 Stephen J. Gaudino
Stephen J. Gaudino Pawan Kumar
Pawan Kumar- Department of Molecular Genetics and Microbiology, Stony Brook University, Stony Brook, NY, United States
Innate immunity is maintained in part by antigen presenting cells (APCs) including dendritic cells, macrophages, and B cells. APCs interact with T cells to link innate and adaptive immune responses. By displaying bacterial and tumorigenic antigens on their surface via major histocompatibility complexes, APCs can directly influence the differentiation of T cells. Likewise, T cell activation, differentiation, and effector functions are modulated by APCs utilizing multiple mechanisms. The objective of this review is to describe how APCs interact with and influence the activation of T cells to maintain innate immunity during exposure to microbial infection and malignant cells. How bacteria and cancer cells take advantage of some of these interactions for their own benefit will also be discussed. While this review will cover a broad range of topics, a general focus will be held around pathogens, cancers, and interactions that typically occur within the gastrointestinal tract.
Introduction to Innate Immunity, APC Activation, and T Cell Function
The immune system is divided into innate and adaptive responses. Adaptive immunity is regulated by B cells and T cells. Maturation of T cells occurs in the thymus and maturation of B cells occurs in the bone marrow. During antigen-dependent activation, B cells can develop into memory cells, which are activated upon subsequent exposure to the contacted antigen, or plasma cells, which secrete antibodies specialized to target that antigen (1). Similarly, T cells can develop into memory cells or effector cells. The two major types of effector T cells produced by the adaptive immune system are helper T cells (Th) and cytotoxic T cells (TC). Th cells are distinguished by their expression of CD4, subset-specific expression of transcription factors (T-bet, GATA3, and RORγt), and the release of cytokines that influence the activation and differentiation of other immune cells. Three major subsets of Th cells exist (Th1, Th2, and Th17), each of which is specialized for protecting against certain infections. Th1 cells primarily secrete interferon-γ (IFN-γ) which is associated with protection against intracellular microbes (predominantly viruses) and the onset of anti- or pro-tumorigenic effects, Th2 cells fight parasitic infections by secreting specific interleukin (IL) proteins including IL-4, IL-5, and IL-13, and Th17 cells fight microbial pathogens by secreting cytokines such as IL-17A, IL-17F, and IL-22 (2–6). TC cells are distinguished by their expression of CD8 and their ability to directly contact and kill transformed and infected cells (1, 7). T cells and B cells act together to establish an immunological memory against individual pathogens or cancer cells. Adaptive immunity can take a few days to fully develop, but once activated allows for the mounting of a rapid immune response upon subsequent exposures to the specific pathogen or cancer cell. Innate immunity, unlike adaptive immunity, is traditionally not based on immunological memory. However, certain innate cells, particularly natural killer (NK) cells, aid in the development of immunological memory against viruses. For instance, Ly49H+ NK cells exposed to mouse cytomegalovirus (MCMV) expand to mount a primary immune response (8). This process is promoted by IL-18 signaling (9). Naïve mice that received an adoptive transfer of memory NK cells were able to mount a strong secondary response upon infection with MCMV (8). Signaling by IL-18, however, is not required for recall responses by memory NK cells (9).
Classical antigen presenting cells (APCs) are dendritic cells (DCs) and B cells (10). To mount an immune response, APCs must first recognize and bind their target. To do so, APCs express antigen-specific surface receptors including pattern recognition receptors (PRRs). PRRs detect pathogen-associated molecular patterns (PAMPs), which are produced by microbes, and damage-associated molecular patterns (DAMPs), which are produced by damaged or mutated host cells (11). Depending on the receptor, expression of PRRs can be constitutive or inducible (12, 13). One major family of PRR is the Toll-like receptors (TLRs). TLRs are typically expressed on the cell surface or within endosomes and are type I transmembrane proteins whose extracellular domains express leucine-rich repeats that are used to recognize and bind to specific PAMPs (14–16). Once the extracellular domain binds its target, the TLR activates a cytosolic signaling cascade which is initiated by an adaptor protein that interacts with the intracellular domain of the TLR. Depending on the TLR, the two adaptor sets that can be activated are TIRAP-MyD88 and TRAM-TRIF (14, 16–18). Another group of PRR is the nucleotide binding oligomerization domain (NOD)-like receptors (NLRs). NLRs are present in the cytoplasm and, like TLRs, initiate signaling cascades upon binding to microbial PAMPs (14, 16). After binding to their appropriate PAMP or DAMP, APCs internalize their target by initiating phagocytosis, pinocytosis, or clathrin-mediated endocytosis. The pathway by which molecules are endocytosed determines how they will be degraded and then displayed by major histocompatibility complex (MHC) for T cell recognition (19, 20).
Two types of MHCs display antigens: class I MHCs and class II MHCs. While MHC I receptors are produced by all nucleated cells and display endogenous antigens to activate CD8+ TC, only APCs produce MHC II receptors to display exogenous antigens and activate CD4+ TH cells. Some APCs including DCs can also present exogenous antigens to the MHC I receptor to activate CD8+ T cells during a process called cross-presentation (20–23). Presentation of antigens by either MHC I or MHC II receptors also depends on antigen composition (particulate vs. soluble), method of endocytosis, and degradation by lysosomal proteases (20). TC and TH cells utilize membrane-bound T cell receptors (TCRs) to bind MHC receptors (1). TCRs consist of two polypeptide chains (alpha and beta) that are linked together by disulfide bonds. T cells also produce co-receptors on their surface which further aid stabilizing interactions with MHCs of APCs. These include CD4 and CD8 (11, 20, 24). Costimulatory interactions between APCs and T cells can also occur, respectively, between B7 and CD28, ICAM-1 and LFA-1, and CD40 and CD40L (25, 26).
Immunity to Bacteria
In this section, innate immunity will be discussed with respect to commensal and pathogenic bacteria. First, immunity will be reviewed within the context of commensals and their influence on altering the activity of Th17 and T regulatory (Treg) cells. Then, APC interactions with T cells, mainly regarding the expression of various TLRs, to fight pathogenic infections will be discussed.
Immunity and Bacterial Commensals
The gut microbiota is composed of all the archaea, fungi, protozoans, viruses, and bacteria that inhabit the gastrointestinal tract. Specifically, the colon of a healthy 29 to 30-year-old male of average weight and height has been estimated to contain ~ 3.8 × 1013 bacteria (27). Commensal bacteria have been shown to modulate immune cell responses in the intestine. Reciprocally, immune cells interact with intestinal epithelial subsets to regulate colonization by commensal bacteria. Altered microbiota composition and aberrant immune responses to commensal bacteria, however, have been thought to play a role in the development of metabolic disorders (obesity and type II diabetes), autoimmune disorders (multiple sclerosis (MS) and type I diabetes), and inflammatory bowel disease (IBD) (28–31).
APCs, specifically DCs and macrophages, utilize PRRs to maintain intestinal homeostasis (discussed in “Immunity and Bacterial Pathogens” section). Macrophages plays an important role in maintaining tolerance against the commensal microbiota and food antigens. Although macrophages display robust bactericidal activity, they are not a key source of major inflammatory cytokines such as TNF-α, IL-1β, IL-6, and IL-23 (32, 33). However, macrophages constitutively produce and respond to IL-10. Macrophage-derived IL-10 is critical for Foxp3+ Treg cell development, maintenance, and expansion (34–36). DCs also maintain tolerogenic responses by interacting with adaptive immune cells. DCs presents luminal antigens in secondary lymphoid organs of the intestine. These include the Peyer's patches (PPs) and mesenteric lymph nodes (MLNs). DCs regulate the homing of lymphocytes to the intestine by inducing expression of gut homing receptors (CCR9 and α4β7) (37, 38). DCs also are the major source of IL-23 which, in combination with other cytokines, influences the differentiation of Th17 cells and promotes the generation of IL-22, a tissue protective cytokine. Defects in NOD2 and autophagy-related genes (ATG16L and IRGM) in DCs of IBD patients reveal compromised antigen presentation, cytokine release, and increased inflammation (39–41).
IBD is marked by chronic gastrointestinal inflammation due to the onset of disorders such as ulcerative colitis (UC) and Crohn's disease (CD). Increased expression of Th1-derived IFN-γ and Th17 associated IL-17A and IL-22 are evident in the inflamed tissue of CD patient (42, 43). Genome-wide association studies show that genes (IL23R and STAT3) encoding Th17 differentiation pathways are associated with increased IBD risk. This suggests a therapeutic significance of targeting IL-23 or IL-17A in IBD (44, 45). Indeed, neutralizing IL-23 has been shown to be effective in reducing intestinal inflammation. Ustekinumab, an antibody directed against the IL-12p40 subunit, is used to block both IL-12 and IL-23 and is approved by the FDA for treatment of moderate to severe CD, whereas antibody targeting IL-23p19 subunit (IL-23) currently is in clinical trial for both CD and UC (46, 47). Interestingly, a clinical trial in which an anti-IL-17A monoclonal antibody was used to treat CD was prematurely ended since treatment with these antibodies was associated with detrimental effects (48). Subsequently we and others have shown that IL-17A is critical for preserving the epithelial barrier and regulating gut microbiota colonization (30, 49, 50). Additionally, it has been shown that IL-23 is not required for IL-17A generation from γδ T cells during chemical-induced colitis (49). This may explain why anti-IL-23 and IL-17A neutralization have counterintuitive outcomes. It remains unclear what regulates IL-17A generation in γδ T cells. It is possible that DC-derived IL-6 or other factors regulate IL-17A responses in the gut since CD103+ CD11b+ DCs have been shown to regulate Th17 differentiation in an IL-16 dependent manner (51).
The gut microbiota and microbial metabolites have been shown to regulate IL-17A responses in the gut. The intestinal microbiota, particularly segmented filamentous bacteria (SFB), has been shown to induce Th17 responses (52–54). Induction of Th17 cell responses by SFB is well-characterized. Monocyte-derived macrophages regulate SFB induction of antigen-specific Th17 cells (55, 56). Furthermore, colonization by SFB is associated with increased expression of IL-21 and isoforms of serum amyloid A (SAA). SAA indirectly promotes differentiation of Th17 cells by acting on DCs (53, 57). Along with SFB, a mixture of twenty bacterial isolates including Clostridium and Bifidobacterium species from an ulcerative colitis patient has been shown to induce Th17 activity (57). Additionally, Escherichia coli Schaedler and Morganella Morganii have been shown to regulate Th1 and Th17 cell differentiation via monocyte-derived DCs (58). Moreover, CD172α+ lamina propria DCs promote microbial antigen-specific Th17 cell differentiation in responses to TLR5 activation (59). The microbiota, including SFB, induces Th17 responses; however, it is poorly understood how immune cells regulate functions of the gut microbiota such as colonization by SFB. We and others have shown that IL-17A and IL-22 regulate the gut microbiota, including SFB colonization (30, 60, 61). Furthermore, we show that intestinal regulation of the gut microbiota by IL-17A modulates systemic autoimmunity suggesting a yin-yang relationship between the gut microbiota and Th17 cell responses (30).
The differentiation of naïve T cells into pathogenic (α/β CD4+ T cells that express high levels of IL-23R, coproduce IL-17A and IFN-γ/GM-CSF and induce autoimmunity) or non-pathogenic (α/β CD4+ T cells that produce IL-17A and IL-17F but do not induce autoimmunity) Th17 cells is influenced by DC-derived cytokines. Naïve T cells exposed to TGF-β1 and IL-6 differentiate into non-pathogenic Th17 cells, but those exposed to TGF-β1, IL-6, and IL-23 or TGF-β3 and IL-6 develop into pathogenic Th17 cells (62). Signaling by IL-23 increases expression of T-bet and production of TGF-β3 by developing Th17 cells. Likewise, IL-23 signaling has been associated with increased expression of RORγt and production of GM-CSF, an essential cytokine for the progression of autoimmunity, by Th17 cells (63). Production of dietary-derived fatty acid metabolites has also been shown to alter the differentiation of T cells (64). For instance, stimulation by long chain fatty acids triggers naïve T cell differentiation into Th1 and Th17 cells via the upregulation of p38-MAPK. This, in turn, promotes the onset of autoimmunity (64).
While SFB have mainly been associated with Th17 cell differentiation, Bacteroides fragilis or Clostridia species have been shown to regulate the induction and activity of Treg cells (65, 66). Polysaccharide A derived from B. fragilis activates DCs in a TLR2-dependent manner to induce Treg cell differentiation and IL-10 generation (66, 67). A mixture of seventeen Clostridia species that induce Treg cell differentiation and function were isolated from a human fecal sample (65). When germ-free mice were inoculated with the mixture, an increase in Treg cell abundance and induction were observed. These changes may be due to an increased production of microbiota-dependent fatty acid metabolites, particularly SCFAs. This study shows that SCFAs stimulate secretion of TGF-β by epithelial cells to promote induction of Treg cells (65). Kashiwagi et al show that TGF-β derived from DCs via TLR2-Smad3 pathways is important for the generation of Treg cells in the lamina propria of mice that were inoculated with Clostridium butyricum (68). Subsequently, the importance of SCFAs particularly butyrate in regulating Treg differentiation has been shown by many studies (69, 70). Butyrate and propionate have been shown to directly modulate Treg generation by promoting histone H3 acetylation of the Foxp3 locus and protein (69, 70). Additionally, butyrate signaling in macrophages and DCs via GPR109a, a receptor for butyrate and niacin, has been shown to promote Treg cell development (71). Mice deficient in GPR109a have fewer IL-10 producing CD4 T cells (71). Colonic Treg cells express TCRs, including CT7, that most likely aid in the recognition of specific antigens derived from the commensal microbiota (72). These TCRs are unique to colonic Treg cells since they are not expressed by Treg cells outside the colon (72).
APCs also modulate commensal microbiota-dependent Th2 cell responses. Mice treated with propionate display enhanced production of macrophage and DC precursors in their bone marrow. However, these DCs are impaired in eliciting effector functions of Th2 cells in a house dust mite extract-dependent allergic inflammation model (73).
Along with Th17 and Treg cells, innate lymphoid cells (ILCs) maintain immunity by interacting with APCs to influence commensal bacteria and T cell effector functions. ILCs are separated into three groups (ILC1, ILC2, and ILC3) based partially on the cytokines they secrete. Similar to Th17 cells, ILC3 cells secrete IL-17A and IL-22 (Figure 1) (74). IL-22 secreted from ILC3 can act on epithelial cells to induce expression of antimicrobial peptides. IL-23 derived from CD103+ CD11b+ DCs has been shown to regulate innate IL-22 responses following administration of bacterial flagellin (75). ILC3s also directly interact with T cells. MHC II receptors are expressed by CCR6+ RORγt+ ILCs which allows for direct binding and presentation of antigens to CD4+ T cells (76). Upon interacting with T cells, intestinal ILCs maintain homeostasis by limiting immune responses against commensal bacteria (76). Mononuclear phagocyte-derived TNF-like ligand 1 (TL1A) has been shown to regulates ILC3-dependent regulation of IL-22 production and mucosal host defense during acute colitis (77). TL1A also regulates expression of costimulatory molecule OX40L in MHC II+ ILC3s. This is required for antigen-specific T cell responses in a chronic colitis model (77).
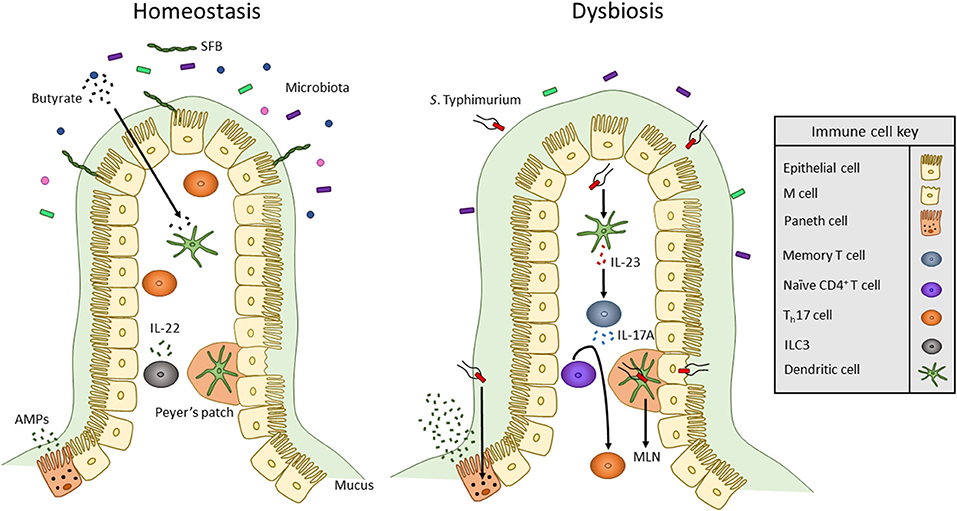
Figure 1. Overview of innate immune interactions under homeostasis and dysbiosis. In healthy individuals, the commensal microbiota secretes products, including SCFAs such as butyrate, that act on DCs to maintain a commensal-tolerant immune system. The presence of commensal microbes has also been associated with an increased presence of Th17 cells. Also, ILCs secrete cytokines to alter T cell function to maintain homeostasis. When dysbiosis is induced by bacterial pathogens such as S. Typhimurium, microbiota composition changes and mucus secretion increases. S. Typhimurium infection occurs via transcytosis by M cells into Peyer's patches where the bacterium can travel through lymphatic vessels to the MLN. To mount an immune response, S. Typhimurium antigens such as flagellin monomers are detected by TLR5 of DCs. DCs then produce IL-23 which promotes the release of IL-17 by memory T cells. IL-17 stimulates the differentiation of naïve CD4+ T cells into Th17 cells. Likewise, TLR9 and NOD2 receptors are activated within Paneth cells to upregulate the secretion of AMPs.
Immunity and Bacterial Pathogens
While commensal bacteria directly or indirectly (via APCs) influence the differentiation of T cells, APCs utilize TLRs to recognize specific microbial markers alter the function of T cells to fight bacterial pathogens. Cell surface TLRs aid in the phagocytosis of microbial pathogens by DCs and macrophages. Binding of surface TLRs to specific microbial PAMPs or whole bacteria such as E. coli or Staphylococcus aureus induces receptor phagocytosis (78). Activation of different APC TLRs results in the production of cytokines that promote the differentiation of naïve T cells into Th1 cells. This is evidenced by the activation of two TLRs, TLR2 and TLR4, that are expressed in immune cells and intestinal epithelial cells (79). TLR2 binds to Gram-positive and Gram-negative bacterial components including lipoteichoic acid, porins, and peptidoglycan, but TLR4 binds only to lipopolysaccharide, a component specific to the Gram-negative outer membrane (79–81). When activated, TLR4 promotes the production of IL-12 p70, the active form of IL-12 which is composed of the p35 and p40 subunits. IL-12 p70 aids in the polarization of naïve T cells into Th1 cells (81). On the other hand, activation of TLR2 results in increased production of IL-12 p40 homodimer which acts as a receptor antagonist of IL-12 (81).
A third TLR expressed by immune cells and the intestinal epithelium is TLR5, a receptor that specifically binds to flagellin monomers of Gram-positive and Gram-negative bacteria (79). Flagella are long whip-like structures utilized by bacteria for motility, adhesion, and secretion of virulence factors. Numerous intestinal pathogens including Listeria monocytogenes, Salmonella typhimurium, and Campylobacter jejuni produce flagella for successful host colonization and invasion (82–84). TLR5 signaling in DCs results in activation of the IL-22/23 axis (85). IL-23 then stimulates the secretion of IL-17A by memory T cells and promotes naïve CD4+ T cells to differentiate into Th17 cells (Figure 1) (86–88). Activation of TLR5 expressed by epithelial cells and DCs plays an important role in clearing pathogenic bacteria such as adherent invasive E. coli. Epithelial TLR5 signaling is important in limiting bacterial adherence in the intestine (85).
TLRs can also interact with and effect the signaling of other innate immune receptors to influence T cell function. This is displayed by interactions that occur between TLRs and NOD2. NOD2 is part of the NLR family, is encoded by the Card15 gene, and recognizes Gram-positive and Gram-negative peptidoglycan-derived peptides including muramyl dipeptide (MDP) (89, 90). Activation of NOD2 is associated with altered TLR2 signaling. As stated previously, TLR2 is activated in response to peptidoglycan. Both NOD2 and TLR2 activation result in NF-κB signaling. When NOD2 signaling is stimulated by MDP, TLR2-induced NF-κB signaling and production of IL-12 are inhibited. However, in Card15−/− APCs, NOD2 signaling does not occur but TLR2 signaling can still occur; when treated with peptidoglycan, these mice display higher production of IL-12 by macrophages due to increased NF-κB signaling. This enhances Th1 responses (production of IL-12, IFN-γ, and IL-18) (91). Besides being expressed by APCs, TLR9 and NOD2 are unique because they are both expressed by Paneth cells, a subset of antimicrobial peptide-producing epithelial cells within the crypts of the small intestine. While many antimicrobial peptides are constitutively expressed, signaling by bacterial antigens including flagellin, peptidoglycan, and lipopolysaccharide can further stimulate their production (92).
Enteric pathogens utilize virulence factors to avoid detection by and alter the response of the innate immune system. Two such pathogens are Salmonella enterica serovar Typhi and Salmonella enterica serovar Typhimurium. While S. Typhi and S. Typhimurium are closely related enteric pathogens, they promote different disease states in humans. S. Typhi causes typhoid fever by crossing the intestine and then spreading to systemic organs; on the other hand, S. Typhimurium infection is restricted to the intestine and typically causes enteritis (93). S. Typhi infection is specific to humans and does not occur in mice (94). This may be due to the ability of mice, but not humans, to produce TLR11 which is activated by flagellin94. Unlike S. Typhi, S. Typhimurium can infect multiple hosts including mice. Therefore, S. Typhimurium is used to study typhoid-like dissemination in susceptible strains of mice (94, 95). Adaptive B and T cell responses are required to provide protection in mouse model of Salmonella infection. In infected mice, S. Typhimurium invades ileal epithelial cells including microfold (M) cells. M cells are specialized epithelial cells that aid in the transcytosis of luminal antigens and microbes into PPs, a region of secondary lymphoid tissue that contains DCs, T cells, and B cells (96). Upon sensing the bacteria, epithelial goblet cells upregulate mucus production, specifically mucin 2, to restrict contact of S. Typhimurium with the epithelial layer (97). To circumvent this, S. Typhimurium expresses flagella and a type III secretion system (T3SS) that is encoded by Salmonella Pathogenicity Island 1 (SPI-1) (84, 93). Effector proteins are injected through the SPI-1 T3SS into epithelial cells to alter their cytoskeletal shape and tight junction integrity. While injection of effectors into M cells promotes optimal bacterial invasion, SPI-1 mutants of S. Typhimurium can still invade M cells but to a lesser extent (Figure 1) (93). Once inside the PP, S. Typhimurium can infect DCs to be transported in a CCR7-dependent manner to MLNs (98). S. Typhimurium then spreads to systemic tissues including the liver and spleen and replicates within phagocytes such as macrophages (93). To avoid degradation within macrophages, S. Typhimurium expresses a second T3SS encoded by Salmonella Pathogenicity Island 2 (99). DCs infected by S. Typhimurium display inhibited antigen presentation which prevents stimulation of naïve T cells (100). S. Typhimurium can also directly infect T cells to inhibit their proliferation and secretion of cytokines including IL-2 and IFN-γ (100).
Another well-studied enteric pathogen, L. monocytogenes, alters APC function to influence T cell function. Similar to S. Typhimurium, L. monocytogenes replicates within macrophages. Infected macrophages release TNF-α and IL-12 which promote the secretion of IFN-γ from NK cells (101). Secreted IFN-γ then activates macrophages to produce reactive oxygen and nitrogen intermediate to prevent the escape of L. monocytogenes from phagosomes and aid in bacterial degradation (102). To promote its growth, L. monocytogenes induces production of type I interferons (IFNs). Escape of L. monocytogenes from macrophage phagosomes (which occurs due to the production of listeriolysin O, a pore-forming toxin) results in the upregulation of IFN-α and IFN-β by the infected macrophage (103, 104). Increased production of type I IFNs has been associated with increased apoptosis of T cells and greater production of IL-10 to favor bacterial proliferation (104).
Memory T cells also aid with combating infection by L. monocytogenes and comprise a major of component of intestinal T cells including tissue effector memory (TEM), central memory (TCM), and resident memory cells (TRM). While TEM and TCM cells both reside in the blood and spleen, TCM can reside within lymphoid tissues and TEM can reside in non-lymphoid tissue (105). TRM cells predominantly reside within the lamina propria and intraepithelial lymphocyte regions of the intestine and provide immune regulation throughout life (106). Upon oral infection with L. monocytogenes, CD103+ DCs acquire and process bacterial antigens. These DCs express CCR7 and travel from the LP via the lymphatics to the MLN (107, 108). In the MLN, DCs present L. monocytogenes antigens to naïve CD8 T cells. Activated T cells proliferate and differentiate into early effector cells that further differentiate into either short-lived effector cells or memory precursor effector cells (108). Memory precursor effector cells can differentiate into TEM or TCM and migrate to the intestine (108). DCs release retinoic acid which binds to the retinoic acid receptor of T cells. Upon binding to RA, T cells exhibit enhanced expression integrin α4β7 and CCR9, both of which direct migration to the LP of the small intestine (105, 109). Once at their site of infection, TEM cells express granzyme B. Expression of granzyme B, however, is downregulated in TCM cells (105).
Enteropathogenic E. coli (EPEC) and Enterohaemorrhagic E. coli (EHEC) are two clinically significant human pathogens of the intestine that are responsible for deaths caused by severe diarrhea. Citrobacter rodentium (C. rodentium) is a mouse pathogen that shares several clinical pathological mechanisms of EPEC and EHEC and, therefore, serves as a useful model to understand the innate and adaptive immune responses in the gut following infection as well as pathogenesis of IBD (110, 111). The attaching and effacing (A/E) lesion formed by EPEC, EHEC, and C. rodentium distinguishes them from other intestinal pathogens and commensal E. coli. A/E lesions form when the pathogenic bacterium binds to the intestinal epithelium, remodels the brush border, and injects effector proteins into the host cell via a T3SS. These effectors then influence the activity of host actin nucleation factors, N-WASP and Arp2/3, to promote actin polymerization. This results in the formation of an actin pedestal that raises the bacterium above neighboring epithelial cells to further assist its pathogenesis (112). Both innate and adaptive immune responses are required to control C. rodentium infection since mice deficient in Rag1 succumb to infection (113). C. rodentium infection leads to microbiota dysbiosis and the development of colitis (114, 115). Interestingly, microbiota dysbiosis is a key factor that influences the susceptibility of C. rodentium infection and immune responses (115, 116). Myd88-dependent TLR2 and TLR4 signaling is stimulated in epithelial and myeloid cells to recognize bacterial PAMPs. DCs and macrophages secrete proinflammatory cytokines including IL-12, IL-6, IL-23, and TNF-α in response to the activation of PRRs. Th17-, Th22-, and ILC3-derived IL-22 plays an important role in regulating C. rodentium infection (117, 118). IL-23 is required for provide protection against C. rodentium infection in a IL-22 dependent manner117. IL-22-dependent induction of antimicrobial peptide Reg3γ has been suggested to regulate C. rodentium infection. However, a recent study shows that Reg3γ−/− mice are equally susceptible to infection. It is possible that another Reg3 family member of antimicrobial peptide compensated for Reg3γ. It remains unclear how IL-22 regulates C. rodentium infection. Since IL-22Ra1 is expressed on absorptive cells (enterocytes), secretory cells (goblet, Paneth), and stem cells of intestine, future work should be directed toward understanding the effects of IL-22 on multiple intestinal cell lineages (119).
Yersinia species are Gram-negative bacterium, among them Y. pestis, Y. pseudotuberculosis, and Y. enterocolitica, are pathogenic to human. Y. pseudotuberculosis and Y. enterocolitica cause yersiniosis which leads to gastroenteritis and mesenteric lymphadenitis and may be fatal if disseminated into liver and spleen. Similar to Salmonella and other pathogens, Yersinia use T3SS to inject toxin (Yersinia outer protein, Yops) to cell cytoplasm. These Yops (YopE, YopJ, YopH, YopM, YopO, and YopT) disrupt intracellular signaling in macrophages which results into inhibition of cytokines (IL-1β, IL-18, required for innate immune cells recruitment) secretion and phagocytosis. Several studies show that YopE contain a dominant CD8 T cells epitope which is required to confer protection (120–124). Furthermore, CD8 T cells response are also required to provide protection from subsequent infection. CXCR1+ macrophages and/or DC-dependent antigen presentation and local inflammatory conditions are critical for the development of heterogenous population (CD103+ or CD103–) of CD8 TRM cells in the intestine (125).
Immunity in Response to Cancer
Colorectal cancer (CRC) consistently accounts for many of the cancer-related deaths in men and women. It is the third most common cancer in men and women and the fourth leading cause of cancer-related deaths. Although there are wide variations in global incidence and mortality rates related to CRC, rapidly transitioning countries (indicated by a medium to high human development index) tend to display increased CRC incidence and mortality (122). Over the past few years, the incidence of CRC has been declining in individuals ≥50 years old but is increasing in young adults (126). The onset of colorectal cancer has been associated with various factors including genetics, intestinal microbiota, and immune activity (127–129). Particularly, the release of cytokines by APCs has been shown to influence the development of cancer cells (87, 130, 131).
CRC cells express tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs). While TSAs are unique to tumors, TAAs can be present on normal cells and tumor cells. Common TAAs and TSAs include carcinoembryonic antigens (CEA), Wilm's tumor gene 1 (WT1), Muc1, Her2, and p53 (132–134). DCs can capture bodies of killed tumor cells, process TAAs, and present these antigens via MHC I or MHC II molecules, respectively, to TCRs of CD4+ T cells or CD8+ T cells (135). APC activation in response to cancer can also occur via the binding of APC-expressed PRRs to tumor-derived DAMPs. DAMPs may be released by tumor cells in response to anti-tumor therapy or stress pathways. With regard to CRC, three of the major DAMPs that stimulate anti-tumorigenic responses of DCs include High Mobility Group Box 1 (HMGB1), extracellular ATP, and calreticulin (CRT). Although typically restricted to the nucleus and cytoplasm, HMGB1 is secreted by necrotic tumor cells. Released HMGB1 can bind to DC-expressed TLR4 to enhance antigen presentation (136, 137). HMGB1 can also signal via DC-expressed RAGE membrane protein to promote NF-κB signaling and DC maturation (138). ATP released from dying tumor cells can bind to the P2XY receptor of DCs. This binding promotes the recruitment of DCs to the tumor stroma. CRT is typically expressed within the endoplasmic reticulum (ER) of tumorigenic cells. Chemotherapeutic agents induce the release of reactive oxygen species and induce ER stress. This promotes the transport of CRT from the ER to the cell surface where CRT serves as a signal for DC-mediated engulfment, degradation, and antigen presentation to cytotoxic CD8+ T cells (139). While DCs are vital for antitumor immune responses, cancer cells utilize various mechanisms to evade immune detection. Cancer cells can downregulate TAAs, modulate antigen processing or presentation pathways, release cytokines that promote Treg function, secrete immunosuppressive factors, and express ligands that block immune checkpoints (140). Additionally, immunosuppressive cells within the tumor microenvironment such as tumor-associated macrophages (TAMs), cancer-associated fibroblasts, and Treg cells have been shown to inhibit antitumor immunity (140).
To promote immunity against tumors, DCs and activated T cells secrete multiple cytokines. Two important anti-tumorigenic cytokines are IL-2 and IL-15. These cytokines can induce similar biological effects, and the receptors for these cytokines share similar structural features including a common γ chain and IL-2/IL-15Rβ chain (141, 142). Binding of IL-2 or IL-15 to their receptor results in the activation of Janus kinases (JAK). JAK1 is activated when IL-2 or IL-15 bind to the β chain of the receptor, but JAK3 is activated when IL-2 or IL-15 bind to the or γ chain of the receptor (141). JAKs bind to their corresponding ligand to promote receptor dimerization. Dimerized JAKs phosphorylate each other, and phosphorylated JAKs phosphorylate a conserved tyrosine residue of signal transducers and activators of transcription (STAT) molecules which then enter the nucleus to regulate transcription (141, 143). While IL-2 is secreted mainly by stimulated CD4+ T cells, it is also produced by effector CD8+ T cells, DCs, and NK cells (141, 144–148). Signaling by IL-2 can influence the functions of CD4+ T cells, CD8+ T cells, and NK cells (141, 149–152). IL-15 is expressed on the surface of a range of cell types including monocytes, macrophages, and DCs and influences the activation of DCs, proliferation of CD8+ T cells, and development of NK cells (142, 153–155). IL-2 and IL-15 may play a role in preventing colorectal cancer and have been studied as potential immunotherapy agents (141, 148, 156). However, treatment with these cytokines may have to be supplemented. For instance, survival of melanoma patients treated with IL-2 and a melanoma vaccine displayed increased survival compared to patients who were only treated with IL-2 (157). Studies have also modified the structure of IL-2 to increase its efficiency as an immunotherapeutic agent (158). IL-2 “superkines” have increased affinity for IL-2Rβ and display increased activation of NK and CD8+ T cells. Mice that were injected with melanoma, colon carcinoma, or lung carcinoma cells and then treated with modified IL-2 displayed decreased tumorigenesis compared to mice treated with unmodified IL-2 (158). The onset of colorectal cancer has commonly been studied by the administration of azoxymethane (AOM) combined with dextran sulfate sodium (DSS) to induce colitis-associated carcinoma in mice. Compared to wild-type mice treated with AOM/DSS, Il15−/− mice treated with AOM/DSS display increased tumorigenesis and decreased survival159. CD11c-Il15 mice display reconstituted production of IL-15 in APCs. Unlike Il15−/− mice which display decreased levels of NK and CD8+ T cells, CD11c-Il15 mice display similar levels of NK and CD8+ T cells as wild-type mice. CD11c-Il15 mice also display reduced AOM/DSS-induced tumorigenesis (159). While IL-2 and IL-15 have been shown to reduce the onset of cancer, other studies have shown that they may have little therapeutic effects or may even help promote tumorigenesis (141, 148). Thus, further studies need to be conducted to elucidate the relationship between these cytokines and the onset of cancer.
The presence of different T cell classes in the tumor microenvironment has been associated with changes in severity of prognosis. High levels of Th17 cells and expression of Th17 genes (including IL17A and Rorc) are associated with a poor prognosis for CRC, but patients with high expression of Th1 cytotoxic genes (including Ccl5, Stat1, and Il27) had improved occurrence of disease-free survival (160, 161). Along with being secreted by T cells, IL-27 is secreted by DCs and macrophages in response to bacterial and parasitic pathogenesis to inhibit Th1, Th2, and Th17 cell development and inflammatory responses (130, 162). Enterocyte-specific knockdown of IL-17RA, a common receptor for IL-17A, IL-17F, and IL-17C resulted in reduced tumor formation, thereby indicating a direct role for IL-17A or other IL-17 family member cytokines in tumorigenesis (163). Upregulation of IL-17A in colon tumors is dependent on upregulated IL-23 signaling by CD11b+ cells (164). This increase in IL-23 by DCs, and subsequent increase in IL-17A, may be due to: (1) the microbiota and (2) an impaired epithelial barrier. Compared to the microbiota of healthy individuals, CRC patients possess an altered microbiota distinguishable by the presence of bacteria, such as Fusobacterium nucleatum, that promote the proliferation of CRC cells (165). Increased IL-23 may be caused by increased TLR/MyD88 signaling induced by the intestinal microbiota since knockout mice in Myd88−/−and triple knockout mice in Tlr2,4,9−/− display reduced tumor growth. Similarly, mice treated with antibiotics to induce a depleted commensal microbiota displayed reduced tumor size and expression of IL-23 and IL-17A (164). CRC cells have also been shown to be “leaky” since they express defective barrier proteins including mucin 2 and junctional adhesion molecules-A and -B (164). This potentially allows for the increased passage of microbes and microbial products into cancer cells to stimulate TLR signaling and, in turn, IL-23 expression by DCs which then activates Th17 cells to release IL-17A (164). Expression of IL-17 has also been linked to tumorigenic processes including the upregulation of proangiogenic factors and neovascularization (Figure 2) (166, 167).
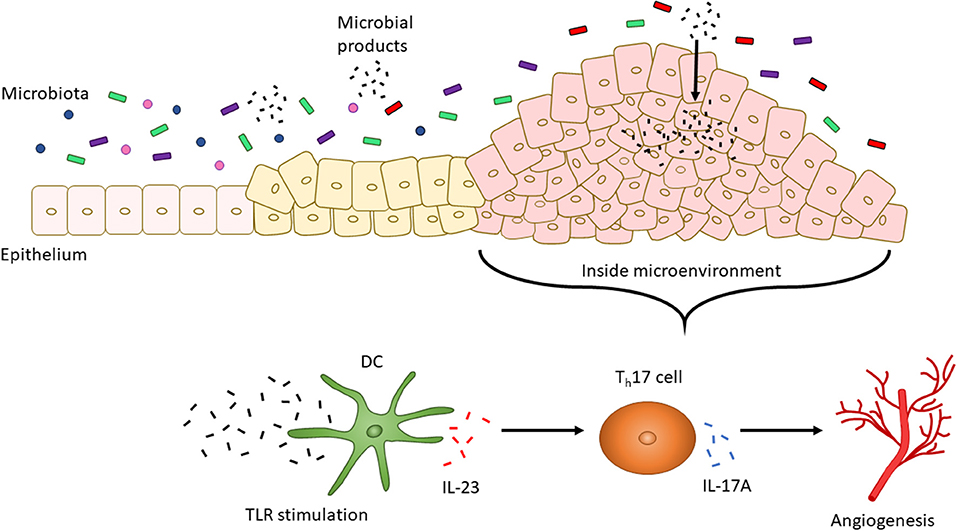
Figure 2. CRC altered microbiota-dependent innate immune signaling facilitates tumorigenesis. The intestinal microbiota changes with the progression of CRC. For instance, increased levels of F. nucleatum have been associated with increased severity of CRC. CRC cells also display a “leaky” epithelial layer which allows for the entrance of microbial products into the tumor microenvironment. These products activate TLR signaling by DCs. IL-23 is then secreted by DCs to promote the secretion of IL-17A by Th17 cells. IL-17A, in turn, is associated with angiogenesis.
IL-6 is another pro-inflammatory cytokine. IL-6 binding to Janus kinase promotes STAT3 activation (168). STAT3 then enters the nucleus to increase the transcription of anti-apoptotic genes including Bcl2 and Mcl1 and metastatic genes including Mmp1 and Mmp2 (169–171). Elevated levels of IL-6 have been associated with various cancers including lung, liver, pancreatic, and colorectal (172). Likewise, IL-6 signaling and the subsequent activation of STAT3 have been associated with the translocation of hMSH3, a DNA mismatch repair protein, from the nucleus into the cytosol (173). This translocation allows for the accumulation of tetranucleotide frameshift mutations to promote CRC (173). Some immune cells that produce IL-6 are DCs, macrophages, and Th17 cells (87, 131, 174). IL-23 has been shown to induce production of IL-6 by Th17 cells (87, 175). In turn, IL-6 aids in the differentiation of Th17 cells by promoting expression of IL-21 which induces production of IL-17A via activation of the transcription factors STAT3 and RORγt (176). IL-6 activation also promotes tumorigenesis by altering gene expression for the induction of cell proliferation, progression of the epithelial to mesenchymal transition, and resistance to anti-cancer drugs such as erlotinib (168). Signaling by IL-11, a member of the IL-6 family of cytokines, also promotes the development of CRC by activating STAT3 (177). IL-11 has been identified as a more dominant activator of STAT3 signaling and inducer of CRC than IL-6. Likewise, inhibition of IL-11 signaling has been linked with reduced tumor growth and burden. However, while hematopoietic cells produce IL-11, the secretion of IL-11 by non-hematopoietic cells is more so responsible for the progression of CRC (177). Along with IL-6 and IL-11, elevated levels of IL-22 have been associated with colon cancer. IL-22 has been shown to activate STAT3 signaling and further enhance the development of colon cancer (178, 179). Knockout mice of IL-22 binding protein, a soluble receptor for IL-22, display IL-22-dependent tumor growth (180). ILC3s have been shown to be a major source of IL-22 in mice for promoting tumorigenesis; however, human studies indicate that CCR6+ Th17 cells are major producers of IL-22 (179, 181). Currently, IL-6 and STAT3 inhibitors are being investigated as potential therapeutic agents to inhibit tumor growth (182).
In addition to STAT3, TNF-α is a critical cytokine for its role in regulating signaling pathways and inflammatory responses, especially in relation to pro- and anti-tumorigenic activity. Like other cytokines, TNF-α is produced by a range of cell types including epithelial cells, macrophages, neutrophils, NK cells, and T cells (183–186). Elevated levels of TNF-α have been associated with increased levels of infiltrating immature myeloid cells which develop into pro-tumorigenic cells such as tumor associated macrophages and neutrophils (TAMs and TANs) once they are exposed to the tumor microenvironment (187, 188). In some cell types, stimulation with TNF-α has been shown to encourage monocyte recruitment to tumors by promoting the expression of monocyte chemoattractant protein-1 (CCL2) (189, 190). Various cell types produce CCL2 including smooth muscle and mesenchymal stromal cells, and other monocytes and macrophages (191–193). TAMs produce cytokines such as IL-10 that inhibit T cell function. IL-10 has been shown to increase the expression of Mgat5, a glycosyltransferase that increases branching of glycoproteins on the surface of CD8+ T cells and has been associated with the progression of cancer (194, 195). Increased glycoprotein branching hinders direct T cell interactions with APCs and decreases TCR signaling to promote poor sensitivity to antigens (194). While increased expression of IL-10 produced by TAMs has been correlated with the progression of certain cancers including non-small cell lung carcinoma, the role of IL-10 in promoting CRC is not fully understood as it displays both protective and inflammatory roles (196–198).
Programmed death 1 (PD-1) is expressed on the surface of T cells and interacts with PD-L1, a ligand mainly expressed by macrophages, activated DCs and cancer cells (199). Likewise, cytotoxic T lymphocyte-associated antigen 4 (CTLA4) is expressed on naïve or memory T cells and interacts with CD80/CD86 on the surface of DCs. Although this review will not cover interactions between PD-1 and PD-L1 or CTLA4 and CD80/86 in depth, interactions between PD-1 or CTLA4 and its ligand act to keep T cell activity in check by preventing overactive immune responses (199). Immunotherapy utilizing anti-PD-1 or anti-CTLA4 monoclonal antibodies is effective in fighting the progression of various cancers, and the intestinal microbiota may influence whether certain individuals respond more positively to therapy (199, 200). For instance, increased levels of Akkermansia muciniphila, a commensal bacterium, were observed in patients who favorably reacted to treatment (200).
Conclusion
While much has been done to determine how APCs interact with T cells, several aspects should be expanded. First, how bacteria influence APC interactions with immune cells to combat microbial infections should be further investigated. The interplay between microbiota-derived signaling and the onset of cancers also requires better understanding. Although not widely covered in this review, potential immunotherapies regarding altering APC or microbiota function should be studied. Likewise, a better understanding of how tumors develop strategies to take advantage of immune cells to promote tumorigenesis and dampen T cell responses, especially regarding the PD-1 system, is required. Lastly, more investigation is needed to determine how factors derived from the microbiota, including SCFAs, and intestinal epithelial cells shape APC and immune cell interactions.
Author Contributions
SG wrote the manuscript and designed the figures. PK edited the text.
Funding
This work was supported by the Crohn's and Colitis Foundation (476637), National Multiple Sclerosis Society (PP-1709-29192), and The Research Foundation of SUNY to PK, and the National Science Foundation Graduate Research Fellowship to SG.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge Michael Beaupre and Xun Lin for their input.
References
1. Bonilla FA, Oettgen HC. Adaptive immunity. J Allergy Clin. Immunol. (2010) 125:S33–40. doi: 10.1016/j.jaci.2009.09.017
2. Kaiko GE, Horvat JC, Beagley KW, Hansbro PM. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology. (2008) 123:326–38. doi: 10.1111/j.1365-2567.2007.02719.x
3. Walker JA, McKenzie ANJ. TH2 cell development and function. Nat Rev Immunol. (2017) 18:121–33. doi: 10.1038/nri.2017.118
4. Bradley LM, Dalton DK, Croft M. A direct role for IFN-gamma in regulation of Th1 cell development. J Immunol Baltim Md 1950. (1996) 157:1350–1358.
5. Castro F, Cardoso AP, Gonçalves RM, Serre, K, Oliveira MJ. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol. (2018) 9:847. doi: 10.3389/fimmu.2018.00847
6. Zhu J, Yamane, H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
7. Halle S, Keyser KA, Stahl FR, Busche A, Marquardt A, Zheng X, et al. In vivo killing capacity of cytotoxic T cells is limited and involves dynamic interactions and T cell cooperativity. Immunity. (2016) 44:233–45. doi: 10.1016/j.immuni.2016.01.010
8. Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. (2009) 457:557–61. doi: 10.1038/nature07665
9. Madera S, Sun JC. Cutting edge: stage-specific requirement of IL-18 for antiviral NK cell expansion. J Immunol Baltim Md 1950. (2015) 194:1408–12. doi: 10.4049/jimmunol.1402001
10. Turvey SE, Broide DH. Innate immunity. J. Allergy Clin Immunol. (2010) 125:S24–32. doi: 10.1016/j.jaci.2009.07.016
11. ten Broeke T, Wubbolts R, Stoorvogel W. MHC class II antigen presentation by dendritic cells regulated through endosomal sorting. Cold Spring Harb Perspect Biol. (2013) 5:a016873. doi: 10.1101/cshperspect.a016873
12. Holmlund U, Cebers G, Dahlfors AR, Sandstedt B, Bremme K, Ekström ES, et al. Expression and regulation of the pattern recognition receptors Toll-like receptor-2 and Toll-like receptor-4 in the human placenta. Immunology. (2002) 107:145–51. doi: 10.1046/j.1365-2567.2002.01491.x
13. Yang H, Wei J, Zhang H, Lin L, Zhang W, He S. Upregulation of Toll-like receptor (TLR) expression and release of cytokines from P815 mast cells by GM-CSF. BMC Cell Biol. (2009) 10:37. doi: 10.1186/1471-2121-10-37
14. Creagh EM, O'Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. (2006) 27:352–7. doi: 10.1016/j.it.2006.06.003
15. Akira S, Takeda, K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. (2001) 2:675–80. doi: 10.1038/90609
16. Fukata M, Vamadevan AS, Abreu MT. Toll-like receptors (TLRs) and Nod-like receptors (NLRs) in inflammatory disorders. Semin Immunol. (2009) 21:242–53. doi: 10.1016/j.smim.2009.06.005
17. Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, et al. LPS-TLR4 Signaling to IRF-3/7 and NF-κB Involves the Toll Adapters TRAM and TRIF. J Exp Med. (2003) 198:1043–55. doi: 10.1084/jem.20031023
18. Verstak B, Nagpal K, Bottomley SP, Golenbock DT, Hertzog PJ, Mansell A. MyD88 adapter-like (Mal)/TIRAP interaction with TRAF6 is critical for TLR2- and TLR4-mediated NF-kappaB proinflammatory responses. J Biol Chem. (2009) 284:24192–203. doi: 10.1074/jbc.M109.023044
19. Burgdorf S, Kautz A, Bohnert V, Knolle PA, Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science. (2007) 316:612–6. doi: 10.1126/science.1137971
20. Burgdorf S, Kurts C. Endocytosis mechanisms and the cell biology of antigen presentation. Curr Opin Immunol. (2008) 20:89–95. doi: 10.1016/j.coi.2007.12.002
21. Delneste Y, Magistrelli G, Gauchat J, Haeuw J, Aubry J, Nakamura K, et al. Involvement of LOX-1 in Dendritic Cell-Mediated Antigen Cross-Presentation. Immunity. (2002) 17:353–62. doi: 10.1016/S1074-7613(02)00388-6
22. Ackerman AL, Cresswell P. Cellular mechanisms governing cross-presentation of exogenous antigens. Nat. Immunol. (2004) 5:678–84. doi: 10.1038/ni1082
23. Wieczorek M, Abualrous ET, Sticht J, Álvaro-Benito M, Stolzenberg S, Noé F, et al. Major Histocompatibility Complex (MHC) Class I and MHC Class II proteins: conformational plasticity in antigen presentation. Front Immunol. (2017) 8:292. doi: 10.3389/fimmu.2017.00292
25. Ni K, O'Neill HC. The role of dendritic cells in T cell activation. Immunol Cell Biol. (1997) 75:223–30.
26. Tai Y, Wang Q, Korner H, Zhang L, Wei W. Molecular mechanisms of t cells activation by dendritic cells in autoimmune diseases. Front. Pharmacol. (2018) 9:642. doi: 10.3389/fphar.2018.00642
27. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. (2016) 14:e1002533. doi: 10.1371/journal.pbio.1002533
28. Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. (2008) 57:1470–81. doi: 10.2337/db07-1403
29. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. (2008) 455:1109–13. doi: 10.1038/nature07336
30. Kumar P, Monin L, Castillo P, Elsegeiny W, Horne W, Eddens T, et al. Intestinal Interleukin-17 receptor signaling mediates reciprocal control of the gut microbiota and autoimmune inflammation. Immunity. (2016) 44:659–71. doi: 10.1016/j.immuni.2016.02.007
31. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. (2008) 105:16731–6. doi: 10.1073/pnas.0804812105
32. Flannigan KL, Geem D, Harusato A, Denning TL. Intestinal antigen-presenting cells: key regulators of immune homeostasis and inflammation. Am J Pathol. (2015) 185:1809–19. doi: 10.1016/j.ajpath.2015.02.024
33. Bain CC, Mowat AM. Macrophages in intestinal homeostasis and inflammation. Immunol. Rev. (2014) 260:102–17. doi: 10.1111/imr.12192
34. Denning TL, Wang Y, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17–producing T cell responses. Nat Immunol. (2007) 8:1086–94. doi: 10.1038/ni1511
35. Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. (2009) 10:1178–84. doi: 10.1038/ni.1791
36. Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity. (2011) 34:237–46. doi: 10.1016/j.immuni.2011.01.016
37. Johansson-Lindbom B, Svensson M, Wurbel MA, Malissen B, Márquez G, Agace W. Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. J. Exp. Med. (2003) 198:963–969. doi: 10.1084/jem.20031244
38. Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, et al. Generation of gut-homing IgA-secreting b cells by intestinal dendritic cells. Science. (2006) 314:1157–60. doi: 10.1126/science.1132742
39. Cho JH, Weaver CT. The genetics of inflammatory bowel disease. Gastroenterology. (2007) 133:1327–39. doi: 10.1053/j.gastro.2007.08.032
40. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. (2010) 16:90–7. doi: 10.1038/nm.2069
41. van Beelen AJ, Zelinkova Z, Taanman-Kueter EW, Muller FJ, Hommes DW, Zaat SA, et al. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity. (2007) 27:660–9. doi: 10.1016/j.immuni.2007.08.013
42. Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte JM, et al. IL-22 is increased in active Crohn's disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am. J. Physiol. Gastrointest. Liver Physiol. (2006) 290:G827–38. doi: 10.1152/ajpgi.00513.2005
43. Hölttä V, Klemetti P, Sipponen T, Westerholm-Ormio M, Kociubinski G, Salo H, et al. IL-23/IL-17 immunity as a hallmark of Crohn's disease: Inflamm Bowel Dis. (2008) 14:1175–84. doi: 10.1002/ibd.20475
44. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. (2006) 314:1461–3. doi: 10.1126/science.1135245
45. Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. (2015) 47:979–86. doi: 10.1038/ng.3359
46. Bilsborough J, Targan SR, Snapper SB. Therapeutic Targets in Inflammatory Bowel Disease: Current and Future. Am J Gastroenterol Suppl. (2016) 3:27–37. doi: 10.1038/ajgsup.2016.18
47. Ma C, Jairath V, Khanna R, Feagan BG. Investigational drugs in phase I and phase II clinical trials targeting interleukin 23 (IL23) for the treatment of Crohn's disease. Exp Opin Invest Drugs. (2018) 1506764:1–12. doi: 10.1080/13543784.2018.1506764
48. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. (2012) 61:1693–700. doi: 10.1136/gutjnl-2011-301668
49. Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, et al. Interleukin-23-Independent IL-17 production regulates intestinal epithelial permeability. Immunity. (2015) 43:727–38. doi: 10.1016/j.immuni.2015.09.003
50. Maxwell JR, Zhang Y, Brown WA, Smith CL, Byrne FR, Fiorino M, et al. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity. (2015) 43:739–50. doi: 10.1016/j.immuni.2015.08.019
51. Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hägerbrand K, Marsal J, et al. IRF4 Transcription-Factor-Dependent CD103+CD11b+ dendritic cells drive mucosal T Helper 17 cell differentiation. Immunity. (2013) 38:958–69. doi: 10.1016/j.immuni.2013.03.009
52. Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. (2011) 108:4615–22. doi: 10.1073/pnas.1000082107
53. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of Intestinal Th17 cells by segmented filamentous bacteria. Cell. (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
54. Gaboriau-Routhiau V, Rakotobe S, Lécuyer E, Mulder I, Lan A, Bridonneau C, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. (2009) 31:677–89. doi: 10.1016/j.immuni.2009.08.020
55. Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, et al. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature. (2014) 510:152–6. doi: 10.1038/nature13279
56. Panea C, Farkas AM, Goto Y, Abdollahi-Roodsaz S, Lee C, Koscsó B, et al. Intestinal monocyte-derived macrophages control commensal-specific Th17 responses. Cell Rep. (2015) 12:1314–24. doi: 10.1016/j.celrep.2015.07.040
57. Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell. (2015) 163:367–80. doi: 10.1016/j.cell.2015.08.058
58. Bene K, Varga Z, Petrov VO, Boyko N, Rajnavolgyi E. Gut microbiota species can provoke both inflammatory and tolerogenic immune responses in human dendritic cells mediated by retinoic acid receptor alpha ligation. Front Immunol. (2017) 8:427. doi: 10.3389/fimmu.2017.00427
59. Liu H, Chen F, Wu W, Cao AT, Xue X, Yao S, et al. TLR5 mediates CD172α+ intestinal lamina propria dendritic cell induction of Th17 cells. Sci Rep. (2016) 6:22040. doi: 10.1038/srep22040
60. Shih VF, Cox J, Kljavin NM, Dengler HS, Reichelt M, Kumar P, et al. Homeostatic IL-23 receptor signaling limits Th17 response through IL-22-mediated containment of commensal microbiota. Proc Natl Acad Sci USA. (2014) 111:13942–7. doi: 10.1073/pnas.1323852111
61. Flannigan KL, Ngo VL, Geem D, Harusato A, Hirota SA, Parkos CA, et al. IL-17A-mediated neutrophil recruitment limits expansion of segmented filamentous bacteria. Mucosal Immunol. (2017) 10:673–84. doi: 10.1038/mi.2016.80
62. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. (2012) 13:991–9. doi: 10.1038/ni.2416
63. Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. (2011) 12:560–7. doi: 10.1038/ni.2027
64. Haghikia A, Jörg S, Duscha A, Berg J, Manzel A, Waschbisch A, et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity. (2015) 43:817–29. doi: 10.1016/j.immuni.2015.09.007
65. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. (2013) 500:232–6. doi: 10.1038/nature12331
66. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci USA. (2010) 107:12204–9. doi: 10.1073/pnas.0909122107
67. Dasgupta S, Erturk-Hasdemir D, Ochoa-Reparaz J, Reinecker H-C, Kasper DL. Plasmacytoid dendritic cells mediate anti-inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host Microbe. (2014) 15:413–23. doi: 10.1016/j.chom.2014.03.006
68. Kashiwagi I, Morita R, Schichita T, Komai K, Saeki K, Matsumoto M, et al. Smad2 and Smad3 Inversely Regulate TGF-β Autoinduction in Clostridium butyricum-activated dendritic cells. Immunity. (2015) 43:65–79. doi: 10.1016/j.immuni.2015.06.010
69. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. (2013) 504:446–50. doi: 10.1038/nature12721
70. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. (2013) 504:451–5. doi: 10.1038/nature12726
71. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. (2014) 40:128–39. doi: 10.1016/j.immuni.2013.12.007
72. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature. (2011) 478:250–4. doi: 10.1038/nature10434
73. Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. (2014) 20:159–66. doi: 10.1038/nm.3444
74. Withers DR. Innate lymphoid cell regulation of adaptive immunity. Immunology. (2016) 149:123–30. doi: 10.1111/imm.12639
75. Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, et al. Interleukin 23 Production by Intestinal CD103+CD11b+ dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. (2012) 36:276–87. doi: 10.1016/j.immuni.2011.12.011
76. Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. (2013) 498:113–7. doi: 10.1038/nature12240
77. Castellanos JG, Woo V, Viladomiu M, Putzel G, Lima S, Diehl GE, et al. Microbiota-Induced TNF-like Ligand 1A drives group 3 innate lymphoid cell-mediated barrier protection and intestinal t cell activation during colitis. Immunity. (2018) 49:1077–89.e5. doi: 10.1016/j.immuni.2018.10.014
78. Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. (2004) 304:1014–8. doi: 10.1126/science.1096158
79. McClure R, Massari, P. TLR-dependent human mucosal epithelial cell responses to microbial pathogens. Front Immunol. (2014) 5:386. doi: 10.3389/fimmu.2014.00386
80. Albiger B, Dahlberg S, Henriques-Normark B, Normark S. Role of the innate immune system in host defence against bacterial infections: focus on the Toll-like receptors. J Intern Med. (2007) 261:511–28. doi: 10.1111/j.1365-2796.2007.01821.x
81. Re F, Strominger JL. Toll-like Receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J Biol Chem. (2001) 276:37692–9. doi: 10.1074/jbc.M105927200
82. Morooka T, Umeda A, Amako K. Motility as an intestinal colonization factor for campylobacter jejuni. Microbiology. (1985) 131:1973–80.
83. O'Neil HS, Marquis, H. Listeria monocytogenes flagella are used for motility, not as adhesins, to increase host cell invasion. Infect Immun. (2006) 74:6675–81. doi: 10.1128/IAI.00886-06
84. Tallant T, Deb A, Kar N, Lupica J, de Veer MJ, DiDonato JA. Flagellin acting via TLR5 is the major activator of key signaling pathways leading to NF-kappa B and proinflammatory gene program activation in intestinal epithelial cells. BMC Microbiol. (2004) 4:33. doi: 10.1186/1471-2180-4-33
85. Chassaing B, Ley RE, Gewirtz AT. Intestinal epithelial cell toll-like receptor 5 regulates the intestinal microbiota to prevent low-grade inflammation and metabolic syndrome in mice. Gastroenterology. (2014) 147:1363–77.e17. doi: 10.1053/j.gastro.2014.08.033
86. Aggarwal S, Ghilardi N, Xie M-H, de Sauvage FJ, Gurney AL. Interleukin-23 Promotes a Distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. (2003) 278:1910–4. doi: 10.1074/jbc.M207577200
87. Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. (2006) 116:1218–22. doi: 10.1172/JCI28508
88. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. (2005) 6:1133–41. doi: 10.1038/ni1261
89. Watanabe T, Kitani A, Strober W. NOD2 regulation of Toll-like receptor responses and the pathogenesis of Crohn's disease. Gut. (2005) 54:1515–8. doi: 10.1136/gut.2005.071795
90. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. (2003) 278:8869–72. doi: 10.1074/jbc.C200651200
91. Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. (2004) 5:800–8. doi: 10.1038/ni1092
92. Lisitsyn NA, Bukurova YA, Nikitina IG, Krasnov GS, Sykulev Y, Beresten SF. Enteric alpha defensins in norm and pathology. Ann Clin Microbiol Antimicrob. (2012) 11:1. doi: 10.1186/1476-0711-11-1
93. Hallstrom K, McCormick BA. Salmonella interaction with and passage through the intestinal mucosa: through the lens of the organism. Front Microbiol. (2011) 2:88. doi: 10.3389/fmicb.2011.00088
94. Mathur R, Oh H, Zhang D, Park SG, Seo J, Koblansky A, et al. A mouse model of Salmonella typhi infection. Cell. (2012) 151:590–602. doi: 10.1016/j.cell.2012.08.042
95. Blackwell JM, Goswami T, Evans CA, Sibthorpe D, Papo N, White JK, et al. SLC11A1 (formerly NRAMP1) and disease resistance. Cell Microbiol. (2001) 3:773–84. doi: 10.1046/j.1462-5822.2001.00150.x
96. Reboldi A, Cyster JG. Peyer's patches: organizing B-cell responses at the intestinal frontier. Immunol Rev. (2016) 271:230–45. doi: 10.1111/imr.12400
97. Zarepour M, Bhullar K, Montero M, Ma C, Huang T, Velcich A, et al. The mucin muc2 limits pathogen burdens and epithelial barrier dysfunction during salmonella enterica serovar typhimurium colitis. Infect Immun. (2013) 81:3672–83. doi: 10.1128/IAI.00854-13
98. Voedisch S, Koenecke C, David S, Herbrand H, Förster R, Rhen M, et al. Mesenteric lymph nodes confine dendritic cell-mediated dissemination of salmonella enterica serovar typhimurium and limit systemic disease in mice. Infect. Immun. (2009) 77:3170–80. doi: 10.1128/IAI.00272-09
99. Cirillo DM, Valdivia RH, Monack DM, Falkow S. Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol Microbiol. (1998) 30:175–88.
100. van der Velden AWM, Copass MK, Starnbach MN. Salmonella inhibit T cell proliferation by a direct, contact-dependent immunosuppressive effect. Proc Natl Acad Sci USA. (2005) 102:17769–74. doi: 10.1073/pnas.0504382102
101. Tripp CS, Wolf SF, Unanue ER. Interleukin 12 and tumor necrosis factor alpha are costimulators of interferon gamma production by natural killer cells in severe combined immunodeficiency mice with listeriosis, and interleukin 10 is a physiologic antagonist. Proc Natl Acad Sci USA. (1993) 90:3725–29.
102. Myers JT, Tsang AW, Swanson JA. Localized reactive oxygen and nitrogen intermediates inhibit escape of listeria monocytogenes from vacuoles in activated macrophages. J Immunol. (2003) 171:5447–53.
103. Beauregard KE, Lee K-D, Collier RJ, Swanson JA. pH-dependent perforation of macrophage phagosomes by Listeriolysin O from Listeria monocytogenes. J Exp Med. (1997) 186:1159–63.
104. Carrero JA, Calderon B, Unanue ER. Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J Exp Med. (2006) 203:933–40. doi: 10.1084/jem.20060045
105. Sheridan BS, Lefrançois L. Regional and mucosal memory T cells. Nat Immunol. (2011) 12:485–91. doi: 10.1038/ni.2029
106. Senda T, Dogra P, Granot T, Furuhashi K, Snyder, MK, Thapa P, et al. Microanatomical dissection of human intestinal T-cell immunity reveals site-specific changes in gut-associated lymphoid tissues over life. Mucosal Immunol. (2018) 12:378–89. doi: 10.1038/s41385-018-0110-8
107. Jang MH, Sougawa N, Tanaka T, Hirata T, Hiroi T, Tohya K, et al. CCR7 is critically important for migration of dendritic cells in intestinal lamina propria to mesenteric lymph nodes. J Immunol Baltim Md 1950. (2006) 176:803–10. doi: 10.4049/jimmunol.176.2.803
108. Qiu Z, Khairallah C, Sheridan BS. Listeria monocytogenes: a model pathogen continues to refine our knowledge of the cd8 t cell response. Pathog Basel Switz. (2018) 7:E55. doi: 10.3390/pathogens7020055
109. Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. (2004) 21:527–38. doi: 10.1016/j.immuni.2004.08.011
110. Silberger DJ, Zindl CL, Weaver CT. Citrobacter rodentium: a model enteropathogen for understanding the interplay of innate and adaptive components of type 3 immunity. Mucosal Immunol. (2017) 10:1108–17. doi: 10.1038/mi.2017.47
111. Koroleva EP, Halperin S, Gubernatorova EO, Macho-Fernandez E, Spencer CM, Tumanov AV. Citrobacter rodentium -induced colitis: a robust model to study mucosal immune responses in the gut. J Immunol Methods. (2015) 421:61–72. doi: 10.1016/j.jim.2015.02.003
112. Lai Y, Rosenshine I, Leong JM, Frankel G. Intimate host attachment: enteropathogenic and enterohaemorrhagic Escherichia coli: Intimate EPEC and EHEC adhesion. Cell Microbiol. (2013) 15:1796–808. doi: 10.1111/cmi.12179
113. Vallance BA, Deng W, Knodler LA, Finlay BB. Mice lacking T and B lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect. Immun. (2002) 70:2070–81. doi: 10.1128/IAI.70.4.2070-2081.2002
114. Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. (2007) 2:119–29. doi: 10.1016/j.chom.2007.06.010
115. Pham TA, Clare S, Goulding D, Arasteh JM, Stares MD, Browne HP, et al. Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe. (2014) 16:504–16. doi: 10.1016/j.chom.2014.08.017
116. Chen J, Waddell A, Lin Y-D, Cantorna MT. Dysbiosis caused by vitamin D receptor deficiency confers colonization resistance to Citrobacter rodentium through modulation of innate lymphoid cells. Mucosal Immunol. (2015) 8:618–26. doi: 10.1038/mi.2014.94
117. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. (2008) 14:282–9. doi: 10.1038/nm1720
118. Basu R, O'Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang W, et al. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity. (2012) 37:1061–75. doi: 10.1016/j.immuni.2012.08.024
119. Hanash AM, Dudakov JA, Hua G, O'Connor MH, Young LF, Singer NV, et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity. (2012) 37:339–50. doi: 10.1016/j.immuni.2012.05.028
120. Bergman MA, Loomis WP, Mecsas J, Starnbach MN, Isberg RR. CD8+ T cells restrict yersinia pseudotuberculosis infection: bypass of anti-phagocytosis by targeting antigen-presenting cells. PLoS Pathog. (2009) 5:e1000573.
121. Zhang Y, Mena P, Romanov G, Lin JS, Smiley ST, Bliska JB. A protective epitope in type iii effector yope is a major CD8 T cell antigen during primary infection with Yersinia pseudotuberculosis. Infect. Immun. (2012) 80:206–14. doi: 10.1128/IAI.05971-11
122. Lin, J.-S., Szaba FM, Kummer LW, Chromy, B. A, Smiley, S. T. Yersinia pestis YopE contains a dominant CD8 T cell epitope that confers protection in a mouse model of pneumonic plague. J Immunol Baltim Md 1950. (2011) 187:897–904.
123. Shen H, Gonzalez-Juarbe N, Blanchette K, Crimmins G, Bergman MA, Isberg RR, et al. CD8 + T cells specific to a single Yersinia pseudotuberculosis epitope restrict bacterial replication in the liver but fail to provide sterilizing immunity. Infect Genet Evol. (2016) 43:289–96. doi: 10.1016/j.meegid.2016.06.008
124. González-Juarbe N, Shen H, Bergman MA, Orihuela CJ, Dube PH. YopE specific CD8+ T cells provide protection against systemic and mucosal Yersinia pseudotuberculosis infection. PLoS ONE. (2017) 12:e0172314. doi: 10.1371/journal.pone.0172314
125. Bergsbaken T, Bevan MJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8+ T cells responding to infection. Nat Immunol. (2015) 16:406–14. doi: 10.1038/ni.3108
126. Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, et al. Colorectal cancer statistics, 2017: colorectal cancer statistics, 2017. CA Cancer J Clin. (2017) 67:177–93. doi: 10.3322/caac.21395
127. Armaghany T, Wilson JD, Chu, Q, Mills G. Genetic alterations in colorectal cancer. Gastrointest Cancer Res. (2012) 5:19–27.
128. Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol. (2014) 12:661–72. doi: 10.1038/nrmicro3344
129. Grizzi F, Bianchi P, Malesci A, Laghi L. Prognostic value of innate and adaptive immunity in colorectal cancer. World J Gastroenterol. (2013) 19:174–84. doi: 10.3748/wjg.v19.i2.174
130. Stumhofer JS, Hunter CA. Advances in understanding the anti-inflammatory properties of IL-27. Immunol Lett. (2008) 117:123–30. doi: 10.1016/j.imlet.2008.01.011
131. Dodge IL, Carr MW, Cernadas, M, Brenner MB. IL-6 production by pulmonary dendritic cells impedes Th1 immune responses. J Immunol Baltim Md 1950. (2003) 170:4457–64. doi: 10.4049/jimmunol.170.9.4457
132. Berinstein NL. Carcinoembryonic antigen as a target for therapeutic anticancer vaccines: a review. J Clin Oncol Off J Am Soc Clin Oncol. (2002) 20:2197–207. doi: 10.1200/JCO.2002.08.017
133. Ajioka Y, Allison LJ, Jass JR. Significance of MUC1 and MUC2 mucin expression in colorectal cancer. J Clin Pathol. (1996) 49:560–4.
134. Speetjens FM, Kuppen PJ, Welters MJ, Essahsah F, Voet van den Brink AM, Lantrua MG, et al. Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clin Cancer Res Off J Am Assoc Cancer Res. (2009) 15:1086–95. doi: 10.1158/1078-0432.CCR-08-2227
135. Nouri-Shirazi M, Banchereau J, Bell D, Burkeholder S, Kraus ET, Davoust J, et al. Dendritic cells capture killed tumor cells and present their antigens to elicit tumor-specific immune responses. J Immunol. (2000) 165:3797–803. doi: 10.4049/jimmunol.165.7.3797
136. Hernandez C, Huebener P, Schwabe RF. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene. (2016) 35:5931–41. doi: 10.1038/onc.2016.104
137. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. (2007) 13:1050–9. doi: 10.1038/nm1622
138. Messmer D, Yang H, Telusma G, Knoll F, Li J, Messmer B, et al. High mobility group box protein 1: an endogenous signal for dendritic cell maturation and th1 polarization. J Immunol. (2004) 173:307–13. doi: 10.4049/jimmunol.173.1.307
139. Zitvogel L, Kepp O, Senovilla L, Menger L, Chaput N, Kroemer G. Immunogenic tumor cell death for optimal anticancer therapy: the calreticulin exposure pathway. Clin Cancer Res. (2010) 16:3100–4. doi: 10.1158/1078-0432.CCR-09-2891
140. Kajihara M, Takakura K, Kanai T, Ito Z, Saito K, Takami S, et al. Dendritic cell-based cancer immunotherapy for colorectal cancer. World J Gastroenterol. (2016) 22:4275–86. doi: 10.3748/wjg.v22.i17.4275
141. Waldmann TA. The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: implications for cancer therapy. Cancer Immunol Res. (2015) 3:219–27. doi: 10.1158/2326-6066.CIR-15-0009
142. Grabstein KH, Eisenman J, Shanebeck K, Rauch C, Srinivasan S, Fung V, et al. Cloning of a T cell growth factor that interacts with the beta chain of the interleukin-2 receptor. Science. (1994) 264:965–8. doi: 10.1126/science.8178155
143. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. (2004) 117:1281–3. doi: 10.1242/jcs.00963
144. Kahan SM, Bakshi RK, Luther R, Harrington LE, Curtis Hendrickson R, Zajac AJ, et al. IL-2 producing and non-producing effector CD8 T cells phenotypically and transcriptionally coalesce to form memory subsets with similar protective properties. J Immunol. (2017) 198:212.6.
145. Sojka DK, Bruniquel D, Schwartz RH, Singh NJ. IL-2 Secretion by CD4+ T Cells in vivo is rapid, transient, and influenced by TCR-specific competition. J Immunol. (2004) 172:6136–43. doi: 10.4049/jimmunol.172.10.6136
146. Granucci F, Vizzardelli C, Pavelka N, Feau S, Persico M, Virzi E, et al. Inducible IL-2 production by dendritic cells revealed by global gene expression analysis. Nat Immunol. (2001) 2:882–8. doi: 10.1038/ni0901-882
147. Yui MA, Sharp LL, Havran WL, Rothenberg EV. Preferential activation of an IL-2 regulatory sequence transgene in TCR gamma delta and NKT cells: subset-specific differences in IL-2 regulation. J Immunol Baltim. Md 1950. (2004) 172:4691–9. doi: 10.4049/jimmunol.172.8.4691
148. Jiang T, Zhou C, Ren S. Role of IL-2 in cancer immunotherapy. Oncoimmunology. (2016) 5:e1163462. doi: 10.1080/2162402X.2016.1163462
149. Henney CS, Kuribayashi K, Kern DE, Gillis S. Interleukin-2 augments natural killer cell activity. Nature. (1981) 291:335–8.
150. Wang KS, Frank DA, Ritz J. Interleukin-2 enhances the response of natural killer cells to interleukin-12 through up-regulation of the interleukin-12 receptor and STAT4. Blood. (2000) 95:3183–90.
151. Kamimura D, Bevan MJ. Naive CD8 + T cells differentiate into protective memory-like cells after IL-2–anti–IL-2 complex treatment in vivo. J Exp Med. (2007) 204:1803–12. doi: 10.1084/jem.20070543
152. Dooms H, Wolslegel K, Lin P, Abbas AK. Interleukin-2 enhances CD4 + T cell memory by promoting the generation of IL-7Rα-expressing cells. J Exp Med. (2007) 204:547–57. doi: 10.1084/jem.20062381
153. Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 Is Expressed by Dendritic Cells in Response to Type I IFN, Double-Stranded RNA, or Lipopolysaccharide and Promotes Dendritic Cell Activation. J Immunol. (2001) 167:1179–87. doi: 10.4049/jimmunol.167.3.1179
154. Flamand L, Stefanescu I, Menezes J. Human herpesvirus-6 enhances natural killer cell cytotoxicity via IL-15. J Clin Invest. (1996) 97:1373–81. doi: 10.1172/JCI118557
155. Doherty TM, Seder RA, Sher A. Induction and regulation of IL-15 expression in murine macrophages. J Immunol. (1996) 156:735–41.
156. Mishra A, Sullivan L, Caligiuri MA. Molecular pathways: interleukin-15 signaling in health and in cancer. Clin Cancer Res. (2014) 20:2044–50. doi: 10.1158/1078-0432.CCR-12-3603
157. Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. (2011) 364:2119–27. doi: 10.1056/NEJMoa1012863
158. Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. (2012) 484:529–33. doi: 10.1038/nature10975
159. Bahri R, Pateras IS, D'Orlando O, Goyeneche-Patino DA, Campbell M, Polansky JK, et al. IL-15 suppresses colitis-associated colon carcinogenesis by inducing antitumor immunity. Oncoimmunology. (2015) 4:e1002721. doi: 10.1080/2162402X.2014.1002721
160. Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, Th2, Treg, Th17) in patients with colorectal cancer. Cancer Res. (2011) 71:1263–71. doi: 10.1158/0008-5472.CAN-10-2907
161. Liu J, Duan Y, Cheng X, Chen X, Xie W, Long H, et al. IL-17 is associated with poor prognosis and promotes angiogenesis via stimulating VEGF production of cancer cells in colorectal carcinoma. Biochem Biophys Res Commun. (2011) 407:348–54. doi: 10.1016/j.bbrc.2011.03.021
162. Robinson CM, Nau GJ. Interleukin-12 and Interleukin-27 regulate macrophage control of Mycobacterium tuberculosis. J Infect Dis. (2008) 198:359–366. doi: 10.1086/589774
163. Wang K, Kim MK, Di Caro G, Wong J, Shalapour S, Wan J, et al. Interleukin-17 receptor A signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity. (2014) 41:1052–63. doi: 10.1016/j.immuni.2014.11.009
164. Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. (2012) 491:254–8. doi: 10.1038/nature11465
165. Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y, et al. Fusobacterium nucleatum Increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear factor–κB, and up-regulating expression of MicroRNA-21. Gastroenterology. (2017) 152:851–66.e24 doi: 10.1053/j.gastro.2016.11.018
166. Numasaki M, Fukushi J, Ono M, Narula SK, Zavodny PJ, Kudo T, et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood. (2003) 101:2620–7. doi: 10.1182/blood-2002-05-1461
167. Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, et al. An interleukin-17–mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med. (2013) 19:1114–23. doi: 10.1038/nm.3291
168. Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell. (2014) 26:207–21. doi: 10.1016/j.ccr.2014.05.019
169. Bhattacharya S, Ray RM, Johnson LR. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem J. (2005) 392:335–44. doi: 10.1042/BJ20050465
170. Xie TX, Wei D, Liu M, Gao AC, Ali-Osman F, Sawaya R, et al. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene. (2004) 23:3550–60. doi: 10.1038/sj.onc.1207383
171. Itoh M, Murata T, Suzuki T, Shindoh M, Nakajima K, Imai K, et al. Requirement of STAT3 activation for maximal collagenase-1 (MMP-1) induction by epidermal growth factor and malignant characteristics in T24 bladder cancer cells. Oncogene. (2006) 25:1195–204. doi: 10.1038/sj.onc.1209149
172. Taniguchi K, Karin M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin Immunol. (2014) 26:54–74. doi: 10.1016/j.smim.2014.01.001
173. Tseng-Rogenski SS, Hamaya Y, Choi DY, Carethers JM. Interleukin 6 alters localization of hMSH3, leading to DNA mismatch repair defects in colorectal cancer cells. Gastroenterology. (2015) 148:579–89. doi: 10.1053/j.gastro.2014.11.027
174. Hirotani T, et al. The nuclear I B protein I BNS selectively inhibits lipopolysaccharide-induced IL-6 production in macrophages of the colonic lamina propria. J Immunol. (2005) 174:3650–7. doi: 10.4049/jimmunol.174.6.3650
175. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
176. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. (2007) 8:967–74. doi: 10.1038/ni1488
177. Putoczki TL, Thiem S, Loving A, Busuttil RA, Wilson NJ, Ziegler PK, et al. Interleukin-11 Is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell. (2013) 24:257–71. doi: 10.1016/j.ccr.2013.06.017
178. Jiang R, Wang H, Deng L, Hou J, Shi R, Yao M, et al. IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer. (2013) 13:59. doi: 10.1186/1471-2407-13-59
179. Kryczek I, Lin Y, Nagarsheth N, Peng D, Zhao L, Zhao E, et al. IL-22+CD4+ T Cells Promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity. (2014) 40:772–84. doi: 10.1016/j.immuni.2014.03.010
180. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. (2012) 491:259–63. doi: 10.1038/nature11535
181. Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med. (2013) 210:917–31. doi: 10.1084/jem.20122308
182. Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. (2018) 15:234–48. doi: 10.1038/nrclinonc.2018.8.
183. Grivennikov SI, Tumanov AV, Liepinsh DJ, Kruglov AA, Marakusha BI, Shakhov AN, et al. Distinct and nonredundant in vivo functions of TNF produced by T cells and macrophages/neutrophils. Immunity. (2005) 22:93–104. doi: 10.1016/j.immuni.2004.11.016
184. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. (2003) 19:59–70. doi: 10.1016/S1074-7613(03)00171-7
185. Wang R, Jaw JJ, Stutzman NC, Zou Z, Sun PD. Natural killer cell-produced IFN-γ and TNF-α induce target cell cytolysis through up-regulation of ICAM-1. J Leukoc Biol. (2012) 91:299–309. doi: 10.1189/jlb.0611308
186. Roulis M, Armaka M, Manoloukos M, Apostolaki M, Kollias G. Intestinal epithelial cells as producers but not targets of chronic TNF suffice to cause murine crohn-like pathology. Proc Natl Acad Sci USA. (2011) 108:5396–401. doi: 10.1073/pnas.1007811108
187. Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-α in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. (2008) 118:560–70. doi: 10.1172/JCI32453
188. Hagerling C, Casbon A-J, Werb Z. Balancing the innate immune system in tumor development. Trends Cell Biol. (2015) 25:214–20. doi: 10.1016/j.tcb.2014.11.001
189. Ho AWY, Wong CK, Lam CWK. Tumor necrosis factor-alpha up-regulates the expression of CCL2 and adhesion molecules of human proximal tubular epithelial cells through MAPK signaling pathways. Immunobiology. (2008) 213:533–44. doi: 10.1016/j.imbio.2008.01.003
190. Ahmad R, Al-Roub A, Kochumon S, Akther N, Thomas R, Kumari M, et al. The synergy between palmitate and TNF-α for CCL2 production is dependent on the TRIF/IRF3 pathway: implications for metabolic inflammation. J Immunol. (2018) 200:3599–611. doi: 10.4049/jimmunol.1701552
191. Cushing SD, Berliner JA, Valente AJ, Territo MC, Navab M, Parhami F, et al. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc Natl Acad Sci USA. (1990) 87:5134–8. doi: 10.1073/pnas.87.13.5134
192. Rafei M, Hsieh J, Fortier S, Li M, Yuan S, Birman E, et al. Mesenchymal stromal cell-derived CCL2 suppresses plasma cell immunoglobulin production via STAT3 inactivation and PAX5 induction. Blood. (2008) 112:4991–8. doi: 10.1182/blood-2008-07-166892
193. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. (2009) 29:313–26. doi: 10.1089/jir.2008.0027
194. Smith LK, Boukhaled GM, Condotta SA, Mazouz S, Guthmiller JJ, Vijay R, et al. Interleukin-10 directly inhibits CD8 + T cell function by enhancing N-glycan branching to decrease antigen sensitivity. Immunity. (2018) 48, 299–312.e5 doi: 10.1016/j.immuni.2018.01.006
195. Granovsky M, Fata J, Pawling J, Muller WJ, Khokha R, Dennis JW. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat Med. (2000) 6:306–12. doi: 10.1038/73163
196. Wang R, Lu M, Zhang J, Chen S, Luo X, Qin Y, et al. Increased IL-10 mRNA expression in tumor-associated macrophage correlated with late stage of lung cancer. J Exp Clin Cancer Res CR. (2011) 30:62. doi: 10.1186/1756-9966-30-62
197. Erreni M, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) and inflammation in colorectal cancer. Cancer Microenviron. (2011) 4:141–54. doi: 10.1007/s12307-010-0052-5
198. Herbeuval J-P, Lelievre E, Lambert C, Dy M, Genin, C. Recruitment of STAT3 for production of IL-10 by colon carcinoma cells induced by macrophage-derived IL-6. J Immunol. (2004) 172:4630–6. doi: 10.4049/jimmunol.172.7.4630
199. Wang Y, Ma R, Liu F, Lee SA, Zhang L. Modulation of gut microbiota: a novel paradigm of enhancing the efficacy of programmed death-1 and programmed death ligand-1 blockade therapy. Front Immunol. (2018) 9:374. doi: 10.3389/fimmu.2018.00374
Keywords: APC, SFB, CRC, SCFA, TLR, PAMP
Citation: Gaudino SJ and Kumar P (2019) Cross-Talk Between Antigen Presenting Cells and T Cells Impacts Intestinal Homeostasis, Bacterial Infections, and Tumorigenesis. Front. Immunol. 10:360. doi: 10.3389/fimmu.2019.00360
Received: 30 August 2018; Accepted: 12 February 2019;
Published: 06 March 2019.
Edited by:
Raghvendra Mohan Srivastava, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Dan Anthony Mitchell, University of Warwick, United KingdomDerek Pociask, Tulane University School of Medicine, United States
Copyright © 2019 Gaudino and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pawan Kumar, cGF3YW4ua3VtYXJAc3Rvbnlicm9vay5lZHU=