 Aude Remot
Aude Remot Emilie Doz
Emilie Doz Nathalie Winter
Nathalie Winter
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 12 March 2019
Sec. Vaccines and Molecular Therapeutics
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00417
This article is part of the Research Topic Innovative Therapeutic and Vaccine Approaches Against Respiratory Pathogens View all 20 articles
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb) is one of the most prevalent lung infections of humans and kills ~1.7 million people each year. TB pathophysiology is complex with a central role played by granuloma where a delicate balance takes place to both constrain bacilli and prevent excessive inflammation that may destroy lung functions. Neutrophils reach the lung in waves following first encounter with bacilli and contribute both to early Mtb elimination and late deleterious inflammation. The hypoxic milieu where cells and bacilli cohabit inside the granuloma favors metabolism changes and the impact on TB infection needs to be more thoroughly understood. At the cellular level while the key role of the alveolar macrophage has long been established, behavior of neutrophils in the hypoxic granuloma remains poorly explored. This review will bring to the front new questions that are now emerging regarding neutrophils activity in TB. Are different neutrophil subsets involved in Mtb infection and how? How do neutrophils and close relatives contribute to shaping the granuloma immune environment? What is the role of hypoxia and hypoxia induced factors inside granuloma on neutrophil fate and functions and TB pathophysiology? Addressing these questions is key to the development of innovative host-directed therapies to fight TB.
Tuberculosis caused by Mycobacterium tuberculosis (Mtb) is present worldwide. With estimated 10.4 million new cases and 1.7 million deaths in 20161, TB remains one of the most devastating respiratory disease of human kind. The key cell in Mtb lung infection is the lung alveolar macrophage (AM) that engulfs the bacilli and orchestrates the adaptive host immune response if bacilli are not eliminated (1). This is the starting point for the granuloma, set as an immune defense mechanism that eventually becomes the pathologic signature of Mtb infection. AM plays major roles in the battle between Mtb and the host and a large body of the literature is devoted to this key cell. However, mature neutrophils circulate in high numbers in blood and are also sequestered in the lung (2). As they are present in the early phase of Mtb infection, before the onset of adaptive immunity, they could play important beneficial protective roles [see extensive review in (3)]. In the zebra-fish (ZF) embryo infected with M. marinum (Mm) as a surrogate of Mtb infection in mammals, neutrophils come in response to signals sent by Mm-infected dying macrophages (MPs). Neutrophils dispose off Mm-infected MPs by efferocytosis in the nascent granuloma and kill bacilli through NADPH oxidase-dependent mechanisms (4). We and others have shown in resistant mouse models that neutrophils come in two different waves after Mtb infection before and after the onset of adaptive immunity (5, 6). While the first wave was found to phagocytose BCG–the vaccine strain used against Mtb–in situ in the lung, the second T-cell dependent wave was seldom associated with bacilli. In response to virulent Mtb, T-cell dependent neutrophils did not control Mtb growth but rather established close contacts with T-cells in the granuloma (6) suggesting their role in regulation of the adaptive immune response. This is in line with their established role in the formation of the structured mature granuloma in mice (7). However, during active TB, it is now consensus that neutrophils are largely responsible for lung destruction (8). They are the most represented cell subset in sputum (9) and drive an interferon-inducible transcriptional signature in blood cells during active TB (10). Several excellent reviews recently covered neutrophils as “good and bad guys” during TB (3, 8, 11, 12). Such opposite roles may depend on several factors including timing and magnitude of neutrophil recruitment or different neutrophils subsets which respective roles in TB remain elusive. Despite the fact that neutrophils are established as key players in the TB granuloma, the impact of hypoxia on their behavior and functions is still poorly explored and we advocate in this review that this should be reconsidered. Moreover, in the granuloma, the influence of the hypoxic milieu on contribution of neutrophils to production of soluble mediators involved in TB pathophysiology needs to be reconsidered. The world is on high demand of host-directed therapies (HDTs) as adjunct to antibiotics to fight against TB and we hope that our mini review will help to design effective strategies by taking into account the impact of hypoxia on neutrophils.
For a long time, the granuloma has been considered as an uniquely host-driven response, set to constrain Mtb and prevent bacilli dissemination. This view was challenged when, in ZF embryo, virulent Mm was shown to disseminate and establish infection by manipulation of the nascent granuloma and adjacent stromal cells (13). Today, the host-pathogen mutual benefit of the granuloma is still a matter of debate (14, 15), as is the role of neutrophils in this structure. Some confounding interpretations may come from animal models, especially the mouse, most extensively used in TB research. In humans, primary TB leads to caseating granulomas that necrotize over time. Cavities, allowing Mtb transmission, represent the most severe signature of the disease (16). Human TB granulomas are hypoxic as demonstrated by Positron Emission Tomography-Computed Tomography (PET-CT) scans using hypoxia-specific tracers in patients with active TB (17). In TB susceptible animals such as the rabbit, the guinea pig, and the non-human primate, hypoxic granulomatous lesions develop in the lung (18). By contrast, the resistant mouse lines C57BL/6 and BALB/c that have been extensively used, do not develop necrotizing hypoxic granulomas which brought the quite general belief that mice are not an adequate model for TB pathophysiology studies (19). However, extremely susceptible mice such as C3HeB/FeJ do develop central caseous necrosis in the lung (20) and these lesions are hypoxic (21). A common feature to all TB susceptible animals that develop hypoxic necrotizing granulomas is the abundance of neutrophils (22, 23). Mtb induces necrosis of human neutrophils, which depends on its main virulence factor, the small protein ESAT-6 secreted by Type VII secretion system. Necrosis is driven by neutrophil-derived Reactive Oxygen Species (ROS) and is required for Mtb growth after uptake of infected neutrophils by human macrophages (24). In C3HeB/FeJ mice, neutrophils dying by necrosis or NETosis rather than apoptosis seem to drive the caseous necrosis and liquefaction process (25). The crucial role of neutrophils and the S100A9 inflammatory protein for granuloma formation is demonstrated (26). Therefore, what “adequate” animal models and available pathology studies in humans teach us is that neutrophils and hypoxia are crucial to the development of lung lesions during TB disease.
However, some clarification is needed regarding the definition of neutrophils. These cells have long been considered as an homogenous population based on their polylobed nucleus and a minimal set of markers: in mice, they are defined by flow cytometry as Gr1, CD11b double positive cells or more recently as CD11b, Ly-6C, Ly-6G triple positive cells. In humans, they are still minimally identified as CD11b+ CD14−CD15+ cells. The picture has become more complex with the description of Myeloid Derived Supressor Cell (MDSCs), which largely share markers with neutrophils. MDSCs are an immature and heterogeneous population present at homeostasis and accumulating in pathological situations. Originally described in cancers, MDSCs are increasingly characterized in inflammatory diseases (27, 28). MDSCs suppress T cell responses, via different mechanisms, including production of ROS, nitric oxide (NO), or arginase 1 (29). MDSCs are present as two main subsets: monocytic MDSCs and granulocytic MDSCs (Gr-MDSCs). The later display the same morphology and phenotype as bona fide neutrophils. They share the Ly-6G or Gr1 markers. Therefore, MDSCs can robustly be distinguished from bona fide neutrophils only based on their suppressive function (30). Expansion of granulocytic and monocytic MDSCs is observed in blood of active TB patients and healthy recently exposed contacts (31, 32). This correlates with enhanced L-arginine catabolism and NO production in plasma from active TB patients (33). In resistant (C57BL/6) or susceptible (129S2) strains MDSCs–defined as Gr1+ cells–are identified in the lung parenchyma during the course of Mtb infection where they suppress T cells (34). They also vigorously ingest Mtb. Interestingly, in susceptible mice, Gr1+ MDSCs cells accumulate in higher numbers and phagocytoze more bacilli as compared to resistant mice. Therefore, MDSCs could represent a niche for Mtb replication, helping the pathogen to escape the immune system (34). MDSCs are also associated with TB progression and lethality (35). These findings emphasize the potential of MDSCs as targets for immunotherapy. However, most studies using depletion antibodies that target the Gr1 or the Ly-6G surface marker, do not allow today a clear distinction of the role of bona fide neutrophils vs. MDSCs in TB pathophysiology. To add to the complexity, the low density neutrophils (LDNs) have recently been described as a new population of neutrophils. LDNs, displaying heterogeneous morphology and containing mature and immature cells, are immunosuppressive via secretion of IL-10 and expression of arginase-1 (36). Interestingly, mature high density neutrophils (HDNs) can switch to LDNs in a TGF-β dependent way, and acquire immunosuppressive functions similar to granulocytic MDSCs (37). First described in cancer (37) and pulmonary pathologies (38), LDNs have also been identified in TB and associated to the severity of the disease. Moreover, Mtb is able to convert HDNs to LDNs in vitro, suggesting manipulation by Mtb (39). Even though LDNs are not yet considered as granulocytic MDSCs, the largely shared purification procedure, analysis methods and markers between these two cell populations suggest possible overlaps (30). Mtb infection in mice recruits an altered “neutrophil” population defined as “Gr-1int/Ly-6Gint” cells with lower levels of Gr1/Ly-6G as compared to classical neutrophils. These immature cells highly express the CD115 and CD135 markers and inhibit T cell proliferation (35). Whether these cells are distinct from granulocytic MDSCs remains to be clarified.
Outside from the TB research field, recent studies identified neutrophils as potential players in inflammatory angiogenesis. Neutrophils store Vascular Endothelial Growth Factor (VEGF), a key player in the process of angiogenesis, that they may release upon stimulation. By recruiting neutrophils, MIP-1α and MIP-2 act in an autocrine loop to promote this process. A new CXCR4high and CD49dhigh neutrophil subset, displays angiogenic properties via secretion of high concentrations of MMP-9 promoting neovascularization (40). In a model of skin hypersensitization, Tan et al. demonstrated that neutrophil MMP9 and heparanase, targeting distinct domains of the extracellular matrix, cooperate to release diverse VEGF isoforms and influence their bioavailability and bioactivity during inflammatory angiogenesis (41). In mice and humans, CD49d+ CXCR4high VEGFR1high neutrophils are recruited specifically in hypoxic ischemic tissues in a VEGFR1 and VEGFR2 dependent way (42). Whether such neutrophils could contribute to formation of the hypoxic TB granuloma remains on open question.
The tremendously exciting field of immunometabolism, which links cellular bioenergetics pathways to immune cell functions, brings new views on the fate of the TB granuloma. In response to inflammatory environment, MPs switch from oxidative phosphorylation–the mitochondrial respiration system that quiescent cells use to generate energy–to aerobic glycolysis. This shift, called the Warburg effect, was discovered in proliferating cancer cells that highly incorporate glucose, that they convert to lactate while producing ATP and cell-building blocks (43). Master regulators of this switch are Hypoxia-Induced–Factors (HIFs), a family of transcription factors that govern cell reprogrammation (44). Under normoxia, the enzymes Prolyl Hydroxylase Domains (PHD) and Factor Inhibiting HIF (FIH) repress HIFs via targeted degradation and transcriptional mechanisms. Under low O2 tension, these enzymes become inactive, HIFs are stabilized and derepressed and activate a transcriptional program to adapt the cell to hypoxia. Other than O2, HIFs respond to a variety of environmental factors. HIF1α−/− mice display enhanced Mtb burden and reduced survival (45). This could be linked to HIFs regulating the bactericidal functions of MPs and neutrophils (46). NF–kb, the master regulator of the inflammatory response, regulates transcription of the hif1a gene encoding one of the HIF subunits (47). In MPs, LPS binding to TLR4 activates HIF-1α that upregulates IL-1β production. The signaling occurs through succinate, one intermediate of the tricarboxylic acid cycle (48) that accumulates upon reprogrammation of the MP toward aerobic glycolysis. However, this effect cannot be generalized to all TLR- signaling pathways (49). Imaging with glucose tracers illustrates high glucose uptake after infection with Mtb in the lungs of C3HeB/FeJ mice (50) non-human primates (51) and humans (52). Aerobic glycolysis is confirmed by NMR-analysis of metabolites in mice (53) and guinea pigs (54), or global transcriptomics of genes encoding glycolytic enzymes in the lungs of mice, rabbits, and humans (55). Reprogramming of the host metabolism translates in coordinated up and down regulation of genes encoding key glycolytic enzymes and glucose transporters, reminiscent of the Warburg effect as well as regulation of HIF-1α (55). While the granuloma becomes necrotic, MPs packed with lipid droplets transform into foamy cells (56) which is driven by reprogrammation of host lipid metabolism in response to Mtb compounds (57). Interestingly, lipid droplets formation in Mtb infected MPs is driven by IFN-γ and requires HIF-1α and its target gene hig2 (58).
Several immunopathology studies demonstrate extensive vascularization of TB granulomas in humans (59) and mice (59, 60) provided that they are not necrotic (61). The link between hypoxia, vascularization, and development of the granuloma was recently established in the ZF infected with Mm (62). In this model, HIF-1α is activated, PHD-3 expression is increased and induces production of the angiogenic factor VGEF-A, which is intimately linked to nascent granuloma formation. In human MPs infected with Mtb, a similar angiogienic signature is observed (63). Moreover, the level of VEGF-A is increased in sputum and peripheral blood of active TB patients and is proposed as a differentiating biomarker for patients progressing to active TB (64–66). Circulating angiogenic factors are markers of disease severity and are associated to the bacterial burden (67). In ZF embryos infected by Mm, CXCR4 signaling is important to initiate angiogenesis for granuloma expansion (68). Surprisingly, despite the established over-representation of neutrophils in TB lesions, little information is available on how these cells behave in face of Mtb in the highly hypoxic and angiogenic granuloma milieu.
Neutrophils are generally seen as short-lived cells. However, the life span of neutrophils is highly increased in hypoxic milieu (69). By high consumption of oxygen during oxidative burst, neutrophils themselves contribute to generate the hypoxic milieu, which may well be the case during active TB when they invade the lung. Prolonged survival is linked to sustained expression of PHD3, in vitro and in vivo, in response to hypoxia and inflammatory stimuli (70). Interestingly, PHD3 is strongly induced in lungs of Mtb infected mice (55). HIF-2α is the most expressed in neutrophils, in contrast to MPs where HIF-1α is the most active. HIF-2α deficiency increases neutrophil apoptosis (71). MIP-1 is also identified as a novel hypoxia stimulated granulocyte survival factor (72).
In the ZF model infected with Mm, neutrophil-specific HIF-1α stabilization decreases bacterial burden via a NO-dependent mechanism. On the contrary, despite also being upregulated, HIF-2α has a negative impact on bacterial burden emphasizing opposite roles of different HIF factors (73). Therefore, it is possible that the hypoxic environment of the TB granuloma that favors extended life-span for neutrophils allows them to actively shape granuloma evolution. On one hand, this may help bacilli control as well as resolution of inflammation, since neutrophils actively participate to MP efferocytosis and the release of lipids such as LXA4. On the other hand, hypoxia increases neutrophil degranulation and confers extended activity to damage lung tissues in a PI3K dependent pathway (74). Hypoxia-induced decrease of neutrophil apoptosis induces a delay in resolution of inflammation by maintaining active neutrophils in the inflamed tissue (75). Moreover, hypoxia impairs the ability of neutrophils to kill certain bacteria (76).
HIF-1α is a major player in an another chronic infection caused by the intracellular parasite Leishmania. HIF-1α crucially enhances immunosuppressive functions of MDSCs and decreases leishmanicidal capacity of myeloid cells (77). Even though to our knowledge no study has tackled the link between HIF-1α and MDSCs in TB, a similar important role could be discovered. Also, since hypoxia and angiogenesis are intimately linked to the granuloma development, another interesting perspective is the possible role of angiogenic neutrophils (40, 42) in the process.
Neutrophils contribute both pro- and anti-inflammatory factors in TB (78, 79). Information on how HIF-1α stabilization in hypoxic environment influences the secretion of critical immune mediators by neutrophils is limited to granule proteases, antimicrobial peptides and TNF (46). Literature on the impact of HIF-1α stabilization on MP-released mediators is more extensive and we consider it as a source of inspiration illustrating the potential role of hypoxia on neutrophil-released mediators (Figure 1). In the following paragraph, we focus on how hypoxia may regulate release by neutrophils of the key mediators in Mtb virulence: ROS, NO, IL-1β, and type I IFN. Some of these mediators may play different roles in humans and animal models and data should sometimes be interpreted with caution. In Mtb infected MPs, HIF-1α is stabilized by IFN-γ and regulates the production of prostaglandins and NO (45). In mice, NO not only acts as an antimicrobial agent and inflammatory mediator but further amplifies myeloid cell bactericidal activity via HIF-1α stabilization. NO modulates the MP response to Mtb through activation of HIF-1α and repression of NF-kB (80). HIF-1α and NO are intrinsically linked: they positively regulate each other, but display distinct roles in the regulation of inflammation. Among the mediators regulated in opposite directions, neutrophil-attracting chemokines, IL-1α and IL-1β, are all down regulated in HIF-1α−/− and upregulated in Nos2−/− Mtb infected and IFN-γ activated BMDMs (80). In the hypoxic granuloma, NO produced by IFNγ-activated MPs restricts neutrophil recruitment to avoid destructive inflammation (80). In Mm infected ZF, HIF-1α stabilization induced IL-1β production by MPs and increased neutrophil NO production that is protective against infection (81). In Mm infected NADPH oxidase 2-deficient mice (Ncf1−/−) mice, ROS-deficiency decreases IL-1β production by MPs but induces early and extensive neutrophilic inflammation, with high elastase activity and IL-1β production (82). This also reveals a novel role for ROS in the early neutrophilic granulomatous inflammation and the importance of neutrophil-driven IL-1β production during mycobacterial infection. In addition to MPs, neutrophils also produce ROS and NOS. Neutrophils are able to discriminate pathogens by differential production and localization of ROS, and tune their own recruitment and distribution to exquisitely tailor the anti-microbial response (83). HIF-1α stabilization in neutrophils induces NO production after infection by Mm (73). NOS and ROS production also influences the secretion of cytokines. NO inhibits NLRP3-dependent IL-1 responses (84). IL-1β signaling is also important for ROS production as Mtb-infected newly recruited neutrophils lacking IL-1R fail to produce ROS, resulting in compromised pathogen control (85). HIF-1α stabilization clearly influences ROS/NOS and IL-1β production by MPs and neutrophils, both factors are important during mycobacterial infection, but their regulation seems different in the two cell types (73, 80–82, 84, 85) (Figure 1). In human neutrophils stimulated with Mtb, hypoxia up-regulates secretion of MMP-8, MMP-9 and neutrophil elastase that are involved in matrix destruction. Hypoxia inhibits NETs formation and both neutrophil apoptosis and necrosis after direct stimulation by Mtb (69).
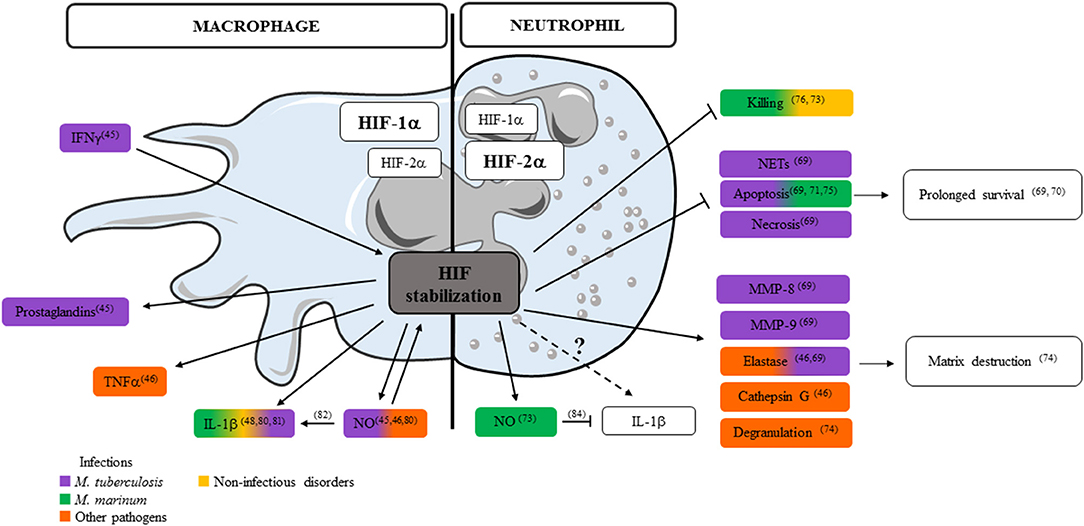
Figure 1. Impact of HIFs on control of key mediators released by neutrophils and macrophages. Key mediators and essential cellular processes controlled by HIF stabilization in macrophage (left) or neutrophil (right) after infections with M. tuberculosis, M. marinum, or other bacteria or during non-infectious disorders are depicted (numbers refer to publications listed in the review).
Type I IFN is a major cytokine in TB pathophysiology. Overproduction of type I IFN (IFN-I) is linked to exacerbated TB in both mouse models and humans. IFN-I triggers immunopathology by increasing the recruitment of inflammatory monocytes and neutrophils to the lung (86). Secretion of IFN-I by MPs and its effect on neutrophils is well-documented (87–89). In MPs, Mtb triggers IFN-I secretion through bacterial DNA release in the cytosol. However, strains display variable ability to activate the IFN-I pathway depending on their effective triggering of mitochondrial stress (87). Host-protective cytokines such as TNF, IL-12, and IL-1β are inhibited by exogenous IFN-I, via production of immunosuppressive IL-10 (88). By contrast, IL-1β suppresses IFN-I through eicosanoid prostaglandin E2 (90). In the inflammatory environment in mouse tumor models, IFN-I shifts neutrophils to antimetastastic phenotype (89). Therefore, IFN-I has multiple and crucial effects on neutrophils, but so far studies on IFN signaling in neutrophils in hypoxic environment are still scarce. As hypoxia leads to accumulation of cytosolic DNA via mitochondrial or nuclear DNA damage, it could favor activation of the cGAS/STING/IRF3 pathway (91). The convergence between hypoxia and IFN-I signaling is suggested by Karuppagounder et al. who identified the effect of Tilorone, a small molecule inducing IFN-I which also triggered hypoxic response in brain cells (91). Another study claims that IFN-I promotes tumorigenic properties through up-regulation of HIF functions in different cancer cell lines (92). Direct IFN-I secretion by neutrophils is proposed, where Sox2 could act as a sequence-specific DNA sensor in neutrophils during microbial infection (93). However, it is unclear if neutrophils can sense DNA via the cGAS/STING pathway (94).
Thus, even though the impact of hypoxic environment encountered in the TB granuloma on the IFN-I pathway is not documented yet, this issue could be of great interest to better understand TB pathophysiology and propose new therapies.
Although it is now consensus that during active TB, neutrophils are the main villains responsible for lung destruction, we–and others (3, 11)–advocate that this narrow vision is revisited. “Neutrophils” encompass different subsets with various functions that remain poorly defined. They come to infectious foci in waves of different magnitude. A better definition of neutrophil subsets, their coordinated dynamics of recruitment to the lung and their associated functions is needed. Neutrophils crosstalk with other cells and secrete a vast number of mediators thus taking full part to the regulation of the immune response against Mtb. They respond to signals sent by their environment, including hypoxia in inflamed tissues. In the hypoxic TB granuloma, light has been recently shed on the fate and behavior of MPs, under the control of the master regulator HIF-1α. However, there is currently scarce information on the fate and behavior of neutrophils in a similar context. How do neutrophils respond to hypoxia in the TB context? How do neutrophils contribute to the shaping of the granuloma? In the future, models allowing development of the hypoxic TB granuloma should be favored. A better definition of mediators released by neutrophils in the hypoxic context of the granuloma is expected. As neutrophils are key players in the game, we believe that these questions need to be solved in order to propose new interventions to fight against TB.
In the era of increasing multidrug resistance of Mtb strains, HDTs sometimes represent the last hope for patients. As the hallmark of destructive inflammation, neutrophils are often considered as potential targets. Inhibiting necrotic neutrophil death could restore Mtb growth control (24). Removing neutrophils by drugs or immunotherapeutic interventions could also alleviate lung tissue destruction.
Recent studies in the TB field shed some light on parallels that could be drawn between the TB granuloma and solid tumors (Table 1), especially regarding the role of neutrophils. Since HDTs are more advanced in the field of cancer than they are in TB, we propose that some tracks well-developed in cancer therapy are explored to advance the field of HDTs for TB patients. Among promising avenues, we propose that metabolic changes occurring in TB granuloma are being considered (113). Modulation of the HIF pathways (114) deserves attention as it could dampen excessive protease secretion (69). PHD3 and HIF-2α that operate in neutrophils under inflammatory or hypoxic conditions (70, 71) represent more attractive targets than largely distributed HIF-1α. In cancer, another active field in the clinics is blocking angiogenesis since this pathway is key to tumor development. Angiogenesis appeared more recently as key to the development of the TB granuloma and it would be interesting to determine whether modulating angiogenesis could bring some benefit to TB patients. Along this line, we believe that recently described angiogenic neutrophils should also be investigated in TB.

Table 1. Impact of neutrophils and close relatives in cancer and TB.
TB still kills 1.7 million people each year and active TB patients continuously spread bacilli that represent threat to human kind. Development of adjunct HDTs is a promising avenue to boost current drug regimen directed against Mtb (115). We believe that addressing the questions that we raised in this review about neutrophils in TB could greatly help in the quest for innovative HDTs.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
AR is supported by a grant from Agence Nationale de la Recherche under the Carnot Program France Futur Elevage.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Pieters J. Mycobacterium tuberculosis and the macrophage: maintaining a balance. Cell Host Microbe. (2008) 3:399–407. doi: 10.1016/j.chom.2008.05.006
2. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
3. Lyadova IV. Neutrophils in tuberculosis: heterogeneity shapes the way? Mediators Inflamm. (2017) 2017:8619307. doi: 10.1155/2017/8619307
4. Yang CT, Cambier CJ, Davis JM, Hall CJ, Crosier PS, Ramakrishnan L. Neutrophils exert protection in the early tuberculous granuloma by oxidative killing of mycobacteria phagocytosed from infected macrophages. Cell Host Microbe. (2012) 12:301–12. doi: 10.1016/j.chom.2012.07.009
5. Appelberg R, Silva M. T cell-dependent chronic neutrophilia during mycobacterial infections. Clin Exp Immunol. (1989) 78:478–83.
6. Lombard R, Doz E, Carreras F, Epardaud M, Le Vern Y, Buzoni-Gatel D, et al. IL-17RA in non-hematopoietic cells controls CXCL-1 and 5 critical to recruit neutrophils to the lung of mycobacteria-infected mice during the adaptive immune response. PLoS ONE. (2016) 11:e0149455. doi: 10.1371/journal.pone.0149455
7. Seiler P, Aichele P, Bandermann S, Hauser A, Lu B, Gerard N, et al. Early granuloma formation after aerosol Mycobacterium tuberculosis infection is regulated by neutrophils via CXCR3-signaling chemokines. Eur J Immunol. (2003) 33:2676–86. doi: 10.1002/eji.200323956
8. Dallenga T, Schaible UE. Neutrophils in tuberculosis–first line of defence or booster of disease and targets for host-directed therapy? Pathog Dis. (2016) 74:ftw012. doi: 10.1093/femspd/ftw012
9. Eum SY, Kong JH, Hong MS, Lee YJ, Kim JH, Hwang SH, et al. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest. (2010) 137:122–8. doi: 10.1378/chest.09-0903
10. Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. (2010) 466:973–7. doi: 10.1038/nature09247
11. Kroon E, Coussens A, Kinnear C, Orlova M, Möller M, Seeger A, et al. Neutrophils: innate effectors of TB resistance? Front Immunol. (2018) 9:2637. doi: 10.3389/fimmu.2018.02637
12. Warren E, Teskey G, Venketaraman V. Effector mechanisms of neutrophils within the innate immune system in response to Mycobacterium tuberculosis infection. J Clin Med. 6:E15. doi: 10.3390/jcm6020015
13. Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol. (2012) 12:352–66. doi: 10.1038/nri3211
14. Ehlers S, Schaible UE. The granuloma in tuberculosis: dynamics of a host-pathogen collusion. Front Immunol. (2012) 3:411. doi: 10.3389/fimmu.2012.00411
15. Pagan AJ, Ramakrishnan L. Immunity and immunopathology in the tuberculous granuloma. Cold Spring Harb Perspect Med. 5:a018499. doi: 10.1101/cshperspect.a018499
16. Hunter RL. Pathology of post primary tuberculosis of the lung: an illustrated critical review. Tuberculosis. (2011) 91:497–509. doi: 10.1016/j.tube.2011.03.007
17. Belton M, Brilha S, Manavaki R, Mauri F, Nijran K, Hong YT, et al. Hypoxia and tissue destruction in pulmonary TB. Thorax. (2016) 71:1145–53. doi: 10.1136/thoraxjnl-2015-207402
18. Via LE, Lin PL, Ray SM, Carrillo J, Allen SS, Eum SY, et al. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect Immun. (2008) 76:2333–40. doi: 10.1128/IAI.01515-07
19. Orme IM, Basaraba RJ. The formation of the granuloma in tuberculosis infection. Semin Immunol. (2014) 26:601–9. doi: 10.1016/j.smim.2014.09.009
20. Pan H, Yan B-S, Shebzukhov YV, Zhou H, Kobzik L. Ipr1 gene mediates innate immunity to tuberculosis. Nature. (2005) 434:767–72. doi: 10.1038/nature03419
21. Harper J, Skerry C, Davis SL, Tasneen R, Weir M, Kramnik I, et al. Mouse model of necrotic tuberculosis granulomas develops hypoxic lesions. J Infect Dis. (2012) 205:595–602. doi: 10.1093/infdis/jir786
22. Mattila JT, Maiello P, Sun T, Via LE, Flynn JL. Granzyme B-expressing neutrophils correlate with bacterial load in granulomas from Mycobacterium tuberculosis-infected cynomolgus macaques. Cell Microbiol. (2015) 17:1085–97. doi: 10.1111/cmi.12428
23. Turner OC, Basaraba RJ, Orme IM. Immunopathogenesis of pulmonary granulomas in the guinea pig after infection with Mycobacterium tuberculosis. Infect Immun. (2003) 71:864–71. doi: 10.1128/IAI.71.2.864-871.2003
24. Dallenga T, Repnik U, Corleis B, Eich J, Reimer R, Griffiths GW, et al. M. tuberculosis-tnduced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe. (2017) 22:519–30.e513. doi: 10.1016/j.chom.2017.09.003
25. Marzo E, Vilaplana C, Tapia G, Diaz J, Garcia V, Cardona PJ. Damaging role of neutrophilic infiltration in a mouse model of progressive tuberculosis. Tuberculosis. (2014) 94:55–64. doi: 10.1016/j.tube.2013.09.004
26. Yoshioka Y, Mizutani T, Mizuta S, Miyamoto A, Murata S, Ano T, et al. Neutrophils and the S100A9 protein critically regulate granuloma formation. Blood Adv. (2016) 1:184–92. doi: 10.1182/bloodadvances.2016000497
27. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. (2008) 181:5791–802. doi: 10.4049/jimmunol.181.8.5791
28. Zhang C, Lei GS, Shao S, Jung HW, Durant PJ, Lee CH. Accumulation of myeloid-derived suppressor cells in the lungs during Pneumocystis pneumonia. Infect Immun. (2012) 80:3634–41. doi: 10.1128/IAI.00668-12
29. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
30. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
31. du Plessis N, Loebenberg L, Kriel M, von Groote-Bidlingmaier F, Ribechini E, Loxton AG, et al. Increased frequency of myeloid-derived suppressor cells during active tuberculosis and after recent Mycobacterium tuberculosis infection suppresses T-cell function. Am J Respir Crit Care Med. (2013) 188:724–32. doi: 10.1164/rccm.201302-0249OC
32. Yang B, Wang X, Jiang J, Zhai F, Cheng X. Identification of CD244-expressing myeloid-derived suppressor cells in patients with active tuberculosis. Immunol Lett. (2014) 158:66–72. doi: 10.1016/j.imlet.2013.12.003
33. El Daker S, Sacchi A, Tempestilli M, Carducci C, Goletti D, Vanini V, et al. Granulocytic myeloid derived suppressor cells expansion during active pulmonary tuberculosis is associated with high nitric oxide plasma level. PLoS ONE. (2015) 10:e0123772. doi: 10.1371/journal.pone.0123772
34. Knaul JK, Jorg S, Oberbeck-Mueller D, Heinemann E, Scheuermann L, Brinkmann V, et al. Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am J Respir Crit Care Med. (2014) 190:1053–66. doi: 10.1164/rccm.201405-0828OC
35. Tsiganov EN, Verbina EM, Radaeva TV, Sosunov VV, Kosmiadi GA, Nikitina IY, et al. Gr-1dimCD11b+ immature myeloid-derived suppressor cells but not neutrophils are markers of lethal tuberculosis infection in mice. J Immunol. (2014) 192:4718–27. doi: 10.4049/jimmunol.1301365
36. Cloke T, Munder M, Taylor G, Muller I, Kropf P. Characterization of a novel population of low-density granulocytes associated with disease severity in HIV-1 infection. PLoS ONE. (2012) 7:e48939. doi: 10.1371/journal.pone.0048939
37. Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. (2015) 10:562–73. doi: 10.1016/j.celrep.2014.12.039
38. Fu J, Tobin MC, Thomas LL. Neutrophil-like low-density granulocytes are elevated in patients with moderate to severe persistent asthma. Ann Allergy Asthma Immunol. (2014) 113:635–40.e632. doi: 10.1016/j.anai.2014.08.024
39. Deng Y, Ye J, Luo Q, Huang Z, Peng Y, Xiong G, et al. Low-density granulocytes are elevated in mycobacterial infection and associated with the severity of tuberculosis. PLoS ONE. (2016) 11:e0153567. doi: 10.1371/journal.pone.0153567
40. Christoffersson G, Vagesjo E, Vandooren J, Liden M, Massena S, Reinert RB, et al. VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood. (2012) 120:4653–62. doi: 10.1182/blood-2012-04-421040
41. Tan KW, Chong SZ, Wong FH, Evrard M, Tan SM, Keeble J, et al. Neutrophils contribute to inflammatory lymphangiogenesis by increasing VEGF-A bioavailability and secreting VEGF-D. Blood. (2013) 122:3666–77. doi: 10.1182/blood-2012-11-466532
42. Massena S, Christoffersson G, Vagesjo E, Seignez C, Gustafsson K, Binet F, et al. Identification and characterization of VEGF-A-responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood. (2015) 126:2016–26. doi: 10.1182/blood-2015-03-631572
43. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
44. Taylor CT, Colgan SP. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat Rev Immunol. (2017) 17:774–85. doi: 10.1038/nri.2017.103
45. Braverman J, Sogi KM, Benjamin D, Nomura DK, Stanley SA. HIF-1α is an essential mediator of IFN-γ-dependent immunity to Mycobacterium tuberculosis. J Immunol. (2016) 197:1287–97. doi: 10.4049/jimmunol.1600266
46. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, et al. HIF-1α expression regulates the bactericidal capacity of phagocytes. J Clin Invest. (2005) 115:1806–15. doi: 10.1172/JCI23865
47. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature. (2008) 453:807–11. doi: 10.1038/nature06905
48. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
49. Lachmandas E, Boutens L, Ratter JM, Hijmans A, Hooiveld GJ, Joosten LA, et al. Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat Microbiol. (2016) 2:16246. doi: 10.1038/nmicrobiol.2016.246
50. Davis SL, Nuermberger EL, Um PK, Vidal C, Jedynak B, Pomper MG, et al. Noninvasive pulmonary [18F]-2-fluoro-deoxy-D-glucose positron emission tomography correlates with bactericidal activity of tuberculosis drug treatment. Antimicrob Agents Chemother. (2009) 53:4879–84. doi: 10.1128/AAC.00789-09
51. Coleman M, Maiello P, Tomko J, Frye L, Fillmore D, Janssen C, et al. Early Changes by (18)Fluorodeoxyglucose positron emission tomography coregistered with computed tomography predict outcome after Mycobacterium tuberculosis infection in cynomolgus macaques. Infect Immun. (2014) 82:2400–4. doi: 10.1128/IAI.01599-13
52. Kim IJ, Lee JS, Kim SJ, Kim YK, Jeong YJ, Jun S, et al. Double-phase 18F-FDG PET-CT for determination of pulmonary tuberculoma activity. Eur J Nucl Med Mol Imaging. (2008) 35:808–14. doi: 10.1007/s00259-007-0585-0
53. Shin JH, Yang JY, Jeon BY, Yoon YJ, Cho SN, Kang YH, et al. (1)H NMR-based metabolomic profiling in mice infected with Mycobacterium tuberculosis. J Proteome Res. (2011) 10:2238–47. doi: 10.1021/pr101054m
54. Somashekar BS, Amin AG, Rithner CD, Troudt J, Basaraba R, Izzo A, et al. Metabolic profiling of lung granuloma in Mycobacterium tuberculosis infected guinea pigs: ex vivo 1H magic angle spinning NMR studies. J Proteome Res. (2011) 10:4186–95. doi: 10.1021/pr2003352
55. Shi L, Eugenin EA, Subbian S. Immunometabolism in tuberculosis. Front Immunol. (2016) 7:150. doi: 10.3389/fimmu.2016.00150
56. Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol. (2009) 10:943–8. doi: 10.1038/ni.1781
57. Kim MJ, Wainwright HC, Locketz M, Bekker LG, Walther GB, Dittrich C, et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med. (2010) 2:258–74. doi: 10.1002/emmm.201000079
58. Knight M, Braverman J, Asfaha K, Gronert K, Stanley S. Lipid droplet formation in Mycobacterium tuberculosis infected macrophages requires IFN-γ/HIF-1α signaling and supports host defense. PLoS Pathog. (2018) 14:e1006874. doi: 10.1371/journal.ppat.1006874
59. Tsai MC, Chakravarty S, Zhu G, Xu J, Tanaka K, Koch C, et al. Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell Microbiol. (2006) 8:218–32. doi: 10.1111/j.1462-5822.2005.00612.x
60. Aly S, Laskay T, Mages J, Malzan A, Lang R, Ehlers S. Interferon-γ-dependent mechanisms of mycobacteria-induced pulmonary immunopathology: the role of angiostasis and CXCR3-targeted chemokines for granuloma necrosis. J Pathol. (2007) 212:295–305. doi: 10.1002/path.2185
61. Ulrichs T, Kosmiadi G, Jörg S, Pradl L, Titukhina M, Mishenko V, et al. Differential organization of the local immune response in patients with active cavitary tuberculosis or with non-progressive tuberculoma. J Infect Dis. (2005) 192:89–97. doi: 10.1086/430621
62. Oehlers SH, Cronan MR, Scott NR, Thomas MI, Okuda KS, Walton EM, et al. Interception of host angiogenic signalling limits mycobacterial growth. Nature. (2015) 517:612–5. doi: 10.1038/nature13967
63. Polena H, Boudou F, Tilleul S, Dubois-Colas N, Lecointe C, Rakotosamimanana N, et al. Mycobacterium tuberculosis exploits the formation of new blood vessels for its dissemination. Sci Rep. (2016) 6:33162. doi: 10.1038/srep33162
64. Abe Y, Nakamura M, Oshika Y, Hatanaka H, Tokunaga T, Ohkubo Y, et al. Serum levels of vascular endothelial growth factor and cavity formation in active pulmonary tuberculosis. Respiration. (2001) 68:496–500. doi: 10.1159/000050557
65. Alatas F, Alatas O, Metintas M, Ozarslan A, Erginel S, Yildirim H. Vascular endothelial growth factor levels in active pulmonary tuberculosis. Chest.(2004) 125:2156–9. doi: 10.1378/chest.125.6.2156
66. Ota M, Mendy J, Donkor S, Togun T, Daramy M, Gomez M, et al. Rapid diagnosis of tuberculosis using ex vivo host biomarkers in sputum. Eur Respir J. (2014) 44:254–7. doi: 10.1183/09031936.00209913
67. Kumar NP, Banurekha VV, Nair D, Babu S. Circulating angiogenic factors as biomarkers of disease severity and bacterial burden in pulmonary tuberculosis. PLoS ONE. (2016) 11:e0146318. doi: 10.1371/journal.pone.0146318
68. Torraca V, Tulotta C, Snaar-Jagalska BE, Meijer AH. The chemokine receptor CXCR4 promotes granuloma formation by sustaining a mycobacteria-induced angiogenesis programme. Sci Rep. (2017) 7:45061. doi: 10.1038/srep45061
69. Ong CWM, Fox K, Ettorre A, Elkington PT, Friedland JS. Hypoxia increases neutrophil-driven matrix destruction after exposure to Mycobacterium tuberculosis. Sci Rep. (2018) 8:11475. doi: 10.1038/s41598-018-29659-1
70. Walmsley SR, Chilvers ER, Thompson AA, Vaughan K, Marriott HM, Parker LC, et al. Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J Clin Invest. (2011) 121:1053–63. doi: 10.1172/JCI43273
71. Thompson AA, Elks PM, Marriott HM, Eamsamarng S, Higgins KR, Lewis A, et al. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood. (2014) 123:366–76. doi: 10.1182/blood-2013-05-500207
72. Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1α-dependent NF-kappaB activity. J Exp Med. (2005) 201:105–15. doi: 10.1084/jem.20040624
73. Elks PM, Brizee S, van der Vaart M, Walmsley SR, van Eeden FJ, Renshaw SA, et al. Hypoxia inducible factor signaling modulates susceptibility to mycobacterial infection via a nitric oxide dependent mechanism. PLoS Pathog. (2013) 9:e1003789. doi: 10.1371/journal.ppat.1003789
74. Hoenderdos K, Lodge KM, Hirst RA, Chen C, Palazzo SG, Emerenciana A, et al. Hypoxia upregulates neutrophil degranulation and potential for tissue injury. Thorax. (2016) 71:1030–8. doi: 10.1136/thoraxjnl-2015-207604
75. Elks PM, van Eeden FJ, Dixon G, Wang X, Reyes-Aldasoro CC, Ingham PW, et al. Activation of hypoxia-inducible factor-1α (Hif-1α) delays inflammation resolution by reducing neutrophil apoptosis and reverse migration in a zebrafish inflammation model. Blood. (2011) 118:712–22. doi: 10.1182/blood-2010-12-324186
76. McGovern NN, Cowburn AS, Porter L, Walmsley SR, Summers C, Thompson AAR, et al. Hypoxia selectively inhibits respiratory burst activity and killing of Staphylococcus aureus in human neutrophils. J Immunol. (2011) 186:453–63. doi: 10.4049/jimmunol.1002213
77. Hammami A, Abidin BM, Charpentier T, Fabie A, Duguay AP, Heinonen KM, et al. HIF-1α is a key regulator in potentiating suppressor activity and limiting the microbicidal capacity of MDSC-like cells during visceral leishmaniasis. PLoS Pathog. (2017) 13:e1006616. doi: 10.1371/journal.ppat.1006616
78. Etna MP, Giacomini E, Severa M, Coccia EM. Pro- and anti-inflammatory cytokines in tuberculosis: a two-edged sword in TB pathogenesis. Semin Immunol. (2014) 26:543–51. doi: 10.1016/j.smim.2014.09.011
79. Domingo-Gonzalez R, Prince O, Cooper A, Khader SA. Cytokines and chemokines in Mycobacterium tuberculosis infection. Microbiol Spectr. (2016) 4:1–37. doi: 10.1128/microbiolspec.TBTB2-0018-2016
80. Braverman J, Stanley SA. Nitric oxide modulates macrophage responses to Mycobacterium tuberculosis infection through activation of HIF-1α and repression of NF-kappaB. J Immunol. (2017) 199:1805–16. doi: 10.4049/jimmunol.1700515
81. Ogryzko NV, Lewis A, Wilson HL, Meijer AH, Renshaw SA, Elks PM. Hif-1α-induced expression of Il-1β protects against mycobacterial infection in zebrafish. J Immunol. (2019) 202:494–502. doi: 10.4049/jimmunol.1801139
82. Chao WC, Yen CL, Hsieh CY, Huang YF, Tseng YL, Nigrovic PA, et al. Mycobacterial infection induces higher interleukin-1β and dysregulated lung inflammation in mice with defective leukocyte NADPH oxidase. PLoS ONE. (2017) 12:e0189453. doi: 10.1371/journal.pone.0189453
83. Warnatsch A, Tsourouktsoglou TD, Branzk N, Wang Q, Reincke S, Herbst S, et al. Reactive oxygen species localization programs inflammation to clear microbes of different size. Immunity. (2017) 46:421–32. doi: 10.1016/j.immuni.2017.02.013
84. Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat Immunol. (2013) 14:52–60. doi: 10.1038/ni.2474
85. Di Paolo NC, Shafiani S, Day T, Papayannopoulou T, Russell DW, Iwakura Y, et al. Interdependence between interleukin-1 and tumor necrosis factor regulates TNF-dependent control of Mycobacterium tuberculosis infection. Immunity. (2015) 43:1125–36. doi: 10.1016/j.immuni.2015.11.016
86. Dorhoi A, Yeremeev V, Nouailles G, Weiner JIII, Jorg S, Heinemann E, et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur J Immunol. (2014) 44:2380–93. doi: 10.1002/eji.201344219
87. Wiens KE, Ernst JD. The mechanism for type I interferon induction by Mycobacterium tuberculosis is bacterial strain-dependent. PLoS Pathog. (2016) 12:e1005809. doi: 10.1371/journal.ppat.1005809
88. McNab FW, Ewbank J, Howes A, Moreira-Teixeira L, Martirosyan A, Ghilardi N, et al. Type I IFN induces IL-10 production in an IL-27-independent manner and blocks responsiveness to IFN-γ for production of IL-12 and bacterial killing in Mycobacterium tuberculosis-infected macrophages. J Immunol. (2014) 193:3600–12. doi: 10.4049/jimmunol.1401088
89. Pylaeva E, Lang S, Jablonska J. The essential role of type I interferons in differentiation and activation of tumor-associated neutrophils. Front Immunol. (2016) 7:629. doi: 10.3389/fimmu.2016.00629
90. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. (2014) 511:99–103. doi: 10.1038/nature13489
91. Karuppagounder S, Zhai Y, Chen Y, He R, Ratan R. The interferon response as a common final pathway for many preconditioning stimuli: unexpected crosstalk between hypoxic adaptation and antiviral defense. Condition. Med. (2018) 1:143–50. Available online at: http://www.conditionmed.org/Data/View/1626
92. Yeh YH, Hsiao HF, Yeh YC, Chen TW, Li TK. Inflammatory interferon activates HIF-1α-mediated epithelial-to-mesenchymal transition via PI3K/AKT/mTOR pathway. J Exp Clin Cancer Res. (2018) 37:70. doi: 10.1186/s13046-018-0730-6
93. Xia P, Wang S, Ye B, Du Y, Huang G, Zhu P, et al. Sox2 functions as a sequence-specific DNA sensor in neutrophils to initiate innate immunity against microbial infection. Nat Immunol. (2015) 16:366–75. doi: 10.1038/ni.3117
94. Yu Z, Chen T, Cao X. Neutrophil sensing of cytoplasmic, pathogenic DNA in a cGAS–STING-independent manner. Cell Mol Immunol. (2015) 13:411–4. doi: 10.1038/cmi.2015.34
95. Zilio S, Serafini P. Neutrophils and granulocytic MDSC: the janus god of cancer immunotherapy. Vaccines. (2016) 4:E31. doi: 10.3390/vaccines4030031
96. Kiss M, Van Gassen S, Movahedi K, Saeys Y, Laoui D. Myeloid cell heterogeneity in cancer: not a single cell alike. Cell Immunol. (2018) 330:188–201. doi: 10.1016/j.cellimm.2018.02.008
97. Martineau AR, Newton SM, Wilkinson KA, Kampmann B, Hall BM, Nawroly N, et al. Neutrophil-mediated innate immune resistance to mycobacteria. J Clin Invest. (2007) 117:1988–94. doi: 10.1172/JCI31097
98. Okamoto Yoshida Y, Umemura M, Yahagi A, O'Brien RL, Ikuta K, Kishihara K, et al. Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J Immunol. (2010) 184:4414–22. doi: 10.4049/jimmunol.0903332
99. Umemura M, Yahagi A, Hamada S, Begum M, Watanabe H, Kawakami K, et al. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J Immunol. (2007) 178:3786–96. doi: 10.4049/jimmunol.178.6.3786
100. Triner D, Xue X, Schwartz AJ, Jung I, Colacino JA, Shah YM. Epithelial hypoxia-inducible factor 2α facilitates the progression of colon tumors through recruiting neutrophils. Mol Cell Biol. 37:e00481–16. doi: 10.1128/MCB.00481-16
101. Blaisdell A, Crequer A, Columbus D, Daikoku T, Mittal K, Dey SK, et al. Neutrophils oppose uterine epithelial carcinogenesis via debridement of hypoxic tumor cells. Cancer Cell. (2015) 28:785–99. doi: 10.1016/j.ccell.2015.11.005
102. Rodriguez P, Quiceno D, Zabaleta J, Ortiz B, Zea A, Piazuelo M, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. (2004) 64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465
103. Hurt B, Schulick R, Edil B, El Kasmi KC, Barnett C Jr. Cancer-promoting mechanisms of tumor-associated neutrophils. Am J Surg. (2017) 214:938–44. doi: 10.1016/j.amjsurg.2017.08.003
104. El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol. (2008) 9:1399–406. doi: 10.1038/ni.1671
105. Monin L, Griffiths KL, Lam WY, Gopal R, Kang DD, Ahmed M, et al. Helminth-induced arginase-1 exacerbates lung inflammation and disease severity in tuberculosis. J Clin Invest. (2015) 125:4699–713. doi: 10.1172/JCI77378
106. Pessanha AP, Martins RA, Mattos-Guaraldi AL, Vianna A, Moreira LO. Arginase-1 expression in granulomas of tuberculosis patients. FEMS Immunol Med Microbiol. (2012) 66:265–8. doi: 10.1111/j.1574-695X.2012.01012.x
107. Bekes EM, Schweighofer B, Kupriyanova TA, Zajac E, Ardi VC, Quigley JP, et al. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am J Pathol. (2011) 179:1455–70. doi: 10.1016/j.ajpath.2011.05.031
108. Deryugina EI, Zajac E, Juncker-Jensen A, Kupriyanova TA, Welter L, Quigley JP. Tissue-infiltrating neutrophils constitute the major in vivo source of angiogenesis-inducing MMP-9 in the tumor microenvironment. Neoplasia. (2014) 16:771–88. doi: 10.1016/j.neo.2014.08.013
109. Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. (2010) 16:219–23. doi: 10.1038/nm.2084
110. Ong CW, Elkington PT, Brilha S, Ugarte-Gil C, Tome-Esteban MT, Tezera LB, et al. Neutrophil-derived MMP-8 drives AMPK-dependent matrix destruction in human pulmonary tuberculosis. PLoS Pathog. (2015) 11:e1004917. doi: 10.1371/journal.ppat.1004917
111. Elkington PT, Ugarte-Gil CA, Friedland JS. Matrix metalloproteinases in tuberculosis. Eur Respir J. (2011) 38:456–64. doi: 10.1183/09031936.00015411
112. Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ, Tanguy M, et al. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood. (2005) 106:1843–50. doi: 10.1182/blood-2005-03-1281
113. Qualls JE, Murray PJ. Immunometabolism within the tuberculosis granuloma: amino acids, hypoxia, and cellular respiration. Semin Immunopathol. (2016) 38:139–52. doi: 10.1007/s00281-015-0534-0
114. Balamurugan K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int J Cancer. (2016) 138:1058–66. doi: 10.1002/ijc.29519
Keywords: neutrophils, Mycobacterium tuberculosis, granuloma, lung, HIF, hypoxia, host-directed therapies
Citation: Remot A, Doz E and Winter N (2019) Neutrophils and Close Relatives in the Hypoxic Environment of the Tuberculous Granuloma: New Avenues for Host-Directed Therapies? Front. Immunol. 10:417. doi: 10.3389/fimmu.2019.00417
Received: 26 November 2018; Accepted: 18 February 2019;
Published: 12 March 2019.
Edited by:
Cynthia Calzas, Institut National de la Recherche Agronomique (INRA), FranceReviewed by:
Max Bastian, Friedrich Loeffler Institut, GermanyCopyright © 2019 Remot, Doz and Winter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathalie Winter, bmF0aGFsaWUud2ludGVyQGlucmEuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.