 Jie Liu1,2
Jie Liu1,2 Qi Zhao
Qi Zhao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 19 March 2019
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00456
This article is part of the Research Topic Current Perspectives, Challenges and Advances in Cell Based Therapies View all 14 articles
Chimeric antigen receptor (CAR) T cells have shown great promise in the treatment of hematological and solid malignancies. However, despite the success of this field, there remain some major challenges, including accelerated T cell exhaustion, potential toxicities, and insertional oncogenesis. To overcome these limitations, recent advances in CRISPR technology have enabled targetable interventions of endogenous genes in human CAR T cells. These CRISPR genome editing approaches have unleashed the therapeutic potential of CAR T cell therapy. Here, we summarize the potential benefits, safety concerns, and difficulties in the generation of gene-edited CAR T cells using CRISPR technology.
Major histocompatibility complex (MHC) molecules play key roles in the surveillance of aberrant proteins of tumor cells. T cell receptors (TCRs) on the surface of T lymphocytes recognize antigenic peptide fragments derived from these aberrant proteins in complex with MHCs (1, 2). The expression of MHC/peptide complexes constitutively occurs on all nucleated cells. Tumor-specific MHC/peptide complexes are considered ideal targets for T cell-based immunotherapies. Diverse strategies have been developed to induce T cell immunity against these tumor epitopes, including cancer vaccination (3), adoptive T cell transfer (4), and TCR engineering (5). In cancer patients, however, tumor cells can effectively escape adoptive immunity via regulatory mechanisms, such as downregulation of MHCs or mutation. Because the presence of relatively fewer MHC molecules on the tumor cell surface limits naive TCR recognition, T cells fail to respond and trigger cascades of immune activation (6).
Recently, the most promising development has been the use of chimeric antigen receptor (CAR) T cell immunotherapy (7). CAR T cell immunotherapy has emerged as a leading curative strategy in the treatment of relapsed hematological malignancies. CAR T cell therapy is based on the immune effect of T cell activation and the principle of transformation through the genetic engineering of T cells. A typical CAR construct comprises a binding domain (single chain antibody fragment, scFv), a transmembrane domain and intracellular signaling domains capable of activating T cells (Figure 1). CARs allow the T cells to be activated independently of MHC. Donor-derived T cells are modified to express multivalent CARs on the cell surface that are responsible for recognizing the tumor-associated antigen (TAA) of tumor cells. Thus, T cells are activated via intracellular signal transduction. CAR designs differ not only in their signaling domains but also in their functional properties. The CAR structures have progressed since the first generation was described in 1989 (8). The first generation of CARs was designed as an scFv linked to the CD3ζ intracellular signaling domain of the TCR through a hinge and a transmembrane domain. Although the CD3ζ signaling domain can trigger activation of T cells, this pattern most likely results in T cell anergy, attenuating T cell activation. Therefore, the first generation of CARs exhibited limited responses in clinical trials (7). To address this limitation, a costimulatory molecule, such as CD28, OX40, or 4-1BB, was incorporated into the intracellular domain for the second generation of CARs. The additional costimulatory domain in the second generation of CARs strikingly improved T-cell proliferation and persistence. To optimize T-cell efficacy, the third generation of CARs has been developed by introducing two costimulatory domains into the CAR structure. Although dual costimulatory domains can enhance the activation and proliferation of T cells, the abundance of cytokines remains to be considered.
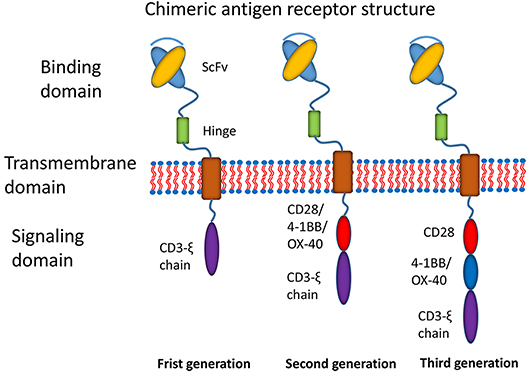
Figure 1. Main structures of chimeric antigen receptors. Three generations of CAR structures. In the first generation of CARs, the binding domain (single chain antibody fragment, scFv) is linked to the signaling domain (CD3ζ) via the transmembrane domain. In the second generation of CARs, the costimulatory molecule (CM1, such as CD28 4-1BB or OX-40) is introduced with the signaling domain (CD3ζ). In the third generation of CARs, the additional costimulatory molecule (CM2) is included.
The CAR T cell approach has provided great advances in the treatment of hematological malignancies. Anti-CD19 CAR T cells have significantly advanced the therapy of human hematological malignancies and were shown to achieve a 90% complete response rate in acute lymphoblastic leukemia (ALL) (9). Tisagenlecleucel, the first anti-CD19 CAR T cell therapy, was approved by the US Food and Drug Administration (US FDA) for the treatment of children and adults with advanced leukemia in 2017 (10, 11). As 2017 ended, there were hundreds of ongoing CAR T cell trials for the treatment of hematologic and solid tumor malignancies (12).
Although most patients infused with CAR T cells show mild or moderate side effects, potentially severe side effects are still challenging. The prominent toxicities include cytokine release syndrome (CRS), insertional oncogenesis, and neurologic toxicity (13, 14).
CRS is an unintended side effect due to overactivation of the host immune system. Severe CRS was observed in some patients who received infusion of CAR T cells (15). An abundance of cytokines is released by either the infused CAR T cells or other polarized immune cells. Several clinical studies indicated that 19–43% of patients exhibited CRS when they were treated with anti-CD19 CAR T cells for relapsed/refractory ALL (13, 16). Clinical features of CRS include high fever, muscle pain, malaise, unstable hypotension, fatigue, ang capillary leakage (17). A wide variety of cytokines can be elevated in the serum of patients. Dramatic elevations of inflammatory cytokines, such as INF-γ, IL-2, IL-6, and IL-10, are observed in CRS (18). Occasionally, neurologic toxicity can be associated with anti-CD19 CAR T cell therapy, probably due to the elevated levels of cytokines (16). The use of the anti-IL-6 receptor antibody tocilizumab was demonstrated to exert curative effects for serious cases of CRS in all patients with a high proliferation of CAR T cells (19).
Continuous CAR expression in T cells relies primarily on the delivery of the CAR gene by integrated gamma retroviral (RV) or lentiviral (LV) vectors. The advantages of both systems are high gene-transfer efficiency and stable expression of the CARs. Although both RV and LV vectors have been shown to be safe in intensive biosafety testing, this safety issue remains a concern. LV- or RV-mediated random and uncontrollable integration in the genome are unpredictable (20). Uncontrollable insertions of CAR genes lead to potential oncogenesis, variegated transgene expression, and transcriptional silencing (21). This possibility poses an oncogenic risk for RV/LV-engineered T cells (22). Although RV-driven oncogenesis has not yet been reported in CAR T cell therapy, this phenomenon was observed in clinical trials of hematopoietic stem cell transplantation (23). Additionally, random integration into the genome causes substantial variations in CAR expression levels in a batch of CAR T cells because of the different copy numbers per cell.
With the gradual initiation of clinical trials, autologous CAR T cells have shown some disadvantages. In infants or adults who are receiving chemotherapy or radiotherapy, it is difficult to harvest sufficient lymphocytes for CAR T cell manufacture. Thus, the quality of CAR T cells for each patient is uncontrollable and unpredictable. The use of allogeneic CAR T cells has become a solution for these problems. Allogeneic CAR T cells can be expanded ex vivo on a large scale and can be reserved to treat multiple patients (24). The concerned with allogeneic infusion is graft-vs.-host disease (GVHD) between the donor cells and recipients. The repertoire of TCRs and MHCs expressed on allogeneic CAR T cells may potentially induce GVHD in recipients who receive donor CAR T cells (25). A study showed that allogeneic anti-CD19-CAR T cells had clinical benefits for relapsed hematologic malignancies (26). No obvious GVHD was observed in these recipients.
Efforts to enhance the efficacy of CAR T cell therapy have been undertaken, including the selection of extracellular receptors (27), optimization of intracellular costimulatory molecules (28), combination with cytokines(29), and improvement of “on-target/off-tumor” toxicity (30). Effective gene-editing technologies have emerged as tools for cell engineering (31). The properties of three gene-editing tools, including CRISPR, zinc-finger nucleases (ZFNs), and transcription activator-like effector nucleases (TALENs), are summarized in Table 1. The use of CRISPR in genome editing is highly efficient and enables a simple and efficient way to multiplex the processing of T cells (32, 33). Both ZFNs and TALENs have also been adopted to modify T cells for clinical applications (34, 35). However, the recognition of the targetable DNA sequences with ZFNs and TALENs in T cells remains complicated and tedious, resulting in a low gene-editing efficiency. The simultaneous multiplexed genetic manipulations of these techniques are challenging (36). CRISPR/Cas9 systems have been used for the knock-out and knock-in of sequences in mammalian genome editing (Figure 2). In principle, a deletion or insertion at a target gene is introduced by a small RNA (sgRNA)-guided Cas9 nuclease that induces a double-stranded DNA break, which is subsequently repaired by non-homologous end joining (NHEJ) (37). Nucleotide insertions or deletions result in non-sense mutations and loss of gene function. In comparison to NHEJ, a relatively large gene sequence can be delivered to a precise locus in the genome through homology directed repair (HDR) after double-stranded DNA is cleaved by sgRNAs (38–40). The HDR process enables precisely targeted nucleotide replacements at the defined site of interest. Currently, several strategies based on CRISPR are being applied to develop next-generation CAR T cells by multiplexed genome editing (41–43). Such approaches include the knockout of endogenous genes (such as TCRs, MHCs, or self-antigens) to build allogeneic universal CAR T cells (41, 44, 45), the disruption of inhibitory receptors (such as CTLA-4, PD-1, or LAG-3) (44, 46, 47), and the integration of the CAR cassette into the endogenous TCR α constant locus (TRAC) (48, 49) or the C-C chemokine receptor type 5 (CCR5) locus (32) (Table 2).
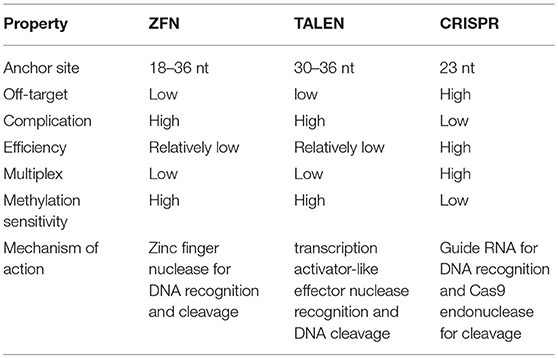
Table 1. Comparison of ZFN, TALEN, and CRISPR.
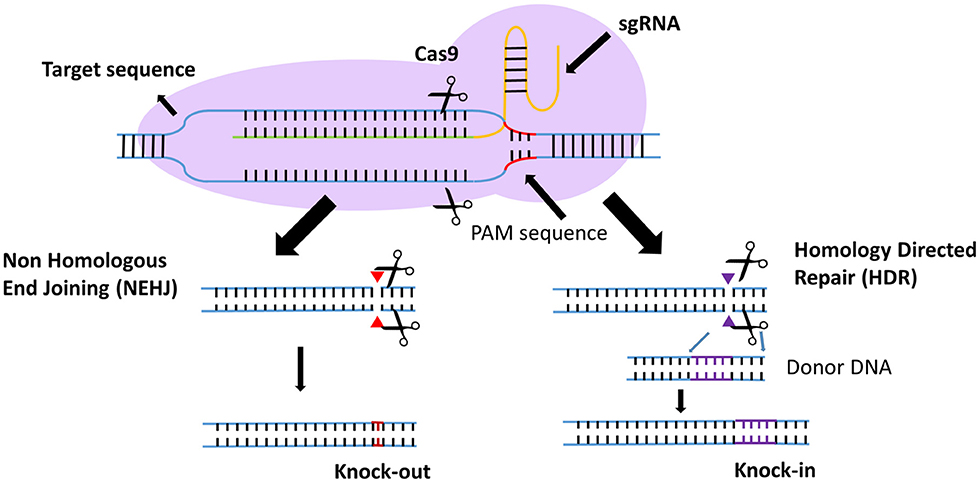
Figure 2. Introduction to the CRISPR gene-editing system. Guided by sgRNAs, the CRISPR-Cas9 nuclease can target short DNA sequences. The PAM specifically creates a sgRNA–target DNA heteroduplex and generates double-strand breaks. Then, the DNA double-strand breaks are repaired by non-homologous end-joining (NHEJ) or homology-directed repair (HDR). In the NHEJ pathway, indels lead to nucleotide deletions or insertions. In the HDR pathway, accessory factors can facilitate genome recombination through the two homology arms, resulting in the knock-in of a gene of interest.
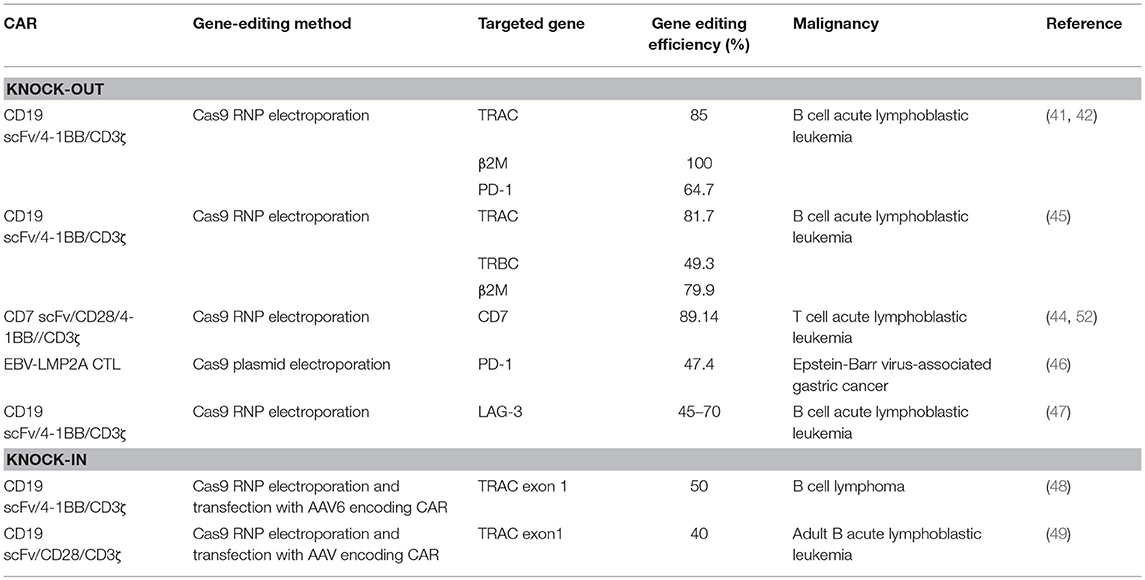
Table 2. Summary of the CAR-T cells modified with gene editing.
Although autologous CAR T cells against B cell malignancies have shown promising results, some clinical studies demonstrated that for some patients, autologous T cells could not be manufactured due to poor lymphocyte counts or low T cell quality and quantity (50). Especially for some patients in infancy, sufficient peripheral blood mononuclear cells (PBMC) cannot be harvested to support T cell manufacture ex vivo. These limitations can be circumvented by utilizing allogeneic T cells. Endogenous TCRs that allogeneic T cells express can recognize the alloantigen of the recipient, resulting in major graft-vs.-host disease (GVHD). Before these allogeneic T cells can be widely used clinically, the issue of GVHD must be resolved (45). Universal allogeneic CAR-T cells are ideal because their manufacture and quality may be more easily controlled and GVHD may be avoided. Several groups have generated allogeneic universal anti-CD19 CAR T cells by deleting multiple genes, such as TRAC, β2M, and MHC, using CRISPR methods (41, 42). Meanwhile, ongoing clinical trials have shown that a suicide gene in the CAR construct can also be used to avoid GVHD after allogeneic CAR T cell injection (25). These results suggest that CAR T cells that utilize multiplexed gene editing generate CAR T cells that are as potent as non-gene-edited T cells.
Until now, most successful CAR T cell therapies have been applied to B cell malignances. For T cell malignances, patients would receive allogeneic T cells rather than autologous CAR T cells. Genomic editing of some antigens, which recognize those “non-self” molecules and are attacked by the host immune system, can broaden the application of CAR T cells. DiPersio et al. reported that fratricide-resistant “off-the-shelf” universal T cells generated with CRISPR gene editing were used for treatment of T-cell malignancies (44). CD7 is a molecule commonly expressed in T lymphocytes. To avoid self-elimination, the CD7 target antigen against malignancies, which is recognized by anti-CD7-CARs, is deleted on CAR T cells (51, 52).
It is widely accepted that the existence of immune checkpoints (such as PD-1, CTLA-4, and LAG-3) can attenuate the activation of CAR T cells and accelerate T cell exhaustion. PD-1 is a primary inhibitory molecule in T cell transduction (53, 54). The PD-1/PD-L1 pathway plays an important role in the regulation of T cell activation and differentiation (55). High expression of PD-1 accelerates T cell tolerance and exhaustion (56–59). Increasing evidence indicates that blocking the PD-1/PD-L1 axis could partially restore the function of exhausted T cells (54, 60). A recent clinical study demonstrated that treatment with anti-CD19 CAR T cells in combination with an anti-PD-1 antibody was effective in patients with relapsed chronic lymphocytic leukemia (CLL) (61). This anti-PD-1 antibody treatment revives the antitumor response of anti-CD19 CAR T cells in patients who fail to respond to CAR T cell treatment (62). In other cases, unanticipated autoimmune responses are associated with anti-PD-1 checkpoint inhibitors (63). Therefore, ablation of PD-1 with gene editing by CRISPR/Cas9 is an alternative to enhance the antitumor response of CAR T cells in anti-CD19 CAR T cell therapy (41, 42). Ren et al. suggested that depletion of PD-1 genes in anti-prostate stem cell antigen (PSCA) CAR T cells with a Cas9/RNP method significantly enhanced T cell immunity in vivo (42). A significant antitumor response was observed after PD-1 was disrupted by genome editing. Controversially, a study indicated that T-cells without PD-1 were susceptible to exhaustion and lacked long-term durability (64). In regard to other checkpoint targets, no obvious improvement was confirmed when LAG-3 genes were deleted in CAR-T cells using CRISPR/Cas9 (47). Nevertheless, these studies still support the promise of checkpoint inhibition in CAR T cell therapy.
Recently, effective homologous recombination was shown to promote the site-specific integration of large transgenes in the T cell genome (65). In this method, after the DNA of the target gene is cleaved using Cas9 RNPs, a gene of interest is subsequently delivered to the cleavage site using adeno-associated viruses (AAVs). Site-specific transgene integration is achieved by HDR. An anti-CD19 CAR gene has been successfully integrated into the TRAC locus using the combined action of Cas9/RNP and AAV donor vectors (49). Targeting the CAR gene to the TRAC locus not only results in uniform CAR expression but also delays effector T-cell differentiation and exhaustion. Moreover, the insertion of a CAR transgene into a defined location avoids the risk of insertional oncogenesis and places CAR expression under the control of endogenous regulatory elements.
To date, although many limitations of conventional CAR T cells have been addressed with CRISPR gene editing, safety issues must be addressed before these gene-edited cells start to move into clinic. Multiple elements, such as off-target effects, Cas9 activity, target site selection, and sgRNA design, and delivery methods, can determine the efficiency and safety of the CRISPR/Cas9 system.
The first concern of CRISPR gene editing is off-target effects (66). These off-target effects might be beneficial to bacteria and archaea (67). However, several recent studies have reported unintentional CRISPR/Cas9-induced large genomic deletions or gene inversions in various species, including mouse, C. elegans, and rabbit (68–70). For human therapies, clinical safety is particularly important. Several recent studies have reported off-target effects of CRISPR in T-cells. Off-target effects introduce random mutations, thus impacting tumor-suppressor genes or activating oncogenes. Off-target effects were also observed when the TRAC or TRBC locus of CAR-T cells was inserted with CRISPR/Cas9 electroporation (42). A controversial study indicated that CRISPR gene editing could cause hundreds of unintended mutations in the genome when whole-genome sequencing was performed on a CRISPR–Cas9-edited mouse (68). Notably, another study showed that CRISPR/Cas9 genome editing resulted in a p53-mediated DNA damage response in human retinal pigment epithelial cells (71). p53 activation may lead to chromosomal rearrangements and other tumorigenic mutations in cells. Although the outcome of CRISPR-induced p53 activation is unconfirmed, it seems to decrease the gene editing efficiency. Therefore, the off-target issues must be considered in the future development of CRISPR/Cas9-edited CAR T cells. Off-target assays during CRISPR target selection may be performed to manage the safety risk of clinical CAR T trials.
Another safety concern is that unpredicted translocations may occur between double-strand breaks when multiple genes are edited (72). Although such events are rare in T cells, transformation analysis should still be performed to ensure the safety of gene-edited CAR-T therapy. In addition to the safety risk of translocations, altered functions of gene-edited CAR-T cells most likely would cause adverse effects in patients. For example, CRISPR gene disruption in CAR T cells can cause unintended innate immune responses (73).
In recent, many antitumor approaches have been developed, including target small molecules (74, 75), antibody drugs (76–84), immune cell therapy (85). Among them, CAR T cell therapy aims to treat cancer through the use of the patient's immune system. This type of therapy has many advantages, such as low toxicity and a long duration (86). However, CAR T cell therapy appears to be effective only in a limited portion of patients with hematological malignancies. CRISPR is a cutting-edge technique that can be used to generate CAR T cells with enhanced potency and safety. Although the clinical use of allogeneic donor CAR cells has been recently reported, their use is highly dependent upon either rigorous patient selection or T cell selection (25). Potential GVHD still limits the wide application of allogeneic CAR cells. Taking advantage of CRISPR, the risk of GVHD may be minimized through the deletion of endogenous TCR and MHC molecules. The additional disruption of PD-1 is believed to optimize the antitumor activities of CAR-T cells through the regulation of T-cell functions (32). The safety of gene-edited CAR T cells is the primarily concern because of notorious off-target effects. To minimize the safety risk of off-target effects, careful selection of the target site combined with prior off-target assays will be required during target site selection of CAR T cells. Although skeptics question whether CRISPR gene-edited T cell therapy is safe and ready for the clinical stage, the first CRISPR gene-editing trial using autologous T cells was initiated to treat patients with melanoma, synovial sarcoma, and multiple myeloma in 2016 (87). These potent T cells have shown merits in preclinical studies. The long-term safety profile of gene-edited CAR-T cells should be further examined in the clinic.
JL, GZ, and LZ wrote part of the manuscript; QZ wrote the manuscript.
This work was supported by the Science and Technology Development Fund of Macau (FDCT/131/2016/A3, FDCT/0015/2018/A1), the Guangzhou Science and Technology Program (201807010004), and Start-up Research Grand (SRG2016-00082-FHS) and the intramural research program of Faculty of Health Sciences, University of Macau, and National Natural Science Foundation of China (31440041).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Santomasso BD, Roberts WK, Thomas A, Williams T, Blachère NE, Dudley ME, et al. A T cell receptor associated with naturally occurring human tumor immunity. Proc Natl Acad Sci USA. (2007) 104:19073–8. doi: 10.1073/pnas.0704336104.
2. Reinherz EL. Revisiting the discovery of the αβ TCR complex and its co-receptors. Front Immunol. (2014) 5:583. doi: 10.3389/fimmu.2014.00583
3. Grenier JM, Yeung ST, Khanna KM. Combination immunotherapy: taking cancer vaccines to the next level. Front Immunol. (2018) 9:610. doi: 10.3389/fimmu.2018.00610
4. Yee C. Adoptive T cell therapy: addressing challenges in cancer immunotherapy. J Transl Med. (2005) 3:17. doi: 10.1186/1479-5876-3-17
5. Liddy N, Bossi G, Adams KJ, Lissina A, Mahon TM, Hassan NJ, et al. Monoclonal TCR-redirected tumor cell killing. Nat Med. (2012) 18:980–7. doi: 10.1038/nm.2764
6. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. (2013) 14:1014–22. doi: 10.1038/ni.2703
7. Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. (2014) 65:333–47. doi: 10.1146/annurev-med-060512-150254
8. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA. (1989) 86:10024–8.
9. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
10. Liu Y, Chen X, Han W, Zhang Y. Tisagenlecleucel, an approved anti-CD19 chimeric antigen receptor T-cell therapy for the treatment of leukemia. Drugs Today. (2017) 53:597–608. doi: 10.1358/dot.2017.53.11.2725754
11. Prasad V. Immunotherapy: tisagenlecleucel–the first approved CAR-T-cell therapy: implications for payers and policy makers. Nat Rev Clin Oncol. (2018) 15:11–2. doi: 10.1038/nrclinonc.2017.156
12. Hartmann J, Schussler-Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med. (2017) 9:1183–97. doi: 10.15252/emmm.201607485
13. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncol. (2016) 3:16011. doi: 10.1038/mto.2016.11
14. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. (2016) 127:3321–30. doi: 10.1182/blood-2016-04-703751
15. Kalaitsidou M, Kueberuwa G, Schutt A, Gilham DE. CAR T-cell therapy: toxicity and the relevance of preclinical models. Immunotherapy. (2015) 7:487–597. doi: 10.2217/imt.14.123
16. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. (2014) 6:224ra25. doi: 10.1126/scitranslmed.3008226
17. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. (2014) 124:188–95. doi: 10.1182/blood-2014-05-552729
18. Smith L, Venella K. Cytokine release syndrome: inpatient care for side effects of CAR T-cell therapy. Clin J Oncol Nursing. (2017) 21(2 Suppl.):29–34. doi: 10.1188/17.cjon.s2.29-34
19. Wang L, Ma N, Okamoto S, Amaishi Y, Sato E, Seo N, et al. Efficient tumor regression by adoptively transferred CEA-specific CAR-T cells associated with symptoms of mild cytokine release syndrome. Oncoimmunology. (2016) 5:e1211218. doi: 10.1080/2162402x.2016.1211218
20. Wang X, Riviere I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncol. (2016) 3:16015. doi: 10.1038/mto.2016.15
21. Ellis J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum Gene Ther. (2005) 16:1241–6. doi: 10.1089/hum.2005.16.1241
22. Jin C, Fotaki G, Ramachandran M, Nilsson B, Essand M, Yu D. Safe engineering of CAR T cells for adoptive cell therapy of cancer using long-term episomal gene transfer. EMBO Mol Med. (2016) 8:702–11. doi: 10.15252/emmm.201505869
23. Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Investig. (2008) 118:3132–42. doi: 10.1172/jci35700
24. Singh N, Barrett DM. Donor-derived CD19 chimeric antigen receptor T cells. Curr Opin Hematol. (2015) 22:503–8. doi: 10.1097/moh.0000000000000179
25. Yang Y, Jacoby E, Fry TJ. Challenges and opportunities of allogeneic donor-derived CAR T cells. Curr Opin Hematol. (2015) 22:509–15. doi: 10.1097/moh.0000000000000181
26. Brudno JN, Somerville RP, Shi V, Rose JJ, Halverson DC, Fowler DH, et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J Clin Oncol. (2016) 34:1112–21. doi: 10.1200/jco.2015.64.5929
27. Zhao Q, Ahmed M, Tassev DV, Hasan A, Kuo TY, Guo HF, et al. Affinity maturation of T-cell receptor-like antibodies for Wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia. (2015) 29:2238–47. doi: 10.1038/leu.2015.125
28. Lai Y, Weng J, Wei X, Qin L, Lai P, Zhao R, et al. Toll-like receptor 2 costimulation potentiates the antitumor efficacy of CAR T Cells. Leukemia. (2018) 32:801–8. doi: 10.1038/leu.2017.249
29. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. (2018) 36:346–51. doi: 10.1038/nbt.4086
30. Xu D, Jin G, Chai D, Zhou X, Gu W, Chong Y, et al. The development of CAR design for tumor CAR-T cell therapy. Oncotarget. (2018) 9:13991–4004. doi: 10.18632/oncotarget.24179
31. Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. (2013) 10:957–63. doi: 10.1038/nmeth.2649
32. Hale M, Lee B, Honaker Y, Leung WH, Grier AE, Jacobs HM, et al. Homology-directed recombination for enhanced engineering of chimeric antigen receptor T cells. Mol Ther Methods Clin Dev. (2017) 4:192–203. doi: 10.1016/j.omtm.2016.12.008
33. Ren J, Zhao Y. Advancing chimeric antigen receptor T cell therapy with CRISPR/Cas9. Protein Cell. (2017) 8:634–43. doi: 10.1007/s13238-017-0410-x
34. Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. (2014) 370:901–10. doi: 10.1056/NEJMoa1300662
35. Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. (2017) 9:eaaj2013. doi: 10.1126/scitranslmed.aaj2013
36. Liu X, Zhao Y. CRISPR/Cas9 genome editing: fueling the revolution in cancer immunotherapy. Curr Res Transl Med. (2018) 66:39–42. doi: 10.1016/j.retram.2018.04.003
37. Ehrke-Schulz E, Schiwon M, Hagedorn C, Ehrhardt A. Establishment of the CRISPR/Cas9 system for targeted gene disruption and gene tagging. Methods Mol. Biol. (2017) 1654:165–76. doi: 10.1007/978-1-4939-7231-9_11
38. Zhang JP, Li XL, Li GH, Chen W, Arakaki C, Botimer GD, et al. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. (2017) 18:35. doi: 10.1186/s13059-017-1164-8
39. Suzuki K, Tsunekawa Y, Hernandez-Benitez R, Wu J, Zhu J, Kim EJ, et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. (2016) 540:144–9. doi: 10.1038/nature20565
40. Bak RO, Porteus MH. CRISPR-mediated integration of large gene cassettes using AAV donor vectors. Cell Rep. (2017) 20:750–6. doi: 10.1016/j.celrep.2017.06.064
41. Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, et al. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. (2017) 27:154–7. doi: 10.1038/cr.2016.142
42. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. (2017) 23:2255–66. doi: 10.1158/1078-0432.CCR-16-1300
43. Seki A, Rutz S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med. (2018) 215:985–97. doi: 10.1084/jem.20171626
44. Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. (2018) 32:1970–83. doi: 10.1038/s41375-018-0065-5
45. Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget. (2017) 8:17002–11. doi: 10.18632/oncotarget.15218
46. Su S, Zou Z, Chen F, Ding N, Du J, Shao J, et al. CRISPR-Cas9-mediated disruption of PD-1 on human T cells for adoptive cellular therapies of EBV positive gastric cancer. Oncoimmunology. (2017) 6:e1249558. doi: 10.1080/2162402X.2016.1249558
47. Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, et al. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front Med. (2017) 11:554–62. doi: 10.1007/s11684-017-0543-6
48. MacLeod DT, Antony J, Martin AJ, Moser RJ, Hekele A, Wetzel KJ, et al. Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T cells. Mol Ther. (2017) 25:949–61. doi: 10.1016/j.ymthe.2017.02.005
49. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. (2017) 543:113–7. doi: 10.1038/nature21405
50. Singh N, Shi J, June CH, Ruella M. Genome-editing technologies in adoptive T cell immunotherapy for cancer. Curr Hematol Malign Rep. (2017) 12:522–9. doi: 10.1007/s11899-017-0417-7
51. Png YT, Vinanica N, Kamiya T, Shimasaki N, Coustan-Smith E, Campana D. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell malignancies. Blood Adv. (2017) 1:2348–60. doi: 10.1182/bloodadvances.2017009928
52. Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. (2017) 130:285–96. doi: 10.1182/blood-2017-01-761320
53. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. (2010) 207:2187–94. doi: 10.1084/jem.20100643
54. Wei F, Zhong S, Ma Z, Kong H, Medvec A, Ahmed R, et al. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci USA. (2013) 110:E2480–9. doi: 10.1073/pnas.1305394110
55. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. (2015) 33:1974–82. doi: 10.1200/jco.2014.59.4358
56. Bai Y, Kan S, Zhou S, Wang Y, Xu J, Cooke JP, et al. Enhancement of the in vivo persistence and antitumor efficacy of CD19 chimeric antigen receptor T cells through the delivery of modified TERT mRNA. Cell Disc. (2015) 1:15040. doi: 10.1038/celldisc.2015.40
57. Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood. (2012) 120:1412–21. doi: 10.1182/blood-2012-02-411678
58. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
59. Mamonkin M, Heslop HE. Exhausting alloreactivity of donor-derived CAR T cells. Nat Med. (2017) 23:147–8. doi: 10.1038/nm.4276
60. Xu-Monette ZY, Zhang M, Li J, Young KH. PD-1/PD-L1 blockade: have we found the key to unleash the antitumor immune response? Front Immunol. (2017) 8:1597. doi: 10.3389/fimmu.2017.01597
61. Ding W, LaPlant BR, Call TG, Parikh SA, Leis JF, He R, et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood. (2017) 129:3419–27. doi: 10.1182/blood-2017-02-765685
62. Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. (2017) 129:1039–41. doi: 10.1182/blood-2016-09-738245
63. Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. (2016) 27:1362. doi: 10.1093/annonc/mdw141
64. Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med. (2015) 212:1125–37. doi: 10.1084/jem.20142237
65. Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, Gate RE, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci USA. (2015) 112:10437–42. doi: 10.1073/pnas.1512503112
66. Peng R, Lin G, Li J. Potential pitfalls of CRISPR/Cas9-mediated genome editing. FEBS J. (2016) 283:1218–31. doi: 10.1111/febs.13586
67. Scherer S, Davis RW. Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc Natl Acad Sci USA. (1979) 76:4951–5.
68. Schaefer KA, Wu WH, Colgan DF, Tsang SH, Bassuk AG, Mahajan VB. Unexpected mutations after CRISPR-Cas9 editing in vivo. Nat Methods. (2017) 14:547–8. doi: 10.1038/nmeth.4293
69. Shin HY, Wang C, Lee HK, Yoo KH, Zeng X, Kuhns T, et al. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat Commun. (2017) 8:15464. doi: 10.1038/ncomms15464
70. Song YN, Lai LX, Li ZJ. Large-scale genomic deletions mediated by CRISPR/Cas9 system. Oncotarget. (2017) 8:5647. doi: 10.18632/oncotarget.14543
71. Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. (2018) 24:927–30. doi: 10.1038/s41591-018-0049-z
72. Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, et al. Multiplex genome-edited T-cell manufacturing platform for “Off-the-Shelf” adoptive T-cell immunotherapies. Cancer Res. (2015) 75:3853–64. doi: 10.1158/0008-5472.Can-14-3321
73. Kim S, Koo T, Jee HG, Cho HY, Lee G, Lim DG, et al. CRISPR RNAs trigger innate immune responses in human cells. Genome Res. (2018) 28:367–73. doi: 10.1101/gr.231936.117
74. Wang Y, Jiang Y, Ding S, Li J, Song N, Ren Y, et al. Small molecule inhibitors reveal allosteric regulation of USP14 via steric blockade. Cell Res. (2018) 28:1186–94. doi: 10.1038/s41422-018-0091-x
75. Zhao Q, Tran H, Dimitrov DS, Cheung NK. A dual-specific anti-IGF-1/IGF-2 human monoclonal antibody alone and in combination with temsirolimus for therapy of neuroblastoma. Int J Cancer. (2015) 137:2243–52. doi: 10.1002/ijc.29588
76. Li DZ, Han BN, Wei R, Yao GY, Chen Z, Liu J, et al. N-terminal alpha-amino group modification of antibodies using a site-selective click chemistry method. mAbs. (2018) 10:712–9. doi: 10.1080/19420862.2018.1463122
77. Chen Z, Liu J, Chu D, Shan Y, Ma G, Zhang H, et al. A dual-specific IGF-I/II human engineered antibody domain inhibits IGF signaling in breast cancer cells. Int J Biol Sci. (2018) 14:799–806. doi: 10.7150/ijbs.25928
78. Chen Z, Wang L, Xu T, Wang Q, Kang L, Zhao Q. Generation of bispecific antibodies by Fc heterodimerization and their application. Curr Pharm Biotechnol. (2016) 17:1324–32. doi: 10.2174/1389201017666161018150553
79. Li D, Gong R, Zheng J, Chen X, Dimitrov DS, Zhao Q. Engineered antibody CH2 domains binding to nucleolin: isolation, characterization and improvement of aggregation. Biochem Biophys Res Commun. (2017) 485:446–53. doi: 10.1016/j.bbrc.2017.02.058
80. Wu Y, Chen Z, Zhang P, Zhou L, Jiang T, Chen H, et al. Recombinant-fully-human-antibody decorated highly-stable far-red AIEdots for in vivo HER-2 receptor-targeted imaging. Chem. Commun. (2018) 54:7314–7. doi: 10.1039/c8cc03037e
81. Xu T, Ying T, Wang L, Zhang XD, Wang Y, Kang L, et al. A native-like bispecific antibody suppresses the inflammatory cytokine response by simultaneously neutralizing tumor necrosis factor-alpha and interleukin-17A. Oncotarget. (2017) 8:81860–72. doi: 10.18632/oncotarget.19899
82. Zhao Q, Ahmed M, Guo HF, Cheung IY, Cheung NK. Alteration of electrostatic surface potential enhances affinity and tumor killing properties of anti-ganglioside GD2 monoclonal antibody hu3F8. J Biol Chem. (2015) 290:13017–27. doi: 10.1074/jbc.M115.650903
83. Zhao Q, Feng Y, Zhu Z, Dimitrov DS. Human monoclonal antibody fragments binding to insulin-like growth factors I and II with picomolar affinity. Mol Cancer Ther. (2011) 10:1677–85. doi: 10.1158/1535-7163.MCT-11-0281
84. Wang Y, Shan Y, Gao X, Gong R, Zheng J, Zhang XD, et al. Screening and expressing HIV-1 specific antibody fragments in Saccharomyces cerevisiae. Mol Immunol. (2018) 103:279–85. doi: 10.1016/j.molimm.2018.10.013
85. Chu D, Zhao Q, Yu J, Zhang F, Zhang H, Wang Z. Nanoparticle targeting of neutrophils for improved cancer immunotherapy. Adv Healthc Mater. (2016) 5:1088–93. doi: 10.1002/adhm.201500998
86. Lipowska-Bhalla G, Gilham DE, Hawkins RE, Rothwell DG. Targeted immunotherapy of cancer with CAR T cells: achievements and challenges. Cancer Immunol Immunother. (2012) 61:953–62. doi: 10.1007/s00262-012-1254-0
Keywords: chimeric antigen receptor, CRISPR, gene editing, immunotherapy, cancer, CAR T
Citation: Liu J, Zhou G, Zhang L and Zhao Q (2019) Building Potent Chimeric Antigen Receptor T Cells With CRISPR Genome Editing. Front. Immunol. 10:456. doi: 10.3389/fimmu.2019.00456
Received: 16 October 2018; Accepted: 20 February 2019;
Published: 19 March 2019.
Edited by:
Conrad Russell Cruz, Children's National Health System, United StatesReviewed by:
Donald R. Shaffer, Jounce Therapeutics, Inc., United StatesCopyright © 2019 Liu, Zhou, Zhang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qi Zhao, cWl6aGFvQHVtLmVkdS5tbw==; emhhb3FpQGFsdW1uaS5jdWhrLm5ldA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.