 Marion Mussbacher1†
Marion Mussbacher1† Manuel Salzmann1†
Manuel Salzmann1† Christine Brostjan2
Christine Brostjan2 Bastian Hoesel1
Bastian Hoesel1 Christian Schoergenhofer3
Christian Schoergenhofer3 Hannes Datler1
Hannes Datler1 Philipp Hohensinner4
Philipp Hohensinner4 José Basílio1
José Basílio1 Peter Petzelbauer5
Peter Petzelbauer5 Alice Assinger1
Alice Assinger1 Johannes A. Schmid1*
Johannes A. Schmid1*- 1Institute of Vascular Biology and Thrombosis Research, Medical University of Vienna, Vienna, Austria
- 2Department of Surgery, General Hospital, Medical University of Vienna, Vienna, Austria
- 3Department of Clinical Pharmacology, Medical University of Vienna, Vienna, Austria
- 4Division of Cardiology, Department of Internal Medicine II, Medical University of Vienna, Vienna, Austria
- 5Skin and Endothelial Research Division, Department of Dermatology, Medical University of Vienna, Vienna, Austria
The transcription factor NF-κB is a central mediator of inflammation with multiple links to thrombotic processes. In this review, we focus on the role of NF-κB signaling in cell types within the vasculature and the circulation that are involved in thrombo-inflammatory processes. All these cells express NF-κB, which mediates important functions in cellular interactions, cell survival and differentiation, as well as expression of cytokines, chemokines, and coagulation factors. Even platelets, as anucleated cells, contain NF-κB family members and their corresponding signaling molecules, which are involved in platelet activation, as well as secondary feedback circuits. The response of endothelial cells to inflammation and NF-κB activation is characterized by the induction of adhesion molecules promoting binding and transmigration of leukocytes, while simultaneously increasing their thrombogenic potential. Paracrine signaling from endothelial cells activates NF-κB in vascular smooth muscle cells and causes a phenotypic switch to a “synthetic” state associated with a decrease in contractile proteins. Monocytes react to inflammatory situations with enforced expression of tissue factor and after differentiation to macrophages with altered polarization. Neutrophils respond with an extension of their life span—and upon full activation they can expel their DNA thereby forming so-called neutrophil extracellular traps (NETs), which exert antibacterial functions, but also induce a strong coagulatory response. This may cause formation of microthrombi that are important for the immobilization of pathogens, a process designated as immunothrombosis. However, deregulation of the complex cellular links between inflammation and thrombosis by unrestrained NET formation or the loss of the endothelial layer due to mechanical rupture or erosion can result in rapid activation and aggregation of platelets and the manifestation of thrombo-inflammatory diseases. Sepsis is an important example of such a disorder caused by a dysregulated host response to infection finally leading to severe coagulopathies. NF-κB is critically involved in these pathophysiological processes as it induces both inflammatory and thrombotic responses.
General Links Between Inflammation and Thrombosis
The close association of inflammatory conditions and coagulatory processes has an evolutionary origin, as injuries require both an efficient blood clotting and an inflammatory immune response against invading pathogens. In this review we focus on the cellular interactions that link inflammation with thrombotic processes, while the plasmatic coagulation cascade is described elsewhere (1, 2). Platelets are the first functional elements that seal damaged blood vessels upon injury by forming aggregates and a subsequent thrombus. They are also the first immunomodulatory cells at the side of injury and inflammation, providing a functional link between host response and coagulation (3). Endothelial cells in an inactivated, quiescent state express potent inhibitors of coagulation and platelet aggregation. However, upon inflammatory stimuli they change their cellular program by expressing leukocytes adhesion molecules to facilitate their entry to sites of inflammation. In addition, they undergo a transition toward a more pro-coagulatory phenotype (4). Furthermore, chronic inflammation causes a phenotypic switch of vascular smooth muscle cells from a contractile to a synthetic phenotype, which is associated with secretion of pro-inflammatory mediators and which can finally result in a macrophage-like state (5). Other cells of the circulation and vasculature are altered by inflammatory conditions toward a pro-thrombotic state, as well. Monocytes and neutrophils contribute to coagulation by expression of tissue factor (6, 7), which is upregulated upon inflammation. Moreover, in their activated state, neutrophils are capable of expelling their DNA in conjunction with histones and other associated proteins thereby forming extracellular DNA designated as neutrophil extracellular traps (NETs), which exert antibacterial functions, but also induce a strong coagulatory response (8). Recent findings indicate that these processes are also a physiological part of an intravascular immunity especially in capillaries causing clinically unnoticed forms of micro-thrombosis that are termed immuno-thrombosis and which have the purpose of immobilizing invaded pathogens (9).
While both physiological hemostasis and immuno-thrombosis represent a normal response to traumas or invading microorganisms, any deregulation of these processes can lead to aberrant intravascular coagulation and a pathological obstruction of the blood flow, which is generally defined as thrombosis. This is often seen in acute inflammatory states, with sepsis representing a clinically weighty example, where patients suffer from anomalous systemic inflammation that is associated with alterations in blood coagulation and microvessel thrombosis in diverse organs (10). Furthermore, the interplay between endothelial cells, smooth muscle cells, platelets, and leukocytes becomes critical under chronic inflammatory conditions, which are a central cause in the pathogenesis of atherosclerosis driving vascular remodeling and plaque formation. Rupture or erosion of the plaques can then cause rapid thrombosis and occlusion of blood vessels that finally leads to myocardial infarction or stroke, the two major reasons of mortality worldwide. Therefore, understanding of the complex interaction between the distinct cell types in inflammation and thrombosis is necessary for prevention or treatment of cardiovascular diseases.
The Transcription Factor NF-κB and its Inhibitors
NF-κB is a central mediator of inflammation and thus fundamentally involved in the molecular links between inflammatory and thrombotic processes. It was first described in 1986 as transcription factor driving the expression of the κ-chain of immunoglobulins in B-cells (11). Thus, the commonly used abbreviation NF-κB stands for: Nuclear Factor of the κ-chain in B-cells. While the name insinuates that this protein is specific for B cells, with the κ-IgG chain being the most important target gene, it is now clear that it is expressed in nearly all cells of the human body and that it regulates the expression of hundreds or thousands of genes (12) involved in a great variety of biological processes. Not even the designation “nuclear” is correct, as this transcription factor is mostly located in the cytosol, as long as it is bound to one of its inhibitors in non-activated cells. Furthermore, NF-κB is not a single factor as implied by the name, but actually a protein family consisting of five members, building homo- or heterodimers via their Rel-homology domain, which is also responsible for DNA binding (Figure 1). Two of the family members (p100 and p105) contain inhibitory domains consisting of ankyrin repeats, which block binding to DNA and constrain nuclear localization. These have to be proteolytically processed by proteasomes for activation of NF-κB and binding to enhancer elements in the promoter regions of target genes (14–16). The processed forms of p100 and p105 (p52 and p50, respectively), do not contain a transactivation domain and need to dimerize with one of the other three family members, RelA (p65), RelB, or c-Rel to function as transcription factors. Dimers of p50 and p52 operate as transcriptional repressors, as they can bind to promoter elements without activation of the transcriptional machinery (17). The other three NF-κB proteins: p65 (RelA), RelB, and c-Rel do not contain these inhibitory domains. However, they bind to inhibitory molecules of the IκB family, which contain ankyrin repeats similar to the inhibitory domains of p100 and p105 and which have to be degraded for release and activation of the transcription factor (17). One of the most common NF-κB forms is a dimer of p65 bound to p50—the processed form of p105, with the dimer again being inactivated by association with a member of the IκB family. Binding of IκB alters the conformation of NF-κB dimers and prevents their association with DNA (18, 19) (Figure 2). Furthermore, it shifts the preferential localization from the nucleus to the cytosol. However, in contrast to the picture that is drawn in most textbooks, IκB molecules do not completely prevent translocation of NF-κB into the nucleus, as a vivid shuttling of NF-κB between cytosol and nucleus can be observed even in the presence of normal levels of IκB—with a halftime of about 7–14 min (21–23). Studies with fluorescently tagged p65 and IκB molecules in non-activated cells revealed that the concentration of nuclear p65 is about 5% of the cytosolic one (21). The basis for this phenomenon seems to be the fact that NF-κB/IκB complexes like most macromolecular complexes are subject to dissociation and re-association, with a certain number of unbound molecules under steady state conditions, which can then be recognized by the nuclear import machinery and translocated to the nucleus. As a consequence of this nucleocytoplasmic shuttling and the dynamics of binding, a low level of NF-κB activity is predicted even in non-activated cells (24). Thus, elevated levels of NF-κB molecules as observed in chronic inflammatory states can contribute to an increased risk of thrombosis even if inhibitory molecules are present.
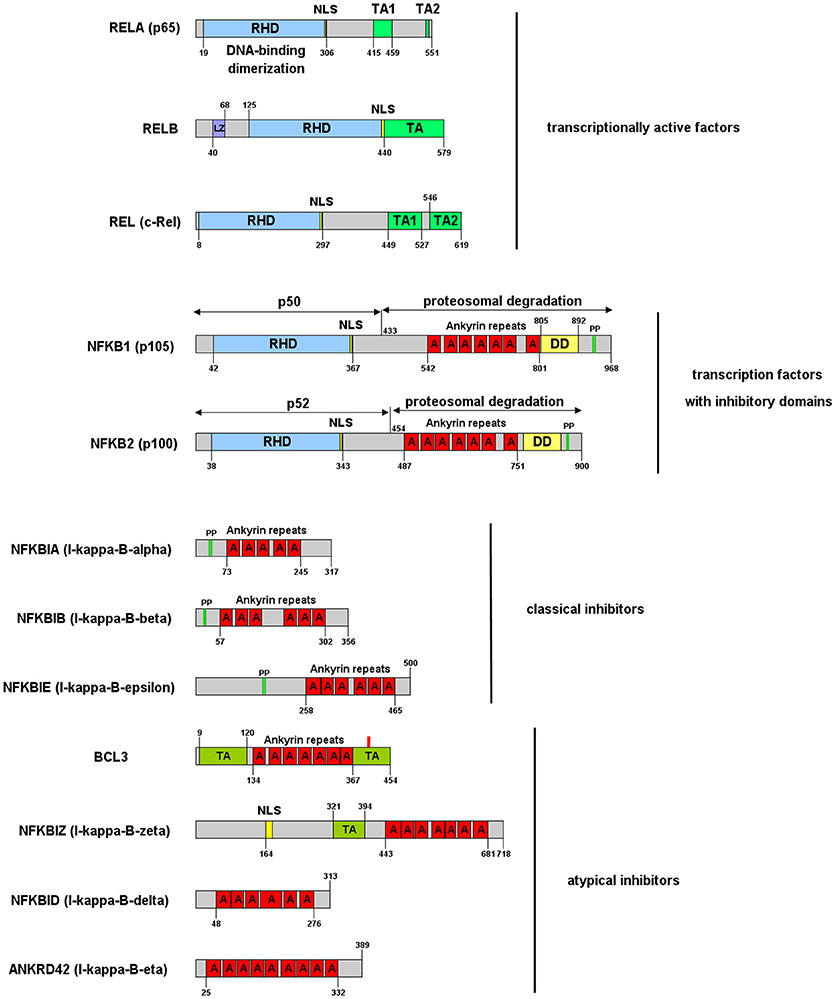
Figure 1. NF-κB and IκB family of proteins and their functional domains. The proteins are designated by their gene symbols with frequently used aliases in brackets. RHD, Rel-homology domain, responsible for DNA binding and dimerization; TA, transactivation domain, responsible for binding of the transcriptional machinery and RNA-polymerase; LZ, leucine zipper; NLS, nuclear localization domain; A, ankyrin repeat; DD, death domain; PP, double-phosphorylation by IκB kinases triggering ubiquitination and proteasomal degradation or processing (in case of NFKB1 and NFKB2). The numbers specify the amino acid borders of domains for human isoforms. Atypical inhibitors are described in more detail in Pettersen et al. (13).
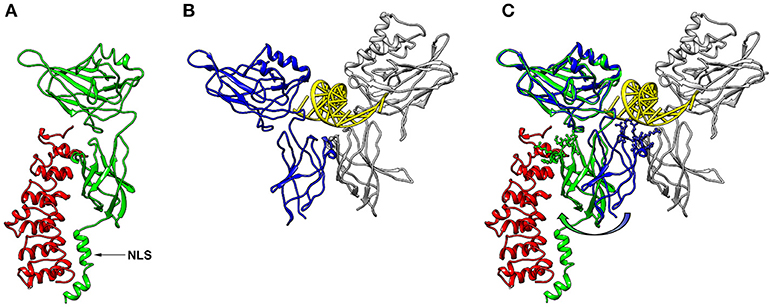
Figure 2. 3D-structures of NF-κB/IκBα and NF-κB/DNA. (A) 3D-Model of a p65-NF-κB fragment (green; amino acids 20–320) bound to IκB (red, amino acids 70–282) generated with Chimera software (20) using the protein database file 1NFI. The position of the nuclear localization sequence (NLS) of p65 is indicated with an arrow. (B) Conformation of a p65 fragment (blue, amino acids 20–291) bound to DNA (yellow) and p50 (gray; amino acids 39–350) forming a characteristic butterfly-like structure (protein database file 1VKX). The p65-fragment, which was crystalized for this structure, lacks the last 29 amino acids of the corresponding structure of (A), but is shown from the same perspective. (C) Superimposed structures of (A, B), illustrating the conformational switch of p65 between the IκB- and the DNA-bound form (green and blue, respectively). The amino acid side chains of the lower p65 wing, which come closer than 0.5 nm to the DNA in the DNA-bound form, are shown in ball-and-stick manner. These side chains are turned away in the IκB-bound form as depicted with an arrow.
NF-κB Signaling Pathways
After the discovery of NF-κB as a crucial transcription factor in inflammation and immunity, great efforts have been undertaken to elucidate the signaling pathways by which it is activated. Quite soon it became clear that NF-κB activity is not only triggered by inflammatory cytokines such as tumor nerosis factor alpha (TNFα) or interleukin 1 (IL-1), but also by bacterial cell wall components like lipopolysaccharides, by viruses and even by physical stress conditions such as gamma- or UV-irradiation (see Table 1 for a more extended list of activating stimuli). The detailed clarification of the receptors that sense the original triggers and the components that transmit and modulate these signals inside the cell took many years and involved the work of numerous research groups [for a review see: (72)]. The variety of individual activation pathways became quite confusing throughout the years, so that some structuring was proposed to group the signaling cascades in a logical way. Since then, most researchers classify the activation in (i) the classical (or canonical) pathway, which is triggered by TNFα, IL-1, or lipopolysaccharide (LPS); (ii) a non-classical (non-canonical or alternative) activation elicited by CD40 ligand (CD40L) or lymphotoxin β (LTbeta); and (iii) atypical signaling pathways such as that initiated by DNA-damage (Figure 3). Yet, it has to be stated that this classification is arbitrary and should not lead to a dogmatic view of NF-κB activation. Furthermore, there appears to be a non-genomic pathway of NF-κB signaling molecules, which will be discussed in the platelet section. Moreover, it has recently been shown that stimulation of the alternative pathway can also activate components of the classical pathway and that the transcriptional responses can be qualitatively very similar (73).
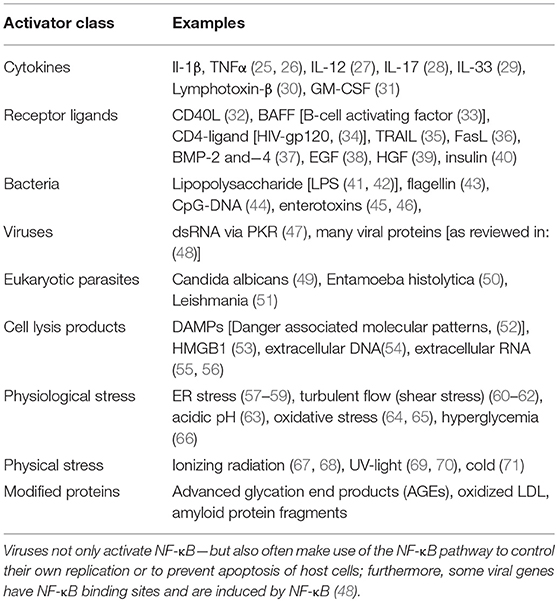
Table 1. Important activators of NF-κB.
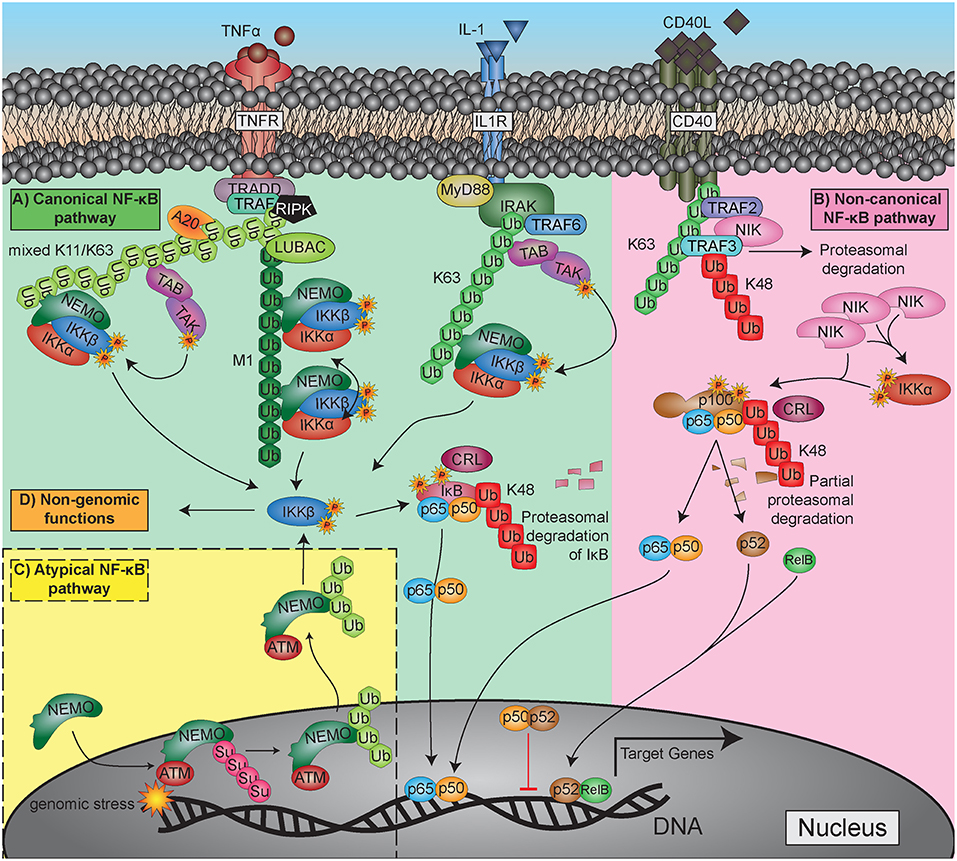
Figure 3. Major NF-κB activation pathways. (A) Canonical (classical) pathway, upper left side: exemplified by TNFα and IL-1 triggered reactions. (B) Non-canonical (alternative, non-classical) pathway, upper-right side: represented with the CD40L-activated pathway. (C) Atypical NF-κB activation pathway triggered by genotoxic stress: lower left side. For more detailed explanation see the text.
Activating ligands usually trigger a conformational change or an oligomerization of receptors, which generates a binding surface for intracellular adaptor proteins. These are then recruiting E3-type ubiquitin-ligases (TRAF and IAP-proteins), which transfer a polyubiquitin chain that has been built up by E1 (ubiquitin-activating) and E2 (ubiquitin-conjugating) enzymes to target proteins such as RIPK1 or other TRAF and IAP proteins. The polyubiquitin-chains that are formed by these enzymes, are linked via lysine-63 (K63) or lysine-11 (K11), creating a structure that serves as binding and signaling platform for downstream adaptor molecules (TAB1/2 and NEMO) and their associated kinases (TAK1 and IκB kinases, IKKα, and IKKβ). Another polyubiquitination structure that activates the NF-κB signaling pathway is formed by linking the carboxy-terminus of a ubiquitin molecule with the amino-terminus of the starting methionine (M1) on the next ubiquitin moiety. This reaction is catalyzed by an enzyme complex termed LUBAC (linear ubiquitin chain assembly complex) and acts primarily on the adapter molecule NEMO or on other polyubiquitin chains as substrates (74–78). Altogether, K11-, K63-, and M1-type polyubiquitin chains generate signaling platforms via adapter molecules and clustering processes, which allow phosphorylation of IκB kinases on their activation loops. This occurs either by upstream signaling kinases such as TAK1 or by proximity-induced auto-phosphorylation in a way, that one IKK molecule activates another one in a dimer or oligomer, a process, which is commonly termed as transphosphorylation (79). Finally, most of the NF-κB-activating signaling pathways converge at the level of IκB kinases (IKKα and IKKβ), which upon activation phosphorylate the inhibitory molecules of the IκB family or the inhibitory domains of p100 and p105 on two adjacent serine residues. This triggers another type of polyubiquitination, which is characterized by linkage of ubiquitin moieties via lysine-48 (K48) and catalyzed by E3-ligases of the CRL (Cullin-Ring ligase) type, also named SCF-type (for the key components Skp1, Cullin, and F-box protein) (80, 81). These K48-linked polyubiquitin chains are recognized by proteasome activators, leading to proteasomal degradation of the inhibitor and release of the NF-κB dimer.
Important feedback mechanisms of NF-κB regulation are found at the level of the activating K63- and M1-type polyubiquitin chains. These can be degraded by specific deubiquitinating enzymes (DUBs), primarily the molecules A20 and CYLD, which act as negative regulators of NF-κB signaling. A20 or an associated E3-ligase have furthermore the capability to catalyze K48-type polyubiquitination of RIPK1, thereby leading to proteasomal degradation of this crucial NF-κB activating effector molecule (82). For a comprehensive overview of the different ubiquitination steps see Figure 3.
The biological response following NF-κB activation is manifold and depends on the cell type, the accessibility of promoter regions, which is regulated by epigenetic mechanisms, and furthermore additional feedback pathways, which intersect with the NF-κB pathway. Basically, we have to view these processes rather as dynamic signaling networks and feedback circuits far beyond the classical cascade scheme of signaling pathways involving significant crosstalk between various upstream and downstream pathways (17), which may have additional implications on the links between inflammation and thrombosis, but which are beyond the scope of this review.
The major biological function of NF-κB is to change cellular programs in all different kinds of stress situations, so that the various cell types can respond to the stress in a way that the organism can cope with the threat, activate defense mechanisms and eliminate or escape the endangering factors with the final aim to re-gain the original physiological state (83). This major biological function of NF-κB signaling explains the various types of target genes that are upregulated or induced after NF-κB activation. As listed more comprehensively in Table 2 and illustrated in Figure 4, these target genes comprise a great variety of cytokines and chemokines, the majority of which is acting in a pro-inflammatory manner, often themselves leading to NF-κB activation and thereby constituting a positive feedback circuit. This is in line with an upregulation of many different immune and chemokine receptors (211). Another set of genes that are induced by NF-κB are adhesion molecules, which are crucial for transmigration of leukocytes through the endothelium, as well as cell-cell interactions that are important for immune defense and platelet function. At the cellular level, NF-κB activation leads to upregulation of anti-apoptotic genes, which supports cell survival under stress condition. However, the same mechanism may contribute to cancer development as high levels of anti-apoptotic genes provide a survival advantage to cells with malignant mutations, which would otherwise die or become senescent (212, 213). Moreover, NF-κB induces cyclin D proteins, which are essential for cell cycle progression (214), as well as the oncogene c-Myc, which upregulates many cell cycle proteins and which is overexpressed in a majority of cancers (215). Apart from c-Myc, various other transcription factors are induced by NF-κB, such as members of the interferon-regulatory IRF family in accordance with immune defense functions, as well as HIF-1α, GATA-3 or LEF1 demonstrating that NF-κB is capable of influencing the cellular transcriptional network in a complex manner involving numerous feedback circuits (17). Additionally, NF-κB up-regulates the transcription of various members of the NF-κB gene family, thereby creating positive feedback loops. However, these are in most cases counteracted by negative feedback mechanisms, which are induced by NF-κB as well. These include the induction of the various IκB family members, which inhibit NF-κB directly, as well as proteins that are removing the activating K63- or M1-linked polyubiquitin chains from NF-κB activating proteins such as A20 or ABIN (216). Finally, the vital role of these feedback inhibitors is to shut off NF-κB activity and to revert the cell to its inactivated state. Impairment of these processes is often the basis for chronic inflammatory diseases. The complexity of all the feedback circuits is further enhanced by NF-κB-dependent upregulation of several miRNAs, which lead to the degradation or reduced translation of many different mRNAs (199).

Table 2. Important target genes of NF-κB.
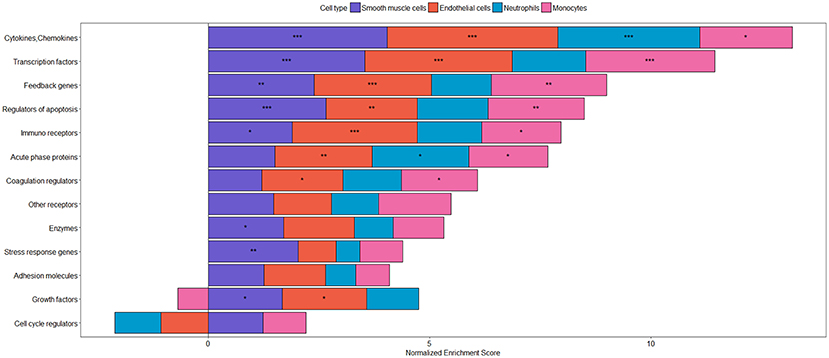
Figure 4. Categories of inflammatory target genes in different cell types. Transcriptional responses after stimulation with TNFα have been analyzed using gene set enrichment analysis (GSEA) with the following datasets: smooth muscle cells: GSE96962; endothelial cells: GSE96962; monocytes: GSE56681; neutrophils: GSE40548. Gene sets were derived from NF-κB target genes as described in Jia et al. (207). As p-value and log fold change (LogFC) are often used to evaluate significant results from differential expression analysis and the up-regulated/down-regulated genes are usually at the top and/ bottom of the ranked gene list, respectively, we used the signed z-value to rank genes, where the sign is from LogFC, as previously described (208). To assess the enrichment of the target genes of NF-kappa B gene sets in the different datasets, the GSEA Preranked tool was used (209). Gene sets showing a significant enrichment are represented by ***(FDR < 0.001), **(FDR < 0.01), and *(FDR < 0.05). The plot was produced using the R package, ggplot2 (210) visualizing the normalized enrichment scores as stacked bars showing differences in the response between different cell types of the vasculature and circulation.
Important NF-κB target genes in the context of inflammation include various enzymes such as cyclooxygenases and lipoxygenases catalyzing the formation of prostaglandins and leukotrienes, as well as NO synthases, which are important for vasodilation and blood pressure regulation (for references see Table 2).
Furthermore, a variety of coagulation factors and regulators are induced by NF-κB, including tissue factor [F3, (121)], factor VIII (122), urokinase-type plasminogen activator (uPA) (123), and Plasminogen activator inhibitor-1 (PAI-1) (124). Thus, NF-κB contributes to coagulatory events not only via cellular activation processes, but also by transcriptional induction of proteins of the plasmatic coagulation cascade. This provides another molecular explanation for the functional links between inflammation and thrombotic processes that contributes to increased cardiovascular risk in situations of acute or chronic inflammation.
Platelets as Mediators Between Inflammation and Thrombosis
Platelets, the cells that build the thrombus in primary hemostasis, are now considered crucial immune-modulatory cells providing essential functional links between inflammatory and thrombotic processes. They are small anucleate cell fragments derived from megakaryocytes with a diameter of 2–4 μm and circulate in the blood for ~7–10 days, where they patrol the endothelial wall, recognizing structures representing vessel damage. Since their discovery by Bizzozero in 1882 they are recognized for their central role in hemostasis (217), preventing blood loss upon injury by formation of platelet-platelet aggregates, which are stabilized by fibrin fibers that are formed by the plasmatic coagulation cascade (218, 219). Negative charges on the surface of activated platelets, which expose phosphatidylserine upon activation-dependent membrane lipid flip-flop, allow for calcium binding and provide the ideal surface for site-specific proteolytic activation of coagulation factors (Figure 5).
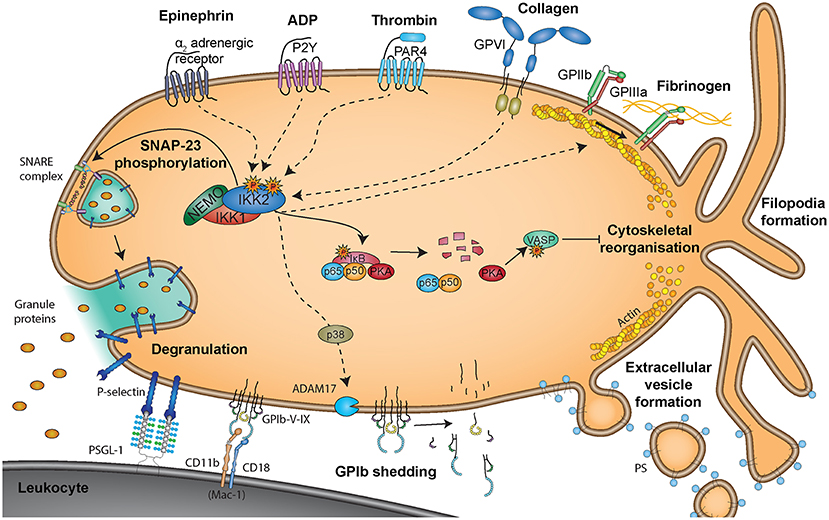
Figure 5. Non-genomic roles of NF-κB signaling molecules in platelets. Non-genomic effects of NF-κB signaling molecules are triggered via binding of epinephrine to α2 adrenergic receptors, ADP to P2Y receptors, thrombin to PAR4 receptors, collagen to glycoprotein VI (GPVI) receptors or fibrinogen to GPIIb/GPIIIa receptors. Degranulation is reported to be mediated via phosphorylation of SNAP-23 by IKK2 (251), representing a positive effect of NF-κB signaling on platelet activation. However, PKA was reported to be present in a complex with NF-κB and IκB and uncoupling of this complex upon IKK2 activation resulted in protein kinase A (PKA) activation, causing phosphorylation of vasodilator-stimulated phosphoprotein (VASP) and inhibition of platelet activity (250). Interaction of platelets with leukocytes is mediated via binding of platelet P-selectin, exposed upon degranulation, to leukocyte PSGL-1, which is supported by platelet GP-Ib-IX binding to Mac-1 on leukocytes.
More and more evidence emerges, that activated platelets not only trigger recruitment and activation of further platelets to the site of injury but that platelets also interact with leukocytes, thereby orchestrating immune responses and mediating wound healing and repair processes via interaction with the endothelium (220–222). Activated platelets and microvesicles bind leukocytes, which leads to mutual activation and rapid, local release of platelet-derived cytokines. Platelets enhance leukocyte extravasation, differentiation and cytokine release. They propagate monocyte differentiation into macrophages and modulate oxidative burst in neutrophils [reviewed in (223)]. Toll-like receptor 4 (TLR-4)-activated platelets bind to neutrophils and initiate neutrophil extracellular trap NET formation (224). Platelets mediate NET formation either via P-selectin-PSGL1 interactions (225), neutrophils integrin αLβ2 [LFA-1 (CD11a/CD18)] (226) or platelet GPIbα (227) resulting in increased bacterial clearance. Additionally, the platelet release products thromboxane (TXA2), platelet factor 4 (CXCL4), von Willebrand factor (vWF) (228), and High mobility group box 1 (HMGB1) (229) trigger NET formation. Activated platelets and platelet microvesicle further present HMGB1 to neutrophils and commit them to autophagy and NET generation, thereby potentially causing thrombo-inflammatory lesions (229–231). Additionally, cleavage of IL-1β by NLRP3-mediated activation of caspase-1 contributes to platelet activation (232) and is associated with acute thrombotic events during hypoxic conditions (233).
Platelets can be activated by vessel injury (e.g., immobilized vWF or collagen exposure) as well as thrombin, which is generated by an activated coagulation cascade. Platelets further release substances that enhance their activation e.g., adenosine diphosphate (ADP) and TXA2. However, more and more evidence emerges that also inflammatory triggers are able to activate platelets. Platelets also express functional TLRs, including TLR2, TLR3, TLR4, TLR7, and TLR9 (234–236). Binding of LPS to platelet TLR4 induces platelet activation (237) to promote microvascular thrombosis (238) as well as platelet and neutrophil sequestration into the lung, liver and spleen (239) as well as formation of NETs.
Upon activation, platelets release their granule content, which comprises over 300 factors, involved in a plethora of processes (221). Platelet dense granules contain ADP, adenosine triphosphate (ATP), serotonin and calcium ions, which are important for activation and recruitment of further platelets. Platelet α-granules contain VWF, Factor V, and Factor VIII and fibrinogen, which can further boost activation of the coagulation cascade. Other α-granule-derived molecules like CXCL4/PF4, chemokine (C-C motif) 4 (CCL4/MIP-1), chemokine (C-C motif) 5 (CCL5/RANTES), CD40L, and P-selectin (CD62P) recruit and/or activate leukocytes, while additional factors such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and transforming growth factor (TGF-β), act on endothelial cells and trigger angiogenesis and wound repair processes (220, 240).
Platelet granule exocytosis, which occurs via fusion of the granule membrane with the plasma membrane, involves a complex interplay of actin polymerization and proteins of the SNARE family (soluble N-ethylmaleimide-sensitive-factor attachment protein receptors), which reside on vesicles (v-SNAREs) and target membranes (t-SNAREs) (241). Synaptosomal-associated protein 23 (SNAP-23), a t-SNARE, is required for release from all three types of granules in platelets (241).
Despite the lack of a nucleus, platelets contain a variety of transcription factors as well as upstream signaling molecules and emerging evidence suggests that these factors trigger non-genomic effects in platelets rather than representing remnants of megakaryocytic packaging. Platelets are further able of shuttling transcription factors to other cells via shedding off transcription factor-laden microvesicles (242), which fulfill various effector functions (243).
Platelets contain the majority of NF-κB signaling proteins (244–249) and activation of the NF-κB/IKK/IκB pathway can be detected in response to platelet stimulation (245, 248–250) (Figure 5). While ADP, collagen, epinephrine, and thrombin all result in NF-κB pathway activation via phosphorylation of IκB and its proteasomal degradation (252), platelet activation in response to arachidonic acid does not seem to involve NF-κB (249). The precise signaling pathways contributing to NF-κB activation in platelets are currently unknown. In thrombin-activated platelets, activation of IκB kinases can be prevented by a neutral sphingomyelinase inhibitor or a p38 MAPK inhibitor downstream of the thrombin receptor protease activated receptor 4 (PAR4) but not PAR1 (253), indicating that these signal mediators are important for distinct pathways of NF-κB activation.
The effects of NF-κB, IκB and IKK on platelet activation were evaluated in vitro and in vivo using genetic ablation or inhibition of different factors of the NF-κB complex. However, these studies do not provide a conclusive picture, so far. Platelets are sensitive to NF-κB inhibitors, but the functional role of NF-κB in platelets is currently still incompletely understood. In vivo experiments revealed, that LDLR knockout-out mice with a platelet-specific genetic ablation of IKKβ show increased neointima formation and enhanced leukocyte adhesion at the injured area due to decreased platelet GPIbβ shedding and prolonged platelet-leukocyte interactions (254). However, another study using IKKβ-deficient platelets postulated that these platelets are unable to degranulate, leading to reduced reactivity and prolonged tail bleeding, which was postulated to be caused by defective SNAP-23 phosphorylation in absence of IKKβ (251).
In vitro studies using pharmacological inhibitors of IKKβ indicated that NF-κB is involved in the activation of platelet fibrinogen receptor GPIIb/IIIa (249), which is important for platelet aggregation and that the NF-κB pathway further participates in lamellipodia formation, clot retraction and stability (249). Inhibition of IKKβ and thus IκBα phosphorylation by BAY-11-7082 or RO-106-9920 suggested a positive role for IKKβ in thrombin- or collagen-induced ATP release, TXA2 formation, P-selectin expression and platelet aggregation (248, 249). Other studies using the NF-κB inhibitor andrographolide were in line with a positive role of NF-κB for platelet activation (255, 256) and it was also reported that platelet vitality may depend on NF-κB, as inhibition with BAY 11-7082 or MLN4924 led to depolarization of mitochondrial membranes, increased Ca2+ levels and ER stress induced apoptosis (257). However, in general it has to be stated that the use of pharmacological inhibitors in platelet function studies in vitro may suffer from artifacts of the assay system, such as inappropriate drug concentrations, which induce off-target effects, or unspecific side effects. It has been reported for instance that the commonly used IKKβ inhibitor BAY-11-7082 can induce apoptosis independent from its effect on NF-κB signaling (258) and that it is an effective and irreversible broad-spectrum inhibitor of protein tyrosine phosphatases (259).
Interestingly, NF-κB activation via IKKβ was also reported to initiate a negative feedback of platelet activation, as the catalytic subunit of PKA is associated with IκBα, from where it is released and activated when IκBα is degraded, followed by the known inhibitory actions of PKA such as VASP phosphorylation (250). This is in line with another report, where NF-κB inhibition in collagen- or thrombin-stimulated platelets led to increased VASP phosphorylation (260). With respect to the role of platelets, certainly further studies are warranted to determine, if increased levels or activity of NF-κB result in increased platelet reactivity and furthermore, how systemic chronic inflammation may affect platelet function differently than the plasmatic phase of coagulation. In general, a better understanding of NF-κB-dependent platelet responses would be crucial to fully understand the effect of NF-κB inhibitors, which are currently used as anti-inflammatory and anti-cancer agents, as they may elicit unintended effects on platelet functions.
Megakaryocytes as Precursors of Platelets
While it is clear that platelets contain basically all upstream signaling molecules of the NF-κB pathway, as well as the transcription factors themselves, they can only respond to inflammatory triggers in a non-genomic manner. In contrast, megakaryocytes (MKs), their progenitors, can convert systemic or local inflammatory conditions to a transcriptional response, which may has consequences on the phenotype of released platelets. Megakaryocytes reside in the vascular niche of the bone marrow where they can sense inflammatory conditions via different receptors, such as TLRs and from where they release platelets into the blood circulation. Interestingly, a recent report has provided evidence that megakaryocytes are also located in the microcirculation and the extravascular space of the lung, contributing up to 50% of the total platelet production (261). At least in the bone marrow, hematopoietic stem cells undergo a unique and remarkable maturation and differentiation process to become megakaryocytes, which involves extensive endomitosis (262, 263). As a result megakaryocytes have a ploidy of up to a 128-fold chromosome-set in one single, giant, poly-lobulated nucleus (264–266), giving megakaryocytes their name. A second distinct feature of megakaryopoiesis is the generation of a complex membrane system, called demarcation membrane system (DMS) or invaginated membrane system (IMS) (264, 267–269), that serves a reservoir for later platelet production (268, 270). The final phase of megakaryocyte maturation includes the formation of proplatelets, in which long branches extend into sinusoidal capillaries allowing proplatelet release into the blood stream. The main driving force of proplatelet elongation is microtubule sliding (271). Finally, due to blood flow, platelets fission from the tips of proplatelets and are released into the blood stream (272). After transfer of the megakaryocyte's cytoplasm and DMS/IMS into platelets, the remaining denuded nucleus is removed by macrophages (273). Interestingly, it seems that apoptosis is a physiological evet for mature megakaryocytes and that peak proplatelet and platelet production is shortly followed by apoptosis (274–276).
Inflammatory cytokines and pathways are involved in various steps of megakaryopoiesis and thrombopoiesis. Megakaryocytes express toll-like receptors (TLRs) (277, 278), tumor necrosis factor receptors (TNFR1 and 2) (279), receptors for IL-1β (280, 281), and IL-6 (282, 283), all of which are important activation pathways of NF-κB. Activity of the IKK complex increases during megakaryopoiesis and decreases during thrombopoiesis, allowing controlled cell death (284). The most important signaling molecule driving differentiation and maturation of megakaryocytes is thrombopoietin (TPO), a glycoprotein primarily produced by liver and kidney. Binding of this protein to its receptor c-Mpl on bone marrow cells is the primary signaling event that promotes and regulates megakaryopoiesis (264, 285, 286). Other cytokines that synergize with TPO include IL-1α, IL-1β, IL-3, IL-6, IL-9, IL-11, and granulocyte-macrophage colony-stimulating factor (GM-CSF) (287–291). However, all of them are dependent on TPO to exert their pro-megakaryopoietic functions (291). Furthermore, immature MKs themselves express IL-1α, IL-1β, IL-3, IL-6, and GM-CSF to stimulate their ploidy via NF-κB and TPO (287–289, 292).
A further link between inflammation and megakaryopoiesis is provided by reactive oxygen species (ROS), which after being released by activated macrophages and neutrophils commit hematopoietic stem cells toward the megakaryocytic lineage (293). Interestingly, a stem cell population was identified, which is already committed to the megakaryocytic lineage and matures rapidly upon inflammatory conditions, to replenish the loss of platelets (294).
One of the most intriguing recent findings was that upon acute inflammation IL-1α leads to rapid, TPO-independent platelet production. IL-1α signaling reduces plasma membrane stability, dysregulates tubulin expression and proplatelet formation, ultimately triggering megakaryocyte rupture and release of enormous amounts of platelets within short time. In this way, platelet loss due to acute injuries, blood loss or infection can be rapidly compensated (281).
To conclude, it can be stated that inflammation in general and NF-κB signaling in particular, does not only directly affect platelets, but also indirectly via modulation of their megakaryocytic progenitors.
Endothelial Cells
The endothelial cell lining of blood vessels represents a selective barrier between the blood stream and the surrounding tissue and exerts a variety of functions that contribute to hemostasis, and inflammatory responses that are related to coagulation (295). Many of these reactions are specific to their localization within the body as endothelial functions vary between different vascular beds. Under homeostatic conditions, endothelial cells constantly secrete nitric oxide, prostacyclin (in large vessels) as well as prostaglandin E2 (in smaller vessels) to suppress platelet adhesion and activation (Figure 6, upper panel) (4, 296). This is additionally supported by negatively charged glycosaminoglycans on the endothelial surface that prevent adhesion of platelets. The NF-κB signaling cascade has a key role in endothelial cells in response to stress situations (Figure 6, lower panel), as it is capable of regulating both pro-inflammatory and coagulatory responses, which are also prone to a significant level of crosstalk (297). In principal, all NF-κB signaling molecules are present in endothelial cells and their activation leads to a pro-adhesive and pro-coagulant phenotype with a concomitant reduction of the barrier function (298). In vitro, the strongest activators of NF-κB in endothelial cells appear to be TNFα and thrombin, but also other cytokines like IFNγ or IL-1β potently activate NF-κB in these cells. One major difference of thrombin- and TNFα-mediated NF-κB activation lies in their respective receptors. Thrombin binds to the extracellular terminus of PAR-1, a member of the G-coupled receptor superfamily, whereas TNFα binds to TNFR-1 and TNFR-2 (299, 300). Both pathways then converge at the level of the IKK complex (76, 301), yet interestingly, thrombin and TNFα appear to induce some overlapping but still differential target gene expression in endothelial cells (302). In addition, there appears to be a synergistic effect of TNFα and thrombin in regulating endothelial permeability (303). Important NF-κB target genes in endothelial cells are adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin that mediate adherence of inflammatory cells including monocytes, neutrophils, lymphocytes, and macrophages to the vascular wall triggering extravasation into tissues (304–307). It has been shown that expression of a constitutively active form of IKKβ, the central activator of NF-κB, in endothelial cells drives full expression of these adhesion molecules in the absence of any cytokine stimulation, indicating that the IKK/IκB/NF-κB axis is essential and sufficient for the pro-inflammatory activation of the endothelium (308). However, in quiescent endothelial cells, the ETS-related gene (ERG) prevents NF-κB p65 binding to DNA, indicating that ERG may compete with p65 for DNA binding under basal conditions (309).
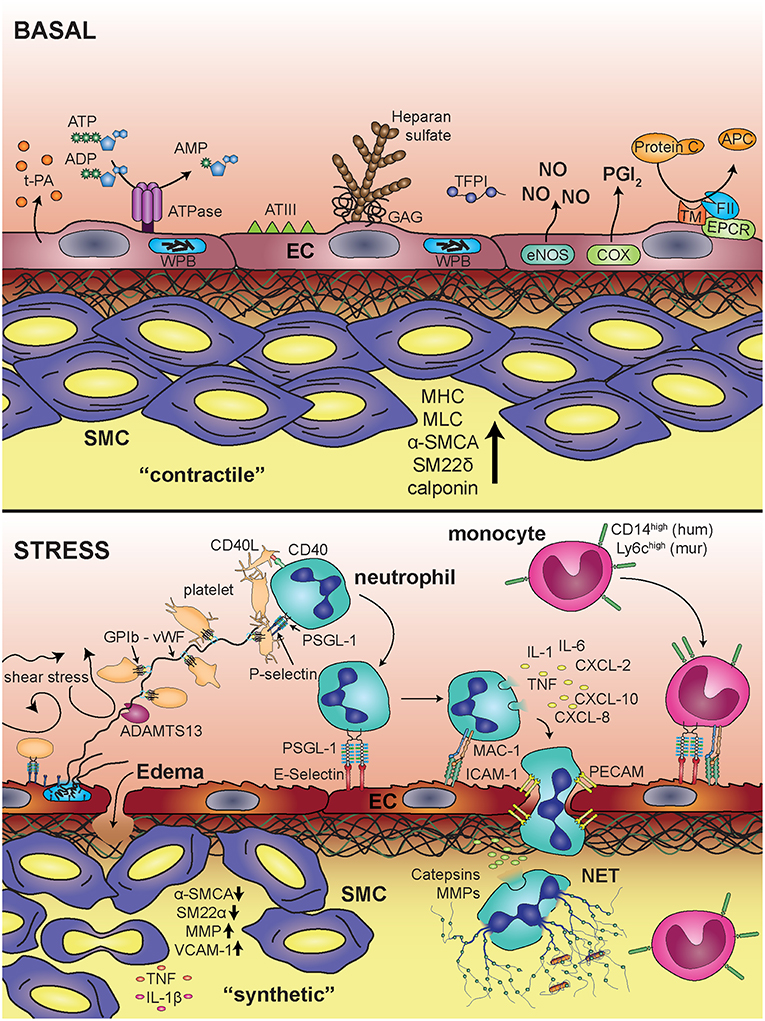
Figure 6. Blood vessel under basal conditions and upon inflammatory stimulation. Under basal conditions the endothelium provides an anti-thrombogenic surface via expression and production of tissue plasminogen activator (t-PA), ATPases, antithrombin III (ATIII), heparan sulfate, glycosaminoglycans (GAGs), tissue factor pathway inhibitor (TFPI), nitric oxide (NO), prostacyclin (PGI2), and endothelial protein C receptor (EPCR). Smooth muscle cells are in a “contractile” state, determined by expression of myosin heavy chain (MHC), myosin light chain kinase (MLCK),α-smooth muscle cell actin (α-SMCA), smooth muscle 22α (SM22α), and calponin. Upon inflammatory stress, endothelial cells release von Willebrand Factor vWF from Weibel Palade bodies (WPB), which triggers platelet string formation via glycoprotein Ib (GPIb). Furthermore, adhesion and transmigration of leukocytes is facilitated by expression of adhesion molecules, like E-selectin and intracellular adhesion molecule 1 (ICAM-1), which bind to PSGL-1 and Mac-1 on leukocytes, respectively. Activation of neutrophils leads to release of inflammatory mediators (IL-1, IL-6, TNFα, CXCL-2, CXCL-10, CXCL-8). Smooth muscle cells change their phenotype toward a “synthetic” state associated with decreased expression of α-SMCA and SM22α and increased expression of matrix metalloproteinases (MMP) and vascular cell adhesion molecule 1 (VCAM-1).
Besides classical activation of endothelial cells by various cytokines, they can also be activated by shear stress, meaning specifically a turbulent blood stream: Unidirectional, laminar shear stress actually limits endothelial activation and is associated with resistance to atherosclerosis (310, 311). In contrast, disturbed flow, such as turbulent or oscillatory conditions (e.g., at sites of vessel branching points, bifurcations, and curvatures) cause physical stress and subsequent pro-inflammatory gene expression that is associated with increased permeability of the cell layer (310, 311). Flow-induced endothelial cell activation is mediated via NF-κB and is integrin-and matrix-dependent (312). Recent studies indicate that focal adhesion kinase regulates NF-κB phosphorylation and transcriptional activity in response to flow (313). Another important aspect refers to the function of PECAM-1, which forms a mechanosensory complex with vascular endothelial cell cadherin and VEGFR2. Together, these receptors confer responsiveness to flow as shown in PECAM-1-knockout mice, which do not activate NF-κB in regions of disturbed flow. This mechano-sensing pathway is required for the earliest-known events in atherogenesis (314).
In addition to NF-κB-driven transcriptional responses to inflammatory states, endothelial cells also react to stress stimuli in other ways. The most prominent one of these is probably the fusion of specific secretory granules designated as Weibel-Palade bodies (WPB) with the cell membrane upon activation by various triggers such as thrombin or histamine. Exocytosis of these granules can also be induced by Toll-like receptors and other activators of the NF-κB pathway such as CD40L implying a role of NF-κB signaling molecules for the degranulation (315–319). Upon membrane fusion, the cargo of the vesicles is released, which includes several proteins that play a role in inflammation and thrombosis such as coagulation factor VIII, vWF, or P-selectin. The latter is exposed on the endothelial cell surface upon fusion of WPBs with the cytoplasmic membrane, triggering the adhesion of leukocytes. vWF is a large glycoprotein, which remains in a folded state in the microenvironment of WPBs, but is unfolded at the neutral pH of the blood circulation. This leads to the formation of ultra-large vWF multimers (ULVWF), which are able to bind platelets via interaction with GPIba or GPIIb (4). ULVWF multimers are subsequently cut by metalloproteinase ADAMTS13 on the endothelial surface providing a feedback mechanism to counteract platelet adhesion (320).
Endothelial cells modulate the balance between coagulation and fibrinolysis also by various other pathways: they counteract coagulation by binding of antithrombin III to the endothelial surface, release of tissue factor pathway inhibitor (TFPI), expression of thrombomodulin, activation of protein C, and synthesis as well as release of tissue plasminogen activator (t-PA) (296). These anti-thrombotic roles are often terminated by inflammatory diseases as for instance in endotoxemia and septic shock where a bacterial infection causes a pro-thrombotic state in endothelial cells by the activation of NF-κB (321). This has been demonstrated in a mouse model of sepsis, where endothelial-selective blockade of NF-κB via transgenic expression of a degradation-resistant form of IκBα resulted in decreased endothelial permeability, reduced infiltration of neutrophils and lower levels of thrombin-antithrombin complexes (322). Other mouse studies confirmed that endothelial NF-κB dampens the thrombomodulin-EPCR anticoagulation pathway (323). In addition to this systemic effect, NF-κB also serves as survival factor for endothelial cells themselves upon LPS-induced acute stress (324). This is also important for the maintenance of the endothelial barrier function as NF-κB inhibits endothelial apoptosis to ensure timely transition from barrier injury to recovery (325). Interestingly, endothelial expression of a degradation-resistant form of IκBα did not affect embryonic development, while endothelial cell-specific knockout of IKKβ resulted in increased embryonic lethality and endothelial apoptosis, which was at least in part mediated by kinase-independent functions of IKKβ (326).
A crucial role of endothelial NF-κB signaling has also been shown in mouse models of atherosclerosis where ablation of canonical NF-κB signaling by endothelial cell-specific deletion of NEMO or overexpression of a dominant-negative variant of IκBα protected ApoE-deficient mice from atherosclerosis induced by a Western-type diet (327). In general, atherosclerosis can be considered as chronic inflammatory disease of the vasculature, which is characterized by a complex crosstalk between different cell types, with endothelial cells constituting a crucial starting point of a vicious cycle, wherein NF-κB activation does not only lead to the expression of adhesion molecules that bind leukocytes, but also causes secretion of inflammatory mediators, which activate smooth muscle cells. This leads to vascular remodeling resulting in the plaque formation and narrowing of the vessel lumen. Furthermore, endothelial cells could undergo a reprogramming process toward a mesenchymal phenotype, designated as endothelial-mesenchymal transition, which is characterized by the expression of smooth muscle actin, various fibroblast markers and collagen (328). This phenotypic shift was reported to be involved in endothelial dysfunction during atherosclerosis. It can be triggered by cytokines such as TGFβ or IL-1, high glucose levels or pressure overload, as well as oxidized LDL (329–331).
Vascular Smooth Muscle Cells
Vascular smooth muscle cells (SMCs) are important players in both inflammatory and thrombotic processes. In general, arteries and veins consist of three layers, the tunica adventitia, largely constituted by connective tissue and fibroblasts, the tunica media mainly containing vascular smooth muscle cells and the tunica intima. Separated from the media by the internal elastic membrane, the intima consists of loose connective tissue intermingled with few SMCs, that is covered by a monolayer of endothelial cells resting on a basal membrane. The main function of SMCs in a blood vessel is to regulate the caliber. In a normal vessel, SMCs are in the contractile phenotype (Figure 6). They have very low cell division rates, a very restricted migratory behavior and express high levels of contractile proteins, such as myosin heavy chain, myosin light chain kinase, calponin, smooth muscle actin, and SM22α. Under conditions of inflammation, SMCs gain plasticity—their phenotype can change from contractile to synthetic; they rearrange their cytoskeleton, loose expression of contractile proteins, and regain their ability to proliferate and migrate. This phenotypic switch is central to many vascular diseases, such as atherosclerosis, re-stenosis, and vascular aging (332). The important role of SMC in stabilizing the cytoskeleton is highlighted in patients with mutations in ACTA2 encoding for smooth muscle actin or its promoter, leading to a higher risk for coronary disease (333). In atherosclerotic plaques, which represent chronically inflamed parts of arteries, SMCs reside predominantly in the superficial parts of lesions. They are mainly locally derived from the vessel wall (334). Phenotyping of the cells within the plaques revealed sizeable populations of SMCs without contractile proteins (335). Of note, also macrophages can express SMC genes such as smooth muscle α-actin and SM22α. Thus, SMC marker–positive cells can be derived from cell types other than SMCs and SMC marker–negative cells can be SMC-derived. Finally, even cells that are positive for CD68—the common macrophage marker, may not be macrophages as SMCs can undergo a cellular transition toward macrophage-like cells while simultaneously losing some of their SMC characteristics. This has been elucidated in more detail by genetic cell tracing approaches, which could show that more than 80% of SMC-derived cells within atherosclerotic lesions lack SMC markers that are commonly used in immuno-histochemical stainings, and that more than 30% of SMC-derived cells express conventional macrophage markers (336, 337). This means that many studies might have misinterpreted cellular markers and that probably many disease processes attributed to macrophages are in fact driven by SMCs that converted their cellular program. An important aspect is that SMC-derived macrophage-like cells are apparently less efficient in phagocytosis of deposits and apoptotic cells within the plaque as compared to “real” macrophages, which exacerbates necrotic core formation rendering the plaque unstable and prone for rupture (338, 339).
Anyway, these cells produce fibrous caps, and SMCs are an important source of collagen (340), which activates platelets, when endothelial cells are lost due to plaque rupture or erosion. The downregulation of SMC contractile genes such as SM22α is a typical phenomenon of atherosclerotic lesions (341). Interestingly, SM22α suppresses NF-κB signaling pathways under inflammatory conditions (342).
SMCs express multiple NF-κB family members and two inhibitor proteins, IκBβ and IκBα. In normal vessels SMCs display no basal NF-κB activity but the latter is readily induced in SMCs within atherosclerotic lesions. Interestingly, exposure to inflammatory cytokines induces prolonged NF-κB activation because of a sustained decrease in the inhibitory subunit IκBβ (343). TNFα appears as a crucial factor for the progression of atherosclerotic lesions as shown in TNFα/ApoE double knock-out mice, which display reduced thickness of vascular walls and reduced sizes of atherosclerotic lesions (344). TNFα binds to TNF receptors expressed on SMCs (345), which then triggers NF-κB via the classical activation pathway. This induces the expression of the pro-coagulatory tissue factor gene (346), as well as pro-inflammatory and matrix-remodeling genes such as MCP-1, matrix metalloproteinase-3 and−9 (MMP3 and MMP9), VCAM-1, and IL-1β, and furthermore potently downregulates SMC contractile genes (smooth muscle actin, SM22α, smooth muscle myosin heavy chain) (347). TNFα decreases expression of these contractile genes through induction of Krüppel-like transcription factor 4 (Klf4), a known regulator of SMC differentiation (348), which seems to be a target gene of NF-κB, based on specific binding sites in its enhancer region (337).
Even though a direct link between the downregulation of SMC contractile genes, NF-κB signaling and an increased risk for plaque rupture and arterial thrombosis has yet not been made, it is clear that elucidating mechanisms of phenotypic changes of SMCs in the course of inflammation seems to be a key in understanding many vascular diseases and may offer new therapeutic approaches.
Neutrophils
Neutrophils are the most abundant leukocyte fraction in humans with a rapid turn-over controlled by constitutive (spontaneous) apoptosis within 24–48 h after release from the bone marrow. Their life-span is markedly extended during inflammatory reactions and coupled to neutrophil activation to promote the inflammatory response (349). Since both, cell survival and pro-inflammatory activation are regulated by NF-κB, this transcription factor is central to neutrophil function and shows a unique expression pattern distinct from other leukocyte subsets (350, 351). In unstimulated neutrophils, NF-κB and in particular IκBα are not restricted to the cytosol as in most other cells but show abundant localization to the cell nucleus, with nuclear IκBα being regarded as a protective mechanism preventing the NF-κB-dependent expression of pro-inflammatory and anti-apoptotic genes (351). Furthermore, the IKK complex is partially localized to the nucleus. Upon neutrophil activation, IKKβ and NEMO are phosphorylated in the cytosol as well as the nucleus while IKKα is entirely lost from both compartments. The subsequent IκBα degradation and phosphorylation of RelA at serine 536 then promote NF-κB target gene expression (352).
Functional dimers of p50 (NFκB1), p65 (RelA), and/or c-Rel are detectable in neutrophils, and their activity is induced by a vast variety of pro-inflammatory mediators (353). While the majority of stimuli including TNFα and LPS trigger DNA binding by p50 and RelA (354), distinct agonists such as G-CSF selectively induce c-Rel activity (355). The first studies showing p50/RelA activation in neutrophils by pathogens, revealed the process of phagocytosis as an important trigger (356, 357). Subsequently, engagement of toll-like receptors (TLRs) by microbial products was identified to regulate NF-κB activity in neutrophilic granulocytes (358), with agonists of TLR4 (359, 360), TLR2 (361, 362) but also TLR7/8 (363) and TLR9 (364, 365) serving as important activators. Apart from TLRs, other pathways for sensing pathogen- or damage-associated molecular patterns [involving e.g., CIRP or Sox2 (366, 367)], as well as pathogen recognition via Fc receptors (368), were more recently identified to control neutrophil activation via NF-κB.
Neutrophil adhesion in the course of an inflammatory reaction is primarily mediated by activated β2 integrins (Mac-1: CD11b/CD18). Integrin binding or aggregation reportedly promotes NF-κB activation to enhance pro-inflammatory and anti-apoptotic gene expression (369). Furthermore, the β2 integrins may function as co-stimulatory signals for cytokines like GM-CSF and IL-8 to activate NF-κB when neutrophils are attached as opposed to suspended (370). Also myeloperoxidase released by these cells may bind to CD11b/CD18 and enhance the activation of NF-κB (371). Engagement of other integrins such as α9β1 by the respective ligand (VCAM-1 on endothelial cells) results in a comparable effect on NF-κB function (372, 373).
In the context of hemostasis and thrombosis, activated platelets expose CD40L at their surface which binds to neutrophil CD40 thereby inducing NF-κB target gene expression via the alternative activation pathway (374). Interestingly, platelet-derived microparticles reportedly transfer glycoprotein IIb/IIIa receptors onto neutrophils, which co-localize with β2-integrins and enhance NF-κB activation (375). Apart from platelets, coagulation factors and derived fragments may function to guide neutrophil activation and extend the neutrophil life-span via NF-κB transcriptional activity. For example, fibrinogen triggers IκBα degradation and NF-κB activation by binding to CD11b/CD18 molecules (376). In addition, the F1 and F2 fragments which are released upon prothrombin processing are known to induce NF-κB activity in neutrophils (377). Furthermore, regulators of plasmin activation (PAI-1 and uPA) may potentiate the polymorphonuclear (PMN) cell response to pro-inflammatory stimuli with respect to NF-κB activation (378).
Moreover, ROS have been implicated in the signaling pathway leading to NF-κB activation (379). However, the impact of ROS such as hydrogen peroxide (H2O2) generated at inflammatory sites has been subject to extensive debate and contradictory reports with respect to NF-κB activation in neutrophils. Direct exposure of neutrophils to H2O2 does not result in NF-κB activity. In contrast, the effect of LPS- or TNFα stimulation are abrogated by H2O2 resulting in decreased IκBα degradation and NF-κB translocation (380, 381). Similarly, when intracellular levels of ROS (superoxide and hydrogen peroxide) are increased by inhibition of catalase or the mitochondrial electron transport chain, the pro-inflammatory activation of NF-κB is inhibited (382–384). However, distinct approaches to raise intra- or extracellular superoxide levels (based on paraquat, nickel or combinations of xanthine oxidase and hypoxanthine or lumazine) showed a promoting rather than inhibiting effect on NF-κB activation (385–387). The controversial results may indicate that ROS regulation of NF-κB activity at inflammatory sites is more complex than previously thought and that ROS may exert both, pro- and anti-inflammatory effects. While low doses of H2O2 seem to trigger NF-κB activation, high oxidative stress does not alter or even adversely affect the NF-κB status (388, 389). Comparably, myeloperoxidase was recently reported to engage in a negative feedback loop of NF-κB downregulation to dampen the pro-inflammatory cytokine response (390). Other inhibitors of NF-κB activation in neutrophils include nitric oxide (391, 392), complement factor C5a (393), and prostaglandin D2 (394).
The target genes regulated by NF-κB in neutrophils can be grouped according to the three major functions of mediating cell adhesion, promoting inflammation, and inhibiting neutrophil apoptosis. In contrast, phagocytosis does not seem to be dependent on NF-κB (395). The induction of integrin CD11b expression requires p65 and promotes the firm adhesion and transmigration of neutrophils (395, 396). Activated PMNs secrete a multitude of pro-inflammatory mediators. Among the NF-κB regulated genes are the cytokines TNFα, IL-1, IL-6 (397, 398), the chemokines CXCL-2,−8, and−10 (360, 387, 397) as well as the TLR4 co-receptor CD14 (399) and the neutrophil gelatinase-associated lipocalin (400). Of interest, NF-κB activation also promotes microparticle release from PMNs (401). While NF-κB is known to exert a negative feedback regulation by inducing transcription of its inhibitor IκBα, an additional feedback mechanism has been identified in neutrophils: Expression of miR-9 is controlled by NF-κB and serves to inhibit the NFκB1 transcript (193).
Importantly, the balance between neutrophil production, survival and cell death is regulated by NF-κB. The mobilization of neutrophils from the bone marrow is subject to control by p50/p65 and seems to involve the NF-κB induced expression of the transcription factor C/EBPα (402, 403). While NF-κB is known to further support neutrophil survival and block spontaneous apoptosis, it may—in turn—facilitate cell death via neutrophil extracellular trap (NET) formation. Thus, NETosis is abrogated in the presence of NF-κB inhibitors such as BAY 11-7082 and Ro 106-9920 (404, 405), although it has to be stated that these inhibitors may also have NF-κB independent effects.
In the context of hemostasis and thrombosis, it was shown that activated platelets promote NET formation by a variety of signals including HMGB1 which induces neutrophil autophagy and subsequent expulsion of DNA NETs (229). It was proposed that autophagy constitutes an essential second step required to trigger NETosis after the initial pro-inflammatory priming of neutrophils (406). Thus, in addition to its role in the inflammatory activation of neutrophils, NF-κB may contribute to further steps of NET induction, as it exerts context-dependent effects on autophagy (407). Importantly, NETs seem to provide a scaffold for platelet, erythrocyte, tissue factor and fibrin deposition, which reportedly promotes arterial and venous thrombosis (227, 408–412). NET-exposed histones as well as neutrophil proteases such as elastase and cathepsin G are known to further enhance platelet activation and to degrade inhibitors of coagulation (413, 414). The detrimental role of NETs in thromboembolic disease has specifically been addressed in the cancer setting (415, 416). Tumor cells were shown to directly trigger NET formation or prime platelets to promote NETosis which results in further platelet activation and release of tissue factor (417, 418). In addition, this process of NET-associated cancer thrombosis is enhanced by tumor-cell derived microparticles (419). Most recently, clinical evidence is corroborating the association between NET formation and thrombosis in cancer patients (420, 421).
The control of neutrophil apoptosis is central to the inflammatory reaction as well as resolution and is primarily dependent on the NF-κB mediated expression of anti-apoptotic genes such as Bcl-x(L), A1, and A20 (363, 422). Thus, unstimulated neutrophils are characterized by the predominant presence of IκBα in the cell nucleus which inhibits NF-κB activity and allows for spontaneous apoptosis and rapid cell turn-over. When the nuclear accumulation of IκBα is artificially increased or when NF-κB activation is blocked, the constitutive apoptosis is accelerated (423, 424). In contrast, the pro-inflammatory activation of neutrophils by e.g., TNFα, LPS, type I interferons, or IL-1β results in IκBα degradation in the cytosol and nucleus and the subsequent liberation of NF-κB to prevent apoptosis (349, 425–428). The signaling pathway of TNFα for NF-κB activation is best characterized in this context. TNFα has a bimodal influence on the rate of neutrophil apoptosis in vitro, causing early acceleration and late inhibition when NF-κB dependent expression of anti-apoptotic proteins is achieved (429). TNF receptor 1 (TNFR-1) mediates activation of PI3 kinase and PKC-delta which results in assembly of the TNFR-1-TRADD-RIP-TRAF2 complex required for anti-apoptotic signaling (430). Apart from pro-inflammatory cytokines, it is the integrin-mediated adhesion and transmigration of neutrophils, which substantially enhances NF-κB mobilization and thereby promotes cell activation and survival in the situation where neutrophils extravasate from blood into tissue to engage at inflammatory sites (373, 431). Importantly, since hemostasis is closely linked to inflammation, the factors of coagulation and fibrinolysis also critically contribute to the localized activation and enhanced life-span of neutrophils. For example, binding of neutrophil surface integrin to fibrinogen activates NF-κB and delays apoptosis (376), and the release of prothrombin fragments or activation of uPA/PAI-1 may similarly enhance NF-κB activity (377, 378).
The shift in balance from spontaneous apoptosis to cell survival is reflected in the expression levels of pro- and anti-apoptotic mediators in PMNs. While pro-apoptotic proteins such as Bad, Bax, Bak, and Bik show stable expression and long half-lives, the NF-κB induced anti-apoptotic regulators like A1 and Mcl-1 are comparably short-lived and seem to transiently tilt the balance toward survival as long as NF-κB remains active (363, 364, 432).
The resolution of these processes at later phases requires the down-modulation of NF-κB activity by the re-expression of IκBα (350) and the induction of counter regulators such as suppressor of cytokine signaling 3 (SOCS3) (433). Failure to downregulate NF-κB results in the inappropriate survival of neutrophils, chronic inflammation, and tissue damage which is associated with neutrophil-mediated inflammatory disorders such as sepsis, rheumatoid arthritis and acute lung injury (349, 434, 435). Furthermore, sustained neutrophil activation and survival via the NF-κB pathway have been shown to promote tumor progression and metastasis by providing a pro-tumorigenic and pro-angiogenic environment (436, 437).
Monocytes
Monocytes contribute essentially to pro-inflammatory immune responses in general. In parallel with neutrophils, monocytes are produced in high numbers in the bone marrow as a response to infections and diseases and are responsible for driving inflammation (438). In addition, monocytes are the main source of circulating TF (439). The myeloid linage gives rise to a variety of functionally diverse cell types and is therefore in need of a tightly regulated differentiation program, which is partly built around the NF-κB pathway (440, 441).
Overall, monocytes can be divided into several subsets. Within the human monocyte compartment, three distinct monocyte populations can be defined according to their expression of CD14 and CD16. Monocytes positive for CD14 and negative for CD16 are termed classical monocytes (CMs) and are the most abundant subset within the human circulation followed by intermediate monocytes (IMs), defined by CD14++CD16+ expression and non-classical monocytes (NCMs), which are CD14+CD16++. The differentiation of monocytes from classical to intermediate and the non-classical phenotype is a linear process. In humans, classical monocytes are the first subset to emerge from the bone marrow, followed by differentiation into intermediate and non-classical monocytes (442). In addition, differentiation of monocytes is connected to cellular aging as NCMs display clear markers of cellular senescence including reduced telomere length and reduced numbers of Ki67-positive cells (443).
CD16+ monocytes overall are more proinflammatory and more procoagulant. In general IMs and NCMs display increased protein levels of p65 (443). When healthy volunteers are infused with low-dose LPS, CD16+ monocytes respond with upregulation of IL-6 and IL-8 which could not be observed in CD16− monocytes (444). Furthermore, in vitro IMs reacted to the alarmin IL-33 with an upregulation of TF via an NF-kB dependent pathway, a pathway probably active also in patients with atherosclerosis as monocyte-derived microvesicles positive for TF were correlated with IL-33 plasma levels (445).
In contrast to human monocytes, mouse monocytes are classified into pro-inflammatory and patrolling monocytes. Even though there are differences between mouse and human monocytes, monocyte subsets within the two species are broadly conserved (446). Pro-inflammatory monocytes are characterized by high expression of Ly6c. This subset of monocytes is strongly associated with encountering infections and driving inflammation. Expression of inflammatory cytokines, chemokines, and ROS production have been observed during heavy recruitment to inflamed tissue in various models (438). Definition and characterization of the Ly6clow CXCR1hi patrolling monocyte subset appears to be more complicated. Their exact role during homeostasis is not completely understood, but it is known that they show features for tissue remodeling and restoration (447). Further they tend to express anti-inflammatory mediators, like IL-10 and arginase (ARG1) (448), which suggest a counterbalancing role against the pro-inflammatory subset.
The balance of murine subsets has been suggested to be mainly defined by GM-CSF and M-CSF stimuli (449, 450), which are both triggering the NF-κB pathway (31, 451). NF-κB itself generates a positive feed-back loop to produce M-CSF (452).
Monocytes require NF-κB for differentiation but also accumulate NF-κB in their cytoplasm during maturation in order to guarantee a rapid NF-κB response upon activation (440). TNFα, which is secreted very early, represents one of the most prominent inflammatory genes, which is induced by the accumulated NF-κB reservoir, subsequently triggering a pro-inflammatory program of monocytes, or macrophages in an autocrine manner.
Importantly, monocytes require growth factors, like M-CSF, not only for differentiation but also for survival. Many of these stimuli are dependent on NF-κB signaling, suggesting a chronical dependence of monocytes on this pathway for survival. This has originally been demonstrated by studies using the NF-κB inhibitor pyrrolidine dithiocarbamate (451, 453) and could be confirmed with other NF-κB inhibitors when studying human monocyte-derived dendritic cells. In this study a role of NF-κB was demonstrated for survival, cytokine production and differentiation (454). More recently, it has been revealed that monocytes require autonomous TNFα to achieve function, survival and maintenance of the Ly6chi subset in an experimental autoimmune encephalomyelitis (EAE) model (455). These findings indicate a critical regulatory function for NF-κB in the autonomous loop of monocytes, as TNFα is driven by NF-κB and, in turn, is a strong inducer of NF-κB by itself (456, 457). Monocyte-specific constitutive activation of NF-κB resulted in a more severe pathogenicity within the EAE model and demonstrated increased levels of inflammatory monocyte-associated cytokines (458). Future studies are required to determine the potential regulatory mechanism of NF-κB in this context.
Interestingly, mouse studies using myeloid-specific deletion of the central NF-κB activator IKKβ revealed an interesting effect on macrophage polarization. Deletion of IKKβ resulted in a shift toward the inflammatory M1 phenotype both in an infection- and a tumor model, indicating a role of IKKβ and NF-κB for polarization toward the M2 phenotype, which decreases inflammation and fosters tissue repair (459, 460).
A disease, where monocytes play a crucial role is atherosclerosis. This complex disorder is orchestrated by multiple variables and cell types, but is fundamentally dependent on infiltrating monocytes (461). In this context, macrophage-specific deletion of IKKβ resulted in an aggravation of atherogenesis in one study (462), while a similar experimental set-up used in another study showed reduced lesion area (463). A protective effect of macrophage IKKβ in the context of atherosclerosis would be in line with the above-mentioned notion that IKKβ deletion or inhibition leads to a shift toward to the M1 phenotype, which is known to drive atherosclerosis. This concept is also supported by the observation that transgenic mice with macrophage-specific upregulation of p65 exhibited reduced atherosclerotic lesion formation and foam cell development (464). In contrast to that, another study with myeloid cell-specific IκB deletion (expected to result in elevated p65 activity) claimed an increase in atherosclerosis (465). Thus, a clear picture on the role of macrophage-specific NF-κB in atherogenesis is still lacking. For atherosclerosis, it is arguable that enhanced NF-κB expression may delay foam cell formation but might have severe consequences in a later stage of the diseases. For instance, increased NF-κB signaling in monocytes also results in a more pronounced expression of tissue factor (466), a critical variable in the pathology of atherothrombosis (467). The relevance of monocyte-derived tissue factor for thrombus formation has been demonstrated in an elegant study of impaired blood flow by von Brühl et al. (227). The authors identified neutrophils and monocytes to be the major leukocyte populations responsible for thrombus development. They found that, besides neutrophil-mediated NETosis, monocyte-derived tissue factor is critical for fibrin generation within the thrombus and contributes fundamentally to thrombus development. This is in line with findings showing a correlation of monocytic NF-κB activity with the occurrence of deep vein thrombosis (DVT) in cancer patients (468). A similar concept has already been suggested based on experimental results describing the necessity of p50 in the pathogenesis of deep vein thrombosis (469).
In conclusion, we know that the NF-κB pathway is involved in multiple aspects of monocyte differentiation and activation, which makes it difficult to distinguish the role of NF-κB in each individual stage of monocytes. It will require elegant inducible gene-manipulation strategies to answer these questions but considering the major influence of NF-κB on monocyte behavior, it might open doors for therapy of a broad spectrum of inflammatory diseases.
Clinical Aspects: Sepsis as an Example of an Acute Thrombo-Inflammatory Disease State
The vasculature and cells of the circulatory system react in a complex manner to inflammatory stress including various feedback circuits and cellular crosstalk coordinating a common systemic response in order to protect the host (Figure 6). However, dysregulation of this subtle balance between physiological inflammation and coagulation causes chronic inflammation and pathological thrombosis. Sepsis is a prime example of such a dysregulated response, which can lead to life-threatening conditions caused by an overshooting host defense (470). In general, the term sepsis denotes a systemic inflammatory response to infection. It is initiated by the activation of innate immune cells via pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS), microbial peptides, cell wall components, or nucleotides, which trigger various receptors on the host cells: C-type lectin receptors; Toll-like receptors (TLRs); RIG-I like receptors, as well as nucleotide-binding oligomerization domain–like receptors (NOD-like receptors). These and similar receptors can also stimulated by so-called danger associated molecular patterns (DAMPs) or “alarmins,” which include various cytosolic proteins, extracellular RNA or DNA that could all be released from damaged cells. In this way, necrosis or physical cell damage as it occurs in course of poly-traumas can trigger sepsis-like processes (commonly termed systemic inflammatory response syndrome, SIRS) in the absence of any infectious pathogen (471). Finally, most of these pattern recognition receptors activate NF-κB, which causes the expression of inflammatory cytokines like IL-1β or TNFα. Since these cytokines are both target genes and triggers of NF-κB, positive feedback loops are initiated, which result in a so-called “cytokine-storm” (472). Furthermore, activation of NF-κB causes not only the release and/or the generation of a multitude of pro-inflammatory mediators, but also the induction of pro-coagulatory mechanisms, which altogether lead to the clinical signs and symptoms of sepsis as well as disseminated intravascular coagulation (DIC) and multiple organ dysfunction (473) (Figure 7). The latter is basically caused by widespread thrombus formation in capillaries and reduced blood pressure causing tissue hypoperfusion. The disseminated coagulation can be explained by NF-κB-mediated upregulation of tissue factor (F III) and F VIII in combination with a reduction of anticoagulatory mechanisms such as Tissue Factor Pathway Inhibitor (TFPI), antithrombin, or thrombomodulin (471). Moreover, inflammatory activation of neutrophils triggers the formation of NETs, which exert not only anti-microbial functions by trapping and killing bacteria, but also initiate the contact pathway of coagulation via F XI and XII (474, 475). Various components of NETs like histones and proteolytic constituents have been identified as crucial regulations of coagulation, which contribute to development of end-organ damage (413). Collaborative interactions between NET-derived histone H4, platelets and inorganic polyphosphates are able to promote disseminated coagulation intendent of the invading pathogen (8). The diminished oxygen supply caused by microvascular thrombi results in deregulation of mitochondria function, which leads to increased formation of ROS thereby aggravating tissue damage and contributing to the release of danger signals.
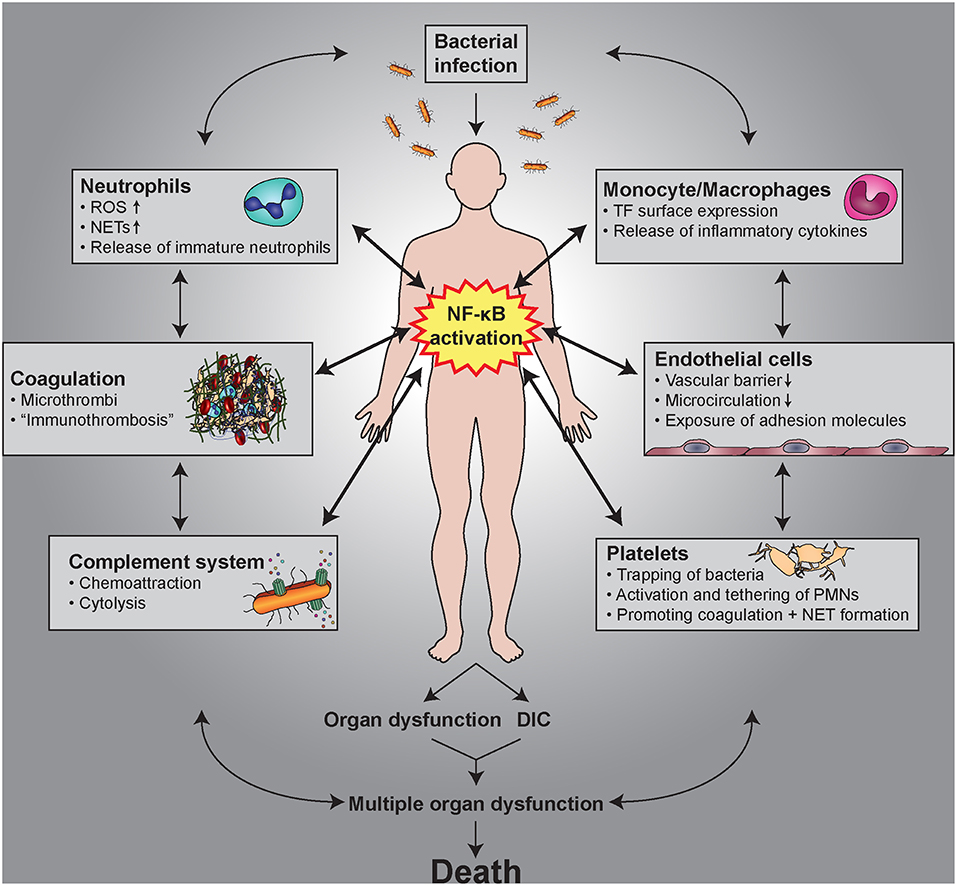
Figure 7. Hallmarks of sepsis as a thrombo-inflammatory disease. Multiple, complex interactions between monocytes/macrophages, endothelial cells, platelets, the complement system, coagulation, and neutrophils are found under septic conditions. Activation of NF-κB causes not only the release and/or the generation of a multitude of pro-inflammatory mediators, but also the induction of pro-coagulatory mechanisms, which lead to the clinical signs and symptoms of sepsis.
Extensive formation of thrombi in the microcirculation causes systemic depletion of coagulation factors and platelets resulting in increased bleeding events at other sites of the organism—a phenomenon generally designated as “coagulopathy.”
This imbalance is not only observed in coagulation—also inflammatory processes are affected. Due to strong, overshooting inflammatory responses in the first phase, counter-acting feedback-mechanism often become predominant at a later stage of the disease resulting in immunosuppression associated with increased risk for secondary or opportunistic infections. Attempts to understand the complex pathogenesis of sepsis included low-dose infusion of LPS into healthy volunteers (476). This revealed that LPS activates the endothelium and the coagulation system, as well as fibrinolysis, accompanied by a pro-inflammatory response (476, 477). Similar to LPS, infusion of the cytokine TNFα into healthy volunteers exerted not only pro-inflammatory actions, but also activated the coagulation cascade (478, 479).
Given the importance of NF-κB for the initiation of the vicious circle of sepsis, its inhibition has often been considered as an interesting therapeutic approach to treat or prevent overshooting immune responses (480). This notion is supported by different animal models of sepsis showing a beneficial effect of NF-κB inhibition (472, 481). However, blocking NF-κB activity is also accompanied by reduced host defense and thus elimination of pathogens—and is therefore contraindicated at the late state of sepsis. Thus, the right balance between positive and negative effects of NF-κB inhibition or the correct timing of blocking NF-κB have not been found, yet. This is reflected by various clinical trials blocking NF-κB or associated inflammatory pathways by treatment with anti-inflammatory substances (as listed in Table 3). These substances included glucocorticoids, which inhibit the NF-κB pathway, as well as non-steroidal anti-inflammatory drugs (NSAIDs) such as acetylsalicylic acid (ASA), which do not only block the synthesis of inflammatory mediators but also inhibit the activity of IKKs (501). Interestingly, ASA and its active metabolite salicylic acid (SA) exert both anti-inflammatory (502) and anti-coagulatory actions (503) and SA naturally occurs in the human body due to up-take of plant-based food and endogenous production (504). Furthermore, a number of antioxidants has been investigated, which indirectly inhibit the NF-κB activation pathway, including vitamin C, vitamin E, β-carotene, N-acetylcysteine, selenium, or omega-3 fatty acids (505–510). However, clinical trials with these antioxidants failed to show any beneficial effect in sepsis (496–500). On the other hand, beneficial effects of anti-inflammatory agents have been reported in a recent systematic meta-analysis showing that anti-TNFα treatment of septic patients slightly reduces mortality with an odds ratio of 0.91 (482). Furthermore, the relevance of LPS as trigger of sepsis could be underlined by studies applying extracorporeal endotoxin elimination devices with promising results (511).
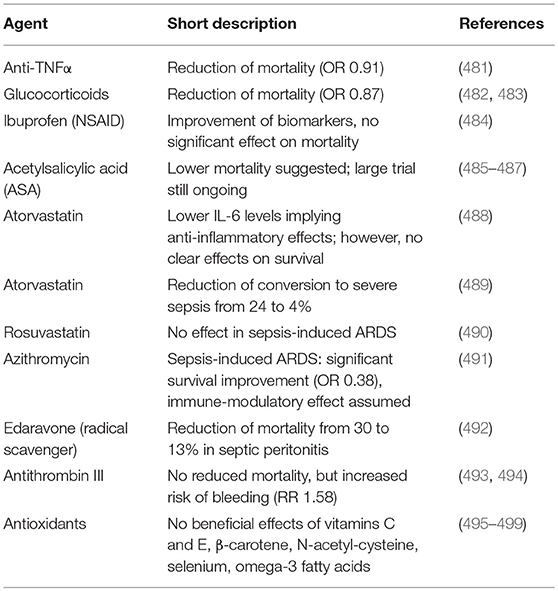
Table 3. Clinical studies targeting the thrombo-inflammatory axis of sepsis.
Nevertheless, the various clinical trials on NF-κB inhibition in sepsis underline the complex role of NF-κB in immune defense, inflammation and coagulation and the difficulty to find the right timing or regimen of treatment. However, concepts of dampening NF-κB activity appear very promising in thrombotic diseases that are characterized by rather low-grade chronic inflammation. This was demonstrated in a recent large clinical trial applying anti-IL-1β antibodies in patients with atherosclerosis and a prior myocardial infarction. The anti-inflammatory effect could be shown by dose-dependent reduction of the CRP level with was associated with an decreased risk to develop a second infarction, non-fatal stroke or cardiovascular death (512). However, as expected anti-IL1β treated patients had a higher risk of infections.
Overall, it is clear that inflammatory processes and thrombotic events are tightly linked on many different levels and that the NF-κB signaling pathway plays a fundamental role in the molecular and cellular linkages. Since NF-κB itself is a central hub in this network of reactions, an unspecific inhibition of this transcription factor might cause unwanted side-effects or be less efficient due to complex feedback circuits. However, considering the diversity of the intracellular as well as intercellular signaling networks that are built around NF-κB, targeting more specific connections between inflammation and coagulation might be very promising to reduce thrombotic morbidities that are associated with numerous chronic inflammatory diseases.
Author Contributions
MM wrote major parts of the manuscript, with an emphasis on endothelial cells, designed figures, and contributed to the overall conception. MS contributed major parts of the platelet- and megakaryocyte section and designed figures. CB wrote the part on neutrophils. BH contributed to the endothelial cell part. CS contributed to the sepsis section and summarized clinical trials targeting inflammation in sepsis. HD wrote major parts of the monocyte/macrophage section. PH wrote major parts of the monocyte/macrophage section. JB performed bioinformatics analysis and designed Figure 4. PP wrote major parts of the smooth muscle cell section. AA contributed major parts to the platelet and megakaryocyte section. JS made the concept for the manuscript, wrote the parts on NF-kappa B, the NF-kappa B signaling pathways, contributed major parts to the sections on endothelial cells, smooth muscle cells, and monocytes, and wrote major parts of the sepsis section, designed Figures 1, 2 as well as parts of Figures 3, 6 and the concept for Figure 4 and also created Tables 1, 2 and contributed to Table 3.
Funding
The authors are funded by the Austrian Science Fund, FWF (special research programme: SFB-F54 and project P-27842). The funding source had no influence on the design of the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Furie B, Furie BC. Molecular and cellular biology of blood coagulation. N Engl J Med. (1992) 326:800–6. doi: 10.1056/nejm199203193261205
2. Spronk HM, Govers-Riemslag JW, Cate Ht. The blood coagulation system as a molecular machine. BioEssays (2003) 25:1220–8. doi: 10.1002/bies.10360
3. Gaertner F, Massberg S. Blood coagulation in immunothrombosis-at the frontline of intravascular immunity. Semin Immunol. (2016) 28:561–9. doi: 10.1016/j.smim.2016.10.010
4. van Hinsbergh VW. Endothelium–role in regulation of coagulation and inflammation. Semin Immunopathol (2012) 34:93–106. doi: 10.1007/s00281-011-0285-5
5. Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. (2014) 115:662–7. doi: 10.1161/CIRCRESAHA.115.304634
6. Eilertsen K-E, Østerud B. Tissue factor: (patho) physiology and cellular biology. Blood Coagul Fibrinolysis (2004) 15:521–38. doi: 10.1097/00001721-200410000-00001
7. Darbousset R, Thomas GM, Mezouar S, Frere C, Bonier R, Mackman N, et al. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood (2012) 120:2133–43. doi: 10.1182/blood-2012-06-437772
8. McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood (2017) 129:1357–67. doi: 10.1182/blood-2016-09-741298
9. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. (2013) 13:34–45. doi: 10.1038/nri3345
10. Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. (2013) 369:2063. doi: 10.1056/NEJMc1312359
11. Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell (1986) 46:705–16. doi: 10.1016/0092-8674(86)90346-6
12. Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene (1999) 18:6853–66. doi: 10.1038/sj.onc.1203239
13. Schuster M, Annemann M, Plaza-Sirvent C, Schmitz I. Atypical IκB proteins – nuclear modulators of NF-κB signaling. Cell Commun Signal. (2013) 11:23. doi: 10.1186/1478-811X-11-23
14. Moorthy AK, Savinova OV, Ho JQ, Wang VY, Vu D, Ghosh G. The 20S proteasome processes NF-kappaB1 p105 into p50 in a translation-independent manner. EMBO J. (2006) 25:1945–56. doi: 10.1038/sj.emboj.7601081
15. Naumann M, Nieters A, Hatada EN, Scheidereit C. NF-kappa B precursor p100 inhibits nuclear translocation and DNA binding of NF-kappa B/rel-factors. Oncogene (1993) 8:2275–81.
16. Liang C, Zhang M, Sun SC. Beta-TrCP binding and processing of NF-kappaB2/p100 involve its phosphorylation at serines 866 and 870. Cell Signal. (2005) 18:1309–17. doi: 10.1016/j.cellsig.2005.10.011
17. Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer (2013) 12:86. doi: 10.1186/1476-4598-12-86
18. Bell S, Matthews JR, Jaffray E, Hay RT. I(kappa)B(gamma) inhibits DNA binding of NF-kappaB p50 homodimers by interacting with residues that contact DNA. Mol Cell Biol. (1996) 16:6477–85. doi: 10.1128/MCB.16.11.6477
19. Nolan GP, Ghosh S, Liou H-C, Tempst P, Baltimore D. DNA binding and IκB inhibition of the cloned p65 subunit of NF-κB, a rel-related polypeptide. Cell (1991) 64:961–9. doi: 10.1016/0092-8674(91)90320-X
20. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. (2004) 25:1605–12. doi: 10.1002/jcc.20084
21. Birbach A, Gold P, Binder BR, Hofer E, de Martin R, Schmid JA. Signaling molecules of the NF-kappa B pathway shuttle constitutively between cytoplasm and nucleus. J Biol Chem. (2002) 277:10842–51. doi: 10.1074/jbc.M112475200
22. Huang TT, Kudo N, Yoshida M, Miyamoto S. A nuclear export signal in the N-terminal regulatory domain of IkappaBalpha controls cytoplasmic localization of inactive NF-kappaB/IkappaBalpha complexes. Proc Natl Acad Sci USA. (2000) 97:1014–9. doi: 10.1073/pnas.97.3.1014
23. Johnson C, Van Antwerp D, Hope TJ. An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IkappaBalpha. Embo J. (1999) 18:6682–93. doi: 10.1093/emboj/18.23.6682
24. Carlotti F, Dower SK, Qwarnstrom EE. Dynamic shuttling of nuclear factor kappa B between the nucleus and cytoplasm as a consequence of inhibitor dissociation. J Biol Chem. (2000) 275:41028–34. doi: 10.1074/jbc.M006179200
25. Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc Natl Acad Sci USA. (1989) 86:2336–40.
26. Israel A, Le Bail O, Hatat D, Piette J, Kieran M, Logeat F, et al. TNF stimulates expression of mouse MHC class I genes by inducing an NF kappa B-like enhancer binding activity which displaces constitutive factors. EMBO J. (1989) 8:3793–800.
27. Grohmann U, Belladonna ML, Bianchi R, Orabona C, Ayroldi E, Fioretti MC, et al. IL-12 acts directly on DC to promote nuclear localization of NF-kappaB and primes DC for IL-12 production. Immunity (1998) 9:315–23.
28. Shalom-Barak T, Quach J, Lotz M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-kappaB. J Biol Chem. (1998) 273:27467–73.
29. Iikura M, Suto H, Kajiwara N, Oboki K, Ohno T, Okayama Y, et al. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab Invest. (2007) 87:971–8. doi: 10.1038/labinvest.3700663
30. Weih F, Caamano J. Regulation of secondary lymphoid organ development by the nuclear factor-kappaB signal transduction pathway. Immunol Rev. (2003) 195:91–105. doi: 10.1034/j.1600-065X.2003.00064.x
31. Ebner K, Bandion A, Binder BR, Martin Rd, Schmid JA. GMCSF activates NF-κB via direct interaction of the GMCSF receptor with IκB kinase β. Blood (2003) 102:192–9. doi: 10.1182/blood-2002-12-3753
32. Berberich I, Shu GL, Clark EA. Cross-linking CD40 on B cells rapidly activates nuclear factor-kappa B. J Immunol. (1994) 153:4357–66.
33. Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol. (2002) 3:958–65. doi: 10.1038/ni842
34. Chirmule N, Kalyanaraman VS, Pahwa S. Signals transduced through the CD4 molecule on T lymphocytes activate NF-kappa B. Biochem Biophys Res Commun. (1994) 203:498–505. doi: 10.1006/bbrc.1994.2210
35. Schneider P, Thome M, Burns K, Bodmer JL, Hofmann K, Kataoka T, et al. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity (1997) 7:831–6.
36. Wajant H, Pfizenmaier K, Scheurich P. Non-apoptotic Fas signaling. Cytokine Growth Factor Rev (2003) 14:53–66. doi: 10.1016/S1359-6101(02)00072-2
37. Mohan RR, Kim WJ, Mohan RR, Chen L, Wilson SE. Bone morphogenic proteins 2 and 4 and their receptors in the adult human cornea. Invest Ophthalmol Vis Sci. (1998) 39:2626–36.
38. Biswas DK, Cruz AP, Gansberger E, Pardee AB. Epidermal growth factor-induced nuclear factor kappa B activation: a major pathway of cell-cycle progression in estrogen-receptor negative breast cancer cells. Proc Natl. Acad. Sci USA. (2000) 97:8542–7. doi: 10.1073/pnas.97.15.8542
39. Yao P, Zhan Y, Xu W, Li C, Yue P, Xu C, et al. Hepatocyte growth factor-induced proliferation of hepatic stem-like cells depends on activation of NF-kappaB. J Hepatol. (2004) 40:391–8. doi: 10.1016/j.jhep.2003.11.001
40. Bertrand F, Philippe C, Antoine PJ, Baud L, Groyer A, Capeau J, et al. Insulin activates nuclear factor kappa B in mammalian cells through a Raf-1-mediated pathway. J Biol Chem. (1995) 270:24435–41.
41. Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell (1986) 47:921–8.
42. Vincenti MP, Burrell TA, Taffet SM. Regulation of NF-kappa B activity in murine macrophages: effect of bacterial lipopolysaccharide and phorbol ester. J Cell Physiol. (1992) 150:204–13. doi: 10.1002/jcp.1041500127
43. Tallant T, Deb A, Kar N, Lupica J, de Veer MJ, DiDonato JA. Flagellin acting via TLR5 is the major activator of key signaling pathways leading to NF-kappa B and proinflammatory gene program activation in intestinal epithelial cells. BMC Microbiol. (2004) 4:33. doi: 10.1186/1471-2180-4-33
44. Hacker H. Signal transduction pathways activated by CpG-DNA. Curr Top. Microbiol. Immunol. (2000) 247:77–92. doi: 10.1007/978-1-59259-305-7_3
45. Wu S, Powell J, Mathioudakis N, Kane S, Fernandez E, Sears CL. Bacteroides fragilis enterotoxin induces intestinal epithelial cell secretion of interleukin-8 through mitogen-activated protein kinases and a tyrosine kinase-regulated nuclear factor-kappaB pathway. Infect Immun. (2004) 72:5832–9. doi: 10.1128/iai.72.10.5832-5839.2004
46. Trede NS, Castigli E, Geha RS, Chatila T. Microbial superantigens induce NF-kappa B in the human monocytic cell line THP-1. J Immunol. (1993) 150:5604–13.
47. Kumar A, Haque J, Lacoste J, Hiscott J, Williams BR. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc Natil Acad Sci USA. (1994) 91:6288–92.
48. Santoro MG, Rossi A, Amici C. NF-kappaB and virus infection: who controls whom. EMBO J. (2003) 22:2552–60. doi: 10.1093/emboj/cdg267
49. Jouault T, Ibata-Ombetta S, Takeuchi O, Trinel PA, Sacchetti P, Lefebvre P, et al. Candida albicans phospholipomannan is sensed through toll-like receptors. J Infect Dis. (2003) 188:165–72. doi: 10.1086/375784
50. Kammanadiminti SJ, Dey I, Chadee K. Induction of monocyte chemotactic protein 1 in colonic epithelial cells by Entamoeba histolytica is mediated via the phosphatidylinositol 3-kinase/p65 pathway. Infect Immun. (2007) 75:1765–70. doi: 10.1128/iai.01442-06
51. Becker I, Salaiza N, Aguirre M, Delgado J, Carrillo-Carrasco N, Kobeh LG, et al. Leishmania lipophosphoglycan (LPG) activates NK cells through toll-like receptor-2. Mol Biochem Parasitol. (2003) 130:65–74. doi: 10.1016/S0166-6851(03)00160-9
52. Sangiuliano B, Perez NM, Moreira DF, Belizario JE. Cell death-associated molecular-pattern molecules: inflammatory signaling and control. Mediators Inflamm. (2014) 2014:821043. doi: 10.1155/2014/821043
53. Silva E, Arcaroli J, He Q, Svetkauskaite D, Coldren C, Nick JA, et al. HMGB1 and LPS induce distinct patterns of gene expression and activation in neutrophils from patients with sepsis-induced acute lung injury. Intensive Care Med. (2007) 33:1829–39. doi: 10.1007/s00134-007-0748-2
54. Sirois CM, Jin T, Miller AL, Bertheloot D, Nakamura H, Horvath GL, et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J Exp Med. (2013) 210:2447–63. doi: 10.1084/jem.20120201
55. Bertheloot D, Naumovski AL, Langhoff P, Horvath GL, Jin T, Xiao TS, et al. RAGE Enhances TLR Responses through Binding and Internalization of RNA. J Immunol. (2016) 197:4118–26. doi: 10.4049/jimmunol.1502169
56. Fischer S, Grantzow T, Pagel JI, Tschernatsch M, Sperandio M, Preissner KT, et al. Extracellular RNA promotes leukocyte recruitment in the vascular system by mobilising proinflammatory cytokines. Thromb Haemost. (2012) 108:730–41. doi: 10.1160/TH12-03-0186
57. Waris G, Tardif KD, Siddiqui A. Endoplasmic reticulum (ER) stress: hepatitis C virus induces an ER-nucleus signal transduction pathway and activates NF-kappaB and STAT-3. Biochem Pharmacol. (2002) 64:1425–30. doi: 10.1016/S0006-2952(02)01300-X
58. Kitamura M. Biphasic, bidirectional regulation of NF-kappaB by endoplasmic reticulum stress. Antioxid Redox Signal. (2009) 11:2353–64. doi: 10.1089/ars.2008.2391
59. Kitamura M. Control of NF-κB and inflammation by the unfolded protein response. Int Rev Immunol. (2011) 30:4–15. doi: 10.3109/08830185.2010.522281
60. Ward AO, Caputo M, Angelini GD, George SJ, Zakkar M. Activation and inflammation of the venous endothelium in vein graft disease. Atherosclerosis 265:266–74. doi: 10.1016/j.atherosclerosis.2017.08.023
61. Ganguli A, Persson L, Palmer IR, Evans I, Yang L, Smallwood R, et al. Distinct NF-kappaB regulation by shear stress through Ras-dependent IkappaBalpha oscillations: real-time analysis of flow-mediated activation in live cells. Circ Res. (2005) 96:626–34. doi: 10.1161/01.res.0000160435.83210.95
62. Lan Q, Mercurius KO, Davies PF. Stimulation of transcription factors NF kappa B and AP1 in endothelial cells subjected to shear stress. Biochem Biophys Res Commun. (1994) 201:950–6.
63. Bischoff DS, Zhu JH, Makhijani NS, Yamaguchi DT. Acidic pH stimulates the production of the angiogenic CXC chemokine, CXCL8 (interleukin-8), in human adult mesenchymal stem cells via the extracellular signal-regulated kinase, p38 mitogen-activated protein kinase, and NF-kappaB pathways. J Cell Biochem. (2008) 104:1378–92. doi: 10.1002/jcb.21714
64. Rupec RA, Baeuerle PA. The genomic response of tumor cells to hypoxia and reoxygenation. Differential activation of transcription factors AP-1 and NF-kappa B. Eur J Biochem. (1995) 234:632–40.
65. Mercurio F, Manning AM. NF-kappaB as a primary regulator of the stress response. Oncogene (1999) 18:6163–71. doi: 10.1038/sj.onc.1203174
66. Pieper GM, Riaz ul H. Activation of nuclear factor-kappaB in cultured endothelial cells by increased glucose concentration: prevention by calphostin C. J Cardiovasc Pharmacol. (1997) 30:528–32.
67. Lee SJ, Dimtchev A, Lavin MF, Dritschilo A, Jung M. A novel ionizing radiation-induced signaling pathway that activates the transcription factor NF-kappaB. Oncogene (1998) 17:1821–6. doi: 10.1038/sj.onc.1202088
68. Li N, Karin M. Ionizing radiation and short wavelength UV activate NF-kappaB through two distinct mechanisms. Proc Natl Acad Sci USA. (1998) 95:13012–7.
69. Legrand-Poels S, Schoonbroodt S, Matroule JY, Piette J. Nf-kappa B: an important transcription factor in photobiology. J Photochem Photobiol B (1998) 45:1–8. doi: 10.1016/s1011-1344(98)00118-3
70. Cooper SJ, Bowden GT. Ultraviolet B regulation of transcription factor families: roles of nuclear factor-kappa B (NF-kappaB) and activator protein-1 (AP-1) in UVB-induced skin carcinogenesis. Curr Cancer Drug Targets (2007) 7:325–34. doi: 10.2174/156800907780809714
71. Fairchild KD, Singh IS, Carter HC, Hester L, Hasday JD. Hypothermia enhances phosphorylation of I{kappa}B kinase and prolongs nuclear localization of NF-{kappa}B in lipopolysaccharide-activated macrophages. Am J Physiol Cell Physiol. (2005) 289:C1114–21. doi: 10.1152/ajpcell.00152.2005
72. Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. (2012) 26:203–34. doi: 10.1101/gad.183434.111
73. Seigner J, Basilio J, Resch U, de Martin R. CD40L and TNF both activate the classical NF-κB pathway, which is not required for the CD40L induced alternative pathway in endothelial cells. Biochem Biophys Res Commun. (2018) 495:1389–94. doi: 10.1016/j.bbrc.2017.11.160
74. Rieser E, Cordier SM, Walczak H. Linear ubiquitination: a newly discovered regulator of cell signalling. Trends Biochem Sci. (2013) 38:94–102. doi: 10.1016/j.tibs.2012.11.007
75. Shimizu Y, Taraborrelli L, Walczak H. Linear ubiquitination in immunity. Immunol Rev. (2015) 266:190–207. doi: 10.1111/imr.12309
76. Iwai K. Diverse ubiquitin signaling in NF-κB activation. Trends Cell Biol. (2012) 22:355–64. doi: 10.1016/j.tcb.2012.04.001
77. Ikeda F. Linear ubiquitination signals in adaptive immune responses. Immunol Rev. (2015) 266:222–36. doi: 10.1111/imr.12300
78. Iwai K. Diverse roles of the ubiquitin system in NF-κB activation. Biochim Biophys Acta (2014) 1843:129–36. doi: 10.1016/j.bbamcr.2013.03.011
79. Schmid JA, Birbach A. IkappaB kinase beta (IKKbeta/IKK2/IKBKB)–a key molecule in signaling to the transcription factor NF-kappaB. Cytokine Growth Fact Rev. (2008) 19:157–65. doi: 10.1016/j.cytogfr.2008.01.006
80. Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. (2000) 18:621–63. doi: 10.1146/annurev.immunol.18.1.621
81. Suzuki H, Chiba T, Kobayashi M, Takeuchi M, Suzuki T, Ichiyama A, et al. IkappaBalpha ubiquitination is catalyzed by an SCF-like complex containing Skp1, cullin-1, and two F-box/WD40-repeat proteins, betaTrCP1 and betaTrCP2. Biochem Biophys Res Commun. (1999) 256:127–32. doi: 10.1006/bbrc.1999.0289
82. Lork M, Verhelst K, Beyaert R. CYLD, A20 and OTULIN deubiquitinases in NF-kappaB signaling and cell death: so similar, yet so different. Cell Death Diff. (2017) 24:1172–83. doi: 10.1038/cdd.2017.46
83. Piva R, Belardo G, Santoro MG. NF-kappaB: a stress-regulated switch for cell survival. Antioxid Redox Signal. (2006) 8:478–86. doi: 10.1089/ars.2006.8.478
84. Mori N, Prager D. Transactivation of the interleukin-1alpha promoter by human T-cell leukemia virus type I and type II Tax proteins. Blood (1996) 87:3410–7.
85. Hiscott J, Marois J, Garoufalis J, D'Addario M, Roulston A, Kwan I, et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol. (1993) 13:6231–40.
86. Lai JH, Horvath G, Subleski J, Bruder J, Ghosh P, Tan TH. RelA is a potent transcriptional activator of the CD28 response element within the interleukin 2 promoter. Mol Cell Biol. (1995) 15:4260–71.
87. Son YH, Jeong YT, Lee KA, Choi KH, Kim SM, Rhim BY, et al. Roles of MAPK and NF-kappaB in interleukin-6 induction by lipopolysaccharide in vascular smooth muscle cells. J Cardiovasc Pharmacol. (2008) 51:71–7. doi: 10.1097/FJC.0b013e31815bd23d
88. Kunsch C, Rosen CA. NF-kappa B subunit-specific regulation of the interleukin-8 promoter. Mol Cell Biol. (1993) 13:6137–46.
89. Murphy TL, Cleveland MG, Kulesza P, Magram J, Murphy KM. Regulation of interleukin 12 p40 expression through an NF-kappa B half-site. Mol Cell Biol. (1995) 15:5258–67.
90. Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J Biol Chem. (2006) 281:24138–48. doi: 10.1074/jbc.M604597200
91. Shakhov AN, Collart MA, Vassalli P, Nedospasov SA, Jongeneel CV. Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J Exp Med. (1990) 171:35–47.
92. Lenardo MJ, Fan CM, Maniatis T, Baltimore D. The involvement of NF-kappa B in beta-interferon gene regulation reveals its role as widely inducible mediator of signal transduction. Cell (1989) 57:287–94.
93. Sica A, Dorman L, Viggiano V, Cippitelli M, Ghosh P, Rice N, et al. Interaction of NF-kappaB and NFAT with the interferon-gamma promoter. J Biol Chem. (1997) 272:30412–20.
94. Wickremasinghe MI, Thomas LH, O'Kane CM, Uddin J, Friedland JS. Transcriptional mechanisms regulating alveolar epithelial cell-specific CCL5 secretion in pulmonary tuberculosis. J Biol Chem. (2004) 279:27199–210. doi: 10.1074/jbc.M403107200
95. Ahn SY, Cho CH, Park KG, Lee HJ, Lee S, Park SK, et al. Tumor necrosis factor-alpha induces fractalkine expression preferentially in arterial endothelial cells and mithramycin A suppresses TNF-alpha-induced fractalkine expression. Am J Pathol. (2004) 164:1663–72. doi: 10.1016/S0002-9440(10)63725-X
96. Anisowicz A, Messineo M, Lee SW, Sager R. An NF-kappa B-like transcription factor mediates IL-1/TNF-alpha induction of gro in human fibroblasts. J Immunol. (1991) 147:520–7.
97. Liu R, Zhao X, Gurney TA, Landau NR. Functional analysis of the proximal CCR5 promoter. AIDS Res Hum Retroviruses (1998) 14:1509–19. doi: 10.1089/aid.1998.14.1509
98. Hopken UE, Foss HD, Meyer D, Hinz M, Leder K, Stein H, et al. Up-regulation of the chemokine receptor CCR7 in classical but not in lymphocyte-predominant Hodgkin disease correlates with distinct dissemination of neoplastic cells in lymphoid organs. Blood (2002) 99:1109–16. doi: 10.1182/blood.V99.4.1109
99. Willard-Gallo KE, Badran BM, Ravoet M, Zerghe A, Burny A, Martiat P, et al. Defective CD3gamma gene transcription is associated with NFATc2 overexpression in the lymphocytic variant of hypereosinophilic syndrome. Exp Hematol. (2005) 33:1147–59. doi: 10.1016/j.exphem.2005.06.027
100. Debnath I, Roundy KM, Weis JJ, Weis JH. Analysis of the regulatory role of BAFF in controlling the expression of CD21 and CD23. Mol Immunol. (2007) 44:2388–99. doi: 10.1016/j.molimm.2006.10.019
101. Hinz M, Loser P, Mathas S, Krappmann D, Dorken B, Scheidereit C. Constitutive NF-kappaB maintains high expression of a characteristic gene network, including CD40, CD86, and a set of antiapoptotic genes in Hodgkin/Reed-Sternberg cells. Blood (2001) 97:2798–807. doi: 10.1182/blood.V97.9.2798
102. Kim JO, Kim HW, Baek KM, Kang CY. NF-kappaB and AP-1 regulate activation-dependent CD137 (4-1BB) expression in T cells. FEBS Lett. (2003) 541:163–70. doi: 10.1016/S0014-5793(03)00326-0
103. Johnson DR, Pober JS. HLA class I heavy-chain gene promoter elements mediating synergy between tumor necrosis factor and interferons. Mol Cell Biol. (1994) 14:1322–32.
104. Gutierrez O, Pipaon C, Inohara N, Fontalba A, Ogura Y, Prosper F, et al. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J Biol Chem. (2002) 277:41701–5. doi: 10.1074/jbc.M206473200
105. Jamieson C, Mauxion F, Sen R. Identification of a functional NF-kappa B binding site in the murine T cell receptor beta 2 locus. J Exp Med. (1989) 170:1737–43.
106. Takeshita F, Suzuki K, Sasaki S, Ishii N, Klinman DM, Ishii KJ. Transcriptional regulation of the human TLR9 gene. J Immunol. (2004) 173:2552–61. doi: 10.4049/jimmunol.173.4.2552
107. Santee SM, Owen-Schaub LB. Human tumor necrosis factor receptor p75/80 (CD120b) gene structure and promoter characterization. J Biol Chem. (1996) 271:21151–9.
108. Zeng H, Ornatowska M, Joo MS, Sadikot RT. TREM-1 expression in macrophages is regulated at transcriptional level by NF-kappaB and PU.1. Eur J Immunol. (2007) 37:2300–8. doi: 10.1002/eji.200737270
109. Morello S, Ito K, Yamamura S, Lee KY, Jazrawi E, Desouza P, et al. IL-1 beta and TNF-alpha regulation of the adenosine receptor (A2A) expression: differential requirement for NF-kappa B binding to the proximal promoter. J Immunol. (2006) 177:7173–83. doi: 10.4049/jimmunol.177.10.7173
110. Cayla C, Heinonen P, Viikari L, Schaak S, Snapir A, Bouloumie A, et al. Cloning, characterisation and identification of several polymorphisms in the promoter region of the human alpha2B-adrenergic receptor gene. Bioch Pharmacol. (2004) 67:469–78. doi: 10.1016/j.bcp.2003.09.029
111. Thornburg NJ, Raab-Traub N. Induction of epidermal growth factor receptor expression by Epstein-Barr virus latent membrane protein 1 C-terminal-activating region 1 is mediated by NF-kappaB p50 homodimer/Bcl-3 complexes. J Virol. (2007) 81:12954–61. doi: 10.1128/jvi.01601-07
112. Li J, Schmidt AM. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem. (1997) 272:16498–506.
113. Whelan J, Ghersa P, Hooft van Huijsduijnen R, Gray J, Chandra G, Talabot F, et al. An NF kappa B-like factor is essential but not sufficient for cytokine induction of endothelial leukocyte adhesion molecule 1 (ELAM-1) gene transcription. Nucleic Acids Res. (1991) 19:2645–53.
114. van de Stolpe A, Caldenhoven E, Stade BG, Koenderman L, Raaijmakers JA, Johnson JP, et al. 12-O-tetradecanoylphorbol-13-acetate- and tumor necrosis factor alpha-mediated induction of intercellular adhesion molecule-1 is inhibited by dexamethasone. Functional analysis of the human intercellular adhesion molecular-1 promoter. J Biol Chem. (1994) 269:6185–92.
115. Norton PA, Reis HM, Prince S, Larkin J, Pan J, Liu J, et al. Activation of fibronectin gene expression by hepatitis B virus x antigen. J Viral Hepat. (2004) 11:332–41. doi: 10.1111/j.1365-2893.2004.00555.x
116. Pan J, McEver RP. Regulation of the human P-selectin promoter by Bcl-3 and specific homodimeric members of the NF-kappa B/Rel family. J Biol Chem. (1995) 270:23077–83.
117. Iademarco MF, McQuillan JJ, Rosen GD, Dean DC. Characterization of the promoter for vascular cell adhesion molecule-1 (VCAM-1). J Biol Chem. (1992) 267:16323–9.
118. Agrawal A, Cha-Molstad H, Samols D, Kushner I. Overexpressed nuclear factor-kappaB can participate in endogenous C-reactive protein induction, and enhances the effects of C/EBPbeta and signal transducer and activator of transcription-3. Immunology (2003) 108:539–47. doi: 10.1046/j.1365-2567.2003.01608.x
119. Basile A, Sica A, d'Aniello E, Breviario F, Garrido G, Castellano M, et al. Characterization of the promoter for the human long pentraxin PTX3. Role of NF-kappaB in tumor necrosis factor-alpha and interleukin-1beta regulation. J Biol Chem. (1997) 272:8172–8.
120. Edbrooke MR, Foldi J, Cheshire JK, Li F, Faulkes DJ, Woo P. Constitutive and NF-kappa B-like proteins in the regulation of the serum amyloid A gene by interleukin 1. Cytokine (1991) 3:380–8.
121. Mackman N, Brand K, Edgington TS. Lipopolysaccharide-mediated transcriptional activation of the human tissue factor gene in THP-1 monocytic cells requires both activator protein 1 and nuclear factor kappa B binding sites. J Exp Med. (1991) 174:1517–26. doi: 10.1084/jem.174.6.1517
122. Begbie M, Notley C, Tinlin S, Sawyer L, Lillicrap D. The Factor VIII acute phase response requires the participation of NFkappaB and C/EBP. Thromb. Haemost. (2000) 84:216–22. doi: 10.1055/s-0037-1613999
123. Novak U, Cocks BG, Hamilton JA. A labile repressor acts through the NFkB-like binding sites of the human urokinase gene. Nucleic Acids Res. (1991) 19:3389–93. doi: 10.1093/nar/19.12.3389
124. Swiatkowska M, Szemraj J, Cierniewski CS. Induction of PAI-1 expression by tumor necrosis factor alpha in endothelial cells is mediated by its responsive element located in the 4G/5G site. FEBS J. (2005) 272:5821–31. doi: 10.1111/j.1742-4658.2005.04979.x
125. Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. J Biol Chem. (1992) 267:17971–6.
126. Grumont RJ, Rourke IJ, Gerondakis S. Rel-dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation-induced apoptosis. Genes Dev. (1999) 13:400–11.
127. Catz SD, Johnson JL. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene (2001) 20:7342–51. doi: 10.1038/sj.onc.1204926
128. Kreuz S, Siegmund D, Scheurich P, Wajant H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol. (2001) 21:3964–73. doi: 10.1128/mcb.21.12.3964-3973.2001
129. Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science (1998) 281:1680–3.
130. Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc Natl Acad Sci. USA. (1997) 94:10057–62.
131. Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J Exp Med. (1998) 188:211–6.
132. Schwenzer R, Siemienski K, Liptay S, Schubert G, Peters N, Scheurich P, et al. The human tumor necrosis factor (TNF) receptor-associated factor 1 gene (TRAF1) is up-regulated by cytokines of the TNF ligand family and modulates TNF-induced activation of NF-kappaB and c-Jun N-terminal kinase. J Biol Chem. (1999) 274:19368–74.
133. Wang Z, Sicinski P, Weinberg RA, Zhang Y, Ravid K. Characterization of the mouse cyclin D3 gene: exon/intron organization and promoter activity. Genomics (1996) 35:156–63. doi: 10.1006/geno.1996.0334
134. Huang Y, Ohtani K, Iwanaga R, Matsumura Y, Nakamura M. Direct trans-activation of the human cyclin D2 gene by the oncogene product Tax of human T-cell leukemia virus type I. Oncogene (2001) 20:1094–102. doi: 10.1038/sj.onc.1204198
135. Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. (1999) 19:5785–99.
136. Duyao MP, Buckler AJ, Sonenshein GE. Interaction of an NF-kappa B-like factor with a site upstream of the c-myc promoter. Proc Natl Acad Sci USA. (1990) 87:4727–31.
137. Yamamoto K, Arakawa T, Ueda N, Yamamoto S. Transcriptional roles of nuclear factor kappa B and nuclear factor-interleukin-6 in the tumor necrosis factor alpha-dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J Biol Chem. (1995) 270:31315–20.
138. Arakawa T, Nakamura M, Yoshimoto T, Yamamoto S. The transcriptional regulation of human arachidonate 12-lipoxygenase gene by NF kappa B/Rel. FEBS Lett. (1995) 363:105–10.
139. Cortese-Krott MM, Kulakov L, Oplander C, Kolb-Bachofen V, Kroncke KD, Suschek CV. Zinc regulates iNOS-derived nitric oxide formation in endothelial cells. Redox Biol. (2014) 2:945–54. doi: 10.1016/j.redox.2014.06.011
140. Nomura Y. NF-kappaB activation and IkappaB alpha dynamism involved in iNOS and chemokine induction in astroglial cells. Life Sci. (2001) 68:1695–701. doi: 10.1016/S0024-3205(01)00967-5
141. Nakata S, Tsutsui M, Shimokawa H, Yamashita T, Tanimoto A, Tasaki H, et al. Statin treatment upregulates vascular neuronal nitric oxide synthase through Akt/NF-kappaB pathway. Arterioscler Thromb Vasc Biol. (2007) 27:92–8. doi: 10.1161/01.atv.0000251615.61858.33
142. Sambamurti K, Kinsey R, Maloney B, Ge YW, Lahiri DK. Gene structure and organization of the human beta-secretase (BACE) promoter. FASEB J. (2004) 18:1034–6. doi: 10.1096/fj.03-1378fje
143. Bien S, Ritter CA, Gratz M, Sperker B, Sonnemann J, Beck JF, et al. Nuclear factor-kappaB mediates up-regulation of cathepsin B by doxorubicin in tumor cells. Mol Pharmacol. (2004) 65:1092–102. doi: 10.1124/mol.65.5.1092
144. Vincenti MP, Coon CI, Brinckerhoff CE. Nuclear factor kappaB/p50 activates an element in the distal matrix metalloproteinase 1 promoter in interleukin-1beta-stimulated synovial fibroblasts. Arthritis Rheum. (1998) 41:1987–94. doi: 10.1002/1529-0131(199811)41:11<1987::aid-art14>3.0.co;2-8
145. Borghaei RC, Rawlings PL Jr Javadi M, Woloshin J. NF-kappaB binds to a polymorphic repressor element in the MMP-3 promoter. Bioch Biophys Res Commun. (2004) 316:182–8. doi: 10.1016/j.bbrc.2004.02.030
146. He C. Molecular mechanism of transcriptional activation of human gelatinase B by proximal promoter. Cancer Lett. (1996) 106:185–91.
147. Xia C, Hu J, Ketterer B, Taylor JB. The organization of the human GSTP1-1 gene promoter and its response to retinoic acid and cellular redox status. Biochem J. (1996) 313 (Pt 1):155–61.
148. Garcia-Nogales P, Almeida A, Fernandez E, Medina JM, Bolanos JP. Induction of glucose-6-phosphate dehydrogenase by lipopolysaccharide contributes to preventing nitric oxide-mediated glutathione depletion in cultured rat astrocytes. J Neurochem. (1999) 72:1750–8.
149. Huang C, Bi E, Hu Y, Deng W, Tian Z, Dong C, et al. A novel NF-kappaB binding site controls human granzyme B gene transcription. J Immunol. (2006) 176:4173–81. doi: 10.4049/jimmunol.176.7.4173
150. Lavrovsky Y, Schwartzman ML, Levere RD, Kappas A, Abraham NG. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc Natl Acad Sci USA. (1994) 91:5987–91.
151. Phi van L. Transcriptional activation of the chicken lysozyme gene by NF-kappa Bp65 (RelA) and c-Rel, but not by NF-kappa Bp50. Biochem J. (1996) 313(Pt 1):39–44.
152. Yang N, Huang J, Greshock J, Liang S, Barchetti A, Hasegawa K, et al. Transcriptional regulation of PIK3CA oncogene by NF-kappaB in ovarian cancer microenvironment. PLoS ONE (2008) 3:e1758. doi: 10.1371/journal.pone.0001758
153. Kaltschmidt B, Ndiaye D, Korte M, Pothion S, Arbibe L, Prullage M, et al. NF-kappaB regulates spatial memory formation and synaptic plasticity through protein kinase A/CREB signaling. Mol Cell Biol. (2006) 26:2936–46. doi: 10.1128/mcb.26.8.2936-2946.2006
154. Suh KS, Tatunchak TT, Crutchley JM, Edwards LE, Marin KG, Yuspa SH. Genomic structure and promoter analysis of PKC-delta. Genomics (2003) 82:57–67. doi: 10.1016/S0888-7543(03)00072-7
155. Couturier C, Brouillet A, Couriaud C, Koumanov K, Bereziat G, Andreani M. Interleukin 1beta induces type II-secreted phospholipase A(2) gene in vascular smooth muscle cells by a nuclear factor kappaB and peroxisome proliferator-activated receptor-mediated process. J Biol Chem. (1999) 274:23085–93.
156. Kim JY, Kim H, Lee SG, Choi BH, Kim YH, Huh PW, et al. Amyloid beta peptide (Abeta42) activates PLC-delta1 promoter through the NF-kappaB binding site. Biochem Biophys Res Commun. (2003) 310:904–9. doi: 10.1016/j.bbrc.2003.09.100
157. Hrdlickova R, Nehyba J, Liss AS, Bose HR Jr. Mechanism of telomerase activation by v-Rel and its contribution to transformation. J Virol. (2006) 80:281–95. doi: 10.1128/jvi.80.1.281-295.2006
158. Mirza A, Liu SL, Frizell E, Zhu J, Maddukuri S, Martinez J, et al. A role for tissue transglutaminase in hepatic injury and fibrogenesis, and its regulation by NF-kappaB. Am J Physiol. (1997) 272(2 Pt 1):G281–8. doi: 10.1152/ajpgi.1997.272.2.G281
159. Ammirante M, Rosati A, Gentilella A, Festa M, Petrella A, Marzullo L, et al. The activity of hsp90 alpha promoter is regulated by NF-kappa B transcription factors. Oncogene (2008) 27:1175–8. doi: 10.1038/sj.onc.1210716
160. Das KC, Lewis-Molock Y, White CW. Activation of NF-kappa B and elevation of MnSOD gene expression by thiol reducing agents in lung adenocarcinoma (A549) cells. Am J Physiol. (1995) 269(5 Pt 1):L588–602. doi: 10.1152/ajplung.1995.269.5.L588
161. Rojo AI, Salinas M, Martin D, Perona R, Cuadrado A. Regulation of Cu/Zn-superoxide dismutase expression via the phosphatidylinositol 3 kinase/Akt pathway and nuclear factor-kappaB. J Neurosci. (2004) 24:7324–34. doi: 10.1523/jneurosci.2111-04.2004
162. Kwak EL, Larochelle DA, Beaumont C, Torti SV, Torti FM. Role for NF-kappa B in the regulation of ferritin H by tumor necrosis factor-alpha. J Biol Chem. (1995) 270:15285–93.
163. Armstrong K, Robson CN, Leung HY. NF-kappaB activation upregulates fibroblast growth factor 8 expression in prostate cancer cells. Prostate (2006) 66:1223–34. doi: 10.1002/pros.20376
164. Nishizawa M, Nagata S. Regulatory elements responsible for inducible expression of the granulocyte colony-stimulating factor gene in macrophages. Mol Cell Biol. (1990) 10:2002–11.
165. Hohensinner PJ, Kaun C, Rychli K, Niessner A, Pfaffenberger S, Rega G, et al. Macrophage colony stimulating factor expression in human cardiac cells is upregulated by tumor necrosis factor-alpha via an NF-kappaB dependent mechanism. J Thromb Haemost. (2007) 5:2520–8. doi: 10.1111/j.1538-7836.2007.02784.x
166. Schreck R, Baeuerle PA. NF-kappa B as inducible transcriptional activator of the granulocyte-macrophage colony-stimulating factor gene. Mol Cell Biol. (1990) 10:1281–6.
167. Heese K, Inoue N, Sawada T. NF-kappaB regulates B-cell-derived nerve growth factor expression. Cell Mol Immunol. (2006) 3:63–6 doi: 10.1242/dev.01702
168. Figueroa YG, Chan AK, Ibrahim R, Tang Y, Burow ME, Alam J, et al. NF-kappaB plays a key role in hypoxia-inducible factor-1-regulated erythropoietin gene expression. Exp Hematol. (2002) 30:1419–27. doi: 10.1016/S0301-472X(02)00934-7
169. Lang CH, Nystrom GJ, Frost RA. Regulation of IGF binding protein-1 in hep G2 cells by cytokines and reactive oxygen species. Am J Physiol. (1999) 276(3 Pt 1):G719–27.
170. Cazals V, Nabeyrat E, Corroyer S, de Keyzer Y, Clement A. Role for NF-kappa B in mediating the effects of hyperoxia on IGF-binding protein 2 promoter activity in lung alveolar epithelial cells. Biochim Biophys Acta (1999) 1448:349–62.
171. Samant RS, Clark DW, Fillmore RA, Cicek M, Metge BJ, Chandramouli KH, et al. Breast cancer metastasis suppressor 1 (BRMS1) inhibits osteopontin transcription by abrogating NF-kappaB activation. Mol. Cancer (2007) 6:6. doi: 10.1186/1476-4598-6-6
172. Chilov D, Kukk E, Taira S, Jeltsch M, Kaukonen J, Palotie A, et al. Genomic organization of human and mouse genes for vascular endothelial growth factor C. J Biol Chem. (1997) 272:25176–83.
173. Zhang L, Charron M, Wright WW, Chatterjee B, Song CS, Roy AK, et al. Nuclear factor-kappaB activates transcription of the androgen receptor gene in Sertoli cells isolated from testes of adult rats. Endocrinology (2004) 145:781–9. doi: 10.1210/en.2003-0987
174. Hannink M, Temin HM. Structure and autoregulation of the c-rel promoter. Oncogene (1990) 5:1843–50.
175. Corn RA, Hunter C, Liou HC, Siebenlist U, Boothby MR. Opposing roles for RelB and Bcl-3 in regulation of T-box expressed in T cells, GATA-3, and Th effector differentiation. J Immunol. (2005) 175:2102–10. doi: 10.4049/jimmunol.175.4.2102
176. Bonello S, Zahringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, et al. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol. (2007) 27:755–61. doi: 10.1161/01.ATV.0000258979.92828.bc
177. Harada H, Takahashi E, Itoh S, Harada K, Hori TA, Taniguchi T. Structure and regulation of the human interferon regulatory factor 1 (IRF-1) and IRF-2 genes: implications for a gene network in the interferon system. Mol Cell Biol. (1994) 14:1500–9.
178. Grumont RJ, Gerondakis S. Rel induces interferon regulatory factor 4 (IRF-4) expression in lymphocytes: modulation of interferon-regulated gene expression by rel/nuclear factor kappaB. J Exp Med. (2000) 191:1281–92. doi: 10.1084/jem.191.8.1281
179. Lu R, Moore PA, Pitha PM. Stimulation of IRF-7 gene expression by tumor necrosis factor alpha: requirement for NFkappa B transcription factor and gene accessibility. J Biol Chem. (2002) 277:16592–8. doi: 10.1074/jbc.M111440200
180. Yun K, Choi YD, Nam JH, Park Z, Im SH. NF-kappaB regulates Lef1 gene expression in chondrocytes. Biochem Biophys Res Commun. (2007) 357:589–95. doi: 10.1016/j.bbrc.2007.03.170
181. Ten RM, Paya CV, Israel N, Le Bail O, Mattei MG, Virelizier JL, et al. The characterization of the promoter of the gene encoding the p50 subunit of NF-kappa B indicates that it participates in its own regulation. EMBO J. (1992) 11:195–203.
182. Lombardi L, Ciana P, Cappellini C, Trecca D, Guerrini L, Migliazza A, et al. Structural and functional characterization of the promoter regions of the NFKB2 gene. Nucleic Acids Res. (1995) 23:2328–36.
183. McEvoy AN, Murphy EA, Ponnio T, Conneely OM, Bresnihan B, FitzGerald O, et al. Activation of nuclear orphan receptor NURR1 transcription by NF-kappa B and cyclic adenosine 5'-monophosphate response element-binding protein in rheumatoid arthritis synovial tissue. J Immunol. (2002) 168:2979–87. doi: 10.4049/jimmunol.168.6.2979
184. Wu H, Lozano G. NF-kappa B activation of p53. A potential mechanism for suppressing cell growth in response to stress. J Biol Chem. (1994) 269:20067–74.
185. Bren GD, Solan NJ, Miyoshi H, Pennington KN, Pobst LJ, Paya CV. Transcription of the RelB gene is regulated by NF-kappaB. Oncogene (2001) 20:7722–33. doi: 10.1038/sj.onc.1204868
186. Hinz M, Lemke P, Anagnostopoulos I, Hacker C, Krappmann D, Mathas S, et al. Nuclear factor kappaB-dependent gene expression profiling of Hodgkin's disease tumor cells, pathogenetic significance, and link to constitutive signal transducer and activator of transcription 5a activity. J Exp Med. (2002) 196:605–17. doi: 10.1084/jem.20020062
187. Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science (1993) 259:1912–5.
188. Tian B, Nowak DE, Jamaluddin M, Wang S, Brasier AR. Identification of direct genomic targets downstream of the nuclear factor-kappaB transcription factor mediating tumor necrosis factor signaling. J Biol Chem. (2005) 280:17435–48. doi: 10.1074/jbc.M500437200
189. Fontalba A, Gutierrez O, Fernandez-Luna JL. NLRP2, an inhibitor of the NF-kappaB pathway, is transcriptionally activated by NF-kappaB and exhibits a nonfunctional allelic variant. J Immunol. (2007) 179:8519–24. doi: 10.4049/jimmunol.179.12.8519
190. Heyninck K, Beyaert R. The cytokine-inducible zinc finger protein A20 inhibits IL-1-induced NF-kappaB activation at the level of TRAF6. FEBS Lett. (1999) 442:147–50.
191. Verstrepen L, Carpentier I, Beyaert R. The biology of A20-binding inhibitors of NF-kappaB activation (ABINs). Adv Exp Med Biol. (2014) 809:13–31. doi: 10.1007/978-1-4939-0398-6_2
192. Hofer-Warbinek R, Schmid JA, Stehlik C, Binder BR, Lipp J, de Martin R. Activation of NF-kappa B by XIAP, the X chromosome-linked inhibitor of apoptosis, in endothelial cells involves TAK1. J Biol Chem. (2000) 275:22064–8. doi: 10.1074/jbc.M910346199
193. Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, et al. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci USA. (2009) 106:5282–7. doi: 10.1073/pnas.0810909106
194. Niu J, Shi Y, Tan G, Yang CH, Fan M, Pfeffer LM, et al. DNA damage induces NF-kappaB-dependent microRNA-21 up-regulation and promotes breast cancer cell invasion. J Biol Chem. (2012) 287:21783–95. doi: 10.1074/jbc.M112.355495
195. Zhang X, Liu S, Hu T, Liu S, He Y, Sun S. Up-regulated microRNA-143 transcribed by nuclear factor kappa B enhances hepatocarcinoma metastasis by repressing fibronectin expression. Hepatology (2009) 50:490–9. doi: 10.1002/hep.23008
196. Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. (2007) 179:5082–9. doi: 10.4049/jimmunol.179.8.5082
197. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. (2006) 103:12481–6. doi: 10.1073/pnas.0605298103
198. Scisciani C, Vossio S, Guerrieri F, Schinzari V, De Iaco R, D'Onorio de Meo P, et al. Transcriptional regulation of miR-224 upregulated in human HCCs by NFkappaB inflammatory pathways. J Hepatol. (2012) 56:855–61. doi: 10.1016/j.jhep.2011.11.017
199. Ma X, Becker Buscaglia LE, Barker JR, Li Y. MicroRNAs in NF-κB signaling. J Mol Cell Biol. (2011) 3:159–66. doi: 10.1093/jmcb/mjr007
200. Williams JL, Garcia J, Harrich D, Pearson L, Wu F, Gaynor R. Lymphoid specific gene expression of the adenovirus early region 3 promoter is mediated by NF-kappa B binding motifs. EMBO J. (1990) 9:4435–42.
201. Sambucetti LC, Cherrington JM, Wilkinson GW, Mocarski ES. NF-kappa B activation of the cytomegalovirus enhancer is mediated by a viral transactivator and by T cell stimulation. EMBO J. (1989) 8:4251–8.
202. Sugano N, Chen W, Roberts ML, Cooper NR. Epstein-Barr virus binding to CD21 activates the initial viral promoter via NF-kappaB induction. J Exp Med. (1997) 186:731–7.
203. Kwon JA, Rho HM. Hepatitis B viral core protein activates the hepatitis B viral enhancer II/pregenomic promoter through the nuclear factor kappaB binding site. Biochem Cell Biol. (2002) 80:445–55. doi: 10.1139/o02-133
204. La Frazia S, Amici C, Santoro MG. Antiviral activity of proteasome inhibitors in herpes simplex virus-1 infection: role of nuclear factor-kappaB. Antivir Ther. (2006) 11:995–1004. Available online at: https://www.intmedpress.com/serveFile.cfm?sUID=36cd9950-354c-4e68-b538-bb03248cda5f&origin=publication_detail
205. Fontaine V, van der Meijden E, de Graaf J, ter Schegget J, Struyk L. A functional NF-kappaB binding site in the human papillomavirus type 16 long control region. Virology (2000) 272:40–9. doi: 10.1006/viro.2000.0363
206. Kanno M, Fromental C, Staub A, Ruffenach F, Davidson I, Chambon P. The SV40 TC-II(kappa B) and the related H-2Kb enhansons exhibit different cell type specific and inducible proto-enhancer activities, but the SV40 core sequence and the AP-2 binding site have no enhanson properties. EMBO J. (1989) 8:4205–14.
207. Gilmore TD. Rel/NF-kappa B Transcription Factors [web page]. [cited 2018]. Available online at: http://people.bu.edu/gilmore/nf-kb/target/index.html.
208. Jia Z, Zhang X, Guan N, Bo X, Barnes MR, Luo Z. Gene Ranking of RNA-Seq Data via Discriminant Non-Negative Matrix Factorization. PLoS ONE (2015) 10:e0137782. doi: 10.1371/journal.pone.0137782
209. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. (2005) 102:15545–50. doi: 10.1073/pnas.0506580102
211. Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta (2014) 1843:2563–82. doi: 10.1016/j.bbamcr.2014.05.014
212. Natarajan V, Komarov AP, Ippolito T, Bonneau K, Chenchik AA, Gudkov AV. Peptides genetically selected for NF-kappaB activation cooperate with oncogene Ras and model carcinogenic role of inflammation. Proc Natl Acad Sci USA. (2014) 111:E474–83. doi: 10.1073/pnas.1311945111
213. Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene (2003) 22:9007. doi: 10.1038/sj.onc.1207261
214. Joyce D, Albanese C, Steer J, Fu M, Bouzahzah B, Pestell RG. NF-kappaB and cell-cycle regulation: the cyclin connection. Cytokine Growth Fact Rev. (2001) 12:73–90. doi: 10.1016/S1359-6101(00)00018-6
215. Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. (2014) 4:a014241. doi: 10.1101/cshperspect.a014241
216. Renner F, Schmitz ML. Autoregulatory feedback loops terminating the NF-kappaB response. Trends Biochem Sci. (2009) 34:128–35. doi: 10.1016/j.tibs.2008.12.003
217. Ribatti D, Crivellato E. Giulio Bizzozero and the discovery of platelets. Leukemia Res. (2007) 31:1339–41. doi: 10.1016/j.leukres.2007.02.008
218. Jurk K, Kehrel BE. Platelets: physiology and biochemistry. Semin Thromb Hemost. (2005) 31:381–92. doi: 10.1055/s-2005-916671
219. Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. (2007) 27:1687–93. doi: 10.1161/ATVBAHA.107.141911
220. von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. (2007) 100:27–40. doi: 10.1161/01.RES.0000252802.25497.b7
221. Karshovska E, Weber C, von Hundelshausen P. Platelet chemokines in health and disease. Thromb Haemost. (2013) 110:894–902. doi: 10.1160/TH13-04-0341
222. Hottz ED, Lopes JF, Freitas C, Valls-de-Souza R, Oliveira MF, Bozza MT, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood (2013) 122:3405–14. doi: 10.1182/blood-2013-05-504449
223. Kral JB, Schrottmaier WC, Salzmann M, Assinger A. Platelet interaction with innate immune cells. Transfus Med Hemother. (2016) 43:78–88. doi: 10.1159/000444807
224. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
225. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science (2014) 346:1234–8. doi: 10.1126/science.1256478
226. McDonald B, Kubes P. Neutrophils and intravascular immunity in the liver during infection and sterile inflammation. Toxicol Pathol. (2012) 40:157–65. doi: 10.1177/0192623311427570
227. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
228. Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, et al. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukoc Biol. (2016) 99:153–62. doi: 10.1189/jlb.3A0415-161R
229. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. (2014) 12:2074–88. doi: 10.1111/jth.12710
230. Maugeri N, Capobianco A, Rovere-Querini P, Ramirez GA, Tombetti E, Valle PD, et al. Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci Transl Med. (2018) 10:eaao3089. doi: 10.1126/scitranslmed.aao3089
231. Vogel S, Bodenstein R, Chen Q, Feil S, Feil R, Rheinlaender J, et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J Clin Invest. (2015) 125:4638–54. doi: 10.1172/JCI81660
232. Murthy P, Durco F, Miller-Ocuin JL, Takedai T, Shankar S, Liang X, et al. The NLRP3 inflammasome and bruton's tyrosine kinase in platelets co-regulate platelet activation, aggregation, and in vitro thrombus formation. Biochem Biophys Res Commun. (2017) 483:230–6. doi: 10.1016/j.bbrc.2016.12.161
233. Gupta N, Sahu A, Prabhakar A, Chatterjee T, Tyagi T, Kumari B, et al. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc Natl Acad Sci USA. (2017) 114:4763–8. doi: 10.1073/pnas.1620458114
234. Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of Toll-like receptor molecules on human platelets. Immunol Cell Biol. (2005) 83:196–8. doi: 10.1111/j.1440-1711.2005.01314.x
235. Anabel AS, Eduardo PC, Pedro Antonio HC, Carlos SM, Juana NM, Honorio TA, et al. Human platelets express Toll-like receptor 3 and respond to poly I:C. Hum Immunol. (2014) 75:1244–51. doi: 10.1016/j.humimm.2014.09.013
236. Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, et al. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood (2014) 124:791–802. doi: 10.1182/blood-2013-11-536003
237. Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, et al. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J Immunol. (2009) 182:7997–8004. doi: 10.4049/jimmunol.0802884
238. Stark RJ, Aghakasiri N, Rumbaut RE. Platelet-derived Toll-like receptor 4 (Tlr-4) is sufficient to promote microvascular thrombosis in endotoxemia. PLoS ONE (2012) 7:e41254. doi: 10.1371/journal.pone.0041254
239. Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional Toll-like receptor-4. Blood (2005) 106:2417–23. doi: 10.1182/blood-2005-03-0916
240. Rondina MT, Weyrich AS, Zimmerman GA. Platelets as cellular effectors of inflammation in vascular diseases. Circ Res. (2013) 112:1506–19. doi: 10.1161/CIRCRESAHA.113.300512
241. Fitch-Tewfik JL, Flaumenhaft R. Platelet granule exocytosis: a comparison with chromaffin cells. Front Endocrinol. (2013) 4:77. doi: 10.3389/fendo.2013.00077
242. Ray DM, Spinelli SL, Pollock SJ, Murant TI, O'Brien JJ, Blumberg N, et al. Peroxisome proliferator-activated receptor gamma and retinoid X receptor transcription factors are released from activated human platelets and shed in microparticles. Thromb Haemost. (2008) 99:86–95. doi: 10.1160/TH07-05-0328
243. Melki I, Tessandier N, Zufferey A, Boilard E. Platelet microvesicles in health and disease. Platelets (2017) 28:214–21. doi: 10.1080/09537104.2016.1265924
244. Berthet J, Damien P, Hamzeh-Cognasse H, Pozzetto B, Garraud O, Cognasse F. Toll-like receptor 4 signal transduction in platelets: novel pathways. Br J Haematol. (2010) 151:89–92. doi: 10.1111/j.1365-2141.2010.08292.x
245. Spinelli SL, Casey AE, Pollock SJ, Gertz JM, McMillan DH, Narasipura SD, et al. Platelets and megakaryocytes contain functional nuclear factor-kappaB. Arterioscler Thromb Vasc Biol. (2010) 30:591–8. doi: 10.1161/atvbaha.109.197343
246. Weyrich AS, Denis MM, Schwertz H, Tolley ND, Foulks J, Spencer E, et al. mTOR-dependent synthesis of Bcl-3 controls the retraction of fibrin clots by activated human platelets. Blood (2007) 109:1975–83. doi: 10.1182/blood-2006-08-042192
247. Weyrich AS, Dixon DA, Pabla R, Elstad MR, McIntyre TM, Prescott SM, et al. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc Natl Acad Sci USA. (1998) 95:5556–61. doi: 10.1073/pnas.95.10.5556
248. Liu F, Morris S, Epps J, Carroll R. Demonstration of an activation regulated NF-kappaB/I-kappaBalpha complex in human platelets. Thromb Res. (2002) 106:199–203. doi: 10.1016/S0049-3848(02)00130-5
249. Malaver E, Romaniuk MA, D'Atri LP, Pozner RG, Negrotto S, Benzadon R, et al. NF-kappaB inhibitors impair platelet activation responses. J Thromb Haemost. (2009) 7:1333–43. doi: 10.1111/j.1538-7836.2009.03492.x
250. Gambaryan S, Kobsar A, Rukoyatkina N, Herterich S, Geiger J, Smolenski A, et al. Thrombin and collagen induce a feedback inhibitory signaling pathway in platelets involving dissociation of the catalytic subunit of protein kinase A from an NFkappaB-IkappaB complex. J Biol Chem. (2010) 285:18352–63. doi: 10.1074/jbc.M109.077602
251. Karim ZA, Zhang J, Banerjee M, Chicka MC, Al Hawas R, Hamilton TR, et al. IkappaB kinase phosphorylation of SNAP-23 controls platelet secretion. Blood (2013) 121:4567–74. doi: 10.1182/blood-2012-11-470468
252. Grundler K, Rotter R, Tilley S, Pircher J, Czermak T, Yakac M, et al. The proteasome regulates collagen-induced platelet aggregation via nuclear-factor-kappa-B (NFkB) activation. Thromb Res. (2016) 148:15–22. doi: 10.1016/j.thromres.2016.10.009
253. Chen Y, Aardema J, Kale S, Whichard ZL, Awomolo A, Blanchard E, et al. Loss of the F-BAR protein CIP4 reduces platelet production by impairing membrane-cytoskeleton remodeling. Blood (2013) 122:1695–706. doi: 10.1182/blood-2013-03-484550
254. Wei S, Wang H, Zhang G, Lu Y, An X, Ren S, et al. Platelet IkappaB kinase-beta deficiency increases mouse arterial neointima formation via delayed glycoprotein Ibalpha shedding. Arterioscler Thromb Vasc Biol. (2013) 33:241–8. doi: 10.1161/ATVBAHA.112.300781
255. Lu WJ, Lee JJ, Chou DS, Jayakumar T, Fong TH, Hsiao G, et al. A novel role of andrographolide, an NF-kappa B inhibitor, on inhibition of platelet activation: the pivotal mechanisms of endothelial nitric oxide synthase/cyclic GMP. J Mol Med. (2011) 89:1261–73. doi: 10.1007/s00109-011-0800-0
256. Lu WJ, Lin KH, Hsu MJ, Chou DS, Hsiao G, Sheu JR. Suppression of NF-kappaB signaling by andrographolide with a novel mechanism in human platelets: regulatory roles of the p38 MAPK-hydroxyl radical-ERK2 cascade. Biochem Pharmacol. (2012) 84:914–24. doi: 10.1016/j.bcp.2012.06.030
257. Paul M, Kemparaju K, Girish KS. Inhibition of constitutive NF-kappaB activity induces platelet apoptosis via ER stress. Biochem Biophys Res Commun. (2017) 493:1471–7. doi: 10.1016/j.bbrc.2017.10.011
258. Rauert-Wunderlich H, Siegmund D, Maier E, Giner T, Bargou RC, Wajant H, et al. The IKK inhibitor Bay 11-7082 induces cell death independent from inhibition of activation of NFκB transcription factors. PLoS ONE (2013) 8:e59292. doi: 10.1371/journal.pone.0059292
259. Krishnan N, Bencze G, Cohen P, Tonks NK. The anti-inflammatory compound BAY 11-7082 is a potent inhibitor of protein tyrosine phosphatases. FEBS J. (2013) 280:2830–41. doi: 10.1111/febs.12283
260. Lee HS, Kim SD, Lee WM, Endale M, Kamruzzaman SM, Oh WJ, et al. A noble function of BAY 11-7082: Inhibition of platelet aggregation mediated by an elevated cAMP-induced VASP, and decreased ERK2/JNK1 phosphorylations. Eur J Pharmacol. (2010) 627:85–91. doi: 10.1016/j.ejphar.2009.11.005
261. Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature (2017) 544:105–9. doi: 10.1038/nature21706
262. Geddis AE, Fox NE, Tkachenko E, Kaushansky K. Endomitotic megakaryocytes that form a bipolar spindle exhibit cleavage furrow ingression followed by furrow regression. Cell Cycle (2007) 6:455–60. doi: 10.4161/cc.6.4.3836
263. Lordier L, Bluteau D, Jalil A, Legrand C, Pan J, Rameau P, et al. RUNX1-induced silencing of non-muscle myosin heavy chain IIB contributes to megakaryocyte polyploidization. Nat Commun. (2012) 3:717. doi: 10.1038/ncomms1704
264. Machlus KR, Thon JN, Italiano JE Jr. Interpreting the developmental dance of the megakaryocyte: a review of the cellular and molecular processes mediating platelet formation. Br J Haematol. (2014) 165:227–36. doi: 10.1111/bjh.12758
265. Zimmet J, Ravid K. Polyploidy: occurrence in nature, mechanisms, and significance for the megakaryocyte-platelet system. Exp Hematol. (2000) 28:3–16. doi: 10.1016/S0301-472X(99)00124-1
266. Odell TT Jr, Jackson CW, Friday TJ. Megakaryocytopoiesis in rats with special reference to polyploidy. Blood (1970) 35:775–82.
267. Radley JM, Haller CJ. The demarcation membrane system of the megakaryocyte: a misnomer? Blood (1982) 60:213–9.
268. Schulze H, Korpal M, Hurov J, Kim SW, Zhang J, Cantley LC, et al. Characterization of the megakaryocyte demarcation membrane system and its role in thrombopoiesis. Blood (2006) 107:3868–75. doi: 10.1182/blood-2005-07-2755
269. Guo T, Wang X, Qu Y, Yin Y, Jing T, Zhang Q. Megakaryopoiesis and platelet production: insight into hematopoietic stem cell proliferation and differentiation. Stem Cell Invest. (2015) 2:3. doi: 10.3978/j.issn.2306-9759.2015.02.01
270. Behnke O. An electron microscope study of the megacaryocyte of the rat bone marrow. I. The development of the demarcation membrane system and the platelet surface coat. J Ultrastruct Res. (1968) 24:412–33. doi: 10.1016/S0022-5320(68)80046-2
271. Bender M, Thon JN, Ehrlicher AJ, Wu S, Mazutis L, Deschmann E, et al. Microtubule sliding drives proplatelet elongation and is dependent on cytoplasmic dynein. Blood (2015) 125:860–8. doi: 10.1182/blood-2014-09-600858
272. Italiano JE Jr, Lecine P, Shivdasani RA, Hartwig JH. Blood platelets are assembled principally at the ends of proplatelet processes produced by differentiated megakaryocytes. J Cell Biol. (1999) 147:1299–312. doi: 10.1083/jcb.147.6.1299
273. Kile BT. The role of apoptosis in megakaryocytes and platelets. Br J Haematol. (2014) 165:217–26. doi: 10.1111/bjh.12757
274. Zauli G, Vitale M, Falcieri E, Gibellini D, Bassini A, Celeghini C, et al. In vitro senescence and apoptotic cell death of human megakaryocytes. Blood (1997) 90:2234–43.
275. Radley JM, Haller CJ. Fate of senescent megakaryocytes in the bone marrow. Br J Haematol. (1983) 53:277–87. doi: 10.1111/j.1365-2141.1983.tb02022.x
276. Falcieri E, Bassini A, Pierpaoli S, Luchetti F, Zamai L, Vitale M, et al. Ultrastructural characterization of maturation, platelet release, and senescence of human cultured megakaryocytes. Anat Rec. (2000) 258:90–9. doi: 10.1002/(SICI)1097-0185(20000101)258:1<90::AID-AR10>3.0.CO;2-G
277. Beaulieu LM, Lin E, Morin KM, Tanriverdi K, Freedman JE. Regulatory effects of TLR2 on megakaryocytic cell function. Blood (2011) 117:5963–74. doi: 10.1182/blood-2010-09-304949
278. Undi RB, Sarvothaman S, Narasaiah K, Gutti U, Gutti RK. Toll-like receptor 2 signalling: Significance in megakaryocyte development through wnt signalling cross-talk and cytokine induction. Cytokine (2016) 83:245–9. doi: 10.1016/j.cyto.2016.05.007
279. Tacchini-Cottier F, Vesin C, Redard M, Buurman W, Piguet PF. Role of TNFR1 and TNFR2 in TNF-induced platelet consumption in mice. J Immunol. (1998) 160:6182–6.
280. Beaulieu LM, Lin E, Mick E, Koupenova M, Weinberg EO, Kramer CD, et al. Interleukin 1 receptor 1 and interleukin 1beta regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arterioscler Thromb Vasc Biol. (2014) 34:552–64. doi: 10.1161/atvbaha.113.302700
281. Nishimura S, Nagasaki M, Kunishima S, Sawaguchi A, Sakata A, Sakaguchi H, et al. IL-1alpha induces thrombopoiesis through megakaryocyte rupture in response to acute platelet needs. J Cell Biol. (2015) 209:453–66. doi: 10.1083/jcb.201410052
282. Nakayama K. Expression of IL-6, IL-6 receptor and its signal transducer gp130 mRNAs in megakaryocytic cell lines. Leuk Lymphoma (1998) 29:399–405. doi: 10.3109/10428199809068576
283. Navarro S, Debili N, Le Couedic JP, Klein B, Breton-Gorius J, Doly J, et al. Interleukin-6 and its receptor are expressed by human megakaryocytes: In vitro effects on proliferation and endoreplication. Blood (1991) 77:461–71.
284. Zhang Y, Sun S, Wang Z, Thompson A, Kaluzhny Y, Zimmet J, et al. Signaling by the Mpl receptor involves IKK and NF-kappaB. J Cell Biochem. (2002) 85:523–35. doi: 10.1002/jcb.10141
285. Angchaisuksiri P, Grigus SR, Carlson PL, Krystal GW, Dessypris EN. Secretion of a unique peptide from interleukin-2-stimulated natural killer cells that induces endomitosis in immature human megakaryocytes. Blood (2002) 99:130–6. doi: 10.1182/blood.V99.1.130
286. Machlus KR, Italiano JE Jr. The incredible journey: from megakaryocyte development to platelet formation. J Cell Biol. (2013) 201:785–96. doi: 10.1083/jcb.201304054
287. Trinh BQ, Barengo N, Kim SB, Lee JS, Zweidler-McKay PA, Naora H. The homeobox gene DLX4 regulates erythro-megakaryocytic differentiation by stimulating IL-1beta and NF-kappaB signaling. J Cell Sci. (2015) 128:3055–67. doi: 10.1242/jcs.168187
288. Wickenhauser C, Lorenzen J, Thiele J, Hillienhof A, Jungheim K, Schmitz B, et al. Secretion of cytokines (interleukins-1 alpha,−3, and−6 and granulocyte-macrophage colony-stimulating factor) by normal human bone marrow megakaryocytes. Blood (1995) 85:685–91.
289. Jiang S, Levine JD, Fu Y, Deng B, London R, Groopman JE, et al. Cytokine production by primary bone marrow megakaryocytes. Blood (1994) 84:4151–6.
290. Zheng C, Yang R, Han Z, Zhou B, Liang L, Lu M. TPO-independent megakaryocytopoiesis. Crit Rev Oncol Hematol. (2008) 65:212–22. doi: 10.1016/j.critrevonc.2007.11.003
291. Avecilla ST, Hattori K, Heissig B, Tejada R, Liao F, Shido K, et al. Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med. (2004) 10:64–71. doi: 10.1038/nm973
292. Chuen CK, Li K, Yang M, Fok TF, Li CK, Chui CM, et al. Interleukin-1beta up-regulates the expression of thrombopoietin and transcription factors c-Jun, c-Fos, GATA-1, and NF-E2 in megakaryocytic cells. J Lab Clin Med. (2004) 143:75–88. doi: 10.1016/j.lab.2003.09.006
293. Ricciardi S, Miluzio A, Brina D, Clarke K, Bonomo M, Aiolfi R, et al. Eukaryotic translation initiation factor 6 is a novel regulator of reactive oxygen species-dependent megakaryocyte maturation. J Thromb Haemost. (2015) 13:2108–18. doi: 10.1111/jth.13150
294. Haas S, Hansson J, Klimmeck D, Loeffler D, Velten L, Uckelmann H, et al. Inflammation-induced emergency megakaryopoiesis driven by hematopoietic stem cell-like megakaryocyte progenitors. Cell Stem Cell (2015) 17:422–34. doi: 10.1016/j.stem.2015.07.007
295. Pries AR, Secomb TW, Gaehtgens P. The endothelial surface layer. Pflugers Arch. (2000) 440:653–66. doi: 10.1007/s004240000307
296. Yau JW, Teoh H, Verma S. Endothelial cell control of thrombosis. BMC Cardiovasc Disord. (2015) 15:130. doi: 10.1186/s12872-015-0124-z
297. Levi M, van der Poll T. Two-way interactions between inflammation and coagulation. Trends Cardiovasc Med. (2005) 15:254–9. doi: 10.1016/j.tcm.2005.07.004
298. Rahman A, Fazal F. Blocking NF-kappaB: an inflammatory issue. Proc Am Thorac Soc. (2011) 8:497–503. doi: 10.1513/pats.201101-009MW
299. Soh UJ, Dores MR, Chen B, Trejo J. Signal transduction by protease-activated receptors. Br J Pharmacol. (2010) 160:191–203. doi: 10.1111/j.1476-5381.2010.00705.x
300. Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. (2001) 11:372–7. doi: 10.1016/S0962-8924(01)02064-5
301. Delekta PC, Apel IJ, Gu S, Siu K, Hattori Y, McAllister-Lucas LM, et al. Thrombin-dependent NF-κB activation and monocyte/endothelial adhesion are mediated by the CARMA3·Bcl10·MALT1 signalosome. J Biol Chem. (2010) 285:41432–42. doi: 10.1074/jbc.M110.158949
302. Luedde T, Assmus U, Wuestefeld T, Zu VA, Roskams T, Schmidt-Supprian M, et al. Deletion of IKK2 in hepatocytes does not sensitize these cells to TNF-induced apoptosis but protects from ischemia/reperfusion-injury. J Clin Invest. (2005). doi: 10.1172/JCI23493
303. Tiruppathi C, Naqvi T, Sandoval R, Mehta D, Malik AB. Synergistic effects of tumor necrosis factor-alpha and thrombin in increasing endothelial permeability. Am J Physiol Lung Cell Mol Physiol. (2001) 281:L958–68. doi: 10.1152/ajplung.2001.281.4.L958
304. Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. (2009) 78:539–52. doi: 10.1016/j.bcp.2009.04.029
305. Rahman A, Anwar KN, True AL, Malik AB. Thrombin-induced p65 homodimer binding to downstream NF-kappa B site of the promoter mediates endothelial ICAM-1 expression and neutrophil adhesion. J Immunol. (1999) 162:5466–76.
306. Sugama Y, Tiruppathi C, offakidevi K, Andersen TT, Fenton JWII, Malik AB. Thrombin-induced expression of endothelial P-selectin and intercellular adhesion molecule-1: a mechanism for stabilizing neutrophil adhesion. J Cell Biol. (1992) 119:935–44. doi: 10.1083/jcb.119.4.935
307. Xue J, Thippegowda PB, Hu G, Bachmaier K, Christman JW, Malik AB, et al. NF-kappaB regulates thrombin-induced ICAM-1 gene expression in cooperation with NFAT by binding to the intronic NF-kappaB site in the ICAM-1 gene. Physiol Genomics (2009) 38:42–53. doi: 10.1152/physiolgenomics.00012.2009
308. Denk A, Goebeler M, Schmid S, Berberich I, Ritz O, Lindemann D, et al. Activation of NF-kappa B via the Ikappa B kinase complex is both essential and sufficient for proinflammatory gene expression in primary endothelial cells. J Biol Chem. (2001) 276:28451–8. doi: 10.1074/jbc.M102698200
309. Dryden NH, Sperone A, Martin-Almedina S, Hannah RL, Birdsey GM, Khan ST, et al. The transcription factor Erg controls endothelial cell quiescence by repressing activity of nuclear factor (NF)-kappaB p65. J Biol Chem. (2012) 287:12331–42. doi: 10.1074/jbc.M112.346791
310. Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol. (2009) 10:53–62. doi: 10.1038/nrm2596
311. Zhou J, Li YS, Chien S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler Thromb Vasc Biol. (2014) 34:2191–8. doi: 10.1161/ATVBAHA.114.303422
312. Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. The subendothelial extracellular matrix modulates NF-kappaB activation by flow: a potential role in atherosclerosis. J Cell Biol. (2005) 169:191–202. doi: 10.1083/jcb.200410073
313. Petzold T, Orr AW, Hahn C, Jhaveri KA, Parsons JT, Schwartz MA. Focal adhesion kinase modulates activation of NF-kappaB by flow in endothelial cells. Am J Physiol Cell Physiol. (2009) 297:C814–22. doi: 10.1152/ajpcell.00226.2009
314. Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature (2005) 437:426. doi: 10.1038/nature03952
315. Nightingale T, Cutler D. The secretion of von Willebrand factor from endothelial cells; an increasingly complicated story. J Thromb Haemost. (2013) 11(Suppl. 1):192–201. doi: 10.1111/jth.12225
316. Möller K, Adolph O, Grünow J, Elrod J, Popa M, Ghosh S, et al. Mechanism and functional impact of CD40 ligand-induced von Willebrand factor release from endothelial cells. Thromb Haemost. (2015) 113:1095–108. doi: 10.1160/th14-04-0336
317. Michels A, Albánez S, Mewburn J, Nesbitt K, Gould TJ, Liaw PC, et al. Histones link inflammation and thrombosis through the induction of Weibel-Palade body exocytosis. J Thromb Haemost. (2016) 14:2274–86. doi: 10.1111/jth.13493
318. Carnevale R, Raparelli V, Nocella C, Bartimoccia S, Novo M, Severino A, et al. Gut-derived endotoxin stimulates factor VIII secretion from endothelial cells. Implications for hypercoagulability in cirrhosis. J Hepatol. (2017) 67:950–6. doi: 10.1016/j.jhep.2017.07.002
319. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood (2014) 123:377–90. doi: 10.1182/blood-2013-04-495887
320. Turner NA, Nolasco L, Ruggeri ZM, Moake JL. Endothelial cell ADAMTS-13 and VWF: production, release, and VWF string cleavage. Blood (2009) 114:5102–11. doi: 10.1182/blood-2009-07-231597
321. Libby P, Simon DI. Inflammation and thrombosis: the clot thickens. Circulation (2001) 103:1718–20. doi: 10.1161/01.CIR.103.13.1718
322. Ye X, Ding J, Zhou X, Chen G, Liu SF. Divergent roles of endothelial NF-kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med. (2008) 205:1303–15. doi: 10.1084/jem.20071393
323. Song D, Ye X, Xu H, Liu SF. Activation of endothelial intrinsic NF-{kappa}B pathway impairs protein C anticoagulation mechanism and promotes coagulation in endotoxemic mice. Blood (2009) 114:2521–9. doi: 10.1182/blood-2009-02-205914
324. Kisseleva T, Song L, Vorontchikhina M, Feirt N, Kitajewski J, Schindler C. NF-kappaB regulation of endothelial cell function during LPS-induced toxemia and cancer. J Clin Invest. (2006) 116:2955–63. doi: 10.1172/JCI27392
325. Liu G, Ye X, Miller EJ, Liu SF. NF-kappaB-to-AP-1 switch: a mechanism regulating transition from endothelial barrier injury to repair in endotoxemic mice. Sci Rep. (2014) 4:5543. doi: 10.1038/srep05543
326. Ashida N, Senbanerjee S, Kodama S, Foo SY, Coggins M, Spencer JA, et al. IKKbeta regulates essential functions of the vascular endothelium through kinase-dependent and -independent pathways. Nat Commun. (2011) 2:318. doi: 10.1038/ncomms1317
327. Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJ, Kardakaris R, et al. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. (2008) 8:372–83. doi: 10.1016/j.cmet.2008.08.016
328. Xiao L, Dudley AC. Fine-tuning vascular fate during endothelial-mesenchymal transition. J Pathol. (2017) 241:25–35. doi: 10.1002/path.4814
329. Jackson AO, Zhang J, Jiang Z, Yin K. Endothelial-to-mesenchymal transition: A novel therapeutic target for cardiovascular diseases. Trends Cardiovasc Med. (2017) 27:383–93. doi: 10.1016/j.tcm.2017.03.003
330. Nie L, Lyros O, Medda R, Jovanovic N, Schmidt JL, Otterson MF, et al. Endothelial-mesenchymal transition in normal human esophageal endothelial cells cocultured with esophageal adenocarcinoma cells: role of IL-1beta and TGF-beta2. Am J Physiol Cell Physiol. (2014) 307:C859–77. doi: 10.1152/ajpcell.00081.2014
331. Liu Y, Hu ZF, Liao HH, Liu W, Liu J, Ma ZG, et al. Toll-like receptor 5 deficiency attenuates interstitial cardiac fibrosis and dysfunction induced by pressure overload by inhibiting inflammation and the endothelial-mesenchymal transition. Biochim Biophys Acta (2015) 1852:2456–66. doi: 10.1016/j.bbadis.2015.08.013
332. Finsterwalder, R, Ganesan, MK, Leb, H, Habertheuer, A, Basílio, J, Lang, I, et al. Hypoxia/reperfusion predisposes to atherosclerosis. PLoS ONE (2018) 13:e0205067. doi: 10.1371/journal.pone.0205067
333. Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. (2009) 84:617–27. doi: 10.1016/j.ajhg.2009.04.007
334. Bentzon JF, Weile C, Sondergaard CS, Hindkjaer J, Kassem M, Falk E. Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler Thromb Vasc Biol. (2006) 26:2696–702. doi: 10.1161/01.ATV.0000247243.48542.9d
335. Gomez D, Shankman LS, Nguyen AT, Owens GK. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat Methods (2013) 10:171–7. doi: 10.1038/nmeth.2332
336. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. (2016) 118:692–702. doi: 10.1161/circresaha.115.306361
337. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. (2015) 21:628–37. doi: 10.1038/nm.3866
338. Tabas I, Garcia-Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. (2015) 209:13–22. doi: 10.1083/jcb.201412052
339. Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. (2012) 95:156–64. doi: 10.1093/cvr/cvs115
340. Durgin BG, Cherepanova OA, Gomez D, Karaoli T, Alencar GF, Butcher JT, et al. Smooth muscle cell-specific deletion of Col15a1 unexpectedly leads to impaired development of advanced atherosclerotic lesions. Am J Physiol Heart Circ Physiol. (2017) 312:H943–58. doi: 10.1152/ajpheart.00029.2017
341. Nguyen AT, Gomez D, Bell RD, Campbell JH, Clowes AW, Gabbiani G, et al. Smooth muscle cell plasticity: fact or fiction? Circ Res. (2013) 112:17–22. doi: 10.1161/CIRCRESAHA.112.281048
342. Dai X, Thiagarajan D, Fang J, Shen J, Annam NP, Yang Z, et al. SM22alpha suppresses cytokine-induced inflammation and the transcription of NF-kappaB inducing kinase (Nik) by modulating SRF transcriptional activity in vascular smooth muscle cells. PLoS ONE (2017) 12:e0190191. doi: 10.1371/journal.pone.0190191
343. Bourcier T, Sukhova G, Libby P. The nuclear factor kappa-B signaling pathway participates in dysregulation of vascular smooth muscle cells In vitro and in human atherosclerosis. J Biol Chem. (1997) 272:15817–24. doi: 10.1074/jbc.272.25.15817
344. Branen L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S. Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. (2004) 24:2137–42. doi: 10.1161/01.ATV.0000143933.20616.1b
345. Maddahi A, Kruse LS, Chen QW, Edvinsson L. The role of tumor necrosis factor-alpha and TNF-alpha receptors in cerebral arteries following cerebral ischemia in rat. J Neuroinflamm. (2011) 8:107. doi: 10.1186/1742-2094-8-107
346. Martinez-Moreno JM, Herencia C, Montes de Oca A, Munoz-Castaneda JR, Rodriguez-Ortiz ME, Diaz-Tocados JM, et al. Vitamin D modulates tissue factor and protease-activated receptor 2 expression in vascular smooth muscle cells. FASEB J. (2016) 30:1367–76. doi: 10.1096/fj.15-272872
347. Ali MS, Starke RM, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, et al. TNF-alpha induces phenotypic modulation in cerebral vascular smooth muscle cells: implications for cerebral aneurysm pathology. J Cereb Blood Flow Metab. (2013) 33:1564–73. doi: 10.1038/jcbfm.2013.109
348. Davis-Dusenbery BN, Chan MC, Reno KE, Weisman AS, Layne MD, Lagna G, et al. Down-regulation of Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-beta and bone morphogenetic protein 4. J Biol Chem. (2011) 286:28097–110. doi: 10.1074/jbc.M111.236950
349. Akgul C, Moulding DA, Edwards SW. Molecular control of neutrophil apoptosis. FEBS Lett. (2001) 487:318–22. doi: 10.1016/S0014-5793(00)02324-3
350. McDonald PP, Bald A, Cassatella MA. Activation of the NF-kappaB pathway by inflammatory stimuli in human neutrophils. Blood (1997) 89:3421–33.
351. Vancurova I, Miskolci V, Davidson D. NF-kappa B activation in tumor necrosis factor alpha-stimulated neutrophils is mediated by protein kinase Cdelta. Correlation to nuclear Ikappa Balpha. J Biol Chem. (2001) 276:19746–52. doi: 10.1074/jbc.M100234200
352. Ear T, Cloutier A, McDonald PP. Constitutive nuclear expression of the I kappa B kinase complex and its activation in human neutrophils. J Immunol. (2005) 175:1834–42. doi: 10.4049/jimmunol.175.3.1834
353. Kato T, Kitagawa S. Regulation of neutrophil functions by proinflammatory cytokines. Int J Hematol. (2006) 84:205–9. doi: 10.1532/ijh97.06141
354. McDonald PP, Bovolenta C, Cassatella MA. Activation of distinct transcription factors in neutrophils by bacterial LPS, interferon-gamma, and GM-CSF and the necessity to overcome the action of endogenous proteases. Biochemistry (1998) 37:13165–73. doi: 10.1021/bi972539o
355. Druker BJ, Neumann M, Okuda K, Franza BR Jr, Griffin JD. rel Is rapidly tyrosine-phosphorylated following granulocyte-colony stimulating factor treatment of human neutrophils. J Biol Chem. (1994) 269:5387–90.
356. McDonald PP, Cassatella MA. Activation of transcription factor NF-kappa B by phagocytic stimuli in human neutrophils. FEBS Lett. (1997) 412:583–6. doi: 10.1016/S0014-5793(97)00857-0
357. Vollebregt M, Hampton MB, Winterbourn CC. Activation of NF-kappaB in human neutrophils during phagocytosis of bacteria independently of oxidant generation. FEBS Lett. (1998) 432:40–4. doi: 10.1016/S0014-5793(98)00821-7
358. Sabroe I, Dower SK, Whyte MK. The role of Toll-like receptors in the regulation of neutrophil migration, activation, and apoptosis. Clin Infect Dis. (2005) 41 Suppl 7:S421–6. doi: 10.1086/431992
359. Nick JA, Avdi NJ, Young SK, Lehman LA, McDonald PP, Frasch SC, et al. Selective activation and functional significance of p38alpha mitogen-activated protein kinase in lipopolysaccharide-stimulated neutrophils. J Clin Invest. (1999) 103:851–8. doi: 10.1172/jci5257
360. Sugita N, Kimura A, Matsuki Y, Yamamoto T, Yoshie H, Hara K. Activation of transcription factors and IL-8 expression in neutrophils stimulated with lipopolysaccharide from Porphyromonas gingivalis. Inflammation (1998) 22:253–67. doi: 10.1023/A:1022344031223
361. Lotz S, Aga E, Wilde I, van Zandbergen G, Hartung T, Solbach W, et al. Highly purified lipoteichoic acid activates neutrophil granulocytes and delays their spontaneous apoptosis via CD14 and TLR2. J Leukoc Biol. (2004) 75:467–77. doi: 10.1189/jlb.0803360
362. Strassheim D, Asehnoune K, Park JS, Kim JY, He Q, Richter D, et al. Phosphoinositide 3-kinase and Akt occupy central roles in inflammatory responses of Toll-like receptor 2-stimulated neutrophils. J Immunol. (2004) 172:5727–33. doi: 10.4049/jimmunol.172.9.5727
363. Francois S, El Benna J, Dang PM, Pedruzzi E, Gougerot-Pocidalo MA, Elbim C. Inhibition of neutrophil apoptosis by TLR agonists in whole blood: involvement of the phosphoinositide 3-kinase/Akt and NF-kappaB signaling pathways, leading to increased levels of Mcl-1, A1, and phosphorylated Bad. J Immunol. (2005) 174:3633–42. doi: 10.4049/jimmunol.174.6.3633
364. El Kebir D, Damlaj A, Filep JG. Toll-like receptor 9 signaling delays neutrophil apoptosis by increasing transcription of Mcl-1. PLoS ONE (2014) 9:e87006. doi: 10.1371/journal.pone.0087006
365. Jozsef L, Khreiss T, El Kebir D, Filep JG. Activation of TLR-9 induces IL-8 secretion through peroxynitrite signaling in human neutrophils. J Immunol. (2006) 176:1195–202. doi: 10.4049/jimmunol.176.2.1195
366. Ode Y, Aziz M, Wang P. CIRP increases ICAM-1(+) phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis. J Leukoc Biol. (2018) 103:693–707. doi: 10.1002/jlb.3a0817-327rr
367. Xia P, Wang S, Ye B, Du Y, Huang G, Zhu P, et al. Sox2 functions as a sequence-specific DNA sensor in neutrophils to initiate innate immunity against microbial infection. Nat Immunol. (2015) 16:366–75. doi: 10.1038/ni.3117
368. Garcia-Garcia E, Rosales C. Nuclear factor activation by FcgammaR in human peripheral blood neutrophils detected by a novel flow cytometry-based method. J Immunol Methods (2007) 320:104–18. doi: 10.1016/j.jim.2006.12.006
369. Kim CH, Lee KH, Lee CT, Kim YW, Han SK, Shim YS, et al. Aggregation of beta2 integrins activates human neutrophils through the IkappaB/NF-kappaB pathway. J Leukoc Biol. (2004) 75:286–92. doi: 10.1189/jlb.0103038
370. Kettritz R, Choi M, Rolle S, Wellner M, Luft FC. Integrins and cytokines activate nuclear transcription factor-kappaB in human neutrophils. J Biol Chem. (2004) 279:2657–65. doi: 10.1074/jbc.M309778200
371. Lau D, Mollnau H, Eiserich JP, Freeman BA, Daiber A, Gehling UM, et al. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci USA. (2005) 102:431–6. doi: 10.1073/pnas.0405193102
372. Ross EA, Douglas MR, Wong SH, Ross EJ, Curnow SJ, Nash GB, et al. Interaction between integrin alpha9beta1 and vascular cell adhesion molecule-1 (VCAM-1) inhibits neutrophil apoptosis. Blood (2006) 107:1178–83. doi: 10.1182/blood-2005-07-2692
373. Saldanha-Gama RF, Moraes JA, Mariano-Oliveira A, Coelho AL, Walsh EM, Marcinkiewicz C, et al. alpha(9)beta(1) integrin engagement inhibits neutrophil spontaneous apoptosis: involvement of Bcl-2 family members. Biochim Biophys Acta (2010) 1803:848–57. doi: 10.1016/j.bbamcr.2010.03.012
374. Jin R, Yu S, Song Z, Zhu X, Wang C, Yan J, et al. Soluble CD40 ligand stimulates CD40-dependent activation of the beta2 integrin Mac-1 and protein kinase C zeda (PKCzeta) in neutrophils: implications for neutrophil-platelet interactions and neutrophil oxidative burst. PLoS ONE (2013) 8:e64631. doi: 10.1371/journal.pone.0064631
375. Salanova B, Choi M, Rolle S, Wellner M, Luft FC, Kettritz R. Beta2-integrins and acquired glycoprotein IIb/IIIa (GPIIb/IIIa) receptors cooperate in NF-kappaB activation of human neutrophils. J Biol Chem. (2007) 282:27960–9. doi: 10.1074/jbc.M704039200
376. Rubel C, Gomez S, Fernandez GC, Isturiz MA, Caamano J, Palermo MS. Fibrinogen-CD11b/CD18 interaction activates the NF-kappa B pathway and delays apoptosis in human neutrophils. Eur J Immunol. (2003) 33:1429–38. doi: 10.1002/eji.200323512
377. Mariano-Oliveira A, De Freitas MS, Monteiro RQ, Barja-Fidalgo C. Prothrombin fragments containing kringle domains induce migration and activation of human neutrophils. Int J Biochem Cell Biol. (2008) 40:517–29. doi: 10.1016/j.biocel.2007.09.002
378. Kwak SH, Wang XQ, He Q, Fang WF, Mitra S, Bdeir K, et al. Plasminogen activator inhibitor-1 potentiates LPS-induced neutrophil activation through a JNK-mediated pathway. Thromb Haemost. (2006) 95:829–35. doi: 10.1160/TH05-12-0782
379. Bubici C, Papa S, Dean K, Franzoso G. Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: molecular basis and biological significance. Oncogene (2006) 25:6731–48. doi: 10.1038/sj.onc.1209936
380. Strassheim D, Asehnoune K, Park JS, Kim JY, He Q, Richter D, et al. Modulation of bone marrow-derived neutrophil signaling by H2O2: disparate effects on kinases, NF-kappaB, and cytokine expression. Am J Physiol Cell Physiol. (2004) 286:C683–92. doi: 10.1152/ajpcell.00296.2003
381. Zmijewski JW, Zhao X, Xu Z, Abraham E. Exposure to hydrogen peroxide diminishes NF-kappaB activation, IkappaB-alpha degradation, and proteasome activity in neutrophils. Am J Physiol Cell Physiol. (2007) 293:C255–66. doi: 10.1152/ajpcell.00618.2006
382. Zmijewski JW, Lorne E, Banerjee S, Abraham E. Participation of mitochondrial respiratory complex III in neutrophil activation and lung injury. Am J Physiol Lung Cell Mol Physiol. (2009) 296:L624–34. doi: 10.1152/ajplung.90522.2008
383. Zmijewski JW, Lorne E, Zhao X, Tsuruta Y, Sha Y, Liu G, et al. Antiinflammatory effects of hydrogen peroxide in neutrophil activation and acute lung injury. Am J Respir Crit Care Med. (2009) 179:694–704. doi: 10.1164/rccm.200806-851OC
384. Zmijewski JW, Lorne E, Zhao X, Tsuruta Y, Sha Y, Liu G, et al. Mitochondrial respiratory complex I regulates neutrophil activation and severity of lung injury. Am J Respir Crit Care Med. (2008) 178:168–79. doi: 10.1164/rccm.200710-1602OC
385. Freitas M, Gomes A, Porto G, Fernandes E. Nickel induces oxidative burst, NF-kappaB activation and interleukin-8 production in human neutrophils. J Biol Inorg Chem. (2010) 15:1275–83. doi: 10.1007/s00775-010-0685-3
386. Lorne E, Zmijewski JW, Zhao X, Liu G, Tsuruta Y, Park YJ, et al. Role of extracellular superoxide in neutrophil activation: interactions between xanthine oxidase and TLR4 induce proinflammatory cytokine production. Am J Physiol Cell Physiol. (2008) 294:C985–93. doi: 10.1152/ajpcell.00454.2007
387. Mitra S, Abraham E. Participation of superoxide in neutrophil activation and cytokine production. Biochim Biophys Acta (2006) 1762:732–41. doi: 10.1016/j.bbadis.2006.06.011
388. Reyes-Quiroz ME, Alba G, Saenz J, Geniz I, Jimenez J, Martin-Nieto J, et al. Platelet-activating factor and hydrogen peroxide exert a dual modulatory effect on the transcription of LXRalpha and its target genes in human neutrophils. Int Immunopharmacol. (2016) 38:357–66. doi: 10.1016/j.intimp.2016.05.001
389. Singh AK, Awasthi D, Dubey M, Nagarkoti S, Kumar A, Chandra T, et al. High oxidative stress adversely affects NFkappaB mediated induction of inducible nitric oxide synthase in human neutrophils: Implications in chronic myeloid leukemia. Nitric Oxide (2016) 58:28–41. doi: 10.1016/j.niox.2016.06.002
390. Endo D, Saito T, Umeki Y, Suzuki K, Aratani Y. Myeloperoxidase negatively regulates the expression of proinflammatory cytokines and chemokines by zymosan-induced mouse neutrophils. Inflamm Res. (2016) 65:151–9. doi: 10.1007/s00011-015-0899-5
391. Fortenberry JD, Owens ML, Chen NX, Brown LA. S-nitrosoglutathione inhibits TNF-alpha-induced NFkappaB activation in neutrophils. Inflamm Res. (2001) 50:89–95. doi: 10.1016/j.bmc.2007.02.004
392. Welters ID, Menzebach A, Goumon Y, Cadet P, Menges T, Hughes TK, et al. Morphine inhibits NF-kappaB nuclear binding in human neutrophils and monocytes by a nitric oxide-dependent mechanism. Anesthesiology (2000) 92:1677–84. doi: 10.1097/00000542-200006000-00027
393. Riedemann NC, Guo RF, Bernacki KD, Reuben JS, Laudes IJ, Neff TA, et al. Regulation by C5a of neutrophil activation during sepsis. Immunity (2003) 19:193–202. doi: 10.1016/S1074-7613(03)00206-1
394. Ward C, Dransfield I, Murray J, Farrow SN, Haslett C, Rossi AG. Prostaglandin D2 and its metabolites induce caspase-dependent granulocyte apoptosis that is mediated via inhibition of I kappa B alpha degradation using a peroxisome proliferator-activated receptor-gamma-independent mechanism. J Immunol. (2002) 168:6232–43. doi: 10.4049/jimmunol.168.12.6232
395. Sokoloski JA, Sartorelli AC, Rosen CA, Narayanan R. Antisense oligonucleotides to the p65 subunit of NF-kappa B block CD11b expression and alter adhesion properties of differentiated HL-60 granulocytes. Blood (1993) 82:625–32.
396. Zhou X, Gao XP, Fan J, Liu Q, Anwar KN, Frey RS, et al. LPS activation of Toll-like receptor 4 signals CD11b/CD18 expression in neutrophils. Am J Physiol Lung Cell Mol Physiol. (2005) 288:L655–62. doi: 10.1152/ajplung.00327.2004
397. Al-Mohanna F, Saleh S, Parhar RS, Collison K. IL-12-dependent nuclear factor-kappaB activation leads to de novo synthesis and release of IL-8 and TNF-alpha in human neutrophils. J Leukoc Biol. (2002) 72:995–1002. doi: 10.1189/jlb.72.5.995
398. Riedemann NC, Guo RF, Hollmann TJ, Gao H, Neff TA, Reuben JS, et al. Regulatory role of C5a in LPS-induced IL-6 production by neutrophils during sepsis. FASEB J. (2004) 18:370–2. doi: 10.1096/fj.03-0708fje
399. Wagner C, Deppisch R, Denefleh B, Hug F, Andrassy K, Hansch GM. Expression patterns of the lipopolysaccharide receptor CD14, and the FCgamma receptors CD16 and CD64 on polymorphonuclear neutrophils: data from patients with severe bacterial infections and lipopolysaccharide-exposed cells. Shock (2003) 19:5–12. doi: 10.1097/00024382-200301000-00002
400. Cowland JB, Borregaard N. Molecular characterization and pattern of tissue expression of the gene for neutrophil gelatinase-associated lipocalin from humans. Genomics (1997) 45:17–23. doi: 10.1006/geno.1997.4896
401. Johnson BL III, Goetzman HS, Prakash PS, Caldwell CC. Mechanisms underlying mouse TNF-alpha stimulated neutrophil derived microparticle generation. Biochem Biophys Res Commun. (2013) 437:591–6. doi: 10.1016/j.bbrc.2013.06.118
402. von Vietinghoff S, Asagiri M, Azar D, Hoffmann A, Ley K. Defective regulation of CXCR2 facilitates neutrophil release from bone marrow causing spontaneous inflammation in severely NF-kappa B-deficient mice. J Immunol. (2010) 185:670–8. doi: 10.4049/jimmunol.1000339
403. Wang D, Paz-Priel I, Friedman AD. NF-kappa B p50 regulates C/EBP alpha expression and inflammatory cytokine-induced neutrophil production. J Immunol. (2009) 182:5757–62. doi: 10.4049/jimmunol.0803861
404. Lapponi MJ, Carestia A, Landoni VI, Rivadeneyra L, Etulain J, Negrotto S, et al. Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J Pharmacol Exp Therap. (2013) 345:430–7. doi: 10.1124/jpet.112.202879
405. Mohammed BM, Fisher BJ, Kraskauskas D, Farkas D, Brophy DF, Fowler AA III, et al. Vitamin C: a novel regulator of neutrophil extracellular trap formation. Nutrients (2013) 5:3131–51. doi: 10.3390/nu5083131
406. Skendros P, Mitroulis I, Ritis K. Autophagy in Neutrophils: From Granulopoiesis to Neutrophil Extracellular Traps. Front Cell Dev Biol. (2018) 6:109. doi: 10.3389/fcell.2018.00109
407. Salminen A, Hyttinen JM, Kauppinen A, Kaarniranta K. Context-dependent regulation of autophagy by IKK-NF-kappaB signaling: impact on the aging process. Int J Cell Biol. (2012) 2012:849541. doi: 10.1155/2012/849541
408. Fuchs T, Brill A, Dürschmied D, Schatzberg D, Hartwig JH, Myers D, et al. Neutrophil extracellular traps induce platelet adhesion and thrombus formation. Blood (2009) 114:1345.
409. Fuchs TA, Brill A, Wagner DD. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler Thromb Vasc Biol. (2012) 32:1777–83. doi: 10.1161/atvbaha.111.242859
410. Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J. (2015) 36:1405–14. doi: 10.1093/eurheartj/ehv007
411. Kimball AS, Obi AT, Diaz JA, Henke PK. The emerging role of NETs in venous thrombosis and immunothrombosis. Front Immunol. (2016) 7:236. doi: 10.3389/fimmu.2016.00236
412. Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood (2014) 123:2768–76. doi: 10.1182/blood-2013-10-463646
413. Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. (2010) 16:887–96. doi: 10.1038/nm.2184
414. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood (2011) 118:1952–61. doi: 10.1182/blood-2011-03-343061
415. Cedervall J, Olsson AK. Immunity gone astray - NETs in cancer. Trends Cancer (2016) 2:633–4. doi: 10.1016/j.trecan.2016.10.012
416. Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci USA. (2012) 109:13076–81. doi: 10.1073/pnas.1200419109
417. Abdol Razak N, Elaskalani O, Metharom P. Pancreatic cancer-induced neutrophil extracellular traps: a potential contributor to cancer-associated thrombosis. Int J Mol Sci. (2017) 18:3. doi: 10.3390/ijms18030487
418. Boone BA, Murthy P, Miller-Ocuin J, Doerfler WR, Ellis JT, Liang X, et al. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer (2018) 18:678. doi: 10.1186/s12885-018-4584-2
419. Leal AC, Mizurini DM, Gomes T, Rochael NC, Saraiva EM, Dias MS, et al. Tumor-derived exosomes induce the formation of neutrophil extracellular traps: implications for the establishment of cancer-associated thrombosis. Sci Rep (2017) 7:6438. doi: 10.1038/s41598-017-06893-7
420. Mauracher LM, Posch F, Martinod K, Grilz E, Daullary T, Hell L, et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J Thromb Haemost. (2018) 16:508–18. doi: 10.1111/jth.13951
421. Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. (2018) 10:436. doi: 10.1126/scitranslmed.aan8292
422. Kupfner JG, Arcaroli JJ, Yum HK, Nadler SG, Yang KY, Abraham E. Role of NF-kappaB in endotoxemia-induced alterations of lung neutrophil apoptosis. J Immunol. (2001) 167:7044–51. doi: 10.4049/jimmunol.167.12.7044
423. Castro-Alcaraz S, Miskolci V, Kalasapudi B, Davidson D, Vancurova I. NF-kappa B regulation in human neutrophils by nuclear I kappa B alpha: correlation to apoptosis. J Immunol. (2002) 169:3947–53. doi: 10.4049/jimmunol.169.7.3947
424. Choi M, Rolle S, Wellner M, Cardoso MC, Scheidereit C, Luft FC, et al. Inhibition of NF-kappaB by a TAT-NEMO-binding domain peptide accelerates constitutive apoptosis and abrogates LPS-delayed neutrophil apoptosis. Blood (2003) 102:2259–67. doi: 10.1182/blood-2002-09-2960
425. Nolan B, Kim R, Duffy A, Sheth K, De M, Miller C, et al. Inhibited neutrophil apoptosis: proteasome dependent NF-kappaB translocation is required for TRAF-1 synthesis. Shock (2000) 14:290–4. doi: 10.1097/00024382-200014030-00008
426. Ward C, Chilvers ER, Lawson MF, Pryde JG, Fujihara S, Farrow SN, et al. NF-kappaB activation is a critical regulator of human granulocyte apoptosis in vitro. J Biol Chem. (1999) 274:4309–18. doi: 10.1074/jbc.274.7.4309
427. Niwa M, Hara A, Kanamori Y, Hatakeyama D, Saio M, Takami T, et al. Nuclear factor-kappaB activates dual inhibition sites in the regulation of tumor necrosis factor-alpha-induced neutrophil apoptosis. Eur J Pharmacol. (2000) 407:211–9. doi: 10.1016/S0014-2999(00)00735-4
428. Wang K, Scheel-Toellner D, Wong SH, Craddock R, Caamano J, Akbar AN, et al. Inhibition of neutrophil apoptosis by type 1 IFN depends on cross-talk between phosphoinositol 3-kinase, protein kinase C-delta, and NF-kappa B signaling pathways. J Immunol. (2003) 171:1035–41. doi: 10.4049/jimmunol.171.2.1035
429. Walmsley SR, Cowburn AS, Sobolewski A, Murray J, Farahi N, Sabroe I, et al. Characterization of the survival effect of tumour necrosis factor-alpha in human neutrophils. Biochem Soc Trans. (2004) 32(Pt3):456–60. doi: 10.1042/bst0320456
430. Kilpatrick LE, Sun S, Korchak HM. Selective regulation by delta-PKC and PI 3-kinase in the assembly of the antiapoptotic TNFR-1 signaling complex in neutrophils. Am J Physiol Cell Physiol. (2004) 287:C633–42. doi: 10.1152/ajpcell.00486.2003
431. Hotta K, Niwa M, Hara A, Ohno T, Wang X, Matsuno H, et al. The loss of susceptibility to apoptosis in exudated tissue neutrophils is associated with their nuclear factor-kappa B activation. Eur J Pharmacol. (2001) 433:17–27. doi: 10.1016/S0014-2999(01)01480-7
432. Moulding DA, Akgul C, Derouet M, White MR, Edwards SW. BCL-2 family expression in human neutrophils during delayed and accelerated apoptosis. J Leukoc Biol. (2001) 70:783–92. doi: 10.1189/jlb.70.5.783
433. Chhabra JK, Chattopadhyay B, Paul BN. SOCS3 dictates the transition of divergent time-phased events in granulocyte TNF-alpha signaling. Cell Mol Immunol. (2014) 11:105–6. doi: 10.1038/cmi.2013.36
434. Miskolci V, Rollins J, Vu HY, Ghosh CC, Davidson D, Vancurova I. NFkappaB is persistently activated in continuously stimulated human neutrophils. Mol Med. (2007) 13:134–42. doi: 10.2119/2006-00072.Miskolci
435. Nolan B, Collette H, Baker S, Duffy A, De M, Miller C, et al. Inhibition of neutrophil apoptosis after severe trauma is NFκβ dependent. J Trauma (2000) 48:599–604. doi: 10.1097/00005373-200004000-00004
436. He M, Peng A, Huang XZ, Shi DC, Wang JC, Zhao Q, et al. Peritumoral stromal neutrophils are essential for c-Met-elicited metastasis in human hepatocellular carcinoma. Oncoimmunology (2016) 5:e1219828. doi: 10.1080/2162402x.2016.1219828
437. Li XF, Chen DP, Ouyang FZ, Chen MM, Wu Y, Kuang DM, et al. Increased autophagy sustains the survival and pro-tumourigenic effects of neutrophils in human hepatocellular carcinoma. J Hepatol. (2015) 62:131–9. doi: 10.1016/j.jhep.2014.08.023
438. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 11:762–74. doi: 10.1038/nri3070
439. Aberg M, Siegbahn A. Tissue factor non-coagulant signaling - molecular mechanisms and biological consequences with a focus on cell migration and apoptosis. J Thromb Haemost. (2013) 11:817–25. doi: 10.1111/jth.12156
440. Takashiba S, Van Dyke TE, Amar S, Murayama Y, Soskolne AW, Shapira L. Differentiation of monocytes to macrophages primes cells for lipopolysaccharide stimulation via accumulation of cytoplasmic nuclear factor κB. Infection Immunity (1999) 67:5573–8.
441. Otero JE, Dai S, Alhawagri MA, Darwech I, Abu-Amer Y. IKKβ Activation is sufficient for RANK-independent osteoclast differentiation and osteolysis. J Bone Miner Res. (2010) 25:1282–94. doi: 10.1002/jbmr.4
442. Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med. 214:1913–23. (2017). doi: 10.1084/jem.20170355
443. Ong S-M, Hadadi E, Dang T-M, Yeap W-H, Tan CT-Y, Ng T-P, et al. The pro-inflammatory phenotype of the human non-classical monocyte subset is attributed to senescence. Cell Death Dis. (2018) 9:266. doi: 10.1038/s41419-018-0327-1
444. Thaler B, Hohensinner PJ, Krychtiuk KA, Matzneller P, Koller L, Brekalo M, et al. Differential in vivo activation of monocyte subsets during low-grade inflammation through experimental endotoxemia in humans. Sci Reports (2016) 6:30162. doi: 10.1038/srep30162
445. Stojkovic S, Thulin A, Hell L, Thaler B, Rauscher S, Baumgartner J, et al. IL-33 stimulates the release of procoagulant microvesicles from human monocytes and differentially increases tissue factor in human monocyte subsets. Thromb Haemost. (2017) 117:1379–90. doi: 10.1160/TH16-10-0784
446. Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood (2010) 115:e10–9. doi: 10.1182/blood-2009-07-235028
447. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. (2007) 204:3037–47. doi: 10.1084/jem.20070885
448. Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, et al. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity (2013) 38:555–69. doi: 10.1016/j.immuni.2013.02.012
449. MacDonald KPA, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. (2010) 116:3955–63. doi: 10.1182/blood-2010-02-266296
450. Overmeire EV, Stijlemans B, Heymann F, Keirsse J, Morias Y, Elkrim Y, et al. M-CSF and GM-CSF receptor signaling differentially regulate monocyte maturation and macrophage polarization in the tumor microenvironment. Cancer Res. (2016) 76:35–42. doi: 10.1158/0008-5472.CAN-15-0869
451. Brach MA, Henschler R, Mertelsmann RH, Herrmann F. Regulation of M-CSF expression by M-CSF: role of protein kinase C and transcription factor NF kappa B. Pathobiology (1991) 59:284–8. doi: 10.1159/000163664
452. Kogan M, Haine V, Ke Y, Wigdahl B, Fischer-Smith T, Rappaport J. Macrophage colony stimulating factor regulation by nuclear factor kappa B: a relevant pathway in human immunodeficiency virus type 1 infected macrophages. DNA Cell Biol. (2012) 31:279–88. doi: 10.1089/dna.2011.1357
453. Pagliari LJ, Perlman H, Liu H, Pope RM. Macrophages require constitutive NF-kappaB activation to maintain A1 expression and mitochondrial homeostasis. Mol Cell Biol. (2000) 20:8855–65. doi: 10.1128/MCB.20.23.8855-8865.2000
454. Laar Lvd, Bosch Avd, Kooij SWvd, Janssen HLA, Coffer PJ, Kooten Cv, et al. A nonredundant role for canonical NF-κB in human myeloid dendritic cell development and function. J Immunol. (2010) 185:7252–61. doi: 10.4049/jimmunol.1000672
455. Wolf Y, Shemer A, Polonsky M, Gross M, Mildner A, Yona S, et al. Autonomous TNF is critical for in vivo monocyte survival in steady state and inflammation. J Exp Med. (2017) 214:905–17. doi: 10.1084/jem.20160499
456. Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. (1995) 9:899–909. doi: 10.1096/fasebj.9.10.7542214
457. Parameswaran N, Patial S. Tumor necrosis factor-α signaling in macrophages. Crit Rev Eukaryot Gene Expr. (2010) 20:87–103. doi: 10.1615/CritRevEukarGeneExpr.v20.i2.10
458. Ellrichmann G, Thöne J, Lee D-H, Rupec RA, Gold R, Linker RA. Constitutive activity of NF-kappa B in myeloid cells drives pathogenicity of monocytes and macrophages during autoimmune neuroinflammation. J Neuroinflamm. (2012) 9:15. doi: 10.1186/1742-2094-9-15
459. Fong CH, Bebien M, Didierlaurent A, Nebauer R, Hussell T, Broide D, et al. An antiinflammatory role for IKKbeta through the inhibition of “classical” macrophage activation. J Exp Med. (2008) 205:1269–76. doi: 10.1084/jem.20080124
460. Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. (2008) 205:1261–8. doi: 10.1084/jem.20080108
461. Hilgendorf I, Swirski FK, Robbins CS. Monocyte fate in atherosclerosis significance. Arterioscler Thromb Vasc Biol. (2015) 35:272–9. doi: 10.1161/ATVBAHA.114.303565
462. Kanters E, Pasparakis M, Gijbels MJ, Vergouwe MN, Partouns-Hendriks I, Fijneman RJ, et al. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J Clin Invest. (2003) 112:1176–85. doi: 10.1172/jci18580
463. Park SH, Sui Y, Gizard F, Xu J, Rios-Pilier J, Helsley RN, et al. Myeloid-specific IkappaB kinase beta deficiency decreases atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. (2012) 32:2869–76. doi: 10.1161/ATVBAHA.112.254573
464. Ye X, Jiang X, Guo W, Clark K, Gao Z. Overexpression of NF-kappaB p65 in macrophages ameliorates atherosclerosis in apoE-knockout mice. Am J Physiol Endocrinol Metab. (2013) 305:E1375–83. doi: 10.1152/ajpendo.00307.2013
465. Goossens P, Vergouwe MN, Gijbels MJ, Curfs DM, van Woezik JH, Hoeksema MA, et al. Myeloid IkappaBalpha deficiency promotes atherogenesis by enhancing leukocyte recruitment to the plaques. PLoS ONE (2011) 6:e22327. doi: 10.1371/journal.pone.0022327
466. Morgan EN, Pohlman TH, Vocelka C, Farr A, Lindley G, Chandler W, et al. Nuclear factor κB mediates a procoagulant response in monocytes during extracorporeal circulation. Journal of Thor Cardiovasc Surg. (2003) 125:165–71. doi: 10.1067/mtc.2003.99
467. Tatsumi K, Mackman N. Tissue factor and atherothrombosis. J Atherosc Thromb. (2015) 22:543–9. doi: 10.5551/jat.30940
468. Malaponte G, Signorelli SS, Bevelacqua V, Polesel J, Taborelli M, Guarneri C, et al. Increased levels of NF-kB-dependent markers in cancer-associated deep venous thrombosis. PLoS ONE 10:e0132496. (2015). doi: 10.1371/journal.pone.0132496
469. Li YD, Ye BQ, Zheng SX, Wang JT, Wang JG, Chen M, et al. NF-kappaB transcription factor p50 critically regulates tissue factor in deep vein thrombosis. J Biol Chem. (2009) 284:4473–83. doi: 10.1074/jbc.M806010200
470. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA (2016) 315:801–10. doi: 10.1001/jama.2016.0287
471. Angus DC, van der Poll T. Severe sepsis and septic shock. New Engl J Med. (2013) 369:840–51. doi: 10.1056/NEJMra1208623
472. Liu SF, Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol. (2006) 290:L622–45. doi: 10.1152/ajplung.00477.2005
473. Levi M, Schultz M, van der Poll T. Sepsis and thrombosis. Semin Thromb Hemost. (2013) 39:559–66. doi: 10.1055/s-0033-1343894
474. Yang S, Qi H, Kan K, Chen J, Xie H, Guo X, et al. Neutrophil extracellular traps promote hypercoagulability in patients with sepsis. Shock (2017) 47:132–9. doi: 10.1097/SHK.0000000000000741
475. Camicia G, Pozner R, de Larranaga G. Neutrophil extracellular traps in sepsis. Shock (2014) 42:286–94. doi: 10.1097/SHK.0000000000000221
476. Mayr FB, Jilma B. Coagulation interventions in experimental human endotoxemia. Transl Res. (2006) 148:263–71. doi: 10.1016/j.trsl.2006.08.002
477. Schoergenhofer C, Matzneller P, Mussbacher M, Schmid JA, Jilma-Stohlawetz P, Zeitlinger M, et al. Colistin dampens fibrinolysis and endothelial activation during endotoxaemia. A randomised, double blind trial. Thromb Haemost. (2017) 117:1714–21. doi: 10.1160/TH17-03-0196
478. Bauer KA, ten Cate H, Barzegar S, Spriggs DR, Sherman ML, Rosenberg RD. Tumor necrosis factor infusions have a procoagulant effect on the hemostatic mechanism of humans. Blood (1989) 74:165–72.
479. van der Poll T, Buller HR, ten Cate H, Wortel CH, Bauer KA, van Deventer SJ, et al. Activation of coagulation after administration of tumor necrosis factor to normal subjects. N Engl J Med. (1990) 322:1622–7. doi: 10.1056/NEJM199006073222302
480. Calzado MA, Bacher S, Schmitz ML. NF-kappaB inhibitors for the treatment of inflammatory diseases and cancer. Curr Med Chem. (2007) 14:367–76. doi: 10.2174/092986707779941113
481. Coldewey SM, Rogazzo M, Collino M, Patel NS, Thiemermann C. Inhibition of IkappaB kinase reduces the multiple organ dysfunction caused by sepsis in the mouse. Dis Model Mech. (2013) 6:1031–42. doi: 10.1242/dmm.012435
482. Lv S, Han M, Yi R, Kwon S, Dai C, Wang R. Anti-TNF-alpha therapy for patients with sepsis: a systematic meta-analysis. Int J Clin Pract. (2014) 68:520–8. doi: 10.1111/ijcp.12382
483. de Kruif MD, Lemaire LC, Giebelen IA, van Zoelen MA, Pater JM, van den Pangaart PS, et al. Prednisolone dose-dependently influences inflammation and coagulation during human endotoxemia. J Immunol. (2007) 178:1845–51.
484. Annane D, Bellissant E, Bollaert PE, Briegel J, Keh D, Kupfer Y. Corticosteroids for treating sepsis. Cochrane Database Syst Rev. (2015) CD002243. doi: 10.1002/14651858.CD002243.pub3
485. Bernard GR, Wheeler AP, Russell JA, Schein R, Summer WR, Steinberg KP, et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group. N Engl J Med. (1997) 336:912–8. doi: 10.1056/NEJM199703273361303
486. Eisen DP, Reid D, McBryde ES. Acetyl salicylic acid usage and mortality in critically ill patients with the systemic inflammatory response syndrome and sepsis. Crit Care Med. (2012) 40:1761–7. doi: 10.1097/CCM.0b013e318246b9df
487. Losche W, Boettel J, Kabisch B, Winning J, Claus RA, Bauer M. Do aspirin and other antiplatelet drugs reduce the mortality in critically ill patients? Thrombosis (2012) 2012:720254. doi: 10.1155/2012/720254
488. Eisen DP, Moore EM, Leder K, Lockery J, McBryde ES, McNeil JJ, et al. AspiriN To Inhibit SEPSIS (ANTISEPSIS) randomised controlled trial protocol. BMJ Open (2017) 7:e013636. doi: 10.1136/bmjopen-2016-013636
489. Kruger P, Bailey M, Bellomo R, Cooper DJ, Harward M, Higgins A, et al. A multicenter randomized trial of atorvastatin therapy in intensive care patients with severe sepsis. Am J Respir Crit Care Med. (2013) 187:743–50. doi: 10.1164/rccm.201209-1718OC
490. Patel JM, Snaith C, Thickett DR, Linhartova L, Melody T, Hawkey P, et al. Randomized double-blind placebo-controlled trial of 40 mg/day of atorvastatin in reducing the severity of sepsis in ward patients (ASEPSIS Trial). Crit Care (2012) 16:R231. doi: 10.1186/cc11895
491. National Heart L, Blood Institute ACTN, Truwit JD, Bernard GR, Steingrub J, Matthay MA, et al. Rosuvastatin for sepsis-associated acute respiratory distress syndrome. N Engl J Med. (2014) 370:2191–200. doi: 10.1056/NEJMoa1401520
492. Kawamura K, Ichikado K, Takaki M, Sakata Y, Yasuda Y, Shingu N, et al. Efficacy of azithromycin in sepsis-associated acute respiratory distress syndrome: a retrospective study and propensity score analysis. Springerplus (2016) 5:1193. doi: 10.1186/s40064-016-2866-1
493. Elbaradey GF, Elshmaa NS, Hodeib H. Role of edaravone in managemant of septic peritonitis. J Anaesthesiol Clin Pharmacol. (2016) 32:465–9. doi: 10.4103/0970-9185.194770
494. Allingstrup M, Wetterslev J, Ravn FB, Moller AM, Afshari A. Antithrombin III for critically ill patients: a systematic review with meta-analysis and trial sequential analysis. Intensive Care Med. (2016) 42:505–20. doi: 10.1007/s00134-016-4225-7
495. Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA (2001) 286:1869–78. doi: 10.1001/jama.286.15.1869
496. Beale RJ, Sherry T, Lei K, Campbell-Stephen L, McCook J, Smith J, et al. Early enteral supplementation with key pharmaconutrients improves Sequential Organ Failure Assessment score in critically ill patients with sepsis: outcome of a randomized, controlled, double-blind trial. Crit Care Med. (2008) 36:131–44. doi: 10.1097/01.CCM.0000297954.45251.A9
497. Najafi A, Mojtahedzadeh M, Ahmadi KH, Abdollahi M, Mousavi M, Chelkeba L, et al. The immunological benefit of higher dose N-acetyl cysteine following mechanical ventilation in critically ill patients. Daru (2014) 22:57. doi: 10.1186/2008-2231-22-57
498. Valenta J, Brodska H, Drabek T, Hendl J, Kazda A. High-dose selenium substitution in sepsis: a prospective randomized clinical trial. Intensive Care Med. (2011) 37:808–15. doi: 10.1007/s00134-011-2153-0
499. Brodska H, Valenta J, Malickova K, Kohout P, Kazda A, Drabek T. Biomarkers in critically ill patients with systemic inflammatory response syndrome or sepsis supplemented with high-dose selenium. J Trace Elem Med Biol. (2015) 31:25–32. doi: 10.1016/j.jtemb.2015.02.005
500. Rice TW, Wheeler AP, Thompson BT, deBoisblanc BP, Steingrub J, Rock P, et al. Enteral omega-3 fatty acid, gamma-linolenic acid, and antioxidant supplementation in acute lung injury. JAMA (2011) 306:1574–81. doi: 10.1001/jama.2011.1435
501. Takada Y, Bhardwaj A, Potdar P, Aggarwal BB. Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-kappaB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation. Oncogene (2004) 23:9247–58. doi: 10.1038/sj.onc.1208169
502. Klessig DF, Tian M, Choi HW. Multiple targets of salicylic acid and its derivatives in plants and animals. Front Immunol. (2016) 7:206. doi: 10.3389/fimmu.2016.00206
503. Mekaj YH, Daci FT, Mekaj AY. New insights into the mechanisms of action of aspirin and its use in the prevention and treatment of arterial and venous thromboembolism. Ther Clin Risk Manag. (2015) 11:1449–56. doi: 10.2147/TCRM.S92222
504. Paterson JR, Baxter G, Dreyer JS, Halket JM, Flynn R, Lawrence JR. Salicylic acid sans aspirin in animals and man: persistence in fasting and biosynthesis from benzoic acid. J Agric Food Chem. (2008) 56:11648–52. doi: 10.1021/jf800974z
505. Glauert HP. Vitamin E and NF-kappaB activation: a review. Vitam Horm. (2007) 76:135–53. doi: 10.1016/S0083-6729(07)76006-5
506. Son EW, Mo SJ, Rhee DK, Pyo S. Vitamin C blocks TNF-alpha-induced NF-kappaB activation and ICAM-1 expression in human neuroblastoma cells. Arch Pharm Res. (2004) 27:1073–9. doi: 10.1007/BF02975434
507. Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. (1991) 10:2247–58.
508. Guruvayoorappan C, Kuttan G. Beta-carotene inhibits tumor-specific angiogenesis by altering the cytokine profile and inhibits the nuclear translocation of transcription factors in B16F-10 melanoma cells. Integr Cancer Ther. (2007) 6:258–70. doi: 10.1177/1534735407305978
509. Youn HS, Lim HJ, Choi YJ, Lee JY, Lee MY, Ryu JH. Selenium suppresses the activation of transcription factor NF-kappa B and IRF3 induced by TLR3 or TLR4 agonists. Int Immunopharmacol. (2008) 8:495–501. doi: 10.1016/j.intimp.2007.12.008
510. Novak TE, Babcock TA, Jho DH, Helton WS, Espat NJ. NF-kappa B inhibition by omega−3 fatty acids modulates LPS-stimulated macrophage TNF-alpha transcription. Am J Physiol Lung Cell Mol Physiol. (2003) 284:L84–9. doi: 10.1152/ajplung.00077.2002
511. Esteban E, Ferrer R, Alsina L, Artigas A. Immunomodulation in sepsis: the role of endotoxin removal by polymyxin B-immobilized cartridge. Mediators Inflamm. (2013) 2013:507539. doi: 10.1155/2013/507539
Keywords: NF-kappa B signaling, inflammation, thrombosis, vasculature, coagulation, sepsis, blood cells
Citation: Mussbacher M, Salzmann M, Brostjan C, Hoesel B, Schoergenhofer C, Datler H, Hohensinner P, Basílio J, Petzelbauer P, Assinger A and Schmid JA (2019) Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 10:85. doi: 10.3389/fimmu.2019.00085
Received: 29 May 2018; Accepted: 11 January 2019;
Published: 04 February 2019.
Edited by:
Fulvio D'Acquisto, University of Roehampton, United KingdomReviewed by:
Michael Thomas Lotze, University of Pittsburgh Cancer Institute, United StatesJens Staal, Flanders Institute for Biotechnology, Belgium
Copyright © 2019 Mussbacher, Salzmann, Brostjan, Hoesel, Schoergenhofer, Datler, Hohensinner, Basílio, Petzelbauer, Assinger and Schmid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johannes A. Schmid, am9oYW5uZXMuc2NobWlkQG1lZHVuaXdpZW4uYWMuYXQ=
†These authors have contributed equally to this work