 Simon Schwager
Simon Schwager Michael Detmar
Michael Detmar- Institute of Pharmaceutical Sciences, Swiss Federal Institute of Technology, ETH Zurich, Zurich, Switzerland
The lymphatic vasculature plays a crucial role in regulating the inflammatory response by influencing drainage of extravasated fluid, inflammatory mediators, and leukocytes. Lymphatic vessels undergo pronounced enlargement in inflamed tissue and display increased leakiness, indicating reduced functionality. Interfering with lymphatic expansion by blocking the vascular endothelial growth factor C (VEGF-C)/vascular endothelial growth factor receptor 3 (VEGFR-3) signaling axis exacerbates inflammation in a variety of disease models, including inflammatory bowel disease (IBD), rheumatoid arthritis and skin inflammation. In contrast, stimulation of the lymphatic vasculature, e.g., by transgenic or viral overexpression as well as local injections of VEGF-C, has been shown to reduce inflammation severity in models of rheumatoid arthritis, skin inflammation, and IBD. Strikingly, the induced expansion of the lymphatic vasculature improves lymphatic function as assessed by the drainage of dyes, fluorescent tracers or inflammatory cells and labeled antigens. The drainage performance of lymphatic vessels is influenced by vascular permeability and pumping activity, which are influenced by VEGF-C/VEGFR-3 signaling as well as several inflammatory mediators, including TNF-α, IL-1β, and nitric oxide. Considering the beneficial effects of lymphatic activation in inflammation, administration of pro-lymphangiogenic factors like VEGF-C, preferably in a targeted, inflammation site-specific fashion, represents a promising therapeutic approach in the setting of inflammatory pathologies.
Introduction
Inflammation is a defensive reaction of the organism against pathogens or irritants. It is characterized by the five cardinal symptoms of rubor (redness), calor (increased heat), tumor (swelling), dolor (pain), and functio laesa (impaired function), which are mostly mediated by the expansion and activation of blood vessels. Inflammation is commonly associated with the formation of new blood (angiogenesis) and lymphatic (lymphangiogenesis) vessels from the pre-existing vascular networks. Interestingly, while the activation of the blood vasculature has been reported to aggravate inflammation severity in a variety of disease models (1–3), lymphatic vessels generally appear to exert beneficial effects, possibly by improving the clearance of extravasated fluid, thus reducing edema formation and levels of pro-inflammatory mediators as well as numbers of immune cells.
This review provides an overview of studies investigating the role of lymphatic expansion and function in common inflammatory diseases such as skin inflammation, inflammatory bowel disease (IBD) and rheumatoid arthritis (RA). In addition, the known effects of inflammatory mediators on the lymphatic vasculature and commonly used mouse models are described.
The lymphatic vasculature is a hierarchically structured, one-way circuit composed of initial capillaries, which lack a continuous basement membrane and smooth muscle cell coverage, draining into larger, smooth muscle cell-covered collectors and ultimately lymph nodes. In the setting of inflammation, the lymphatic system is critically important, as it is needed to ensure tissue fluid homeostasis by draining the larger amounts of extravasated fluid originating from increasingly leaky, inflammatory blood vessels. Indeed, an increased interstitial fluid pressure has been found to lead to the dilation of initial lymphatic vessels, thus facilitating the entry of fluid and inflammatory cells into the lymphatic vasculature and thereby removal from the inflamed tissue (4). In addition, lymphatic vessels are crucial for immune surveillance, as they serve as main transport routes for cells and inflammatory mediators to lymph nodes, where immune responses are mounted.
The most-thoroughly characterized signaling axis involved in lymphatic expansion and development consists of the vascular endothelial growth factor receptor 3 (VEGFR-3) and its ligands VEGF-C and VEGF-D. VEGFR-3 is part of the receptor tyrosine kinase family and is expressed widely in vascular endothelial cells during embryonic development, but becomes strongly restricted to lymphatic endothelial cells (LECs) in the adult organism under physiological conditions (5).
VEGF-C is the main ligand of VEGFR-3 and induces proliferation and migration of endothelial cells (6, 7). It undergoes extensive post-translational proteolytic processing, which also regulates the molecule's binding properties. Fully processed VEGF-C binds VEGFR-3 and, albeit with a lower affinity, VEGFR-2 (8). A mutated form of VEGF-C in which the cysteine 156 is replaced with a serine (VEGF-C156Ser) selectively binds VEGFR-3 (9).
VEGF-D has been reported to induce proliferation of endothelial cells (10). In mice, VEGF-D exclusively binds VEGFR-3, while fully processed human VEGF-D may also bind VEGFR-2 (11).
In order to study the role of lymphatic vessels in different pathologies, various mouse lines with a modified VEGFR-3 signaling axis have been generated. In K14-VEGF-C mice, VEGF-C is overexpressed under the control of the keratin-14 promoter, resulting in elevated levels of the growth factor in the skin and an enlarged dermal lymphatic vascular network (12). A similar lymphatic hyperplasia has been observed in mice transgenic for VEGF-D (K14-VEGF-D) (13). In contrast, mice overexpressing a soluble form of VEGFR-3 in the skin (K14-VEGFR-3-Ig mice) lack dermal lymphatic vessels and develop edema in the feet and skin (14).
Apart from promoting or inhibiting lymphatic vascular expansion, the clearance capacity of lymphatic vessels is subject to regulation by various signals. Drainage performance is influenced by vascular permeability and pumping activity of lymphatic vessels. Mediators inducing increased lymphatic vessel permeability include TNF-α, IL-1β, histamine, and the VEGF-C/VEGFR-3 axis (15–17). Lymphatic contractions and thereby pumping are negatively regulated by various inflammatory mediators, including prostaglandins, histamine, and nitric oxide (NO), while VEGF-C has enhancing effects (18–21). NO regulates lymphatic vessel function via its effects on lymphatic smooth muscle cells leading to vasodilation. It is produced constitutively by the endothelial nitric oxide synthase (eNOS) under physiological conditions. In inflammation, however, its levels are elevated due to the higher expression of inducible nitric oxide synthase (iNOS) on immune cells and inflamed endothelium, which has been linked to reduced lymphatic contraction frequency (22).
The Lymphatic Vasculature in Inflammatory Diseases
Skin Inflammation
A wide range of skin pathologies including psoriasis, atopic dermatitis, rosacea, and UV damage are characterized by pronounced and often prolonged inflammation. The lymphatic vasculature is often aberrant in inflamed skin; in human psoriatic plaques for example, lymphatic vessels are dilated and tortuous (23–25). Nevertheless, lymphatic dysregulation in the human disease has attracted comparatively little attention.
Multiple mouse models have been established to facilitate the study of these diseases in general and the role of the lymphatic vasculature in particular. A common model are K14-VEGF-A transgenic mice which overexpress VEGF-A under the control of the keratin-14 promoter, resulting in chronically elevated levels of said growth factor in the skin and a concomitant expanded, leaky blood vasculature. Homozygous mice spontaneously develop a chronic skin inflammation at the age of 6 months (26). In hemizygous mice, a contact sensitizer (e.g., oxazolone) can be used to trigger a contact hypersensitivity reaction (CHS), leading to a similar chronic inflammatory skin disease (24).
In wild-type mice, skin inflammation may be elicited by inducing CHS, exposure to UVB radiation, injection of bacterial antigens like LPS or application of pro-inflammatory agents such as tetradecanoylphorbolacetate (TPA) or imiquimod (27).
Using these models, skin inflammation has been extensively studied in mice and the lymphatic vasculature has been demonstrated to be functionally impeded in UVB-irradiated, chronically inflamed ear skin. Evans blue injected into the inflamed skin stained strongly dilated lymphatic vessels that were extremely leaky, indicating reduced drainage capacity (1).
Stimulation of Lymphatic Vessels in Skin Inflammation
Activating the lymphatic vasculature in the setting of skin inflammation has been associated with reduced disease severity (summarized in Table 1). In K14-VEGF-A mice that had been crossed with K14-VEGF-C mice and were undergoing chronic CHS of the ear skin, the lymphatic vasculature was expanded and the inflammation, as assessed by edema formation, inflammatory cell infiltrate, and altered epidermal proliferation or differentiation, was significantly reduced compared to control inflamed K14-VEGF-A mice. Strikingly, the vascular expansion was accompanied by an improved lymphatic clearance function. Local injections of VEGF-C156Ser had similar disease-alleviating effects, indicating that VEGFR-3- rather than VEGFR-2-mediated signaling is mainly responsible for the observed anti-inflammatory effects (28). In agreement with this observation, local injections of VEGF-C156Ser also triggered a strong lymphangiogenic response and reduced inflammatory ear swelling and CD11b-positive immune cell infiltration in UVB-irradiated ear skin inflammation (32).
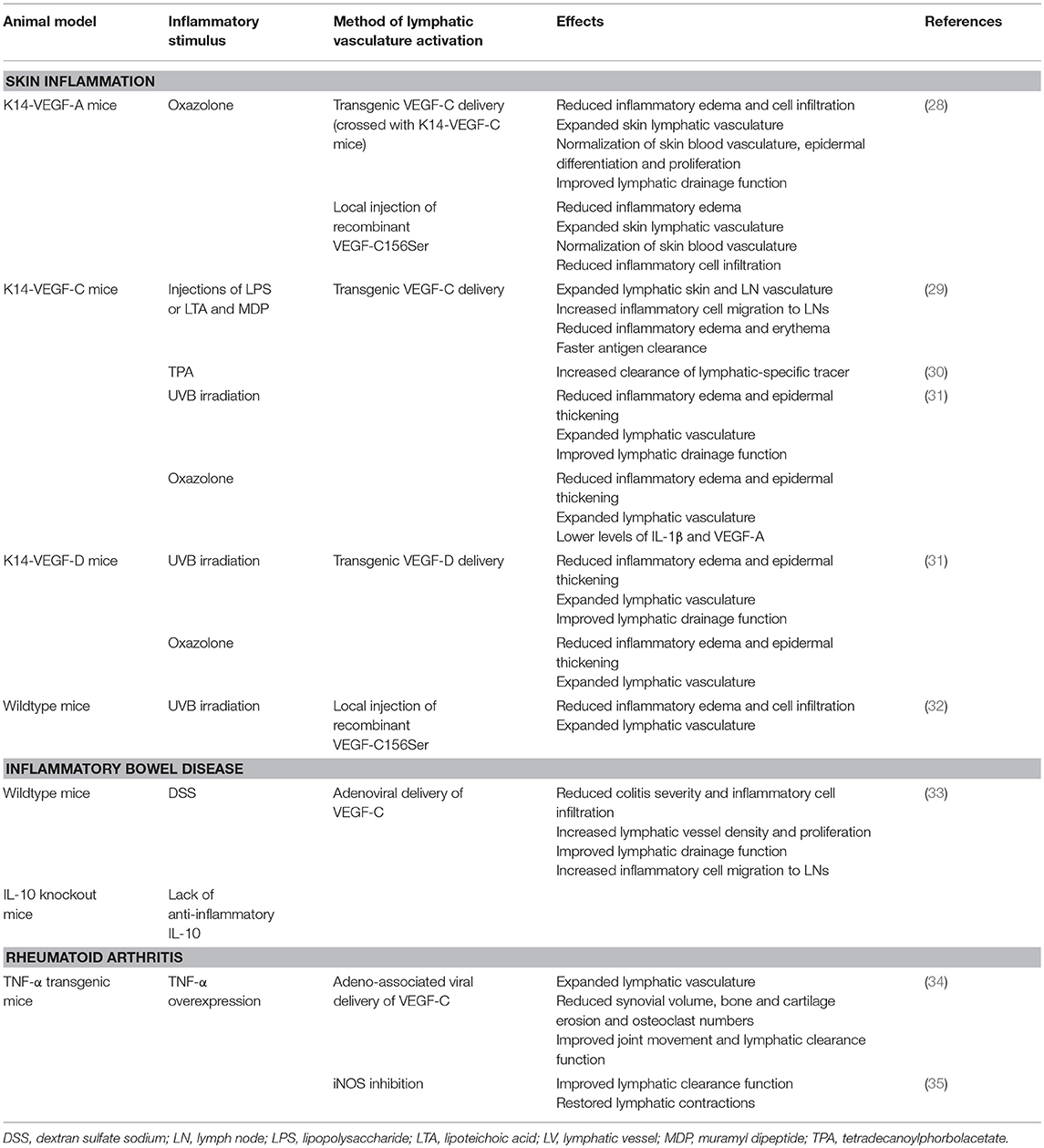
Table 1. Effects of lymphatic vessel stimulation in inflammatory diseases.
These findings are in line with a different study investigating the role of macrophages and lymphatic vessels in cutaneous inflammation. K14-VEGF-C mice that were subjected to lipopolysaccharide (LPS)- or lipoteichoic acid (LTA)/muramyl dipeptide (MDP)-induced skin inflammation presented with an expanded dermal and lymph node lymphatic vasculature. In addition, inflammatory tissue swelling and skin reddening were reduced. While no difference in FITC-dextran clearance was found, inflammatory cell migration to the draining lymph nodes and the drainage of fluorescently labeled antigen was significantly accelerated in K14-VEGF-C mice. These effects appeared to be dependent on macrophages, as clodronate-mediated depletion of these cells reduced lymphangiogenesis and delayed inflammation resolution (29). An enhanced lymphatic drainage function due to lymphatic stimulation has also been reported in other studies, e.g., after repeated application of TPA to the back skin of K14-VEGF-C transgenic mice, in which a lymphatic-specific, near-infrared tracer was cleared more rapidly than in wild-type mice (30). Similarly, in a study of acute skin inflammation, both K14-VEGF-C and, to a lesser extent, K14-VEGF-D transgenic mice had improved clearance of Evans blue out of UVB-irradiated ear skin (31). Moreover, these mice also had less inflammatory edema and reduced epidermal thickening in oxazolone- and UVB-induced skin inflammation. The reduction in inflammation was generally more pronounced in VEGF-C transgenic mice than in VEGF-D transgenic animals, indicating stronger anti-inflammatory effects of VEGF-C (31).
Inhibition of Lymphatic Vessels in Skin Inflammation
In contrast to stimulation of the lymphatic vasculature, inhibiting lymphatic vessels has been shown to aggravate skin inflammation in several studies (summarized in Table 2). Antibody-mediated blocking of VEGFR-3 strikingly reduced the number of lymphatic vessels in the inflamed ear skin of K14-VEGF-A mice during a CHS reaction. At the same time, tissue swelling, epidermal thickening, keratinocyte proliferation and the numbers of CD8- and CD11b-positive cells were significantly increased, indicating a more severe inflammatory phenotype. Interestingly, blocking VEGFR-2 alone or in combination with VEGFR-3 alleviated inflammation, indicating that VEGFR-2-mediated inhibition of blood vessels is beneficial in skin inflammation and outweighs the detrimental effects of VEGFR-3 inhibition (28). Similarly, adenoviral overexpression of a soluble VEGFR-3 strongly reduced lymphangiogenesis in mice undergoing LPS- or LTA/MDP-induced skin inflammation, resulting in delayed inflammation-resolution, slower clearance of FITC-dextran as well as FITC-labeled LPS, and reduced migration of inflammatory cells from the skin to the draining lymph nodes (29). Systemic, antibody-mediated inhibition of VEGFR-3 also led to increased edema formation and CD11b-positive cell numbers in UVB-irradiated ear skin (36).
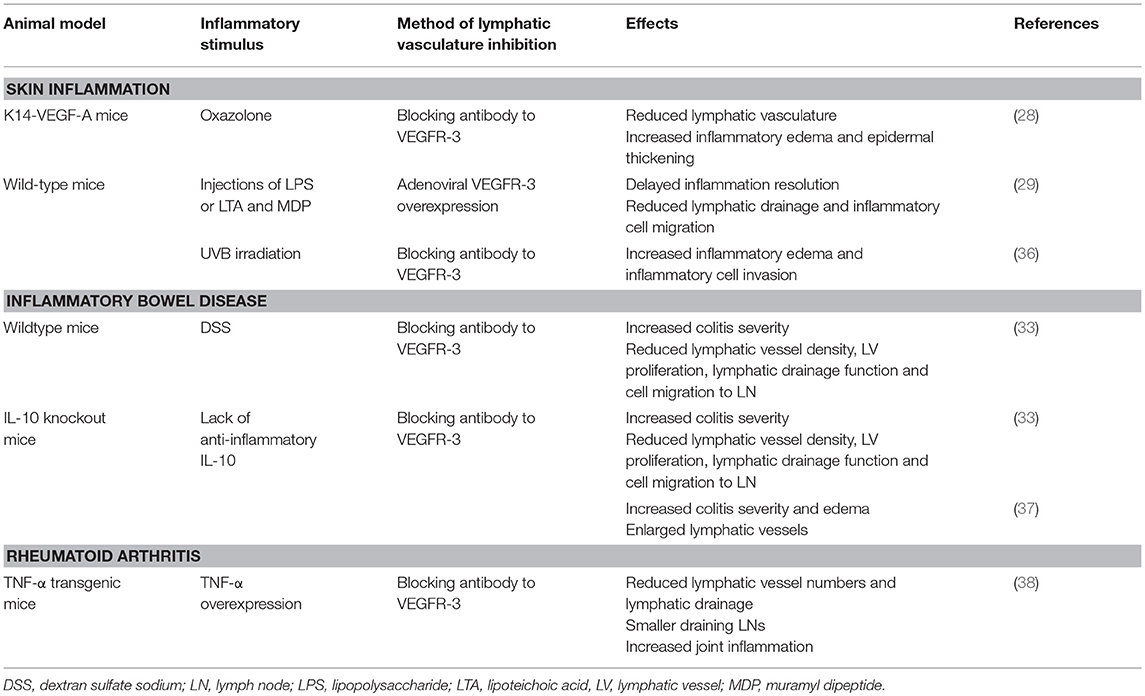
Table 2. Effects of lymphatic vessel inhibition in inflammatory diseases.
Inflammatory Bowel Disease
The term inflammatory bowel disease (IBD) comprises Crohn's disease (CD) and ulcerative colitis (UC), which are characterized by a chronic inflammation of the digestive tract. While UC generally affects the colon and presents with superficial ulcerations of the mucosa and submucosa, CD may occur at any location in the gastrointestinal tract and often causes transmural inflammation. As in the case of skin inflammation, research has long been focused on changes in the blood vasculature and VEGF-A has been suggested as an important mediator of IBD (39, 40).
In human patients suffering from IBD, lymphangiogenesis, lymphatic vessel obstruction, dilation, and submucosal edema are commonly observed (41–44) and abnormalities in the lymphatic vasculature had already been recognized during the original characterization of CD (45). In addition to morphological alterations, the functionality of IBD-associated lymphatic vessels is reduced. A study in patients with CD employed injections of the lymphatic-vessel-staining Patent Blue V dye in the inflamed colon and demonstrated morphological aberrations and functional impairment of the lymphatic vasculature, which could be correlated with disease severity. Strikingly, following surgical intervention and inflammation regression, lymphatic vessel appearance reverted back to normal, indicating that lymphatic vessel function may be involved in IBD pathogenesis in humans (46). In line with this, a lower density of lymphatic vessels could be linked to an increased risk of CD recurrence (47).
A multitude of studies have been performed in mouse models of IBD, the two most-commonly used being IL-10 knockout mice and dextran sulfate sodium (DSS)-induced colitis. IL-10-deficient mice spontaneously develop colitis at the age of 10–12 weeks, most likely due to the lacking anti-inflammatory and immunosuppressive activity of IL-10 (48, 49).
DSS-induced colitis relies on administration of the detergent DSS in drinking water, which damages the intestinal epithelium, most strongly in the distal colon, and compromises its barrier function, making the underlying tissue accessible to bacteria and associated substances. In order to model acute inflammation, mice are commonly given DSS for a certain amount of time (e.g., a week), for chronic inflammation, mice receive multiple cycles of DSS and intermittent regular drinking water (50, 51).
Stimulation of Lymphatic Vessels in Inflammatory Bowel Disease
Akin to skin inflammation, inducing the lymphatic vasculature is generally correlated with a reduction in inflammation severity (summarized in Table 1).
In IL-10 knockout mice as well as in animals undergoing DSS-induced colitis, adenoviral delivery of VEGF-C significantly increased lymphatic vessel density and was associated with a reduction in bodyweight loss and disease severity as assessed by stool consistency and presence or absence of fecal blood. Moreover, histological analyses revealed decreased submucosal tissue edema and inflammatory cell infiltration, while the proliferation of LECs was greatly increased. Quantification of Evans blue clearance out of inflamed distal colon tissue revealed an enhanced lymphatic drainage function, which was also reflected in an improved clearance of fluorescently labeled antigen-coated beads and an augmented inflammatory cell migration from the inflamed tissue to the draining lymph nodes. Similar to the observations in skin inflammation, depletion of macrophages by clodronate largely abolished the protective effects of VEGF-C (33). It has been suggested that VEGF-C may influence the cytokine balance in the inflamed colon. Indeed, in vitro experiments have shown VEGF-C to induce the upregulation of IL-10 by bone marrow-derived macrophages (33). In line with this, increased levels of IL-10 in combination with a reduction of IL-9, which is associated with intestinal barrier disruption, have been reported upon treatment with adenovirally delivered VEGF-C in mice undergoing DSS-induced colitis (52, 53).
Inhibition of Lymphatic Vessels in Inflammatory Bowel Disease
Blocking VEGFR-3 resulted in a worsened colitis in IL-10 knockout mice as well as DSS-treated animals in terms of the histological score (summarized in Table 2). Animals of both models presented with strongly reduced lymphatic vessel density and LEC proliferation upon VEGFR-3 inhibition. At the same time, lymphatic clearance of Evans blue and bacterial antigen as well as inflammatory cell mobilization to the draining lymph nodes were significantly reduced (33).
In a different, independent study, IL-10 knockout mice were treated with a blocking antibody to VEGFR-3. This resulted in enlarged and tortuous lymphatic vessels in the colon, increased submucosal edema and a higher leukocyte infiltration in the inflamed tissue as well as a higher disease severity score (37).
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a chronic inflammatory disease affecting the joints and characterized by episodic flares (54). In its chronic stage, RA is commonly associated with lymphadenopathy and a decrease in lymphatic drainage function, as shown for example by tracking the drainage of intradermally-injected, radioactively labeled albumin from the forearm (55). Lymphangiogenesis is also commonly observed in the joints of human RA patients and has been reproduced in mouse models of the disease (56, 57).
Commonly used mouse models of rheumatoid arthritis include TNF-α transgenic mice and K/B × N mice. The former overexpress human TNF-α and spontaneously develop chronic progressive joint inflammation at the age of ~4 weeks (58). K/B × N mice model the autoimmunity aspect of RA and are based on a mouse line transgenic for a T cell receptor specific for bovine ribonuclease. After breeding onto the NOD background, accidental recognition of a NOD-derived antigen triggers the onset of joint inflammation at 4 weeks after birth (59).
Lymphatic function has mainly been studied in these animals and a two-phase model has been proposed [reviewed in (60)]. In an initial “expansion” phase during joint inflammation, lymphangiogenesis and popliteal lymph node expansion with or without increased lymphatic vessel contractions limit the inflammatory response (57, 61, 62). During the following “collapse” phase, popliteal lymph nodes shrink and lymphatic vessel contractions as well as lymphatic drainage function decrease significantly. At the same time, the joint inflammation increases in severity (61, 63–65). Blocking TNF-α signaling resulted in increased lymphatic contractions and reduced joint inflammation (66).
Similar changes in lymph node characteristics have also been reported in human patients, where lymph node hypertrophy could be observed in the vast majority of patients suffering from active RA, while healthy individuals and patients in remission showed no lymph node alterations (67).
Stimulation of Lymphatic Vessels in Rheumatoid Arthritis
Stimulating the lymphatic vasculature has been associated with reduced disease severity in animal models of RA (summarized in Table 1). Adeno-associated viral (AAV) delivery of VEGF-C in the inflamed ankle joints of 6-week-old TNF-α transgenic mice partially reversed the inflammation-associated increase in synovial volume and significantly improved leg mobility. Histological analyses revealed that mice treated with VEGF-C had less cartilage and bone destruction than animals injected with a control vector. In chronic arthritis (mice at 5 months of age), lymphatic drainage of indocyanine green (ICG) out of the footpad was strongly decreased in TNF-α transgenic compared to wild-type mice. AAV-mediated delivery of VEGF-C significantly improved the clearance of ICG out of the paws and increased the number of lymphatic vessels in the pannus of the inflamed joint (34).
In an alternative approach, based on the observation that increased levels of NO in inflammation reduce lymphatic pumping, lymphatic vessel function was studied using inhibition of NOS. Local application of L-N6-(1-iminoethyl)lysine 5-tetrazole-amide (L-NIL), a moderately selective inhibitor of iNOS (68), in TNF-α transgenic mice with collapsed lymph nodes restored lymphatic contractions and strongly improved ICG transport from the footpad to popliteal lymph nodes, while Nω-nitro-l-arginine methyl ester (L-NAME), an unspecific inhibitor of both eNOS and iNOS was not associated with beneficial effects (35). Although the impact on disease severity in these mice was not assessed in the study, it provides evidence that selective inhibition of iNOS might offer an alternative and clinically relevant approach for RA therapy.
Inhibition on Lymphatic Vessels in Rheumatoid Arthritis
Inhibiting the lymphatic vasculature led to worsened inflammation in mouse models of arthritis (summarized in Table 2). Injecting TNF-α transgenic mice that had developed joint inflammation with a VEGFR-3-blocking antibody for 2 months significantly reduced the number of lymphatic capillaries in the draining popliteal lymph nodes and inflamed ankles. Blocking VEGFR-3 also aggravated inflammation of the knee and ankle joints, as the increase in synovial volume over time as well as its absolute size were elevated in these animals compared to IgG-treated controls. Similarly, histological analyses of hematoxylin-eosin-stained sections revealed exacerbated inflammation after VEGFR-3 inhibition. Akin to the effects observed in chronic skin inflammation, blocking VEGFR-2 was associated with a reduced inflammatory reaction, as assessed by synovial volume and histological scoring. Lymphatic drainage function, as assessed by tracking the ICG signal in paws and draining popliteal lymph nodes following injection into the footpad, was dramatically reduced upon blocking VEGFR-3 (38).
The Effect of Inflammatory Mediators on the Lymphatic Vasculature
Inflammatory lymphangiogenesis is mostly mediated by VEGF-A and VEGF-C which are produced by keratinocytes and stromal cells like fibroblasts as well as immune cells, most importantly macrophages (69–71). Indeed, several inflammatory mediators have been found to induce VEGF-C transcription (72–74).
Macrophages are of critical importance, as demonstrated in a model of IBD and LPS-induced skin inflammation, where depletion of macrophages aggravated the inflammation (29, 33). While VEGFs are important for inflammation-induced lymphangiogenesis, there are many additional factors at play. IL-17, a crucial cytokine in the pathogenesis of psoriasis for example, has been shown to induce lymphangiogenesis in vitro and in cornea micropocket assays (75), and IL-8 promoted lymphangiogenesis in cell culture experiments and in an animal model of lymphedema (76). Similarly, inhibition of TGF-β, which mediates anti-inflammatory effects, supported lymphangiogenesis in a mouse model of peritonitis and in lymphedema (77, 78). In line with this, cytokines characteristic for TH2 cells like IL-4 and IL-13, which are often linked to inflammation resolution, inhibited lymphangiogenesis (79). Interestingly, several inflammatory mediators have anti-lymphangiogenic activity. Interferon-γ (IFN-γ), which is produced by activated T cells, decreased lymphatic vessel formation of both human and murine lymphatic endothelial cells in vitro as well as in mouse lymph nodes (15, 80). Likewise, TNF-α inhibited capillary formation and proliferation of mouse LECs, while IL-1β had no consistent effects on proliferation, but reduced barrier function of LECs (15). Indeed, inflammatory mediators not only influence lymphangiogenesis, but also impact lymphatic function more directly. Prostaglandins, IL-1β, IL-6, and TNF-α reduced lymphatic pumping frequency (81, 82). Similarly, inflammatory mediators affect lymphatic vessel permeability, as demonstrated in vitro by assessing the effect of a wide array of inflammatory mediators on rat lymphatic endothelial cell monolayers, where IL-6, TNF-α, and IFN-γ strikingly increased the permeability, probably by reducing vascular endothelial (VE)-cadherin expression (83). Few studies have addressed lymphatic vessel permeability in vivo, but results of those that have showed impaired barrier function as well as pronounced leakiness and reported VEGF-A as important mediator of these effects, possibly by signaling via VEGFR-2 (1, 84).
It is important to consider that cytokines and growth factors often have pleiotropic effects, making it challenging to distinguish between direct and indirect mechanisms. IL-17 for example has been reported to induce VEGF-D expression, thereby triggering lymphangiogenesis indirectly (75). The wide array of signaling molecules involved in inflammation as well as their different and often pleiotropic effects on the lymphatic vasculature result in a highly complex network of signals which is still incompletely understood.
Conclusions
The lymphatic vasculature represents a crucial, although often under-appreciated, player in inflammation. Lymphatic vessels serve as the main transport route for inflammatory mediators, fluid, antigen and immune cells, thus playing a pivotal role in inflammation initiation and resolution. Indeed, it has been controversial whether expansion of the lymphatic endothelium contributes to inflammation by facilitating transport of leukocytes to lymph nodes and mounting of immune responses, or whether lymphatic vessels support inflammation resolution by draining inflammatory mediators and cells from the site of inflammation. However, in recent years, a number of studies detailed above have reported alleviated inflammation severity following activation and/or expansion of the lymphatic vasculature (depicted for skin inflammation in Figure 1), thus indicating that promoting the lymphatic vasculature supports inflammation resolution and may represent a valid therapeutic approach. It should be considered, however, that VEGF-C/VEGFR-3 signaling itself might also account for some of the anti-inflammatory effects observed in VEGF-C transgenic mice, as it has been shown to reduce the production of pro-inflammatory cytokines and protect mice from septic shock (85).
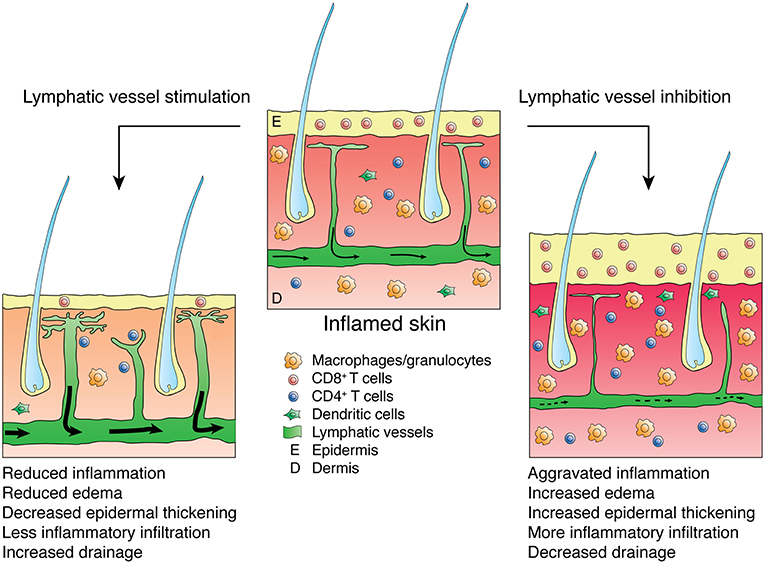
Figure 1. Effects of lymphatic vessel stimulation or inhibition on skin inflammation. Inflamed skin presents with epidermal thickening, edema and infiltration by inflammatory leukocytes (e.g., CD8-positive cells or macrophages and granulocytes). Stimulation of the lymphatic vasculature alleviates inflammation, reducing edema, epidermal thickening and inflammatory infiltration while improving lymphatic drainage, thus lowering the numbers of inflammatory cells in the inflamed skin. Inhibition of the lymphatic vasculature aggravates inflammation and reduces lymphatic clearance.
Interestingly, the lymphatic vasculature is also affected by established standard therapies used for the treatment of inflammatory diseases, e.g., in RA, where blocking TNF-α resulted in an increased lymphangiogenic response and increased lymphatic contractions in the inflamed tissue (66, 86). Other therapies aimed at blocking certain cytokines (e.g., IL-17 in psoriasis) may also exert parts of their anti-inflammatory effects by modulating the lymphatic vasculature. Curiously, some anti-inflammatory agents have been associated with anti-lymphangiogenic activity. Glucocorticoids reduced lymphangiogenesis in cornea inflammation and chronic airway inflammation mediated by M. pulmonis infection (87, 88). In addition, prostaglandin E2, whose biosynthesis is inhibited by cyclooxygenase (COX)-blocking non-steroidal anti-inflammatory drugs (NSAIDs), has been reported to induce VEGF-C expression and lymphangiogenesis in the setting of lung cancer (73). Coherently, inhibition of COX-2 reduced tumor-induced lymphangiogenesis (89). A possible explanation for these findings could be that potent therapeutic agents inhibit inflammation strongly enough to also reduce the concomitant inflammation-induced lymphangiogenesis. Moreover, while prostaglandin E2 interferes with lymphatic expansion, it has also been reported to inhibit lymphatic function (81). Therefore, glucocorticoids and NSAIDs may improve lymphatic clearance despite reducing lymphangiogenesis. However, further studies are needed to thoroughly investigate these possibilities.
It is important to consider that immunomodulatory properties of the lymphatic endothelium, which have received increasing attention over the last decade, may explain the observed anti-inflammatory effects of lymphatic vessel induction at least partially. A good example is the receptor D6, which is highly expressed by lymphatic endothelial cells and scavenges inflammatory cytokines. Mice deficient for D6 suffered from more severe skin inflammation and colitis compared to wild-type animals (90, 91), hence, lymphatic expansion may increase the levels of D6 and accordingly lower the levels of inflammatory mediators in the inflamed tissue, resulting in reduced disease severity. However, the immunomodulatory roles of the lymphatic vasculature are outside the scope of this review.
Although VEGF-C has been associated with anti-inflammatory effects in a variety of diseases as described above, its biological roles are highly complex and may be organ- and disease-dependent. In the setting of experimental obesity for example, transgenically overexpressed VEGF-C induced pro-inflammatory macrophage chemotaxis, increased weight gain and worsened metabolic parameters such as insulin resistance (92). In contrast, blockade of VEGF-C and VEGF-D by overexpression of a soluble form of VEGFR-3 reduced macrophage infiltration and improved insulin sensitivity in diet-induced obesity (93). Similarly, in tumor studies, VEGF-C has been reported to induce tumor lymphangiogenesis and stimulate the migration of macrophages (94), which may explain the observed increase in tumor metastasis in VEGF-C transgenic mice (95).
Applying VEGF-C in these diseases might be counter-productive and these findings therefore highlight the complexity of VEGF-C biology and emphasize the necessity of thoroughly evaluating possible beneficial and detrimental effects of VEGF-C in individual pathologies.
Considering all available data, the induction of lymphangiogenesis and activation of the lymphatic vasculature in the setting of inflammation appears to represent a potent therapeutic approach. It is therefore striking that this strategy has not been explored more thoroughly, let alone exploited clinically. A major obstacle has been the lack of clinically feasible delivery systems of lymphangiogenic factors. In a recent study, however, a targeted F8-VEGF-C fusion protein that specifically accumulates in the inflamed tissue was characterized and shown to reduce inflammation in two mouse models of skin inflammation, possibly filling this therapeutic gap (96).
Author Contributions
SS designed and wrote the manuscript. MD designed and revised the manuscript.
Funding
Work in the authors' laboratory was supported by Swiss National Science Foundation grant 310030_166490, and Advanced European Research Council Grant LYVICAM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kajiya K, Hirakawa S, Detmar M. Vascular endothelial growth factor-A mediates ultraviolet B-induced impairment of lymphatic vessel function. Am J Pathol. (2006) 169:1496–503. doi: 10.2353/ajpath.2006.060197
2. Chidlow JH, Langston W, Greer JJM, Ostanin D, Abdelbaqi M, Houghton J, et al. Differential angiogenic regulation of experimental colitis. Am J Pathol. (2006) 169:2014–30. doi: 10.2353/ajpath.2006.051021
3. Elshabrawy HA, Chen Z, Volin MV, Ravella S, Virupannavar S, Shahrara S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis (2015) 18:433–48. doi: 10.1007/s10456-015-9477-2
4. Skobe M, Detmar M. Structure, function, and molecular control of the skin lymphatic system. J Investig Dermatol Symp Proc. (2000) 5:14–9. doi: 10.1046/j.1087-0024.2000.00001.x
5. Kaipainen A, Korhonen J, Mustonen T, van Hinsbergh VW, Fang GH, Dumont D, et al. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci USA. (1995) 92:3566–70. doi: 10.1073/pnas.92.8.3566
6. Joukov V, Pajusola K, Kaipainen A, Chilov D, Lahtinen I, Kukk E, et al. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. (1996) 15:1751. doi: 10.1002/j.1460-2075.1996.tb00359.x
7. Makinen T, Veikkola T, Mustjoki S, Karpanen T, Catimel B, Nice EC, et al. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. (2001) 20:4762–73. doi: 10.1093/emboj/20.17.4762
8. Joukov V, Sorsa T, Kumar V, Jeltsch M, Claesson-Welsh L, Cao Y, et al. Proteolytic processing regulates receptor specificity and activity of VEGF-C. Embo J. (1997) 16:3898–911. doi: 10.1093/emboj/16.13.3898
9. Joukov V, Kumar V, Sorsa T, Arighi E, Weich H, Saksela O, et al. A recombinant mutant vascular endothelial growth factor-C that has lost vascular endothelial growth factor receptor-2 binding, activation, and vascular permeability activities. J Biol Chem. (1998) 273:6599–602. doi: 10.1074/jbc.273.12.6599
10. Achen MG, Jeltsch M, Kukk E, Makinen T, Vitali A, Wilks AF, et al. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci USA. (1998) 95:548–53. doi: 10.1073/pnas.95.2.548
11. Baldwin ME, Catimel B, Nice EC, Roufail S, Hall NE, Stenvers KL, et al. The specificity of receptor binding by vascular endothelial growth factor-d is different in mouse and man. J Biol Chem. (2001) 276:19166–71. doi: 10.1074/jbc.M100097200
12. Jeltsch M, Kaipainen A, Joukov V, Meng X, Lakso M, Rauvala H, et al. Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science (1997) 276:1423–5. doi: 10.1126/science.276.5317.1423
13. Veikkola T, Jussila L, Makinen T, Karpanen T, Jeltsch M, Petrova TV, et al. Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. Embo J. (2001) 20:1223–31. doi: 10.1093/emboj/20.6.1223
14. Makinen T, Jussila L, Veikkola T, Karpanen T, Kettunen MI, Pulkkanen KJ, et al. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat Med. (2001) 7:199–205. doi: 10.1038/84651
15. Chaitanya GV, Franks SE, Cromer W, Wells SR, Bienkowska M, Jennings MH, et al. Differential cytokine responses in human and mouse lymphatic endothelial cells to cytokines in vitro. Lymphat Res Biol. (2010) 8:155–64. doi: 10.1089/lrb.2010.0004
16. Breslin JW, Yuan SY, Wu MH. VEGF-C alters barrier function of cultured lymphatic endothelial cells through a VEGFR-3-dependent mechanism. Lymphat Res Biol. (2007) 5:105–13. doi: 10.1089/lrb.2007.1004
17. Breslin JW. ROCK and cAMP promote lymphatic endothelial cell barrier integrity and modulate histamine and thrombin-induced barrier dysfunction. Lymphat Res Biol. (2011) 9:3–11. doi: 10.1089/lrb.2010.0016
18. Kurtz KH, Moor AN, Souza-Smith FM, Breslin JW. Involvement of H1 and H2 receptors and soluble guanylate cyclase in histamine-induced relaxation of rat mesenteric collecting lymphatics. Microcirculation (2014) 21:593–605. doi: 10.1111/micc.12138
19. Nizamutdinova IT, Maejima D, Nagai T, Bridenbaugh E, Thangaswamy S, Chatterjee V, et al. Involvement of histamine in endothelium-dependent relaxation of mesenteric lymphatic vessels. Microcirculation (2014) 21:640–8. doi: 10.1111/micc.12143
20. Gasheva OY, Zawieja DC, Gashev AA. Contraction-initiated NO-dependent lymphatic relaxation: a self-regulatory mechanism in rat thoracic duct. J Physiol. (2006) 575:821–32. doi: 10.1113/jphysiol.2006.115212
21. Breslin JW, Gaudreault N, Watson KD, Reynoso R, Yuan SY, Wu MH. Vascular endothelial growth factor-C stimulates the lymphatic pump by a VEGF receptor-3-dependent mechanism. Am J Physiol Heart Circ Physiol. (2007) 293:H709–H718. doi: 10.1152/ajpheart.00102.2007
22. Liao S, Cheng G, Conner DA, Huang Y, Kucherlapati RS, Munn LL, et al. Impaired lymphatic contraction associated with immunosuppression. Proc Natl Acad Sci USA. (2011) 108:18784–9. doi: 10.1073/pnas.1116152108
23. Braverman IM. Electron microscopic studies of the microcirculation in psoriasis. J Invest Dermatol. (1972) 59:91–8. doi: 10.1111/1523-1747.ep12625852
24. Kunstfeld R. Induction of cutaneous delayed-type hypersensitivity reactions in VEGF-A transgenic mice results in chronic skin inflammation associated with persistent lymphatic hyperplasia. Blood (2004) 104:1048–57. doi: 10.1182/blood-2003-08-2964
25. Henno A, Blacher S, Lambert C, Colige A, Seidel L, Noël A, et al. Altered expression of angiogenesis and lymphangiogenesis markers in the uninvolved skin of plaque-type psoriasis. Br J Dermatol. (2009) 160:581–90. doi: 10.1111/j.1365-2133.2008.08889.x
26. Xia Y-P, Li B, Hylton D, Detmar M, Yancopoulos GD, Rudge JS. Transgenic delivery of VEGF to mouse skin leads to an inflammatory condition resembling human psoriasis. Blood (2003) 102:161–8. doi: 10.1182/blood-2002-12-3793
27. van der Fits L, Mourits S, Voerman JSA, Kant M, Boon L, Laman JD, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. (2009) 182:5836–45. doi: 10.4049/jimmunol.0802999
28. Huggenberger R, Ullmann S, Proulx ST, Pytowski B, Alitalo K, Detmar M. Stimulation of lymphangiogenesis via VEGFR-3 inhibits chronic skin inflammation. J Exp Med. (2010) 207:2255–69. doi: 10.1084/jem.20100559
29. Kataru RP, Jung K, Jang C, Yang H, Schwendener RA, Baik JE, et al. Critical role of CD11b+ macrophages and VEGF in inflammatory lymphangiogenesis, antigen clearance, and inflammation resolution. Blood (2009) 113:5650–9. doi: 10.1182/blood-2008-09-176776
30. Christiansen AJ, Dieterich LC, Ohs I, Bachmann SB, Bianchi R, Proulx ST, et al. Lymphatic endothelial cells attenuate inflammation via suppression of dendritic cell maturation. Oncotarget (2016) 7:39421–35. doi: 10.18632/oncotarget.9820
31. Huggenberger R, Siddiqui SS, Brander D, Ullmann S, Zimmermann K, Antsiferova M, et al. An important role of lymphatic vessel activation in limiting acute inflammation. Blood (2011) 117:4667–78. doi: 10.1182/blood-2010-10-316356
32. Kajiya K, Sawane M, Huggenberger R, Detmar M. Activation of the VEGFR-3 pathway by VEGF-C attenuates UVB-induced edema formation and skin inflammation by promoting lymphangiogenesis. J Invest Dermatol. (2009) 129:1292–8. doi: 10.1038/jid.2008.351
33. D'Alessio S, Correale C, Tacconi C, Gandelli A, Pietrogrande G, Vetrano S, et al. VEGF-C–dependent stimulation of lymphatic function ameliorates experimental inflammatory bowel disease. J Clin Invest. (2014) 124:3863–78. doi: 10.1172/JCI72189
34. Zhou Q, Guo R, Wood R, Boyce BF, Liang Q, Wang Y-J, et al. Vascular endothelial growth factor C attenuates joint damage in chronic inflammatory arthritis by accelerating local lymphatic drainage in mice. Arthritis Rheum. (2011) 63:2318–28. doi: 10.1002/art.30421
35. Liang Q, Ju Y, Chen Y, Wang W, Li J, Zhang L, et al. Lymphatic endothelial cells efferent to inflamed joints produce iNOS and inhibit lymphatic vessel contraction and drainage in TNF-induced arthritis in mice. Arthritis Res Ther. (2016) 18:62. doi: 10.1186/s13075-016-0963-8
36. Kajiya K, Detmar M. An important role of lymphatic vessels in the control of UVB-induced edema formation and inflammation. J Invest Dermatol. (2006) 126:919–21. doi: 10.1038/sj.jid.5700126
37. Jurisic G, Sundberg JP, Detmar M. Blockade of VEGF receptor-3 aggravates inflammatory bowel disease and lymphatic vessel enlargement. Inflamm Bowel Dis. (2013) 19:1983–9. doi: 10.1097/MIB.0b013e31829292f7
38. Guo R, Zhou Q, Proulx ST, Wood R, Ji R-C, Ritchlin CT, et al. Inhibition of lymphangiogenesis and lymphatic drainage via vascular endothelial growth factor receptor 3 blockade increases the severity of inflammation in a mouse model of chronic inflammatory arthritis. Arthritis Rheum. (2009) 60:2666–76. doi: 10.1002/art.24764
39. Scaldaferri F, Vetrano S, Sans M, Arena V, Straface G, Stigliano E, et al. VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology (2009) 136:585–95.e585. doi: 10.1053/j.gastro.2008.09.064
40. Chidlow JH, Glawe JD, Pattillo CB, Pardue S, Zhang S, Kevil CG. VEGF164 isoform specific regulation of T-cell-dependent experimental colitis in mice. Inflamm Bowel Dis. (2011) 17:1501–12. doi: 10.1002/ibd.21525
41. Geleff S, Schoppmann SF, Oberhuber G. Increase in podoplanin-expressing intestinal lymphatic vessels in inflammatory bowel disease. Virchows Arch. (2003) 442:231–7. doi: 10.1007/s00428-002-0744-4
42. Pedica F, Ligorio C, Tonelli P, Bartolini S, Baccarini P. Lymphangiogenesis in Crohn's disease: an immunohistochemical study using monoclonal antibody D2-40. Virchows Arch. (2008) 452:57–63. doi: 10.1007/s00428-007-0540-2
43. Heatley RV, Bolton PM, Hughes LE, Owen EW. Mesenteric lymphatic obstruction in Crohn's disease. Digestion (1980) 20:307–13. doi: 10.1159/000198452
44. Van Kruiningen HJ, Hayes AW, Colombel JF. Granulomas obstruct lymphatics in all layers of the intestine in Crohn's disease. Apmis (2014) 122:1125–9. doi: 10.1111/apm.12268
45. Crohn BB, Ginzburg L, Oppenheimer GD. Regional ileitis: a pathologic and clinical entity. JAMA (1932) 99:1323–9. doi: 10.1001/jama.1932.02740680019005
46. Tonelli F, Giudici F, Liscia G. Is lymphatic status related to regression of inflammation in Crohn's disease? World J Gastrointest Surg. (2012) 4:228–33. doi: 10.4240/wjgs.v4.i10.228
47. Rahier J-F, Dubuquoy L, Colombel J-F, Jouret-Mourin A, Delos M, Ferrante M, et al. Decreased lymphatic vessel density is associated with postoperative endoscopic recurrence in Crohn's disease. Inflamm Bowel Dis. (2013) 19:2084–90. doi: 10.1097/MIB.0b013e3182971cec
48. Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell (1993) 75:263–74.
49. Spencer DM, Veldman GM, Banerjee S, Willis J, Levine AD. Distinct inflammatory mechanisms mediate early versus late colitis in mice. Gastroenterology (2002) 122:94–105. doi: 10.1053/gast.2002.30308
50. Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology (1990) 98:694–702. doi: 10.1016/0016-5085(90)90290-H
51. Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol. (2014) 104:Unit15.25.−15.25.14. doi: 10.1002/0471142735.im1525s104
52. Vyas SP, Goswami R. A Decade of Th9 Cells: Role of Th9 Cells in Inflammatory Bowel Disease. Front Immunol (2018) 9:1139. doi: 10.3389/fimmu.2018.01139
53. Wang X, Zhao J, Qin L. VEGF-C mediated enhancement of lymphatic drainage reduces intestinal inflammation by regulating IL-9/IL-17 balance and improving gut microbiota in experimental chronic colitis. Am J Transl Res. (2017) 9:4772–84. Available online at: http://www.ajtr.org/files/ajtr0064922.pdf
54. Firestein GS. Evolving concepts of rheumatoid arthritis. Nature (2003) 423:356–61. doi: 10.1038/nature01661
55. Jayson MI, Cavill I, Barks JS. Lymphatic clearance rates in rheumatoid arthritis. Ann Rheum Dis. (1971) 30:638–9. doi: 10.1136/ard.30.6.638
56. Xu H, Edwards J, Banerji S, Prevo R, Jackson DG, Athanasou NA. Distribution of lymphatic vessels in normal and arthritic human synovial tissues. Ann Rheum Dis. (2003) 62:1227–9. doi: 10.1136/ard.2003.005876
57. Zhang Q, Lu Y, Proulx ST, Guo R, Yao Z, Schwarz EM, et al. Increased lymphangiogenesis in joints of mice with inflammatory arthritis. Arthritis Res Ther. (2007) 9:R118. doi: 10.1186/ar2326
58. Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. Embo J. (1991) 10:4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x
59. Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell (1996) 87:811–22. doi: 10.1016/S0092-8674(00)81989-3
60. Bouta EM, Bell RD, Rahimi H, Xing L, Wood RW, Bingham CO, et al. Targeting lymphatic function as a novel therapeutic intervention for rheumatoid arthritis. Nat Rev Rheumatol. (2018) 14:94–106. doi: 10.1038/nrrheum.2017.205
61. Proulx ST, Kwok E, You Z, Beck CA, Shealy DJ, Ritchlin CT, et al. MRI and quantification of draining lymph node function in inflammatory arthritis. Ann NY Acad Sci USA. (2007) 1117:106–23. doi: 10.1196/annals.1402.016
62. Zhou Q, Wood R, Schwarz EM, Wang Y-J, Xing L. Near-infrared lymphatic imaging demonstrates the dynamics of lymph flow and lymphangiogenesis during the acute versus chronic phases of arthritis in mice. Arthritis Rheum. (2010) 62:1881–9. doi: 10.1002/art.27464
63. Proulx ST, Kwok E, You Z, Papuga MO, Beck CA, Shealy DJ, et al. Longitudinal assessment of synovial, lymph node, and bone volumes in inflammatory arthritis in mice by in vivo magnetic resonance imaging and microfocal computed tomography. Arthritis Rheum. (2007b) 56:4024–37. doi: 10.1002/art.23128
64. Bouta EM, Ju Y, Rahimi H, de Mesy-Bentley KL, Wood RW, Xing L, et al. Power Doppler ultrasound phenotyping of expanding versus collapsed popliteal lymph nodes in murine inflammatory arthritis. PLoS ONE (2013) 8:e73766. doi: 10.1371/journal.pone.0073766
65. Bouta EM, Wood RW, Brown EB, Rahimi H, Ritchlin CT, Schwarz EM. In vivo quantification of lymph viscosity and pressure in lymphatic vessels and draining lymph nodes of arthritic joints in mice. J Physiol. (2014) 592:1213–23. doi: 10.1113/jphysiol.2013.266700
66. Bouta EM, Kuzin I, de Mesy Bentley K, Wood RW, Rahimi H, Ji R-C, et al. Brief report: treatment of tumor necrosis factor-transgenic mice with anti-tumor necrosis factor restores lymphatic contractions, repairs lymphatic vessels, and may increase monocyte/macrophage egress. Arthr Rheumatol. (2017) 69:1187–93. doi: 10.1002/art.40047
67. Manzo A, Caporali R, Vitolo B, Alessi S, Benaglio F, Todoerti M, et al. Subclinical remodelling of draining lymph node structure in early and established rheumatoid arthritis assessed by power Doppler ultrasonography. Rheumatology (2011) 50:1395–400. doi: 10.1093/rheumatology/ker076
68. Víteček J, Lojek A, Valacchi G, Kubala L. Arginine-based inhibitors of nitric oxide synthase: therapeutic potential and challenges. Mediators Inflamm. (2012) 2012:318087–22. doi: 10.1155/2012/318087
69. Detmar M, Brown LF, Claffey KP, Yeo KT, Kocher O, Jackman RW, et al. Overexpression of vascular permeability factor/vascular endothelial growth factor and its receptors in psoriasis. J Exp Med. (1994) 180:1141–6. doi: 10.1084/jem.180.3.1141
70. Kim KE, Koh Y-J, Jeon B-H, Jang C, Han J, Kataru RP, et al. Role of CD11b+ macrophages in intraperitoneal lipopolysaccharide-induced aberrant lymphangiogenesis and lymphatic function in the diaphragm. Am J Pathol. (2009) 175:1733–45. doi: 10.2353/ajpath.2009.090133
71. Cursiefen C, Chen L, Borges LP, Jackson D, Cao J, Radziejewski C, et al. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J Clin Invest (2004) 113:1040–50. doi: 10.1172/JCI20465
72. Ristimäki A, Narko K, Enholm B, Joukov V, Alitalo K. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J Biol Chem. (1998) 273:8413–8.
73. Su J-L, Shih J-Y, Yen M-L, Jeng Y-M, Chang C-C, Hsieh C-Y, et al. Cyclooxygenase-2 induces EP1- and HER-2/Neu-dependent vascular endothelial growth factor-C up-regulation: a novel mechanism of lymphangiogenesis in lung adenocarcinoma. Cancer Res. (2004) 64:554–64. doi: 10.1158/0008-5472.CAN-03-1301
74. Cha H-S, Bae E-K, Koh J-H, Chai J-Y, Jeon CH, Ahn K-S, et al. Tumor necrosis factor-alpha induces vascular endothelial growth factor-C expression in rheumatoid synoviocytes. J Rheumatol. (2007) 34:16–9. Available online at: http://www.jrheum.org/content/34/1/16.long
75. Chauhan SK, Jin Y, Goyal S, Lee HS, Fuchsluger TA, Lee HK, et al. A novel pro-lymphangiogenic function for Th17/IL-17. Blood (2011) 118:4630–4. doi: 10.1182/blood-2011-01-332049
76. Choi I, Lee YS, Chung HK, Choi D, Ecoiffier T, Lee HN, et al. Interleukin-8 reduces post-surgical lymphedema formation by promoting lymphatic vessel regeneration. Angiogenesis (2013) 16:29–44. doi: 10.1007/s10456-012-9297-6
77. Oka M, Iwata C, Suzuki HI, Kiyono K, Morishita Y, Watabe T, et al. Inhibition of endogenous TGF-beta signaling enhances lymphangiogenesis. Blood (2008) 111:4571–9. doi: 10.1182/blood-2007-10-120337
78. Avraham T, Daluvoy S, Zampell J, Yan A, Haviv YS, Rockson SG, et al. Blockade of transforming growth factor-beta1 accelerates lymphatic regeneration during wound repair. Am J Pathol. (2010) 177:3202–14. doi: 10.2353/ajpath.2010.100594
79. Savetsky IL, Ghanta S, Gardenier JC, Torrisi JS, García Nores GD, Hespe GE, et al. Th2 cytokines inhibit lymphangiogenesis. PLoS ONE (2015) 10:e0126908. doi: 10.1371/journal.pone.0126908
80. Kataru RP, Kim H, Jang C, Choi DK, Koh BI, Kim M, et al. T lymphocytes negatively regulate lymph node lymphatic vessel formation. Immunity (2011) 34:96–107. doi: 10.1016/j.immuni.2010.12.016
81. Rehal S, Blanckaert P, Roizes S, von der Weid PY. (2009). Characterization of biosynthesis and modes of action of prostaglandin E2 and prostacyclin in guinea pig mesenteric lymphatic vessels. Br. J. Pharmacol. 158, 1961–1970. doi: 10.1111/j.1476-5381.2009.00493.x
82. Aldrich MB, Sevick-Muraca EM. Cytokines are systemic effectors of lymphatic function in acute inflammation. Cytokine (2013) 64:362–9. doi: 10.1016/j.cyto.2013.05.015
83. Cromer WE, Zawieja SD, Tharakan B, Childs EW, Newell MK, Zawieja DC. The effects of inflammatory cytokines on lymphatic endothelial barrier function. Angiogenesis (2014) 17:395–406. doi: 10.1007/s10456-013-9393-2
84. Nagy JA, Vasile E, Feng D, Sundberg C, Brown LF, Detmar MJ, et al. Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J Exp Med. (2002) 196:1497–506. doi: 10.1084/jem.20021244
85. Zhang Y, Lu Y, Ma L, Cao X, Xiao J, Chen J, et al. Activation of vascular endothelial growth factor receptor-3 in macrophages restrains TLR4-NF-κB signaling and protects against endotoxin shock. Immunity (2014) 40:501–14. doi: 10.1016/j.immuni.2014.01.013
86. Polzer K, Baeten D, Soleiman A, Distler J, Gerlag DM, Tak PP, et al. Tumour necrosis factor blockade increases lymphangiogenesis in murine and human arthritic joints. Ann Rheum Dis. (2008) 67:1610–6. doi: 10.1136/ard.2007.083394
87. Hos D, Saban DR, Bock F, Regenfuss B, Onderka J, Masli S, et al. Suppression of inflammatory corneal lymphangiogenesis by application of topical corticosteroids. Arch Ophthalmol. (2011) 129:445–52. doi: 10.1001/archophthalmol.2011.42
88. Yao L-C, Baluk P, Feng J, McDonald DM. Steroid-resistant lymphatic remodeling in chronically inflamed mouse airways. Am J Pathol. (2010) 176:1525–41. doi: 10.2353/ajpath.2010.090909
89. Iwata C, Kano MR, Komuro A, Oka M, Kiyono K, Johansson E, et al. Inhibition of cyclooxygenase-2 suppresses lymph node metastasis via reduction of lymphangiogenesis. Cancer Res. (2007) 67:10181–9. doi: 10.1158/0008-5472.CAN-07-2366
90. Jamieson T, Cook DN, Nibbs RJB, Rot A, Nixon C, McLean P, et al. The chemokine receptor D6 limits the inflammatory response in vivo. Nat Immunol. (2005) 6:403–11. doi: 10.1038/ni1182
91. Vetrano S, Borroni EM, Sarukhan A, Savino B, Bonecchi R, Correale C, et al. The lymphatic system controls intestinal inflammation and inflammation-associated Colon Cancer through the chemokine decoy receptor D6. Gut (2010) 59:197–206. doi: 10.1136/gut.2009.183772
92. Karaman S, Hollmén M, Yoon SY, Alkan HF, Alitalo K, Wolfrum C, et al. Transgenic overexpression of VEGF-C induces weight gain and insulin resistance in mice. Sci Rep. (2016) 6:31566. doi: 10.1038/srep31566
93. Karaman S, Hollmén M, Robciuc MR, Alitalo A, Nurmi H, Morf B, et al. Blockade of VEGF-C and VEGF-D modulates adipose tissue inflammation and improves metabolic parameters under high-fat diet. Mol Metab. (2015) 4:93–105. doi: 10.1016/j.molmet.2014.11.006
94. Skobe M, Hamberg LM, Hawighorst T, Schirner M, Wolf GL, Alitalo K, et al. Concurrent induction of lymphangiogenesis, angiogenesis, and macrophage recruitment by vascular endothelial growth factor-C in melanoma. Am J Pathol. (2001) 159:893–903. doi: 10.1016/S0002-9440(10)61765-8
95. Hirakawa S, Brown LF, Kodama S, Paavonen K, Alitalo K, Detmar M. VEGF-C-induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood (2007) 109:1010–7. doi: 10.1182/blood-2006-05-021758
Keywords: lymphatic vessels, lymphangiogenesis, inflammation, inflammatory bowel disease, arthritis, psoriasis, skin, inflammatory mediators
Citation: Schwager S and Detmar M (2019) Inflammation and Lymphatic Function. Front. Immunol. 10:308. doi: 10.3389/fimmu.2019.00308
Received: 01 November 2018; Accepted: 06 February 2019;
Published: 26 February 2019.
Edited by:
Beth Ann Tamburini, University of Colorado Denver, United StatesReviewed by:
Sophia Ran, Southern Illinois University School of Medicine, United StatesGirdhari Lal, National Centre for Cell Science (NCCS), India
Copyright © 2019 Schwager and Detmar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Detmar, bWljaGFlbC5kZXRtYXJAcGhhcm1hLmV0aHouY2g=