 Bipandeep Dhillon1†
Bipandeep Dhillon1† Fatemah Aleithan
Fatemah Aleithan Zahi Abdul-Sater
Zahi Abdul-Sater Ali A. Abdul-Sater
Ali A. Abdul-Sater- 1School of Kinesiology and Health Science, Muscle Health Research Centre, York University, Toronto, ON, Canada
- 2Department of Basic Sciences, Phoenicia University, Mazraat El Daoudiyeh, Lebanon
TRAFs [tumor necrosis factor (TNF) receptor associated factors] are a family of signaling molecules that function downstream of multiple receptor signaling pathways and play a pivotal role in the biology of innate, and adaptive immune cells. Following receptor ligation, TRAFs generally function as adapter proteins to mediate the activation of intracellular signaling cascades. With the exception of TRAF1 that lacks a Ring domain, TRAFs have an E3 ubiquitin ligase activity which also contributes to their ability to activate downstream signaling pathways. TRAF-mediated signaling pathways culminate in the activation of several transcription factors, including nuclear factor-κB (NF-κB), mitogen-activated protein kinases (MAPKs; e.g., ERK-1 and ERK-2, JNK, and p38), and interferon-regulatory factors (IRF; e.g., IRF3 and IRF7). The biological role of TRAFs is largely due to their ability to positively or negatively regulate canonical and non-canonical NF-κB signaling. While TRAF-mediated signaling regulates various immune cell functions, this review is focused on the recent advances in our knowledge regarding the molecular mechanisms through which TRAF proteins regulate, positively and negatively, inflammatory signaling pathways, including Toll–IL-1 receptors, RIG-I like receptors, and Nod-like receptors. The review also offers a perspective on the unanswered questions that need to be addressed to fully understand how TRAFs regulate inflammation.
Introduction
The Tumor-Necrosis Factor (TNF) Receptor Associated Factor (TRAF) family is comprised of cytoplasmic adaptor proteins involved in transducing downstream effects of a variety of receptors (1). TRAF1 and TRAF2 were first discovered through their association with TNF-R2 (2). Since then four other members have been identified, thus, a total of six known members exist (TRAF1 to TRAF6) (3, 4). The TRAF domain can be divided into a N-terminal coiled-coil region (TRAF-N) and a highly conserved C-terminal Beta-sandwich domain (MATH Domain) (4, 5). It is the MATH domain which allows TRAF molecules to form dimers and recruit downstream effectors to receptors (1). With the exception of TRAF1, all other TRAF members, contain a N-terminal RING finger, followed by a variable number of zinc fingers (1, 4, 6). The RING finger motif allows TRAF molecules to act as E3 ubiquitin ligases (5, 6). As adaptor proteins and E3 ubiquitin ligases involved in several immune pathways, TRAFs ultimately lead to the activation of transcription factors, such as nuclear factor-κB (NF-κB), mitogen-activated protein kinases (MAPKs; e.g., ERK-1 and ERK-2, JNK, and p. 38), and interferon-regulatory factors (IRF; e.g., IRF3 and IRF7) (5, 6). In addition, TRAF proteins play important roles in embryonic development, tissue homeostasis, stress response, and bone metabolism (3, 6). Since being discovered in TNF receptor signaling, TRAFs' role has expanded to include involvement in many other inflammatory signaling pathways such as toll-like receptors (TLRs), nucleotide binding-oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), and cytokine receptors (4, 6). Aberrant and prolonged activation of inflammation following the activation of these receptors has been associated with debilitating diseases including cancer, atherosclerosis, type II diabetes, and autoimmune diseases (7). Therefore, a number of mechanisms have evolved to negatively regulate these pathways (8). This review is focused on recent studies that identified new roles for TRAF proteins in activating and inhibiting TLR, RLR, and NLR signaling, and emphasizes newly discovered mechanisms of regulating these pathways by targeting TRAF expression and function.
The role of TRAFs in Toll-Like Receptor signaling
Toll-like receptors (TLRs) are a family of transmembrane receptors lining both cellular and endosomal membranes that sense various pathogen-associated-molecular patterns (PAMPs), and danger-associated molecular patterns (DAMPS) (6, 9–11). There are 10 known TLRs in humans that either exist as homo or heterodimers (11). TLRs are characterized by an extracellular ectodomain comprised of leucine-rich repeats (LRRs), which senses the corresponding PAMP or DAMP, a transmembrane domain, and an intracellular Toll/IL-1 receptor (TIR) domain, which induces the downstream response (9, 12). Upon stimulation, TLRs oligomerize, and recruit MyD88, with the exception of TLR3, which recruits TRIF through TIR domain interaction (12). TLR4 can uniquely induce both MyD88-dependent signaling when it's on the plasma membrane and TRIF-mediated signaling when translocated to the endosomal compartment. Subsequently, a signaling cascade is initiated which results in the activation of transcription factors like NF-κB, MAPKs, and IRFs. This ultimately leads to the production of chemokines, cytokines, and other inflammatory mediators, which initiate the innate immune response and prime the adaptive immune response (Figure 1) (6, 9, 13).
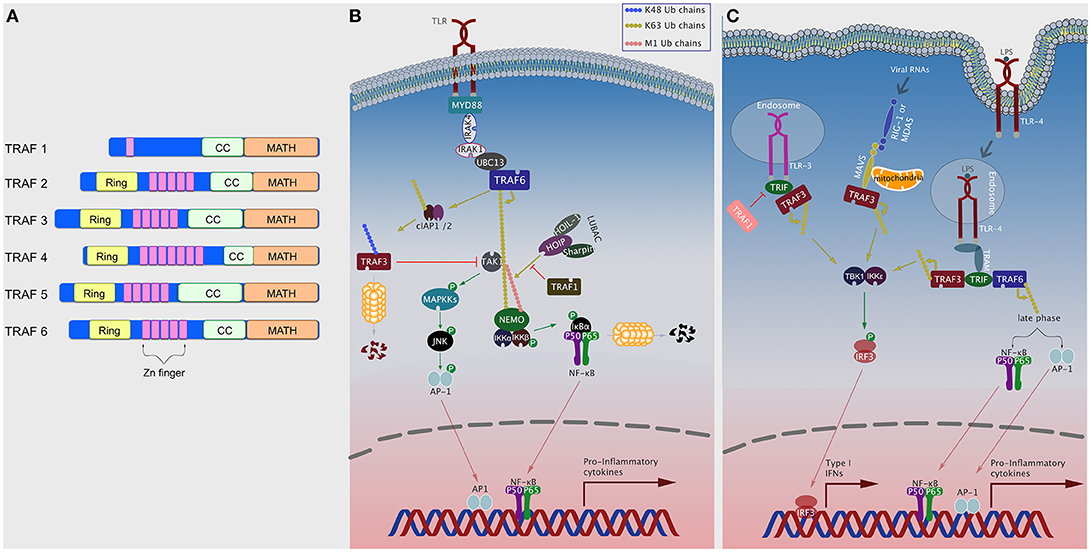
Figure 1. TRAFs in TLR signaling pathways. (A) Domain organization of TRAF proteins. Domains shown include Ring, Zinc (Zn) finger, coiled-coil (CC; TRAF-N), and MATH (TRAF-C) (B) Plasma membrane TLRs, upon ligand binding, recruit various intracellular signaling elements including TRAF6 to activate NF-κB and MAPK pathways. TRAF1, TRAF3, TRAF4, and TRAF5 can negatively regulate this pathway by different means. (C) Following ligand binding, TLR4 translocates to the endosomal compartment and recruits TRIF and TRAF3 to induce the TBK1/IKKε/IRF3 axis, or TRAF6 to induce NF-κB and AP-1 via late phase (slower) kinetics. TLR3 and RLRs can also induce the TBK1/IKKε/IRF3 axis by recruiting TRAF3.
MyD88-dependent signaling is initiated with the recruitment of the IL-1 receptor-associated kinase (IRAK) 4 which, in turn, recruits and activates, through phosphorylation, IRAK1 and IRAK2 (14). IRAK1/2 recruit TRAF6, which functions as an E3 ubiquitin ligase following its oligomerization via the CC domains (15). This also allows TRAF6 to associate with the E2 ubiquitin complex Uev1A:Ubc13, which together then catalyze the K63-linked polyubiquitination to TRAF6 and other substrates, including TAK1, TAB1, TAB2, and NEMO (IKKγ) (16). This activates TAK1 which co-localizes with the IKK complex and activate IKKβ via phosphorylation (9, 17). Importantly, optimal activation of the IKK complex requires the linear ubiquitination of NEMO (M1-linked) (18–20). This is mediated by a ubiquitin ligase complex termed the Linear UBiquitin chain Assembly Complex (LUBAC) and consists of heme-oxidized IRP2 ubiquitin ligase-1 (HOIL-1), HOIL-1–interacting protein (HOIP), and the Shank-associated RH domain interactor (SHARPIN) (21). Activation of the IKK complex leads to phosphorylation and subsequent degradation of the inhibitor of κB, IκBα, which eventually leads to NF-κB activation (12, 13). TAK1 also induces activation of MAPKs, such as ERK1/2, P38, and JNK, through phosphorylation leading to activation of transcription factors like AP-1 (Figure 1A) (12).
In TRIF-mediated signaling, TRIF recruits TRAF3, which catalyzes its own K63-linked polyubiquitination. This leads to the activation of the TBK1 and the non-canonical IKK, IKKε, which in turn phosphorylates IRF3 resulting in its nuclear translocation and the subsequent induction of type 1 IFNs (IFN-Is) (22, 23). With slower kinetics (i.e., late phase), TRIF can also form a complex with TRAF6 and RIP1, to induce the TAK1/IKK axis and the subsequent activation of NF-κB (Figure 1B) (11). IFN-Is can also be induced following TLR7 and TLR9 stimulation through the MyD88-dependent pathway. MyD88 forms a complex with TRAF3 which then recruits and activates an IRAK-IKKα complex, which in turn phosphorylates IRF7 resulting in its translocation into the nucleus to induce interferon production (Figure 1C) (9).
TRAFs Negatively Regulate TLR Signaling
In addition to activating TLRs, TRAFs can also function as negative regulators of TLR signaling. TRAF3 negatively regulates TLR-mediated MAPK activity, possibly by preventing the release of the TRAF6:TAB1/2:TAK1 complex, but the negative regulation is inhibited by cIAP1/2 which catalyze K48-polyubiquitinated degradation of TRAF3 (Figure 1A) (11, 23, 24). Under specific conditions, TRAF2 can dampen TLR mediated cytokine production by causing proteasome-dependent degradation of c-Rel, a member of the NF-κB family, in a mechanism that also requires TRAF3 and cIAP1/2 (25). TRAF5 has also been shown to inhibit TLR-stimulated cytokine production by preventing the interaction between TRAF6 and TAB2 (26). TRAF4 associates with p47phox, a component of cytosolic NADPH oxidase, to negatively regulate TLR signaling by interacting with TRAF6 and TRIF and disrupting their functions (27). Recently, TRAF1 has been shown to attenuate TLR-induced NF-κB signaling by interfering with LUBAC-mediated linear ubiquitination of NEMO (Figure 1A) (28). Interestingly, downstream of TLR3 signaling, TRAF1 inhibits TRIF mediated activation of NF-κB, ISRE, and the IFN-β promoter independent of IRF-3 (Figure 1B) (29).
Negative Regulation of TLR Signaling by Targeting TRAFs
TLR signaling can also be regulated by targeting the function, expression, or catalytic activity of certain TRAF proteins. Several deubiquitinases (DUB) have been shown to negatively regulate TLR signaling by removing ubiquitin chains from TRAFs or their targets. For instance, A20 is a key regulator of TLR signaling, whereby it targets several aspects of the signaling cascade. It can accomplish this, in part, by directly deubiquitinating TRAF6 (30). Monocyte chemotactic protein-induced protein 1 (MCPIP1) is another DUB that negatively regulates JNK and NF-κB signaling by deubiquitinating TRAF2, TRAF3, and TRAF6 (31). Recently, peroxiredoxin-1 (PRDX1) has been shown to directly interact with TRAF6 ring finger motif and inhibit its ubiquitin-ligase activity, which diminishes NF-κB activation downstream of TLR4 stimulation [(32), p. 1]. Several members of the NLR family, discussed below, have been shown to regulate TLR signaling by targeting TRAF proteins. NLRC3 can attenuate TLR-mediated NF-κB activation by reducing K63-linked polyubiquitination of TRAF6 (33). NLRX1 can interact with TRAF6 to reduce canonical NF-κB activation through the TLR4 mediated pathway [(34), p. 1]. Under normal conditions, NLRX1 associates with TRAF6, but upon TLR4 stimulation, NLRX1 dissociates from TRAF6 and binds to NEMO preventing TRAF6 recruitment of the IKK complex, and subsequent NF-κB activation (35). It's important to note, however, that some of those findings have been controversial in the field as other studies were not able to reach a similar conclusion [(36, 37); (38), p. 1]. NLRP11 inhibits TLR signaling by recruiting RNF19A, an ubiquitin ligase, to catalyze K48-linked polyubiquitination and the subsequent degradation of TRAF6 [(39), p. 11]. NLRP12 reduces non-canonical NF-κB stimulation by interacting with TRAF3 and NIK causing NIK is degraded preventing/reducing cleavage of p100 to p52 (40).
The role of TRAFs in NOD-Like Receptor signaling
NOD-like receptors (NLRs) are a family of cytosolic receptors that sample intracellular PAMPs and DAMPs (41–44). These receptors participate in a plethora of cellular processes including: inflammasome assembly, pyroptosis, activation of NF-κB and MAPK pathways, autophagy, IFN signaling, antigen processing and presentation, and ROS production (6, 43, 45). These receptors are characterized by a central nucleotide binding (NOD, also known as NACHT) domain, which allows oligomerization, followed by a C-terminal leucine-rich repeat (LRR) domain, which detects PAMPs and DAMPs and a variable N-terminal domains, which helps induce the downstream response (41–44, 46).
NOD1 and NOD2 are the most studied members of the NLR family [reviewed in Motta et al. (47)]. Upon activation, these receptors oligomerize through their NACHT domains and form a complex with RIPK2, through homotypic CARD-CARD interactions (44). In this complex, RIP2 is associated with multiple E3 ligases, including cIAP1/2, xIAP, TRAF2, and TRAF5, but only cIAP1/2 catalyzes its K63-linked polyubiquitination (44, 48–50). TRAF2 and TRAF5 serve as adaptor molecules to facilitate interaction of cIAP1/2 with RIP2 in this complex (48). RIPK2 then induces K63-linked polyubiquitination of TAK1 and NEMO, which recruits the IKK complex to the platform leading to IKKβ phosphorylation by TAK1 (43, 44). TRAF6 and CARD9 serve as adaptors which allow NOD1 and NOD2 to induce MAPKs and subsequently activate AP-1 transcription factor (51, 52). In addition to NF-κB and MAPK activation, NOD1 and NOD2, induce IFN-I production by forming a complex with RIPK2. This results in the recruitment of TRAF3 leading to the activation of TBK1/IKKε/IRF7 axis (Figure 2) (42, 53).
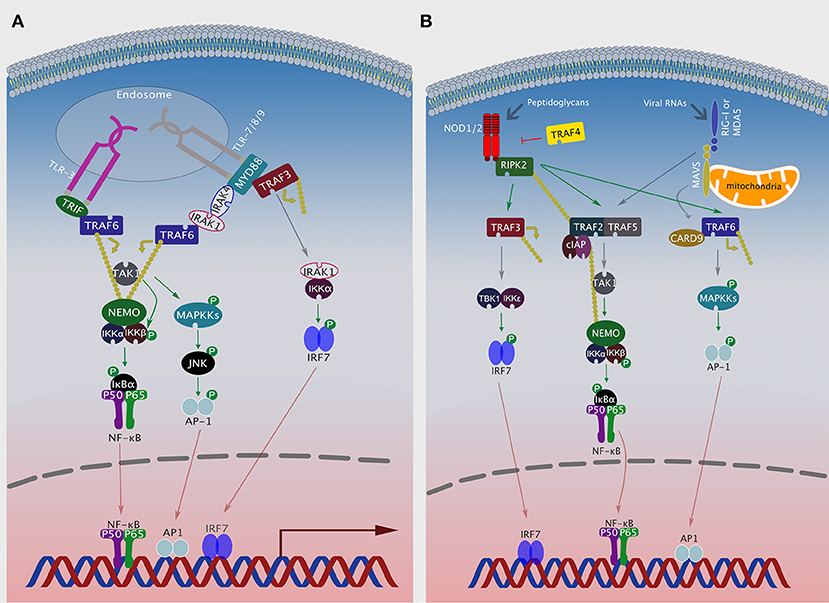
Figure 2. TRAFs in NLR and RLR signaling pathways. (A) After ligand recognition, endosomal TLRs recruit TRAF6, either via TRIF (TLR3) or via MyD88/IRAK1/IRAK4 (TLR7, 8 or 9) to activate NF-κB. Additionally, TLR 7, 8, or 9 can recruit MyD88, TRAF3, IRAK1, and IKKα to activate IRF7. (B) Ligand activated NOD1 or NOD2 associate with RIPK2, which can then recruit either TRAF3 to activate the TBK1/IKKε/IRF7 axis, TRAF2, and TRAF5 to activate NF-κB or TRAF6 and CARD9 to activate MAPK signaling. Viral RNAs activate RIG-I or MDA5, which then associate with the mitochondrial protein MAVS and activate NF-κB or MAPK signaling by recruiting TRAF2/5 or TRAF6/CARD9, respectively.
A few NLR family members (e.g., NLRP1, NLRP3, NLRP6, NLRC4, and NLRC5) are capable of activating inflammasomes (41, 54). Inflammasomes are multimeric protein complexes that play a key role in regulating the secretion of potent cytokines like IL-1β and IL-18. Most inflammasomes are composed of an NLR protein, the zymogen pro-caspase-1, and the adapter protein, apoptosis-associated speck-like protein containing a CARD (ASC) (55, 56). Intriguingly, TRAF3 has been recently shown to play a role in NLRP3 inflammasome activation, as it catalyzes K63-linked polyubiquitination of ASC in order to induce ASC speck formation and inflammasome activation (57). In addition, TRAF2, along with cIAP1/2, mediates K63-linked polyubiquitination of caspase-1 for optimum activity (58). However, TRAF2−/− bone marrow-derived macrophages (BMDMs) show normal inflammasome activation, suggesting that TRAF2 may not even be involved in this pathway (59). Following TLR signaling, TRAF6 promotes the non-transcriptional priming of NLRP3 by inducing its oligomerization and association with ASC (60).
TRAFs Negatively Regulate NLR Signaling
TRAF4 has been shown to act as a negative regulator in NOD2-mediated NF-κB signaling by direct interaction with NOD2. This interaction then allows IKKα to phosphorylate TRAF4, which results in its dissociation from NOD2 and inhibition of NOD2 signaling (61). NLRC3 has been shown to attenuate NLRP3 inflammasome by with ASC for pro-caspase-1 binding (62).
The role of TRAFs in RIG-I-Like Receptor signaling
RIG-I-Like receptors (RLRs) are a family of DEAD box helicases that play a crucial role in the innate immune response to viral infections by detecting the presence of viral RNA in the cytosol. RIG-I and MDA5 are the two prototypical members of the RLR family (63). Upon sensing viral RNAs, RLRs dimerize and interact with mitochondrial antiviral signaling adaptor (MAVS, a.k.a. IPS-1, or VISA), with the subsequent formation of a complex that includes among others TRAF2, TRAF3, TRAF5, and TRAF6 [(64); (65), p. 3; (66)]. TRAF proteins then recruit various downstream signaling proteins that culminate in the activation of several transcription factors, including IRF3, NF-κB, and MAPKs.
RIG-I/MDA5 employ TRAF3 to induce IRF3-mediated IFN-I production. Mechanistically, TRAF3 is recruited by MAVS, where it catalyzes its own K63 polyubiquitination followed by recruitment and subsequent activation of TBK1 and IKKε (Figure 1B) (67). TRAF2 and TRAF5 play a crucial role in mediating NF-κB activation after RLRs bind their viral PAMPs, albeit the mechanism remains poorly understood (64, 68). TRAF6 can also be recruited via MAVS, where it activates the TBK1/IKKε/IRF7 as well as the MAPKs/AP-1 signaling axes (Figure 2) (66, 69). Intriguingly, the RIG-I-MAVS-TRAF6 signaling axis leads to IKKβ-dependent phosphorylation of NF-κB (70). Furthermore, RIG-I-MAVS-TRAF6 signaling induces K63-ubiquitination of Beclin-1, a critical step for inducing autophagy (71). Finally, TRAF6 interacts with Ubiquitin-specific protease 4 (USP4) to induce NF-κB activation following RLR-simulation (72). This is achieved via targeting of TRAF6 for K48-linked deubiquitination.
Regulation of RLR Signaling by Modulating TRAF Function or Its Interactions
During bacterial infections, the E3 ligase HCTD3 adds K63-linked ubiquitin chains to TRAF3, which enhances the activation of the TBK1/IKKε complex and subsequent production of IFN-Is (73). Conversely, several deubiquitinases, including OTUB1, OTUB2, DUBA, and HSCARG, have been shown to downregulate RLR-mediated IFN-I production by removing K63-linked polyubiquitin chains from TRAF3 or TRAF6 [(74, 75), p. 1; (76)]. MCPIP1, which is known to inhibit JNK and NF-κB signaling by deubiquitinating several TRAFs [see above; (31)], has been recently shown to negatively regulate IFNβ production. Overexpression studies showed that MCPIP1 disrupts the interaction between TRAF3, TBK1, and IKKε, as shown by co-immunoprecipitation, and thereby inhibiting the phosphorylation and translocation of IRF3 into the nucleus (77). There was no evidence to show that this process requires the deubiquitinase activity of MCPIP1. Another deubiquitinase, OTU deubiquitinase 1 (OTUD1), has also been demonstrated to attenuate IFN-I production following RIG-I activation by viral RNAs (78). Mechanistically, OTUD1 deubiquitinates and stabilizes the ubiquitin ligase, Smurf1, which then targets the MAVS/TRAF3/TRAF6 signalosome by mediating K48-linked polyubiquitination and the subsequent degradation of MAVS, TRAF3, and TRAF6 (78). Parkin is another ubiquitin ligase that targets RLR signaling by promoting K48-linked polyubiquitination of TRAF3 and reducing its stability (79). An interesting study demonstrated that linear ubiquitination of NEMO promotes its interaction with TRAF3, which in turn, disrupts the recruitment of TRAF3 to the RIG-I/MAVS complex leading to diminished IFN-I expression (80).
The role of TRAFs in STING signaling
In addition to cytosolic sensors of RNA, DNA sensors in the cytosol are equally crucial in detecting and mounting an inflammatory response against viral and bacterial pathogens. Stimulator of Interferon Genes (STING) is activated directly by second messengers like bacterial cyclic dinucleotides (e.g., c-diAMP and c-diGMP) (81–83) or by cellular cyclic GMP-AMP (cGAMP), which is produced by cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS) upon sensing cytosolic DNA (84). Activation of STING leads to an effective inflammatory response which include the activation of the TBK-1/IRF3 and NF-κB axes. TRAF3 and TRAF6 have been shown to enhance STING-mediated NF-κB and IFN-β promoter activity, albeit in an overexpression system in 293 cells (85). Both TRAF3 and TRAF6 appear to interact with STING (85, 86). Recently, an alternative STING pathway has been revealed in keratinocytes in response to DNA damage. This alternative STING signaling complex includes the tumor suppressor p53 and TRAF6, whereby TRAF6 catalyzes K63-polyubiquitination of STING and activates NF-κB (87). An elegant study by Genhong Cheng's group recently showed that the alternative NF-κB inducing kinase (NIK) can associate with STING and enhance its activation via an alternative NF-κB pathway-independent mechanism (88). Interestingly, they showed that TRAF3, unlike its positive role in RNA-induced IFN-I production, plays an opposite role in the DNA pathway and inhibit IFN-I production by suppressing NIK expression (88).
Perspectives and Future Directions
There continues to be a great interest in understanding how TRAFs regulate innate immune signaling. Specifically, novel mechanisms have been recently identified to regulate TLR, NLR, and RLR pathways by modulating the ubiquitination status of TRAFs, their stability or their function. However, as most of these regulators seem to be non-redundant, investigating additional novel regulators, and their mechanism of action remains an active area of investigation.
Each individual TRAF protein plays several, sometimes contradictory, roles that are pathway and/or cell specific. For example, a particular TRAF protein might induce lymphocyte survival and maturation while inhibiting a certain inflammatory pathway. Furthermore, most TRAFs function as E3 ligases as well as adapter proteins. Therefore, TRAFs are poor candidates for novel therapies since targeting a TRAF protein could lead to unintended consequences. For these reasons, future studies should focus on assessing the various roles of each TRAF protein in isolation from its other functions. This is especially important when designing therapies for complex inflammatory and autoimmune diseases by targeting TRAFs.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication. BD, FA and AA-S wrote the text. A-AS edited the manuscript and conceptualized the figures. ZA-S created the figures.
Funding
Our research in this area is funded by the Banting Research Foundation, Grant # 2018-1395 and York University Startup funds.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Arch RH, Gedrich RW, Thompson CB. Tumor necrosis factor receptor-associated factors (TRAFs)—a family of adapter proteins that regulates life and death. Genes Dev. (1998) 12:2821–30. doi: 10.1101/gad.12.18.2821
2. Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell (1994) 78:681–92. doi: 10.1016/0092-8674(94)90532-0
3. Chung JY, Park YC, Ye H, Wu H. All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci. (2002) 115:679–88.
4. Wajant H, Henkler F, Scheurich P. The TNF-receptor-associated factor family: scaffold molecules for cytokine receptors, kinases and their regulators. Cell Signal. (2001) 13:389–400. doi: 10.1016/S0898-6568(01)00160-7
5. Lalani AI, Zhu S, Gokhale S, Jin J, Xie P. TRAF molecules in inflammation and inflammatory diseases. Curr Pharmacol Rep. (2018) 4:64–90. doi: 10.1007/s40495-017-0117-y
6. Xie P. TRAF molecules in cell signaling and in human diseases. J Mol Signal. (2013) 8:7. doi: 10.1186/1750-2187-8-7
7. Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol. (2009) 27:621–68. doi: 10.1146/annurev.immunol.25.022106.141627
8. Kondo T, Kawai T, Akira S. Dissecting negative regulation of toll-like receptor signaling. Trends Immunol. (2012) 33:449–58. doi: 10.1016/j.it.2012.05.002
9. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
10. Lin Q, Li M, Fang D, Fang J, Su SB. The essential roles of Toll-like receptor signaling pathways in sterile inflammatory diseases. Int Immunopharmacol. (2011) 11:1422–32. doi: 10.1016/j.intimp.2011.04.026
11. Ostuni R, Zanoni I, Granucci F. Deciphering the complexity of Toll-like receptor signaling. Cell Mol Life Sci. (2010) 67:4109–34. doi: 10.1007/s00018-010-0464-x
12. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
13. Keating SE, Bowie AG. Role of non-degradative ubiquitination in interleukin-1 and toll-like receptor signaling. J Biol Chem. (2009) 284:8211–5. doi: 10.1074/jbc.R800038200
14. Kollewe C, Mackensen A-C, Neumann D, Knop J, Cao P, Li S, et al. Sequential autophosphorylation steps in the interleukin-1 receptor-associated kinase-1 regulate its availability as an adapter in interleukin-1 signaling. J Biol Chem. (2004) 279:5227–36. doi: 10.1074/jbc.M309251200
15. Hu L, Xu J, Xie X, Zhou Y, Tao P, Li H, et al. Oligomerization-primed coiled-coil domain interaction with Ubc13 confers processivity to TRAF6 ubiquitin ligase activity. Nat Commun. (2017) 8:814. doi: 10.1038/s41467-017-01290-0
16. Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell (2000) 103:351–61. doi: 10.1016/S0092-8674(00)00126-4
17. Xia Z-P, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, et al. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature (2009) 461:114–9. doi: 10.1038/nature08247
18. Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature (2011) 471:591–6. doi: 10.1038/nature09816
19. Ikeda F, Deribe YL, Skanland SS, Stieglitz B, Grabbe C, Franz-Wachtel M, et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature (2011) 471:637–41. doi: 10.1038/nature09814
20. Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata S, et al. SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature (2011) 471:633–6. doi: 10.1038/nature09815
21. Rittinger K, Ikeda F. Linear ubiquitin chains: enzymes, mechanisms and biology. Open Biol. (2017) 7:170026. doi: 10.1098/rsob.170026
22. Häcker H, Tseng P-H, Karin M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol. (2011) 11:457–68. doi: 10.1038/nri2998
23. Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines., Different modes of TRAF3 ubiquitination selectively activate type I interferon and proinflammatory cytokine expression. Nat Immunol. (2010) 70:70–5. doi: 10.1038/ni.1819:10.1038/ni.1819
24. Matsuzawa A, Tseng P-H, Vallabhapurapu S, Luo J-L, Zhang W, Wang H, et al. Essential cytoplasmic translocation of a cytokine receptor–assembled signaling complex. Science (2008) 321:663–8. doi: 10.1126/science.1157340
25. Jin J, Xiao Y, Hu H, Zou Q, Li Y, Gao Y, et al. Proinflammatory TLR signalling is regulated by a TRAF2-dependent proteolysis mechanism in macrophages. Nat Commun. (2015) 6:5930. doi: 10.1038/ncomms6930
26. Buchta CM, Bishop GA. TRAF5 negatively regulates Toll-like receptor signaling in B lymphocytes. J Immunol. (2014) 192:145–50. doi: 10.4049/jimmunol.1301901
27. Takeshita F, Ishii KJ, Kobiyama K, Kojima Y, Coban C, Sasaki S, et al. TRAF4 acts as a silencer in TLR-mediated signaling through the association with TRAF6 and TRIF. Eur J Immunol. (2005) 35:2477–85. doi: 10.1002/eji.200526151
28. Abdul-Sater AA, Edilova MI, Clouthier DL, Mbanwi A, Kremmer E, Watts TH. The signaling adaptor TRAF1 negatively regulates Toll-like receptor signaling and this underlies its role in rheumatic disease. Nat Immunol. (2017) 18:26–35. doi: 10.1038/ni.3618
29. Su X, Li S, Meng M, Qian W, Xie W, Chen D, et al. TNF receptor-associated factor-1 (TRAF1) negatively regulates Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF)-mediated signaling. Eur J Immunol. (2006) 36:199–206. doi: 10.1002/eji.200535415
30. Boone DL, Turer EE, Lee EG, Ahmad R-C, Wheeler MT, Tsui C, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. (2004) 5:1052–60. doi: 10.1038/ni1110
31. Liang J, Saad Y, Lei T, Wang J, Qi D, Yang Q, et al. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-kappaB signaling. J Exp Med. (2010) 207:2959–73. doi: 10.1084/jem.20092641
32. Min Y, Kim M-J, Lee S, Chun E, Lee K-Y. Inhibition of TRAF6 ubiquitin-ligase activity by PRDX1 leads to inhibition of NFKB activation and autophagy activation. Autophagy (2018) 14:1347–58. doi: 10.1080/15548627.2018.1474995
33. Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen H, et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-κB. Nat Immunol. (2012) 13:823–31. doi: 10.1038/ni.2378
34. Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-κB signaling pathways. Immunity (2011) 34:854–65. doi: 10.1016/j.immuni.2011.03.026
35. Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, et al. NLRX1 negatively regulates TLR-induced NF-κB signaling by targeting TRAF6 and IKK. Immunity (2011) 34:843–53. doi: 10.1016/j.immuni.2011.02.022
36. Ling A, Soares F, Croitoru DO, Tattoli I, Carneiro LAM, Boniotto M, et al. Post-transcriptional inhibition of luciferase reporter assays by the Nod-like receptor proteins NLRX1 and NLRC3. J Biol Chem. (2012) 287:28705–16. doi: 10.1074/jbc.M111.333146
37. Soares F, Tattoli I, Wortzman ME, Arnoult D, Philpott DJ, Girardin SE. NLRX1 does not inhibit MAVS-dependent antiviral signalling. Innate Immun. (2013) 19:438–48. doi: 10.1177/1753425912467383
38. Tattoli I, Carneiro LA, Jehanno M, Magalhaes JG, Shu Y, Philpott DJ, et al. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. (2008) 9:293–300. doi: 10.1038/sj.embor.7401161
39. Wu C, Su Z, Lin M, Ou J, Zhao W, Cui J, et al. NLRP11 attenuates Toll-like receptor signalling by targeting TRAF6 for degradation via the ubiquitin ligase RNF19A. Nat Commun. (2017) 8:1977. doi: 10.1038/s41467-017-02073-3
40. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity (2012) 36:742–54. doi: 10.1016/j.immuni.2012.03.012
41. Coutermarsh-Ott S, Eden K, Allen IC. Beyond the inflammasome: regulatory NOD-like receptor modulation of the host immune response following virus exposure. J Gen Virol. (2016) 97:825–38. doi: 10.1099/jgv.0.000401
42. Elinav E, Strowig T, Henao-Mejia J, Flavell RA. Regulation of the antimicrobial response by NLR proteins. Immunity (2011) 34:665–79. doi: 10.1016/j.immuni.2011.05.007
43. Kersse K, Bertrand MJM, Lamkanfi M, Vandenabeele P. NOD-like receptors and the innate immune system: coping with danger, damage and death. Cytokine Growth Factor Rev. (2011) 22:257–76. doi: 10.1016/j.cytogfr.2011.09.003
44. Tigno-Aranjuez JT, Abbott DW. Ubiquitination and phosphorylation in the regulation of NOD2 signaling and NOD2-mediated disease. BBA Mol Cell Res. (2012) 1823:2022–8. doi: 10.1016/j.bbamcr.2012.03.017
45. Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol. (2014) 14:9–23. doi: 10.1038/nri3565
46. Brodsky IE, Monack D. NLR-mediated control of inflammasome assembly in the host response against bacterial pathogens. Semin Immunol. (2009) 21:199–207. doi: 10.1016/j.smim.2009.05.007
47. Motta V, Soares F, Sun T, Philpott DJ. NOD-like receptors: versatile cytosolic sentinels. Physiol Rev. (2015) 95:149–78. doi: 10.1152/physrev.00009.2014
48. Bertrand MJM, Doiron K, Labbé K, Korneluk RG, Barker PA, Saleh M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity (2009) 30:789–801. doi: 10.1016/j.immuni.2009.04.011
49. Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Núñez G, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-κB activation. EMBO J. (2008) 27:373–83. doi: 10.1038/sj.emboj.7601962
50. Yang Y, Yin C, Pandey A, Abbott D, Sassetti C, Kelliher MA. NOD2 pathway activation by MDP or Mycobacterium tuberculosis infection involves the stable polyubiquitination of Rip2. J Biol Chem. (2007) 282:36223–9. doi: 10.1074/jbc.M703079200
51. Abbott DW, Yang Y, Hutti JE, Madhavarapu S, Kelliher MA, Cantley LC. Coordinated regulation of toll-like receptor and NOD2 signaling by K63-linked polyubiquitin chains. Mol Cell Biol. (2007) 27:6012–25. doi: 10.1128/MCB.00270-07
52. Zhong X, Chen B, Yang L, Yang Z. Molecular and physiological roles of the adaptor protein CARD9 in immunity. Cell Death Dis. (2018) 9:52. doi: 10.1038/s41419-017-0084-6
53. Watanabe T, Asano N, Kitani A, Fuss IJ, Chiba T, Strober W. Activation of type I IFN signaling by NOD1 mediates mucosal host defense against Helicobacter pylori infection. Gut Microbes (2011) 2:61–5. doi: 10.4161/gmic.2.1.15162
54. Chen G, Shaw MH, Kim Y-G, Nuñez G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol. (2009) 4:365–98. doi: 10.1146/annurev.pathol.4.110807.092239
55. Abdul Sater A, Philpott D. Inflammasomes. In: Ratcliffe MJH, editor. Encyclopedia of Immunobiology. Vol. 2. Toronto, ON: University of Toronto and Trinity College (2016). p. 447–53.
56. Abdul-Sater AA, Said-Sadier N, Ojcius DM, Yilmaz O, Kelly KA. Inflammasomes bridge signaling between pathogen identification and the immune response. Drugs Today (2009) 45 (Suppl. B) 105–112.
57. Guan K, Wei C, Zheng Z, Song T, Wu F, Zhang Y, et al. MAVS promotes inflammasome activation by targeting ASC for K63-linked ubiquitination via the E3 ligase TRAF3. J Immunol. (2015) 194:4880–90. doi: 10.4049/jimmunol.1402851
58. Labbé K, McIntire CR, Doiron K, Leblanc PM, Saleh M. cellular inhibitors of apoptosis proteins cIAP1 and cIAP2 are required for efficient Caspase-1 activation by the inflammasome. Immunity (2011) 35:897–907. doi: 10.1016/j.immuni.2011.10.016
59. Vince JE, Wong WW-L, Gentle I, Lawlor KE, Allam R, O'Reilly L, et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity (2012) 36:215–27. doi: 10.1016/j.immuni.2012.01.012
60. Xing Y, Yao X, Li H, Xue G, Guo Q, Yang G, et al. Cutting edge: TRAF6 mediates TLR/IL-1R signaling–induced nontranscriptional priming of the NLRP3 inflammasome. J Immunol. (2017) 199:1561–6. doi: 10.4049/jimmunol.1700175
61. Marinis JM, Hutti JE, Homer CR, Cobb BA, Cantley LC, McDonald C, et al. IκB kinase α phosphorylation of TRAF4 downregulates innate immune signaling. Mol Cell Biol. (2012) 32:2479–89. doi: 10.1128/MCB.00106-12
62. Eren E, Berber M, Özören N. NLRC3 protein inhibits inflammation by disrupting NALP3 inflammasome assembly via competition with the adaptor protein ASC for pro-caspase-1 binding. J Biol Chem. (2017) 292:12691–701. doi: 10.1074/jbc.M116.769695
63. Yoo J-S, Kato H, Fujita T. Sensing viral invasion by RIG-I like receptors. Curr Opin Microbiol. (2014) 20:131–8. doi: 10.1016/j.mib.2014.05.011
64. Mikkelsen SS, Jensen SB, Chiliveru S, Melchjorsen J, Julkunen I, Gaestel M, et al. RIG-I-mediated activation of p38 MAPK is essential for viral induction of interferon and activation of dendritic cells: dependence on TRAF2 and TAK1. J Biol Chem. (2009) 284:10774–82. doi: 10.1074/jbc.M807272200
65. Saha SK, Pietras EM, He JQ, Kang JR, Liu SY, Oganesyan G, et al. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. (2006) 25:3257–63. doi: 10.1038/sj.emboj.7601220
66. Yoshida R, Takaesu G, Yoshida H, Okamoto F, Yoshioka T, Choi Y, et al. TRAF6 and MEKK1 play a pivotal role in the RIG-I-like helicase antiviral pathway. J Biol Chem. (2008) 283:36211–20. doi: 10.1074/jbc.M806576200
67. Loo YM, Gale M. Immune signaling by RIG-I-like receptors. Immunity (2011) 34:680–92. doi: 10.1016/j.immuni.2011.05.003
68. Tang ED, Wang C-Y. TRAF5 is a downstream target of MAVS in antiviral innate immune signaling. PLoS ONE (2010) 5:e9172. doi: 10.1371/journal.pone.0009172
69. Konno H, Yamamoto T, Yamazaki K, Gohda J, Akiyama T, Semba K, et al. TRAF6 establishes innate immune responses by activating NF-kappaB and IRF7 upon sensing cytosolic viral RNA and DNA. PLoS ONE (2009) 4:e5674. doi: 10.1371/journal.pone.0005674
70. Yoboua F, Martel A, Duval A, Mukawera E, Grandvaux N. Respiratory syncytial virus-mediated NF-kappa B p65 phosphorylation at serine 536 is dependent on RIG-I, TRAF6, and IKK beta. J Virol. (2010) 84:7267–77. doi: 10.1128/JVI.00142-10
71. Lee NR, Ban J, Lee NJ, Yi CM, Choi JY, Kim H, et al. Activation of RIG-I-mediated antiviral signaling triggers autophagy through the MAVS-TRAF6-Beclin-1 signaling axis. Front Immunol. (2018) 9:2096. doi: 10.3389/fimmu.2018.02096
72. Xu C, Peng Y, Zhang Q, Xu XP, Kong XM, Shi WF. USP4 positively regulates RLR-induced NF-κB activation by targeting TRAF6 for K48-linked deubiquitination and inhibits enterovirus 71 replication. Sci Rep. (2018) 8:13418. doi: 10.1038/s41598-018-31734-6
73. Li F, Li Y, Liang H, Xu T, Kong Y, Huang M, et al. HECTD3 mediates TRAF3 polyubiquitination and type I interferon induction during bacterial infection. J Clin Invest. (2018) 128:4148–62. doi: 10.1172/JCI120406
74. Kayagaki N, Phung Q, Chan S, Chaudhari R, Quan C, O'Rourke KM, et al. DUBA: a deubiquitinase that regulates type I interferon production. Science (2007) 318:1628–32. doi: 10.1126/science.1145918
75. Li S, Zheng H, Mao A-P, Zhong B, Li Y, Liu Y, et al. Regulation of virus-triggered signaling by OTUB1- and OTUB2-mediated deubiquitination of TRAF3 and TRAF6. J Biol Chem. (2010) 285:4291–7. doi: 10.1074/jbc.M109.074971
76. Peng Y, Xu R, Zheng X. HSCARG negatively regulates the cellular antiviral RIG-I like receptor signaling pathway by inhibiting TRAF3 ubiquitination via recruiting OTUB1. PLoS Pathog. (2014) 10:e1004041. doi: 10.1371/journal.ppat.1004041
77. Chen X, Zhao Q, Xie Q, Xing Y, Chen Z. MCPIP1 negatively regulate cellular antiviral innate immune responses through DUB and disruption of TRAF3-TBK1-IKKε complex. Biochem Biophys Res Commun. (2018) 503:830–6. doi: 10.1016/j.bbrc.2018.06.083
78. Zhang L, Liu J, Qian L, Feng Q, Wang X, Yuan Y, et al. Induction of OTUD1 by RNA viruses potently inhibits innate immune responses by promoting degradation of the MAVS/TRAF3/TRAF6 signalosome. PLoS Pathog. (2018) 14:e1007067. doi: 10.1371/journal.ppat.1007067
79. Xin D, Gu H, Liu E, Sun Q. Parkin negatively regulates the antiviral signaling pathway by targeting TRAF3 for degradation. J Biol Chem. (2018) 293:11996–2010. doi: 10.1074/jbc.RA117.001201
80. Belgnaoui SM, Paz S, Samuel S, Goulet ML, Sun Q, Kikkert M, et al. Linear ubiquitination of NEMO negatively regulates the interferon antiviral response through disruption of the MAVS-TRAF3 complex. Cell Host Microbe (2012) 12:211–22. doi: 10.1016/j.chom.2012.06.009
81. Abdul-Sater AA, Grajkowski A, Erdjument-Bromage H, Plumlee C, Levi A, Schreiber MT, et al. The overlapping host responses to bacterial cyclic dinucleotides. Microbes Infect. (2012) 14:188–97. doi: 10.1016/j.micinf.2011.09.002
82. Abdul-Sater AA, Tattoli I, Jin L, Grajkowski A, Levi A, Koller BH, et al. Cyclic-di-GMP and cyclic-di-AMP activate the NLRP3 inflammasome. EMBO Rep. (2013) 14:900–6. doi: 10.1038/embor.2013.132
83. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature (2011) 478:515–8. doi: 10.1038/nature10429
84. Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. (2018) 215:1287–99. doi: 10.1084/jem.20180139
85. Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol. (2014) 88:5328–41. doi: 10.1128/JVI.00037-14
86. Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity (2008) 29:538–50. doi: 10.S1074-7613(08)00406-8[pii]10.1016/j.immuni.2008.09.003
87. Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol Cell (2018) 71:745–760.e5. doi: 10.1016/j.molcel.2018.07.034
Keywords: inflammation, innate immunity, TRAF, TLRs, NLR, RLR, STING, TNFR
Citation: Dhillon B, Aleithan F, Abdul-Sater Z and Abdul-Sater AA (2019) The Evolving Role of TRAFs in Mediating Inflammatory Responses. Front. Immunol. 10:104. doi: 10.3389/fimmu.2019.00104
Received: 02 October 2018; Accepted: 14 January 2019;
Published: 04 February 2019.
Edited by:
Angela Ianaro, University of Naples Federico II, ItalyReviewed by:
Shi Yue, University of Southern California, United StatesMichael Paul Gantier, Hudson Institute of Medical Research, Australia
Copyright © 2019 Dhillon, Aleithan, Abdul-Sater and Abdul-Sater. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ali A. Abdul-Sater, YWFzYXRlckB5b3JrdS5jYQ==
†These authors have contributed equally to this work