 Yi Miao1,2,3†
Yi Miao1,2,3† Hua-Yuan Zhu1,2,3†
Hua-Yuan Zhu1,2,3† Chun Qiao1,2,3
Chun Qiao1,2,3 Yi Xia1,2,3
Yi Xia1,2,3 Yiling Kong1,2,3
Yiling Kong1,2,3 Yi-Xin Zou1,2,3
Yi-Xin Zou1,2,3 Yu-Qing Miao1,2,3
Yu-Qing Miao1,2,3 Xiao Chen1,2,3
Xiao Chen1,2,3 Lei Cao1,2,3Wei Wu1,2,3
Lei Cao1,2,3Wei Wu1,2,3 Jin-Hua Liang1,2,3
Jin-Hua Liang1,2,3 Jia-Zhu Wu1,2,3
Jia-Zhu Wu1,2,3 Li Wang1,2,3Lei Fan1,2,3
Li Wang1,2,3Lei Fan1,2,3 Wei Xu1,2,3*
Wei Xu1,2,3* Jian-Yong Li1,2,3*
Jian-Yong Li1,2,3*- 1Department of Hematology, The First Affiliated Hospital of Nanjing Medical University, Jiangsu Province Hospital, Nanjing, China
- 2Key Laboratory of Hematology, Nanjing Medical University, Nanjing, China
- 3Collaborative Innovation Center for Cancer Personalized Medicine, Nanjing Medical University, Nanjing, China
Hemophagocytic lymphohistiocytosis (HLH) can be classified into primary HLH and secondary HLH. Primary HLH usually occurs in infants and children with an underlying genetic defect, and there are also teens and occasional adults with primary HLH. Most cases with secondary HLH are adult patients with secondary triggers including infections, malignancies, and autoimmune diseases. The distinction between primary HLH and secondary HLH seems to be less straightforward, as patients with secondary HLH may also have genetic defects while primary HLH can be triggered by secondary causes. In this study, using amplicon-based targeted gene sequencing (TGS), we sequenced eighteen HLH-related genes in 112 adult HLH cases, which were mostly secondary HLH. Mutations or rare variants were identified in 48 cases (42.9%). All the variants except one were missense variants, and biallelic gene mutations were identified in 3 cases in which only one case harbored homogenous missense mutation. Recurrent variants including UNC13D p.G863D and AP3B1 p.T359A are much more prevalent in our cohort than in normal East Asian population, and in silico analysis predicted pathogenicity of these variants. In conclusion, according to our study, genetic defects may also contribute to the development of adult HLH cases or secondary HLH cases.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is characterized by overwhelming inflammatory cytokine storm. Patients with HLH usually present with fever, hepatosplenomegaly and cytopenia (1). Traditionally, HLH can be classified into primary and secondary HLH according to the underlying etiologies. Primary HLH is caused by genetic defects and always present in young children, and increasing evidence suggests that primary HLH could also occur in teens and young adults (2, 3). Secondary HLH, mainly identified in adults, is mostly triggered extrinsic factors including infections, malignancies or autoimmune diseases (2). Suppressing cytokine storm using HLH-94 or HLH-2004 regimen is essential in the induction treatment of HLH, however, the subsequent treatments vary dependent on the underlying etiologies (4). For patients with primary HLH, allogeneic hematopoietic stem cell transplantation needs to be considered, while for secondary HLH, treatments focused on eliminating the secondary triggers are of vital importance (4).
In recent years, increasing evidence suggests that the distinction between primary HLH and secondary HLH may be less clear. HLH could be result of the combination of inherited genetic mutations, susceptibility loci and extrinsic triggers. The cases with so-called “secondary” HLH may also have genetic defects in cytotoxic lymphocyte function. Recently, adult cases with HLH caused by influenza A (H1N1) infection were found to have heterozygous LYST mutations or PRF1 p.A91V mutation, which led to deceased NK cell cytolytic function (5). Gene mutations related to HLH were also identified in patients with macrophage activation syndrome (MAS)/HLH, which was secondary to rheumatic diseases (6, 7). The study by Zhang et al. showed that adults with HLH might also harbor missense or splice-site variants in familial HLH associated genes, however, in this study only 3 genes were studied by Sanger sequencing (3). To better understand the genetic background of adult HLH, we performed TGS of 18 genes related to HLH in a cohort of 112 adult patients with HLH.
Methods
Patients
This study included 112 patients diagnosed as HLH. The diagnosis of HLH was based on HLH-2004 criteria, and only cases that fulfilled 5 or more HLH-2004 criteria were diagnosed as HLH and included in our study (8). Every participant provided informed consent according to the Declaration of Helsinki. This study was approved by the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University.
Targeted Gene Sequencing
A targeted gene sequencing (TGS) panel consisting of 18 HLH-associated genes was designed (Table 1). We used genome build hg19 as reference and included all the exons in the above genes into this panel. The genomic DNA was isolated using peripheral blood or bone marrow samples using QIAamp DNA Blood Mini Kit (Qiagen). Amplification was performed with 20 ng gDNA using Ion AmpliSeq Library Kit 2.0 for each multiplex PCR reaction (Ion Torrent, Thermo Fisher Scientific). Libraries were then constructed using KAPA HyperPlus Kits (Kapa Biosystems, Wilmington, MA, USA). After purification using microbeads (Agencourt AMPure XP reagent, Beckman Coulter, Brea, CA, USA), libraries were quantified by Qubit 2.0 Fluorometer (Life Technologies) and diluted to a concentration of 1 ng/μl. The libraries were pooled and denatured, and further diluted to 12 pmol/L. The pooled libraries (600 μl) were then sequenced on Illumina Miseq sequencing platform or Illumina Hiseq sequencing platform. The median value of the mean coverage for each sample was 4,712 (range: 1,694–14,368).
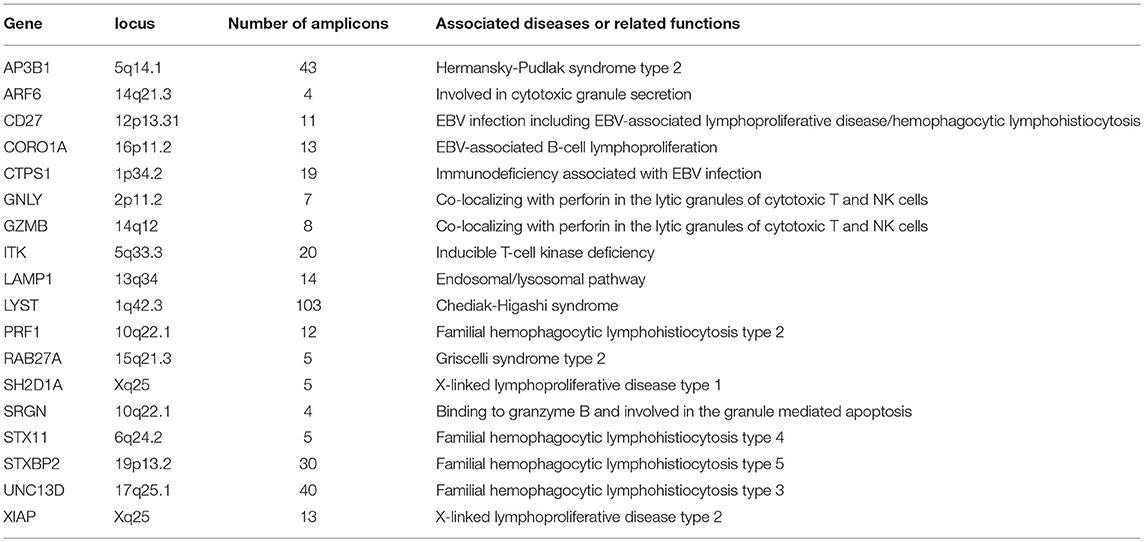
Table 1. Genes included in targeted sequencing panel.
Bioinformatics and Statistical Analyses
We used Burrows-Wheeler Aligner to map the sequencing reads to hg19 genome (9). Only bases that were covered by at least 50 reads were qualified for variant calling. Variant calling was performed using the SAMtools software (10). Integrative Genomics Viewer (IGV) version 2.3.32 was used for visualization of sequencing reads and visual assessment of detected variants (11). To confirm a heterozygous variant, variant alleles were required to be present in more than 15% of mapped reads. The Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/) was referred to extract the frequencies of variants in normal Eastern Asian population. And 1,000 Genomes Project (http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/) was referred to evaluate the frequencies of variants in the whole population. Variants those with mean allele frequencies lower than 1% in normal Eastern Asia population were included into further analysis. In silico analysis of candidate variants was carried out using SIFT (12), Polyphen-2 (13), and MutationTaster (14).
Student t-test was used to compare continuous variables and fisher exact test or chi-square test was used to compare categorical variables. Kaplan-Meier method was used to construct survival curves and log-rank test was used to compare the difference. The primary endpoint in our study was survival at 8 weeks after admission to our hospital. Statistical analyses were performed using Graphpad Prism 6 (GraphPad Software, San Diego, CA) software. P ≤ 0.05 (2-sided) was considered statistically significant.
Results
Mutations or Rare Variants Identified by TGS
Totally, 112 cases were analyzed in this study, which included 66 cases of malignancy-associated HLH, 23 infection-associated HLH, 4 autoimmune disease-associated HLH as well as 19 HLH with unknown origin. Almost all the cases of malignancy-associated HLH were lymphoma related (including mature lymphoid malignancies in leukemic phase), except two cases. The median age was 41.5 and the male/female ratio was about 3:2. There are more male patients in our cohort, as most of patients are lymphoma patients (64/112, 57.1%). It is known that, in major subtypes of lymphoma, including diffuse large B cell lymphoma, peripheral T cell lymphoma and extranodal NK/T cell lymphoma, there are more male patients (15).
Forty-eight patients (48/112, 42.9%) harbored mutations or rare variants in genes studied in the TGS panel. Except for one nonsense mutation, all the mutations were missense base substitutions. One case had homozygous mutation (P6, UNC13D p.G863D) and two cases had compound heterozygous mutations (P38, CORO1A p.L235V and p.P277R; P24, PRF1 p.R33H, and p.R4C). Three patients were double heterozygous. In cases with double heterozygous mutations, P21 had heterozygous STX11 p.F281C mutation and LYST p.D2665G mutation, and STX11 and LYST are both involved in the degranulation of cytotoxic cells, suggesting these two mutations may cooperate in the development of HLH. P32 had a single ITK p.R581W mutation and a single STX11 p.F281C mutation. And P40 had a CORO1A p.A88T mutation and a LYST p.E1036A mutation. The remaining cases (42/48, 87.5%) carried only one mutated allele in one of these genes.
Of 18 genes tested in our cohort, 14 genes were found to be mutated at least one time, and UNC13D was the most frequently mutated gene (Figure 1). Thirty-nine mutations or rare variants were identified in this cohort, and in silico analysis and frequencies in normal Eastern Asian population (from ExAC database) and 1,000 genomes of these variants were summarized in Table 2. Some of the variants were previously reported in cases of HLH and the references were provided in Table 2 (16–25). And the conditions and clinical significance of variants documented in ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar) were also listed in Table 2. We further classified these variants based on the pathogenicity predicted by the algorithms. These variants that were predicted to be pathogenic (including possibly pathogenic) or not pathogenic by all three algorithms were classified as “pathogenic variants” (n = 12) or “benign variants” (n = 7), respectively. The remaining variants were classified as “variants of uncertain significance” (n = 20). The variants involving UNC13D, LYST, STXBP2, ITK, and AP3B1 were depicted in Figure 2.
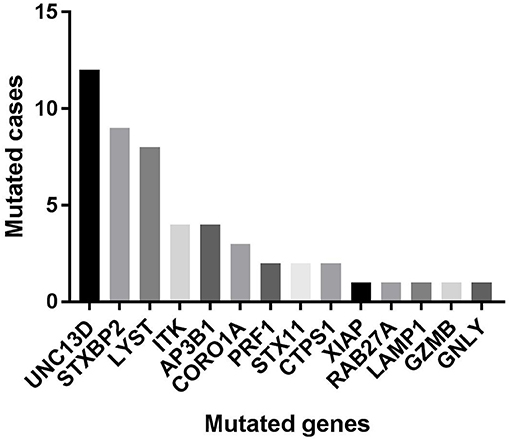
Figure 1. The numbers of cases with each mutated gene in our cohort.
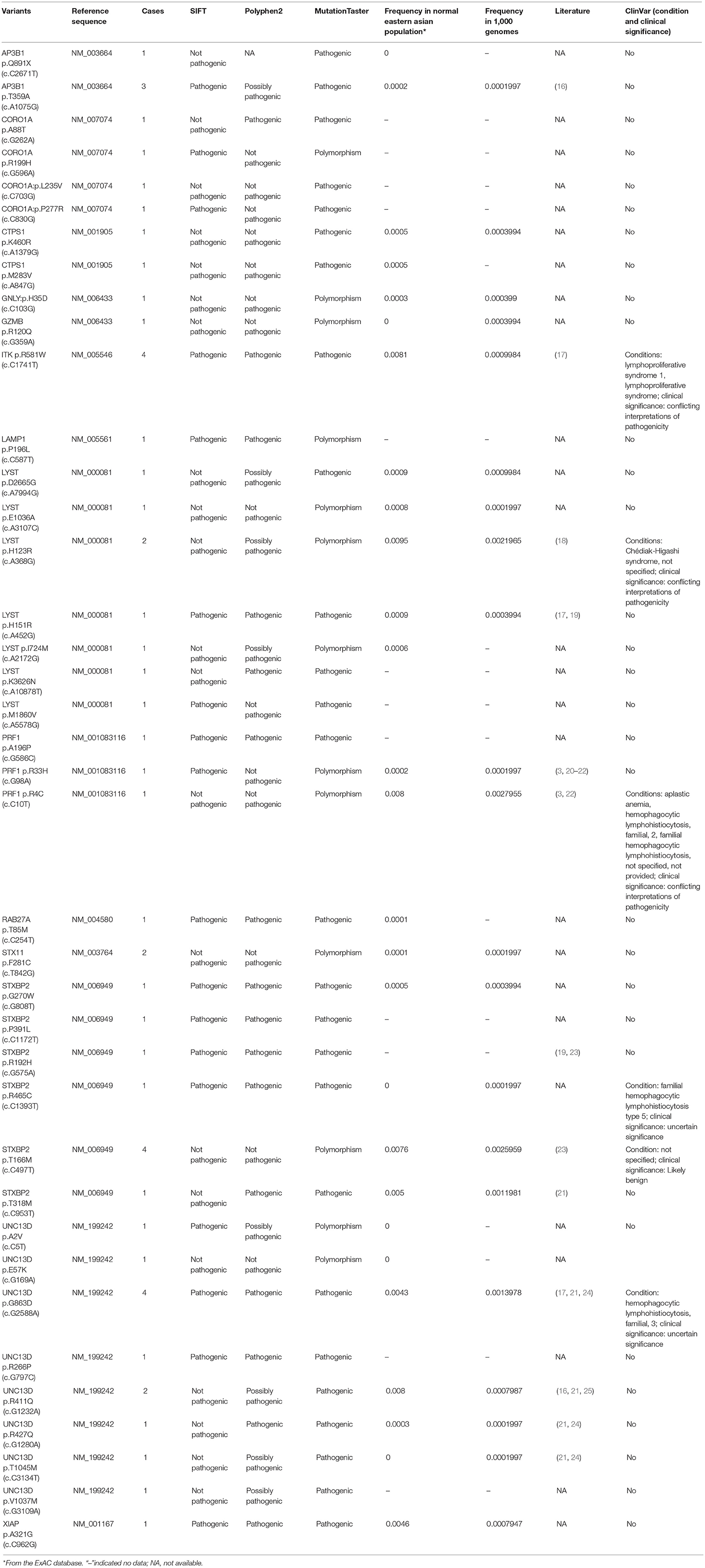
Table 2. Summary of variants detected in our cohort.
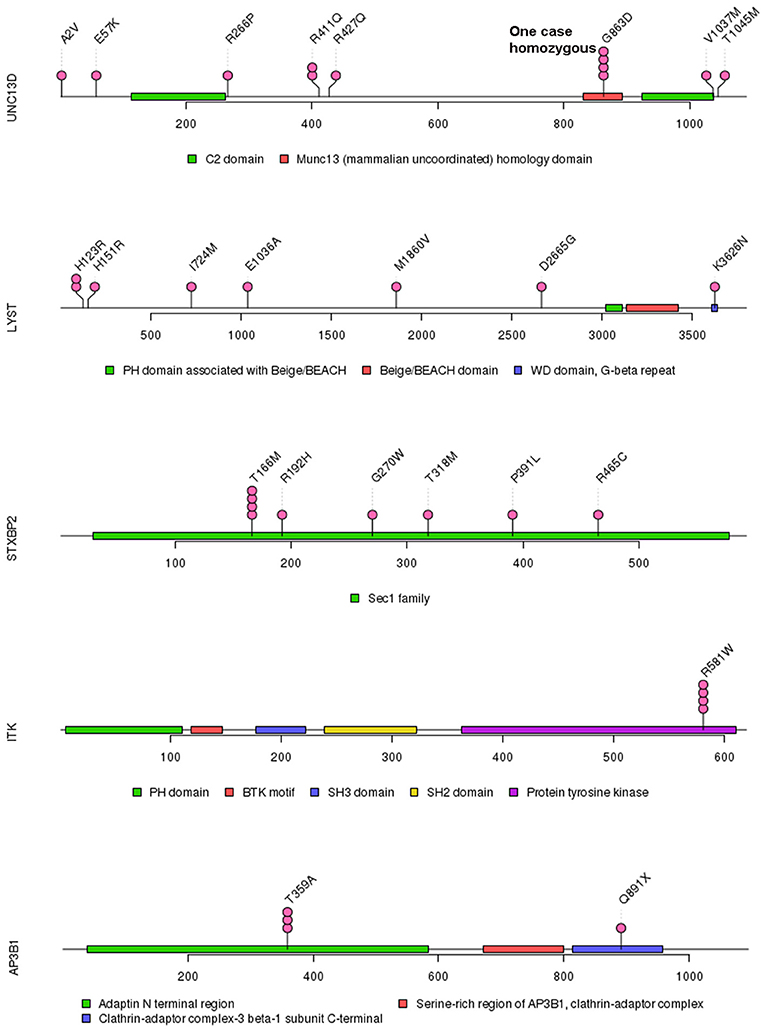
Figure 2. Mutations in UNC13D, LYST, STXBP2, ITK, and AP3B1.
Of these variants, seven variants were recurrently identified (Table 2). UNC13D p.G863D was identified in four patients, including one patient with homozygous mutation. The allele frequency of UNC13D p.G863D in our cohort was higher than that in normal East Asian population [5/224 [2.2%] vs. 37/8638 [0.4%]; odds ratio [OR] 5.307, 95%CI 2.066–13.64, p = 0.0007]. In silico analysis with three algorithms revealed that the functional consequence of UNC13D p.G863D missense mutation was damaging, suggesting UNC13D p.G863D was pathogenic in the development of HLH in these four cases. Recurrent ITK p.R581W mutation occurred in 4 cases, and the allele frequency of this mutation was also higher than that in normal East Asian population [4/224 [1.8%] vs. 70/8652 [0.8%], OR 2.229, 95% CI 0.8065–6.161; p = 0.1165], although not statistically significant. Prediction using three algorithms all suggested this amino acid substitution produced damaging effects to ITK protein. AP3B1 p.T359A occurred more frequently in our cohort as compared with normal East Asian population [3/224 [1.4%] vs. 2/8648 [0.02%], OR 58.68, 95% CI 9.753-353.1, p = 0.0002], meanwhile, in silico analysis also confirmed the pathogenicity of this mutation. Four cases had the rare variant STXBP2 p.T166M, and the frequency of this variant in the HLH cohort did not differ with that in normal East Asian population [4/224 [1.8%] vs. 5/660 [0.75%], OR 2.382, 95% CI 0.6338–8.951, p = 0.2422], and in silico prediction showed that this variant was benign to the function of the protein, suggesting this variant may not carry pathological significance in HLH.
The Association of Variants With Clinical Variables and Short-Term Prognosis
The age of cases with identified variants was similar to those without identified variants (median: 46.5 years vs. 48 years, p = 0.3310). The frequency of cases with mutations or variants did not differ among four groups, which were classified by underlying causes (Figure 3A). For genes mutated in > = 3 cases, the mutation rate of individual gene did not vary among these four groups (Table 3). The frequency of mutation in at least one of these genes involved in the degranulation pathway including UNC13D, STXBP2, LYST, and AP3B1 did not differ significantly among these four groups (Table 3). There was no association between the presence of gene mutations or variants and EBV infection (EBV+ 21/52 vs. EBV− 27/60, p = 0.7030). However, while considering specific gene mutations, we found there was a trend that cases of EBV infection had more frequent CORO1A mutations than the rest of our cohort [3/52 [6.4%] vs. 0/60 [0.0%], OR 8.556, 95% CI: 0.4313–169.7, p = 0.0970], which suggested that CORO1A mutation may lead to an increased risk of EBV infection.
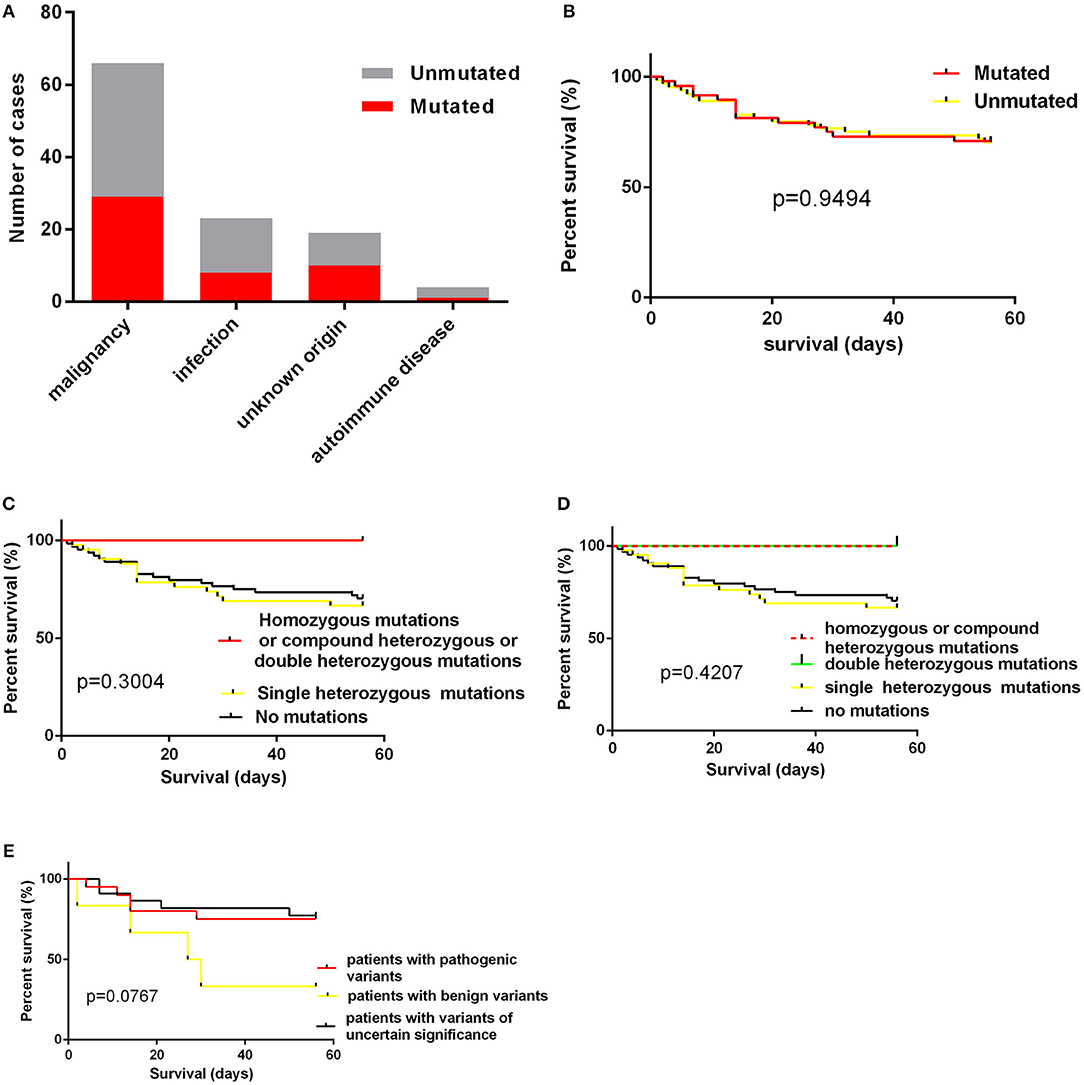
Figure 3. (A) the numbers of cases with mutations according to underlying causes; (B) the impact of presence of mutations on short-term survival; (C,D) the impact of homozygous/compound heterozygous mutations or double heterozygous mutations on short-term survival; (E) the short term survival of patients with pathogenic variants, patients with benign variants and patients with variants of uncertain significance.
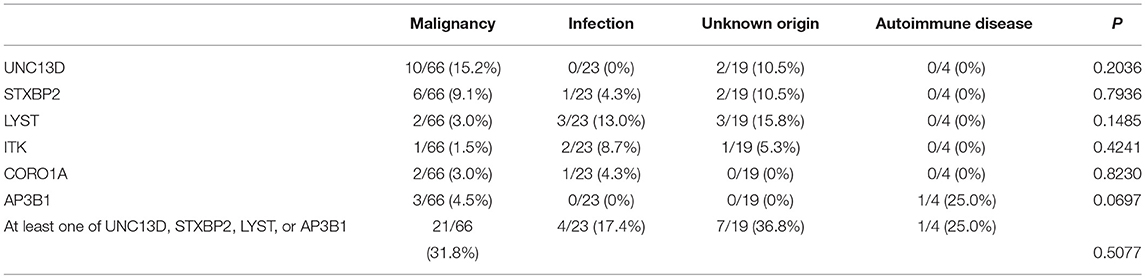
Table 3. Mutation rate of UNC13D, STXBP2, LYST, ITK, CORO1A, and AP3B1 in different groups.
We also examined the impact of the presence of gene mutations on short-term survival of HLH patients and found that the detection of HLH-associated gene mutations did not confer a worse short-term prognosis (Figure 3B).The outcome of patients with homozygous or compound heterozygous mutations (n = 3) or patients with double heterozygous mutations (n = 3) was not significantly different from that of others (Figures 3C,D). And we classified patients with variants or mutations into three groups: patients with pathogenic variants (at least one pathogenic variant) (n = 20), patients with benign variants (for cases with two variants, both two variants predicted to be benign) (n = 6), and patients with variants of uncertain significance (n = 22). The underlying causes did not differ among these three groups. We also found that there was no significant difference in short-term survival among these three groups. There was a trend that patients with benign variants had a worse short-term outcome (Figure 3E, p = 0.0767). However, it is hard to draw a conclusion as there were only six patients in this group.
Discussion
The advent of high-throughput sequencing allows sequencing of hundreds of genes at one time. In the present study, we sequenced 18 HLH-related genes in 112 cases adults HLH patients using TGS. These 18 genes included genes that have been reported to be related to the pathogenesis of HLH as well as those involved in cytotoxicity of NK cells or T cells, which may potentially contribute to the development of HLH. Mutations or rare variants in HLH-related genes were uncovered in cases with or without underlying causes. In contrast to primary HLH, in which homozygous mutations or compound heterozygous mutations were always identified, most of patients in our cohort with mutations only harbored one mutated allele (26). Only one case with homozygous mutation and two cases of compound heterozygous mutations were identified. Three cases with double heterozygous mutations were identified, suggesting mutations of two genes may cooperate in the pathogenesis of HLH. Additionally, almost all the variants were missense mutations in our cohort, which is different from that in pediatric cases in which nonsense and frame-shift mutations are much more prevalent (26), suggesting that most of variants identified in adult HLH may partly impair rather than completely abrogate the function of affected proteins.
In our study, the most frequently mutated gene was UNC13D, which was different from a previous study by Wang et al. in which PRF1 was the most mutated gene (20). A possible explanation for this discrepancy is different populations included in two studies (20). Other reasons could be also possible. Several recurrent gene mutations were identified. The most frequent recurrent variant in our cohort was UNC13D p.G863D. Interestingly, homozygous UNC13D p.G863D mutation has been previously reported in a male adult HLH patient and his asymptomatic sister also harbored this mutation (27). Both of them displayed impaired NK cell activity and decreased CD107a expression, suggesting the pathogenic role of UNC13D p.G863D mutation in HLH (27). UNC13D p.G863D mutation is in the MHD2 protein domain of UNC13D, which mediates localization on recycling endosomes and lysosome (28). Homozygous missense ITK mutations have been linked to fatal EBV-associated lymphoproliferation (29). In our cohort, ITK p.R581W was found to be potentially associated with HLH and ITK p.R581W mutation has be reported to be associated with EBV associated T/NK-cell lymphoproliferative diseases and familial HLH (17, 30). However, we did not confirm any significant association between ITK p.R581W mutation and EBV infection status in our cohort, suggesting that further functional studies to characterize the biological significance of ITK p.R581W is necessary. Another important recurrent variant identified in our cohort is AP3B1 p.T359A, which has been previously reported in a case of adult hemophagocytic lymphohistiocytosis (16). Although some variants in our cohort are not recurrent, the absence of these variants in normal East Asian population and in silico analysis of them suggest the variants are possibly pathogenic in HLH.
Our results suggest that, adult cases with HLH including those with an identified second cause could have possible genetic defects. These genetic defects, mostly heterozygous missense mutations, do not lead to HLH in normal conditions. When a second trigger such as lymphoma occurs, these cases with genetic defects have an increased risk of developing HLH, when compared with those cases without genetic defects. Cases with these mutations show a similar clinical outcome to those without mutations, suggest these mutations may not confer a poor prognosis. Therefore, these heterozygous mutations, which are different from those in pediatric familial HLH cases, could be classified as hypomorphic mutations (3). Regarding therapeutic options, these cases with heterozygous mutations may not need to receive frontline allogenic stem cell transplantation, which are used to treat pediatric cases, since the outcome of these cases are dependent on the underlying causes (31).
As this is a retrospective study, we are not able to conduct immunologic experiments including NK cell cytotoxicity, perforin or CD107a expression tests in our patients, making it less possible for us to analyze the association between those phenotypic characteristics and genetic variants. Although TGS enables simultaneous sequencing of multiple genes, in some cases, it remains insufficient for exploring the mechanisms underlying development of HLH. According to a study in pediatric cases (32), whole-exome sequencing (WES) can help find molecular explanations for the pathogenesis of HLH when target gene sequencing fails. This may also apply to adult cases with HLH.
In conclusion, targeted gene sequencing of HLH-related genes in adult HLH patients revealed a possible role of genetic factors in the pathogenesis of adult HLH. Further studies, which include immunologic tests and genetic testing, especially WES, will be helpful in characterizing the role of genetic factors in the development of adult HLH.
Author Contributions
J-YL and WX designed and supervised the study. YM, H-YZ, J-YL, and WX analyzed the data and drafted the manuscript. YX, YK, Y-XZ, Y-QM, XC, CQ, and LC performed the experiments and collected the data. J-HL, J-ZW, LW, WW, and LF collected the clinical data. All authors read and approved the final manuscript.
Funding
Supported by National Natural Science Foundation of China (81720108002), Project of National Key Clinical Specialty, Excellent Youth Foundation Project of Jiangsu Province (BK20160099), Jiangsu Province's Medical Elite Program (ZDRCA2016022), Project of National Key Clinical Specialty, Jiangsu Provincial Special Program of Medical Science (BL2014086 and BE2017751) and National Science and Technology Major Project (2018ZX09734007).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Brisse E, Matthys P, Wouters CH. Understanding the spectrum of haemophagocytic lymphohistiocytosis: update on diagnostic challenges and therapeutic options. Br J Haematol. (2016) 174:175–87. doi: 10.1111/bjh.14144
2. Wysocki CA. Comparing hemophagocytic lymphohistiocytosis in pediatric and adult patients. Curr Opin Allergy Clin Immunol. (2017) 17:405–13. doi: 10.1097/ACI.0000000000000405
3. Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood. (2011) 118:5794–8. doi: 10.1182/blood-2011-07-370148
4. Wang Y, Wang Z. Treatment of hemophagocytic lymphohistiocytosis. Curr Opin Hematol. (2017) 24:54–8. doi: 10.1097/MOH.0000000000000302
5. Schulert GS, Zhang M, Fall N, Husami A, Kissell D, Hanosh A, et al. Whole-exome sequencing reveals mutations in genes linked to hemophagocytic lymphohistiocytosis and macrophage activation syndrome in fatal cases of H1N1 influenza. J Infect Dis. (2016) 213:1180–8. doi: 10.1093/infdis/jiv550
6. Filocamo G, Petaccia A, Torcoletti M, Sieni E, Ravelli A, Corona F. Recurrent macrophage activation syndrome in spondyloarthritis and monoallelic missense mutations in PRF1: a description of one paediatric case. Clin Exp Rheumatol. (2016) 34:719.
7. Zhang M, Behrens EM, Atkinson TP, Shakoory B, Grom AA, Cron RQ. Genetic defects in cytolysis in macrophage activation syndrome. Curr Rheumatol Rep. (2014) 16:439. doi: 10.1007/s11926-014-0439-2
8. Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2007) 48:124–31. doi: 10.1002/pbc.21039
9. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
10. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. (2009) 25:2078–9. doi: 10.1093/bioinformatics/btp352
11. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. (2011) 29:24–6. doi: 10.1038/nbt.1754
12. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. (2003) 31:3812–4. doi: 10.1093/nar/gkg509
13. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
14. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. (2010) 7:575–6. doi: 10.1038/nmeth0810-575
15. Xu W, Fan L, Miao Y, Xu H, Yu L, Xu X, et al. [Distribution of lymphomas subtypes in Jiangsu Province: a multicenter analysis of 5 147 cases]. Zhonghua Xue Ye Xue Za Zhi. (2014) 35:300–3. doi: 10.3760/cma.j.issn.0253-2727.2014.04.011
16. Gao L, Zhu L, Huang L, Zhou J. Synergistic defects of UNC13D and AP3B1 leading to adult hemophagocytic lymphohistiocytosis. Int J Hematol. (2015) 102:488–92. doi: 10.1007/s12185-015-1807-z
17. Li CR, Gao LL, Yang L, Xiao M, Zhou JF, Zhang YC. Clinical and genetic features of Epstein-Barr virus-triggered late-onset primary hemophagocytic lymphohistiocytosis: 10 pedigrees study. Blood. (2018) 132(Suppl. 1):3903. doi: 10.1182/blood-2018-99-114009
18. Mukda E, Trachoo O, Pasomsub E, Tiyasirichokchai R, Iemwimangsa N, Sosothikul D, et al. Exome sequencing for simultaneous mutation screening in children with hemophagocytic lymphohistiocytosis. Int J Hematol. (2017) 106:282–90. doi: 10.1007/s12185-017-2223-3
19. Xu XJ, Wang HS, Ju XL, Xiao PF, Xiao Y, Xue HM, et al. Clinical presentation and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in China: a retrospective multicenter study. Pediatr Blood Cancer. (2017) 64:e26264. doi: 10.1002/pbc.26264
20. Wang Y, Wang Z, Zhang J, Wei Q, Tang R, Qi J, et al. Genetic features of late onset primary hemophagocytic lymphohistiocytosis in adolescence or adulthood. PLoS ONE. (2014) 9:e107386. doi: 10.1371/journal.pone.0107386
21. Chen X, Wang F, Zhang Y, Teng W, Wang M, Nie D, et al. Genetic variant spectrum in 265 Chinese patients with hemophagocytic lymphohistiocytosis: molecular analyses of PRF1, UNC13D, STX11, STXBP2, SH2D1A, and XIAP. Clin Genet. (2018) 94:200–12. doi: 10.1111/cge.13363
22. Zhou XH, Luo JM, Bin Q, Huang XH. [Expression of porforin and granzyme B in familial hemophagocytic lymphohistiocytosis]. Zhonghua Xue Ye Xue Za Zhi. (2016) 37:227–32. doi: 10.3760/cma.j.issn.0253-2727.2016.03.010
23. Chen Y, Wang Z, Luo Z, Zhao N, Yang S, Tang Y. Comparison of Th1/Th2 cytokine profiles between primary and secondary haemophagocytic lymphohistiocytosis. Ital J Pediatr. (2016) 42:50. doi: 10.1186/s13052-016-0262-7
24. Jin Z, Wang Y, Wang J, Zhang J, Wu L, Gao Z, et al. Primary hemophagocytic lymphohistiocytosis in adults: the utility of family surveys in a single-center study from China. Orphanet J Rare Dis. (2018) 13:17. doi: 10.1186/s13023-017-0753-7
25. Zhang K, Chandrakasan S, Chapman H, Valencia CA, Husami A, Kissell D, et al. Synergistic defects of different molecules in the cytotoxic pathway lead to clinical familial hemophagocytic lymphohistiocytosis. Blood. (2014) 124:1331–4. doi: 10.1182/blood-2014-05-573105
26. Zur Stadt U, Beutel K, Kolberg S, Schneppenheim R, Kabisch H, Janka G, et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat. (2006) 27:62–8. doi: 10.1002/humu.20274
27. Wang Z. [How to make the diagnosis of hemophagocytic lymphohistiocytosis]. Zhonghua Xue Ye Xue Za Zhi. (2016) 37:550–3. doi: 10.3760/cma.j.issn.0253-2727.2016.07.002
28. Holt OJ, Gallo F, Griffiths GM. Regulating secretory lysosomes. J Biochem. (2006) 140:7–12. doi: 10.1093/jb/mvj126
29. Huck K, Feyen O, Niehues T, Ruschendorf F, Hubner N, Laws HJ, et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest. (2009) 119:1350–8. doi: 10.1172/JCI37901
30. Zhang W, Xiao M, Zhou JF. Genetic defects affecting lymphocyte cytotoxicity in Epstein-Barr virus-associated T/NK-cell lymphoproliferative diseases. Blood. (2017) 130(Suppl 1):4002.
31. Cattaneo C, Oberti M, Skert C, Passi A, Farina M, Re A, et al. Adult onset hemophagocytic lymphohistiocytosis prognosis is affected by underlying disease and coexisting viral infection: analysis of a single institution series of 35 patients. Hematol Oncol. (2017) 35:828–34. doi: 10.1002/hon.2314
Keywords: hemophagocytic lymphohistiocytosis, targeted gene sequencing, mutation, UNC13D, AP3B1, ITK, LYST, PRF1
Citation: Miao Y, Zhu H-Y, Qiao C, Xia Y, Kong Y, Zou Y-X, Miao Y-Q, Chen X, Cao L, Wu W, Liang J-H, Wu J-Z, Wang L, Fan L, Xu W and Li J-Y (2019) Pathogenic Gene Mutations or Variants Identified by Targeted Gene Sequencing in Adults With Hemophagocytic Lymphohistiocytosis. Front. Immunol. 10:395. doi: 10.3389/fimmu.2019.00395
Received: 25 November 2018; Accepted: 14 February 2019;
Published: 07 March 2019.
Edited by:
Guzide Aksu, Ege University, TurkeyReviewed by:
Grant Schulert, Cincinnati Children's Hospital Medical Center, United StatesKimberly Gilmour, Great Ormond Street Hospital for Children NHS Foundation Trust, United Kingdom
Copyright © 2019 Miao, Zhu, Qiao, Xia, Kong, Zou, Miao, Chen, Cao, Wu, Liang, Wu, Wang, Fan, Xu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Xu, eHV3ZWkxMDAwMEBob3RtYWlsLmNvbQ==
Jian-Yong Li, bGlqaWFueW9uZ2xtQDEyNi5jb20=
†These authors have contributed equally to this work