 Anna Grohová
Anna Grohová Klára Dáňová
Klára Dáňová Radek Špíšek
Radek Špíšek Lenka Palová-Jelínková
Lenka Palová-Jelínková
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 05 February 2019
Sec. Immunological Tolerance and Regulation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00079
This article is part of the Research Topic Tolerogenic Dendritic Cells and Regulatory T Cells as Therapeutics for Immune-Mediated Disorders View all 29 articles
Diabetes mellitus is characterized by long standing hyperglycemia leading to numerous life-threatening complications. For type 1 diabetes mellitus, resulting from selective destruction of insulin producing cells by exaggerated immune reaction, the only effective therapy remains exogenous insulin administration. Despite accurate compliance to treatment of certain patients, transient episodes of hyperglycemia cannot be completely eliminated by this symptomatic treatment. Novel immunotherapeutic approaches based on tolerogenic dendritic cells, T regulatory cells and mesenchymal stem cells (MSCs) have been tested in clinical trials, endeavoring to directly modulate the autoimmune destruction process in pancreas. However, hyperglycemia itself affects the immune system and the final efficacy of cell-based immunotherapies could be affected by the different glycemic control of enrolled patients. The present review explores the impact of hyperglycemia on immune cells while providing greater insight into the molecular mechanisms of high glucose action and subsequent metabolic reprogramming of different immune cells. Furthermore, over-production of mitochondrial reactive oxygen species, formation of advanced glycation end products as a consequence of hyperglycemia and their downstream signalization in immune cells are also discussed. Since hyperglycemia in patients with type 1 diabetes mellitus might have an impact on immune-interventional treatment, the maintenance of a tight glucose control seems to be beneficial in patients considered for cell-based therapy.
Diabetes mellitus (DM) is a complex metabolic disorder resulting from immune-mediated destruction of insulin-producing cells in the islets of Langerhans (type 1 diabetes mellitus—T1D) or from the combination of insulin resistance and relative insulin deficiency (type 2 diabetes mellitus—T2D). Both types are associated with severe complications stemming from chronic hyperglycemia, the hallmark of diabetes mellitus (1). Hyperglycemia develops in T1D due to insufficient insulin production by pancreatic β cells. Transient episodes of hyperglycemia create an abnormal metabolic environment in various cell types leading to cell and tissue-specific metabolic reprogramming with subsequent macro- and microvascular complication in diabetic patients (2). Besides that, hyperglycemia also causes cumulative changes in long-lived macromolecules, which persist despite restoration of normoglycemia. This phenomenon was described as a metabolic memory, meaning that early high glucose environment is remembered by the cells (3).
Regarding the immune system, defects in the immune defense against miscellaneous pathogens were detected in patients with DM (4). Hyperglycemia is associated with decreased function of innate immunity such as lower complement levels, impaired chemotaxis, phagocytosis, and decrease in diapedesis of polymorphonuclear cells and monocytes/macrophages (5, 6). There is also considerable evidence that hyperglycemia contributes to the partial breakdown of peripheral tolerance (7), modulates patients' leukocyte profile (8), affects the function of antigen presenting cells (9), facilitates higher proinflammatory Th1/Th17 cells differentiation and suppresses regulatory T cells (Tregs) (10–12).
The adaptive immunity plays an essential role in the pathogenesis of T1D, particularly due to the imbalance between the autoaggressive effector T cells and Tregs (13, 14). In recent years, huge effort has been undertaken to suppress the inadvertent immune response in T1D. Several clinical trials and in vitro studies focused on cell-based therapy were launched with the goal to directly modulate the autoimmune destruction process of pancreatic β cells and to regenerate lost islets (15–18). Tolerogenic dendritic cells (tolDCs) and Tregs especially represent a new promising therapeutic strategy, either alone or in combinatorial therapies. Next, human stem cell (SCs) therapy represent another therapeutic approach for both inducing tolerance and islet cell regeneration (19). Current status of cell-based therapy is summarized in Table 1. However, little is known about the impact of the patient's glucose level on the potential cell-based vaccine's functional characteristics and efficacy. The initial immune cells isolated from hyperglycemic patient for the vaccine generation could exhibit different properties compared to those ones from euglycemic patients. Thus, the subsequent cell-based vaccine may exhibit different tolerogenic properties than in euglycemic subjects and the autoimmune destruction process in pancreas might be more difficult to suppress in patients with suboptimal glycemic control.

Table 1. Clinical studies (completed and with published results) for T1D treatment based on cells with regulatory properties including Tregs, tolerogenic DCs, and some examples of SCs.
This review will focus on the regulating effect of hyperglycemia on immune cells, with a particular emphasis on tolerogenic DCs, T cells and Tregs. In addition, we will more specifically explore the subsequent changes in immune cell metabolism, initiation of alternative metabolic pathways such as the advanced glycation pathway, the process of advanced glycation end products (AGEs) formation and their molecular signaling in different immune cells.
Hyperglycemia is associated with increased oxidative stress caused by over-production of nicotinamide adenindinucleotide (NADH) and mitochondrial reactive oxygen species (ROS), that inhibit glucose metabolism via glycolysis and tricarboxylic acid (TCA) cycle and consequently activate alternative glucose metabolic pathways including polyol pathway and hexosamine biosynthetic pathway (HBP). All these alternative pathways lead to increased ROS production, thus completing the vicious circle of cellular oxidative stress. Hyperglycemia-induced activation of protein kinase C (PKC) isoforms also strongly contributes to cellular and tissue damage by induction of proinflammatory gene expression and further ROS increase (30–32).
Last but not least, the formation of advanced glycation products (AGEs) is an equally important mechanism in the processes of tissue-damaging effects of hyperglycemia. AGEs are heterogeneous compounds arising from irreversible non-enzymatic glycation of proteins, nucleic acids and lipids (33). One of the major products is glycated hemoglobin (HbA1c) that has been used as a biomarker for diabetes because it reflects long-term glycemia (34). Formation of AGEs occurs in several steps. First, the condensation of carbonyl group with a free amino group of proteins forms Schiff bases, which transform to more stable covalently bound Amadori products. The final irreversible products of glycation—AGEs arise from slow Amadori product rearrangements (35). In diabetic patients, chronic hyperglycemia-driven accumulation of AGEs and its cognate receptor for advanced glycation end-product (RAGE) are involved in the pathogenesis of both micro and macrovascular complications (36). Indeed, the formation of AGEs interferes with cell integrity by irreversible glycation of various proteins. Furthermore, signaling via RAGE-mediated pathways increases ROS production via the activation of an NADPH oxidase system which contributes to further mitochondrial protein damage and DNA damage (37). RAGE consists of 5 domains (three extracellular, one transmembrane and one intracellular domain) (38). The intracellular domain is important for the activation of the transcriptional factor nuclear factor-κB (NF-κB), which leads to expression of growth factors, cytokines and RAGE itself in miscellaneous cells (39). In endothelial cells, downstream signaling via AGEs-RAGE receptor complex activates the signaling pathways of glycogen synthase kinase 3β (GSK3β), p38 mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinase 1 and 2 (ERK1/2), c-Jun amino-terminal kinase (JNK) and NF-κB, all of which lead to endothelial cell dysfunction and diabetic vascular disease. Moreover, AGEs play an important role in β-cell failure by activating NADPH oxidase with a consequence of increased ROS generation and induction of β-cell apoptosis through the PKCβ2 pathway (40, 41) (Figure 1).
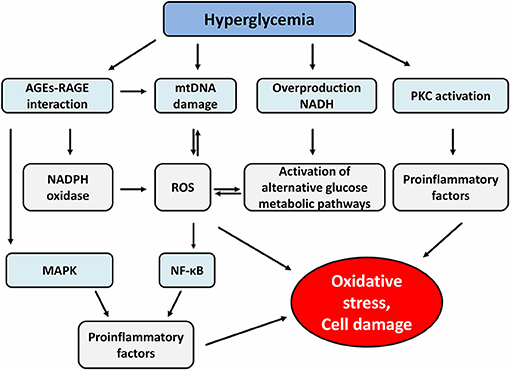
Figure 1. Effect of hyperglycemia and advanced glycation end products on different signaling pathways. AGEs, advanced glycation end products; RAGE, receptor for advanced glycation end product; mtDNA, mitochondrial DNA; NADH, nicotinamide adenine dinucleotide; PKC, protein kinase c; NADPH, nicotinamide adenine dinucleotide phosphate; ROS, reactive oxygen species; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor-κB.
RAGE receptors are expressed on the surface membrane of various immune cells and can be found also in the cytosol. Several findings have been recently reported that AGEs-RAGE mediated signaling causes the induction of dendritic cell (DCs) maturation, Th1/Th17 polarization from naïve CD4+ T cells, activation and maturation of B cells and the reduction of the suppressor function of Tregs (9, 42–44).
Dendritic cells, the most potent antigen-presenting cells (APC), are crucial for the induction of the pathological process in autoimmune DM. TolDCs represent immature or semi-mature DCs capable to promote immune tolerance by various mechanisms such as induction of autoreactive T cell hyporesponsiveness, anergy, or apoptosis and by induction of various types of T and B regulatory cells. These essential properties determine tolDCs as a new promising strategy for prevention of autoimmune DM in the subjects who are at risk or even for treatment of T1D. A human phase I trial of autologous monocyte-derived tolDCs in T1D has been completed (16, 45) and another trial based on proinsulin-loaded tolDCs has been recently opened (46).
Dendritic cell immune-metabolic state dictates the balance between immunity and tolerance. Immature DCs utilize oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) as their main energy sources, while DC maturation is followed by a pronounced switch to aerobic glycolysis (47, 48). TolDCs show a distinct metabolic profile. Generally, they exhibit catabolic and highly energetic profile favoring OXPHOS and FAO; however, metabolic plasticity can be observed regarding the protocol used for tolDCs generation (49, 50). Vitamin D3 (VitD3) and its derivatives, immunosuppressive drug dexamethasone (DEX) or Vit/DEX combination are widely used for tolDC generation (51, 52). Malinarich et al. reported high mitochondrial activity, increased OXPHOS, a shift in redox state and high glycolytic capacity as the metabolic signatures of VitD3/DEX generated tolDCs. In this study, inhibition of FAO caused increased expression of maturation markers on tolDCs and partially restored their T cell stimulatory capacity, suggesting their dependence on FAO (53). Ferreira et al. showed that VitD3–induced tolDCs exhibited increased glucose metabolism, high mitochondrial activity and elevated OXPHOS profile with glucose as the main oxidative fuel compared to mature DCs. Interestingly, PI3K/Akt/mTOR driven glycolysis, but not glucose oxidation, was crucial for the maintenance of tolerogenic phenotype of VitD3-modulated tolDCs (54). Next, we documented in our study that increased glycolysis contributes to the stability of VitD2/DEX generated tolDCs in the inflammatory environment (55). Another study pointed out the profound metabolic changes toward increased OXPHOS and lipid metabolism while reducing amino acids and fatty acid synthesis in VitD3/DEX generated tolDCs compared to mature DCs (50). Such catabolic metabolic reprogramming with nutrient deprivation supports regulatory T cell induction (56).
The studies assessing the potential effect of hyperglycemia, oxidative stress and AGEs in diabetic patients on tolDCs-based vaccine generation and its functional properties are limited. In our study we analyzed the impact of the metabolic state on functional properties of tolDCs generated from patients with T1DM, aiming to investigate the potential group of patients targeted for tolDCs immunotherapy. We showed that monocyte-derived tolDCs generated by DEX and VitD2 displayed better regulatory properties when prepared from the group of T1D patients with tight glucose control. TolDCs generated from T1D patients with a suboptimal level of glycated hemoglobin (HbA1c) exhibited down-regulation of immunoregulatory molecules PD-L1 and IL-T3 in comparison with tolDCs prepared from patients with optimal HbA1c level. Tolerogenic effect of tolDCs was better in normoglycemic patients in terms of more efficient suppression of Th1 and Th17 related cytokines production. Indeed, autologous primary culture of T cells and tolDCs from patients with optimal glucose control showed lower supernatant concentration of Th1-related cytokines IFN-γ and TNF-α and Th-17 related cytokine IL-17A, IL-23 and IL-9 and upregulated levels of IL-10 compared to cultures with control DCs. However, in patients with high level of HbA1c, we detected significantly lower levels of IFN-γ, TNF-α, and IL-9, but not IL-17A and IL-23 in comparison to control DCs. Overall, the ability of tolDCs to induce hyporesponsiveness of autologous T cells accompanied by a reduction of Th1 and Th17 cytokines was better in patients with tight glucose control. Importantly, improvement of glycemic control in T1D patients restores the ability of tolDCs to tolerize their autoreactive T cells (12).
One of the potential explanations for better effectiveness of DEX/VitD2-generated tolDCs from patients with tight glucose control is that hyperglycemia might attenuate the expression or function of vitamin D receptor (VDR) on monocytes used for generation of tolDCs. Indeed, hyperglycemia inhibited VDR expression in human vascular smooth cells, induced glycosylation of VDR in human monocytes and macrophages due to hexosamine pathway activation and interacted with VDR to impair its DNA binding and function. Thus, the high glucose level might exacerbate the function of VitD as a crucial tolerogenic agent indispensable for IL-T3 and PD-L1 expression on tolDCs (57–61). Next, high glucose, ROS, and AGEs have been previously shown to have an effect on in vitro DC generation from blood monocytes. Indeed, high glucose impaired in vitro differentiation of monocytes from healthy donors into DCs by inducing ROS, activating Wnt/β-catenin pathway and p38MAPK (62). Moreover, AGEs treatment led to persistent NF-κB activation and abnormal NF-κB function observed in T1D monocytes (63, 64). As Dex or Vitamin D receptor agonists have been described to generate tolDCs through NF-κB down-regulation, it is possible that well-controlled patients have a better capacity to overcome sustained hyperglycemia driven NF-κB activation in the process of tolDCs generation.
Once the immature or semimature tolDCs are applied to the patients' body, they will experience proinflammatory environment and high glucose milieu. Although the stability of various tolDCs in the proinflammatory environment is well documented, the data assessing the effect of high glucose are scarce (55, 65, 66). Regarding the effect of high glucose on immature DCs, short-term (24–48 h) high glucose treatment of monocyte-derived immature DCs generated from healthy donors accelerated the expression of co-stimulatory molecules, such as CD83 and CD86, and induced proinflammatory cytokine profile with up-regulation of IL-6 and IL-12 while the level of IL-10 was diminished (9, 67). Additionally, high glucose enhanced up-regulation of several DCs scavenger receptors, probably via increased production of intracellular ROS, and the activation of p38 MAPK pathway (67). Other studies demonstrated that AGE-modified serum molecules augmented the capacity of DCs to stimulate T cell proliferation and T cell cytokine secretion possibly through the up-regulation of RAGE on DCs. The subsequent activation of MAPK pathways and NF-κB was crucial for this phenomenon (68, 69). Buttari et al. documented that polyphenolic antioxidant resveratrol prevented the immature DC maturation, IL-12, IL-1β, TNF-α production and diminished the allostimulatory capacity of AGEs-treated DCs via abrogation of MAPK and NF-κB activation (70). Overall, these findings highlight the role of ROS, MAPK, and NF-κB as signaling molecules mediating the activating effect of high glucose in monocyte-derived DCs. Thus, the possibility exists, that tolDCs activated by high glucose conditions or AGEs might modify their tolerogenic profile into more matured and less potent phenotype due to the augmented DCs activation, presence of maturation markers and favorable cytokine profile. However, further studies are needed to fully elucidate the effect of high glucose levels, oxidative stress, and ROS on the stability of tolDCs.
So far, we can just speculate whether and how hyperglycemia can modulate bioenergetics and metabolism of tolDCs once they experience hyperglycemic conditions in T1D patients. As discussed above, hyperglycemia drives dysregulation of glycolysis as well as mitochondrial TCA cycle leading to mitochondrial respiratory chain complex dysfunction and the production of increased levels of ROS. Moreover, hyperglycemia driven AGEs formation and hexosamine biosynthetic pathway activation participate on the post-translational modification of protein subunits of mitochondrial electron transport chain (ETC) complexes leading to impaired mitochondrial function (31). Given the critical role of metabolic pathways in sustaining tolDCs function, exposure to hyperglycemia might affect the behavior of tolDCs and the outcome of the tolDCs therapy. On the other hand, some data suggest that once the metabolic reprogramming takes place in particular tolDCs, they sustain their tolerogenic phenotype independently of the actual glycemia level (Figure 2).
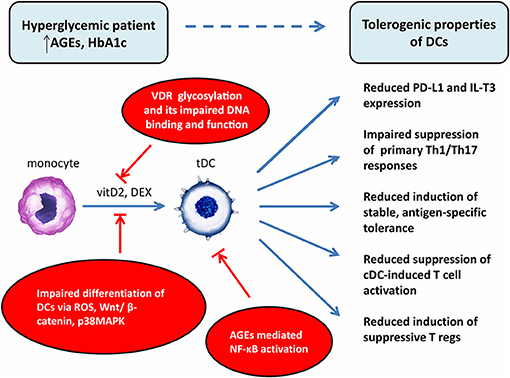
Figure 2. Potential explanations of impaired effect of tolerogenic DCs in vaccine generated from patients with chronic hyperglycemia. DCs, dendritic cells; cDC, control dendritic cells; tDC, tolerogenic dendritic cells; T regs, T regulatory cells; AGEs, advanced glycation end products; HbA1c, glycated hemoglobin; VDR, vitamin D receptor; vitD2, vitamin D2; DEX, dexamethasone; ROS, reactive oxygen species; NF-κB, nuclear factor-κB; Wnt, wingless/integrated signaling pathway; p38MAPK, p38 mitogen-activated protein kinases; PD-L1, programmed death-ligand 1; IL-T3, immunoglobulin-like transcript 3.
Metabolically aberrant microenvironment caused by hyperglycemia in cooperation with chronic low-grade inflammation might affect T cell metabolism and immunological phenotype. Naïve T cells are in metabolically dormant state and primarily rely on OXPHOS and FAO to meet their bioenergetics and biosynthetic demands (71, 72). However, upon activation naïve T cells rapidly switch to high rates of glycolysis, glutaminolysis, and OXPHOS for cell growth and proliferation (73). Hyperglycemia drives increase in glycolysis in activated T cells thus leading to robust IFN-γ production by preventing glycolytic enzyme glyceradehydphosphate dehydrogenase (GAPDH) from binding to and inhibiting the translation of IFN-γ mRNA (2, 74) and thus driving inflammation in a non-antigen specific manner. Moreover, hyperglycemia-driven oxidative stress exerts a global effect on T cells as shown by T cell hyperresponsiveness (75), induction of proinflammatory Th17 cells (10, 11) and diminished regulation of IL-7-mediated T cell survival and homeostatic expansion (76). Thus, it is plausible that “pre-activated” phenotype of T-cells detected in diabetic subjects (75) may affect the ability of immunoregulatory cell vaccine to induce the generation of antigen-specific tolerance in T cells.
In our study we reported that DEX/VitD2-generated tolDCs from patients with euglycemia, but not hyperglycemia, markedly reduced not only primary Th1/Th17 responses, but also mature DC-induced proinflammatory IFN- γ, IL-17, TNF-α, and IL-9 production. Suppressive effect of tolDCs on mature DC-induced cytokine production correlated negatively with HbA1c level. Interestingly, T cells form hyperglycemic patients exhibited higher basal Th1/Th17 cytokine production and higher basal proliferation (antigen- independent) alongside with lower responses to antigen stimulation in primary cultures (12). The similar observation was reported on IL-10/TGF-β-induced tolDCs from T1DM patients (15, 77), where tolerance induction was dependent on the current activation state of T cells in each patient. In Segovia-Gamboa's study, antigen-specific T cell tolerance was lost in patients with high homeostatic T cell proliferation and low autoantigen-specific T cell response and the magnitude of T cell suppression inversely correlated with the level of HbA1c (15). These data suggest that hyperglycemia reduces the ability of tolDCs to induce stable antigen-specific T cell tolerance by making autoreactive T cells more activated and thus less prone to be tolerized.
Nevertheless, the mechanism of increased T cell activation/proliferation in hyperglycemic diabetic patients is not fully understood. It has been assumed that hyperglycemia driven oxidative stress, AGEs formation, and AGEs-RAGE interaction is one of the possible cause as RAGE ligation affects T cell activation and controls T cell differentiation (44, 78). Mechanistically, Kumar et al. showed that hyperglycemia drives the expression of proinflammatory cytokines and chemokines, especially IL-6 and IL-17 family members transcriptionally through oxidative stress and NF-κB activation via PKC and p38MAPK pathway in blood lymphocytes from diabetic subjects (11). Another study reported that AGEs dose-dependently promoted RAGE expression and induced differentiation of naïve CD4+ T cells into Th1/Th17 phenotype via down-regulation of transcription factor peroxisome proliferator-activated receptor gamma (PPAR-γ). RAGE knock-down abolished the AGEs-induced Th1/Th17 differentiation (10). Interestingly, RAGE is constitutively present intracellularly in both CD4+ and CD8+ T cells from diabetic subjects compared to healthy donors where RAGE was found to be expressed only following TCR activation. Moreover, RAGE+ T cells in diabetic patients display proinflammatory gene expression profile and express high levels of IL-17A. Elevated RAGE expression in diabetes is most likely due to the abundant overproduction of RAGE ligands including AGEs in diabetic patients (79) since it was detected on T cells in diabetic patients but not in T cells from patients with other autoimmune diseases such as Sjogren's syndrome and rheumatoid arthritis (80). Despite no correlation of RAGE expression with the level of glycemia in this study, the abundant overproduction of RAGE ligands in diabetic patients supports the hypothesis that glycated molecules forming AGEs could be contributing factor for exaggerated activation of T cells. On the other hand, higher expression of intracellular RAGE was shown also in high-risk euglycemic relatives (81). Thus, the direct impact of primary impaired T cell activation pathway in autoimmune diabetes cannot be ruled out.
Chronic hyperglycemia-induced “pre-activated” T cells phenotype was presented also in the mouse with streptozotocin-induced diabetes. Naïve T cells from hyperglycemic mice showed functional marks of antigen-experienced T cells with increased Th1, Th2, and Th17 cytokine production and higher proliferation upon TCR stimulation compared to euglycemic control, despite having the low CD44 expression. Simultaneously, they exhibited considerable chromatin decondensation which resulted in the facilitation of the transport of transcriptional factors to the DNA. These findings could be a consequence of inner modulation of TCR downstream regulators in high glucose milieu and are likely to be RAGE-dependent as RAGE deficiency reversed the hyperactivated phenotype of T cells in hyperglycemic mice. Of particular interest, all hyperglycemia-induced changes persisted after adoptive transfer into euglycemic hosts (75). This points to the phenomenon of metabolic memory and partly explains why the activating phenotype of T cells may persist even after glucose normalization.
Both naturally occurring Tregs arising from thymus (nTregs) as well as induced T regs (iTregs) developing in the periphery are the key negative regulators of the immune response and play a crucial role in the maintenance of the peripheral tolerance. Administration of ex vivo expanded Tregs has been considered as another interesting strategy for the treatment of T1D. Early clinical studies utilizing polyclonally expanded autologous CD4+CD25+FoxP3+ Tregs demonstrated the safety and feasibility of this approach and supported their therapeutic potential for inhibition or delay of the destruction of pancreatic β cells in T1D patients (17, 20).
Recent studies utilizing more precise Tregs markers such as low CD127 expression or the selective demethylation of certain regions of the Foxp3 locus (82) show that T1D patients appear to have normal T regs frequencies compared to healthy donors, but they are often poorly functioning (83–85). The lower suppressive capacity of Tregs from T1D patients, their phenotypic shift accompanied by decreased production of anti-inflammatory cytokines IL-10, IL-35 and TGF-β and increased production of inflammatory cytokines IFN-γ and IL-17 (84, 86–89), unstable FoxP3 expression (90), and higher susceptibility to apoptosis were documented (91–95).
Whether the functional defect stems from the inner modulation of Tregs population in a subject with an autoimmune disease or whether it is partly caused or deepened by hyperglycemia remains to be clarified (96). Glycolysis was shown to orchestrate the generation and suppressive function of human iTregs by controlling of FoxP3 splicing variants through the glycolytic enzyme enolase-1 (97) that can bind DNA and regulate gene expression. Glycolysis was also shown to affect expression of Tregs' cell inhibitory and regulatory molecules CTLA-4, PD-1, GITR, CD37, and CD71 (97). Thus, impaired glycolysis observed in T1D might participate on defective induction/function of Tregs (98). Next, hyperglycemia contributes to increased expression of IL-6 and TGF-β (11, 99) that synergistically down regulate FoxP3 at the post-translational level by promoting FoxP3 protein degradation in Tregs (100).
The negative effect of hyperglycemia on Tregs phenotype and function was observed also in streptozotocin (STZ) induced hyperglycemic mice, where long-term hyperglycemia changed the phenotype of CD4+CD25+FoxP3+ Tregs into activated/memory phenotype with lower CD62L, CD45RB, GITR, and higher CTLA-4 expression and reduced their suppressive effect on effector T cells proliferation. These properties were unambiguously reversed by insulin administration (101). In human studies from healthy donors, glucose-induced AGEs also reduced the suppressive function of Tregs and decreased the ratio of Tregs/Th17 cells (10). In T1D patients, the frequency of Tregs correlated negatively with the level of HbA1c (102–104).
Next, Tregs defective function and number might result from defective signaling from dendritic cells which under hyperglycemia might exhibit weaker tolerogenic potential. Indeed, in our study, co-cultures of naïve T cells and antigen-loaded tolDCs from patients with high HbA1c levels led to the induction of lower levels of proliferating CD4+CD25highCD127lowFoxP3+ Tregs producing low IL-10 levels compared to euglycemic patients. Of a great interest, Tregs expanded by tolDCs from patients with high HbA1c levels exhibited weaker suppressive abilities (12). In this case, it seems that reduced surface expression of ILT-3 and PD-L1 on dexamethasone/VitD2-generated tolDCs from poorly compensated T1D patients might play a role in defective Tregs induction and function (12).
From the present clinical studies is not clear whether the glycemia level on diabetic subjects influents the quality of resulting vaccine or not. Okubo et al. demonstrated in their study that T1D patients with tight glucose control (based on the HbA1c level) showed higher percentage of activated Tregs (CD4+CD25+FoxP3+CD45RA+) in peripheral blood and lower percentage of resting Tregs (CD4+CD25+FoxP3+CD45RO+). As activated Tregs are the most suppressive Tregs, their higher percentage in primary culture could increase suppressive effect of the resulting vaccine in patient with optimal glucose control (105).
Stem cells (SCs) are multipotent, self-renewing cells, with anti-inflammatory and immunomodulatory properties. Implantation of SCs to T1D patients might regulate ongoing autoimmune process by inhibition of T and B cell activation, DCs differentiation and NK cell activity, while induction of T cell anergy and T regs generation (106, 107). Moreover, SCs can differentiate into insulin-producing cells and revitalize the damaged pancreatic β-cells. Sources for SCs therapies in diabetes mellitus can be multiple, including embryonic stem cells (ESCs), cord blood stem cells, induced pluripotent stem cells (iPSCs), and adult stem cells derived from adult tissues. Among all kinds of SCs, mesenchymal stem cells (MSCs) derived from the bone marrow or other sources, have been shown as an interesting viable approach for treatment of T1D and tested in human clinical studies aiming to prevent or arrest the onset and progression of T1D, inhibit β-cell destruction, and restore glycometabolic and immune homeostasis. Despite the favorable results indicating potential efficacy of MSCs to preserve β-cell function in some T1D patients demonstrated by the higher C-peptide level, decreased insulin doses and improved HbA1c levels, there are still doubts on the long-term effectiveness of MSCs for the management of T1D (108, 109).
Regarding the hampering effect of hyperglycemia on MSCs therapies, several studies reported that hyperglycemic state and underlying defective microenvironment in diabetic patients impair SCs function. Indeed, high glucose can induce senescence of MSCs via Akt/mTOR signaling (110). Next, hyperglycemia and subsequent oxidative stress triggered by high glucose or chronic RAGE signaling might have a large negative effect on the differentiation, proliferation and regeneration capability of MSCs by affecting Wnt/β-catenin pathway, PI3K/Akt pathway, MAPK signaling, PKC pathway or micro RNA expression (111–116). Study by Kornicka et al. documented that MSCs isolated from patients with T2D exhibited increased apoptosis, autophagy, ROS accumulation and mitochondria deterioration (117). In another study, hyperglycemic conditions in embryos hindered differentiation of human embryonic stem cells by changing their histone methylation pattern leading to pancreatic malfunction (118). Thus, it seems that hyperglycemic conditions in T1D patients might limit therapeutic potential of MSCs-based therapies.
In summary, new emerging evidence suggests that chronic hyperglycemia in patients with diabetes markedly influences their immune system. The rising incidence of T1D calls for new therapeutic options and the immune suppression strategy based on the tolerogenic DCs or T regs vaccines seems to be very promising. The purpose of our review was to summarize the effect of hyperglycemia on various cells of adaptive immunity aiming to clarify the link between the patient's metabolic status and different efficacy of immune interventional treatment. One of the important mechanisms mediating the effect of chronic high glucose level is AGEs formation and their signaling through a specific receptor—RAGE and other groups of scavenger receptors. In essence, AGEs-RAGE interaction leads to activation of transcription factor NF-κB and ROS generation via diverse downstream signaling pathways including MAPK kinase signaling. Therefore, it is believed that this mechanism could contribute to hyperglycemia-induced proinflammatory environment in the body.
For the purpose of cell-based vaccine, tolDCs are generated in autologous system from monocytes of the peripheral blood of patients. Thus, the different status of patient's metabolic control may provide a different input for the cell-based vaccine resulting in a different final quality. Taking into account the metabolic memory phenomenon together with the fact, that even transient episodes of hyperglycemia were associated with epigenetic changes in several cells including progenitor cells (119), the very early tight glucose control seems to be essential for adequate effect of resulting tolerogenic vaccination therapy. In fact, resistance to a further maturation stimulus such as proinflammatory environment and high glucose milieu is a prerequisite for tolerogenic DC clinical application, since a potential transformation of immature or semi-mature DCs into fully mature DCs would lead to the acquisition of the capacity to promote immunogenic instead of protective immune responses and thereby to exacerbation of patient's autoimmune condition. Similarly, stem-cell based vaccines might lose their immunomodulatory potential by facing hyperglycemia conditions at the time of cell administration. Controversially, our previous study showed that alteration of some immune cells (specifically tolDCs) caused by hyperglycemia may be reversed after improvement of glycemic control (12). Thus, the equal importance should be considered for optimal vaccine timing, preferably in the period of ideal metabolic control.
Not only patient's metabolic status, but also appropriate manufacturing protocols for generation of cell-based vaccines, especially with regard to glucose concentration, has to be managed as the applied culture media components may influence the tolerogenic properties of generated immune cells. Indeed, concentration of glucose in culture media above 10 mM are analogous to hyperglycemic conditions in diabetic patients and the cells growing under high-glucose conditions are modified by processes of glycation, glyoxidation, and subsequent oxidative stress. Several important media in bio-manufacturing contain diabetic levels of glucose supplementation (for example DMEM (Hi), GMEM, and IMDM media all contain 25 mM levels of D-glucose). For tolDCs manufacturing and/or T regs expansion Cell Gro GMP medium or X-VIVO 15 medium are widely used, however the precise glucose concentration in those media is unknown and should have been tested in house to avoid hyperglycemic culture conditions (21, 120, 121).
The final question remains, how to circumvent the hyperglycemia issues in cell-based therapy for T1D. Based on the data mentioned above, special attention should be paid on managing patient's metabolic status. Precise adjustment of insulin regimen or conversion to insulin pump for tight glucose controlling seems to be the first step in consideration of cell-based therapy. Managing manufacturing process for generation or expansion of immune cells (appropriate culture conditions) and adjusting precise timing of cell-based vaccine application (euglycemia in patients) are equally important. Furthermore, it remains to be tested whether application of oxidative stress-reducing compounds such as resveratrol during in vitro immune cells generation /or expansion might in consequence improve the possible dysfunctionality of patient's immune cells and the efficacy of cell-based therapies (122). Regarding MSCs, considering allogeneic healthy donors as a source of MSCs for cell based therapies in diabetic patients might be another option.
To sum up, the precise effect of hyperglycemia on immune cells is not fully clear. More detailed studies should be performed to distinguish the genuine effect of high glucose from the changes in immune cells caused by autoimmune disease itself in T1D patients. Nevertheless, based on the recent knowledge, hyperglycemia in diabetic patients may have an impact on immune-interventional treatment and maintaining of a tight glucose control seems to be beneficial in patients considered for cell-based therapy.
AG and LP-J wrote the manuscript and KD and RŠ edited while adding additional insights. The final version was proofread and edited by RS.
The manuscript was supported by SOTIO a.s. only.
LP-J, KD, and RS are named inventor in a related patent, Tolerogenic Dendritic cells, Methods of Producing the Same, and Uses Thereof PCT/EP2015/074536 which describes methods for the preparation of stable semi-mature tolDCs.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Skyler JS, Bakris GL, Bonifacio E, Darsow T, Eckel RH, Groop L, et al. Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes (2017) 66:241–55. doi: 10.2337/db16-0806
2. Amersfoort J, Kuiper J. T cell metabolism in metabolic disease-associated autoimmunity. Immunobiology (2017) 222:925–36. doi: 10.1016/j.imbio.2017.03.001
3. Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. (2005) 353:2643–53. doi: 10.1056/NEJMoa052187
4. Shah BR, Hux JE. Quantifying the risk of infectious diseases for people with diabetes. Diabetes Care (2003) 26:510–3. doi: 10.2337/diacare.26.2.510
5. Geerlings SE, Hoepelman AI. Immune dysfunction in patients with diabetes mellitus (DM). FEMS Immunol Med Microbiol. (1999) 26:259–65.
6. Jafar N, Edriss H, Nugent K. The effect of short-term hyperglycemia on the innate immune system. Am J Med Sci. (2016) 351:201–11. doi: 10.1016/j.amjms.2015.11.011
7. Yip L, Fuhlbrigge R, Taylor C, Creusot RJ, Nishikawa-Matsumura T, Whiting CC, et al. Inflammation and hyperglycemia mediate Deaf1 splicing in the pancreatic lymph nodes via distinct pathways during type 1 diabetes. Diabetes (2015) 64:604–17. doi: 10.2337/db14-0803
8. Menart-Houtermans B, Rutter R, Nowotny B, Rosenbauer J, Koliaki C, Kahl S, et al. Leukocyte profiles differ between type 1 and type 2 diabetes and are associated with metabolic phenotypes: results from the German Diabetes Study (GDS). Diabetes Care (2014) 37:2326–33. doi: 10.2337/dc14-0316
9. Yao K, Ge JB, Sun AJ, Hong XW, Shi HY, Huang RC, et al. Effects and mechanism of hyperglycemia on development and maturation and immune function of human monocyte derived dendritic cells. Zhonghua Xin Xue Guan Bing Za Zhi (2006) 34:60–4.
10. Han XQ, Gong ZJ, Xu SQ, Li X, Wang LK, Wu SM, et al. Advanced glycation end products promote differentiation of CD4(+) T helper cells toward pro-inflammatory response. J Huazhong Univ Sci Technolog Med Sci. (2014) 34:10–7. doi: 10.1007/s11596-014-1224-1
11. Kumar P, Natarajan K, Shanmugam N. High glucose driven expression of pro-inflammatory cytokine and chemokine genes in lymphocytes: molecular mechanisms of IL-17 family gene expression. Cell Signal (2014) 26:528–39. doi: 10.1016/j.cellsig.2013.11.031
12. Dáňová K, Grohova A, Strnadova P, Funda DP, Sumnik Z, Lebl J, et al. Tolerogenic dendritic cells from poorly compensated type 1 diabetes patients have decreased ability to induce stable antigen-specific T cell hyporesponsiveness and generation of suppressive regulatory T cells. J Immunol. (2017) 198:729–40. doi: 10.4049/jimmunol.1600676
13. Roep BO, Tree TI. Immune modulation in humans: implications for type 1 diabetes mellitus. Nat Rev Endocrinol. (2014) 10:229–42. doi: 10.1038/nrendo.2014.2
14. Arif S, Moore F, Marks K, Bouckenooghe T, Dayan CM, Planas R, et al. Peripheral and islet interleukin-17 pathway activation characterizes human autoimmune diabetes and promotes cytokine-mediated beta-cell death. Diabetes (2011) 60:2112–9. doi: 10.2337/db10-1643
15. Segovia-Gamboa N, Rodriguez-Arellano ME, Rangel-Cruz R, Sanchez-Diaz M, Ramirez-Reyes JC, Faradji R, et al. Tolerogenic dendritic cells induce antigen-specific hyporesponsiveness in insulin- and glutamic acid decarboxylase 65-autoreactive T lymphocytes from type 1 diabetic patients. Clin Immunol. (2014) 154:72–83. doi: 10.1016/j.clim.2014.06.009
16. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care (2011) 34:2026–32. doi: 10.2337/dc11-0472
17. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juscinska J, et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin Immunol. (2014) 153:23–30. doi: 10.1016/j.clim.2014.03.016
18. Creusot RJ, Giannoukakis N, Trucco M, Clare-Salzler MJ, Fathman CG. It's time to bring dendritic cell therapy to type 1 diabetes. Diabetes (2014) 63:20–30. doi: 10.2337/db13-0886
19. Farooq T, Rehman K, Hameed A, Akash MSH. Stem cell therapy and type 1 diabetes mellitus: treatment strategies and future perspectives. Adv Exp Med Biol. (2018). doi: 10.1007/5584_2018_195. [Epub ahead of print].
20. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
21. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, et al. Administration of CD4+CD25highCD127- regulatory T cells preserves beta-cell function in type 1 diabetes in children. Diabetes Care (2012) 35:1817–20. doi: 10.2337/dc12-0038
22. Marek-Trzonkowska N, Mysliwiec M, Iwaszkiewicz-Grzes D, Gliwinski M, Derkowska I, Zalinska M, et al. Factors affecting long-term efficacy of T regulatory cell-based therapy in type 1 diabetes. J Transl Med. (2016) 14:332. doi: 10.1186/s12967-016-1090-7
23. Carlsson PO, Schwarcz E, Korsgren O, Le Blanc K. Preserved beta-cell function in type 1 diabetes by mesenchymal stromal cells. Diabetes (2015) 64:587–92. doi: 10.2337/db14-0656
24. Cai J, Wu Z, Xu X, Liao L, Chen J, Huang L, et al. Umbilical cord mesenchymal stromal cell with autologous bone marrow cell transplantation in established type 1 diabetes: a pilot randomized controlled open-label clinical study to assess safety and impact on insulin secretion. Diabetes Care (2016) 39:149–57. doi: 10.2337/dc15-0171
25. Haller MJ, Wasserfall CH, Hulme MA, Cintron M, Brusko TM, McGrail KM, et al. Autologous umbilical cord blood transfusion in young children with type 1 diabetes fails to preserve C-peptide. Diabetes Care (2011) 34:2567–9. doi: 10.2337/dc11-1406
26. Haller MJ, Wasserfall CH, McGrail KM, Cintron M, Brusko TM, Wingard JR, et al. Autologous umbilical cord blood transfusion in very young children with type 1 diabetes. Diabetes Care (2009) 32:2041–6. doi: 10.2337/dc09-0967
27. Zhao Y, Jiang Z, Zhao T, Ye M, Hu C, Yin Z, et al. Reversal of type 1 diabetes via islet beta cell regeneration following immune modulation by cord blood-derived multipotent stem cells. BMC Med. (2012) 10:3. doi: 10.1186/1741-7015-10-3
28. Delgado E, Perez-Basterrechea M, Suarez-Alvarez B, Zhou H, Revuelta EM, Garcia-Gala JM, et al. Modulation of autoimmune T-cell memory by stem cell educator therapy: phase 1/2 clinical trial. EBioMedicine (2015) 2:2024–36. doi: 10.1016/j.ebiom.2015.11.003
29. Dave SD, Vanikar AV, Trivedi HL, Thakkar UG, Gopal SC, Chandra T. Novel therapy for insulin-dependent diabetes mellitus: infusion of in vitro-generated insulin-secreting cells. Clin Exp Med. (2015) 15:41–5. doi: 10.1007/s10238-013-0266-1
30. Ceriello A. Hypothesis: the “metabolic memory”, the new challenge of diabetes. Diabetes Res Clin Pract. (2009) 86(Suppl. 1):S2–6. doi: 10.1016/S0168-8227(09)70002-6
31. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes (2005) 54:1615–25. doi: 10.2337/diabetes.54.6.1615
32. Yan LJ. Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. J Diabetes Res. (2014) 2014:137919. doi: 10.1155/2014/137919
33. Singh R, Barden A, Mori T, Beilin L. Advanced glycation end-products: a review. Diabetologia (2001) 44:129–46. doi: 10.1007/s001250051591
34. Gholap NN, Davies MJ, Mostafa SA, Khunti K. Diagnosing type 2 diabetes and identifying high-risk individuals using the new glycated haemoglobin (HbA1c) criteria. Br J Gen Pract. (2013) 63:e165–7. doi: 10.3399/bjgp13X663244
35. Vistoli G, De Maddis D, Cipak A, Zarkovic N, Carini M, Aldini G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): an overview of their mechanisms of formation. Free Radic Res. (2013) 47(Suppl. 1):3–27. doi: 10.3109/10715762.2013.815348
36. Ramasamy R, Yan SF, Schmidt AM. Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis of diabetes and its complications. Ann N Y Acad Sci. (2011) 1243:88–102. doi: 10.1111/j.1749-6632.2011.06320.x
37. Ihnat MA, Thorpe JE, Kamat CD, Szabo C, Green DE, Warnke LA, et al. Reactive oxygen species mediate a cellular ‘memory' of high glucose stress signalling. Diabetologia (2007) 50:1523–31. doi: 10.1007/s00125-007-0684-2
38. Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. (1992) 267:14998–5004.
39. Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem. (1999) 274:19919–24.
40. Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol. (2014) 2:411–29. doi: 10.1016/j.redox.2013.12.016
41. Wang M, Zhang W, Xu S, Peng L, Wang Z, Liu H, et al. TRB3 mediates advanced glycation end product-induced apoptosis of pancreatic beta-cells through the protein kinase C beta pathway. Int J Mol Med. (2017) 40:130–36. doi: 10.3892/ijmm.2017.2991
42. Brisslert M, Amu S, Pullerits R. Intra-peritoneal sRAGE treatment induces alterations in cellular distribution of CD19(+), CD3 (+) and Mac-1 (+) cells in lymphoid organs and peritoneal cavity. Cell Tissue Res. (2013) 351:139–48. doi: 10.1007/s00441-012-1508-3
43. Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med. (2003) 198:1507–15. doi: 10.1084/jem.20030800
44. Chen Y, Akirav EM, Chen W, Henegariu O, Moser B, Desai D, et al. RAGE ligation affects T cell activation and controls T cell differentiation. J Immunol. (2008) 181:4272–8. doi: 10.4049/jimmunol.181.6.4272
45. Giannoukakis N. Tolerogenic dendritic cells for Type 1 diabetes. Immunotherapy (2013) 5:569–71. doi: 10.2217/imt.13.50
46. Roep B. Immune Intervention With Tolerogenic Dendritic Cells in Type 1 Diabetes. A Phase 1 Safety Study Called D-sense. Clinical Trial no NTR5542 (2015) Available online at: http://www.trialregister.nl/trialreg/admin/rctview.asp?TC=5542
47. Everts B, Amiel E, van der Windt GJ, Freitas TC, Chott R, Yarasheski KE, et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood (2012) 120:1422–31. doi: 10.1182/blood-2012-03-419747
48. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood (2010) 115:4742–9. doi: 10.1182/blood-2009-10-249540
49. Sim WJ, Ahl PJ, Connolly JE. Metabolism is central to tolerogenic dendritic cell function. Mediators Inflamm. (2016) 2016:2636701. doi: 10.1155/2016/2636701
50. Nikolic T, Woittiez NJC, van der Slik A, Laban S, Joosten A, Gysemans C, et al. Differential transcriptome of tolerogenic versus inflammatory dendritic cells points to modulated T1D genetic risk and enriched immune regulation. Genes Immun. (2017) 18:176–83. doi: 10.1038/gene.2017.18
51. Adorini L, Penna G. Induction of tolerogenic dendritic cells by vitamin D receptor agonists. Handb Exp Pharmacol. (2009) 188:251–73.
52. van Kooten C, Stax AS, Woltman AM, Gelderman KA. Handjournal of experimental pharmacology “dendritic cells”: the use of dexamethasone in the induction of tolerogenic DCs. Handb Exp Pharmacol. (2009) 233–49. doi: 10.1007/978-3-540-71029-5_11
53. Malinarich F, Duan K, Hamid RA, Bijin A, Lin WX, Poidinger M, et al. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J Immunol. (2015) 194:5174–86. doi: 10.4049/jimmunol.1303316
54. Ferreira GB, Vanherwegen AS, Eelen G, Gutierrez AC, Van Lommel L, Marchal K, et al. Vitamin D3 induces tolerance in human dendritic cells by activation of intracellular metabolic pathways. Cell Rep. (2015) 10:711–25. doi: 10.1016/j.celrep.2015.01.013
55. Dáňová K, Klapetkova A, Kayserova J, Sediva A, Spisek R, Jelinkova LP. NF-kappaB, p38 MAPK, ERK1/2, mTOR, STAT3 and increased glycolysis regulate stability of paricalcitol/dexamethasone-generated tolerogenic dendritic cells in the inflammatory environment. Oncotarget (2015) 6:14123–38. doi: 10.18632/oncotarget.4234
56. Procaccini C, Galgani M, De Rosa V, Matarese G. Intracellular metabolic pathways control immune tolerance. Trends Immunol. (2012) 33:1–7. doi: 10.1016/j.it.2011.09.002
57. Patel SR, Xu Y, Koenig RJ, Hsu CH. Effect of glucose on the function of the calcitriol receptor and vitamin D metabolism. Kidney Int. (1997) 52:79–86.
58. Unger WW, Laban S, Kleijwegt FS, van der Slik AR, Roep BO. Induction of Treg by monocyte-derived DC modulated by vitamin D3 or dexamethasone: differential role for PD-L1. Eur J Immunol. (2009) 39:3147–59. doi: 10.1002/eji.200839103
59. Waschbisch A, Sanderson N, Krumbholz M, Vlad G, Theil D, Schwab S, et al. Interferon beta and vitamin D synergize to induce immunoregulatory receptors on peripheral blood monocytes of multiple sclerosis patients. PLoS ONE (2014) 9:e115488. doi: 10.1371/journal.pone.0115488
60. Hernandez-Sanchez F, Guzman-Beltran S, Herrera MT, Gonzalez Y, Salgado M, Fabian G, et al. High glucose induces O-GlcNAc glycosylation of the vitamin D receptor (VDR) in THP1 cells and in human macrophages derived from monocytes. Cell Biol Int. (2017) 41:1065–74. doi: 10.1002/cbin.10827
61. Somjen D, Knoll E, Sharon O, Many A, Stern N. Calciotrophic hormones and hyperglycemia modulate vitamin D receptor and 25 hydroxyy vitamin D 1-alpha hydroxylase mRNA expression in human vascular smooth muscle cells. J Steroid Biochem Mol Biol. (2015) 148:210–3. doi: 10.1016/j.jsbmb.2014.11.007
62. Gilardini Montani MS, Granato M, Cuomo L, Valia S, Di Renzo L, D'Orazi G, et al. High glucose and hyperglycemic sera from type 2 diabetic patients impair DC differentiation by inducing ROS and activating Wnt/beta-catenin and p38 MAPK. Biochim Biophys Acta (2016) 1862:805–13. doi: 10.1016/j.bbadis.2016.01.001
63. Mollah ZU, Pai S, Moore C, O'Sullivan BJ, Harrison MJ, Peng J, et al. Abnormal NF-kappa B function characterizes human type 1 diabetes dendritic cells and monocytes. J Immunol. (2008) 180:3166–75. doi: 10.4049/jimmunol.180.5.3166
64. Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes (2001) 50:2792–808. doi: 10.2337/diabetes.50.12.2792
65. Harry RA, Anderson AE, Isaacs JD, Hilkens CM. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann Rheum Dis. (2010) 69:2042–50. doi: 10.1136/ard.2009.126383
66. Boks MA, Kager-Groenland JR, Haasjes MS, Zwaginga JJ, van Ham SM, ten Brinke A. IL-10-generated tolerogenic dendritic cells are optimal for functional regulatory T cell induction–a comparative study of human clinical-applicable DC. Clin Immunol. (2012) 142:332–42. doi: 10.1016/j.clim.2011.11.011
67. Lu H, Yao K, Huang D, Sun A, Zou Y, Qian J, et al. High glucose induces upregulation of scavenger receptors and promotes maturation of dendritic cells. Cardiovasc Diabetol. (2013) 12:80. doi: 10.1186/1475-2840-12-80
68. Buttari B, Profumo E, Capozzi A, Facchiano F, Saso L, Sorice M, et al. Advanced glycation end products of human beta(2) glycoprotein I modulate the maturation and function of DCs. Blood (2011) 117:6152–61. doi: 10.1182/blood-2010-12-325514
69. Ge J, Jia Q, Liang C, Luo Y, Huang D, Sun A, et al. Advanced glycosylation end products might promote atherosclerosis through inducing the immune maturation of dendritic cells. Arterioscler Thromb Vasc Biol. (2005) 25:2157–63. doi: 10.1161/01.ATV.0000181744.58265.63
70. Buttari B, Profumo E, Facchiano F, Ozturk EI, Segoni L, Saso L, et al. Resveratrol prevents dendritic cell maturation in response to advanced glycation end products. Oxid Med Cell Longev. (2013) 2013:574029. doi: 10.1155/2013/574029
71. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. (2010) 185:1037–44. doi: 10.4049/jimmunol.0903586
72. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38:225–36. doi: 10.1016/j.immuni.2012.10.020
73. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
74. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
75. Martinez N, Vallerskog T, West K, Nunes-Alves C, Lee J, Martens GW, et al. Chromatin decondensation and T cell hyperresponsiveness in diabetes-associated hyperglycemia. J Immunol. (2014) 193:4457–68. doi: 10.4049/jimmunol.1401125
76. Monti P, Brigatti C, Krasmann M, Ziegler AG, Bonifacio E. Concentration and activity of the soluble form of the interleukin-7 receptor alpha in type 1 diabetes identifies an interplay between hyperglycemia and immune function. Diabetes (2013) 62:2500–8. doi: 10.2337/db12-1726
77. Torres-Aguilar H, Sanchez-Torres C, Jara LJ, Blank M, Shoenfeld Y. IL-10/TGF-beta-treated dendritic cells, pulsed with insulin, specifically reduce the response to insulin of CD4+ effector/memory T cells from type 1 diabetic individuals. J Clin Immunol. (2010) 30:659–68. doi: 10.1007/s10875-010-9430-5
78. Ramasamy R, Yan SF, Herold K, Clynes R, Schmidt AM. Receptor for advanced glycation end products: fundamental roles in the inflammatory response: winding the way to the pathogenesis of endothelial dysfunction and atherosclerosis. Ann N Y Acad Sci. (2008) 1126:7–13. doi: 10.1196/annals.1433.056
79. Jaisson S, Souchon PF, Desmons A, Salmon AS, Delemer B, Gillery P. Early formation of serum advanced glycation end-products in children with type 1 diabetes mellitus: relationship with glycemic control. J Pediatr. (2016) 172:56–62. doi: 10.1016/j.jpeds.2016.01.066
80. Akirav EM, Preston-Hurlburt P, Garyu J, Henegariu O, Clynes R, Schmidt AM, et al. RAGE expression in human T cells: a link between environmental factors and adaptive immune responses. PLoS ONE (2012) 7:e34698. doi: 10.1371/journal.pone.0034698
81. Durning SP, Preston-Hurlburt P, Clark PR, Xu D, Herold KC, Type 1 Diabetes TrialNet Study Group. The Receptor for Advanced Glycation Endproducts Drives T Cell Survival and Inflammation in Type 1 Diabetes Mellitus. J Immunol. (2016) 197:3076–85. doi: 10.4049/jimmunol.1600197
82. Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. (2009) 182:259–73. doi: 10.4049/jimmunol.182.1.259
83. Jeker LT, Bour-Jordan H, Bluestone JA. Breakdown in peripheral tolerance in type 1 diabetes in mice and humans. Cold Spring Harb Perspect Med. (2012) 2:a007807. doi: 10.1101/cshperspect.a007807
84. Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes (2005) 54:92–9. doi: 10.2337/diabetes.54.1.92
85. Zhang Y, Bandala-Sanchez E, Harrison LC. Revisiting regulatory T cells in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. (2012) 19:271–8. doi: 10.1097/MED.0b013e328355a2d5
86. Brusko T, Wasserfall C, McGrail K, Schatz R, Viener HL, Schatz D, et al. No alterations in the frequency of FOXP3+ regulatory T-cells in type 1 diabetes. Diabetes (2007) 56:604–12. doi: 10.2337/db06-1248
87. Brusko TM, Wasserfall CH, Clare-Salzler MJ, Schatz DA, Atkinson MA. Functional defects and the influence of age on the frequency of CD4+ CD25+ T-cells in type 1 diabetes. Diabetes (2005) 54:1407–14. doi: 10.2337/diabetes.54.5.1407
88. Haseda F, Imagawa A, Murase-Mishiba Y, Terasaki J, Hanafusa T. CD4(+) CD45RA(-) FoxP3high activated regulatory T cells are functionally impaired and related to residual insulin-secreting capacity in patients with type 1 diabetes. Clin Exp Immunol. (2013) 173:207–16. doi: 10.1111/cei.12116
89. Singh K, Kadesjo E, Lindroos J, Hjort M, Lundberg M, Espes D, et al. Interleukin-35 administration counteracts established murine type 1 diabetes–possible involvement of regulatory T cells. Sci Rep. (2015) 5:12633. doi: 10.1038/srep12633
90. Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S, et al. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes (2010) 59:407–15. doi: 10.2337/db09-0694
91. McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol. (2011) 186:3918–26. doi: 10.4049/jimmunol.1003099
92. Marwaha AK, Crome SQ, Panagiotopoulos C, Berg KB, Qin H, Ouyang Q, et al. Cutting edge: Increased IL-17-secreting T cells in children with new-onset type 1 diabetes. J Immunol. (2010) 185:3814–8. doi: 10.4049/jimmunol.1001860
93. Hull CM, Nickolay LE, Estorninho M, Richardson MW, Riley JL, Peakman M, et al. Generation of human islet-specific regulatory T cells by TCR gene transfer. J Autoimmun. (2017) 79:63–73. doi: 10.1016/j.jaut.2017.01.001
94. Glisic-Milosavljevic S, Wang T, Koppen M, Kramer J, Ehlenbach S, Waukau J, et al. Dynamic changes in CD4+ CD25+(high) T cell apoptosis after the diagnosis of type 1 diabetes. Clin Exp Immunol. (2007) 150:75–82. doi: 10.1111/j.1365-2249.2007.03475.x
95. Jailwala P, Waukau J, Glisic S, Jana S, Ehlenbach S, Hessner M, et al. Apoptosis of CD4+ CD25(high) T cells in type 1 diabetes may be partially mediated by IL-2 deprivation. PLoS ONE (2009) 4:e6527. doi: 10.1371/journal.pone.0006527
96. Tan T, Xiang Y, Chang C, Zhou Z. Alteration of regulatory T cells in type 1 diabetes mellitus: a comprehensive review. Clin Rev Allergy Immunol. (2014) 47:234–43. doi: 10.1007/s12016-014-8440-0
97. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol. (2015) 16:1174–84. doi: 10.1038/ni.3269
98. Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, et al. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity (2016) 44:406–21. doi: 10.1016/j.immuni.2016.01.028
99. Devaraj S, Venugopal SK, Singh U, Jialal I. Hyperglycemia induces monocytic release of interleukin-6 via induction of protein kinase c-{alpha} and -{beta}. Diabetes (2005) 54:85–91. doi: 10.2337/diabetes.54.1.85
100. Gao Z, Gao Y, Li Z, Chen Z, Lu D, Tsun A, et al. Synergy between IL-6 and TGF-beta signaling promotes FOXP3 degradation. Int J Clin Exp Pathol. (2012) 5:626–33.
101. Zhen Y, Sun L, Liu H, Duan K, Zeng C, Zhang L, et al. Alterations of peripheral CD4+CD25+Foxp3+ T regulatory cells in mice with STZ-induced diabetes. Cell Mol Immunol. (2012) 9:75–85. doi: 10.1038/cmi.2011.37
102. Ryba M, Rybarczyk-Kapturska K, Zorena K, Mysliwiec M, Mysliwska J. Lower frequency of CD62L(high) and higher frequency of TNFR2(+) Tregs are associated with inflammatory conditions in type 1 diabetic patients. Mediators Inflamm. (2011) 2011:645643. doi: 10.1155/2011/645643
103. Stechova K, Sklenarova-Labikova J, Kratzerova T, Pithova P, Filipp D. Not only glycaemic but also other metabolic factors affect T regulatory cell counts and proinflammatory cytokine levels in women with type 1 diabetes. J Diabetes Res. (2017) 2017:5463273. doi: 10.1155/2017/5463273
104. El-Samahy MH, Adly AA, Ismail EA, Salah NY. Regulatory T cells with CD62L or TNFR2 expression in young type 1 diabetic patients: relation to inflammation, glycemic control and micro-vascular complications. J Diabetes Complications (2015) 29:120–6. doi: 10.1016/j.jdiacomp.2014.07.004
105. Okubo Y, Torrey H, Butterworth J, Zheng H, Faustman DL. Treg activation defect in type 1 diabetes: correction with TNFR2 agonism. Clin Transl Immunol. (2016) 5:e56. doi: 10.1038/cti.2015.43
106. El-Badri N, Ghoneim MA. Mesenchymal stem cell therapy in diabetes mellitus: progress and challenges. J Nucleic Acids (2013) 2013:194858. doi: 10.1155/2013/194858
107. Katuchova J, Harvanova D, Spakova T, Kalanin R, Farkas D, Durny P, et al. Mesenchymal stem cells in the treatment of type 1 diabetes mellitus. Endocr Pathol. (2015) 26:95–103. doi: 10.1007/s12022-015-9362-y
108. Hashemian SJ, Kouhnavard M, Nasli-Esfahani E. Mesenchymal stem cells: rising concerns over their application in treatment of type one diabetes mellitus. J Diabetes Res. (2015) 2015:675103. doi: 10.1155/2015/675103
109. Cho J, D'Antuono M, Glicksman M, Wang J, Jonklaas J. A review of clinical trials: mesenchymal stem cell transplant therapy in type 1 and type 2 diabetes mellitus. Am J Stem Cells (2018) 7:82–93.
110. Zhang D, Lu H, Chen Z, Wang Y, Lin J, Xu S, et al. High glucose induces the aging of mesenchymal stem cells via Akt/mTOR signaling. Mol Med Rep (2017) 16:1685–90. doi: 10.3892/mmr.2017.6832
111. Zhang X, Meng K, Pu Y, Wang C, Chen Y, Wang L. Hyperglycemia altered the fate of cardiac stem cells to adipogenesis through inhibiting the beta-catenin/TCF-4 pathway. Cell Physiol Biochem. (2018) 49:2254–63. doi: 10.1159/000493828
112. Deng X, Xu M, Shen M, Cheng J. Effects of type 2 diabetic serum on proliferation and osteogenic differentiation of mesenchymal stem cells. J Diabetes Res. (2018) 2018:5765478. doi: 10.1155/2018/5765478
113. Denu RA, Hematti P. Effects of oxidative stress on mesenchymal stem cell biology. Oxid Med Cell Longev. (2016) 2016:2989076. doi: 10.1155/2016/2989076
114. Aikawa E, Fujita R, Asai M, Kaneda Y, Tamai K. Receptor for advanced glycation end products-mediated signaling impairs the maintenance of bone marrow mesenchymal stromal cells in diabetic model mice. Stem Cells Dev. (2016) 25:1721–32. doi: 10.1089/scd.2016.0067
115. Zhu G, Chai J, Ma L, Duan H, Zhang H. Downregulated microRNA-32 expression induced by high glucose inhibits cell cycle progression via PTEN upregulation and Akt inactivation in bone marrow-derived mesenchymal stem cells. Biochem Biophys Res Commun. (2013) 433:526–31. doi: 10.1016/j.bbrc.2013.03.018
116. Saki N, Jalalifar MA, Soleimani M, Hajizamani S, Rahim F. Adverse effect of high glucose concentration on stem cell therapy. Int J Hematol Oncol Stem Cell Res (2013) 7:34–40.
117. Kornicka K, Houston J, Marycz K. Dysfunction of mesenchymal stem cells isolated from metabolic syndrome and type 2 diabetic patients as result of oxidative stress and autophagy may limit their potential therapeutic use. Stem Cell Rev. (2018) 14:337–45. doi: 10.1007/s12015-018-9809-x
118. Chen ACH, Lee YL, Fong SW, Wong CCY, Ng EHY, Yeung WSB. Hyperglycemia impedes definitive endoderm differentiation of human embryonic stem cells by modulating histone methylation patterns. Cell Tissue Res. (2017) 368:563–78. doi: 10.1007/s00441-017-2583-2
119. Berezin A. Metabolic memory phenomenon in diabetes mellitus: achieving and perspectives. Diabetes Metab Syndr. (2016) 10:S176–83. doi: 10.1016/j.dsx.2016.03.016
120. Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA, et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheum Dis. (2017) 76:227–34. doi: 10.1136/annrheumdis-2015-208456
121. Putnam AL, Brusko TM, Lee MR, Liu W, Szot GL, Ghosh T, et al. Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes (2009) 58:652–62. doi: 10.2337/db08-1168
Keywords: dendritic cells, immune tolerance, cell-based therapy, diabetes mellitus, hyperglycemia
Citation: Grohová A, Dáňová K, Špíšek R and Palová-Jelínková L (2019) Cell Based Therapy for Type 1 Diabetes: Should We Take Hyperglycemia Into Account? Front. Immunol. 10:79. doi: 10.3389/fimmu.2019.00079
Received: 25 October 2018; Accepted: 11 January 2019;
Published: 05 February 2019.
Edited by:
Djordje Miljkovic, University of Belgrade, SerbiaReviewed by:
Julien Diana, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2019 Grohová, Dáňová, Špíšek and Palová-Jelínková. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lenka Palová-Jelínková, cGFsb3ZhQHNvdGlvLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.