Immunol. (2016) 13:251–62. doi: 10.1038/cmi.2015.09
113. Johansen FE, Pekna M, Norderhaug IN, Haneberg B, Hietala MA, Krajci P, et al. Absence of epithelial immunoglobulin A transport, with increased mucosal leakiness, in polymeric immunoglobulin receptor/secretory component-deficient mice. J Exp Med. (1999) 190:915–22. doi: 10.1084/jem.190.7.915
114. Uren TK, Johansen FE, Wijburg OL, Koentgen F, Brandtzaeg P, Strugnell RA. Role of the polymeric Ig receptor in mucosal B cell homeostasis. J Immunol. (2003) 170:2531–9. doi: 10.4049/jimmunol.170.5.2531
115. Hansen IS, Hoepel W, Zaat SAJ, Baeten DLP, den Dunnen J. Serum IgA immune complexes promote proinflammatory cytokine production by human macrophages, monocytes, and kupffer cells through FcalphaRI-TLR cross-talk. J Immunol. (2017) 199:4124–31. doi: 10.4049/jimmunol.1700883
116. Mestecky J, Hammarstrom L. IgA-associated diseases. In: Kaetzel CS, editor. Mucosal Immune Defense: Immunoglobulin A [Internet]. Springer (2007). p. 321–44. doi: 10.1007/978-0-387-72232-0_13
117. Fadlallah J, El Kafsi H, Sterlin D, Juste C, Parizot C, Dorgham K, et al. Microbial ecology perturbation in human IgA deficiency. Sci Transl Med. (2018) 10:eaan1217. doi: 10.1126/scitranslmed.aan1217
118. Harriman GR, Bogue M, Rogers P, Finegold M, Pacheco S, Bradley A, et al. Targeted deletion of the IgA constant region in mice leads to IgA deficiency with alterations in expression of other Ig isotypes. J Immunol. (1999) 162:2521–9.
119. Arnaboldi PM, Behr MJ, Metzger DW. Mucosal B cell deficiency in IgA-/- mice abrogates the development of allergic lung inflammation. J Immunol. (2005) 175:1276–85. doi: 10.4049/jimmunol.175.2.1276
120. Yazdani R, Azizi G, Abolhassani H, Aghamohammadi A. Selective IgA deficiency: epidemiology, pathogenesis, clinical phenotype, diagnosis, prognosis and management. Scand J Immunol. (2017) 85:3–12. doi: 10.1111/sji.12499
121. Lemarquis AL, Einarsdottir HK, Kristjansdottir RN, Jonsdottir I, Ludviksson BR. Transitional B cells and TLR9 responses are defective in selective IgA deficiency. Front Immunol. (2018) 9:909. doi: 10.3389/fimmu.2018.00909
122. Hammarstrom L, Lonnqvist B, Ringden O, Smith CI, Wiebe T. Transfer of IgA deficiency to a bone-marrow-grafted patient with aplastic anaemia. Lancet. (1985) 1:778–81. doi: 10.1016/S0140-6736(85)91446-1
123. Singh K, Chang C, Gershwin ME. IgA deficiency and autoimmunity. Autoimmun Rev. (2014) 13:163–77. doi: 10.1016/j.autrev.2013.10.005
124. Catanzaro JR, Strauss JD, Bielecka A, Porto AF, Lobo FM, Urban A, et al. IgA-deficient humans exhibit gut microbiota dysbiosis despite production of compensatory IgM. bioRxiv. 2018:446724. doi: 10.1101/446724
125. Woof JM, Kerr MA. IgA function–variations on a theme. Immunology. (2004) 113:175–7. doi: 10.1111/j.1365-2567.2004.01958.x
126. Ludvigsson JF, Neovius M, Hammarstrom L. IgA deficiency and mortality: a population-based cohort study. J Clin Immunol. (2013) 33:1317–24. doi: 10.1007/s10875-013-9948-4
127. Hauge SC, Jensen CK, Nielsen LK, Pedersen OB, Sorensen E, Thorner LW, et al. The association of IgA deficiency on infection rate, self-perceived health, and levels of C-reactive protein in healthy blood donors. APMIS. (2018) 126:248–56. doi: 10.1111/apm.12807
128. Ludvigsson JF, Neovius M, Hammarstrom L. Risk of infections among 2100 individuals with IgA deficiency: a nationwide cohort study. J Clin Immunol. (2016) 36:134–40. doi: 10.1007/s10875-015-0230-9
129. Papadopoulou A, Mermiri D, Taousani S, Triga M, Nicolaidou P, Priftis KN. Bronchial hyper-responsiveness in selective IgA deficiency. Pediatr Allergy Immunol. (2005) 16:495–500. doi: 10.1111/j.1399-3038.2005.00316.x
130. Price P, Witt C, Allcock R, Sayer D, Garlepp M, Kok CC, et al. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol Rev. (1999) 167:257–74. doi: 10.1111/j.1600-065X.1999.tb01398.x
131. Aghamohammadi A, Cheraghi T, Gharagozlou M, Movahedi M, Rezaei N, Yeganeh M, et al. IgA deficiency: correlation between clinical and immunological phenotypes. J Clin Immunol. (2009) 29:130–6. doi: 10.1007/s10875-008-9229-9
132. Jacob CM, Pastorino AC, Fahl K, Carneiro-Sampaio M, Monteiro RC. Autoimmunity in IgA deficiency: revisiting the role of IgA as a silent housekeeper. J Clin Immunol. (2008) 28 (Suppl. 1):S56–61. doi: 10.1007/s10875-007-9163-2
133. Lilic D, Sewell WA. IgA deficiency: what we should-or should not-be doing. J Clin Pathol. (2001) 54:337–8. doi: 10.1136/jcp.54.5.337
134. Hammarstrom L, Persson MA, Smith CI. Anti-IgA in selective IgA deficiency. In vitro effects and Ig subclass pattern of human anti-IgA. Scand J Immunol. (1983) 18:509–13. doi: 10.1111/j.1365-3083.1983.tb00885.x
135. Stone KD, Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. (2010) 125 (2 Suppl. 2):S73–80. doi: 10.1016/j.jaci.2009.11.017
136. Pascal M, Perez-Gordo M, Caballero T, Escribese MM, Lopez Longo MN, Luengo O, et al. Microbiome and allergic diseases. Front Immunol. (2018) 9:1584. doi: 10.3389/fimmu.2018.01584
137. Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. (2001) 292:1115–8. doi: 10.1126/science.1058709
138. Ludviksson BR, Eiriksson TH, Ardal B, Sigfusson A, Valdimarsson H. Correlation between serum immunoglobulin A concentrations and allergic manifestations in infants. J Pediatr. (1992) 121:23–7. doi: 10.1016/S0022-3476(05)82535-1
139. Dzidic M, Abrahamsson TR, Artacho A, Bjorksten B, Collado MC, Mira A, et al. Aberrant IgA responses to the gut microbiota during infancy precede asthma and allergy development. J Allergy Clin Immunol. (2017) 139:1017–25 e14. doi: 10.1016/j.jaci.2016.06.047
140. Kim WJ, Choi IS, Kim CS, Lee JH, Kang HW. Relationship between serum IgA level and allergy/asthma. Korean J Intern Med. (2017) 32:137–45. doi: 10.3904/kjim.2014.160
141. Balzar S, Strand M, Nakano T, Wenzel SE. Subtle immunodeficiency in severe asthma: IgA and IgG2 correlate with lung function and symptoms. Int Arch Allergy Immunol. (2006) 140:96–102. doi: 10.1159/000092252
142. Nahm DH, Kim HY, Park HS. Elevation of specific immunoglobulin A antibodies to both allergen and bacterial antigen in induced sputum from asthmatics. Eur Respir J. (1998) 12:540–5. doi: 10.1183/09031936.98.12030540
143. Aghayan-Ugurluoglu R, Ball T, Vrtala S, Schweiger C, Kraft D, Valenta R. Dissociation of allergen-specific IgE and IgA responses in sera and tears of pollen-allergic patients: a study performed with purified recombinant pollen allergens. J Allergy Clin Immunol. (2000) 105:803–13. doi: 10.1067/mai.2000.104782
144. Bartemes KR, Cooper KM, Drain KL, Kita H. Secretory IgA induces antigen-independent eosinophil survival and cytokine production without inducing effector functions. J Allergy Clin Immunol. (2005) 116:827–35. doi: 10.1016/j.jaci.2005.07.014
145. Karimifar M, Moussavi H, Babaei M, Akbari M. The association of immunoglobulin A, immunoglobulin G and anti-cyclic citrullinated peptide antibodies with disease activity in seronegative rheumatoid arthritis patients. J Res Med Sci. (2014) 19:823–6.
146. Jonsson T, Arinbjarnarson S, Thorsteinsson J, Steinsson K, Geirsson AJ, Jonsson H, et al. Raised IgA rheumatoid factor (RF) but not IgM RF or IgG RF is associated with extra-articular manifestations in rheumatoid arthritis. Scand J Rheumatol. (1995) 24:372–5. doi: 10.3109/03009749509095183
147. Bobbio-Pallavicini F, Caporali R, Alpini C, Moratti R, Montecucco C. Predictive value of antibodies to citrullinated peptides and rheumatoid factors in anti-TNF-alpha treated patients. Ann NY Acad Sci. (2007) 1109:287–95. doi: 10.1196/annals.1398.034
148. Anquetil F, Clavel C, Offer G, Serre G, Sebbag M. IgM and IgA rheumatoid factors purified from rheumatoid arthritis sera boost the Fc receptor- and complement-dependent effector functions of the disease-specific anti-citrullinated protein autoantibodies. J Immunol. (2015) 194:3664–74. doi: 10.4049/jimmunol.1402334
149. Aleyd E, Al M, Tuk CW, van der Laken CJ, van Egmond M. IgA complexes in plasma and synovial fluid of patients with rheumatoid arthritis induce neutrophil extracellular traps via FcalphaRI. J Immunol. (2016) 197:4552–9. doi: 10.4049/jimmunol.1502353
150. Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol. (2014) 10:593–601. doi: 10.1038/nrrheum.2014.80
151. Czerkinsky C, Koopman WJ, Jackson S, Collins JE, Crago SS, Schrohenloher RE, et al. Circulating immune complexes and immunoglobulin A rheumatoid factor in patients with mesangial immunoglobulin A nephropathies. J Clin Invest. (1986) 77:1931–8. doi: 10.1172/JCI112522
152. Allen AC, Willis FR, Beattie TJ, Feehally J. Abnormal IgA glycosylation in Henoch-Schonlein purpura restricted to patients with clinical nephritis. Nephrol Dial Transplant. (1998) 13:930–4. doi: 10.1093/ndt/13.4.930
153. Chen A, Yang SS, Lin TJ, Ka SM. IgA nephropathy: clearance kinetics of IgA-containing immune complexes. Semin Immunopathol. (2018) 40:539–43. doi: 10.1007/s00281-018-0708-7
154. Novak J, Julian BA, Tomana M, Mestecky J. IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin Nephrol. (2008) 28:78–87. doi: 10.1016/j.semnephrol.2007.10.009
155. Novak J, Julian BA, Tomana M, Mesteck J. Progress in molecular and genetic studies of IgA nephropathy. J Clin Immunol. (2001) 21:310–27. doi: 10.1023/A:1012284402054
156. Xu L, Li B, Huang M, Xie K, Li D, Li Y, et al. Critical role of Kupffer cell CD89 expression in experimental IgA nephropathy. PLoS ONE. (2016) 11:e0159426. doi: 10.1371/journal.pone.0159426
157. Berthelot L, Papista C, Maciel TT, Biarnes-Pelicot M, Tissandie E, Wang PH, et al. Transglutaminase is essential for IgA nephropathy development acting through IgA receptors. J Exp Med. (2012) 209:793–806. doi: 10.1084/jem.20112005
158. Launay P, Grossetete B, Arcos-Fajardo M, Gaudin E, Torres SP, Beaudoin L, et al. Fcalpha receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger's disease). Evidence for pathogenic soluble receptor-Iga complexes in patients and CD89 transgenic mice. J Exp Med. (2000) 191:1999–2009. doi: 10.1084/jem.191.11.1999
159. Moura IC, Arcos-Fajardo M, Sadaka C, Leroy V, Benhamou M, Novak J, et al. Glycosylation and size of IgA1 are essential for interaction with mesangial transferrin receptor in IgA nephropathy. J Am Soc Nephrol. (2004) 15:622–34. doi: 10.1097/01.ASN.0000115401.07980.0C
160. Moresco RN, Speeckaert MM, Zmonarski SC, Krajewska M, Komuda-Leszek E, Perkowska-Ptasinska A, et al. Urinary myeloid IgA Fc alpha receptor (CD89) and transglutaminase-2 as new biomarkers for active IgA nephropathy and henoch-Schonlein purpura nephritis. BBA Clin. (2016) 5:79–84. doi: 10.1016/j.bbacli.2016.02.002
161. Saulsbury FT. Henoch-Schonlein purpura. Curr Opin Rheumatol. (2010) 22:598–602. doi: 10.1097/BOR.0b013e32833af608
162. Heineke MH, Ballering AV, Jamin A, Ben Mkaddem S, Monteiro RC, Van Egmond M. New insights in the pathogenesis of immunoglobulin A vasculitis (Henoch-Schonlein purpura). Autoimmun Rev. (2017) 16:1246–53. doi: 10.1016/j.autrev.2017.10.009
163. Davin JC, Ten Berge IJ, Weening JJ. What is the difference between IgA nephropathy and Henoch-Schonlein purpura nephritis? Kidney Int. (2001) 59:823–34. doi: 10.1046/j.1523-1755.2001.059003823.x
164. Lau KK, Wyatt RJ, Moldoveanu Z, Tomana M, Julian BA, Hogg RJ, et al. Serum levels of galactose-deficient IgA in children with IgA nephropathy and Henoch-Schonlein purpura. Pediatr Nephrol. (2007) 22:2067–72. doi: 10.1007/s00467-007-0623-y
165. Berthelot L, Jamin A, Viglietti D, Chemouny JM, Ayari H, Pierre M, et al. Value of biomarkers for predicting immunoglobulin A vasculitis nephritis outcome in an adult prospective cohort. Nephrol Dial Transplant. (2017) 33:1579–90. doi: 10.1093/ndt/gfx300
166. Pillebout E, Jamin A, Ayari H, Housset P, Pierre M, Sauvaget V, et al. Biomarkers of IgA vasculitis nephritis in children. PLoS ONE. (2017) 12:e0188718. doi: 10.1371/journal.pone.0188718
167. Yang YH, Wang SJ, Chuang YH, Lin YT, Chiang BL. The level of IgA antibodies to human umbilical vein endothelial cells can be enhanced by TNF-alpha treatment in children with Henoch-Schonlein purpura. Clin Exp Immunol. (2002) 130:352–7. doi: 10.1046/j.1365-2249.2002.01964.x
168. Yang YH, Lai HJ, Huang CM, Wang LC, Lin YT, Chiang BL. Sera from children with active Henoch-Schonlein purpura can enhance the production of interleukin 8 by human umbilical venous endothelial cells. Ann Rheum Dis. (2004) 63:1511–3. doi: 10.1136/ard.2003.016196
169. Yang YH, Huang YH, Lin YL, Wang LC, Chuang YH, Yu HH, et al. Circulating IgA from acute stage of childhood Henoch-Schonlein purpura can enhance endothelial interleukin (IL)-8 production through MEK/ERK signalling pathway. Clin Exp Immunol. (2006) 144:247–53. doi: 10.1111/j.1365-2249.2006.03076.x
170. Besbas N, Saatci U, Ruacan S, Ozen S, Sungur A, Bakkaloglu A, et al. The role of cytokines in Henoch Schonlein purpura. Scand J Rheumatol. (1997) 26:456–60. doi: 10.3109/03009749709065719
171. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. (2010) 28:573–621. doi: 10.1146/annurev-immunol-030409-101225
172. Arsenescu R, Bruno ME, Rogier EW, Stefka AT, McMahan AE, Wright TB, et al. Signature biomarkers in Crohn's disease: toward a molecular classification. Mucosal Immunol. (2008) 1:399–411. doi: 10.1038/mi.2008.32
173. Bakema JE, Tuk CW, van Vliet SJ, Bruijns SC, Vos JB, Letsiou S, et al. Antibody-opsonized bacteria evoke an inflammatory dendritic cell phenotype and polyfunctional Th cells by cross-talk between TLRs and FcRs. J Immunol. (2015) 194:1856–66. doi: 10.4049/jimmunol.1303126
174. Mitsuyama K, Niwa M, Takedatsu H, Yamasaki H, Kuwaki K, Yoshioka S, et al. Antibody markers in the diagnosis of inflammatory bowel disease. World J Gastroenterol. (2016) 22:1304–10. doi: 10.3748/wjg.v22.i3.1304
175. Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. (2014) 158:1000–10. doi: 10.1016/j.cell.2014.08.006
176. van der Waaij LA, Kroese FG, Visser A, Nelis GF, Westerveld BD, Jansen PL, et al. Immunoglobulin coating of faecal bacteria in inflammatory bowel disease. Eur J Gastroenterol Hepatol. (2004) 16:669–74. doi: 10.1097/01.meg.0000108346.41221.19
177. Apperloo-Renkema HZ, Wilkinson MH, van der Waaij D. Circulating antibodies against faecal bacteria assessed by immunomorphometry: combining quantitative immunofluorescence and image analysis. Epidemiol Infect. (1992) 109:497–506. doi: 10.1017/S0950268800050494
178. Green PH, Lebwohl B, Greywoode R. Celiac disease. J Allergy Clin Immunol. (2015) 135:1099–106; quiz 107. doi: 10.1016/j.jaci.2015.01.044
179. Papista C, Berthelot L, Monteiro RC. Dysfunctions of the Iga system: a common link between intestinal and renal diseases. Cell Mol Immunol. (2011) 8:126–34. doi: 10.1038/cmi.2010.69
180. De Re V, Magris R, Cannizzaro R. New insights into the pathogenesis of celiac disease. Front Med. (2017) 4:137. doi: 10.3389/fmed.2017.00137
181. Sardy M, Karpati S, Merkl B, Paulsson M, Smyth N. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J Exp Med. (2002) 195:747–57. doi: 10.1084/jem.20011299
182. Smith AD, Streilein RD, Hall RP III. Neutrophil CD11b, L-selectin and Fc IgA receptors in patients with dermatitis herpetiformis. Br J Dermatol. (2002) 147:1109–17. doi: 10.1046/j.1365-2133.2002.05004.x
183. Coppo R, Amore A, Roccatello D. Dietary antigens and primary immunoglobulin A nephropathy. J Am Soc Nephrol. (1992) 2(10 Suppl.):S173–80.
184. Papista C, Lechner S, Ben Mkaddem S, LeStang MB, Abbad L, Bex-Coudrat J, et al. Gluten exacerbates IgA nephropathy in humanized mice through gliadin-CD89 interaction. Kidney Int. (2015) 88:276–85. doi: 10.1038/ki.2015.94
185. Sitaru C, Zillikens D. Mechanisms of blister induction by autoantibodies. Exp Dermatol. (2005) 14:861–75. doi: 10.1111/j.1600-0625.2005.00367.x
186. van der Steen LP, Bakema JE, Sesarman A, Florea F, Tuk CW, Kirtschig G, et al. Blocking Fcalpha receptor I on granulocytes prevents tissue damage induced by IgA autoantibodies. J Immunol. (2012) 189:1594–601. doi: 10.4049/jimmunol.1101763
187. Williams A, Reljic R, Naylor I, Clark SO, Falero-Diaz G, Singh M, et al. Passive protection with immunoglobulin A antibodies against tuberculous early infection of the lungs. Immunology. (2004) 111:328–33. doi: 10.1111/j.1365-2567.2004.01809.x
188. Balu S, Reljic R, Lewis MJ, Pleass RJ, McIntosh R, van Kooten C, et al. A novel human IgA monoclonal antibody protects against tuberculosis. J Immunol. (2011) 186:3113–9. doi: 10.4049/jimmunol.1003189
189. Bioley G, Monnerat J, Lotscher M, Vonarburg C, Zuercher A, Corthesy B. Plasma-derived polyreactive secretory-like IgA and IgM opsonizing Salmonella enterica typhimurium reduces invasion and gut tissue inflammation through agglutination. Front Immunol. (2017) 8:1043. doi: 10.3389/fimmu.2017.01043
190. Renegar KB, Small PA Jr. Passive transfer of local immunity to influenza virus infection by IgA antibody. J Immunol. (1991) 146:1972–8.
191. Renegar KB, Small PA Jr, Boykins LG, Wright PF. Role of IgA versus IgG in the control of influenza viral infection in the murine respiratory tract. J Immunol. (2004) 173:1978–86. doi: 10.4049/jimmunol.173.3.1978
192. Planque S, Salas M, Mitsuda Y, Sienczyk M, Escobar MA, Mooney JP, et al. Neutralization of genetically diverse HIV-1 strains by IgA antibodies to the gp120-CD4-binding site from long-term survivors of HIV infection. AIDS. (2010) 24:875–84. doi: 10.1097/QAD.0b013e3283376e88
193. Wills S, Hwang KK, Liu P, Dennison SM, Tay MZ, Shen X, et al. HIV-1-specific IgA monoclonal antibodies from an HIV-1 vaccinee mediate galactosylceramide blocking and phagocytosis. J Virol. (2018) 92:e01552-17. doi: 10.1128/JVI.01552-17
194. Tomaras GD, Ferrari G, Shen X, Alam SM, Liao HX, Pollara J, et al. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc Natl Acad Sci USA. (2013) 110:9019–24. doi: 10.1073/pnas.1301456110
195. Rochereau N, Pavot V, Verrier B, Ensinas A, Genin C, Corthesy B, et al. Secretory IgA as a vaccine carrier for delivery of HIV antigen to M cells. Eur J Immunol. (2015) 45:773–9. doi: 10.1002/eji.201444816
196. Rochereau N, Pavot V, Verrier B, Jospin F, Ensinas A, Genin C, et al. Delivery of antigen to nasal-associated lymphoid tissue microfold cells through secretory IgA targeting local dendritic cells confers protective immunity. J Allergy Clin Immunol. (2016) 137:214–22 e2. doi: 10.1016/j.jaci.2015.07.042
197. Buisman AM, Abbink F, Schepp RM, Sonsma JA, Herremans T, Kimman TG. Preexisting poliovirus-specific IgA in the circulation correlates with protection against virus excretion in the elderly. J Infect Dis. (2008) 197:698–706. doi: 10.1086/527487
198. Herremans MM, van Loon AM, Reimerink JH, Rumke HC, van der Avoort HG, Kimman TG, et al. Poliovirus-specific immunoglobulin A in persons vaccinated with inactivated poliovirus vaccine in The Netherlands. Clin Diagn Lab Immunol. (1997) 4:499–503.
199. Herremans TM, Reimerink JH, Buisman AM, Kimman TG, Koopmans MP. Induction of mucosal immunity by inactivated poliovirus vaccine is dependent on previous mucosal contact with live virus. J Immunol. (1999) 162:5011–8.
200. Hutchings AB, Helander A, Silvey KJ, Chandran K, Lucas WT, Nibert ML, et al. Secretory immunoglobulin A antibodies against the sigma1 outer capsid protein of reovirus type 1 Lang prevent infection of mouse Peyer's patches. J Virol. (2004) 78:947–57. doi: 10.1128/JVI.78.2.947-957.2004
201. Tamura S, Funato H, Hirabayashi Y, Suzuki Y, Nagamine T, Aizawa C, et al. Cross-protection against influenza A virus infection by passively transferred respiratory tract IgA antibodies to different hemagglutinin molecules. Eur J Immunol. (1991) 21:1337–44. doi: 10.1002/eji.1830210602
202. Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. (2010) 10:317–27. doi: 10.1038/nri2744
203. Bakema JE, van Egmond M. Immunoglobulin A: a next generation of therapeutic antibodies? MAbs. (2011) 3:352–61. doi: 10.4161/mabs.3.4.16092
204. Valerius T, Stockmeyer B, van Spriel AB, Graziano RF, van den Herik-Oudijk IE, Repp R, et al. FcalphaRI (CD89) as a novel trigger molecule for bispecific antibody therapy. Blood. (1997) 90:4485–92.
205. van Egmond M, Bakema JE. Neutrophils as effector cells for antibody-based immunotherapy of cancer. Semin Cancer Biol. (2013) 23:190–9. doi: 10.1016/j.semcancer.2012.12.002
206. Otten MA, Bakema JE, Tuk CW, Glennie MJ, Tutt AL, Beelen RH, et al. Enhanced FcalphaRI-mediated neutrophil migration towards tumour colonies in the presence of endothelial cells. Eur J Immunol. (2012) 42:1815–21. doi: 10.1002/eji.201141982
207. Heemskerk N, van Egmond M. Monoclonal antibody-mediated killing of tumour cells by neutrophils. Eur J Clin Invest. (2018) 48 (Suppl. 2):e12962. doi: 10.1111/eci.12962
208. Bakema JE, Ganzevles SH, Fluitsma DM, Schilham MW, Beelen RH, Valerius T, et al. Targeting FcalphaRI on polymorphonuclear cells induces tumor cell killing through autophagy. J Immunol. (2011) 187:726–32. doi: 10.4049/jimmunol.1002581
209. Dechant M, Valerius T. IgA antibodies for cancer therapy. Crit Rev Oncol Hematol. (2001) 39:69–77. doi: 10.1016/S1040-8428(01)00105-6
210. Boross P, Lohse S, Nederend M, Jansen JH, van Tetering G, Dechant M, et al. IgA EGFR antibodies mediate tumour killing in vivo. EMBO Mol Med. (2013) 5:1213–26. doi: 10.1002/emmm.201201929
211. Meyer S, Nederend M, Jansen JH, Reiding KR, Jacobino SR, Meeldijk J, et al. Improved in vivo anti-tumor effects of IgA-Her2 antibodies through half-life extension and serum exposure enhancement by FcRn targeting. MAbs. (2016) 8:87–98. doi: 10.1080/19420862.2015.1106658
212. Pascal V, Laffleur B, Debin A, Cuvillier A, van Egmond M, Drocourt D, et al. Anti-CD20 IgA can protect mice against lymphoma development: evaluation of the direct impact of IgA and cytotoxic effector recruitment on CD20 target cells. Haematologica. (2012) 97:1686–94. doi: 10.3324/haematol.2011.061408
213. Lohse S, Loew S, Kretschmer A, Jansen JHM, Meyer S, Ten Broeke T, et al. Effector mechanisms of IgA antibodies against CD20 include recruitment of myeloid cells for antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity. Br J Haematol. (2018) 181:413–7. doi: 10.1111/bjh.14624
214. Rouwendal GJ, van der Lee MM, Meyer S, Reiding KR, Schouten J, de Roo G, et al. A comparison of anti-HER2 IgA and IgG1 in vivo efficacy is facilitated by high N-glycan sialylation of the IgA. MAbs. (2016) 8:74–86. doi: 10.1080/19420862.2015.1102812
215. Hart F, Danielczyk A, Goletz S. Human cell line-derived monoclonal IgA antibodies for cancer immunotherapy. Bioengineering. (2017) 4:42. doi: 10.3390/bioengineering4020042
216. Lohse S, Meyer S, Meulenbroek LA, Jansen JH, Nederend M, Kretschmer A, et al. An Anti-EGFR IgA that displays improved pharmacokinetics and myeloid effector cell engagement in vivo. Cancer Res. (2016) 76:403–17. doi: 10.1158/0008-5472.CAN-15-1232
217. Ben Mkaddem S, Rossato E, Heming N, Monteiro RC. Anti-inflammatory role of the IgA Fc receptor (CD89): from autoimmunity to therapeutic perspectives. Autoimmun Rev. (2013) 12:666–9. doi: 10.1016/j.autrev.2012.10.011
218. Rossato E, Ben Mkaddem S, Kanamaru Y, Hurtado-Nedelec M, Hayem G, Descatoire V, et al. Reversal of arthritis by human monomeric IgA through the receptor-mediated SH2 domain-containing phosphatase 1 inhibitory pathway. Arthritis Rheumatol. (2015) 67:1766–77. doi: 10.1002/art.39142
219. Kanamaru Y, Pfirsch S, Aloulou M, Vrtovsnik F, Essig M, Loirat C, et al. Inhibitory ITAM signaling by Fc alpha RI-FcR gamma chain controls multiple activating responses and prevents renal inflammation. J Immunol. (2008) 180:2669–78. doi: 10.4049/jimmunol.180.4.2669
220. Watanabe T, Kanamaru Y, Liu C, Suzuki Y, Tada N, Okumura K, et al. Negative regulation of inflammatory responses by immunoglobulin A receptor (FcalphaRI) inhibits the development of Toll-like receptor-9 signalling-accelerated glomerulonephritis. Clin Exp Immunol. (2011) 166:235–50. doi: 10.1111/j.1365-2249.2011.04452.x
221. Heineke MH, van der Steen LPE, Korthouwer RM, Hage JJ, Langedijk JPM, Benschop JJ, et al. Peptide mimetics of immunoglobulin A (IgA) and FcalphaRI block IgA-induced human neutrophil activation and migration. Eur J Immunol. (2017) 47:1835–45. doi: 10.1002/eji.201646782
Keywords: IgA, CD89, mucosa, autoimmunity, IgA deficiency, microbiome, vaccination, therapy
Citation: Breedveld A and van Egmond M (2019) IgA and FcαRI: Pathological Roles and Therapeutic Opportunities. Front. Immunol. 10:553. doi: 10.3389/fimmu.2019.00553
Received: 21 December 2018; Accepted: 01 March 2019;
Published: 22 March 2019.
Edited by:
Mark S. Cragg, University of Southampton, United KingdomReviewed by:
Laureline Berthelot, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCedric Vonarburg, CSL Behring AG, Switzerland
Copyright © 2019 Breedveld and van Egmond. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marjolein van Egmond, bS52YW5lZ21vbmRAdnVtYy5ubA==