 Bhargavi Duvvuri
Bhargavi Duvvuri Christian Lood
Christian Lood- Division of Rheumatology, Department of Medicine, University of Washington, Seattle, WA, United States
Endogenous DNA is primarily found intracellularly in nuclei and mitochondria. However, extracellular, cell-free (cf) DNA, has been observed in several pathological conditions, including autoimmune diseases, prompting the interest of developing cfDNA as a potential biomarker. There is an upsurge in studies considering cfDNA to stratify patients, monitor the treatment response and predict disease progression, thus evaluating the prognostic potential of cfDNA for autoimmune diseases. Since the discovery of elevated cfDNA levels in lupus patients in the 1960s, cfDNA research in autoimmune diseases has mainly focused on the overall quantification of cfDNA and the association with disease activity. However, with recent technological advancements, including genomic and methylomic sequencing, qualitative changes in cfDNA are being explored in autoimmune diseases, similar to the ones used in molecular profiling of cfDNA in cancer patients. Further, the intracellular origin, e.g., if derived from mitochondrial or nuclear source, as well as the complexing with carrier molecules, including LL-37 and HMGB1, has emerged as important factors to consider when analyzing the quality and inflammatory potential of cfDNA. The clinical relevance of cfDNA in autoimmune rheumatic diseases is strengthened by mechanistic insights into the biological processes that result in an enhanced release of DNA into the circulation during autoimmune and inflammatory conditions. Prior work have established an important role of accelerated apoptosis and impaired clearance in leakage of nucleic acids into the extracellular environment. Findings from more recent studies, including our own investigations, have demonstrated that NETosis, a neutrophil cell death process, can result in a selective extrusion of inflammatory mitochondrial DNA; a process which is enhanced in patients with lupus and rheumatoid arthritis. In this review, we will summarize the evolution of cfDNA, both nuclear and mitochondrial DNA, as biomarkers for autoimmune rheumatic diseases and discuss limitations, challenges and implications to establish cfDNA as a biomarker for clinical use. This review will also highlight recent advancements in mechanistic studies demonstrating mitochondrial DNA as a central component of cfDNA in autoimmune rheumatic diseases.
Introduction
In 1948, Mandel and Metais were the first to report on the presence of cfDNA in human plasma (1). However, it was not until the discovery of high levels of cfDNA in systemic lupus erythematosus (SLE) patients in 1966 (2), that the interest in cfDNA as a potential biomarker for autoimmune diseases started. Early reports were flawed by leukocyte release of DNA during coagulation, with a major breakthrough in the field upon recognizing plasma as a better source of pathological cfDNA, with undetectable levels in healthy volunteers (3–5). These early observations were followed by the demonstration of cfDNA in other autoimmune diseases including rheumatoid arthritis (RA) (6). However, with the advent of more sensitive methods, cfDNA was also detected in plasma of healthy individuals, albeit at very low levels. This observation, together with the temporal association of the increased levels of cfDNA with disease activity in patients with SLE and RA, led to the proposition that cfDNA can be a potential biomarker for autoimmune diseases. Subsequent, technological advancements such as fluorometric assays and real-time PCR led to the simple and rapid quantification of cfDNA, also providing information on the intracellular origin of cfDNA, such as the mitochondria. Despite the initial excitement and technological advancements in cfDNA quantification, there was not a substantial interest in cfDNA as a biomarker for autoimmune diseases, until more recently with the increasing understanding into the role of DNA-sensing receptors in inflammation and autoimmunity, especially in SLE and RA. In this review, we will give an overview of basic biology of cfDNA, followed by evolution of cfDNA in SLE and RA as a biomarker of diagnosis, disease activity and progression, and as a prognostic marker of treatment response.
Mechanisms of cfDNA Release
Though our understanding of mechanisms contributing to the generation of cfDNA is evolving with several novel pathways described in recent years (7), it is still unclear which, if any, of the current models account for the elevated levels of cfDNA observed in patients with rheumatic disease. Beneath we will highlight key cell death processes and active release mechanisms, and their potential implication in rheumatic disease.
Apoptosis
Apoptosis, also known as programmed cell death, is an essential part of physiological maintenance of cellular homeostasis that eliminates unwanted and damaged cells. Apoptosis is executed by effector caspases that are activated in extrinsic and intrinsic-pathways triggered by death-receptors and intracellular stimuli such as oxidative stress and DNA damage, respectively. Activation of caspases, a mark of an irreversible commitment to apoptosis, results in a proteolytic cascade leading to several characteristic morphological and biochemical changes in apoptotic cells that include cell and nuclear shrinkage, DNA fragmentation and lipid re-distribution (8, 9). Taking cues from these alterations as find-me signals, especially upon the exposure of phosphatidylserine onto the cell surface, apoptotic debris under normal conditions, is promptly recognized and cleared by phagocytes in a non-inflammatory process called efferocytosis (10). The clearance of apoptotic cells exerts powerful anti-inflammatory and immunosuppressive effects (11). In contrast, impaired clearance of apoptotic material and/or increased cell death process lead to an accumulation of intracellular antigens and DNA extracellularly, which long-term can lead to autoinflammatory responses (12–14). Inter-nucleosomal fragmentation of DNA into double-stranded DNA fragments of 180–200 bp by calcium-dependent endonucleases is the biochemical hallmark of apoptosis. The fragmented DNA is detectable as a ladder pattern when subjected to gel electrophoresis (15). Multiple characteristics of cfDNA suggest that it is reminiscent of apoptotic DNA. cfDNA, like apoptotic DNA, is a highly fragmented, low molecular weight double stranded DNA with an average size of ~150–200 bp in length, a size corresponding to nucleosomal DNA, and exhibits a ladder pattern on gel electrophoresis as multiples of nucleosomal units (16). SLE, a disease characterized by increased apoptosis and impaired clearance of apoptotic cells (17, 18), shows evidence of low molecular weight cfDNA with an apoptosis-like size distribution pattern. DNA purified from SLE plasma formed discrete bands, predominantly with a unit size of ~200 bp, characteristic of DNA found in oligonucleosomes (19). In another study that isolated DNA from the DNA-anti-DNA antibody immune complexes in sera of SLE patients, a strong correlation was observed between low molecular weight DNA sizes (both 30–50 and 150–200 bp), disease activity, and the frequency of renal disease (20). Genome-wide sequencing identified that plasma DNA in SLE patients exhibit size shortening (≤115 bp in length) that correlated with SLEDAI and anti-double stranded DNA (dsDNA) antibody level. In addition, IgG-bound DNA fragments of SLE patients are shorter (≤115 bp) than non-IgG bound DNA (19).
Necrosis
The presence of also high molecular weight cfDNA led to the proposition that necrosis could be the release mechanism (21, 22). Necrosis is an accidental form of cell death in response to physical or chemical injury characterized by cell swelling and rapid loss of plasma membrane integrity, leading to the release of intracellular contents. Necrosis results in non-specific digestion of chromatin, thus enabling release of high molecular weight DNA of many kilo base pairs (23). Necrotic release of cfDNA could be relevant in conditions including trauma, injury and sepsis, where levels of cfDNA were associated with the severity of trauma and post-traumatic complications (24, 25), injury (26, 27), and mortality in patients with sepsis (28, 29). Although the role of necrosis in the elevated levels of cfDNA observed in patients with rheumatic disease has not been carefully investigated, studies in SLE patients suggest that necrotic cell death can be a major source of cfDNA. Intracellular ATP concentration is one of the factors that determines the cell's fate to undergo cell death via apoptosis or necrosis. Interestingly, CD4+ T cells from SLE patients are characterized by ATP depletion due to persistent mitochondrial hyperpolarization, which subsequently results in the uncoupling of oxidative phosphorylation i.e., continued production of reactive oxygen intermediates in the absence of ATP synthesis. This ATP depletion results in a diminished activation-induced apoptosis and sensitization of CD4+ T cells to undergo necrosis, thus enabling the release of cellular contents, including cfDNA, into the extracellular space (30, 31).
NETosis
In response to microbes, as well as sterile inflammation, neutrophils can undergo a unique form of programmed cell death known as NETosis. It results in the extrusion of a web-like structure of nuclear-derived decondensed DNA coated with histones, granular proteins, and cytoplasmic proteins into the extracellular space. Since the extracellular chromatin fibrils could entangle microbes, the structures were named neutrophil extracellular traps (NETs) (32). Unlike suicidal NETosis described above, which results in the death of neutrophils, neutrophils may undergo vital NETosis, a process in which they only extrude a small amount of DNA, preferentially mitochondrial DNA (mtDNA), allowing for the neutrophil to remain alive and continue to exert antimicrobial actions (32, 33). Following the discovery of NETosis, and extrusion of DNA by neutrophils, several other cell types have been identified to release extracellular traps (34–37), a process termed ETosis. Considering dsDNA being the structural backbone of NETs, in the conditions of excessive NETosis (38) or impaired clearance (39, 40), remnants of NETs could account for elevated levels of circulating cfDNA. Consistent with this proposition, NET deposition was found to be associated with levels of cfDNA in various pathological conditions including SLE, rheumatoid arthritis, cancer, and transfusion-related acute lung injury (38, 41–44).
Pyroptosis
Pyroptosis is a lytic form of inflammatory cell death induced by inflammasome activation in response to diverse pathogen and host-derived danger signals (45). Inflammasome activation leads to the processing and activation of inflammatory caspases, which in turn mediate the downstream inflammatory processes that include the processing of pro-inflammatory cytokines, IL-1β and IL-18, and lytic events associated with pyroptosis (46–48). Early during this cell death, pores are formed in the cell membrane in a caspase-dependent manner ultimately resulting in cell lysis and the release of intracellular inflammatory contents, including IL-1β and IL-18 (49). An unidentified-caspase1-activated nuclease leads to DNA fragmentation during pyroptosis (49). Hence, in conditions associated with caspase-mediated induction of IL-1β and IL-18, the disruption of cell membrane can contribute to release of cfDNA. Tan et al. (50) demonstrated that HIV patients who develop tuberculosis-associated immune reconstitution inflammatory syndrome following cART therapy exhibit increased inflammasome activation, represented by the increased plasma levels of IL-18 that correlate significantly with plasma levels of cell-free mtDNA (cf-mtDNA), a finding suggestive of pyroptosis in cfDNA release (51).
Active Secretion
Other than cell death, cells could actively secrete DNA in the form of extracellular vehicles (EVs), including exosomes and microparticles (microparticles). This cfDNA, present in membrane-bound EVs, may be protected from degradation by nucleases and can be released through the breakdown of EVs. Exosomes are small 30–100 nm vesicles released from the fusion of multivesicular bodies of endosomal origin with plasma membranes. The composition of exosomes includes nucleic acids, proteins, lipids and other metabolites (20). Through their biologically active components, including DNA, exosomes were shown to modulate various physiological and pathological processes (52, 53). A recent study by Fernando et al. (54), demonstrated that more than 93% of amplifiable plasma cfDNA is present in exosomes and the size of the majority of exosomal DNA is <200 bp, consistent with the size of cfDNA reported in plasma from patients with rheumatic disease. Alternatively, a study by Kahlert et al. (55) demonstrated that exosomes could also be the source of high molecular weight DNA (>10 kb). Interestingly, they could not detect any PCR amplifiable products in serum depleted of exosomes; also suggesting that the majority of cfDNA is present in exosomes. These findings suggest that the content of exosomes could vary depending on their cellular origin and the stimuli modulating their release from cells. Microparticles (MPs) or shedding vesicles, a small membrane-bound 100–1,000 nm vesicles can be released from apoptotic cells as blebs or can be actively secreted from living cells. DNA from MPs shows laddering pattern, resembling nucleolytic cleavage of apoptotic cells (56). In general, SLE patients have a higher frequency of pro-inflammatory MPs in the circulation (57, 58). MPs, in particular if released from activated platelets, could also be the source of cf-mtDNA (57, 59, 60). Importantly, upon platelet activation, mitochondria (either naked or localized within MPs) are extruded together with the bactericidal enzyme phospholipase A2, enabling digestion of membranes, allowing the pro-inflammatory mtDNA to escape unto the extracellular space (59). Indeed, levels of mtDNA increase concomitantly with levels of phospholipase A2 in platelet storage bags, with mtDNA levels being associated with adverse transfusion reactions (59).
Clearance of cfDNA
We have so far only considered the extrusion of cfDNA. Another important component in generating elevated levels of circulating cfDNA is impaired clearance mechanisms. A rapid clearance of cfDNA is critical to prevent not only inflammation, but also the potential development of autoimmunity toward DNA, as seen in SLE. Early studies on cfDNA clearance revealed that under physiological conditions, cfDNA is rapidly degraded by endonucleases and eliminated from the circulation through several organ systems, including the spleen, liver, and kidney (51, 61–63). Many factors can influence the ability of DNases to clear cfDNA, including whether the cfDNA is complexed with proteins, nucleosomes and/or antibodies, as well as whether the cfDNA is in free circulating form or encapsulated within membrane-enclosed particles, including exosomes, MPs and apoptotic bodies. Further, based on its intracellular origin, e.g., either nuclear or mitochondrial, cfDNA can exhibit different structural characteristics and stability (64–66).
Efficient degradation of free and protein-bound DNA i.e., nucleosomal DNA in plasma/serum is carried out by extracellular nuclease homologs, DNase I and DNase I-like III (DNase I L3), respectively (67, 68). While, DNase I efficiently cleaves free DNA, the digestion of nucleosomal DNA present in extracellular space and/or sequestered in MPs is majorly performed by DNase I L3 (69, 70). These specific activities of DNase I L3 are attributed to the presence of short, positively charged peptide in the carboxyl-terminal of DNase I L3 (71). Given the importance of DNase I and DNase I L3 in the degradation of circulating DNA, several studies have investigated the role of these nucleases in the context of SLE, a disease characterized by reduced ability to clear cellular debris. Abnormalities of DNase I activity reported in lupus include low serum DNase I activity particularly in patients with renal disease (72, 73), increased serum levels of DNase I inhibitors like G-actin that associated with the high titers of antinuclear antibodies (74), and novel mutations in the enzyme accompanied by elevated titers of anti-dsDNA antibodies (75, 76). Autoantibodies, including anti-dsDNA antibodies, can protect DNA from DNase I digestion (40, 77). Further, anti-DNase antibodies as observed in SLE, were shown to interfere with the enzyme activity, leading to low serum DNase activity (78). As briefly mentioned above, molecules interacting with the DNA may also affect the ability of the DNA to be recognized and degraded by DNase I. Cationic proteins like cathelicidin LL37, human neutrophil peptides and IL-26, protect cfDNA by forming insoluble aggregates through their charge interactions (79, 80) and mitochondrial transcription factor A (TFAM), a mitochondrial packaging protein is involved in the protection of mtDNA from nuclease degradation (81). In addition, defects in the cofactors that promote the DNase I activity (82–85) can also cause or perpetuate the decreased DNase I activity. For instance, complement component C1q, a deficiency of which is strongly associated with the genetic susceptibility of SLE, was shown to promote DNase I activity in degrading necrotic cell-derived chromatin (82). These cofactors likely promote the DNase I activity either by displacing DNA binding proteins from chromatin thus allowing the access to cleavage sites on DNA or by stabilizing DNase I on the target. With regards to DNase I L3, a homozygous loss-of-function variant mutation in DNASE1L3 gene, identified in several families of pediatric-onset SLE patients, was found to be associated with a higher frequency of anti-dsDNA antibodies and lupus nephritis (86). Another study reported two unique DNASE1L3 gene mutations in families with autosomal-recessive hypocomplementemic urticarial vasculitis syndrome (HUVS) (87). Incidentally, HUVS is more often associated with SLE, with >50% of HUVS patients often developing SLE (87). In this particular study, 3 of 5 children with HUVS carrying a homozygous frame-shift mutation in DNASE1L3 gene developed severe symptoms of SLE accompanied by anti-dsDNA antibodies (87). In addition to extracellular nucleases, TREX1, a major mammalian intracellular DNase with a preference for single-stranded DNA (ssDNA) substrates, can be involved in the degradation of cfDNA that translocate to the cytosol through carrier proteins. TREX1 is defective in the degradation of oxidized substrates such as oxidized mtDNA, which are preferentially from SLE neutrophils (38, 88). Hence, in conditions like lupus, the persistent presence of oxidized cf-mtDNA in the cytosol of immune cells can potentially activate inflammatory pathways. TREX1 variant mutations are also reported in SLE (89, 90). Finally, complement C1q, as well as other complement components also play an important role in opsonizing dead cells or extracellular debris for phagocytosis, thus efficiently removing cfDNA from the circulation (82, 91). Other opsonins, including serum amyloid P component (92), IgM (93, 94), C-reactive protein (CRP) (95, 96), and Mannan Binding Lectin (97) serve similar functions in clearance of dying cells, with deficiencies in either one of the opsonins commonly leading to accumulation of cfDNA (98).
Inflammatory Potential of cfDNA
Under physiological conditions, cfDNA is normally not inflammatory due to its rapid degradation as well as its inability to access intracellular DNA sensors. Consistent with this proposition, cfDNA failed to induce immune responses from plasmacytoid dendritic cells (pDCs), which are otherwise potent responders to microbial nucleic acids (79, 99, 100). Initially, this tolerance to self-DNA was thought to be due to the sequence composition differences between self- and microbial DNA. However, numerous studies have shown that self-DNA can be immunostimulatory provided it has access to intracellular DNA sensors (101–103). These carrier proteins, often elevated in inflammatory conditions (79, 104), can facilitate the uptake of DNA and also protect the DNA from degradation, thus promoting the induction of pro-inflammatory responses.
Based on Complexation With Carrier Proteins
In SLE, anti-dsDNA autoantibodies are one of the prominent carrier molecules of cfDNA into cells. Among others, anti-dsDNA antibodies, through their interaction with Fcγ receptor II (FcγRII) facilitate the receptor-mediated endocytosis of DNA into the TLR9-containing endosomal compartments of pDCs, eliciting a robust induction of interferon (IFN)-α (IFN-α), a cytokine markedly elevated in SLE and associated with disease activity (105). In an attempt to understand the role of the anti-dsDNA antibodies in promoting DNA-immune complex (IC)-mediated inflammation, Means et al. (104) undertook a series of experiment to dissect the role of the autoantibodies. Whereas, neutralization of FcgRII abrogated the immune reactivity of ICs, this could be rescued through a liposomal carrier. In all, these elegant experiments suggested that the primary role of IgG autoantibodies is to facilitate the entry of DNA into cells and are not obligatory for the immunogenicity of DNA (104). Later, a study by Lande et al. (79), provided evidence that cationic microbial peptides that are released in the context of NETosis, confer the immunogenicity to DNA-ICs by protecting DNA from nuclease degradation and facilitating uptake. Further, their data suggest that complexes of self-DNA-antimicrobial peptides (i.e., LL37) constitute the immunogenic core of the DNA-ICs in SLE, since anti-DNA antibodies alone were not sufficient to confer immunogenicity to DNA. LL37 is highly expressed in the circulation of SLE patients (106). LL37 stably binds to DNA through charge interactions between the unique cationic α-helical residues of LL37 and anionic phosphate backbone of DNA, thus forming insoluble DNA aggregates that are resistant to nuclease degradation (107). Antimicrobial peptides, including human neutrophil peptides, seem to function synergistically with LL37 to promote pDC activation by DNA-ICs (79). IL-26, a cationic amphipathic cytokine secreted by Th17 cells seems to stabilize and thereby promote the immunogenicity of extracellular DNA (80). IL-26, through its clusters of cationic charges, binds, and aggregates human DNA, thus forming insoluble particles that are resistant to extracellular degradation. Further, Meller et al. (80) showed that IL-26-DNA complexes could induce IFN-α secretions from pDCs in TLR9-dependent manner, and the internalization of complexes is mediated by the attachment of IL-26 cationic residues to heparan-sulfate proteoglycan on the cell membrane. Later, Poli et al. (108) showed that IL-26 shuttles different forms of extracellular DNA (genomic DNA, mitochondrial DNA, and NETs) into the cytosol of monocytes and promotes cyclic GMP-AMP synthase (cGAS)-STING- and inflammasome-dependent pro-inflammatory responses. Further, high levels of IL-26-DNA complexes have been found in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis (108), RA (109), psoriasis (110), and Crohn's disease (111). cfDNA can also be translocated into cell when bound to DNA-binding high mobility group box proteins, like high mobility group 1 (HMGB1) and TFAM that are released as DAMPs into extracellular space from damaged and dying cells and also during inflammation (112, 113). These molecules can specifically engage receptor for advanced glycation end products (RAGE) on pDCs to elicit TLR9-dependent IFN-α secretions for DNA ligands (114, 115). Further, DNA bound by these molecules can presumably undergo conformational changes that allow them to bind and activate a cytosolic DNA sensor, cGAS to initiate STING-mediated type-I IFNs (116).
Based on Oxidative Status of cfDNA
Another important aspect that can render cfDNA inflammatory is the presence of oxidized nucleotides, as DNA oxidation, whether bound to cationic peptides or not, is recognized as a danger associated molecular pattern. Further, the immunogenicity of DNA seems to depend on the degree of oxidation (117). Elevated levels of oxidized cfDNA is observed in various inflammatory conditions (118), supporting the role of oxidative status in promoting the inflammatory potential of DNA. DNA can be oxidized by free oxygen radicals or reactive oxygen species (ROS) that are generated during cell death processes (119), thus resulting in the release of oxidized cfDNA. We have shown that neutrophil cell death i.e., NETosis in response to ribonucleoprotein-containing ICs (RNP ICs), is dependent on mitochondrial ROS and the released NETs are enriched in oxidized interferogenic mtDNA (38). Alternatively, cfDNA can be oxidized by free oxygen radicals or ROS that are released by activated phagocytic cells at sites of inflammation (120). Oxidized DNA undergo structural changes due to the base modifications introduced by free radicals. As a result, the oxidized DNA is more resistant to nuclease degradation, as compared to unmodified DNA, thus making the oxidized DNA available to initiate pro-inflammatory responses (117). Although ROS can cause diverse arrays of DNA base modifications, a lesion to guanine residue identified as 8-hydroxy-2′-deoxyguanosine (8-OHdG) remains the most abundant and well-characterized DNA lesion (121). In fact, 8-OHdG was identified as a marker to determine the oxidative stress in various pathological conditions (122). Oxidized DNA was shown to activate various inflammatory pathways including, cGAS-STING, TLR9, and NLRP3 inflammasome pathways (38, 117, 123, 124). Further, oxidation by ROS increases the antigenicity of DNA, as demonstrated by the enhanced reactivity of SLE sera with oxidized DNA compared to native DNA (88, 125, 126). Consistent with the above observation, DNA contained within SLE immune complexes show an accumulation of 8-OHdG, indicating that oxygen radicals play a key role in the SLE pathology by modulating the antigenic nature of DNA in circulation (125, 126).
Based on Intracellular Origin
The intracellular origin of cfDNA, e.g., either from nucleus or mitochondria, can also influence the inflammatory potential of cfDNA. MtDNA, unlike nuclear DNA, is a potent inflammatory trigger (26, 38, 123, 127–130). MtDNA, due to its prokaryotic origin, holds many features that are similar to bacterial DNA, including the presence of a relatively high content of unmethylated CpG motifs, which are rarely observed in nuclear DNA (131). The unmethylated CpG motifs are of particular importance as TLR9, the only endolysosomal DNA-sensing receptor, has a unique specificity for unmethylated CpG DNA. As such, mtDNA was shown to activate neutrophils through TLR9 engagement (26, 103). Thus, unless coupled to carrier proteins, mtDNA, but not nuclear DNA, can be recognized as a danger-associated molecular pattern inducing pro-inflammation through TLR9. Collins et al. (129), reported that intra-articular injection of mtDNA induces arthritis in vivo, proposing a direct role of mtDNA extrusion in the disease pathogenesis of RA. Also, the oxidative status of mtDNA makes it highly inflammatory (123, 127). MtDNA, in contrast to nuclear DNA, is characterized by elevated basal levels of 8-OHdG, a marker of oxidative damage (66). The high content of oxidative damage in mtDNA is primarily attributed to the close proximity of mtDNA to ROS and relatively inefficient DNA repair mechanisms that can lead to the accumulation of DNA lesions (132, 133). We, as well as others, have shown that oxidative burst during NETosis can oxidize mtDNA and the released oxidized mtDNA by itself, or in complex with TFAM, can generate prominent induction of type I IFNs (38, 88, 117). Oxidative status of mtDNA renders its resistant to degradation by DNases such as TREX1, enabling it to activate multiple pro-inflammatory pathways (95). Oxidized mtDNA generated during programmed cell death is not limited to activate TLR9, but was shown to also engage the NRLP3 inflammasome, leading to the production of pro-inflammatory cytokines, IL-1β, and IL-18 (123, 127, 128, 130). MtDNA can also be recognized by cyclic GMP-AMP synthase (cGAS), a cytosolic dsDNA sensor to initiate a STING-IRF3-dependent pathway that in turn orchestrates the production of type I IFNs (134).
DNase I as a Therapeutic Agent
Considering the role of DNase I in promoting the clearance of autoantigenic DNA and perhaps even the destruction of DNA in ICs, the therapeutic potential of DNase I has been explored. Initial studies by Johnson and colleagues in the 1950s demonstrated that bovine pancreatic DNase I is harmless to humans and is a very poor antigen (135). It was used as a liquefaction agent to treat disease conditions associated with exudative responses to inflammation and infection (136). These studies by Johnson et al., (135, 136) laid basis for the first therapeutic application of bovine DNase I to treat SLE where patients injected with DNase I showed improvement in clinical symptoms, including a rapid fall in the ESR and also in the levels of autoantibodies specific to DNA-containing antigens but not in other autoantibodies. However, contrary to the initial reports, bovine DNase I was found to be antigenic and patients developed antibodies toward it 4–6 weeks following the administration, thus precluding the therapeutic usage of DNase I (137). Meanwhile, animal studies with recombinant mouse DNase I to treat SLE in NZB/W F1 hybrid mice prompted further interest in DNase as therapeutic agent for SLE (138). Mice treated with DNase I from the early phases of disease development (4 months of age) had a prolonged survival of about a month that was paralleled by the delay in the selective formation of anti-DNA antibodies but not in other autoantibodies or total IgG levels. Further, a concomitant rise in anti-DNA antibodies was observed in urine suggesting the destruction of DNA-containing ICs in kidney by DNase treatment. The DNase-treated mice developed less severe glomerulonephritis compared to control mice. Anti-DNase antibodies were formed in all DNase-treated mice, although they did not rise and remained low throughout the treatment period. The effect of these anti-DNase antibodies on the DNase function was not clear. In addition, it was not clear why sustained effect of DNase could not be seen and if more enzyme could have been beneficial given the rise in DNase inhibitors with inflammation. Nevertheless, much more dramatic changes in clinical course were observed in a group of mice treated with DNase for 3 weeks at the peak of their disease activity (7 months of age). DNase-treated mice displayed significantly lower levels of serum creatinine and less proteinuria compared to controls accompanied by remarkably less severe histopathological changes in the kidney, suggesting that DNA-containing ICs might still be involved in the advanced stage of disease course. Since animals were killed at the end of experiment, the long-term effect of DNase treatment is not clear. The study also suggests that destruction of established ICs might be an effective therapeutic option for SLE, thus preventing the stimulation of autoreactive B cells. Investigations into the human usage of DNase I in SLE were prompted by the discovery of recombinant human DNase I (rhDNase) as a potential agent to attenuate the sputum viscosity in cystic fibrosis patients (139). RhDNase administered by an inhalation method was found to be safe and well-tolerated in patients with cystic fibrosis (140–142). Subsequently, a 40-day, phase 1b placebo-controlled clinical trial conducted in SLE patients with lupus nephritis showed that rhDNase I is safe in SLE patients, and that the treatment was not associated with the development of anti-rhDNase antibodies. Patients were given an initial single dose (25 or 125 μg/kg) of intravenous injection followed by 10 subcutaneous injections of rhDNAse I. However, the treatment did not result in a significant improvement in markers of disease activity including the levels of serum dsDNA antibodies, levels of complement components C3 and C4, levels of circulating immune complexes, serum cytokines levels (IL-6, IL-10, and TNF-α) and there was no change in the immune complex deposition in skin biopsies pre- and post-treatment. This lack of clinical efficacy could partly be explained by the observation that rhDNase at bioactive serum concentrations was maintained for only a few hours at both doses, and thus future studies should investigate the effect of dose or regimen that allows the maintenance of rhDNase at concentrations capable of serum hydrolytic activity for prolonged time periods. The absence of neutralizing antibodies to rhDNase unlike to bovine DNase I suggests the potential possibility for long-term therapy (143).
Cell-Free DNA in Rheumatic Diseases
Systemic Lupus Erythematosus
SLE is a prototype autoimmune disease that effects multiple tissues and organs systems, including skin, joints, and kidney. Though known to have both environmental and genetic components, the etiology of the disease is not fully understood. Among several markers implicated in the SLE pathogenesis, the role of dsDNA is especially interesting. In the majority of the SLE literature, the pathological role of dsDNA in the disease pathogenesis is discussed from the perspective of dsDNA-containing immune complexes, frequently found in SLE patients, partaking in the development of lupus nephritis. The prevailing idea is that these immune complexes activate complement factors and Fc gamma receptor-bearing cells to initiate pathological inflammatory responses. In fact, anti-dsDNA antibodies are listed among the classification criteria for diagnosing SLE in accordance with the American College of Rheumatology, and the Systemic Lupus International Collaborating Clinics classification criteria (144, 145). Other than the diagnostic utility, increasing research into the potential role of DNA sensing receptors (146) to initiate multiple pro-inflammatory pathways resulting in the secretion of SLE-associated type I interferons, brought dsDNA back into the center stage of the SLE pathogenesis. Subsequently, the interest to detect, quantify and/or characterize cfDNA in plasma/sera of SLE patients emerged. In the section below, we will present a glimpse of how the research into the field of cfDNA has evolved in SLE, with initial studies mainly focusing on the detection and quantification of cfDNA followed by studies to associate cfDNA levels to disease activity, progression and/or for monitoring treatment response. Finally, we will touch upon the application of advanced sequencing techniques to determine characteristics of plasma DNA, all with a common goal to explore the diagnostic and prognostic potential of cfDNA for SLE. Major findings of cfDNA in SLE as discussed in the following sections are summarized and listed in Table 1.
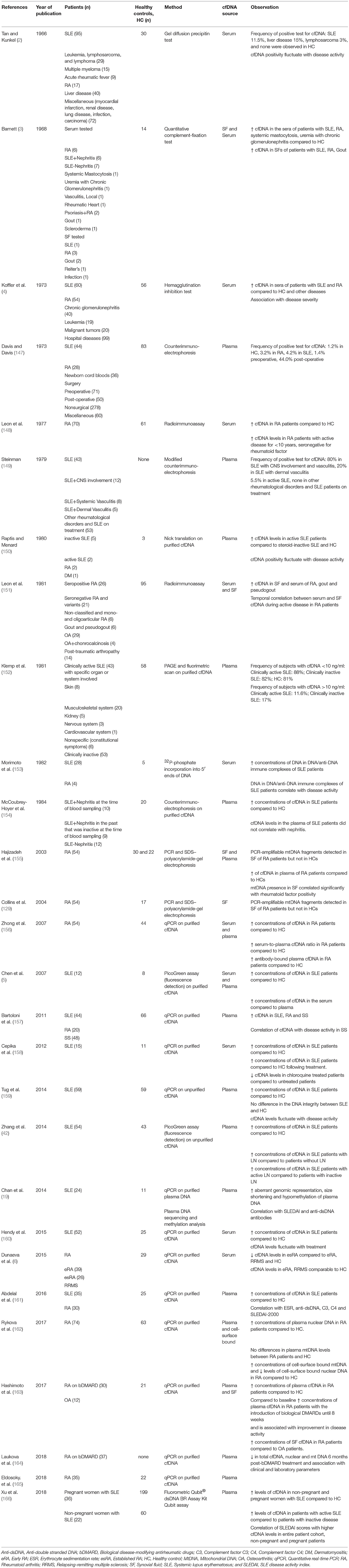
Table 1. Cell-free DNA research in Systemic lupus erythematosus and Rheumatoid arthritis.
Early Reports of cfDNA Detection in SLE
In 1948, Mandel and Metais were the first to report on the presence of cfDNA in human plasma (1). However, it was not until the discovery of high levels of cfDNA in SLE patients in 1966 by Tan et al. (2), that the interest in cfDNA as a potential biomarker for autoimmune diseases started. Tan et al. (2) used SLE sera with a precipitating antibody to DNA in a gel diffusion method to detect the presence of DNA in pathological sera. By this method, native (ds) DNA could be detected in sera of some SLE patients (11/95) and in patients with other disease conditions but not in the sera of healthy controls. Gel precipitation has a detection limit of 1 μg/ml; hence it is possible that samples with lower levels of cfDNA, especially of healthy controls could have gone undetected. Later, in 1968 Barnett used complement-fixation, to demonstrate the presence of cfDNA in normal and pathologic human sera and synovial fluids (3). Sera of patients positive for precipitating antibodies to DNA and having complement-fixing antibodies to DNA were used to detect DNA in human samples. Unlike Tan et al. (2), small but detectable amounts of DNA could be measured in normal sera. However, the levels of cfDNA were markedly elevated in pathologic sera and synovial fluid. In 1973, Koffler et al. (4), by hemagglutinin inhibition test, reported that about 50% of SLE sera are positive for the presence of single-stranded DNA (ssDNA) as an antigen. In contrast, ssDNA appeared infrequently in the sera of healthy controls (4% incidence). Almost at the same time, Davis and Davis (147), used a counterimmunoelectrophoresis (CIE) method to detect cfDNA in the plasma of patients with various illnesses including SLE. The number of positive samples in SLE patients (2/47, 4.2%) were not significantly different from healthy controls (1/83, 1.2%).
The Controversy of Detection Methods and Serum vs. Plasma Utility for cfDNA Measurement
At the time when the clinical importance of cfDNA was being actively pursued, conflicting data on the detection of cfDNA in the circulation of healthy individuals led to controversies on the detection methods and on the significance of serum and plasma cfDNA levels for disease. In addition, a consensus was yet to be reached on the levels of cfDNA in plasma for normal vs. pathological scenarios. Davis and Davis (147), with their method of CIE, were the first to hint that cfDNA in sera, at least in part, could be an artifact of the method, i.e., DNA that is sporadically released into sera during blood clotting. This observation was confirmed by Steinman (149) in an elegant study that employed four different techniques with improved sensitivities and/or specificities to detect cfDNA in normal serum and plasma samples. In that study, plasma cfDNA remained undetectable by all four methods including a highly sensitive CIE method. Further, it was reported that the cfDNA measurements in plasma by ethidium bromide and diphenylamine assays, are mainly due to interfering substances (non-DNase sensitive substances) in plasma rather than true DNA. In contrast, cfDNA could be detected in serum samples by a CIE method. Based on these findings, it was concluded that cfDNA detection in plasma is pathological and levels >50 ng/ml for dsDNA and 100 ng/ml for ssDNA in plasma are abnormal. These findings were later confirmed by Dennin (167), who, by employing CsCl-buoyant density centrifugation found that in healthy adults the concentration of plasma cfDNA ranged from 3 to 11 ng/ml (167). In more recent times, Chen et al. (5), showed that serum samples from lupus patients had higher levels of cfDNA than the corresponding plasma samples by using a fluorochrome PicoGreen assay on purified cfDNA. A higher sera-to-plasma cfDNA ratio suggests that white blood cells from SLE patients are fragile and/or damaged and thus are prone to undergo disruption during coagulation releasing DNA. These studies strengthened the view that cfDNA levels from serum samples should be interpreted with caution especially when employing sensitive detection methods and if possible, should be replaced by carefully collected plasma samples.
cfDNA and Disease Activity in SLE
The majority of studies reported an association between cfDNA and disease activity in lupus. However, there are a few studies with conflicting data on the link between cfDNA and disease activity (152, 154, 160, 168). Early reports by serial sampling of cfDNA from sera demonstrated that an increased appearance of cfDNA in SLE patients is associated with the exacerbation of disease and interestingly becomes undetectable following the clinical improvement (2, 4, 153). Koffler et al. (4) in a serial study of 18 SLE patients, followed for periods of 6–51 months observed that in certain patients, cfDNA (ssDNA) can reach extreme high levels in the range of 125–250 μg/ml. Similar to Tan et al. (2), ssDNA antigen appearance was associated with episodes of clinical exacerbations and patients with prolonged presence of ssDNA (4–8 months) reported progressive renal disease (4). Evidence for in vivo antigen-antibody formation was demonstrated where ssDNA antigen appearance alternated or occurred simultaneously with anti-ssDNA antibodies. Morimoto et al. (153) observed a highly significant correlation between DNA derived from circulating immune complexes and disease activity index in SLE patients. A serial study of two patients demonstrated that the levels of cfDNA remain elevated (52 ng/ml) during the episodes of active disease and glomerulonephritis (100 ng/ml) and return to lower levels during clinical remission (10 ng/ml) and further becoming non-detectable with treatment (<1 ng/ml). Steinman (149), observed that majority of SLE patients (80%) with central nervous system involvement and/or systemic vasculitis have a persistent presence of cfDNA in plasma. Longitudinal studies in four SLE patients also confirmed this association with CNS and/or vasculitis, where only episodes associated with these manifestations are characterized by cfDNA appearance. Raptis et al. (150), showed that cfDNA exists in much higher levels in the plasma of untreated SLE patients with active disease compared to plasma of patients with corticosteroid-induced disease remission and healthy controls. A serial determination of plasma in SLE demonstrated that cfDNA levels are elevated at disease onset and diminished considerably when disease has stabilized accompanied by a concomitant decrease in the serum dsDNA binding activity. Tug et al. (159), though not observing a clear link between SLE disease activity index (SLEDAI) and cfDNA concentrations, found a significant correlation between fluctuations in cfDNA levels and transition from remission to deteriorating status. This study suggested that changes in disease state, in particular deterioration status, could be reflected by fluctuations in cfDNA (159). Zhang et al., (42), investigated the proposition that elevated levels of plasma cfDNA in SLE are related to lupus nephritis (LN). cfDNA concentrations were significantly higher in SLE patients with LN than in patients without LN and further subgrouping analysis revealed that patients with active LN had significantly elevated cfDNA concentrations compared to patients with inactive LN. Studies in SLE patients also reported that cfDNA levels correlate significantly with SLEDAI (53, 166). A recent study by Xu et al. (166), showed that median cfDNA levels are significantly higher in SLE patients with active disease compared to patients with inactive disease.
cfDNA and Treatment Response in SLE
Considering that inflammation can cause the release of cfDNA and the fact that cfDNA itself can perpetuate ongoing inflammation leading to a vicious feedback loop, a drug treatment that reduces systemic inflammation or specifically antagonizes receptors that recognize DNA, would likely affect cfDNA levels as well. Currently, literature demonstrating the dynamics of cfDNA levels in SLE patients with treatment is sparse, but promising. Hendy et al. (160) observed that SLE patients following specific therapy with cytotoxic drugs show a significant reduction in serum cfDNA levels compared to pre-treatment and this was accompanied by a concomitant reduction in anti-dsDNA levels and anti-nucleosome antibodies. Cepika et al. (158) showed that sera cfDNA levels in SLE patients decrease significantly following treatment with chloroquine, a drug known to block the DNA-sensing TLR9 pro-inflammatory pathway. Although not significant, corticosteroid (CS) treatment also decreased serum cfDNA levels, suggesting that reduction in systemic inflammation decreased cfDNA levels. In contrast, in another study (157), the type of treatment, CS and/or immunosuppressive (IS) did not seem to affect the levels of plasma cfDNA in SLE patients.
cfDNA and Association With Inflammatory Markers in SLE
cfDNA has been receiving increasing attention as an inflammatory marker with the advent of new mechanistic studies highlighting the role of DNA sensing receptors in SLE pathogenesis (146). Hence, attempts were made to evaluate the relationship of cfDNA with other known markers of inflammation. Overall, although limited by the number of studies, it can be concluded that cfDNA levels in SLE associate well with several markers of inflammation. One study found a significant positive correlation between serum cfDNA levels and a generic marker of inflammation, CRP (149). NETs released from neutrophils and low-density granules (LDGs), could also be the source of cfDNA in SLE. Consistent with this assumption, Zhang et al. (42) found a highly significant positive correlation between levels of plasma cfDNA and the percentage of LDGs and neutrophil levels, suggesting that LDGs and neutrophils, through NET formation, can contribute to cfDNA in SLE patients. However, the authors never confirmed whether cfDNA levels correlated with presence of circulating NETs in these patients. In addition to the abnormal production of NETs, impaired clearance of NETs can also lead to elevated levels of cfDNA in SLE. Further analysis demonstrated a significantly lower DNase I activity in SLE patients compared to healthy controls, although no significant correlation could be observed between DNase I activity and cfDNA levels (42). Studies found contrasting associations for the levels of complement factors and cfDNA in SLE patients. Tug et al. (159) and Abdelal et al. (161) found a positive correlation between the levels of complement factors and plasma cfDNA levels in SLE patients. This observation was surprising given that complement consumption is commonly seen in active SLE patients. However, the authors speculated that this positive relationship between complement levels and cfDNA might be due to the increase in complement levels as a part of an acute phase response that can obscure the complement consumption. In contrast, a study by Hendy et al. (160) found a significant negative correlation between levels of serum cfDNA and C3 in SLE patients.
Based on the central role of anti-nuclear antibodies in SLE, and the possibility that high levels of cfDNA in circulation might initiate and/or perpetuate the production of anti-dsDNA antibodies, a positive correlation between cfDNA levels and anti-dsDNA antibodies is expected. However, in contrast, studies reported either a lack of or negative correlation between cfDNA and anti-dsDNA antibody levels. McCoubrey et al. (154) and Hendy et al. (160) observed an inverse correlation between levels of plasma DNA in SLE patients and titers of antibody for DNA. In another study (157), cfDNA in plasma did not correlate with titers of anti-dsDNA antibodies. This situation can likely arise due to the accelerated tissue deposition of immune complexes during active disease. A serial sampling might provide a better picture of the association between levels of cfDNA and anti-dsDNA antibodies.
Qualitative Features of Plasma cfDNA in SLE
The SLE genome exhibits distinct qualitative features such as, a higher frequency of CpG dinucleotides (169), increased hypomethylation (170, 171), and increased oxidation (172). Interestingly, all these qualitative features promote the immune stimulatory properties of DNA to be recognized by DNA sensing receptors and subsequently induction of pro-inflammatory responses. Hence, it appears that cfDNA released from SLE patients is inherently proinflammatory and, therefore analyzing the qualitative changes of cfDNA might provide more in-depth understanding on the association of cfDNA with SLE pathogenesis and eventually it's utility as a biomarker for SLE. However, in SLE literature only one study has been reported investigating the qualitative changes of plasma cfDNA (19), warranting the need for more studies in this area of cfDNA research. Chan et al. (19) by a parallel genomic and methylomic sequencing observed various abnormalities in plasma DNA from SLE patients including aberrant measured genomic representations (MGRs), size shortening, fragments of <115 bp in size, and hypomethylation. Very interestingly all these plasma DNA abnormalities, as discussed below, were seemed to be modulated by anti-dsDNA antibodies, pathological circulating markers of SLE. It was observed that the frequency of aberrant MGRs correlated with the levels of serum anti-dsDNA antibodies. Subsequent experiments demonstrated that aberrant MGRs DNAs had increased binding affinity to anti-dsDNA antibodies. Thus, given the ability of IgG to protect DNA from subsequent degradation, DNA molecules with increased dsDNA antibody binding, such as aberrant MGR DNA, may have increased representation in cfDNA analyses due to their reduced clearance. The percentage of plasma DNA shortening in SLE correlated positively with SLEDAI and the anti-dsDNA antibody, suggesting that there is either an increased release or decreased clearance of short fragments in SLE. This observation aligns well with the evidence of increased apoptosis as well as defective clearance in SLE patients (17, 173). Further, IgG-bound DNA was enriched in short fragments (<115 bp), strengthening the proposition that IgG antibody has a preferential binding to short DNA fragments, that in turn protect them from degradation. Plasma DNA molecules from active SLE patients were more hypomethylated compared to inactive SLE and healthy controls. In addition, the degree of hypomethylation correlated with SLEDAI and anti-dsDNA antibody levels. Based on size distribution profiles and methylation density, it was suggested that plasma DNA in SLE patients exhibit size shortening with hypomethylation and are protected from degradation by antibody-binding.
Summary of cfDNA Research in SLE
Overall, SLE patients show elevated levels of cfDNA that fluctuate concomitantly with disease activity, inflammatory markers and to some extent with therapeutic interventions. The association of cfDNA with existing SLE diagnostic marker, anti-dsDNA antibodies, is unclear with conflicting data, suggesting that the dynamics of cfDNA in SLE is independent of anti-dsDNA antibodies. cfDNA also seems to reflect the genomic modifications characteristic of SLE disease. However, there are many factors that needs to be addressed in order to establish cfDNA as a biomarker for SLE from a clinical standpoint. cfDNA quantification as a diagnostic marker for SLE is promising but lacks clinical specificity since it is detected in other diseases albeit at lower levels. Findings from epigenomic research on cfDNA are critical in establishing the clinical biomarker specificity of cfDNA for SLE. Mechanistic studies of SLE pathogenesis have demonstrated an enhanced and preferential release of proinflammatory oxidized mitochondrial DNA into circulation by SLE neutrophils (38). Interestingly, none of the SLE-cfDNA studies have analyzed mitochondrial cfDNA in SLE patients. Given the preferential release of mitochondrial DNA in SLE, the quantification and characterization of mitochondrial DNA, and its relative abundance to nuclear DNA, will likely provide disease specific information on cfDNA in SLE. Apart from being a diagnostic marker, cfDNA might also function as a broad disease management marker for SLE, such as a marker of prognosis for remission, flare and/or treatment response. However, it requires a rigorous evaluation of cfDNA in longitudinal studies with large cohorts of patients and careful comparison with existing inflammatory and clinical makers of disease.
Rheumatoid Arthritis
RA is, similar to SLE, an autoimmune rheumatic disease primarily affecting joints, with severe and disabling erosion (174). Though not as frequent as in SLE, also RA patients have been reported to develop anti-DNA antibodies (175). This has significance given the ability of DNA immune complexes to engage both antigen receptor and TLR9 simultaneously, inducing B cell proliferation and antibody secretion as seen in rheumatoid factor (RF) expressing B cells (176). Further, mtDNA and/or oxidized nucleic acid material was able to induce arthritis in mice (129). In addition to the above-mentioned evidence of DNA in RA, the potential release of cfDNA in general during inflammation, have led to many studies exploring the potential of cfDNA as a biomarker of diagnosis, disease activity and progression, and/or treatment response in RA. Major findings of cfDNA studies in RA are summarized and listed in Table 1.
cfDNA and RA
Similar to SLE, the majority of studies reported elevated levels of circulating cfDNA in RA patients compared to controls (3, 4, 155, 156, 161–165). Cell-free nuclear and mtDNA content of synovial fluid in RA patients was reported to be many folds higher than corresponding plasma levels and was exclusive to patients, suggesting that cfDNA release in RA patients is mainly localized to the joints and is pathologically relevant (151, 155, 163). Previous investigations of Rykova et al. (162) in cancer, demonstrated that cfDNA levels in whole blood are a result of continuous exchange between free DNA and cell-surface bound (csb) DNA, and that free DNA binds to cells via direct binding to cell surface proteins and through plasma proteins. In their study on RA patients, Rykova et al. (162) found contrasting dynamics of free- and csb-DNA forms for nuclear and mtDNA compared to controls. While plasma mtDNA levels were not significantly different between RA and healthy controls, csb-mtDNA levels were significantly elevated in RA patients. In contrast, plasma nuclear DNA levels were significantly elevated in RA patients, with a significant decrease in the csb-nuclear DNA levels. The finding on csb-mtDNA is interesting given its role in inflammation, and the decrease in the levels of csb-nuclear DNA could be due to disease-induced changes in the composition of circulating nuclear DNA-protein complexes in RA patients, that might have influenced the nuclear DNA binding to cells (162). In contrast to prior studies, Dunaeva et al. (6) reported that serum cfDNA levels were comparable between patients with early RA (eRA) and healthy controls. Furthermore, patients with established disease (esRA) have significantly lower levels of serum cfDNA compared to patients with eRA and healthy controls. Levels of cfDNA in serum did not correlate with serum DNase activities, suggesting that lower cfDNA levels in esRA is not due to elevated DNase activities. Lower levels of cfDNA in esRA patients could be due to treatment with disease-modifying drugs that are known to reduce systemic inflammation and subsequent cfDNA release. Nevertheless, this study suggested that serum cfDNA can be used as a disease progression marker in RA patients (177).
High Serum-to-Plasma cfDNA Ratio in RA
Consistent with the prior observations in SLE, RA patients also exhibited a higher serum-to-plasma cfDNA ratio compared to healthy controls (156), implying that leukocytes in pathological conditions, in general, have an altered susceptibility to undergo cellular death and cfDNA release upon clotting. In an independent study authors evaluated if NETs are the source of higher cfDNA levels in sera of RA patients, based on their in vitro findings that neutrophils from RA patients were prone to undergo excessive NETosis. Accordingly, analysis of sera and corresponding plasma samples revealed that, sera from RA patients and not the plasma, have increased concentrations of cfDNA and NET-derived components compared to healthy controls. This observation suggested that coagulation during serum preparation triggers an extensive NETosis in RA neutrophils releasing NETs that in turn contribute to the elevated levels of cfDNA in serum (41).
cfDNA and Serological Parameters in RA
Evidence for the association of cfDNA with serological parameters of RA, including RF and anti-citrullinated protein antibodies (ACPA), is sparse, with conflicting data. Leon et al. demonstrated that higher levels of serum cfDNA in RA patients correlate with seronegativity (148). Consistent with the susceptibility of mtDNA to undergo oxidation, Hajizadeh et al. (155) found that patients that were positive for mtDNA in SF also had high levels of 8-OHdG, a marker of oxidative status. In contrast to Leon et al. (148) both cell-free mtDNA positivity and levels of 8-OHdG correlated significantly with rheumatoid factor positivity. Rykova et al. (162) found a negative correlation between ACPA and plasma mtDNA levels in RA patients. These data suggest that cfDNA and ACPA/RF could be independent circulating makers of RA development and their combination might result in an improved diagnostic tool for RA.
cfDNA and Disease Activity in RA
In a majority of studies, cfDNA levels in RA patients were reported to be associated with disease activity and markers of inflammation (148, 151, 156, 161). DNA levels measured in paired samples of serum and synovial fluid (SF) from patients with arthritides [seropositive RA, seronegative RA variants including psoriatic arthropathy, ankylosing spondylitis and juvenile RA, gout, pseudogout, osteoarthritis (OA), and post-traumatic arthritis (TRA)], demonstrated that the RA patients, as well as patients with gout and pseudogout had the highest levels of cfDNA in SF and serum. Very low levels of cfDNA were seen in patients with OA and TRA. A serial determination of cfDNA revealed a temporal correlation between the elevated levels of DNA in serum and SF and parameters of disease activity and inflammation in some RA patients (151). Abdelal et al. (161) showed that elevated levels of plasma cfDNA in RA patients correlated significantly with erythrocyte sedimentation rate (ESR), CRP and Disease Activity Score-28 (DAS28, suggesting that plasma cfDNA can be a potential marker of disease activity in RA patients. In contrast, Hajizadeh et al. (155) did not find any obvious association between cell-free mtDNA in SF and markers of disease activity and severity, including extra-articular manifestations, erosion, leukocyte counts, CRP levels, or disease duration. Similar to SLE, the majority of plasma cfDNA in RA patients was found to be associated with antibody (156), suggesting the role of DNA-anti-dsDNA immune complexes in RA pathogenesis. More recently, Eldosky et al. (165) found that in RA patients, cfDNA levels are significantly higher in active disease group compared to control group, while the levels are comparable between remission and control groups. ROC curve analysis revealed a sensitivity and specificity of 86 and 84%, to differentiate active and remission states of RA. Correlation analysis in all RA patients showed that cfDNA levels correlate significantly with DAS28-ESR, a marker of disease activity. In addition, cfDNA showed an inverse correlation with absolute lymphocyte count, suggesting a possible role of enhanced lymphocyte death in RA patients (178) including NETosis, in the formation of cfDNA. Overall, the study suggested that cfDNA can be potential marker of disease activity progression in RA (165).
cfDNA and Treatment Response in RA
In RA patients, changes in cfDNA levels following the treatment with biological disease-modifying anti-rheumatic drugs (bDMARDs), were associated with the improvement in disease activity. Hashimoto et al. (163) reported that in a subset of RA patients, plasma cfDNA can be a predictor of early therapeutic response of bDMARDs. Specifically, a cfDNA elevation at 8 weeks can predict the therapeutic response of biological bDMARDs from 12 to 24 weeks. At baseline, plasma cfDNA levels in RA patients were significantly higher than the healthy controls. After the introduction of DMARDs, the cfDNA increase at 8 weeks was associated with a concomitant improvement in average SDAI score and disease activity. A study by Laukova et al. (164), further investigated the effect of bDMARDs on plasma cfDNA with regards to it subcellular origin, nuclear, and mitochondrial, respectively. This study, in contrast to Hashimoto et al. (163) demonstrated that plasma cfDNA decreases following dDMARDs therapy. Plasma samples were analyzed for cfDNA content from RA patients at baseline as well as 3- and 6-months post-bDMARD treatment. There was a clear improvement in clinical (DAS28, swollen and tender joints) and laboratory parameters (ESR, CRP) in all patients by 3 months following bDMARD therapy. Although the levels of total cfDNA, nuclear, and mitochondrial cfDNA started to decrease by 3 months, significant differences from baseline were observed only 6 months post-treatment, where the concentrations decreased by half. These observations were in contrast to Hashimoto et al. (163), where an increase in plasma cfDNA was observed until 8 weeks post-bDMARD treatment. However, in the absence of additional time points between baseline and 3 months, it is not possible to rule out the dynamics of cfDNA levels in the Laukova et al (164), study as well. Laukova et al. (164) further observed that in RA patients, lower concentration of total cfDNA, correlated positively with DAS28, ESR and CRP. No differences were found between good responders and moderate responders in the levels of total cfDNA, nuclear and mitochondrial cfDNA pre- and post-bDMARDs treatment. In good responders, the concentration of total cfDNA and nuclear cfDNA decreased significantly 6 months from the baseline. Since the decrease in cfDNA levels following bDMARDs therapy is much slower in comparison to other routinely measured laboratory and clinical parameters, the decrease in cfDNA was interpreted as a consequence of reduced inflammation due to treatment. On similar lines, Zhong et al. (156), observed that plasma cfDNA levels were markedly changed (either increase or decrease) in 7 out of 10 patients, 1 h after infusion of infliximab. In another study, a group of RA patients treated with rituximab showed a tendency of lower cell-surface bound mtDNA levels than a group treated with methotrexate and etoricoxib, although the difference did not reach statistical significance (156).
Summary of cfDNA Research in RA
To summarize, RA patients in general have elevated levels of circulating cfDNA and in SF, cfDNA is found at concentrations many times higher than in circulation, suggesting the role of localized inflammation in the release of cfDNA. Further, the detection of cfDNA exclusively in the joints (SF) of RA, strengthens the arthritic potential of cfDNA. Association of cfDNA with seropositivity is unclear with conflicting results and, the quantitative changes in cfDNA seem to reflect the disease progression and treatment response in RA. Data suggest that dynamics of cfDNA in RA patients is independent of existing diagnostic markers, ACPA and RF, and cfDNA in combination with ACPA/RF might form an improved diagnostic tool. Although, the role of mtDNA in arthritis is demonstrated (129), only two studies have quantified the levels of mtDNA in the circulation of RA patients, highlighting the need for more research to explore the role of circulating mtDNA as a biomarker for RA. While, the studies demonstrate the biomarker potential of cfDNA in RA, longitudinal studies with large cohorts of patients are needed to capture the dynamics of cfDNA in RA with disease progression and drug effects.
Comparison of cfDNA Observations Between SLE and RA
Considering the many technical and sampling variations in different studies, direct comparisons of cfDNA levels between SLE and RA patients can only be made from studies where both patient cohorts were analyzed simultaneously. In general, SLE patients have higher concentrations of sera cfDNA (ssDNA) along with higher positivity for anti-ssDNA antibodies as compared to patients with RA (4). The association of cfDNA levels with serological parameters in both diseases, e.g., anti-dsDNA in SLE (154, 160) and ACPA and RF in RA (148, 155, 162), suggest that cfDNA may reflect common processes involved in both diseases, including inflammation and cell death. RA patients exhibit increased levels of mtDNA in plasma and SF (155, 162). Given recent studies implicating mitochondrial extrusion in the SLE pathogenesis (38, 88), we expect upcoming studies to demonstrate increased mtDNA levels also in SLE patients. cfDNA from both RA and SLE exhibit increased oxidation (125, 155), possibly a consequence of inflammation. Contrasting dynamics of cfDNA was observed with treatment for SLE and RA with cfDNA levels decreasing in SLE patients upon treatment (158, 160). In RA, however, cfDNA levels initially increase, where after they decrease at later time-points (163, 164). Finally, cfDNA levels associate with markers of disease activity and inflammation in both RA and SLE (2, 4, 42, 148–151, 153, 156–158, 161).
Recent Advances in Analysis of cfDNA in Other Medical Fields
cfDNA is not only used for chronic inflammatory rheumatic diseases, but also in several other conditions, including non-invasive prenatal testing to detect fetal aneuploidies (179), cancers (180), as an early marker of allograft rejection and graft damage (181) and for tracking microbial infection, including the detection of oncogenic viral DNA (182). Methodological advancements in the analysis of cfDNA are comprehensively addressed in recent reviews (183, 184). In brief, emerging targeted approaches to screen for specific mutations of cfDNA include; digital droplet PCR (ddPCR), a highly sensitive method that allows the identification of rare targets based on the partitioning of samples into water-into-oil droplets; BEAMing (beads, emulsification, amplification, and magnetics) a combination of emulsion PCR and flow cytometry to achieve higher sensitivity; next generation sequencing (NGS), a powerful technique that allows the screening of both targeted and untargeted mutations; and methylated CpG tandem amplification and sequencing (MCTA-Seq) to identify genome-wide hypermethylated CpG regions. Other cutting-edge technologies that allow personalized therapies include targeted plasma re-sequencing (TAm-Seq) and personalized analysis of rearranged ends (PARE) which is based on the identification of disease-specific somatic rearrangements. Fluorescence correlation spectroscopy (FCS), a method based on the fluctuations of fluorescence due to the Brownian movement of fluorescence molecules, allows a rapid and sensitive detection of single molecules, including the size determination of DNA (185). FCS can effectively complement existing methods of cfDNA detection in autoimmune diseases.
Closing Remarks and Future Directions
The relevance of DNA to the disease pathology of lupus is undisputed and, there is an increasing attention in RA as well. In contrast to non-specific markers of inflammation, cfDNA can be pathologically relevant to autoimmune rheumatic diseases given the role of DNA-sensing receptors in inflammation and autoimmunity. cfDNA allows a rapid, easy, non-invasive and repetitive method of sampling. A combination of these biological features and technical feasibility of sampling, position cfDNA as a potential biomarker of enormous utility for autoimmune rheumatic diseases. However, there are many issues that needs to be addressed toward this goal. It should be acknowledged that the underlying heterogeneity of autoimmune disease by itself, can contribute to a considerable amount of variation in the levels of cfDNA, and hence adequate measures must be taken to minimize the variations at the level of cfDNA sampling. Notably, there is a lack of uniformity on the type of sample (plasma/serum/synovial fluid), methods of sample collection/processing, free or cell-surface bound DNA, cfDNA extraction and cfDNA quantification, and also in the presentation and interpretation of quantitative cfDNA findings. Additional, complexity is brought by the advent of qualitative research of cfDNA, which needs to be standardized as well. Given this lack of homogeneity, it is not surprising that consensus is yet to be reached on cfDNA levels in healthy individuals. Further, the majority of studies have been cross-sectional, and were limited by sample sizes. However, in order to fully understand the biomarker potential of cfDNA in autoimmune rheumatic diseases, a systematic scientific framework with collaborative efforts is needed to conduct large, multicenter trials with prospective analyses.
Author Contributions
BD and CL conceptualized the manuscript idea. BD conducted the literature search and drafted the manuscript. CL provided critical revisions. Both BD and CL edited the manuscript.
Funding
This work was supported by the Lupus Research Alliance (519414).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mandel P, Metais P. Comptes rendus des seances de la Societe de biologie et de ses filiales. C R Seances Soc Biol Fil. (1948) 142:241–3.
2. Tan EM, Kunkel HG. Characteristics of a soluble nuclear antigen precipitating with sera of patients with systemic lupus erythematosus. J Immunol. (1966) 96:464–71.
3. Barnett EV. Detection of nuclear antigens (DNA) in normal and pathologic human fluids by quantitative complement fixation. Arthritis Rheum. (1968) 11:407–17. doi: 10.1002/art.1780110306
4. Koffler D, Agnello V, Winchester R, Kunkel HG. The occurrence of single-stranded DNA in the serum of patients with systemic lupus erythematosus and other diseases. J Clin Invest. (1973) 52:198–204. doi: 10.1172/JCI107165
5. Chen JA, Meister S, Urbonaviciute V, Rodel F, Wilhelm S, Kalden JR, et al. Sensitive detection of plasma/serum DNA in patients with systemic lupus erythematosus. Autoimmunity. (2007) 40:307–10. doi: 10.1080/08916930701356317
6. Dunaeva M, Buddingh BC, Toes RE, Luime JJ, Lubberts E, Pruijn GJ. Decreased serum cell-free DNA levels in rheumatoid arthritis. Auto Immun Highlights. (2015) 6:23–30. doi: 10.1007/s13317-015-0066-6
7. Aucamp J, Bronkhorst AJ, Badenhorst CPS, Pretorius PJ. The diverse origins of circulating cell-free DNA in the human body: a critical re-evaluation of the literature. Biol Rev Camb Philos Soc. (2018) 93:1649–83. doi: 10.1111/brv.12413
8. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. (2007) 35:495–516. doi: 10.1080/01926230701320337
9. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. (1972) 26:239–57. doi: 10.1038/bjc.1972.33
10. Fadok VA, Bratton DL, Frasch SC, Warner ML, Henson PM. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ. (1998) 5:551–62. doi: 10.1038/sj.cdd.4400404
11. Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. (2000) 407:784–8. doi: 10.1038/35037722
12. Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. (2002) 2:965–75. doi: 10.1038/nri957
13. Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. (2004) 304:1147–50. doi: 10.1126/science.1094359
14. Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. (2010) 6:280–9. doi: 10.1038/nrrheum.2010.46
15. Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. (1980) 284:555–6. doi: 10.1038/284555a0
16. Giacona MB, Ruben GC, Iczkowski KA, Roos TB, Porter DM, Sorenson GD. Cell-free DNA in human blood plasma: length measurements in patients with pancreatic cancer and healthy controls. Pancreas. (1998) 17:89–97. doi: 10.1097/00006676-199807000-00012
17. Courtney PA, Crockard AD, Williamson K, Irvine AE, Kennedy RJ, Bell AL. Increased apoptotic peripheral blood neutrophils in systemic lupus erythematosus: relations with disease activity, antibodies to double stranded DNA, and neutropenia. Ann Rheum Dis. (1999) 58:309–14. doi: 10.1136/ard.58.5.309
18. Chan EY, Ko SC, Lau CS. Increased rate of apoptosis and decreased expression of bcl-2 protein in peripheral blood lymphocytes from patients with active systemic lupus erythematosus. Asian Pac J Allergy Immunol. (1997) 15:3–7.
19. Chan RW, Jiang P, Peng X, Tam LS, Liao GJ, Li EK, et al. Plasma DNA aberrations in systemic lupus erythematosus revealed by genomic and methylomic sequencing. Proc Natl Acad Sci USA. (2014) 111:5302. doi: 10.1073/pnas.1421126111
20. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. (2014) 30:255–89. doi: 10.1146/annurev-cellbio-101512-122326
21. Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. (2001) 61:1659–65.
22. Wang BG, Huang HY, Chen YC, Bristow RE, Kassauei K, Cheng CC, et al. Increased plasma DNA integrity in cancer patients. Cancer Res. (2003) 63:3966–8.
23. Rock KL, Kono H. The inflammatory response to cell death. Ann Rev Pathol. (2008) 3:99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456
24. Lam NY, Rainer TH, Chan LY, Joynt GM, Lo YM. Time course of early and late changes in plasma DNA in trauma patients. Clin Chem. (2003) 49:1286–91. doi: 10.1373/49.8.1286
25. Lo YM, Zhang J, Leung TN, Lau TK, Chang AM, Hjelm NM. Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet. (1999) 64:218–24. doi: 10.1086/302205
26. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) 464:104–7. doi: 10.1038/nature08780
27. Macher H, Egea-Guerrero J, Revuelto-Rey J, Gordillo-Escobar E, Enamorado-Enamorado J, Boza A, et al. Role of early cell-free DNA levels decrease as a predictive marker of fatal outcome after severe traumatic brain injury. Clin Chim Acta. (2012) 414:12–7. doi: 10.1016/j.cca.2012.08.001
28. Rhodes A, Wort SJ, Thomas H, Collinson P, Bennett ED. Plasma DNA concentration as a predictor of mortality and sepsis in critically ill patients. Crit Care. (2006) 10:R60. doi: 10.1186/cc4894
29. Avriel A, Paryente Wiessman M, Almog Y, Perl Y, Novack V, Galante O, et al. Admission cell free DNA levels predict 28-day mortality in patients with severe sepsis in intensive care. PLoS ONE. (2014) 9:e100514. doi: 10.1371/journal.pone.0100514
30. Gergely P Jr., Niland B, Gonchoroff N, Pullmann R, Jr., Phillips PE, Perl A. Persistent mitochondrial hyperpolarization, increased reactive oxygen intermediate production, and cytoplasmic alkalinization characterize altered IL-10 signaling in patients with systemic lupus erythematosus. J Immunol. (2002) 169:1092–101. doi: 10.4049/jimmunol.169.2.1092
31. Gergely P Jr, Grossman C, Niland B, Puskas F, Neupane H, Allam F, et al. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. (2002) 46:175–90. doi: 10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H
32. Yipp BG, Kubes P. NETosis: how vital is it? Blood. (2013) 122:2784–94. doi: 10.1182/blood-2013-04-457671
33. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
34. Granger V, Faille D, Marani V, Noel B, Gallais Y, Szely N, et al. Human blood monocytes are able to form extracellular traps. J Leukoc Biol. (2017) 102:775–81. doi: 10.1189/jlb.3MA0916-411R
35. Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. (2008) 14:949–53. doi: 10.1038/nm.1855
36. Yousefi S, Morshed M, Amini P, Stojkov D, Simon D, von Gunten S, et al. Basophils exhibit antibacterial activity through extracellular trap formation. Allergy. (2015) 70:1184–8. doi: 10.1111/all.12662
37. von Kockritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, et al. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood. (2008) 111:3070–80. doi: 10.1182/blood-2007-07-104018
38. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
39. Leffler J, Martin M, Gullstrand B, Tyden H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. (2012) 188:3522–31. doi: 10.4049/jimmunol.1102404
40. Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. (2010) 107:9813–8. doi: 10.1073/pnas.0909927107
41. Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther. (2014) 16:R122. doi: 10.1186/ar4579
42. Zhang S, Lu X, Shu X, Tian X, Yang H, Yang W, et al. Elevated plasma cfDNA may be associated with active lupus nephritis and partially attributed to abnormal regulation of neutrophil extracellular traps (NETs) in patients with systemic lupus erythematosus. Intern Med. (2014) 53:2763–71. doi: 10.2169/internalmedicine.53.2570
43. Oklu R, Sheth RA, Wong KHK, Jahromi AH, Albadawi H. Neutrophil extracellular traps are increased in cancer patients but does not associate with venous thrombosis. Cardiovasc Diagn Ther. (2017) 7:S149. doi: 10.21037/cdt.2017.08.01
44. Thomas GM, Carbo C, Curtis BR, Martinod K, Mazo IB, Schatzberg D, et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood. (2012) 119:6335–43. doi: 10.1182/blood-2012-01-405183
45. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. (2009) 7:99–109. doi: 10.1038/nrmicro2070
46. van dV, Netea MG, Dinarello CA, Joosten LA. Inflammasome activation and IL-1beta and IL-18 processing during infection. Trends Immunol. (2011) 32:110–6. doi: 10.1016/j.it.2011.01.003
47. Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
48. Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med. (2016) 213:2113–28. doi: 10.1084/jem.20151613
49. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. (2006) 8:1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
50. Tan HY, Yong YK, Shankar EM, Paukovics G, Ellegard R, Larsson M, et al. Aberrant inflammasome activation characterizes tuberculosis-associated immune reconstitution inflammatory syndrome. J Immunol. (2016) 196:4052–63. doi: 10.4049/jimmunol.1502203
51. Tsumita T, Iwanaga M. Fate of injected deoxyribonucleic acid in mice. Nature. (1963) 198:1088–9. doi: 10.1038/1981088a0
52. Li Z, Wang Y, Xiao K, Xiang S, Li Z, Weng X. Emerging Role of Exosomes in the Joint Diseases. Cell Physiol Biochem. (2018) 47:2008–17. doi: 10.1159/000491469
53. Takahashi A, Okada R, Nagao K, Kawamata Y, Hanyu A, Yoshimoto S, et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat Commun. (2017) 8:15287. doi: 10.1038/ncomms15287
54. Fernando MR, Jiang C, Krzyzanowski GD, Ryan WL. New evidence that a large proportion of human blood plasma cell-free DNA is localized in exosomes. PLoS ONE. (2017) 12:e0183915. doi: 10.1371/journal.pone.0183915
55. Kahlert C, Melo SA, Protopopov A, Tang J, Seth S, Koch M, et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem. (2014) 289:3869–75. doi: 10.1074/jbc.C113.532267
56. Reich CF III, Pisetsky DS. The content of DNA and RNA in microparticles released by Jurkat and HL-60 cells undergoing in vitro apoptosis. Exp Cell Res. (2009) 315:760–8. doi: 10.1016/j.yexcr.2008.12.014
57. Mobarrez F, Vikerfors A, Gustafsson JT, Gunnarsson I, Zickert A, Larsson A, et al. Microparticles in the blood of patients with systemic lupus erythematosus (SLE): phenotypic characterization and clinical associations. Sci Rep. (2016) 6:36025. doi: 10.1038/srep36025
58. Pereira J, Alfaro G, Goycoolea M, Quiroga T, Ocqueteau M, Massardo L, et al. Circulating platelet-derived microparticles in systemic lupus erythematosus. Association with increased thrombin generation and procoagulant state. Thromb Haemost. (2006) 95:94–9. doi: 10.1160/TH05-05-0310
59. Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. (2014) 124:2173–83. doi: 10.1182/blood-2014-05-573543
60. Marcoux G, Duchez AC, Rousseau M, Levesque T, Boudreau LH, Thibault L, et al. Microparticle and mitochondrial release during extended storage of different types of platelet concentrates. Platelets. (2017) 28:272–80. doi: 10.1080/09537104.2016.1218455
61. Emlen W, Mannik M. Kinetics and mechanisms for removal of circulating single-stranded DNA in mice. J Exp Med. (1978) 147:684–99. doi: 10.1084/jem.147.3.684
62. Chused TM, Steinberg AD, Talal N. The clearance and localization of nucleic acids by New Zealand and normal mice. Clin Exp Immunol. (1972) 12:465–76.
63. Gosse C, Le Pecq JB, Defrance P, Paoletti C. Initial degradation of deoxyribonucleic acid after injection in mammals. Cancer Res. (1965) 25:877–83.
64. Ljungman M, Hanawalt PC. Efficient protection against oxidative DNA damage in chromatin. Mol Carcinog. (1992) 5:264–9. doi: 10.1002/mc.2940050406
65. Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA. (1997) 94:514–9. doi: 10.1073/pnas.94.2.514
66. Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci USA. (1988) 85:6465–7. doi: 10.1073/pnas.85.17.6465
67. Mannherz HG, Peitsch MC, Zanotti S, Paddenberg R, Polzar B. A new function for an old enzyme: the role of DNase I in apoptosis. Curr Top Microbiol Immunol. (1995) 198:161–74. doi: 10.1007/978-3-642-79414-8_10
68. Rodriguez AM, Rodin D, Nomura H, Morton CC, Weremowicz S, Schneider MC. Identification, localization, and expression of two novel human genes similar to deoxyribonuclease I. Genomics. (1997) 42:507–13. doi: 10.1006/geno.1997.4748
69. Napirei M, Wulf S, Eulitz D, Mannherz HG, Kloeckl T. Comparative characterization of rat deoxyribonuclease 1 (Dnase1) and murine deoxyribonuclease 1-like 3 (Dnase1l3). Biochem J. (2005) 389(Pt 2):355–64. doi: 10.1042/BJ20042124
70. Sisirak V, Sally B, D'Agati V, Martinez-Ortiz W, Ozcakar ZB, David J, et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell. (2016) 166:88–101. doi: 10.1016/j.cell.2016.05.034
71. Wilber A, Lu M, Schneider MC. Deoxyribonuclease I-like III is an inducible macrophage barrier to liposomal transfection. Mol Ther. (2002) 6:35–42. doi: 10.1006/mthe.2002.0625
72. Chitrabamrung S, Rubin RL, Tan EM. Serum deoxyribonuclease I and clinical activity in systemic lupus erythematosus. Rheumatol Int. (1981) 1:55–60. doi: 10.1007/BF00541153
73. Leffler J, Ciacma K, Gullstrand B, Bengtsson AA, Martin M, Blom AM. A subset of patients with systemic lupus erythematosus fails to degrade DNA from multiple clinically relevant sources. Arthritis Res Ther. (2015) 17:205. doi: 10.1186/s13075-015-0726-y
74. Frost PG, Lachmann PJ. The relationship of desoxyribonuclease inhibitor levels in human sera to the occurrence of antinuclear antibodies. Clin Exp Immunol. (1968) 3:447–55.
75. Bodano A, Amarelo J, Gonzalez A, Gomez-Reino J, Conde C. Novel DNASE I mutations related to systemic lupus erythematosus. Arthritis Rheum. (2004) 50:4070–1. doi: 10.1002/art.20721
76. Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet. (2001) 28:313–4. doi: 10.1038/91070
77. Emlen W, Ansari R, Burdick G. DNA-anti-DNA immune complexes. Antibody protection of a discrete DNA fragment from DNase digestion in vitro. J Clin Invest. (1984) 74:185–90. doi: 10.1172/JCI111400
78. Yeh TM, Chang HC, Liang CC, Wu JJ, Liu MF. Deoxyribonuclease-inhibitory antibodies in systemic lupus erythematosus. J Biomed Sci. (2003) 10:544–51. doi: 10.1007/BF02256116
79. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra19. doi: 10.1126/scitranslmed.3001180
80. Meller S, Di Domizio J, Voo KS, Friedrich HC, Chamilos G, Ganguly D, et al. T(H)17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol. (2015) 16:970–9. doi: 10.1038/ni.3211
81. Fisher RP, Topper JN, Clayton DA. Promoter selection in human mitochondria involves binding of a transcription factor to orientation-independent upstream regulatory elements. Cell. (1987) 50:247–58. doi: 10.1016/0092-8674(87)90220-0
82. Gaipl US, Beyer TD, Heyder P, Kuenkele S, Bottcher A, Voll RE, et al. Cooperation between C1q and DNase I in the clearance of necrotic cell-derived chromatin. Arthritis Rheum. (2004) 50:640–9. doi: 10.1002/art.20034
83. Napirei M, Wulf S, Mannherz HG. Chromatin breakdown during necrosis by serum Dnase1 and the plasminogen system. Arthritis Rheum. (2004) 50:1873–83. doi: 10.1002/art.20267
84. Butler PJ, Tennent GA, Pepys MB. Pentraxin-chromatin interactions: serum amyloid P component specifically displaces H1-type histones and solubilizes native long chromatin. J Exp Med. (1990) 172:13–8. doi: 10.1084/jem.172.1.13
85. Stephan F, Marsman G, Bakker LM, Bulder I, Stavenuiter F, Aarden LA, et al. Cooperation of factor VII-activating protease and serum DNase I in the release of nucleosomes from necrotic cells. Arthritis Rheumatol. (2014) 66:686–93. doi: 10.1002/art.38265
86. Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. (2011) 43:1186–8. doi: 10.1038/ng.975
87. Ozcakar ZB, Foster J II, Diaz-Horta O, Kasapcopur O, Fan YS, Yalcinkaya F, et al. DNASE1L3 mutations in hypocomplementemic urticarial vasculitis syndrome. Arthritis Rheum. (2013) 65:2183–9. doi: 10.1002/art.38010
88. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. (2016) 213:697–713. doi: 10.1084/jem.20151876
89. Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, et al. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. (2007) 39:1065–7. doi: 10.1038/ng2091
90. Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. (2011) 12:270–9. doi: 10.1038/gene.2010.73
91. Erlendsson K, Traustadottir K, Freysdottir J, Steinsson K, Jonsdottir I, Valdimarsson H. Reciprocal changes in complement activity and immune-complex levels during plasma infusion in a C2-deficient SLE patient. Lupus. (1993) 2:161–5. doi: 10.1177/096120339300200306
92. Gillmore JD, Hutchinson WL, Herbert J, Bybee A, Mitchell DA, Hasserjian RP, et al. Autoimmunity and glomerulonephritis in mice with targeted deletion of the serum amyloid P component gene: SAP deficiency or strain combination? Immunology. (2004) 112:255–64. doi: 10.1111/j.1365-2567.2004.01860.x
93. Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc Natl Acad Sci USA. (2000) 97:1184–9. doi: 10.1073/pnas.97.3.1184
94. Ogden CA, Kowalewski R, Peng Y, Montenegro V, Elkon KB. IGM is required for efficient complement mediated phagocytosis of apoptotic cells in vivo. Autoimmunity. (2005) 38:259–64. doi: 10.1080/08916930500124452
95. Du Clos TW, Zlock LT, Rubin RL. Analysis of the binding of C-reactive protein to histones and chromatin. J Immunol. (1988) 141:4266–70.
96. Janko C, Franz S, Munoz LE, Siebig S, Winkler S, Schett G, et al. CRP/anti-CRP antibodies assembly on the surfaces of cell remnants switches their phagocytic clearance toward inflammation. Front Immunol. (2011) 2:70. doi: 10.3389/fimmu.2011.00070
97. Saevarsdottir S, Steinsson K, Ludviksson BR, Grondal G, Valdimarsson H. Mannan-binding lectin may facilitate the clearance of circulating immune complexes–implications from a study on C2-deficient individuals. Clin Exp Immunol. (2007) 148:248–53. doi: 10.1111/j.1365-2249.2007.03349.x
98. Mahajan A, Herrmann M, Munoz LE. Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front Immunol. (2016) 7:35. doi: 10.3389/fimmu.2016.00035
99. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. (2007) 449:564–9. doi: 10.1038/nature06116
100. Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. (2002) 20:709–60. doi: 10.1146/annurev.immunol.20.100301.064842
101. Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. (2005) 23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633
102. Yasuda K, Yu P, Kirschning CJ, Schlatter B, Schmitz F, Heit A, et al. Endosomal translocation of vertebrate DNA activates dendritic cells via TLR9-dependent and -independent pathways. J Immunol. (2005) 174:6129–36. doi: 10.4049/jimmunol.174.10.6129
103. Stacey KJ, Young GR, Clark F, Sester DP, Roberts TL, Naik S, et al. The molecular basis for the lack of immunostimulatory activity of vertebrate DNA. J Immunol. (2003) 170:3614–20. doi: 10.4049/jimmunol.170.7.3614
104. Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. (2005) 115:407–17. doi: 10.1172/JCI23025
105. Ronnblom L, Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus. (2008) 17:394–9. doi: 10.1177/0961203308090020
106. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. (2003) 197:711–23. doi: 10.1084/jem.20021553
107. Kubo T, Fujii M. Specific binding and stabilization of DNA and phosphorothioate DNA by amphiphilic alpha-helical peptides. Nucleoside, Nucleotides Nucleic Acids. (2001) 20:1313–6. doi: 10.1081/NCN-100002544
108. Poli C, Augusto JF, Dauve J, Adam C, Preisser L, Larochette V, et al. IL-26 confers proinflammatory properties to extracellular DNA. J Immunol. (2017) 198:3650–61. doi: 10.4049/jimmunol.1600594
109. Corvaisier M, Delneste Y, Jeanvoine H, Preisser L, Blanchard S, Garo E, et al. IL-26 is overexpressed in rheumatoid arthritis and induces proinflammatory cytokine production and Th17 cell generation. PLoS Biol. (2012) 10:e1001395. doi: 10.1371/journal.pbio.1001395
110. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. (2007) 8:950–7. doi: 10.1038/ni1497
111. Dambacher J, Beigel F, Zitzmann K, De Toni EN, Goke B, Diepolder HM, et al. The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut. (2009) 58:1207–17. doi: 10.1136/gut.2007.130112
112. Parisi MA, Clayton DA. Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science. (1991) 252:965–9. doi: 10.1126/science.2035027
113. Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta. (2010) 1799:101–13. doi: 10.1016/j.bbagrm.2009.09.008
114. Julian MW, Shao G, Bao S, Knoell DL, Papenfuss TL, VanGundy ZC, et al. Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. J Immunol. (2012) 189:433–43. doi: 10.4049/jimmunol.1101375
115. Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. (2007) 8:487–96. doi: 10.1038/ni1457
116. Andreeva L, Hiller B, Kostrewa D, Lassig C, de OM, Jan Drexler D, et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature. (2017) 549:394–8. doi: 10.1038/nature23890
117. Gehrke N, Mertens C, Zillinger T, Wenzel J, Bald T, Zahn S, et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity. (2013) 39:482–95. doi: 10.1016/j.immuni.2013.08.004
118. Ermakov AV, Konkova MS, Kostyuk SV, Izevskaya VL, Baranova A, Veiko NN. Oxidized extracellular DNA as a stress signal in human cells. Oxidat Med Cell Longev. (2013) 2013:649747. doi: 10.1155/2013/649747
119. Kannan K, Jain SK. Oxidative stress and apoptosis. Pathophysiology. (2000) 7:153–63. doi: 10.1016/S0928-4680(00)00053-5
120. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radical Biol Med. (2010) 49:1603–16. doi: 10.1016/j.freeradbiomed.2010.09.006
121. van Loon B, Markkanen E, Hubscher U. Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair. (2010) 9:604–16. doi: 10.1016/j.dnarep.2010.03.004
122. Valavanidis A, Vlachogianni T, Fiotakis C. 8-hydroxy-2' -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. (2009) 27:120–39. doi: 10.1080/10590500902885684
123. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
124. Yoshida H, Nishikawa M, Kiyota T, Toyota H, Takakura Y. Increase in CpG DNA-induced inflammatory responses by DNA oxidation in macrophages and mice. Free Radical Biol Med. (2011) 51:424–31. doi: 10.1016/j.freeradbiomed.2011.04.035
125. Lunec J, Herbert K, Blount S, Griffiths HR, Emery P. 8-Hydroxydeoxyguanosine. A marker of oxidative DNA damage in systemic lupus erythematosus. FEBS Lett. (1994) 348:131–8. doi: 10.1016/0014-5793(94)00583-4
126. Blount S, Griffiths HR, Lunec J. Reactive oxygen species induce antigenic changes in DNA. FEBS Lett. (1989) 245:100–4. doi: 10.1016/0014-5793(89)80200-5
127. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. (2018) 560:198–203. doi: 10.1038/s41586-018-0372-z
128. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222–30. doi: 10.1038/ni.1980
129. Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol. (2004) 75:995–1000. doi: 10.1189/jlb.0703328
130. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. (2012) 485:251–5. doi: 10.1038/nature10992
131. Yang D, Oyaizu Y, Oyaizu H, Olsen GJ, Woese CR. Mitochondrial origins. Proc Natl Acad Sci USA. (1985) 82:4443–7. doi: 10.1073/pnas.82.13.4443
132. Clayton DA, Doda JN, Friedberg EC. Absence of a pyrimidine dimer repair mechanism for mitochondrial DNA in mouse and human cells. Basic Life Sci. (1975) 5B:589–91. doi: 10.1007/978-1-4684-2898-8_26
133. Clayton DA, Doda JN, Friedberg EC. The absence of a pyrimidine dimer repair mechanism in mammalian mitochondria. Proc Natl Acad Sci USA. (1974) 71:2777–81. doi: 10.1073/pnas.71.7.2777
134. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. (2015) 520:553–7. doi: 10.1038/nature14156
135. Johnson AJ, Goger PR, Tillett WS. The intravenous injection of bovine crystalline pancreatic desoxyribonuclease into patients. J Clin Invest. (1954) 33:1670–86. doi: 10.1172/JCI103048
136. Johnson AJ, Ayvazian JH, Tillett WS. Crystalline pancreatic desoxyribonuclease as an adjunct to the treatment of pneumococcal meningitis. N Engl J Med. (1959) 260:893–900. doi: 10.1056/NEJM195904302601801
137. Lachmann PJ. Allergic reactions, connective tissue, and disease. Sci Basis Med Annu Rev. (1967) 36–58.
138. Macanovic M, Sinicropi D, Shak S, Baughman S, Thiru S, Lachmann PJ. The treatment of systemic lupus erythematosus (SLE) in NZB/W F1 hybrid mice; studies with recombinant murine DNase and with dexamethasone. Clin Exp Immunol. (1996) 106:243–52. doi: 10.1046/j.1365-2249.1996.d01-839.x
139. Shak S, Capon DJ, Hellmiss R, Marsters SA, Baker CL. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc Natl Acad Sci USA. (1990) 87:9188–92. doi: 10.1073/pnas.87.23.9188
140. Wilmott RW, Amin RS, Colin AA, DeVault A, Dozor AJ, Eigen H, et al. Aerosolized recombinant human DNase in hospitalized cystic fibrosis patients with acute pulmonary exacerbations. Am J Respir Crit Care Med. (1996) 153(6 Pt 1):1914–7. doi: 10.1164/ajrccm.153.6.8665055
141. Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med. (1994) 331:637–42. doi: 10.1056/NEJM199409083311003
142. Shah PI, Bush A, Canny GJ, Colin AA, Fuchs HJ, Geddes DM, et al. Recombinant human DNase I in cystic fibrosis patients with severe pulmonary disease: a short-term, double-blind study followed by six months open-label treatment. Eur Respir J. (1995) 8:954–8.
143. Davis JC, Jr., Manzi S, Yarboro C, Rairie J, McInnes I, Averthelyi D, et al. Recombinant human Dnase I (rhDNase) in patients with lupus nephritis. Lupus. (1999) 8:68–76. doi: 10.1191/096120399678847380
144. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. (1982) 25:1271–7. doi: 10.1002/art.1780251101
145. Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. (2012) 64:2677–86. doi: 10.1002/art.34473
146. Elkon KB. Review: Cell death, nucleic acids, and immunity: inflammation beyond the grave. Arthritis Rheumatol. (2018) 70:805–16. doi: 10.1002/art.40452
147. Davis GL Jr, Davis JSt. Detection of circulating DNA by counterimmunoelectrophoresis (CIE). Arthritis Rheum. (1973) 16:52–8. doi: 10.1002/art.1780160108
148. Leon SA, Ehrlich GE, Shapiro B, Labbate VA. Free DNA in the serum of rheumatoid arthritis patients. J Rheumatol. (1977) 4:139–43.
149. Steinman CR. Circulating DNA in systemic lupus erythematosus. Association with central nervous system involvement and systemic vasculitis. Am J Med. (1979) 67:429–35. doi: 10.1016/0002-9343(79)90789-7
150. Raptis L, Menard HA. Quantitation and characterization of plasma DNA in normals and patients with systemic lupus erythematosus. J Clin Invest. (1980) 66:1391–9. doi: 10.1172/JCI109992
151. Leon SA, Revach M, Ehrlich GE, Adler R, Petersen V, Shapiro B. DNA in synovial fluid and the circulation of patients with arthritis. Arthritis Rheum. (1981) 24:1142–50. doi: 10.1002/art.1780240905
152. Klemp P, Meyers OL, Harley EH. Measurement of plasma DNA by a physiochemical method: relevance in SLE. Ann Rheum Dis. (1981) 40:593–9. doi: 10.1136/ard.40.6.593
153. Morimoto C, Sano H, Abe T, Homma M, Steinberg AD. Correlation between clinical activity of systemic lupus erythematosus and the amounts of DNA in DNA/anti-DNA antibody immune complexes. J Immunol. (1982) 129:1960–5.
154. McCoubrey-Hoyer A, Okarma TB, Holman HR. Partial purification and characterization of plasma DNA and its relation to disease activity in systemic lupus erythematosus. Am J Med. (1984) 77:23–34. doi: 10.1016/0002-9343(84)90431-5
155. Hajizadeh S, DeGroot J, TeKoppele JM, Tarkowski A, Collins LV. Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res Ther. (2003) 5:234. doi: 10.1186/ar787
156. Zhong XY, von Muhlenen I, Li Y, Kang A, Gupta AK, Tyndall A, et al. Increased concentrations of antibody-bound circulatory cell-free DNA in rheumatoid arthritis. Clin Chem. (2007) 53:1609–14. doi: 10.1373/clinchem.2006.084509
157. Bartoloni E, Ludovini V, Alunno A, Pistola L, Bistoni O, Crino L, et al. Increased levels of circulating DNA in patients with systemic autoimmune diseases: a possible marker of disease activity in Sjogren's syndrome. Lupus. (2011) 20:928–35. doi: 10.1177/0961203311399606
158. Cepika AM, Soldo Juresa D, Morovic Vergles J, Malenica B, Santak M, Kapitanovic S, et al. Decrease in circulating DNA, IL-10 and BAFF levels in newly-diagnosed SLE patients after corticosteroid and chloroquine treatment. Cell Immunol. (2012) 276:196–203. doi: 10.1016/j.cellimm.2012.05.009
159. Tug S, Helmig S, Menke J, Zahn D, Kubiak T, Schwarting A, et al. Correlation between cell free DNA levels and medical evaluation of disease progression in systemic lupus erythematosus patients. Cell Immunol. (2014) 292:32–9. doi: 10.1016/j.cellimm.2014.08.002
160. Hendy MO, Motalib AT, Shafie AEM, Khalaf AF, Kotb ES, Khalil A, et al. Circulating cell free DNA as a predictor of systemic lupus erythematosus severity and monitoring of therapy. Egypt J Med Hum Genet. (2016) 17:79–85. doi: 10.1016/j.ejmhg.2015.07.001
161. Abdelal TI, Zakaria AM, Sharaf MD, Elakadc MG. Levels of plasma cell-free DNA and its correlation with disease activity in rheumatoid arthritis and systemic lupus erythematosus patients. Egypt Rheumatol. (2016) 38:295–300. doi: 10.1016/j.ejr.2016.06.005
162. Rykova E, Sizikov A, Roggenbuck D, Antonenko O, Bryzgalov L, Morozkin E, et al. Circulating DNA in rheumatoid arthritis: pathological changes and association with clinically used serological markers. Arthritis Res Ther. (2017) 19:85. doi: 10.1186/s13075-017-1295-z
163. Hashimoto T, Yoshida K, Hashimoto N, Nakai A, Kaneshiro K, Suzuki K, et al. Circulating cell free DNA: a marker to predict the therapeutic response for biological DMARDs in rheumatoid arthritis. Int J Rheum Dis. (2017) 20:722–30. doi: 10.1111/1756-185X.12959
164. Laukova L, Konecna B, Vlkova B, Mlynarikova V, Celec P, Stenova E. Anti-cytokine therapy and plasma DNA in patients with rheumatoid arthritis. Rheumatol Int. (2018) 38:1449–54. doi: 10.1007/s00296-018-4055-8
165. Eldosoky MAER, Hammad RHM, Kotb HG, Eldesoky NA-R, Elabd HA. Cell free DNA and CD38 expression on T helper cells as biomarkers of disease activity in rheumatoid arthritis. Ame J Biochem. (2018) 8:60–8. doi: 10.5923/j.ajb.20180803.03
166. Xu Y, Song Y, Chang J, Zhou X, Qi Q, Tian X, et al. High levels of circulating cell-free DNA are a biomarker of active SLE. Eur J Clin Invest. (2018) 48:e13015. doi: 10.1111/eci.13015
167. Dennin RH. DNA of free and complexed origin in human plasma: concentration and length distribution. Klinische Wochenschrift. (1979) 57:451–6. doi: 10.1007/BF01477498
168. Atamaniuk J, Hsiao YY, Mustak M, Bernhard D, Erlacher L, Fodinger M, et al. Analysing cell-free plasma DNA and SLE disease activity. Eur J Clin Invest. (2011) 41:579–83. doi: 10.1111/j.1365-2362.2010.02435.x
169. Sano H, Takai O, Harata N, Yoshinaga K, Kodama-Kamada I, Sasaki T. Binding properties of human anti-DNA antibodies to cloned human DNA fragments. Scand J Immunol. (1989) 30:51–63. doi: 10.1111/j.1365-3083.1989.tb01188.x
170. Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. (1990) 33:1665–73. doi: 10.1002/art.1780331109
171. Isenberg DA, Ehrenstein MR, Longhurst C, Kalsi JK. The origin, sequence, structure, and consequences of developing anti-DNA antibodies. A human perspective. Arthritis Rheum. (1994) 37:169–80. doi: 10.1002/art.1780370204
172. Cooke MS, Mistry N, Wood C, Herbert KE, Lunec J. Immunogenicity of DNA damaged by reactive oxygen species–implications for anti-DNA antibodies in lupus. Free Radical Biol Med. (1997) 22:151–9. doi: 10.1016/S0891-5849(96)00283-3
173. Perniok A, Wedekind F, Herrmann M, Specker C, Schneider M. High levels of circulating early apoptic peripheral blood mononuclear cells in systemic lupus erythematosus. Lupus. (1998) 7:113–8. doi: 10.1191/096120398678919804
174. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
175. Bell C, Talal N, Schur PH. Antibodies to DNA in patients with rheumatoid arthritis and juvenile rheumatoid arthritis. Arthritis Rheum. (1975) 18:535–40. doi: 10.1002/art.1780180602
176. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. (2002) 416:603–7. doi: 10.1038/416603a
177. Smolenska Z, Kaznowska Z, Zarowny D, Simmonds HA, Smolenski RT. Effect of methotrexate on blood purine and pyrimidine levels in patients with rheumatoid arthritis. Rheumatology. (1999) 38:997–1002. doi: 10.1093/rheumatology/38.10.997
178. Moodley D, Mody GM, Chuturgoon AA. Initiation but no execution - modulation of peripheral blood lymphocyte apoptosis in rheumatoid arthritis - a potential role for heat shock protein 70. J Inflamm. (2011) 8:30. doi: 10.1186/1476-9255-8-30
179. Allyse MA, Wick MJ. Noninvasive prenatal genetic screening using cell-free DNA. JAMA. (2018) 320:591–2. doi: 10.1001/jama.2018.9418
180. Fettke H, Kwan EM, Azad AA. Cell-free DNA in cancer: current insights. Cell Oncol. (2019) 42:13–28. doi: 10.1007/s13402-018-0413-5
181. Gielis EM, Ledeganck KJ, De Winter BY, Del Favero J, Bosmans JL, Claas FH, et al. Cell-Free DNA: An upcoming biomarker in transplantation. Am J Transplant. (2015) 15:2541–51. doi: 10.1111/ajt.13387
182. Mutirangura A, Pornthanakasem W, Theamboonlers A, Sriuranpong V, Lertsanguansinchi P, Yenrudi S, et al. Epstein-Barr viral DNA in serum of patients with nasopharyngeal carcinoma. Clin Cancer Res. (1998) 4:665–9.
183. Han X, Wang J, Sun Y. Circulating tumor DNA as biomarkers for cancer detection. Genomics Proteomics Bioinformatics. (2017) 15:59–72. doi: 10.1016/j.gpb.2016.12.004
184. Neumann MHD, Bender S, Krahn T, Schlange T. ctDNA and CTCs in liquid biopsy - current status and where we need to progress. Comput Struct Biotechnol J. (2018) 16:190–5. doi: 10.1016/j.csbj.2018.05.002
Keywords: cell-free DNA, biomarker, autoimmune rheumatic diseases, systemic lupus erythematosus, rheumatoid arthritis
Citation: Duvvuri B and Lood C (2019) Cell-Free DNA as a Biomarker in Autoimmune Rheumatic Diseases. Front. Immunol. 10:502. doi: 10.3389/fimmu.2019.00502
Received: 13 December 2018; Accepted: 25 February 2019;
Published: 19 March 2019.
Edited by:
David Stephen Pisetsky, Duke University, United StatesReviewed by:
Andras Perl, Upstate Medical University, United StatesAnne Cooke, University of Cambridge, United Kingdom
Copyright © 2019 Duvvuri and Lood. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Lood, bG9vZGNAdXcuZWR1