 Lotte Nijhuis
Lotte Nijhuis Janneke G. C. Peeters
Janneke G. C. Peeters Jorg van Loosdregt
Jorg van Loosdregt- Laboratory of Translational Immunology, Department of Pediatric Immunology & Rheumatology, University Medical Center Utrecht, University of Utrecht, Utrecht, Netherlands
Juvenile Idiopathic Arthritis (JIA) is characterized by a loss of immune tolerance. Here, the balance between the activity of effector T (Teff) cells and regulatory T (Treg) cells is disturbed resulting in chronic inflammation in the joints. Presently, therapeutic strategies are predominantly aimed at suppressing immune activation and pro-inflammatory effector mechanisms, ignoring the opportunity to also promote tolerance by boosting the regulatory side of the immune balance. Histone deacetylases (HDACs) can deacetylate both histone and non-histone proteins and have been demonstrated to modulate epigenetic regulation as well as cellular signaling in various cell types. Importantly, HDACs are potent regulators of both Teff cell and Treg cell function and can thus be regarded as attractive therapeutic targets in chronic inflammatory arthritis. HDAC inhibitors (HDACi) have proven therapeutic potential in the cancer field, and are presently being explored for their potential in the treatment of autoimmune diseases. Specific HDACi have already been demonstrated to reduce the secretion of pro-inflammatory cytokines by Teff cells, and promote Treg numbers and suppressive capacity in vitro and in vivo. In this review, we outline the role of the different classes of HDACs in both Teff cell and Treg cell function. Furthermore, we will review the effect of different HDACi on T cell tolerance and explore their potential as a therapeutic strategy for the treatment of oligoarticular and polyarticular JIA.
Introduction
Juvenile Idiopathic Arthritis (JIA) is the most common rheumatic disease in children and an important cause of short- and long-term disability (1, 2). It includes several different entities and has an intriguing heterogeneity in disease course and outcome. Oligo-articular JIA (oJIA) has a relatively mild course, with lasting medication-free remission in approximately half of the children, while poly-articular JIA (pJIA) more often is non-remitting and can lead to severe disability (2, 3). The main pathophysiological concept of JIA is that the immunological balance is disturbed resulting in loss of immune tolerance (1). A unique subtype of JIA is systemic-onset JIA (sJIA), involving 10% of all JIA patients, and in contrast to oJIA and pJIA, sJIA is mainly characterized as an autoinflammatory disease instead of an autoimmune disease. In the pathogenesis of sJIA there is a key role for cells of the innate immune system including monocytes and neutrophils (4, 5). This is illustrated by the high incidence of macrophage activation syndrome (MAS) in patients with sJIA (6, 7). Although cells from the innate immune system play an essential role in the pathogenesis of oJIA and pJIA as well, it is generally thought that activation of autoreactive CD4+ T cells, leading to a T cell-driven immune response is a key manifestation in the pathogenesis of oJIA and pJIA. This subsequently results in recruitment of other immune cells, including innate immune cells and the production of several pro-inflammatory cytokines including tumor necrosis factor (TNF)α, interleukin (IL)-6, IL-17, and interferon (IFN) γ which collectively leads to joint inflammation. Innate immune cells, such as neutrophils and monocytes/macrophages to the site of inflammation (8). The pathogenic T cells present within the synovial compartment are predominantly Thelper (Th) 1 and Th17 cells (9, 10).
Regulatory T (Treg) cells are key players in maintaining immunological balance and tolerance (11, 12). The transcription factor forkhead box P3 (FOXP3) is crucial for Treg cell development and function and mutations in the FOXP3 gene can result in severe dysregulation of the immune system due to a Treg cell deficiency (13, 14). Treg cell numbers and function have also been implicated in complex autoimmune diseases including rheumatoid arthritis (RA) and JIA, and in fact the first data on CD4+ Treg cells in human chronic arthritis comes from JIA patients (15, 16). Treg cells can be identified by the high expression of several markers, such as (but not limited to) FOXP3, CD25high, cytotoxic T lymphocyte associated protein (CTLA)-4 and low expression of CD127. Treg cells can adapt to local environment (tissues) and acquire additional characteristics in inflammatory conditions (12, 17). They seem to exert their regulatory or suppressive actions both cell-contact dependent and independent via the secretion of anti-inflammatory cytokines such as Transforming Growth Factor beta (TGF)β and IL-10 (18). In JIA, the balance between pro-inflammatory Teff cells and anti-inflammatory Treg cells can be associated with the course of the disease (16, 19–22). For instance, higher numbers of Treg and lower numbers of Teff cells (Th17 and Th1) at the site of inflammation have been correlated to a more favorable course and outcome in JIA (16, 20–22). These observations support the concept that treatment may be aimed to restore the immunological imbalance between effector mechanisms and regulatory mechanism in children with JIA.
Current treatment of JIA, consisting of intra-articular corticosteroids, disease modifying anti-rheumatic drugs (DMARDs) and biologicals, such as anti-TNFα, seem primarily directed at the effector side of the immunological imbalance (23–26). In the past two decades, biologicals are increasingly being used in JIA. They certainly have been a major- breakthrough in the treatment of JIA, but even today, a significant percentage of patients do not respond to therapy or only show partial response. Furthermore, after achieving clinical inactive disease on therapy, many patients suffer from relapse when treatment is discontinued (27, 28). Therefore, there is still a need for improved treatment strategies in chronic inflammatory diseases such as JIA. Restoring tolerance, either by; decreasing Teff cell function, increasing Treg cell function or preferentially both, might be a promising therapeutic strategy.
Histone deacetylases (HDACs) are a novel class of therapeutic targets that are being explored for the treatment of autoimmune disease. These enzymes can modulate epigenetic regulation and important cellular functions in many different cell types, including T cells by the deacetylation of both histone and non-histone proteins. In other diseases and research fields, mainly cancer research, HDAC inhibitors (HDACi) have already demonstrated therapeutic potential (29). Interestingly, in the context of autoimmune disease, HDAC inhibition proved to influence both the innate immune system and Teff cell and Treg cell function, potentially restoring immunological tolerance. We here provide an overview and focus on the role of the different types of HDACs in CD4+ Teff cells and Treg cells, and explore the potential of specific HDACi as a therapeutic strategy for the treatment of autoimmune diseases, in specific oJIA and pJIA.
Histone Acetylation as Regulatory Mechanism of Immune Activation
The function of many intracellular proteins, particularly transcription factors, and histones, can be altered by post-translational modifications. Here, one or more amino acids are covalently modified, often modulating subcellular localization, activation state, interaction with other proteins or protein turnover/degradation. Acetylation is one of the most prominent post-translational modifications. The majority of literature on acetylation is directed at its role in epigenetic regulation, which refers to changes in gene expression without altering the genetic code. In the nucleus, DNA is tightly wrapped around histones to form a nucleosome (30) which controls the accessibility of DNA binding sequence to their transcription factors (31). An important epigenetic mechanism that affects this accessibility is the post-translational modification of histones by acetylation (32), a process which is reciprocally regulated by lysine acetyl transferases (HATs) and lysine deacetylases (HDACs) (33–35) (Figure 1). In general, histone acetylation is associated with transcriptional activation by rendering the DNA more accessible to transcription factors (32, 36). The reverse process, deacetylation by HDACs, can therefore lead to condensation of chromatin structure and inhibition of gene transcription. However, deacetylation is also associated with activation of genes, and the inhibition of HDACs in fact results in both upregulation and downregulation of genes in equivalent percentages (37–41).
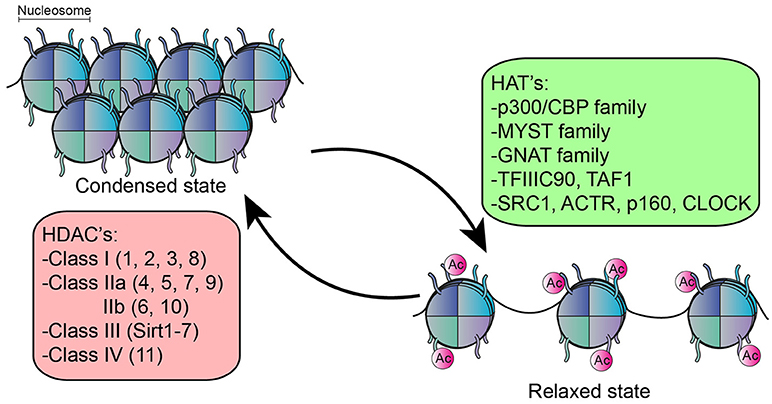
Figure 1. Function of HDACs an HATs. HATs acetylate the lysine residue on histones resulting in relaxation of chromatin structure, rendering the DNA more accessible for transcription factors. The reverse process, deacetylation by HDACs results in condensation of chromatin structure. 5 families of HATs have been described and 18 different HDACs, divided into 4 classes.
There are 18 different HDAC enzymes, which can be divided into 4 classes based upon homology to yeast HDACs and their function. Class I (HDAC1, 2, 3, and 8), Class II divided into class IIa (HDAC4, 5, 7, and 9) and IIb (HDAC6, 10) and class IV (HDAC11) are all Zinc-dependent and are considered classical HDACs. Class III consist of the Sirtuin family (Sirtuin 1–7) and are NAD+ dependent (42).
Histone modifications are widely associated with human disease, including malignancies and autoimmune disease such as RA, systemic lupus erythematosus (SLE) and JIA (35, 43–46). For instance, autoimmune disease associated single nucleotide polymorphisms (SNP's) are significantly enriched in regions with high acetylation of lysine 27 on histone 3 (H3K27) (46–48). Accordingly, increased regions of H3K27 acetylation in CD4+ T cells of JIA patients corresponded with increased expression of pro-inflammatory genes in these patients. Furthermore, in SLE, global acetylation of histone H3 and H4 in CD4+ T cells was reduced in patients compared to healthy controls and the degree of histone H3 acetylation negatively correlated with diseases activity (43). This demonstrates an important role for histone acetylation in autoimmune disease.
Next to histones, HDACs can also deacetylate non-histone proteins, hereby affecting their localization in the cell, stability and function (49, 50). For example, the activity of the transcription factors FOXP3 and Rar-related orphan receptor gamma (RORγt), key regulators of T cell function, is directly regulated by acetylation. Numerous studies have directly assessed the role of acetylation in modulating immune responses. Although there is a clear role for HDACs in the regulation of innate immune responses, reviewed by others (51, 52), we will focus on the role of the different HDACs in T cells and discuss their potential implications in oJIA and pJIA.
HDAC Function in CD4+ T Cells
Several classes of HDACs were demonstrated to have an important function in CD4+T cell development and function (Figures 2, 3).
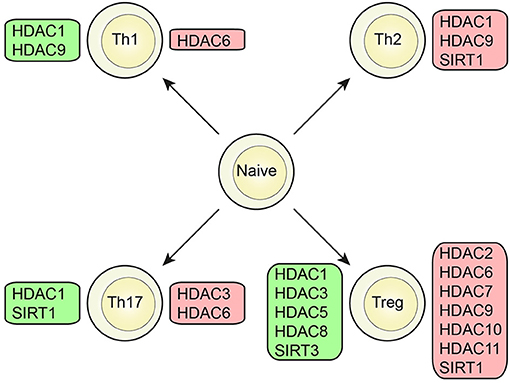
Figure 2. Role of HDAC members in different T cell subsets. Specific HDAC members which was demonstrated to either promote (green box) or inhibit (red box) function or differentiation of CD4+ T cells or the CD4+ T cell subsets; Th1, Th2, Th17, and Treg cells.
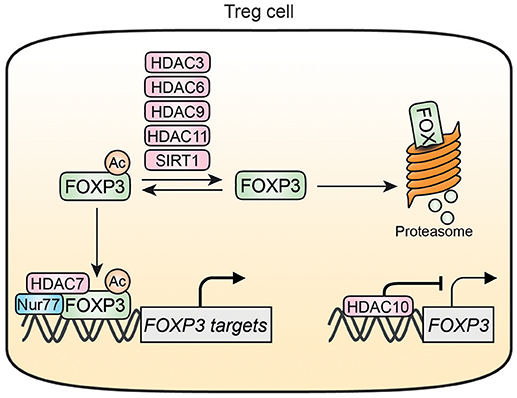
Figure 3. HDAC-mediated regulation of FOXP3 in Treg cells. HDAC3, HDAC6, HDAC9, HDAC11, and SIRT1 can deacetylate FOXP3 thereby increasing FOXP3 susceptibility for degradation. Acetylation of FOXP3 also promotes associations with DNA where it can form a transcriptional complex with HDAC7 and Nur77. HDAC10 can bind to and inhibit the FOXP3 promotor resulting in decreased FOXP3 expression. Nur77 is inhibited by HDAC7 and interacts with the HDAC7/FOXP3 transcriptional complex.
Class I HDACs; HDAC 1, 2, 3, and 8
Several members of Class I HDACs are essential for T cell development and differentiation in mice. In mice, T cell-specific knock-out of Hdac1 resulted in normal T cell numbers of both CD4+ and CD8+ T cells. However, these mice displayed enhanced Th2 responses, characterized by increased production of Th2 associated cytokines such as IL-4 and IL-5 combined with airway inflammation (53). In addition, Hdac1 knock-out mice were resistant to the induction of experimental autoimmune encephalomyelitis (EAE), a multiple sclerosis animal model which is associated with Th1 and Th17 responses (54). These data suggest that HDAC1 can skew T cell responses by impairing Th2 function and potentiating Th1 and Th17 activity. Furthermore, HDAC1 can also modulate Treg function. In a mouse cardiac transplant model, the deletion of HDAC1 in FOXP3+ Treg cells resulted in an impaired function of these cells, combined with increased secretion of the pro-inflammatory cytokines IL-2, IL-17, IFNγ, and decreased cardiac allograft survival (55). This demonstrates that HDAC1 contributes to Treg cell suppressive capacity. While also T cell-specific deletion of HDAC2 did not affect T cell numbers, combined deletion of both HDAC1 and HDAC2 resulted in severe defects in mature T cell development, especially in the CD4+ T cell lineage. This indicates that HDAC1 and HDAC2 have overlapping functions in T cell development. In a combined Hdac1/2 T cell knock-out mouse there was a decreased differentiation from double negative to double positive T cells (CD4+/CD8+) accompanied with a 5-fold decrease in thymocyte cellularity indicating a block in T cell development (56). Hdac1/2 knock-out was proposed to result in defective propagation of T cell receptor (TCR) signaling. In a later stage of T cell development, the combined knock-out of Hdac1/2 in CD4+ T cells specifically resulted in an increased CD8 surface expression by these CD4+ T cells and a decrease in peripheral T cell numbers (55). It was demonstrated that HDAC1 and HDAC2 maintain CD4 T cell integrity via repressing Runx-CBFß, a heterodimeric transcription factor required for the appropriate expression of CD4 and CD8. Furthermore, in T cells a dose dependent tumor suppressor function of HDAC1/2 was observed (56, 57). In Treg cells, the inhibition or deletion of HDAC2 specifically resulted in an increased Treg function, promoting cardiac allograft survival. This indicates that selective inhibition of HDAC2 in contrast to HDAC1 could serve a protective role against graft rejection (55). Combined targeting of both HDAC1 and 2 could result in severe defects in T cell development and therefore seems not suited for the treatment of JIA based upon data from mouse knock-out studies.
Comparable to the developmental defects observed in Hdac1/2 knock-outs, HDAC3 also was shown to be essential for T cell development and maturation (58–60). Knock-out of Hdac3 in mice resulted in a block in T cell development during positive selection in the thymus resulting in a strong reduction in CD4+ and CD8+ peripheral T cells. Within the CD4+ T cells present in the peripheral blood there was an increase in RORyt and IL-17 producing cells, indicating a skewing toward the Th17 phenotype (58). Furthermore, it was demonstrated that HDAC3 is an important mediator of the development and function of both induced and natural Treg cells. Interestingly, HDAC3 can directly associate with FOXP3, resulting in reduced IL-2 production by Treg cells. Moreover, mice with a FOXP3 cell-specific deletion of HDAC3 died within weeks from severe autoimmunity (61). For HDAC8, ongoing studies indicate that deletion of HDAC8 in Treg cells results in impaired Treg cell function (62). Together these data indicate that next to combined HDAC1/2 targeting, the selective inhibition of HDAC3 or HDAC8 could either result in T cell development defects or impaired Treg cell function and therefore does not seem to be of any therapeutic potential in a chronic autoimmune disease such as JIA.
Class IIa HDACs; HDAC4, 5, 7, and 9
Most members from Class IIa HDACs are protective for the development of autoimmunity in mice. HDAC4 has been mainly assessed in the nervous system due to its high expression in brain and skeletal tissue and little is known about its function in T cells. Although HDAC4 is expressed in all T cell subsets, CD4+ T cells specific Hdac4 knock-out mice display normal T cell numbers and function (63). A role for HDAC4 in CD4+ T cells was suggested based upon hypermethylation of the HDAC4 region of the DNA of CD4+ T cells from RA patients. These data indicate that HDAC4 expression would be decreased in these patients, but gene expression or function of HDAC4 was not assessed in this study (64). Therefore, the exact role of HDAC4 in T cell and in autoimmune diseases is currently unknown.
Similarly, Hdac5 knock-out mice display a normal CD4+ T cell development and function. In these mice however, Treg cell displayed impaired suppressive capacity (65). In addition, IFNγ production was decreased in CD8+ T cells, indicating that HDAC5 perhaps has a more profound role in CD8+ T cells and Treg cells compared to CD4+ Teff cells (65). In contrast to HDAC4 and HDAC5, HDAC7 is an important regulator of T cell development by regulating both positive and negative selection in the thymus (66–68). Knock down of Hdac7 in mice T cells results in a defect in positive selection in the thymus with a decrease in T cell survival and TCR repertoire (66). The effect of HDAC7 on negative thymocyte selection was shown to be via inhibition of the expression of the transcription factor; Nur77 (69). Nur77 promotes thymocyte apoptosis during negative selection of autoreactive thymocytes (69) and the overexpression of Nur77 significantly decreases numbers of peripheral CD4+ and CD8+ T cells in mice (70). It was demonstrated that HDAC7 is recruited to the Nur77 promotor via interaction with the transcription factor MEF2D resulting in decreased Nur77 expression. HDAC7 is exported out of the nucleus during T cell activation resulting in increased Nur77 expression. Blocking the nuclear export of HDAC7 in T cells in mice resulted in a block in negative selection in the thymus, promoted survival of auto-reactive T cells and was accompanied with the development of autoimmunity and a decreased life span in mice (67). The exact mechanism of inhibition of expression of Nur77 by HDAC7 remains unknown, but it is demonstrated to be dependent on its deacetylase activity (69). A profound role for HDAC7 in Treg function was also shown. Overexpression of Nur77 in mice T cells was associated with an increase in Treg cell percentages and cardiac allograft survival. It was demonstrated that Nur77 can interact with a HDAC7/FOXP3 transcriptional complex in Treg cells and that Nur77 overexpression resulted in increased expression of Treg associated genes, including Foxp3, Foxp1, TIP60 (70). Implicating, that Nur77 affects the balance between Teff and Treg cells, favoring Treg cell survival. Taken together, HDAC7 plays an essential role in negative T cell selection in the thymus as an inhibitor of Nur77. Both in Teff and Treg cells a protective role of Nur77 with respect to development of autoimmunity is suggested making Nur77 an interesting treatment target. However, the importance of HDAC7 in the positive selection of T cells in the thymus and TCR repertoire formation and therefore T cell development makes HDAC7 an undesirable target for the treatment of autoimmune diseases.
A role for HDAC9 has been implicated in several T cell subsets, including Treg, Th1, and Th2 cells both in mice and humans. HDAC9 expression was found to be increased in different subsets of CD4+ T cells of SLE patients and the autoimmune prone MLR/lpr mice. In addition, HDAC9 deficiency was associated with hyperacetylation of several lysine residues of histone H3 in mice (71). In the MLR/lpr mouse, knock-out of Hdac9 resulted in a prolonged survival and decrease in autoimmune disease progression. This was determined by smaller lymph nodes and spleen and a decreased percentage of activated CD4+ T cells and double negative T cells. Furthermore, knock-out of Hdac9 resulted in an inhibition of T cell activation in vitro and a decrease in Th1 and increase in Th2 cytokine production in vivo (71). Therefore, HDAC9 appears to promote skewing toward Th1 subsets. In addition, there is a differential expression of HDAC9 in different T cell subsets. HDAC9 is higher expressed in Treg cells compared to non-Treg cells and HDAC9 expression is markedly decreased in non-Treg T cells after stimulation in contrast to Treg cells (72). This could implicate an important role for HDAC9 in Treg function or development. Indeed, knock-out of HDAC9 in mice resulted in increased Treg numbers with enhanced suppressive function (73) and predisposition to iTreg development (74). This was confirmed by in vitro knock-down of Hdac9 in mice Treg cells which resulted in increased FOXP3 expression and increased suppressive function (73). Furthermore, the induction of colitis in mice was associated with increased HDAC9 expression, while the Hdac9 knock-out mice were resistant to development of colitis (73). Taken together, HDAC9 showed to promote the development of autoimmune disease via its function in both Teff and Treg cells.
In summary class IIa HDACs in CD4+ T cells seem to be protective against the development of autoimmunity or essential for T cell development, with the exception of HDAC9. The inhibition of HDAC9 could therefore be of therapeutic interest in the context of autoimmune diseases such as JIA, however it's specific role in arthritis had not been investigated yet.
Class IIb; HDAC6, 10
The two members of class IIb HDACs; HDAC6, and HDAC10 have both been assessed for their therapeutic potential as a new treatment of autoimmune disease. Importantly, in CD4+ specific Hdac6 knock-out mice, CD4+ and CD8+ T-cell development and function was described to be normal (74–76). However, in these mice the population of IL-17 producing gamma delta (γδ) T cells was increased which was accompanied by a decreased expression of the transcription factor SOX4. This was confirmed in vitro, by treating a murine lymphoma cell line (EL4) with the two different HDAC6 inhibitors; tubacin, and tubastatin. HDAC6 inhibition in these cells resulted in a concentration-dependent increase of IL-17 expression (75). Since IL-17 production is associated with autoimmunity, these observations implicate a protective role for HDAC6 in the development of autoimmune disease via inhibition of Th17 cell differentiation. However, in Treg cells, it has been shown that HDAC6 is involved in deacetylation of FOXP3 and selective inhibition of HDAC6 enhanced the suppressive function of Treg cells (76–78). This was confirmed in an arthritis mouse model were HDAC6 inhibition resulted in decreased arthritis scores in mice (79). Furthermore, in human peripheral blood mononuclear cells (PBMCs) from RA patients, in vitro HDAC6 inhibition resulted in a decreased expression of the pro-inflammatory cytokines TNFα and IL-1β and increased the anti-inflammatory cytokine IL-10 (79). These observations indicate that HDAC6 can exert different effects in different T cell subsets, both pro-inflammatory and anti-inflammatory. Due to the observation that HDAC6 inhibition in Treg cells resulted in enhanced suppressive function and a decrease in arthritis development in a mouse model, there is great interest in the potential application of specific HDAC6i immune suppression therapy in for example transplant recipients. (62) However, more research is needed to determine the potential risk of HDAC6i by promoting autoimmune disease by increasing IL-17 production.
For HDAC10, it was reported very recently that it can bind to the FOXP3 promotor and inhibit its transcriptional activity (80). Treg cells from HDAC10 knock-out mice showed increased expression of FOXP3 accompanied by increased Treg suppressive function. Furthermore, transfer of Treg cells from HDAC10 knock-out mice in a colitis mouse model resulted in a reduced induction of colitis compared to transfer of wild-type (WT) Treg cells (80).
Altogether, both HDAC6 and HDAC10, as class IIb HDACs, seem to exert negative effects on Treg cells and the inhibition of class IIb HDACs could therefore be beneficial in inducing Treg cell function. However, for HDAC6 there is a differential effect on different Th subsets and a pro-inflammatory effect has also been demonstrated via the induction of IL-17 production.
Class IV; HDAC11
HDAC11, the only member of class IV HDACs is suggested to have an inhibitory role in maintaining immune tolerance. For Treg cells it was demonstrated in a human T cell line that HDAC11 can associate with and deacetylate FOXP3 (81). Knock-out of Hdac11 in Treg cells resulted in an increased expression of FOXP3 and TGF-β, and an increased suppressive capacity in vitro. This was confirmed in vivo by an increased cardiac allograft survival in Hdac11 knock-out mice (81). In mice, in vitro activation of Teff cells resulted in a downregulation of HDAC11 expression. In addition, Hdac11 knock-out in CD4+ and CD8+ T cells resulted in an increased proliferation and pro-inflammatory cytokine production after activation indicating an inhibitory role for HDAC11 in Teff cell activation. Interestingly, the Hdac11 knock-out CD4+ T cells were resistant to the in vivo induction of tolerance via the injection of a tolerogenic dose of ovalbumin (82). In summary, Hdac11 knock-out has a differential effect on the different T cell subsets with an increased Teff function and decreased tolerance induction after HDAC11 knock-out in all T cells. In contrast, Treg cell-specific knock-out of HDAC11 results in the opposite effect with an increased suppressive function of Treg cells and improved immune tolerance. The effect of HDAC11 inhibition in vivo therefore seems to depend on which cell type is affected most. To our knowledge, no specific inhibitor for HDAC11 is available yet and therefore the effect of HDAC11 inhibition in vivo remains incompletely understood.
Class III; Sirtuins1-7
The most extensively studied member of the sirtuin family, SIRT1, is an important player in chronic inflammation. The exact function of SIRT1 is under debate and both a pro-inflammatory and an anti-inflammatory role of SIRT1 have been described in human disease. It was demonstrated that SIRT1 is upregulated in the synovial tissue and PBMC from patients with RA compared to patients with osteoarthritis. In this study, a pro-inflammatory role for SIRT1 was implied in monocytes with a reduction in lipopolysaccharide (LPS) induced TNFα production in vitro after inhibition of SIRT1, either by incubation with SIRT1 inhibitors or siRNA targeting (83). However, the opposing effect has also been demonstrated where a protective role for SIRT1 against inflammation and tissue destruction is suggested in chondrocytes and osteoblasts via the deacetylation and therefore inactivation of Nuclear factor-kappa B (NF-κB) (84, 85). Part of this controversy may be explained by differences in experimental setup, cell type and readout.
In mice T cells, the germline knock-out of Sirt1 resulted in an increased T cell proliferation and expression of IL-2, IFNγ, and IL-5 compared to WT upon in vitro activation. In these mice, this decreased T cell tolerance resulted in increased development of EAE (86). In contrast to what is found in the germline knock-out mice, T cell development, cytokine expression and proliferation upon in vitro activation is normal in CD4+ T cell specific Sirt1 knock-out mice (87). These observations indicate that the role of SIRT1 varies within different T cell subsets and different stages of T cell development. This is in line with the finding that basal sirtuin/SIRT1 levels differ between different T cell subsets. For instance, in mice, all sirtuin members are expressed in higher levels in Treg cells compared to Teff cells. Interestingly, upon in vitro activation via the T cell receptor there is a markedly increased expression of all sirtuin members in mouse Teff cells, but a downregulation of some sirtuins, including SIRT1, in Treg cells (76). Furthermore, SIRT1 plays an important role in the function of Treg cells via its effect on FOXP3. Both FOXP3 activity and stability is dependent on its acetylation status and it was demonstrated that SIRT1 associates with and deacetylates FOXP3, resulting in its degradation (55, 88–90). This was confirmed in vivo by Treg-specific Sirt1 knock-out in mice where there was increased expression of FOXP3 (87). In addition, the increase in FOXP3 expression was accompanied by an increased suppressive capacity of Treg cells. Transfer of Treg cells from both Cd4+ and Foxp3+ specific knock-out mice into immune-deficient mice showed a more potent suppressive capacity compared to WT Treg cells (87). Moreover, in a colitis mouse model, the adoptive transfer of Teff cells isolated from CD4+ specific Sirt1 knock-out mice resulted in a nearly 3-fold increase in iTreg formation compared with mice receiving WT Teff cells. This correlated with reduced weight loss and reduced development of colitis (91).
In other T cell subsets, treatment with an siRNA for Sirt1, resulted in increased of IL-9 production in vitro by both mouse and human CD4+ T cells which was accompanied with an increase in allergic airway inflammation in mice (89). In Th17 cells, Sirt1 knock-out was demonstrated to inhibit Th17 differentiation via RORyt hyperacetylation and showed to be protective in an EAE mouse model (92). IL-9 producing Th9 cells are associated with a Th2 type response while Th17 cells are important pro-inflammatory players in autoimmune disease.
Altogether, the function of SIRT1 can exert varying effects on different T cell subsets and has been linked to both protective and aggravating effects in disease models. Overall, most studies suggest a pro-inflammatory role for SIRT1 in CD4+ T cells, and especially Treg cells, in the setting of autoimmune diseases.
The role of SIRT2, SIRT4, SIRT5, SIRT6, and SIRT7 in CD4+ T cells has not been studied to our knowledge. Germline knock-out of SIRT3 resulted in a normal T cell development and response to bacterial and fungal infections in mice (93). Treg cells from FOXP3 cell-specific Sirt3 knock-out in mice had impaired suppressive function in vitro which resulted in an increased cardiac allograft rejection and chronic graft injury in vivo. These findings suggest a protective role for SIRT3 in the function of Treg cells (61). Collectively, these data demonstrate that, although opposite effects, both SIRT1 and 3 can be regarded as a potential therapeutic target in autoimmune disease.
HDAC Inhibition Based Therapy for Chronic Inflammatory Diseases
Acetylation has directly been implicated in the control of cell cycle arrest and apoptosis, making modulators of acetylation such as HATs and HDACs interesting targets for treatment of various diseases, especially cancer. Presently, the therapeutic potential of various HDACi is being tested in clinical trials involving several different malignancies (29, 94–97). For example, the tumor suppressor gene p53, a master coordinator of crucial cellular functions such as apoptosis and genomic stability, is deacetylated by several members of class I and class III HDACs, thereby decreasing its activity (98–102). This underlies the therapeutic potential of HDACi in cancer treatment. HDACi are generally well-tolerated compared to other drugs used in cancer therapy and the most commonly described side effects are gastro-intestinal complaints and fatigue (97, 103). Serious adverse events described include bone marrow depression, liver toxicity, electrolyte disturbances and electrocardiogram (ECG) changes. However, bone marrow depression was shown to be reversible after cessation of the therapeutic agent (97, 104) and intensive monitoring of ECG changes in clinical trials did not show an increase in cardiac adverse events, but long-term follow up is needed (97, 103). Furthermore, of the many HDACi tested in phase I/II clinical trials just a few have been approved for clinical use, which could be caused by absence of selectivity and unclear mechanism of action of many HDACi (97). The broad impact of HDACs on major cell functions, including on CD4+ T cell function (both Teff and Treg), suggests that HDACs could be a potential therapeutic target as well for non-malignant diseases like chronic inflammatory diseases. However, especially for non-lethal chronic diseases such as JIA, potential side effects should be carefully studied, monitored and balanced against the possible or expected benefits.
Although on a different scale compared to cancer research, the role of HDACi in the context of autoimmune diseases has also been investigated (Table 1). HDACi were demonstrated to suppress key players of the innate immune system (115–121). For example, Trichostatin A (TSA) and nicotinamide, HDACi inhibiting class I/II or class III HDACs, respectively, decreased the in vitro production of IL-6 and TNFα by macrophages from healthy donors and patients with RA after stimulation with TNFα or LPS (115). In mice, oral treatment with the pan-HDACi; suberoylanilide hydroxamic acid (SAHA), also known as Vorinostat, reduced the circulating levels of the pro-inflammatory cytokines TNFα, IL-1β, IL-6, and IFNγ after LPS stimulation (116). Moreover, in a murine lethal LPS-induced septic shock model, treatment with SAHA improved survival by attenuation of several inflammatory markers including neutrophil infiltration in the lungs (117). Furthermore, in LPS stimulated cultured human PBMCs, the class I/II HDACi: ITF2357 (Givinostat) reduced the production of the pro-inflammatory cytokines TNFα, IL-1α, IL-1β, and IFNγ. This in vitro data was confirmed in an in vivo mouse model where oral treatment with Givinostat reduced LPS-induced serum TNFα and IFNγ by more than 50% (118). Next, HDAC inhibition in RA fibroblast like synoviocytes (FLS) suppressed inflammatory gene expression, including type I IFNγ, IL-6, IL-8 (120, 121) via regulation of cytokine mRNA stability (121). Accordingly, Etinostat (MS275), a class I HDAC inhibitor, selectively affecting HDAC1-3, showed to decrease cell proliferation and secretion of the pro-inflammatory cytokines IL-6 and IL-18 and nitric oxide in cultured human fibroblastic cells from RA patients (119). The nuclear accumulation of NFκ-B was decreased in this model indicating that the anti-inflammatory effect could be mediated via increased acetylation of NFκ-B. A more detailed overview of HDAC inhibition in innate immune cells can be found in literature (51, 52) and is beyond the scope of this review. These data demonstrate that HDAC inhibition of various classes can result in reduced pro-inflammatory cytokine production by innate immune cells, both in vitro and in vivo.

Table 1. Effect of HDAC inhibition on T cell subsets.
HDACi Affecting Class I and II HDAC
Next to suppressive effects on innate inflammation, HDACi could be of great interest as therapeutic strategy by suppressing key players of the adaptive immune system. Of special interest for this review is the effect of various HDACi on FOXP3 expression and Treg cell mediated suppression. Treatment of WT mice with the pan-HDACi TSA resulted in an increase of absolute numbers and percentages of CD4+FOXP3+ cells in lymphoid tissue by increased production in the thymus (72). This was accompanied by an increase in FOXP3 acetylation and expression of Treg associated genes, including Foxp3, Ctla-4, and Il-10 (72). In addition, in a mouse colitis model, the pan HDACi TSA and SAHA inhibited the development of colitis, defined by weight loss, diarrhea, bleeding, and histological findings. In line with previous results, this was accompanied by an increase in CD4+FOXP3+ cells in both absolute numbers and percentages in the lymphoid tissue of these mice (73). Furthermore, in a mouse cardiac transplant model, SAHA treatment prolonged cardiac allograft survival which was associated with an increased percentage of FOXP3+ cells in the thymus, lymph nodes and spleen (105). This was combined with an improved suppressive capacity of the Treg cells. However, depending on the dose used, different effects of SAHA on Treg cells were observed. A low dose of SAHA selectively promoted Teff cell apoptosis and hereby increased the relative percentage of Treg cells, while a high dose suppressed the generation of FOXP3+ cells (105). Perhaps, this could be explained by differential effects of SAHA concentrations on the various types of HDACs (122). In an arthritis mouse model, Valproic acid (VPA), a strong class I but also class II HDACi, decreased the incidence and disease activity of collagen induced arthritis. In addition, there was an increase in the suppressive capacity and numbers of Treg cells (112). In contrast to TSA and SAHA, Etinostat, a selective inhibitor of HDAC1-3 from class I, had no effect on the development of colitis or on the percentage CD4+FOXP3+ cells in the colitis mouse model (73). Since TSA is an inhibitor of Class I and II and SAHA of class I, II, and IV HDACs this implies that the inhibition of one or more HDACs from class II are responsible for the increase in Treg cell numbers in this study. As we described above, an important role for HDAC9 in the development of autoimmune disease was demonstrated in several studies. Therefore, HDAC9 inhibition could potentially be responsible for the increased Treg cell numbers and the protective effect in colitis development in these mice treated with TSA and SAHA. HDAC9 inhibition could therefore be of important therapeutic potential in the treatment of autoimmune diseases such as JIA.
In humans, a phase I/II clinical trial aiming to reduce the incidence of graft vs. host disease in patients receiving allogeneic hematopoietic cell transplantation, add-on treatment with Vorinostat (SAHA) reduced pro-inflammatory cytokine levels in plasma and increased Treg cell numbers and suppressive capacity (106, 107).
Although non-specific, a compound known to exert an effect on T cells via HDAC8 inhibition is butyrate (113). Butyrate treatment resulted in a reduction in disease severity in a collagen induced arthritis mouse model. This was associated with decreased expression levels of pro-inflammatory cytokines and a reduction in inflammation scores, bone damage and cartilage damage scores (113). Inhibition of HDAC8 in T cells by butyrate resulted in decreased Th17 cell number and increased number of Treg cells in these mice. However, the effect of butyrate on the immune system seems to be very broad, with the potential to inhibit Class I, IIa, and IV HDACs affecting several cell types, and being not restricted to HDAC inhibition (123–125).
Givinostat, another class I and II HDACi, is together with SAHA one of the few HDACi that has been investigated for its potential therapeutic effect in autoimmune disease in a clinical trial. Both in vitro (human primary cells) and in in vivo mouse models, Givinostat was demonstrated to have a strong anti-inflammatory effect by affecting key players of the innate and the adaptive immune system (108, 109, 118). The SLE prone NZB/W mice treated with Givinostat showed decreased SLE disease activity and increased survival (108). This was defined by a decrease in anti-nuclear antibodies and immune complex deposition, improvement of renal histopathology, decrease of the pro-inflammatory cytokine IL-1β and increase in the anti-inflammatory cytokine tumor growth factor (TGF)-β. In addition, an increased percentage of Treg cells and a decreased number of IL-17 producing cells was observed in the spleen (108). Furthermore, in an arthritis mouse model, Givinostat treatment showed to decrease joint swelling and cell influx into the joint cavity (109). A reduced production of the pro-inflammatory cytokines TNFα and IL-1β by synovial tissue was demonstrated which resulted in strong inhibition of bone resorption (109). These promising results from pre-clinical (animal) studies resulted in further exploring Givinostat as a potential therapeutic treatment in human autoimmune disease. First, oral treatment with Givinostat was proven to be safe in a phase I trial involving healthy volunteers (110). In addition, the ex vivo stimulation of the peripheral blood from these volunteers with LPS showed a reduction in pro-inflammatory cytokine production. Next, in a small phase II trial involving 17 children with systemic onset JIA (sJIA), oral treatment with Givinostat during 12 weeks proved to be relatively safe, with only mild adverse events. Although 4/17 patients discontinued treatment for safety reasons this was reported to be non-drug related. Interestingly, although not set-up and powered to demonstrate efficacy, possible therapeutic effects of treatment were suggested. In the per-protocol treated group, 5/9 patients showed a relevant decrease in disease activity scores (ACRPed50%) at 12 weeks. Moreover, some patients showed a decrease in neutrophil count and a decrease in pro-inflammatory cytokines such as CD40L, IL-1α, and IFNγ in whole blood lysates (111). As this was only a small phase II trial, the therapeutic potential of Givinostat in the treatment of sJIA or other forms of JIA needs to be explored further.
HDACi Affecting Class III HDAC
The pro-inflammatory role of SIRT1, at least partially caused by an inhibitory effect on FOXP3 stability and function, makes SIRT1 a potentially interesting target for the treatment of autoimmune disease. In line with what was demonstrated in Sirt1 knock-out mice, selective inhibition of SIRT1 with small-molecule inhibitors promoted Treg cell numbers and function in in vitro studies using primary human cells and in in vivo animal models for autoimmune diseases (87, 88, 91). Mouse Treg cells treated in vitro with the selective SIRT1 inhibitor EX-527 showed a more potent suppressive capacity (87). This was confirmed in vivo in a kidney transplant model where mice treated with EX-527 showed increased survival and improved kidney function (114). In addition, in a mouse colitis model, EX-527 treatment resulted in reduced weight loss and promoted the development of induced Treg cells (91). These studies outline the therapeutic potential of SIRT1 inhibition with EX-527 in autoimmune diseases although its safety and efficacy needs to be further investigated in humans.
Even more interesting for potential future use in patients is the relatively specific SIRT1 inhibitor nicotinamide, also known as Vitamin B3 and well-known for many years as a food additive. SIRT1 inhibition via nicotinamide proved to increase percentages of FOXP3+ cells in vitro in primary human cells (88). Furthermore, both nicotinamide and EX-527 dose dependently decreased the production of IL-17 during ex vivo induction of mouse Th17 cells, indicating an important role for SIRT1 in Th17 differentiation (92). Therefore, SIRT1 inhibition, via nicotinamide or EX-527, has the potential to both induce Treg cells and inhibit Th17 cell differentiation, affecting both sides of the disturbed immune balance in autoimmune diseases such as JIA. Importantly, the potential therapeutic effects of nicotinamide in autoimmune diseases is not a new concept, as it has been studied in humans in a variety of diseases for over more than 50 years (126–134). In both adults and children, maintenance therapy with nicotinamide has been associated with improvement of beta-cell function and reduction in pancreatic inflammation in Type 1 diabetes (128, 131, 132, 134, 135). However, results proved variable in other studies and the effects on either Treg or Teff cells in these studies has not been investigated (127, 129, 130, 136). Importantly, in particular for intended future development of nicotinamide maintenance therapy in JIA, the long term use of high dose nicotinamide treatment proved to be safe in multiple clinical trials involving large numbers of adults and children from the age of 5 (130, 132, 136, 137).
Discussion
In JIA, the distorted immunological balance results in chronic, sometimes lifelong inflammatory arthritis and imposes a significant risk for restricted mobility and even disability in children. Although therapy with DMARDs and biologicals has proven to be very successful in inducing remission in JIA, there is a high percentage of patients that relapse after tapering and stop of maintenance immunosuppressive treatment. Therefore, there is still a medical need for novel treatment strategies that focus on restoring immune tolerance and the prevention of relapses in these children. As increased insight of the mechanisms underlying JIA has revealed that the balance between anti-inflammatory mechanisms such as Treg cells and pro-inflammatory mechanisms such as Teff cells can determine the course of the disease, this balance represents therefore a promising therapeutic target.
HDACs have been demonstrated to directly regulate the differentiation, proliferation and function of CD4+ Teff and Treg cells. Therefore, HDACi harbor therapeutic potential to restore immune tolerance and inhibit activation in autoimmune diseases such as JIA. Currently however, literature on HDAC function in T cells and autoimmune disease is mainly focused on mice. HDAC function in humans and autoimmune disease in specific, needs to be further investigated before conclusions about the therapeutic potential of HDACi can be drawn. These mice knock-out studies however help us to better understand the function of specific HDACs in the different T cell subtypes and indicate which HDACs have the best potential as a therapeutic target in autoimmune disease. As HDACs, specifically from class I, have shown to be crucial for the development, differentiation and function of CD4+ T cells, the inhibition of some of these HDACs could potentially lead to severe dysregulation of the immune response. This indicates that, in order to use HDACs in the treatment of human autoimmune disease, it is extremely important that HDACi will selectively target specific HDACs. Several pan HDACi, which have a broad effect and target different HDACs and different HDAC classes, have shown a great potential for the treatment of numerous malignancies and pronounced anti-inflammatory effects by affecting both the innate and adaptive immune system both in vitro and in vivo without affecting general T cell differentiation or development. However, the broad and pronounced anti-inflammatory effect of these pan HDACi certainly hinders the applicability in chronic autoimmune diseases due to the potential side effects. Some of the HDACi however, may show the required selectivity that is needed for treating chronic inflammatory diseases. For example, the class I/II HDACi; Givinostat, was demonstrated to be safe in a small first clinical trial and data from this phase II trial suggested anti-inflammatory effects in sJIA patients, a disease characterized by hyperactivated (innate) inflammatory pathways. In this study, Givinostat seemed to mainly act on effector mechanisms, comparable to the current available treatment options for sJIA. Although promising in this small initial prove of concept trial, the effect of Givinostat treatment in other forms of JIA and the long-term side effects needs to be explored in more extent before it could be considered as part of a potential treatment strategy.
Other HDACi, targeting HDAC6, HDAC9, and SIRT1 not only show suppressive effects on effector mechanisms in inflammation, but may have promising anti-inflammatory effects via improving Treg cell numbers and function as well. The selective and specific inhibition of these HDACs is therefore currently being explored as potential treatment for use in transplant patients suffering GVHD and could be attractive candidates as part of the treatment regimens in chronic autoimmune diseases such as JIA. Although selective deletion of HDAC9 showed promising anti-inflammatory effects, to our knowledge no specific inhibitor of HDAC9 is available yet. In contrast, specific inhibition of HDAC6 is possible and clinical trials involving specific HDAC6 inhibition to prevent allograft rejection in transplant patients are expected in the near future. Since HDAC6i mainly modulate Treg cell numbers and function, the expansion of treatment with specific HDAC6i to other autoimmune diseases seems a logical next step provided that these HDACi have been demonstrated to be safe and in these patients.
Another promising candidate for the treatment of chronic inflammatory diseases such as JIA is the specific inhibition of SIRT1, which showed an anti-inflammatory effect in several models both in vitro and in vivo. The beneficial effect of SIRT1 inhibition was shown to be the result of induction of Treg cells (numbers) and function as well as inhibition of Th17 function. This means that SIRT1 inhibition positively affects the disturbed immune balance in diseases like JIA in both ways. As nicotinamide is a selective inhibitor of SIRT1, and has already proven to be safe in multiple trials for a range of chronic (inflammatory) diseases, nicotinamide could therefore be considered an attractive compound, even in long term use for human adults and children. This is specifically of interest when used as part of tapering and stop regimens for already proven and effective therapies with either DMARDS and/or biologicals. Long term treatment with both DMARDS and biologicals have potential side effects and can be a burden for both patients (as weekly or bi-weekly injections and for example intolerance complaints for MTX) and society (high costs). Development of novel stop-strategies, directed to decrease the chance of relapse of disease once DMARD and/or biologicals are stopped, by introducing maintenance therapy with nicotinamide as HDACi, are therefore both interesting and attractive in chronic disease such as JIA.
In conclusion, there seems to be potential for HDACi, in particularly for specific HDACi, in restoring immunological tolerance in JIA and other autoimmune diseases. However, the data on HDACi in arthritis and specifically JIA is still very limited and needs to be further explored. When considered (nicotinamide) or proven (other HDACi) safe, these agents could first be potentially used as adjuvant agents in stopping and tapering strategies for conventional immunosuppressive treatment with DMARDS/biologicals. First, the effectivity of HDACi in such strategies need to be tested in double blind randomized controlled clinical trial.
Author Contributions
LN, JP, SV, and JvL wrote the manuscript. LN, JP, and JvL made figures and table. SV and JvL edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet (2011) 377:2138–49. doi: 10.1016/S0140-6736(11)60244-4
2. Lovell DJ. Update on treatment of arthritis in children: New treatments, new goals. Bull NYU Hosp Jt Dis. (2006) 64:72–6.
3. Griffin TA, Barnes MG, Ilowite NT, Olson JC, Sherry DD, Gottlieb BS, et al. Gene expression signatures in polyarticular juvenile idiopathic arthritis demonstrate disease heterogeneity and offer a molecular classification of disease subsets. Arthritis Rheum. (2009) 60:2113–23. doi: 10.1002/art.24534
4. Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. Nat Rev Rheumatol. (2011) 7:416–26. doi: 10.1038/nrrheum.2011.68
5. Schneider R, Laxer RM. Systemic onset juvenile rheumatoid arthritis. Baillieres Clin Rheumatol. (1998) 12:245–71.
6. Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. (2007) 34:1133–8.
7. Minoia F, Davi S, Horne A, Demirkaya E, Bovis F, Li C, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol. (2014) 66:3160–9. doi: 10.1002/art.38802
8. Lin Y, Wang C, Gershwin ME, Chiang B. Autoimmunity reviews the pathogenesis of oligoarticular/polyarticular vs systemic juvenile idiopathic arthritis. Autoimmun Rev. (2011) 10:482–9. doi: 10.1016/j.autrev.2011.02.001
9. Nistala K, Moncrieffe H, Newton KR, Varsani H, Hunter P, Wedderburn LR. Interleukin-17–producing T cells are enriched in the joints of children with arthritis, but have a reciprocal relationship to regulatory T cell numbers. Arthritis Rheum. (2008) 58:875–87. doi: 10.1002/art.23291
10. Wedderburn LR, Robinson N, Patel A, Varsani H, Woo P. Selective recruitment of polarized T cells expressing CCR5 and CXCR3 to the inflamed joints of children with juvenile idiopathic arthritis. Arthritis Rheum. (2000) 43:765. doi: 10.1002/1529-0131(200004)43:4<765::AID-ANR7>3.0.CO;2-B
11. Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell (2000) 101:455–8. doi: 10.1016/S0092-8674(00)80856-9
12. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. (1995) 155:1151–64.
13. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. (2001) 27:20–1. doi: 10.1038/83713
14. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. (2001) 27:18–20. doi: 10.1038/83707
15. de Kleer I. Autologous stem cell transplantation for autoimmunity induces immunologic self-tolerance by reprogramming autoreactive T cells and restoring the CD4+CD25+ immune regulatory network. Blood (2006) 107:1696–702. doi: 10.1182/blood-2005-07-2800
16. de Kleer IM, Wedderburn LR, Taams LS, Patel A, Varsani H, Klein M, et al. CD4+CD25bright Regulatory T cells actively regulate inflammation in the joints of patients with the remitting form of juvenile idiopathic arthritis. J Immunol. (2004) 172:6435–43. doi: 10.4049/jimmunol.172.10.6435
17. Kitagawa Y, Sakaguchi S. Molecular control of regulatory T cell development and function. Curr Opin Immunol. (2017) 49:64–70. doi: 10.1016/j.coi.2017.10.002
18. Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, et al. Foxp3+CD25+CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. (2006) 212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x
19. Wehrens EJ, Prakken BJ, Van Wijk F. T cells out of control-impaired immune regulation in the inflamed joint. Nat Rev Rheumatol. (2013) 9:34–42. doi: 10.1038/nrrheum.2012.149
20. Nistala K, Wedderburn LR. Th17 and regulatory T cells: rebalancing pro- and anti-inflammatory forces in autoimmune arthritis. Rheumatology (2009) 48:602–6. doi: 10.1093/rheumatology/kep028
21. Wehrens EJ, Mijnheer G, Duurland CL, Klein M, Meerding J, Van Loosdregt J, et al. Functional human regulatory T cells fail to control autoimmune inflammation due to PKB/c-akt hyperactivation in effector cells. Blood (2011) 118:3538–48. doi: 10.1182/blood-2010-12-328187
22. Prakken B, Ellen Wehrens, Van Wijk F. Editorial: Quality or quantity? unraveling the role of treg cells in rheumatoid arthritis. Arthritis Rheum. (2013) 65:552–4. doi: 10.1002/art.37831
23. Bulatović Calasan M, Vastert SJ, Scholman RC, Verweij F, Klein M, Wulffraat NM, et al. Methotrexate treatment affects effector but not regulatory T cells in juvenile idiopathic arthritis. Rheumatology (2015) 54: 1724–34. doi: 10.1093/rheumatology/kev101
24. Petrelli A, Wehrens EJ, Scholman RC, Prakken BJ, Vastert SJ, van Wijk F. Self-Sustained resistance to suppression of CD8+ Teff Cells at the site of autoimmune inflammation can be reversed by tumor necrosis factor and interferon-gamma blockade. Arthritis Rheumatol. (2016) 68:229–36. doi: 10.1002/art.39418
25. van Loosdregt J, van Wijk F, Prakken B, Vastert B. Update on research and clinical translation on specific clinical areas from biology to bedside: unpacking the mysteries of juvenile idiopathic arthritis pathogenesis. Best Pract Res Clin Rheumatol. (2017) 31:460–75. doi: 10.1016/j.berh.2018.02.003
26. Wehrens EJ, Vastert SJ, Mijnheer G, Meerding J, Klein M, Wulffraat NM, et al. Brief report: anti-tumor necrosis factor α targets protein kinase B/c-akt-induced resistance of effector cells to suppression in juvenile idiopathic arthritis. Arthritis Rheum. (2013) 65:3279–84 doi: 10.1002/art.38132
27. Foell D, Wulffraat N, Wedderburn LR, Wittkowski H, Frosch M, Gerss J, et al. Methotrexate withdrawal at 6 vs 12 months in juvenile idiopathic arthritis in remission: a randomized clinical trial. JAMA (2010) 303:1266–73. doi: 10.1001/jama.2010.375
28. Guzman J, Oen K, Huber AM, Watanabe Duffy K, Boire G, Shiff N, et al. The risk and nature of flares in juvenile idiopathic arthritis: results from the ReACCh-Out cohort. Ann Rheum Dis. (2016) 75:1092–8. doi: 10.1136/annrheumdis-2014-207164
29. Lin H-Y, Chen C-S, Lin S-P, Weng J-R, Chen C-S. Targeting histone deacetylase in cancer therapy. Med Res Rev. (2006) 26:397–413. doi: 10.1002/med.20056
30. Kornberg RD. Structure of chromatin. Annu Rev Biochem. (1977) 46:931–54. doi: 10.1146/annurev.bi.46.070177.004435
31. Zlatanova J. Histone H1 and the regulation of transcription of eukaryotic genes. Trends Biochem Sci. (1990) 15:273–276.
32. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. New York, NY: Garland Science (2002).
33. Clayton AL, Hazzalin CA, Mahadevan LC. enhanced histone acetylation and transcription: a dynamic perspective. Mol Cell. (2006) 23:289–96. doi: 10.1016/j.molcel.2006.06.017
34. Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. (2007) 76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114
35. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. (2009) 10:32–42. doi: 10.1038/nrg2485
36. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature (2004) 429:457–463. doi: 10.1038/nature02625
37. Nusinzon I, Horvath CM. Positive and negative regulation of the innate antiviral response and beta interferon gene expression by deacetylation. Mol Cell Biol. (2006) 26:3106–13. doi: 10.1128/MCB.26.8.3106-3113.2006
38. Glaser KB, Staver MJ, Waring JF, Stender J, Ulrich RG, Davidsen SK. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol Cancer Ther. (2003) 2:151–163.
39. Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. (2001) 7:437–43. doi: 10.1038/86507
40. Nair AR, Boersma LJ, Schiltz L, Chaudhry MA, Muschel RJ. Paradoxical effects of trichostatin a: inhibition of NF-Y-associated histone acetyltransferase activity, phosphorylation of hGCN5 and downregulation of cyclin A and B1 mRNA. Cancer Lett. (2001) 166:55–64. doi: 10.1016/S0304-3835(01)00418-9
41. Tong X, Yin L, Giardina C. Butyrate suppresses Cox-2 activation in colon cancer cells through HDAC inhibition. Biochem Biophys Res Commun. (2004) 317:463–71. doi: 10.1016/j.bbrc.2004.03.066
42. de Ruijter AJM, van Gennip AH, Caron HN, Kemp S, van Kuilenburg ABP. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. (2003) 370:737–49. doi: 10.1042/BJ20021321
43. Hu N, Qiu X, Luo Y, Yuan J, Li Y, Lei W, et al. Abnormal histone modification patterns in lupus CD4+ T cells. J Rheumatol. (2008) 35:804–10.
44. Horiuchi M, Morinobu A, Chin T, Sakai Y, Kurosaka M, Kumagai S. Expression and function of histone deacetylases in rheumatoid arthritis synovial fibroblasts. J Rheumatol. (2009) 36:1580–9. doi: 10.3899/jrheum.081115
45. Gillespie J, Savic S, Wong C, Hempshall A, Inman M, Emery P, et al. Histone deacetylases are dysregulated in rheumatoid arthritis and a novel histone deacetylase 3-selective inhibitor reduces interleukin-6 production by peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Rheum. (2012) 64:418–22. doi: 10.1002/art.33382
46. Peeters JGC, Vervoort SJ, Tan SC, Mijnheer G, de Roock S, Vastert SJ, et al. Inhibition of super-enhancer activity in autoinflammatory site-derived t cells reduces disease-associated gene expression. Cell Rep. (2015) 12:1986–96. doi: 10.1016/j.celrep.2015.08.046
47. Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature (2015) 518:337–43. doi: 10.1038/nature13835
48. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science (2012) 337:1190–5. doi: 10.1126/science.1222794
49. Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. (2009) 41:185–98. doi: 10.1016/j.biocel.2008.08.027
50. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science (2009) 325:834–40. doi: 10.1126/science.1175371
51. Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. (2011) 32:335–43. doi: 10.1016/j.it.2011.04.001
52. Grabiec AM, Tak PP, Reedquist KA. Function of histone deacetylase inhibitors in inflammation. Crit Rev Immunol. (2011) 31:233–263.
53. Grausenburger R, Bilic I, Boucheron N, Zupkovitz G, El-Housseiny L, Tschismarov R, et al. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J Immunol. (2010) 185:3489–97. doi: 10.4049/jimmunol.0903610
54. Goschl L, Preglej T, Hamminger P, Bonelli M, Andersen L, Boucheron N, et al. A T cell-specific deletion of HDAC1 protects against experimental autoimmune encephalomyelitis. J Autoimmun. (2018) 86:51–61. doi: 10.1016/j.jaut.2017.09.008
55. Wang L, Samanta A, Levine M, Beier U, Han R, Kalin J, et al. Vital Role of CoREST Complex as a master regulator of Foxp3+ T-regulatory cell gene expression and suppressive function. Am J Transpl. (2017) 17(Suppl. 3).
56. Dovey OM, Foster CT, Conte N, Edwards SA, Edwards JM, Singh R, et al. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood (2013) 121:1335–44. doi: 10.1182/blood-2012-07-441949
57. Heideman MR, Wilting RH, Yanover E, Velds A, de Jong J, Kerkhoven RM, et al. Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood (2013) 121:2038–50. doi: 10.1182/blood-2012-08-450916
58. Philips RL, Chen MW, McWilliams DC, Belmonte PJ, Constans MM, Shapiro VS. HDAC3 is required for the downregulation of RORgammat during thymocyte positive selection. J Immunol. (2016) 197:541–54. doi: 10.4049/jimmunol.1502529
59. Stengel KR, Zhao Y, Klus NJ, Kaiser JF, Gordy LE, Joyce S, et al. Histone deacetylase 3 is required for efficient T cell development. Mol Cell Biol. (2015) 35:3854–65. doi: 10.1128/MCB.00706-15
60. Hsu F-C, Belmonte PJ, Constans MM, Chen MW, McWilliams DC, Hiebert SW, et al. Histone deacetylase 3 is required for T cell maturation. J Immunol. (2015) 195:1578–90. doi: 10.4049/jimmunol.1500435
61. Wang L, Liu Y, Han R, Beier UH, Bhatti TR, Akimova T, et al. FOXP3+ regulatory T cell development and function require histone/protein deacetylase 3. J Clin Invest. (2015) 125:1111–23. doi: 10.1172/JCI77088
62. Wang L, Beier UH, Akimova T, Dahiya S, Han R, Samanta A, et al. Histone/protein deacetylase inhibitor therapy for enhancement of Foxp3+ T-regulatory cell function posttransplantation. Am J Transl. (2018) 18:1596–603. doi: 10.1111/ajt.14749
63. Liu Q, Zhang X, Yin C, Chen X, Zhang Z, Brown S, et al. HDAC4 is expressed on multiple T cell lineages but dispensable for their development and function. Oncotarget (2017) 8:17562–72. doi: 10.18632/oncotarget.15077
64. Guo S, Zhu Q, Jiang T, Wang R, Shen Y, Zhu X, et al. Genome-wide DNA methylation patterns in CD4+ T cells from Chinese Han patients with rheumatoid arthritis. Mod Rheumatol. (2017) 27:441–7. doi: 10.1080/14397595.2016.1218595
65. Xiao H, Jiao J, Wang L, O'Brien S, Newick K, Wang L-CS, et al. HDAC5 controls the functions of Foxp3(+) T-regulatory and CD8(+) T cells. Int J Cancer (2016) 138:2477–86. doi: 10.1002/ijc.29979
66. Kasler HG, Young BD, Mottet D, Lim HW, Collins AM, Olson EN, et al. Histone deacetylase 7 regulates cell survival and TCR signaling in CD4/CD8 double-positive thymocytes. J Immunol. (2011) 186:4782–93. doi: 10.4049/jimmunol.1001179
67. Kasler HG, Lim HW, Mottet D, Collins AM, Lee IS, Verdin E. Nuclear export of histone deacetylase 7 during thymic selection is required for immune self-tolerance. EMBO J. (2012) 31:4453–65. doi: 10.1038/emboj.2012.295
68. Kasler HG, Verdin E. Histone deacetylase 7 functions as a key regulator of genes involved in both positive and negative selection of thymocytes. Mol Cell Biol. (2007) 27:5184–200. doi: 10.1128/MCB.02091-06
69. Dequiedt F, Kasler H, Fischle W, Kiermer V, Weinstein M, Herndier BG, et al. HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity (2003) 18:687–698. doi: 10.1016/S1074-7613(03)00109-2
70. Tao R, Hancock WW. Resistance of Foxp3+ regulatory T cells to Nur77-induced apoptosis promotes allograft survival. PLoS ONE (2008) 3:e2321. doi: 10.1371/journal.pone.0002321
71. Yan K, Cao Q, Reilly CM, Young NL, Garcia BA, Mishra N. Histone deacetylase 9 deficiency protects against effector T cell-mediated systemic autoimmunity. J Biol Chem. (2011) 286:28833–43. doi: 10.1074/jbc.M111.233932
72. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. (2007) 13:1299–307. doi: 10.1038/nm1652
73. de Zoeten EF, Wang L, Sai H, Dillmann WH, Hancock WW. Inhibition of HDAC9 Increases T regulatory cell function and prevents colitis in Mice. Gastroenterology (2010) 138:583–94 doi: 10.1053/j.gastro.2009.10.037
74. Beier UH, Wang L, Han R, Akimova T, Liu Y, Hancock WW. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci Signal (2012) 5:ra45. doi: 10.1126/scisignal.2002873
75. Yan B, Liu Y, Bai H, Chen M, Xie S, Li D, et al. HDAC6 regulates IL-17 expression in T lymphocytes: implications for HDAC6-targeted therapies. Theranostics (2017) 7:1002–9. doi: 10.7150/thno.17615
76. de Zoeten EF, Wang L, Butler K, Beier UH, Akimova T, Sai H, et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol Cell Biol. (2011) 31:2066–78. doi: 10.1128/MCB.05155-11
77. Kalin JH, Butler KV, Akimova T, Hancock WW, Kozikowski AP. Second-generation histone deacetylase 6 inhibitors enhance the immunosuppressive effects of Foxp3+ T-regulatory cells. J Med Chem. (2012) 55:639–51. doi: 10.1021/jm200773h
78. Segretti MCF, Vallerini GP, Brochier C, Langley B, Wang L, Hancock WW, et al. Thiol-based potent and selective HDAC6 inhibitors promote tubulin acetylation and t-regulatory cell suppressive function. ACS Med Chem Lett. (2015) 6:1156–61. doi: 10.1021/acsmedchemlett.5b00303
79. Oh BR, Suh D, Bae D, Ha N, Choi Y Il, Yoo HJ, et al. Therapeutic effect of a novel histone deacetylase 6 inhibitor, CKD-L, on collagen-induced arthritis in vivo and regulatory T cells in rheumatoid arthritis in vitro. Arthritis Res Ther. (2017) 19:154. doi: 10.1186/s13075-017-1357-2
80. Dahiya S, Wang L, Beier ulf H, Han R, Hancock WW. HDAC10 Targeting Regulates Foxp3 Promoter, enhances T-regulatory (Treg) function and suppresses autoimmune colitis. J.Immunol. (2018) 200 (Suppl. 1) 54.11.
81. Huang J, Wang L, Dahiya S, Beier UH, Han R, Samanta A, et al. Histone/protein deacetylase 11 targeting promotes Foxp3+ Treg function. Sci Rep. (2017) 7:8626. doi: 10.1038/s41598-017-09211-3
82. Woods DM, Woan KV, Cheng F, Sodre AL, Wang D, Wu Y, et al. T cells lacking HDAC11 have increased effector functions and mediate enhanced alloreactivity in a murine model. Blood (2017) 130:146–55. doi: 10.1182/blood-2016-08-731505
83. Niederer F, Ospelt C, Brentano F, Hottiger MO, Gay RE, Gay S, et al. SIRT1 overexpression in the rheumatoid arthritis synovium contributes to proinflammatory cytokine production and apoptosis resistance. Ann Rheum Dis. (2011) 70:1866–73. doi: 10.1136/ard.2010.148957
84. Moon M-H, Jeong J-K, Lee Y-J, Seol J-W, Jackson CJ, Park S-Y. SIRT1, a class III histone deacetylase, regulates TNF-alpha-induced inflammation in human chondrocytes. Osteoarthr Cartil. (2013) 21:470–80. doi: 10.1016/j.joca.2012.11.017
85. Huang W, Shang W, Wang H, Wu W, Hou S. Sirt1 overexpression protects murine osteoblasts against TNF-alpha-induced injury in vitro by suppressing the NF-kappaB signaling pathway. Acta Pharmacol Sin. (2012) 33:668–74. doi: 10.1038/aps.2011.189
86. Zhang J, Lee S-M, Shannon S, Gao B, Chen W, Chen A, et al. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J Clin Invest. (2009) 119:3048–58. doi: 10.1172/JCI38902
87. Beier UH, Wang L, Bhatti TR, Liu Y, Han R, Ge G, Hancock WW. Sirtuin-1 targeting promotes Foxp3+ T-regulatory cell function and prolongs allograft survival. Mol Cell Biol. (2011) 31:1022–9. doi: 10.1128/MCB.01206-10
88. Van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YYJ, Beekman JM, Van Beekum O, et al. Regulation of treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood (2010) 115:965–74. doi: 10.1182/blood-2009-02-207118
89. Wang Y, Bi Y, Chen X, Li C, Li Y, Zhang Z, et al. Histone deacetylase SIRT1 negatively regulates the differentiation of interleukin-9-producing CD4(+) T Cells. Immunity (2016) 44:1337–49. doi: 10.1016/j.immuni.2016.05.009
90. Li B, Samanta A, Song X, Iacono KT, Bembas K, Tao R, et al. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci USA. (2007) 104:4571–6. doi: 10.1073/pnas.0700298104
91. Akimova T, Xiao H, Liu Y, Bhatti TR, Jiao J, Eruslanov E, et al. Targeting sirtuin-1 alleviates experimental autoimmune colitis by induction of Foxp3+ T-regulatory cells. Mucosal Immunol. (2014) 7:1209–20. doi: 10.1038/mi.2014.10
92. Lim HW, Kang SG, Ryu JK, Schilling B, Fei M, Lee IS, et al. SIRT1 deacetylates RORgammat and enhances Th17 cell generation. J Exp Med. (2015) 212:607–17. doi: 10.1084/jem.20132378
93. Ciarlo E, Heinonen T, Lugrin J, Acha-Orbea H, Le Roy D, Auwerx J, et al. Sirtuin 3 deficiency does not alter host defenses against bacterial and fungal infections. Sci Rep. (2017) 7:3853. doi: 10.1038/s41598-017-04263-x
94. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov (2006) 5:769–84. doi: 10.1038/nrd2133
95. Weidle UH, Grossmann A. Inhibition of histone deacetylases: a new strategy to target epigenetic modifications for anticancer treatment. Anticancer Res. (2000) 20:1471–85.
96. McGovern JL, Nguyen DX, Notley CA, Mauri C, Isenberg DA, Ehrenstein MR. Th17 cells are restrained by treg cells via the inhibition of interleukin-6 in patients with rheumatoid arthritis responding to anti-tumor necrosis factor antibody therapy. Arthritis Rheum (2012) doi: 10.1002/art.34565
97. Subramanian S, Bates SE, Wright JJ, Espinoza-delgado I, Piekarz RL. Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals (2010) 3:2751–67. doi: 10.3390/ph3092751
98. Juan LJ, Shia WJ, Chen MH, Yang WM, Seto E, Lin YS, et al. Histone deacetylases specifically down-regulate p53-dependent gene activation. J Biol Chem. (2000) 275:20436–43. doi: 10.1074/jbc.M000202200
99. Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature (2000) 408:377–81. doi: 10.1038/35042612
100. Ito A, Kawaguchi Y, Lai C-H, Kovacs JJ, Higashimoto Y, Appella E, et al. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. (2002) 21:6236–45. doi: 10.1093/emboj/cdf616
101. Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell (2001) 107:149–59.10. doi: 10.1016/S0092-8674(01)00527-X
102. Kuo Y-H, Qi J, Cook GJ. Regain control of p53: Targeting leukemia stem cells by isoform-specific HDAC inhibition. Exp Hematol. (2016) 44:315–21. doi: 10.1016/j.exphem.2016.02.007
103. Kim M, Thompson LA, Wenger SD, O'Bryant CL. Romidepsin: a histone deacetylase inhibitor for refractory cutaneous T-cell lymphoma. Ann Pharmacother. (2012) 46:1340–8. doi: 10.1345/aph.1R036
104. Bishton MJ, Harrison SJ, Martin BP, McLaughlin N, James C, Josefsson EC, et al. Deciphering the molecular and biologic processes that mediate histone deacetylase inhibitor-induced thrombocytopenia. Blood (2011) 117:3658–68. doi: 10.1182/blood-2010-11-318055
105. Zhang X, Han S, Kang Y, Guo M, Hong S, Liu F, et al. SAHA, an HDAC inhibitor, synergizes with tacrolimus to prevent murine cardiac allograft rejection. Cell Mol Immunol. (2012) 9:390–8. doi: 10.1038/cmi.2012.28
106. Choi SW, Gatza E, Hou G, Sun Y, Whitfield J, Song Y, et al. Histone deacetylase inhibition regulates inflammation and enhances Tregs after allogeneic hematopoietic cell transplantation in humans. Blood (2015) 125:815–9. doi: 10.1182/blood-2014-10-605238
107. Choi SW, Braun T, Chang L, Ferrara JLM, Pawarode A, Magenau JM, et al. Vorinostat plus tacrolimus and mycophenolate to prevent graft-versus-host disease after related-donor reduced-intensity conditioning allogeneic haemopoietic stem-cell transplantation: a phase 1/2 trial. Lancet Oncol. (2014) 15:87–95. doi: 10.1016/S1470-2045(13)70512-6
108. Regna NL, Chafin CB, Hammond SE, Puthiyaveetil AG, Caudell DL, Reilly CM. Class I and II histone deacetylase inhibition by ITF2357 reduces SLE pathogenesis in vivo. Clin Immunol. (2014) 151:29–42. doi: 10.1016/j.clim.2014.01.002
109. Joosten LAB, Leoni F, Meghji S, Mascagni P. Inhibition of HDAC activity by ITF2357 ameliorates joint inflammation and prevents cartilage and bone destruction in experimental arthritis. Mol Med. (2011) 17:391–6. doi: 10.2119/molmed.2011.00058
110. Furlan A, Monzani V, Reznikov LL, Leoni F, Fossati G, Modena D, et al. Pharmacokinetics, safety and inducible cytokine responses during a phase 1 trial of the oral histone deacetylase inhibitor ITF2357 (givinostat). Mol Med. (2011) 17:353–62. doi: 10.2119/molmed.2011.00020
111. Vojinovic J, Damjanov N. HDAC inhibition in rheumatoid arthritis and juvenile idiopathic arthritis. Mol Med (2011) 17:397–403. doi: 10.2119/molmed.2011.00030
112. Saouaf SJ, Li B, Zhang G, Shen Y, Furuuchi N, Hancock WW, et al. Deacetylase inhibition increases regulatory T cell function and decreases incidence and severity of collagen-induced arthritis. Exp Mol Pathol. (2009) 87:99–104. doi: 10.1016/j.yexmp.2009.06.003
113. Kim DS, Kwon J-E, Lee SH, Kim EK, Ryu J-G, Jung K-A, et al. Attenuation of Rheumatoid Inflammation by Sodium Butyrate Through Reciprocal Targeting of HDAC2 in Osteoclasts and HDAC8 in T Cells. Front Immunol. (2018) 9:1525. doi: 10.3389/fimmu.2018.01525
114. Levine MH, Wang Z, Xiao H, Jiao J, Wang L, Bhatti TR, et al. Targeting Sirtuin-1 prolongs murine renal allograft survival and function. Kidney Int. (2016) 89:1016–26. doi: 10.1016/j.kint.2015.12.051
115. Grabiec AM, Krausz S, de Jager W, Burakowski T, Groot D, Sanders ME, et al. Histone deacetylase inhibitors suppress inflammatory activation of rheumatoid arthritis patient synovial macrophages and tissue. J Immunol. (2010) 184:2718–28. doi: 10.4049/jimmunol.0901467
116. Leoni F, Zaliani A, Bertolini G, Porro G, Pagani P, Pozzi P, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci USA. (2002) 99:2995–3000. doi: 10.1073/pnas.052702999
117. Li Y, Liu B, Zhao H, Sailhamer EA, Fukudome EY, Zhang X, et al. Protective effect of suberoylanilide hydroxamic acid against LPS-induced septic shock in rodents. Shock (2009) 32:517–23. doi: 10.1097/SHK.0b013e3181a44c79
118. Leoni F, Fossati G, Lewis EC, Lee J-K, Porro G, Pagani P, et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med. (2005) 11:1–15. doi: 10.2119/2006-00005.Dinarello
119. Choo Q-Y, Ho PC, Tanaka Y, Lin H-S. Histone deacetylase inhibitors MS-275 and SAHA induced growth arrest and suppressed lipopolysaccharide-stimulated NF-kappaB p65 nuclear accumulation in human rheumatoid arthritis synovial fibroblastic E11 cells. Rheumatology (2010) 49:1447–60. doi: 10.1093/rheumatology/keq108
120. Angiolilli C, Kabala PA, Grabiec AM, Van Baarsen IM, Ferguson BS, Garcia S, et al. Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann Rheum Dis. (2017) 76:277–85. doi: 10.1136/annrheumdis-2015-209064
121. Angiolilli C, Kabala PA, Grabiec AM, Rossato M, Lai WS, Fossati G, et al. Control of cytokine mRNA degradation by the histone deacetylase inhibitor ITF2357 in rheumatoid arthritis fibroblast-like synoviocytes: beyond transcriptional regulation. Arthritis Res Ther. (2018) 20:148. doi: 10.1186/s13075-018-1638-4
122. Huber K, Doyon G, Plaks J, Fyne E, Mellors JW, Sluis-Cremer N. Inhibitors of histone deacetylases: correlation between isoform specificity and reactivation of HIV type 1 (HIV-1) from latently infected cells. J Biol Chem. (2011) 286:22211–8. doi: 10.1074/jbc.M110.180224
123. Berni Canani R, Di Costanzo M, Leone L. The epigenetic effects of butyrate: potential therapeutic implications for clinical practice. Clin Epigenet. (2012) 4:4. doi: 10.1186/1868-7083-4-4
124. Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr. (2003) 133:2485S−93S. doi: 10.1093/jn/133.7.2485S
125. Meijer K, de Vos P, Priebe MG. Butyrate and other short-chain fatty acids as modulators of immunity: what relevance for health? Curr Opin Clin Nutr Metab Care (2010) 13:715–21. doi: 10.1097/MCO.0b013e32833eebe5
126. Carlson LA. Nicotinic acid: the broad-spectrum lipid drug. a 50th anniversary review. J Intern Med. (2005) 258:94–114. doi: 10.1111/j.1365-2796.2005.01528.x
127. Visalli N, Cavallo MG, Signore A, Baroni MG, Buzzetti R, Fioriti E, et al. A multi-centre randomized trial of two different doses of nicotinamide in patients with recent-onset type 1 diabetes (the IMDIAB VI). Diabetes Metab Res Rev. (1999) 15:181–5.
128. Chianelli M, Parisella MG, Visalli N, Mather SJ, D'Alessandria C, Pozzilli P, et al. Pancreatic scintigraphy with 99mTc-interleukin-2 at diagnosis of type 1 diabetes and after 1 year of nicotinamide therapy. Diabetes Metab Res Rev. (2008) 24:115–22. doi: 10.1002/dmrr.767
129. Lampeter EF, Klinghammer A, Scherbaum WA, Heinze E, Haastert B, Giani G, et al. The Deutsche Nicotinamide Intervention Study: an attempt to prevent type 1 diabetes. DENIS Group. Diabetes (1998) 47:980–4.
130. Gale EAM, Bingley PJ, Emmett CL, Collier T. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet (2004) 363:925–31. doi: 10.1016/S0140-6736(04)15786-3
131. Pozzilli P, Browne PD, Kolb H. Meta-analysis of nicotinamide treatment in patients with recent-onset IDDM. The Nicotinamide Trialists. Diabetes Care (1996) 19:1357–63.
132. Crino A, Schiaffini R, Manfrini S, Mesturino C, Visalli N, Beretta Anguissola G, et al. A randomized trial of nicotinamide and vitamin E in children with recent onset type 1 diabetes (IMDIAB IX). Eur J Endocrinol. (2004) 150:719–24.
133. HOFFER A. Treatment of arthritis by nicotinic acid and nicotinamide. Can Med Assoc J. (1959) 81:235–238.
134. Kamal M, Abbasy A-J Al, Muslemani A Al, Bener A. Effect of nicotinamide on newly diagnosed type 1 diabetic children. Acta Pharmacol Sin. (2006) 27:724–27. doi: 10.1111/j.1745-7254.2006.00313.x
135. Olmos PR, Hodgson MI, Maiz A, Manrique M, De Valdes MD, Foncea R, et al. Nicotinamide protected first-phase insulin response (FPIR) and prevented clinical disease in first-degree relatives of type-1 diabetics. Diabetes Res Clin Pract. (2006) 71:320–33. doi: 10.1016/j.diabres.2005.07.009
136. Pitocco D, Crino A, Di Stasio E, Manfrini S, Guglielmi C, Spera S, et al. The effects of calcitriol and nicotinamide on residual pancreatic beta-cell function in patients with recent-onset Type 1 diabetes (IMDIAB XI). Diabet Med. (2006) 23:920–3. doi: 10.1111/j.1464-5491.2006.01921.x
Keywords: juvenile idiopathic arthritis (JIA), acetylation, HDAC inhibitor (histone deacetylase inhibitor), T cells, tolerance
Citation: Nijhuis L, Peeters JGC, Vastert SJ and van Loosdregt J (2019) Restoring T Cell Tolerance, Exploring the Potential of Histone Deacetylase Inhibitors for the Treatment of Juvenile Idiopathic Arthritis. Front. Immunol. 10:151. doi: 10.3389/fimmu.2019.00151
Received: 01 November 2018; Accepted: 17 January 2019;
Published: 07 February 2019.
Edited by:
David Bending, University of Birmingham, United KingdomReviewed by:
Wayne Hancock, University of Pennsylvania, United StatesMichael Francis McDermott, University of Leeds, United Kingdom
Copyright © 2019 Nijhuis, Peeters, Vastert and van Loosdregt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jorg van Loosdregt, ai52YW5sb29zZHJlZ3RAdW1jdXRyZWNodC5ubA==
†These authors have contributed equally to this work