 Fernando Alvarez1,2,3
Fernando Alvarez1,2,3 Jörg H. Fritz
Jörg H. Fritz Ciriaco A. Piccirillo
Ciriaco A. Piccirillo- 1Department of Microbiology and Immunology, McGill University, Montréal, QC, Canada
- 2Program in Infectious Diseases and Immunology in Global Health, Centre for Translational Biology, The Research Institute of the McGill University Health Center, Montréal, QC, Canada
- 3Centre of Excellence in Translational Immunology, Montréal, QC, Canada
- 4McGill University Research Center on Complex Traits, McGill University, Montréal, QC, Canada
IL-33, a member of the IL-1 family of cytokines, was originally described in 2005 as a promoter of type 2 immune responses. However, recent evidence reveals a more complex picture. This cytokine is released locally as an alarmin upon cellular damage where innate cell types respond to IL-33 by modulating their differentiation and influencing the polarizing signals they provide to T cells at the time of antigen presentation. Moreover, the prominent expression of the IL-33 receptor, ST2, on GATA3+ T helper 2 cells (TH2) demonstrated that IL-33 could have a direct impact on T cells. Recent observations reveal that T-bet+ TH1 cells and Foxp3+ regulatory T (TREG) cells can also express the ST2 receptor, either transiently or permanently. As such, IL-33 can have a direct effect on the dynamics of T cell populations. As IL-33 release was shown to play both an inflammatory and a suppressive role, understanding the complex effect of this cytokine on T cell homeostasis is paramount. In this review, we will focus on the factors that modulate ST2 expression on T cells, the effect of IL-33 on helper T cell responses and the role of IL-33 on TREG cell function.
Multi-Faceted Functions of IL-33
Barrier sites are exposed to varying levels of danger at every moment, which requires the constant involvement of the local immune system to maintain epithelial function and immune homeostasis. As such, many foreign and self-derived warning signals dictate the response of these immune cells. The molecules that provide these signals are classified as pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs). However, some specialized endogenous molecules, released upon cellular damage, were improperly organized using these definitions. Thus, a new concept was introduced during the EMBO Workshop on Innate Danger Signals and HMGB1 in February 2006, which would separate PAMPs from self-signals. Joost Oppenheim introduced at that meeting what he coined “alarmins,” self-molecules released upon cellular damage that play a role in modulating the immune response (1, 2). The proposed description classifies “alarmins” as molecules that (1) are released upon non-programmed cells death; (2) can be produced by immune cells without dying; (3) can recruit and activate receptor-expressing immune cells; and (4) can contribute to the restoration of immune homeostasis and epithelial repair mechanisms (1). In recent years, several examples of dysregulated expression or activity of alarmins were associated with immune-related pathologies in many diseases. Thus, alarmins can play pro-inflammatory or regulatory roles at the site of inflammation (3).
Of the many members of alarmins, the IL-1 family, comprised of 11 members, was introduced early in this classification (4). IL-1 family members include IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β, IL-36γ, and IL-37 which possess agonist properties and IL-1Ra, IL-36Ra, and IL-38, which possess antagonist properties on their respective receptors (5). A unique feature of this family, with the exception of IL-1Ra, is their capacity to accumulate as pro-cytokines and possess enzymatic cleavage sites in their sequence (6). However, cleavage is not always required for these pro-cytokines to bind and activate their respective receptors. For example, as caspase 1 and caspase 8 are required for the activation of IL-1β and IL-18, pro-IL-33 does not require enzymatic processing to exert its biological activity (6). However, processing by neutrophils proteases, notably cathepsin G and elastase, and proteases brought by airway allergens were shown to enhance IL-33 activity (6, 7). This peculiarity reveals that IL-33, as opposed to IL-1β or IL-18, exerts most of its effect in a caspase-independent manner (6). Thus, IL-33 possesses intrinsic biomolecular peculiarities that dictate its role at mucosal sites and its effect on the innate and adaptive immune system.
Expression of ST2 was first described in CD4+ TH2 cells (8). However, a wide range of immune cells has been described to respond to IL-33 directly. A functional ST2 receptor was notably described in eosinophils (9), basophils (10), natural killer (NK), and NK-T cells (11, 12), as well as group 2 innate lymphoid cells (ILC2s) (13). In eosinophils, IL-33 was shown to directly facilitate their maturation through enhanced survival, activation and adhesion (14). Similarly, IL-33 potentiates adhesion and histamine release in basophils (15). IL-33 is also known to facilitate the maturation, migration from the bone marrow and local functions of ILC2s in the lungs (13, 16). Furthermore, dendritic cells (DCs) can respond to IL-33 directly to polarize naïve T cells into TH2 or facilitate TREG proliferation (17, 18). Interestingly, although the effect of IL-33 was originally thought to be a determinant of type 2 immune responses, it was shown to also favor the expansion of NK and NK T cells during viral infections (11, 12). Thus, IL-33 has pleiotropic functions in directing the innate immune response, a feature that is also found in its effect on adaptive immunity, most notably in the function and differentiation of CD4+ T cells.
In mammals, T cells are critical members of the immune system and play a pivotal role in all aspects of immune responses from the effective clearance of pathogens to the establishment of a memory response and the quick return to immune homeostasis. CD4+ T cells are characterized by their ability to recognize antigens through their T cell specific receptor (TCR), upon which they undergo rapid clonal expansion and differentiate into functionally distinct TH subsets. These subsets then migrate and orchestrate the immune response at inflammatory sites. It is of no surprise that the distinct subsets of helper CD4+ T cells, TH1, TH2, and TH17 cells, respond to alarmins of the IL-1 family in order to proliferate and function locally (19). However, the categorization of T cells by their master transcription factors like T-bet, GATA3, RORγT or Foxp3, does not reflect the high level of T cell plasticity observed in vivo. For example, T cells expressing both GATA3 and T-bet were observed in the lung during infection with parasites (20). Similarly, although ST2 is strongly associated with the function of TH2 T cells (8), TH1 cells can also transiently express it (21). Moreover, it was shown that Foxp3+ TREG cells and TH17 cells signal through IL-33 to modulate their respective functions (22, 23).
In this review, we will focus on the effects of IL-33 on CD4+ T cell responses. We will highlight recent advances in our understanding of the IL-33 pathway and its impact on T cell differentiation and effector functions, including the modulatory role of IL-33 on Foxp3+ TREG cells, in both autoimmune and infectious diseases.
Regulation of IL-33 Expression and Secretion
IL-33 is constitutively expressed as a nuclear protein in epithelial and endothelial cells. Body-wide analysis through immunohistochemistry, mRNA transcripts and a unique il-33-LacZ reporter mouse line revealed that IL-33 is constitutively expressed in secondary lymphoid tissues, but more prominently found at mucosal sites like the gut and lungs, as well as in the brain and adipose tissues (24). However, although humans and mice share most of the constitutive expression of IL-33, species-specific differences exist. For example, it was shown that murine keratinocytes express IL-33 constitutively whereas human keratinocytes required prior IFNγ stimulation (25). Thus, conclusions derived from mouse models must be corroborated with human samples.
Many biological mechanisms regulate the half-life and activity of IL-33. On one hand, pro-IL-33, a 31 kDa protein, does not require enzymatic cleavage to exert its biological functions, although these can be potentiated by the action of self and non-self proteases and elastases that cut it down to a more potent 20 kDa protein (6, 7, 26). On the other hand, the activity of IL-33 is known to be reduced by: (1) cleavage of IL-33 after Asp178 by caspases 3 and 7 (27); (2) upregulation of the LMP2 proteasome by IFNγ during type 1 immune responses (28); (3) extracellular cysteine oxidation that cause the formation of two disulfide bridges on IL-33 and disrupts its binding to ST2 (29), and (4) the extracellular release of the soluble ST2 (sST2), that acts as a decoy receptor for IL-33 (30, 31). Furthermore, IL-33 lacks a conventional signal sequence or any non-canonical export pathway and thus requires either cellular death by necrosis or necroptosis of endothelial and epithelial cells or a still unknown excretory mechanism by innate immune cells to be released in the extracellular milieu (5, 30). In fact, the full-length IL-33 was shown to bind to chromatin causing it to be 10 times slower than IL-1α (32). This novel post-translational mechanism of cytokine release, along with the many enzymatic and environmental processes described, reveals the fine control of the activity of IL-33 at mucosal surfaces and illustrates the evolutionary control of these immunomodulatory signals.
IL-33 Signaling
ST2 was first described as an orphan receptor until the discovery of IL-33 (31). A member of the Toll-like/Interleukine-1 receptor superfamily, it was shown that it forms a heterodimer with the ubiquitous IL1R accessory protein (IL1RAcp) at the membrane surface in order to bind IL-33. Interestingly, all the members of the IL-1 family share a common intracellular Toll/IL-1 receptor (TIR) domain. However, four distinct isoforms of ST2 were described: (1) the membrane-bound ST2 (ST2L or ST2), which provides the activation pathway; (2) the soluble ST2 (sST2)–that originates from another promoter region of the il1rl1 gene and lacks the transmembrane and cytoplasmic domains of ST2–acts as a decoy for IL-33, and is notably used as a biological marker of cardiac injury (31, 33); the latter two forms are splice variants identified in a tumor cells line 3) ST2V (34), which possesses a hydrophobic tail at the C-terminal; and 4) in chicken, ST2LV (35), which lacks the transmembrane domain of ST2 and whose function remains to be elucidated.
IL-33 binds specifically to ST2, which in turn associates to the IL1RAcP to form a heterodimeric receptor that leads to the dimerization of the TIR domain with the TIR domain of cytosolic adaptor protein myeloid differentiation factor 88 (MyD88). In turn, the N-terminal death domain (28) of MyD88 recruits the IL-1-associated kinase 1 (36) and 4 (37). The IRAK1/4 complex can then activate the downstream mitogen-activated protein kinase (MAPK) through the TNF receptor-associated factor 6 (TRAF6). TRAF6 does not possess enzymatic activity but plays a critical role through its ubiquitin E3 ligase (38). TRAF6 is thus required for the induction of several kinase cascades such as NF-kB, JNK, p38, and PI3K. Interestingly, IL-33 can activate ERK even in TRAF6-deficient cells, indicating a parallel activation cascade upon signaling (38). In fact, IL-33 could still induce the expression of ST2L in TRAF6-deficient embryonic fibroblasts (38), indicating the presence of distinct pathways in the IL-33 cascade. However, most of these analyses were conducted using non-T cell lines, and studies in primary immune cells are warranted (39, 40).
TRAF6 Activation in T Cells
In T cells, TRAF6 is known to regulate TCR signaling via ubiquitination at Lys(88) of the LAT adapter and phosphorylation of the IKK/NEMO complex (41). Interestingly, TRAF6 deficiency leads to a hyperactivation of the PI3K-AKT pathway in T cells and to TH2 polarization in mice (42). Furthermore, TRAF6 is essential for the survival and proliferation of TREG cells that suppress TH2 type autoimmunity (43, 44). As such, TRAF6 is required for the maintenance of peripheral tolerance and control of T cell hyper-reactivity. The downstream targets of TRAF6 include the phosphorylation of JNK1/2 (38). JNK1/2 activation is required for T cell differentiation, but not activation, as the lack of JNK leads to a decrease in inflammatory cytokine production, but not proliferation or IL-2 production (45). In fact, the p38-MAPK pathway plays a non-redundant role on memory ST2+ TH2 cells, since selective inhibition of p38, but not JNK, PI3K or ERK, leads to a decrease in IL-5 production in these cells upon IL-33 stimulation (46). Thus, although TRAF6 deficiency leads to increased TH2 differentiation and a lack of TREG-mediated suppression, IL-33 signaling is required for TH2 function, illustrating the complexity of this signal in T cells.
ERK Activation in T Cells
Biochemical dissection of the IL-33/ST2 pathway in mammalian cell lines was performed using data mined through an extensive survey of the literature (40). This model includes the phosphorylation and activation of ERK1/2, JNK1/2, p38, and PI3K/AKT downstream of IL-33. However, the underlying processes affected by these changes remain unknown. This is likely due to the large heterogeneity of the recipient cells and their varied epigenetic status. In T cells, ERK activity is notably linked to a reduction in the TCR activation threshold, as it delays the binding of the inhibitory protein SHP-1 to the complex, leading to the activation of T cells under suboptimal stimulation (47). ERK1 is particularly required for TH2 but not TH1 proliferation and function and plays a major role in a model of experimental asthma (48). On the other hand, lack of ERK2 inhibits TH1 and TH17 T cell differentiation and function (49, 50). This was shown to occur notably through the control of the master transcription factors of these subsets, as ERK2 suppresses the transcription of Foxp3 (TREG) and GATA3 (TH2) and favors the expression of T-bet (TH1) (49). Interestingly, although the lack of either ERK2 or ERK1 does not hinder the suppressive ability of TREG cells (49), it favors the TGFβ-mediated induction of Foxp3 (50). Thus, ERK1/2 activation is a major pathway involved in the control of the function of TH1, TH17, TH2, and TREG cells at mucosal sites. Further investigation into the T cell-intrinsic modulation of ERK1 and ERK2 by IL-33 might reveal how the distinct T cell subsets respond to this alarmin.
On the other hand, p38, composed of four known members (α, β, γ, δ), plays key roles in T cell activation and proliferation. Constitutive activation of p38α and p38β (p38αβY323F) was shown to skew T cell differentiation toward TH1 and TH17 cells (51), whereas knock-down of p38 α/β led to increased TREG cells (52). Interestingly, the IL-33-p38 pathway was shown to be directly linked to the function of ST2+ TH2 cells, as inhibition of p38-MAPK, but not JNK or PI3K, resulted in a lack of IL-5 production by TH2 cells upon IL-33 stimulation (46). Finally, although we know little about the role of JNK activation by IL-33 on T cells, JNK1/2 was shown to play a critical role in T cell function but not activation (45).
While some signaling pathways downstream of IL-33 are known, the transcriptional targets downstream of IL-33 depend largely on the state of the recipient T cell and the environmental context. Thus, in order to fully understand the role of IL-33 on T cells, assessing the effects of IL-33 on the functions of TH cell subsets is required.
Effect of IL-33 on TH Cell Responses
Regulation of ST2 Expression
In an early study, inflammatory factors such as tumor necrosis factor (TNF), IL-1α, IL-1β or Phorbol 12-myristate 13-acetate were shown to be required for the upregulation of the membrane-bound ST2 on responding cells (53). TH2 cells were the first to be shown to express ST2 (8). TH2 polarizing conditions, involving both STAT5 (IL-2) and STAT6 (IL-4) activation, were shown to induce ST2 on T cells in vitro, although multiple rounds of polarization were required (54). In fact, the transcription factor GATA3, associated with the development and function of TH2 cells, was necessary for the selective upregulation of ST2 in vitro, as genetic deficiency of GATA3 abrogated ST2 expression in TH2 cells (55). GATA3 binds an enhancer region situated 12kb up-stream of the transcription start site of il1rl1 (ST2) (55, 56), a finding confirmed through genome-wide mapping of GATA3 binding (57). Under the same conditions, the expression of ST2 was also dependent on the binding of STAT5 to the intron 7 of il1rl1 (55) which also leads to the production of IL-13 and IL-5, but not IL-4, in vitro, suggesting that STAT5-activating signals, such as IL-2, IL-7 or TSLP are required for the upregulation of ST2 in T H2 cells (Figure 1).
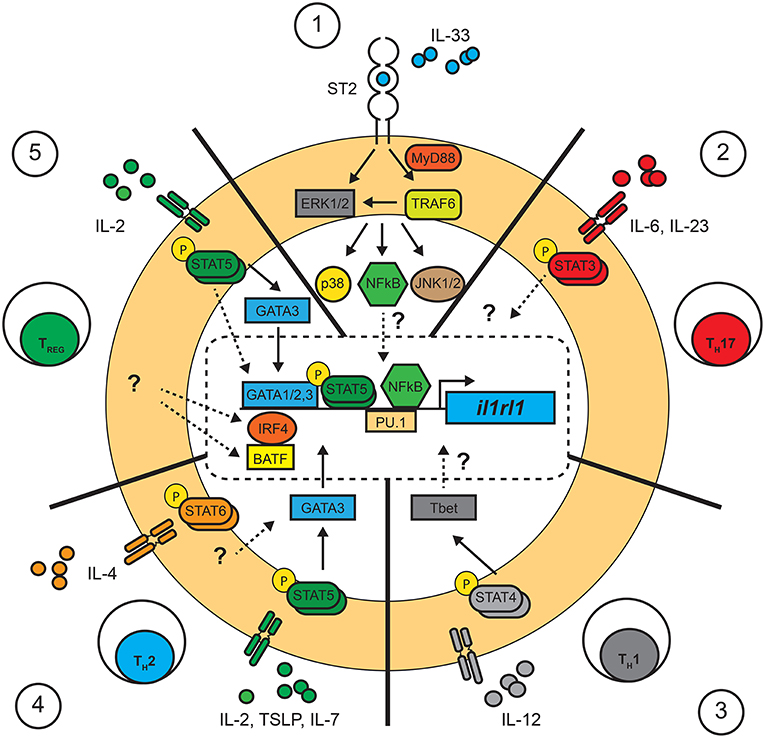
Figure 1. Pathways involved in the control of il1rl1 (ST2) transcription in T cells. Summary of the transcription factors confirmed or suggested to interact with the promoter region of il1rl1 (ST2) in T cells. Confirmed (full lines), suggested (dotted lines) and unknown (?) pathways involved in the control of il1rl1 in T cells. (1) IL-33 is required for the maintained expression of ST2 in T cells in TH2, TREG, and TH1 through unknown mechanisms. This process has been suggested to require the involvement of NF-κB translocation and its binding to a consensus sequence in the promoter region of il1rl1. (2) TH17 cells: The molecular pathways involved in the expression of ST2 in TH17 T cells remains largely unknown, although the transcription factor STAT3 was shown to bind the promoter region of il1rl1 in fibroblast cell lines. (3) TH1 cells: Expression of ST2 in TH1 cells was shown to be dependent on a STAT4 signal leading to T-bet expression, although the molecular interaction with il1rl1 remains unknown. (4) TH2 cells: It has been suggested that expression of ST2 requires STAT5 signals through the upregulation of GATA3 in conjunction with IL-33 stimulation. Although a STAT6 signal is not necessary, little is known about its role in the maintenance of ST2. (5) TREG cells: Expression of ST2 on TREG cells follows a similar pathway as in TH2 cells, requiring a STAT5 signal and IL-33 activation for the upregulation of GATA3 and ST2. The transcription factors IRF4 and BAFT were also shown to promote expression of ST2 by TREG cells although little is known about the upstream signals involved.
Interestingly, the expression of ST2 is particularly enhanced by the provision of exogenous IL-33 in CD4+ T cell cultures, illustrating that IL-33, in a positive feedback loop, is directly involved in the up-regulation of its own receptor (55). It has been suggested that IL-33 potentiates STAT5 signaling in T cells since in vitro polarized TH2 cells show increased STAT5 phosphorylation when exposed to IL-33 (55). On the other hand, a consensus site for NF-κB was found in the Il1rl1 promoter region (58), revealing a potential mechanism by which IL-33 could regulate its own expression. Nonetheless, further investigations are required in order to understand why and how IL-33 is required for the expression of its own receptor. Thus, both a STAT5 signal (IL-2, IL-7 or TSLP) and IL-33 are sufficient to upregulate ST2 on TH2 cells (55) and TREG cells (23) (Figure 1).
The involvement of a STAT6 signal in the development of ST2+ TH2 cells remains to be understood. In early experiments, IL-4 was required for the polarization of TH2 cells, and thus was involved, amid indirectly, in the cells' responsiveness to IL-33. On a molecular level, STAT6 is not known to bind the promoter region of ST2 but does bind to the distal promoter of gata3 (59). Yet, STAT6, but not GATA3, is necessary for binding the locus control region (LCR) inside the TH2 cytokine gene cluster of il4, il5, and il13 (60). As such, STAT6 remodels the LCR, whereas GATA3 acts as a local promoter of these genes. Nonetheless, forced expression of a constitutively activated form of STAT5A (STAT5A1*6) through retroviral transduction in T cells revealed that a STAT6 signal was not essential to differentiate TH2 cells (61). The binding sites of STAT5 on the gene cluster differs from STAT6 and could illustrate a parallel evolutionary mechanism in the polarization of TH2 cells (61). Interestingly, even in these conditions, co-expression of a constitutive GATA3 potentiated the effect of STAT5 in TH2 cell development (61). Thus, although STAT5 plays a significant role, a co-stimulatory STAT6 signal is required to potentiate GATA3 expression and leads to the full differentiation of TH2 cells.
Apart from GATA3, other transcription factors were shown to bind to the distal promoter site of il1rl1 (ST2). Four GATA1 binding sites were identified within 1,001 bp of the distal promoter region of il1rl1 in human and murine cells lines (62, 63). GATA2 and PU.1 were further identified to exert key roles in the expression of ST2 in mast cells and basophils as they bind the distal promoter region of the il1rl1 gene (64, 65). Interestingly, while GATA1 acted as a repressor, GATA2 provided a transactivation signal for the expression of il1rl1 (64). Little is known as to the role of GATA1 and GATA2 in the later stages of T cell polarization and function. A report demonstrated that GATA1 possesses a degree of redundancy with GATA3 in T cells, as it suppresses TH1 differentiation and functions in a similar, yet less efficient, manner as GATA3 (66). More recent evidence points to a possible role of PU.1 in the regulation of GATA3 expression in T cell differentiation. PU.1 is required for the development of T cells in the thymus (67), and is expressed in TH9 and not in TH1 cells (68). Interestingly, PU.1 can alter GATA3 promoter regions in dendritic and T cells and was found to facilitate the expression of il5 and il13, but not il4 (68, 69). Although yet unknown, the role of PU.1 in ST2+ TH2 might reveal why these cells respond to IL-33 by expressing IL-5 and IL-13, but not IL-4 (55). A recent report identifies the transcription factors IRF4 and BATF as binding the il1rl1 loci in TREG cells (70) (Figure 1). In fact, a reduced expression of BATF lead to a decrease in ST2 expression in TREG cells (71). Thus, TREG cells may possess distinct mechanisms to control the transcription of il1rl1.
Surprisingly, ST2 can be transiently expressed by TH1 cells (21). In these reports, the upregulation of ST2 was significantly lower and short lived when compared to ST2+ TH2 cells and was dependent on the expression of the transcription factor T-bet and the IL-12-dependent STAT4 signal (21, 72). Interestingly, co-stimulation with IL-33 was also required for the expression of ST2. Although these observations were corroborated in vivo with STAT4−/− and Tbet−/− mice during the course of a lymphocytic choriomeningitis virus (LCMV) infection (21), it was suggested that these cells might represent a hybrid T-bet+GATA3+ cell subset as low levels of GATA3 were found to be upregulated in a subset of Tbet+ cells (20, 56). Nonetheless, further investigations are required to understand the transcriptional mechanisms by which TH1 cells express ST2.
Finally, TH17 cell, expressing the transcription factor RORγT and producing IL-17A, could, under strong TCR stimuli, express ST2 in the small intestine (22). The process by which TH17 cells upregulate ST2 remains unclear. However, it is well-known that STAT3 signaling plays a major role in the development and cytokine expression of TH17 T cells (73). Recently, it was show that STAT3, along with ERK, had the potential to upregulate the proximal promoter region of ST2 in both human and murine fibroblastic cells lines (74). Although the proximal region of ST2 results in the truncated soluble form of ST2 (sST2), an analog mechanism involving the distal promoter might be found in ST2+ TH17 cells.
TH2 Cell Development and Function
Since the discovery of IL-33, progress has been made to identify its multifunctional roles. Initially, IL-33 was described for its role in promoting type 2 immunity in infectious and allergic diseases (75). Polymorphisms in the il1rl1 or il33 genes are found in patients suffering from exacerbated type 2 immune responses, notably severe atopic dermatitis and asthma, illustrating the important role of these genes in the susceptibility to allergic diseases (36, 76). IL-33 administration in the airways of mice enhances TH2-associated cytokine production in the lungs, increases mucus production and causes a severe type 2 airway hyper-reactivity that mimics the pathophysiology of asthma (37). These reports highlight the role of IL-33 in the differentiation, and function of TH2 cells (77–79).
Naïve T cell express little to no ST2 on their surface. ST2 is expressed in vitro when T cells receive TCR activation in combination with cytokine polarization that drives TH2 T cell differentiation (55, 80). Thus, unique to T cells, TCR engagement is, along with STAT5 and IL-33, a critical signal for naïve T cells to upregulate the ST2 receptor. Conversely, differentiated TH2 cells maintain the ability, long after TCR stimuli, to respond to IL-33 and produce IL-5 and IL-13 (55). Thus, T cells must undergo a round of activation to upregulate the receptor but, once activated remain capable of responding to IL-33. Human CD4+ T cells also express the ST2 receptor in vitro upon TH2, but not TH1, differentiation, although IL-4, a STAT6 inducer, was used in these assays (81).
In vitro, IL-33 enhances IL-5 and IL-13 production, but not IL-4, in TH2 polarized cells (55, 80). This phenotype is unusual, as IL-4 expression was long thought to be the hallmark of differentiated TH2 cells (82) and hinted that in vitro IL-33-responding TH2 cells may have undergone further epigenetic modifications (83). In contrast, IL-33 administration leads to the accumulation of IL-4+ TH2 cells in the lungs and lymph nodes of treated mice (37, 84). This discrepancy could be due to the effect of IL-33 on APCs, directly involved in T cell differentiation. In fact, IL-33 modulates the differentiation and maturation of DCs as they polarize naïve T cells into ST2+ TH2 cells (18). Similarly, IL-33, in conjunction with TGFβ, can facilitate IL-9 production in both mouse and human T cells (85, 86). Thus, the effect of IL-33 signaling on the cytokine production of T cells is highly dependent on the cytokine microenvironment (Figure 2).
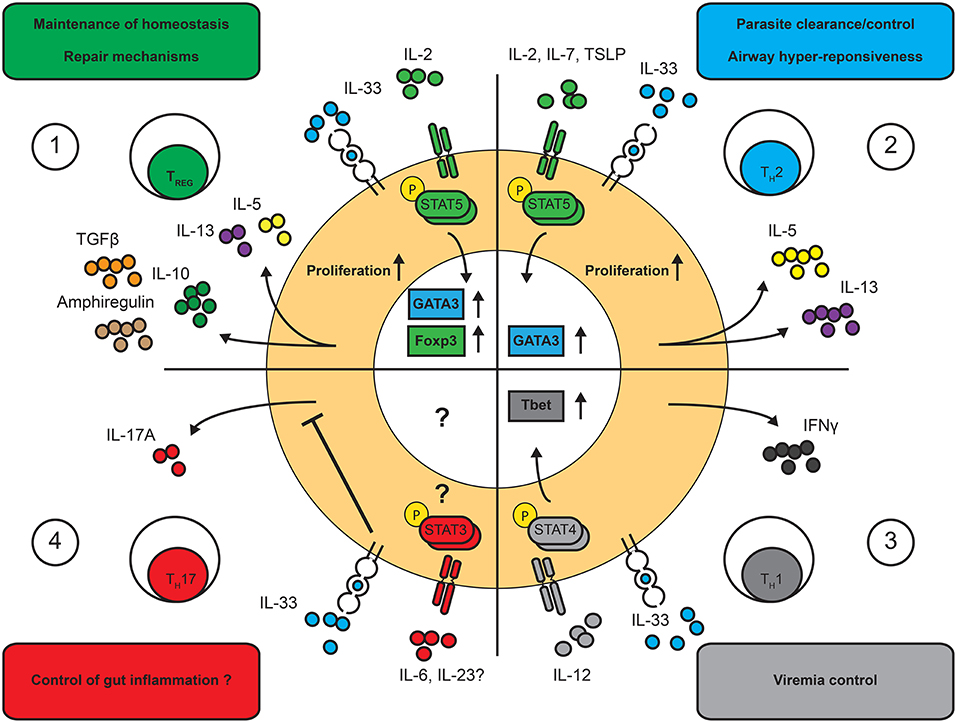
Figure 2. Effects of IL-33 on T cell functions. IL-33 is a multi-faceted cytokine regulating distinct T cell functions and in a highly context-dependent manner. Known functional outcomes of IL-33 on T cell driven immune responses. (1) TREG cells: IL-33 increases proliferation of TREG cells and facilitates the production of amphiregulin, IL-10 and TGFβ as well as low levels of IL-5 and IL-13 in a STAT5-dependent manner. (2) TH2 cells: IL-33 enhances the proliferation and the expression of IL-5 and IL-13 in TH2 cells in a STAT5-dependent manner. (3) TH1 cells: IL-33 was shown to enhance IFNγ production in TH1 cells in a STAT4-dependent manner. (4) TH17 cells: IL-33 was shown to inhibit IL-17 production in TH17 cells. The effects on other T cell functions remains to be assessed.
IL-33 plays an important role in the pathology of asthma and the TH2 cell differentiation in vivo. Immunization of mice to a single dose of ovalbumin (OVA) (5) together with IL-33 induces long-lasting memory TH2 cells that leads to severe asthma-like pathology in the lungs. These IL-33-induced OVA-specific TH2 cells produce particularly high levels of IL-5 and IL-13 upon re-stimulation with OVA, a phenomenon not seen in memory TH2 cells of mice were immunized with OVA alone (84). Furthermore, when mice are exposed to airway antigens, ST2−/− TH2 cells produce less IL-13, while ST2−/− ILC2 functions remain unaffected (87). Concomitantly, memory IL-5–secreting ST2+ TH2 cells have been isolated from patients suffering from eosinophilic chronic rhinosinusitis, a common allergic condition (46).
However, once ST2+ TH2 cells are developed, unexpected outcomes have been observed in response to IL-33. When in vitro polarized OVA-specific, ST2-deficient (OVA-Tg/ST2−/−) or WT (OVA-Tg) TH2 cells are donor ST2−/− TH2, not WT TH2, cells expressed higher levels of IL-5 production concomitant with a more severe cellular infiltrations in the lungs (88). Similarly, when ST2−/− mice were exposed to extracts containing ragweed, dust mite and Aspergillus fumigatus, a more severe form of airway hyper-reactivity was observed compared to WT mice (87); an observation that correlated with a reduction of TREG cells in the lungs of ST2−/− mice. Thus, although IL-33 enhances TH2 responses, it is not essential for the development of airway hyper-reactivity but seems to play a prominent role in TREG cell homeostasis. Overall, these data are in contrast to the known in vitro effects of IL-33 and illustrates the multifaceted roles of IL-33 in both enhancing or dampening TH2 cell responses in a context-dependent manner.
TH1 and TH17 Cell Differentiation and Function
Recent experimental evidence revealed that IL-33 plays a role in the development and maintenance of type 1 immune responses. When studying the T cell response to a systemic LCMV infection, Baumann et al. identified a prominent subset of T-bet+ ST2+ T cells within the antigen-specific memory T cell pool (21). In contrast to ST2+ TH2 cells, TH1 cells expressed ST2 transiently. Interestingly, after injecting LCMV-TCR specific T cells in infected mice, WT, but not Tbet- or STAT4- deficient T cells were able to express ST2, demonstrating that during strong type 1 immunity, TH1 cells can upregulate ST2. Furthermore, ST2−/− T cells failed to expand and produce high levels of IFNγ, TNFα or IL-2 after transfer in LCMV-infected mice (21), suggesting that TH1 cells require ST2 in order to optimally expand and function during the course of LCMV. Interestingly, a similar observation was revealed upon influenza infection, where the rapid release of IL-33 correlated with enhanced IFNγ and TNFα production (89). In fact, IL-33 was shown to potentiate in vitro the action of IL-12, a STAT4 inducer, in TH1 cells, resulting in increased production of IFNγ (72). Similarly, CD8+ T cells were also shown to transiently express the ST2 receptor. IL-33 enhanced the clonal expansion of activated CD8+ T cells and was necessary for the effective control of LCMV infection (90). These observations demonstrate a role of IL-33 to enhance IFNγ through the action of IL-12 without affecting TH1 polarization (Figure 2).
Finally, a recent account suggests a possible role of IL-33 in TH17 cell differentiation (22). These cells express the transcription factor RORγT and release IL-17A and IL-17F. Upon anti-CD3 treatment in vivo, ST2 surface-expression was observed by IL-17-producing T cells in the gut (22). However, IL-33 inhibited the proliferation and pro-inflammatory cytokine production of TH17 cells both in vivo and in vitro. Here, contrarily to TH2 and TH1 cells, IL-33 signaling controlled the exacerbated inflammatory response by TH17 cells, although further work is required to understand the full extent of the role of IL-33 on these cells. In summary, many T cell subsets can respond to IL-33, making the modulation of T cells responses by IL-33 complex and context-dependent.
IL-33-Mediated Regulation of T Cells in Infection
A way to dissect the distinct roles of IL-33 on T cells is to study its effect in distinct infectious diseases. Little is known about the role of IL-33 in human diseases, as there is currently a lack of tools to identify and follow human ST2+ T cells, yet important progress has been made in the field through rodent models of infectious disease. IL-33 most likely plays a key role in human disease, as evidenced by increased levels of the cytokine or its decoy receptor sST2 during both viral (91–93) and bacterial (94) infections. In rodent models, IL-33 was shown to play both protective and deleterious roles during the course of infection(95).
This is seen in models of viral infections, where IL-33 plays ambiguous roles on the T cell response. In certain cases, viral virulence is linked to enhanced IL-33 release, as observed upon infection with respiratory syncytial virus (RSV) in both human and mice (96). When mice are infected with RSV, IL-33 is rapidly released in the early phases of viral infection in the lung (97). Antibody-mediated blockade of ST2 leads to a decrease in IL-13 production and eosinophil recruitment but does not affect viral growth or clearance of RSV by type 1 immune responses (97). Concomitantly, anti-IL-33 therapy was shown to mitigate the establishment of the deleterious type 2 memory response during a Rhinovirus infection that promotes airway hyper-reactivity (98). On the other hand, IL-33 was also shown to contribute to the clearance of LCMV and Coxsackievirus-B5 systemic viral infections through enhanced TH1 and CD8+ T cell responses (90, 99).
IL-33 can also play an important role in the control and clearance of parasites. In a model of intestinal infection with Nippostrongylus brasiliensis, a mouse-pathogenic hookworm, clearance of the parasite and the establishment of a T cell memory response required IL-33 (100). Interestingly, IL-4+ TH2 cells–as well as high levels of IgE, basophils and mast cells responses—were readily detected in infected mice lacking ST2 (ST2−/−) yet insufficient IL-13+ TH2 cells and ILC2s lead to a failure to clear the parasite (100). Similarly, mice infected with Trichuris muris or Strongyloides venezuelensis require IL-33 signaling for the effective control of the parasite (101, 102). On the other hand, in a model of visceral Leishmania donovani infection, IL-33 was shown to be deleterious to the host, as it inhibited the TH1 response necessary for the clearance of this parasite (103). This was attributed to a skewed ST2+ TH2 immune response, as these cells accumulated in the chronic lesion of Leishmania (104). Similarly, lack of ST2 in mice infected with the protozoa Toxoplasma gondii, lead to a more severe form of encephalitis, characterized by increased levels of TNFα and IFNγ (105). Finally, lack of ST2 signaling leads to a better control of the fungus Cryptococcus neoformans, characterized by a significant reduction in IL-5 and IL-13 production by TH2 cells, but no difference in the level of expression of IFNγ and IL-17A (106). Importantly, the effect of IL-33 on the skewing of T cell responses may play a major role in predisposing to virus-induced asthma through the differentiation of pathogenic TH2 cells over anti-viral T cells (98). These experiments provide further evidence that IL-33 influences the function of T cells in disease and this effect is highly dependent on the target tissue of infection and type of pathogen. Furthermore, IL-33 modulates important functions in other compartments of the immune system, notably the innate immune response, which was not addressed here but contributes to the overall response against pathogens (107).
Regulatory T Cells
TREG cells are an important immunosuppressive subset of CD4+ T cells characterized by the expression of the transcription factor Foxp3, the key master regulator that enforces the transcriptional program global phenotype and function of TREG cells (108). However, TREG cells can also undergo distinct epigenetic modifications and co-express transcription factors in order to acquire effector functions enabling them to migrate, survive and suppress in inflammatory sites, particularly at mucosal surfaces (109, 110). This particular ability enables them to adapt to specific environmental conditions. IL-33 was recently identified as one of the signals involved in the maintenance of Foxp3+ TREG cell homeostasis at mucosal sites. At the steady-state, ST2+ TREG cells represent the majority of ST2-expressing CD4+ T cells and are notably found in the gut (23) and lungs (111).
Phenotypic Characteristics of ST2+ TREG
CD4+ TREG cells, including those found at mucosal surfaces, originate from the thymus (thymic-derived tTREG) or develop de novo from polarizing signals in the periphery (peripherally-induced pTREG). Both tTREG and pTREG cells effectively suppress innate and adaptive responses including a variety of effector T cell functions (112). Interestingly, tTREG and pTREG were shown to play non-redundant functions in the suppression of the adaptive immune response, as both of these subsets are required to maintain immune homeostasis in the mucosa. Although surface markers capable of distinguishing them remain poorly defined, tTREG generally have a fully demethylated TREG-specific demethylated region (TSDR), located in the foxp3 locus, compared to pTREG cells (113, 114). Helios, a transcription factor that is prominently expressed in tTreg cells, is frequently regarded as a marker of TREG cells of thymic origin (115) but is also contested (116). Both Helios+ and Helios− TREG cells isolated from the lamina propria of the gut express ST2 (23), while the vast majority of Helios+ TREG cells express ST2 in secondary lymphoid organs and in the lungs (17), all-the-while expressing high levels of other proposed markers of tTREG, such as Neuropilin 1 and TIGIT (unpublished observations). Interestingly, the expression of Helios was recently associated with distinct TREG cell functions in the periphery (117) as well as the stability of foxp3 expression on TREG cells (118). Similarly, IL-33 signaling on TREG cells was shown to play an important role in enhancing the stability of Foxp3 in TREG cells and is notably necessary for these cells to prevent T cell-mediated colitis (23). However, the molecular relationship between IL-33 signaling and Helios expression in TREG cells remains to be understood.
On the other hand, ST2+ TREG cells also express the transcription factor GATA3. Upon IL-33 stimulation, GATA3 is rapidly phosphorylated in TREG cells (23), in turn enhancing the expression of its own receptor. Expression of GATA3, like ST2, was identified in TREG cells in the gut (119) where it plays a central role in (1) the maintenance of immune homeostasis (120), (2) in the stability of foxp3 and (3) is critical for TREG cells to prevent T cell mediated colitis (119). Thus, ST2 and GATA3 follow a similar pattern of expression and play similar functional roles in TREG cells, indicating a strong inter-relationship between the two in orchestrating TREG adaptation in the mucosa.
Finally, the STAT5 signaling pathway can be triggered by IL-2, IL-7, IL-15, or TSLP. TREG cells constitutively express high levels of the IL-2 receptor α chain (CD25), as they are highly dependent on exogeneous IL-2 for survival, function and proliferation (121, 122). In contrast, TREG cells express little IL-7R outside of the thymus in human and mice (123), yet IL-7 could play a role on the expansion of TREG cells at mucosal sites (124). Although there is little information on the role of IL-15 on TREG cells, a recent account reveals that gut-resident T cells depend on IL-15 to enhance Foxp3 over RORγT expression and block a Th17-driven inflammatory bowel disease (125). Finally, TREG cells in the lungs were recently shown to express the TSLP receptor (126). So far, however, only IL-2, in the presence of IL-33, was shown to facilitate the expression of the ST2 receptor on TREG cells (23). Thus, further investigation into the role of the cytokines involved in STAT5 signaling is required.
Role of IL-33 on TREG Function
IL-33 can support many aspects of TREG cell functions. IL-33 facilitates the selective expansion of TREG cells in vitro in a MyD88-dependent manner (127, 128). Moreover, ST2+ TREG cells show increased suppressive capacity in vitro and in vivo in the presence of IL-33 (127, 129, 130), although this was recently contested (131). However, the techniques used by these groups differed and this might provide insight into the modulation of the suppressive ability of ST2+ TREG cells.
Moreover, in vivo, the increased fitness and suppressive function of ST2+ TREG cells is also highlighted by the effect of IL-33 on the maintenance of foxp3 expression in the gut and their ability to suppress T-cell mediated colitis (23). Concomitantly, ST2+ TREG cells readily expand in the mucosa during the course of distinct infectious diseases (111, 129), where they resist the expression of pro-inflammatory cytokines like IFNγ, even strong polarizing conditions (129). IL-33-responsive TREG cells are also endowed with unique cytokine production potential. For example, ST2+ TREG cells were found to produce high levels of IL-10, TGFβ and amphiregulin, which favor a tolerogenic environment and the establishment of tissue repair mechanisms (111, 129) (Figure 2). On the other hand, ST2+ TREG cells can also express type 2 cytokines, like IL-5 and IL-13, when stimulated in vitro in the presence of IL-33 (129). Similarly, in mice exposed to airway allergens in combination with IL-33, WT, but not ST2−/−, TREG cells express high levels of IL-5 and IL-13 (131). Thus, there are reports of both highly suppressive and pro-inflammatory ST2+ TREG cells. To answer this disparity, it was proposed that IL-33 could facilitate the transition from suppressive to dysregulated TREG cells in a dose-dependent manner, although more investigations are required (56). On the other hand, we know little about the potential effect of secondary signals on ST2+ TREG cells, as these cells could have acquired the ability to respond to other environmental cues.
ST2+ TREG in Disease
We do not know the full extent of the role of ST2+ TREG cells in infectious diseases. Nonetheless, ST2+ TREG cells were shown to (1) promote the establishment of memory T cells, (2) control the expansion of inflammatory TH1 and TH17 cells, and (3) promote epithelial cell repair (23, 111, 129). The role of IL-33 on TREG cells has been studied in several infectious and non-infectious inflammatory models. In models that elicit prominent TH1 or TH17 responses, the role of IL-33 on TREG cells was shown to be protective. For example, during Influenza infection, ST2+ TREG cells accumulate in the lung where they produce amphiregulin, a cytokine involved in tissue repair (111). Throughout infection, ST2+ TREG cells are refractory to inflammatory signals and resist the production of inflammatory cytokines. Moreover, in a mouse model of T-cell induced colitis, ST2 expression by TREG cells was shown to be critical to prevent the onset of disease in the gut (23). Moreover, ST2+ TREG cells are induced upon cytomegalovirus (CMV) infection in mice where they play a critical role in dampening liver damage (132). Finally, we recently observed that in chronic infection with Cryptococcus neoformans, which leads to a prominent TH17 response, ST2+ TREG cells resist the up-regulation of RORγT and the production of IL-17 (133). However, this suppressive function of TREG cells could have a negative impact, as it was shown that in helminth infections ST2+ TREG cells, but not ST2−, suppress TH2 cells and facilitate helminth fecundity (134). Similarly, a recent account revealed that the tumor-specific release of IL-33 can promote the accumulation of TREG cells at the site where they contribute to tumor growth and immune evasion (135). Thus, the effect of IL-33 was suggested to be generally protective and promote immune regulation, notably through an enhanced suppressive ability of TREG cells. However, this effect seems to be context-dependent, as recent evidence reveals that IL-33 can also fuel inflammatory responses (131, 136).
Role of IL-33 in Autoimmune Diseases
IL-33 was shown to play important roles in either driving or dampening dysregulated T cells responses in autoimmune diseases. Polymorphisms in the Il33 gene are detected in patients with Alzheimer's disease (137) and Inflammatory Bowel disease (IBD) (138) suggesting that a complete or partial loss of function leads to exacerbated disease (139). In addition, increased levels of IL-33 are detected in patients with multiple sclerosis (MS) (140), systemic lupus erythematous (SLE) (141), type 1 diabetes (T1D) (142) and rheumatoid arthritis (RA) (143). At the steady-state, high levels of IL-33 are produced in the central nervous system (CNS), where it favors the release of IL-1β and IL-10 (144). Expectedly, IL-33 is a major component of the global inflammatory process within the CNS. In experimental autoimmune encephalitis (EAE), a mouse model for multiple sclerosis (MS), IL-33 plays a protective role by dampening the generation of inflammatory astrocytes and the expansion of effector T cells, while enhancing TREG and TH2 responses (145). IL-33 directly attenuates the production of IL-17 and IFNγ by pathogenic TH17 or TH1 cells (146). Moreover, adoptive transfer of MOG-specific T cells from ST2−/− but not ST2+/+ mice fail to prevent EAE onset in BALB/c mice, a strain that is naturally resistant to the disease (147). On the other hand, administration of recombinant IL-33 (rIL-33) is shown to exacerbate EAE in C57BL/6 mice while anti-IL-33 therapy attenuates IL-17 and IFNγ production in situ (148). This strain-specific difference may to be due to a time or context-dependent effect of IL-33, as signaling during the onset of disease is most likely protective, while IL-33 activity in the later stages likely exacerbates TH1 and TH17 responses (149).
A similarly complex role of IL-33 is found in rheumatoid arthritis (RA). While IL-33 is produced at high levels in joints during both RA in human and in experimental arthritis in mice, anti-ST2 therapy significantly attenuates the progression of disease (150). However, while ST2−/− mice show reduced disease severity, IL-33−/− mice do not (151), although the reasons for this difference remain unknown. Similarly, the attenuating effect of IL-33 in the onset of disease was also shown in mouse models of uveitis (152) and T1D (153), although this observation is yet to be described in human disease.
Finally, IL-33 is closely associated to asthma, since it is increased in asthmatic patients (154) and was shown to potentiate airway hyper-reactivity (136). Notably, IL-33 was shown to directly impair TREG cell function during antigen-driven type 2 airway hyper-reactivity (131) and enhance TH2 differentiation through enhanced OX40 ligand interaction (155). Interestingly, this unexpected effect of IL-33 on TREG cells differed from prior reports showing that IL-33 facilitated the suppressive function of TREG cells. Future experiments will have to address these controversial observations.
Clinical Implications of IL-33 and Related Therapeutics
The immunomodulatory functions of IL-33 are being exploited to develop novel therapeutic avenues. The IL-33/ST2 axis is currently being targeted in pre-clinical studies [reviewed by Chen et colleagues (156)]. Among the latest strategies developed to inhibit IL-33 signaling in exacerbated type 2 immune responses are monoclonal antibodies against IL-33 that mimic the capturing effect of the sST2, as they bind the biologically active IL-33 and prevent its association with the membrane receptor (157). Similarly, the use of IL-33 traps, using the extracellular domains of ST2 and IL1RAcP, and blocking the membrane-bound ST2 are strategies currently being investigated with drugs in Phase I or II clinical trials (156).
Although the rationale for the use of inhibitory drugs is mostly based on the effects of IL-33 on innate immune responses, the use of drugs or biologics that enhance the IL-33 signaling pathway generally aims to target the adaptive immune response. One notable exception is the use of IL-33 blockade in tumor microenvironments. For example, it was recently shown that monoclonal antibody blockade of IL-33 in mice xenografted with human non-small-cell lung carcinoma (NSCLC) decreased the accumulation of TREG cells and reduced macrophage M2 polarization, leading to the efficient inhibition of tumor growth (158). Similarly, neutralization of IL-33 inhibits the development of colorectal cancer in mice (135), as IL-33 promotes TREG cell accumulation. On the other hand, an engineered IL-2-IL-33 fusion protein was developed to reduce renal injury in mice by targeting and enhancing TREG cells homeostasis and proliferation in situ (159). Moreover, administration of IL-33 during the recovery phase of DSS-induced colitis in mice was shown to enhance recovery, by skewing the accumulation of TH2 and TREG cells over TH1/TH17 responses in the gut (160). Finally, it was recently suggested to use IL-33 to potentiate highly suppressive TREG cells ex vivo, as adoptive transfer of these cells attenuates disease progression in a model of type 1 diabetes (130). However, care must be taken when considering the use of drugs that influence the IL-33 axis. For example, local IL-33 production in a mouse model of hepatocellular carcinoma was shown to enhance CD4+ and CD8+ anti-tumor activity (135), warranting a re-evaluation of the use of IL-33-neutralizing drugs in tumor models.
Conclusion
T cell function at mucosal sites is intimately linked to the processes of antigen presentation, polarizing cytokine signaling, migration to inflamed sites and the subsequent adaptation to local conditions. The role of “alarmins” in the modulation of mucosal T cell function is yet to be fully understood. Nonetheless, IL-33 was shown to play a major role in this process, illustrating the potential for other, less studied, alarmins to play similar roles.
We focused this review on the recent advances in IL-33 and T cells, but the complexity of the relationship between the adaptive and the innate immune response dictate further investigation into the effect of IL-33 on APC-T cell activation. Notably, little is known about the effect of IL-33 on the modulation of Notch signaling, a key component of T cell differentiation.
In T cells, IL-33 plays a major role in cytokine production, cell proliferation and immune regulation. However, many aspects of T cell responses to IL-33 remain to be elucidated. Notably, many reports have shown that GATA3 and STAT5 play clear roles in promoting the transcription of il1rl1, yet the role of T-bet, STAT4, and STAT3 remain obscure. Thus, we need more insights into the factors that influence IL-33 signaling, from the transcription of the receptor to its effect on the function of T cells. The discovery that IL-33 could directly impact distinct T cell subset differentiation and effector functions is of particular interest, as favoring a given type of response might alter the proper course of immune control and cause irreparable damage to the host. Further investigation into co-stimulatory factors might reveal how distinct alarmins influence each other. Multiple factors may compete, synergize or otherwise influence each other in the inflammatory “soup” to which T cells are exposed to. Finally, a thorough understanding of the kinetics of each alarmin might reveal the intrinsic mechanism by which competing alarmins orchestrate the balance between inflammation and tolerance.
In summary, IL-33 can play both inflammatory and regulatory roles during the evolution of an immune response. A deeper understanding of the effects of IL-33 will undoubtedly open the door toward the generation of unique therapeutic approaches. In fact, the use of a chimeric IL2/IL33 protein was shown to be protective in renal injury (159) and monoclonal anti-IL-33 antibodies where shown to excert promising effects in the control of atopic dermatitis. (161). However, when considering a therapeutic modulation of IL-33 signaling, care must be observed in light of the multifaceted roles of IL-33.
Data Availability
No datasets were generated in this study.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Financial support came from the Fonds de partenariat pour un Québec innovant et en santé (FPQIS) (CP), Canadian Institutes of Health Research (CIHR) operating grant (PJT-148821 to CP), and the Canada Research Chair program (CP). CP is supported by the Anna Maria Solinas Laroche Career Award in Immunology. Financial support also came from a CIHR Foundation grant (#354133 to JF) and a Leaders Opportunity Fund infrastructure grant from the Canadian Foundation of Innovation (CFI) (JF). JF is further supported by a Junior 1 and 2 Investigator Award by the Fonds de recherche santé (FRQS).
References
1. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. (2007) 81:1–5. doi: 10.1189/jlb.0306164
2. Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol. (2005) 17:359–65. doi: 10.1016/j.coi.2005.06.002
3. Matta BM, Reichenbach DK, Blazar BR, Turnquist HR. Alarmins and their receptors as modulators and indicators of alloimmune responses. Am J Transplant. (2017) 17:320–7. doi: 10.1111/ajt.13887
4. Kim B, Lee Y, Kim E, Kwak A, Ryoo S, Bae SH, et al. The interleukin-1alpha precursor is biologically active and is likely a key alarmin in the IL-1 family of cytokines. Front Immunol. (2013) 4:391. doi: 10.3389/fimmu.2013.00391
5. Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family:back to the future. Immunity. (2013) 39:1003–18. doi: 10.1016/j.immuni.2013.11.010
6. Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines:variations on a common theme. Immunity. (2015) 42:991–1004. doi: 10.1016/j.immuni.2015.06.003
7. Scott IC, Majithiya JB, Sanden C, Thornton P, Sanders PN, Moore T, et al. Interleukin-33 is activated by allergen- and necrosis-associated proteolytic activities to regulate its alarmin activity during epithelial damage. Sci Rep. (2018) 8:3363. doi: 10.1038/s41598-018-21589-2
8. Lohning M, Stroehmann A, Coyle AJ, Grogan JL, Lin S, Gutierrez-Ramos JC, et al. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc Natl Acad Sci USA. (1998) 95:6930–5. doi: 10.1073/pnas.95.12.6930
9. Suzukawa M, Koketsu R, Iikura M, Nakae S, Matsumoto K, Nagase H, et al. Interleukin-33 enhances adhesion, CD11b expression and survival in human eosinophils. Lab Invest. (2008) 88:1245–53. doi: 10.1038/labinvest.2008.82
10. Suzukawa M, Iikura M, Koketsu R, Nagase H, Tamura C, Komiya A, et al. An IL-1 cytokine member, IL-33, induces human basophil activation via its ST2 receptor. J Immunol. (2008) 181:5981–9. doi: 10.4049/jimmunol.181.9.5981
11. Nabekura T, Girard JP, Lanier LL. IL-33 receptor ST2 amplifies the expansion of NK cells and enhances host defense during mouse cytomegalovirus infection. J Immunol. (2015) 194:5948–52. doi: 10.4049/jimmunol.1500424
12. Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol. (2008) 20:1019–30. doi: 10.1093/intimm/dxn060
13. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. (2011) 12:1045–54. doi: 10.1038/ni.2131
14. Johnston LK, Bryce PJ. Understanding interleukin 33 and its roles in eosinophil development. Front Med. (2017) 4:51. doi: 10.3389/fmed.2017.00051
15. Valent P. Interleukin-33:a regulator of basophils. Blood. (2009) 113:1396–7. doi: 10.1182/blood-2008-11-189811
16. Stier MT, Zhang J, Goleniewska K, Cephus JY, Rusznak M, Wu L, et al. IL-33 promotes the egress of group 2 innate lymphoid cells from the bone marrow. J Exp Med. (2018) 215:263–81. doi: 10.1084/jem.20170449
17. Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, et al. IL-33 is an unconventional Alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2+ regulatory T cells. J Immunol. (2014) 193:4010–20. doi: 10.4049/jimmunol.1400481
18. Rank MA, Kobayashi T, Kozaki H, Bartemes KR, Squillace DL, Kita H. IL-33-activated dendritic cells induce an atypical TH2-type response. J Allergy Clin Immunol. (2009) 123:1047–54. doi: 10.1016/j.jaci.2009.02.026
19. Sims JE, Smith DE. The IL-1 family:regulators of immunity. Nat Rev Immunol. (2010) 10:89–102. doi: 10.1038/nri2691
20. Peine M, Rausch S, Helmstetter C, Frohlich A, Hegazy AN, Kuhl AA, et al. Stable T-bet(+)GATA-3(+) Th1/Th2 hybrid cells arise in vivo, can develop directly from naive precursors, and limit immunopathologic inflammation. PLoS Biol. (2013) 11:e1001633. doi: 10.1371/journal.pbio.1001633
21. Baumann C, Bonilla WV, Frohlich A, Helmstetter C, Peine M, Hegazy AN, et al. T-bet- and STAT4-dependent IL-33 receptor expression directly promotes antiviral Th1 cell responses. Proc Natl Acad Sci USA. (2015) 112:4056–61. doi: 10.1073/pnas.1418549112
22. Pascual-Reguant A, Bayat Sarmadi J, Baumann C, Noster R, Cirera-Salinas D, Curato C, et al. TH17 cells express ST2 and are controlled by the alarmin IL-33 in the small intestine. Mucosal Immunol. (2017) 10:1431–42. doi: 10.1038/mi.2017.5
23. Schiering C, Krausgruber T, Chomka A, Frohlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. (2014) 513:564–8. doi: 10.1038/nature13577
24. Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues:in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J Immunol. (2012) 188:3488–95. doi: 10.4049/jimmunol.1101977
25. Sundnes O, Pietka W, Loos T, Sponheim J, Rankin AL, Pflanz S, et al. Epidermal expression and regulation of interleukin-33 during homeostasis and Inflammation: Strong species differences. J Invest Dermatol. (2015) 135:1771–80. doi: 10.1038/jid.2015.85
26. Lefrancais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard JP, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA. (2012) 109:1673–8. doi: 10.1073/pnas.1115884109
27. Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. (2009) 31:84–98. doi: 10.1016/j.immuni.2009.05.007
28. Kopach P, Lockatell V, Pickering EM, Haskell RE, Anderson RD, Hasday JD, et al. IFN-gamma directly controls IL-33 protein level through a STAT1- and LMP2-dependent mechanism. J Biol Chem. (2014) 289:11829–43. doi: 10.1074/jbc.M113.534396
29. Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, England E, et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun. (2015) 6:8327. doi: 10.1038/ncomms9327
30. Bandara G, Beaven MA, Olivera A, Gilfillan AM, Metcalfe DD. Activated mast cells synthesize and release soluble ST2-a decoy receptor for IL-33. Eur J Immunol. (2015) 45:3034–44. doi: 10.1002/eji.201545501
31. Tominaga S. A putative protein of a growth specific cDNA from BALB/c-3T3 cells is highly similar to the extracellular portion of mouse interleukin 1 receptor. FEBS Lett. (1989) 258:301–4. doi: 10.1016/0014-5793(89)81679-5
32. Travers J, Rochman M, Miracle CE, Habel JE, Brusilovsky M, Caldwell JM, et al. Chromatin regulates IL-33 release and extracellular cytokine activity. Nat Commun. (2018) 9:3244. doi: 10.1038/s41467-018-05485-x
33. Rehman SU, Mueller T, Januzzi JL Jr. Characteristics of the novel interleukin family biomarker ST2 in patients with acute heart failure. J Am Coll Cardiol. (2008) 52:1458–65. doi: 10.1016/j.jacc.2008.07.042
34. Tominaga S, Kuroiwa K, Tago K, Iwahana H, Yanagisawa K, Komatsu N. Presence and expression of a novel variant form of ST2 gene product in human leukemic cell line UT-7/GM. Biochem Biophys Res Commun. (1999) 264:14–8. doi: 10.1006/bbrc.1999.1469
35. Iwahana H, Hayakawa M, Kuroiwa K, Tago K, Yanagisawa K, Noji S, et al. Molecular cloning of the chicken ST2 gene and a novel variant form of the ST2 gene product, ST2LV. Biochim Biophys Acta. (2004) 1681:1–14. doi: 10.1016/j.bbaexp.2004.08.013
36. Shimizu M, Matsuda A, Yanagisawa K, Hirota T, Akahoshi M, Inomata N, et al. Functional SNPs in the distal promoter of the ST2 gene are associated with atopic dermatitis. Hum Mol Genet. (2005) 14:2919–27. doi: 10.1093/hmg/ddi323
37. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
38. Funakoshi-Tago M, Tago K, Hayakawa M, Tominaga S, Ohshio T, Sonoda Y, et al. TRAF6 is a critical signal transducer in IL-33 signaling pathway. Cell Signal. (2008) 20:1679–86. doi: 10.1016/j.cellsig.2008.05.013
39. Pinto SM, Nirujogi RS, Rojas PL, Patil AH, Manda SS, Subbannayya Y, et al. Quantitative phosphoproteomic analysis of IL-33-mediated signaling. Proteomics. (2015) 15:532–44. doi: 10.1002/pmic.201400303
40. Pinto SM, Subbannayya Y, Rex DAB, Raju R, Chatterjee O, Advani J, et al. A network map of IL-33 signaling pathway. J Cell Commun Signal. (2018) 12:615–24. doi: 10.1007/s12079-018-0464-4
41. Xie JJ, Liang JQ, Diao LH, Altman A, Li Y. TNFR-associated factor 6 regulates TCR signaling via interaction with and modification of LAT adapter. J Immunol. (2013) 190:4027–36. doi: 10.4049/jimmunol.1202742
42. King CG, Kobayashi T, Cejas PJ, Kim T, Yoon K, Kim GK, et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat Med. (2006) 12:1088–92. doi: 10.1038/nm1449
43. Chiffoleau E, Kobayashi T, Walsh MC, King CG, Walsh PT, Hancock WW, et al. TNF receptor-associated factor 6 deficiency during hemopoiesis induces Th2-polarized inflammatory disease. J Immunol. (2003) 171:5751–9. doi: 10.4049/jimmunol.171.11.5751
44. Muto G, Kotani H, Kondo T, Morita R, Tsuruta S, Kobayashi T, et al. TRAF6 is essential for maintenance of regulatory T cells that suppress Th2 type autoimmunity. PLoS ONE. (2013) 8:e74639. doi: 10.1371/journal.pone.0074639
45. Dong C, Yang DD, Tournier C, Whitmarsh AJ, Xu J, Davis RJ, et al. JNK is required for effector T-cell function but not for T-cell activation. Nature. (2000) 405:91–4. doi: 10.1038/35011091
46. Endo Y, Hirahara K, Iinuma T, Shinoda K, Tumes DJ, Asou HK, et al. The interleukin-33-p38 kinase axis confers memory T helper 2 cell pathogenicity in the airway. Immunity. (2015) 42:294–308. doi: 10.1016/j.immuni.2015.01.016
47. Singh K, Deshpande P, Pryshchep S, Colmegna I, Liarski V, Weyand CM, et al. ERK-dependent T cell receptor threshold calibration in rheumatoid arthritis. J Immunol. (2009) 183:8258–67. doi: 10.4049/jimmunol.0901784
48. Goplen N, Karim Z, Guo L, Zhuang Y, Huang H, Gorska MM, et al. ERK1 is important for Th2 differentiation and development of experimental asthma. FASEB J. (2012) 26:1934–45. doi: 10.1096/fj.11-196477
49. Chang CF, D'Souza WN, Ch'en IL, Pages G, Pouyssegur J, Hedrick SM. Polar opposites:Erk direction of CD4 T cell subsets. J Immunol. (2012) 189:721–31. doi: 10.4049/jimmunol.1103015
50. Liu H, Yao S, Dann SM, Qin H, Elson CO, Cong Y. ERK differentially regulates Th17- and Treg-cell development and contributes to the pathogenesis of colitis. Eur J Immunol. (2013) 43:1716–26. doi: 10.1002/eji.201242889
51. Jirmanova L, Giardino Torchia ML, Sarma ND, Mittelstadt PR, Ashwell JD. Lack of the T cell-specific alternative p38 activation pathway reduces autoimmunity and inflammation. Blood. (2011) 118:3280–9. doi: 10.1182/blood-2011-01-333039
52. Hayakawa M, Hayakawa H, Petrova T, Ritprajak P, Sutavani RV, Jimenez-Andrade GY, et al. Loss of functionally redundant p38 isoforms in T cells enhances regulatory T cell induction. J Biol Chem. (2017) 292:1762–72. doi: 10.1074/jbc.M116.764548
53. Kumar S, Tzimas MN, Griswold DE, Young PR. Expression of ST2, an interleukin-1 receptor homologue, is induced by proinflammatory stimuli. Biochem Biophys Res Commun. (1997) 235:474–8. doi: 10.1006/bbrc.1997.6810
54. Meisel C, Bonhagen K, Lohning M, Coyle AJ, Gutierrez-Ramos JC, Radbruch A, et al. Regulation and function of T1/ST2 expression on CD4+ T cells: induction of type 2 cytokine production by T1/ST2 cross-linking. J Immunol. (2001) 166:3143–50. doi: 10.4049/jimmunol.166.5.3143
55. Guo L, Wei G, Zhu J, Liao W, Leonard WJ, Zhao K, et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci USA. (2009) 106:13463–8. doi: 10.1073/pnas.0906988106
56. Peine M, Marek RM, Lohning M. IL-33 in T cell differentiation, function, and immune homeostasis. Trends Immunol. (2016) 37:321–33. doi: 10.1016/j.it.2016.03.007
57. Wei G, Abraham BJ, Yagi R, Jothi R, Cui K, Sharma S, et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity. (2011) 35:299–311. doi: 10.1016/j.immuni.2011.08.007
58. Weinberg EO, Shimpo M, De Keulenaer GW, MacGillivray C, Tominaga S, Solomon SD, et al. Expression and regulation of ST2, an interleukin-1 receptor family member, in cardiomyocytes and myocardial infarction. Circulation. (2002) 106:2961–6. doi: 10.1161/01.CIR.0000038705.69871.D9
59. Scheinman EJ, Avni O. Transcriptional regulation of GATA3 in T helper cells by the integrated activities of transcription factors downstream of the interleukin-4 receptor and T cell receptor. J Biol Chem. (2009) 284:3037–48. doi: 10.1074/jbc.M807302200
60. Lee DU, Rao A. Molecular analysis of a locus control region in the T helper 2 cytokine gene cluster: a target for STAT6 but not GATA3. Proc Natl Acad Sci USA. (2004) 101:16010–5. doi: 10.1073/pnas.0407031101
61. Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity. (2003) 19:739–48. doi: 10.1016/S1074-7613(03)00292-9
62. Gachter T, Werenskiold AK, Klemenz R. Transcription of the interleukin-1 receptor-related T1 gene is initiated at different promoters in mast cells and fibroblasts. J Biol Chem. (1996) 271:124–9. doi: 10.1074/jbc.271.1.124
63. Iwahana H, Yanagisawa K, Ito-Kosaka A, Kuroiwa K, Tago K, Komatsu N, et al. Different promoter usage and multiple transcription initiation sites of the interleukin-1 receptor-related human ST2 gene in UT-7 and TM12 cells. Eur J Biochem. (1999) 264:397–406. doi: 10.1046/j.1432-1327.1999.00615.x
64. Baba Y, Maeda K, Yashiro T, Inage E, Kasakura K, Suzuki R, et al. GATA2 is a critical transactivator for the human IL1RL1/ST2 promoter in mast cells/basophils: opposing roles for GATA2 and GATA1 in human IL1RL1/ST2 gene expression. J Biol Chem. (2012) 287:32689–96. doi: 10.1074/jbc.M112.374876
65. Baba Y, Maeda K, Yashiro T, Inage E, Niyonsaba F, Hara M, et al. Involvement of PU.1 in mast cell/basophil-specific function of the human IL1RL1/ST2 promoter. Allergol Int. (2012) 61:461–7. doi: 10.2332/allergolint.12-OA-0424
66. Sundrud MS, Vancompernolle SE, Eger KA, Bruno TC, Subramaniam A, Mummidi S, et al. Transcription factor GATA-1 potently represses the expression of the HIV-1 coreceptor CCR5 in human T cells and dendritic cells. Blood. (2005) 106:3440–8. doi: 10.1182/blood-2005-03-0857
67. Spain LM, Guerriero A, Kunjibettu S, Scott EW. T cell development in PU.1-deficient mice. J Immunol. (1999) 163:2681–7.
68. Chang HC, Zhang S, Thieu VT, Slee RB, Bruns HA, Laribee RN, et al. PU.1 expression delineates heterogeneity in primary Th2 cells. Immunity. (2005) 22:693–703. doi: 10.1016/j.immuni.2005.03.016
69. Yashiro T, Kubo M, Ogawa H, Okumura K, Nishiyama C. PU.1 Suppresses Th2 cytokine expression via silencing of GATA3 transcription in dendritic cells. PLoS ONE. (2015) 10:e0137699. doi: 10.1371/journal.pone.0137699
70. Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol. (2015) 16:276–85. doi: 10.1038/ni.3085
71. Hayatsu N, Miyao T, Tachibana M, Murakami R, Kimura A, Kato T, et al. Analyses of a mutant Foxp3 allele reveal BATF as a critical transcription factor in the differentiation and accumulation of tissue regulatory T cells. Immunity. (2017) 47:268–283e9. doi: 10.1016/j.immuni.2017.07.008
72. Komai-Koma M, Wang E, Kurowska-Stolarska M, Li D, McSharry C, Xu D. Interleukin-33 promoting Th1 lymphocyte differentiation dependents on IL-12. Immunobiology. (2016) 221:412–7. doi: 10.1016/j.imbio.2015.11.013
73. Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. (2007) 282:9358–63. doi: 10.1074/jbc.C600321200
74. Tago K, Ohta S, Funakoshi-Tago M, Aoki-Ohmura C, Matsugi J, Tominaga SI, et al. STAT3 and ERK pathways are involved in cell growth stimulation of the ST2/IL1RL1 promoter. FEBS Open Bio. (2017) 7:293–302. doi: 10.1002/2211-5463.12192
75. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. (2016) 16:676–89. doi: 10.1038/nri.2016.95
76. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. (2010) 363:1211–21. doi: 10.1056/NEJMoa0906312
77. Greenfeder S, Umland SP, Cuss FM, Chapman RW, Egan RW. Th2 cytokines and asthma. The role of interleukin-5 in allergic eosinophilic disease. Respir Res. (2001) 2:71–9. doi: 10.1186/rr41
78. Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, et al. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. (1992) 326:298–304. doi: 10.1056/NEJM199201303260504
79. Steinke JW, Borish L. Th2 cytokines and asthma. Interleukin-4:its role in the pathogenesis of asthma, and targeting it for asthma treatment with interleukin-4 receptor antagonists. Respir Res. (2001) 2:66–70. doi: 10.1186/rr40
80. Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. (2008) 181:4780–90. doi: 10.4049/jimmunol.181.7.4780
81. Komai-Koma M, Xu D, Li Y, McKenzie AN, McInnes IB, Liew FY. IL-33 is a chemoattractant for human Th2 cells. Eur J Immunol. (2007) 37:2779–86. doi: 10.1002/eji.200737547
82. Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol. (2010) 10:225–35. doi: 10.1038/nri2735
83. Zhu J. Transcriptional regulation of Th2 cell differentiation. Immunol Cell Biol. (2010) 88:244–9. doi: 10.1038/icb.2009.114
84. Kobayashi T, Iijima K, Checkel JL, Kita H. IL-1 family cytokines drive Th2 and Th17 cells to innocuous airborne antigens. Am J Respir Cell Mol Biol. (2013) 49:989–98. doi: 10.1165/rcmb.2012-0444OC
85. Blom L, Poulsen BC, Jensen BM, Hansen A, Poulsen LK. IL-33 induces IL-9 production in human CD4+ T cells and basophils. PLoS ONE. (2011) 6:e21695. doi: 10.1371/journal.pone.0021695
86. Ramadan A, Griesenauer B, Adom D, Kapur R, Hanenberg H, Liu C, et al. Specifically differentiated T cell subset promotes tumor immunity over fatal immunity. J Exp Med. (2017) 214:3577–96. doi: 10.1084/jem.20170041
87. Verma M, Liu S, Michalec L, Sripada A, Gorska MM, Alam R. Experimental asthma persists in IL-33 receptor knockout mice because of the emergence of thymic stromal lymphopoietin-driven IL-9(+) and IL-13(+) type 2 innate lymphoid cell subpopulations. J Allergy Clin Immunol. (2018) 142:793–803e8. doi: 10.1016/j.jaci.2017.10.020
88. Mangan NE, Dasvarma A, McKenzie AN, Fallon PG. T1/ST2 expression on Th2 cells negatively regulates allergic pulmonary inflammation. Eur J Immunol. (2007) 37:1302–12. doi: 10.1002/eji.200636520
89. Le Goffic R, Arshad MI, Rauch M, L'Helgoualc'h A, Delmas B, Piquet-Pellorce C, et al. Infection with influenza virus induces IL-33 in murine lungs. Am J Respir Cell Mol Biol. (2011) 45:1125–32. doi: 10.1165/rcmb.2010-0516OC
90. Bonilla WV, Frohlich A, Senn K, Kallert S, Fernandez M, Johnson S, et al. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science. (2012) 335:984–9. doi: 10.1126/science.1215418
91. Becerra A, Warke RV, de Bosch N, Rothman AL, Bosch I. Elevated levels of soluble ST2 protein in dengue virus infected patients. Cytokine. (2008) 41:114–20. doi: 10.1016/j.cyto.2007.11.001
92. Wu X, Li Y, Song CB, Chen YL, Fu YJ, Jiang YJ, et al. Increased expression of sST2 in early HIV infected patients attenuated the IL-33 induced T cell responses. Front Immunol. (2018) 9:2850. doi: 10.3389/fimmu.2018.02850
93. Wang J, Cai Y, Ji H, Feng J, Ayana DA, Niu J, et al. Serum IL-33 levels are associated with liver damage in patients with chronic hepatitis B. J Interferon Cytokine Res. (2012) 32:248–53. doi: 10.1089/jir.2011.0109
94. Lv R, Zhao J, Lei M, Xiao D, Yu Y, Xie J. IL-33 Attenuates sepsis by inhibiting IL-17 receptor signaling through upregulation of SOCS3. Cell Physiol Biochem. (2017) 42:1961–72. doi: 10.1159/000479836
95. Rostan O, Arshad MI, Piquet-Pellorce C, Robert-Gangneux F, Gangneux JP, Samson M. Crucial and diverse role of the interleukin-33/ST2 axis in infectious diseases. Infect Immun. (2015) 83:1738–48. doi: 10.1128/IAI.02908-14
96. Becker Y. Respiratory syncytial virus (RSV) evades the human adaptive immune system by skewing the Th1/Th2 cytokine balance toward increased levels of Th2 cytokines and IgE, markers of allergy–a review. Virus Genes. (2006) 33:235–52. doi: 10.1007/s11262-006-0064-x
97. Zeng S, Wu J, Liu J, Qi F, Liu B. IL-33 receptor (ST2) signalling is important for regulation of Th2-mediated airway inflammation in a murine model of acute Respiratory syncytial virus infection. Scand J Immunol. (2015) 81:494–501. doi: 10.1111/sji.12284
98. Werder RB, Zhang V, Lynch JP, Snape N, Upham JW, Spann K, et al. Chronic IL-33 expression predisposes to virus-induced asthma exacerbations by increasing type 2 inflammation and dampening antiviral immunity. J Allergy Clin Immunol. (2018) 141:1607–19e9. doi: 10.1016/j.jaci.2017.07.051
99. Sesti-Costa R, Silva GK, Proenca-Modena JL, Carlos D, Silva ML, Alves-Filho JC, et al. The IL-33/ST2 pathway controls coxsackievirus B5-induced experimental pancreatitis. J Immunol. (2013) 191:283–92. doi: 10.4049/jimmunol.1202806
100. Hung LY, Lewkowich IP, Dawson LA, Downey J, Yang Y, Smith DE, et al. IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proc Natl Acad Sci USA. (2013) 110:282–7. doi: 10.1073/pnas.1206587110
101. Humphreys NE, Xu D, Hepworth MR, Liew FY, Grencis RK. IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J Immunol. (2008) 180:2443–9. doi: 10.4049/jimmunol.180.4.2443
102. Yasuda K, Muto T, Kawagoe T, Matsumoto M, Sasaki Y, Matsushita K, et al. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc Natl Acad Sci USA. (2012) 109:3451–6. doi: 10.1073/pnas.1201042109
103. Rostan O, Gangneux JP, Piquet-Pellorce C, Manuel C, McKenzie AN, Guiguen C, et al. The IL-33/ST2 axis is associated with human visceral leishmaniasis and suppresses Th1 responses in the livers of BALB/c mice infected with Leishmania donovani. MBio. (2013) 4:e00383–13. doi: 10.1128/mBio.00383-13
104. Kropf P, Bickle Q, Herath S, Klemenz R, Muller I. Organ-specific distribution of CD4+ T1/ST2+ Th2 cells in Leishmania major infection. Eur J Immunol. (2002) 32:2450–2459. doi: 10.1002/1521-4141(200209)32:9<2450::AID-IMMU2450>3.0.CO;2-O
105. Jones LA, Roberts F, Nickdel MB, Brombacher F, McKenzie AN, Henriquez FL, et al. IL-33 receptor (T1/ST2) signalling is necessary to prevent the development of encephalitis in mice infected with Toxoplasma gondii. Eur J Immunol. (2010) 40:426–36. doi: 10.1002/eji.200939705
106. Flaczyk A, Duerr CU, Shourian M, Lafferty EI, Fritz JH, Qureshi ST. IL-33 signaling regulates innate and adaptive immunity to Cryptococcus neoformans. J Immunol. (2013) 191:2503–13. doi: 10.4049/jimmunol.1300426
107. Cayrol C, Girard JP. IL-33:an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. (2014) 31:31–7. doi: 10.1016/j.coi.2014.09.004
108. Bin Dhuban K, Kornete M, Mason SE, Piccirillo CA. Functional dynamics of Foxp3(+) regulatory T cells in mice and humans. Immunol Rev. (2014) 259:140–58. doi: 10.1111/imr.12168
109. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
110. Rothstein DM, Camirand G. New insights into the mechanisms of Treg function. Curr Opin Organ Transplant. (2015) 20:376–84. doi: 10.1097/MOT.0000000000000212
111. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell. (2015) 162:1078–89. doi: 10.1016/j.cell.2015.08.021
112. Schmitt EG, Williams CB. Generation and function of induced regulatory T cells. Front Immunol. (2013) 4:152. doi: 10.3389/fimmu.2013.00152
113. Toker A, Engelbert D, Garg G, Polansky JK, Floess S, Miyao T, et al. Active demethylation of the Foxp3 locus leads to the generation of stable regulatory T cells within the thymus. J Immunol. (2013) 190:3180–8. doi: 10.4049/jimmunol.1203473
114. Hori S. Lineage stability and phenotypic plasticity of Foxp3(+) regulatory T cells. Immunol Rev. (2014) 259:159–72. doi: 10.1111/imr.12175
115. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. (2010) 184:3433–41. doi: 10.4049/jimmunol.0904028
116. Szurek E, Cebula A, Wojciech L, Pietrzak M, Rempala G, Kisielow P, et al. Differences in expression level of helios and neuropilin-1 do not distinguish thymus-derived from extrathymically-induced CD4+Foxp3+ regulatory T cells. PLoS ONE. (2015) 10:e0141161. doi: 10.1371/journal.pone.0141161
117. Sebastian M, Lopez-Ocasio M, Metidji A, Rieder SA, Shevach EM, Thornton AM. Helios controls a limited subset of regulatory T cell functions. J Immunol. (2016) 196:144–55. doi: 10.4049/jimmunol.1501704
118. Nakagawa H, Sido JM, Reyes EE, Kiers V, Cantor H, Kim HJ. Instability of Helios-deficient Tregs is associated with conversion to a T-effector phenotype and enhanced antitumor immunity. Proc Natl Acad Sci USA. (2016) 113:6248–53. doi: 10.1073/pnas.1604765113
119. Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, et al. GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J Clin Invest. (2011) 121:4503–15. doi: 10.1172/JCI57456
120. Wang Y, Su MA, Wan YY. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity. (2011) 35:337–48. doi: 10.1016/j.immuni.2011.08.012
121. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol. (2016) 17:1322–33. doi: 10.1038/ni.3540
122. Mahmud SA, Manlove LS, Farrar MA. Interleukin-2 and STAT5 in regulatory T cell development and function. JAKSTAT. (2013) 2:e23154. doi: 10.4161/jkst.23154
123. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. (2006) 203:1701–11. doi: 10.1084/jem.20060772
124. Simonetta F, Gestermann N, Bloquet S, Bourgeois C. Interleukin-7 optimizes FOXP3+CD4+ regulatory T cells reactivity to interleukin-2 by modulating CD25 expression. PLoS ONE. (2014) 9:e113314. doi: 10.1371/journal.pone.0113314
125. Tosiek MJ, Fiette L, El Daker S, Eberl G, Freitas AA. IL-15-dependent balance between Foxp3 and RORgammat expression impacts inflammatory bowel disease. Nat Commun. (2016) 7:10888. doi: 10.1038/ncomms10888
126. Nguyen KD, Vanichsarn C, Nadeau KC. TSLP directly impairs pulmonary Treg function: association with aberrant tolerogenic immunity in asthmatic airway. Allergy Asthma Clin Immunol. (2010) 6:4. doi: 10.1186/1710-1492-6-4
127. Matta BM, Turnquist HR. Expansion of regulatory T cells in vitro and in vivo by IL-33. Methods Mol Biol. (2016) 1371:29–41. doi: 10.1007/978-1-4939-3139-2_3
128. Xu L, Li W, Wang X, Zhang L, Qi Q, Dong L, et al. The IL-33-ST2-MyD88 axis promotes regulatory T cell proliferation in the murine liver. Eur J Immunol. (2018) 48:1302–7. doi: 10.1002/eji.201747402
129. Siede J, Frohlich A, Datsi A, Hegazy AN, Varga DV, Holecska V, et al. IL-33 receptor-expressing regulatory T cells are highly activated, Th2 biased and suppress CD4 T cell proliferation through IL-10 and TGFbeta Release. PLoS ONE. (2016) 11:e0161507. doi: 10.1371/journal.pone.0161507
130. Ryba-Stanislawowska M, Buksa L, Brandt A, Juhas U, Mysliwiec M. IL-33 improves the suppressive potential of regulatory T cells in patients with type 1 diabetes. Diabetes Res Clin Pract. (2017) 128:67–73. doi: 10.1016/j.diabres.2017.04.011
131. Chen CC, Kobayashi T, Iijima K, Hsu FC, Kita H. IL-33 dysregulates regulatory T cells and impairs established immunologic tolerance in the lungs. J Allergy Clin Immunol. (2017) 140:1351–1363.e7. doi: 10.1016/j.jaci.2017.01.015
132. Popovic B, Golemac M, Podlech J, Zeleznjak J, Bilic-Zulle L, Lukic ML, et al. IL-33/ST2 pathway drives regulatory T cell dependent suppression of liver damage upon cytomegalovirus infection. PLoS Pathog. (2017) 13:e1006345. doi: 10.1371/journal.ppat.1006345
133. Alvarez F, Istomine R, Shourian M, Pavey N, Al-Aubodah T, Qureshi S, et al. The alarmins IL-1 and IL-33 differentially regulate the functional specialization of Foxp3+ regulatory T cells during mucosal inflammation. Mucosal Immunol. (2019). doi: 10.1038/s41385-019-0153-5. [Epub ahead of print].
134. Obata-Ninomiya K, Ishiwata K, Nakano H, Endo Y, Ichikawa T, Onodera A, et al. CXCR6(+)ST2(+) memory T helper 2 cells induced the expression of major basic protein in eosinophils to reduce the fecundity of helminth. Proc Natl Acad Sci USA. (2018) 115:E9849–58. doi: 10.1073/pnas.1714731115
135. Zhou Y, Ji Y, Wang H, Zhang H, Zhou H. IL-33 Promotes the development of colorectal cancer through inducing tumor-infiltrating ST2L(+) regulatory T cells in mice. Technol Cancer Res Treat. (2018) 17:1533033818780091. doi: 10.1177/1533033818780091
136. Sjoberg LC, Nilsson AZ, Lei Y, Gregory JA, Adner M, Nilsson GP. Interleukin 33 exacerbates antigen driven airway hyperresponsiveness, inflammation and remodeling in a mouse model of asthma. Sci Rep. (2017) 7:4219. doi: 10.1038/s41598-017-03674-0
137. Chapuis J, Hot D, Hansmannel F, Kerdraon O, Ferreira S, Hubans C, et al. Transcriptomic and genetic studies identify IL-33 as a candidate gene for Alzheimer's disease. Mol Psychiatry. (2009) 14:1004–16. doi: 10.1038/mp.2009.10
138. Latiano A, Palmieri O, Pastorelli L, Vecchi M, Pizarro TT, Bossa F, et al. Associations between genetic polymorphisms in IL-33, IL1R1 and risk for inflammatory bowel disease. PLoS ONE. (2013) 8:e62144. doi: 10.1371/journal.pone.0062144
139. Seo DH, Che X, Kwak MS, Kim S, Kim JH, Ma HW, et al. Interleukin-33 regulates intestinal inflammation by modulating macrophages in inflammatory bowel disease. Sci Rep. (2017) 7:851. doi: 10.1038/s41598-017-00840-2
140. Christophi GP, Gruber RC, Panos M, Christophi RL, Jubelt B, Massa PT. Interleukin-33 upregulation in peripheral leukocytes and CNS of multiple sclerosis patients. Clin Immunol. (2012) 142:308–19. doi: 10.1016/j.clim.2011.11.007
141. Yang Z, Liang Y, Xi W, Li C, Zhong R. Association of increased serum IL-33 levels with clinical and laboratory characteristics of systemic lupus erythematosus in Chinese population. Clin Exp Med. (2011) 11:75–80. doi: 10.1007/s10238-010-0115-4
142. Shruthi S, Mohan V, Amutha A, Aravindhan V. Increased serum levels of novel T cell cytokines IL-33, IL-9 and IL-17 in subjects with type-1 diabetes. Cytokine. (2016) 86:6–9. doi: 10.1016/j.cyto.2016.07.007
143. Xu D, Jiang HR, Kewin P, Li Y, Mu R, Fraser AR, et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc Natl Acad Sci USA. (2008) 105:10913–8. doi: 10.1073/pnas.0801898105
144. Yasuoka S, Kawanokuchi J, Parajuli B, Jin S, Doi Y, Noda M, et al. Production and functions of IL-33 in the central nervous system. Brain Res. (2011) 1385:8–17. doi: 10.1016/j.brainres.2011.02.045
145. Xiao Y, Lai L, Chen H, Shi J, Zeng F, Li J, et al. Interleukin-33 deficiency exacerbated experimental autoimmune encephalomyelitis with an influence on immune cells and glia cells. Mol Immunol. (2018) 101:550–63. doi: 10.1016/j.molimm.2018.08.026
146. Jiang HR, Milovanovic M, Allan D, Niedbala W, Besnard AG, Fukada SY, et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-gamma production and inducing alternatively activated macrophages. Eur J Immunol. (2012) 42:1804–14. doi: 10.1002/eji.201141947
147. Milovanovic M, Volarevic V, Ljujic B, Radosavljevic G, Jovanovic I, Arsenijevic N, et al. Deletion of IL-33R (ST2) abrogates resistance to EAE in BALB/C mice by enhancing polarization of APC to inflammatory phenotype. PLoS ONE. (2012) 7:e45225. doi: 10.1371/journal.pone.0045225
148. Li M, Li Y, Liu X, Gao X, Wang Y. IL-33 blockade suppresses the development of experimental autoimmune encephalomyelitis in C57BL/6 mice. J Neuroimmunol. (2012) 247:25–31. doi: 10.1016/j.jneuroim.2012.03.016
149. Pei C, Barbour M, Fairlie-Clarke KJ, Allan D, Mu R, Jiang HR. Emerging role of interleukin-33 in autoimmune diseases. Immunology. (2014) 141:9–17. doi: 10.1111/imm.12174
150. Palmer G, Talabot-Ayer D, Lamacchia C, Toy D, Seemayer CA, Viatte S, et al. Inhibition of interleukin-33 signaling attenuates the severity of experimental arthritis. Arthritis Rheum. (2009) 60:738–49. doi: 10.1002/art.24305
151. Martin P, Talabot-Ayer D, Seemayer CA, Vigne S, Lamacchia C, Rodriguez E, et al. Disease severity in K/BxN serum transfer-induced arthritis is not affected by IL-33 deficiency. Arthritis Res Ther. (2013) 15:R13. doi: 10.1186/ar4143
152. Barbour M, Allan D, Xu H, Pei C, Chen M, Niedbala W, et al. IL-33 attenuates the development of experimental autoimmune uveitis. Eur J Immunol. (2014) 44:3320–9. doi: 10.1002/eji.201444671
153. Miller AM, Asquith DL, Hueber AJ, Anderson LA, Holmes WM, McKenzie AN, et al. Interleukin-33 induces protective effects in adipose tissue inflammation during obesity in mice. Circ Res. (2010) 107:650–8. doi: 10.1161/CIRCRESAHA.110.218867
154. Momen T, Ahanchian H, Reisi M, Shamsdin SA, Shahsanai A, Keivanfar M. Comparison of interleukin-33 serum levels in asthmatic patients with a control group and relation with the severity of the disease. Int J Prev Med. (2017) 8:65. doi: 10.4103/ijpvm.IJPVM_179_16
155. Murakami-Satsutani N, Ito T, Nakanishi T, Inagaki N, Tanaka A, Vien PT, et al. IL-33 promotes the induction and maintenance of Th2 immune responses by enhancing the function of OX40 ligand. Allergol Int. (2014) 63:443–55. doi: 10.2332/allergolint.13-OA-0672
156. Chen WY, Tsai TH, Yang JL, Li LC. Therapeutic strategies for targeting IL-33/ST2 signalling for the treatment of inflammatory diseases. Cell Physiol Biochem. (2018) 49:349–58. doi: 10.1159/000492885
157. Xi H, Katschke KJ Jr., Li Y, Truong T, Lee WP, Diehl L, et al. IL-33 amplifies an innate immune response in the degenerating retina. J Exp Med. (2016) 213:189–207. doi: 10.1084/jem.20150894
158. Wang K, Shan S, Yang Z, Gu X, Wang Y, Wang C, et al. IL-33 blockade suppresses tumor growth of human lung cancer through direct and indirect pathways in a preclinical model. Oncotarget. (2017) 8:68571–82. doi: 10.18632/oncotarget.19786
159. Stremska ME, Jose S, Sabapathy V, Huang L, Bajwa A, Kinsey GR, et al. IL233, a novel IL-2 and IL-33 hybrid cytokine, ameliorates renal injury. J Am Soc Nephrol. (2017) 28:2681–93. doi: 10.1681/ASN.2016121272
160. Duan L, Chen J, Zhang H, Yang H, Zhu P, Xiong A, et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3(+) regulatory T-cell responses in mice. Mol Med. (2012) 18:753–61. doi: 10.2119/molmed.2011.00428
Keywords: T cell differentiation, Th17 and Tregs cells, th1/th2 balance, infection, immunoregulation, IL-33, ST2
Citation: Alvarez F, Fritz JH and Piccirillo CA (2019) Pleiotropic Effects of IL-33 on CD4+ T Cell Differentiation and Effector Functions. Front. Immunol. 10:522. doi: 10.3389/fimmu.2019.00522
Received: 23 November 2018; Accepted: 26 February 2019;
Published: 20 March 2019.
Edited by:
Jose Carlos Alves-Filho, University of São Paulo, BrazilReviewed by:
Mariola Stefania Kurowska-Stolarska, University of Glasgow, United KingdomMiodrag L. Lukic, University of Kragujevac, Serbia
Copyright © 2019 Alvarez, Fritz and Piccirillo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ciriaco A. Piccirillo, Q2lyby5waWNjaXJpbGxvQG1jZ2lsbC5jYQ==