 Mohib Uddin
Mohib Uddin Henrik Watz3,4
Henrik Watz3,4 Anna Malmgren
Anna Malmgren Frauke Pedersen
Frauke Pedersen- 1Respiratory Global Medicines Development, AstraZeneca, Gothenburg, Sweden
- 2Respiratory, Inflammation and Autoimmunity, IMED Biotech Unit, AstraZeneca, Gothenburg, Sweden
- 3Pulmonary Research Institute at LungenClinic, Großhansdorf, Germany
- 4Airway Research Center North (ARCN), German Center for Lung Research (DZL), Großhansdorf, Germany
- 5LungenClinic, Großhansdorf, Germany
Neutrophils play a central role in innate immunity, inflammation, and resolution. Unresolving neutrophilia features as a disrupted inflammatory process in the airways of patients with chronic obstructive pulmonary disease (COPD) and severe asthma. The extent to which this may be linked to disease pathobiology remains obscure and could be further confounded by indication of glucocorticoids or concomitant respiratory infections. The formation of neutrophil extracellular traps (NETs) represents a specialized host defense mechanism that entrap and eliminate invading microbes. NETs are web-like scaffolds of extracellular DNA in complex with histones and neutrophil granular proteins, such as myeloperoxidase and neutrophil elastase. Distinct from apoptosis, NET formation is an active form of cell death that could be triggered by various microbial, inflammatory, and endogenous or exogenous stimuli. NETs are reportedly enriched in neutrophil-dominant refractory lung diseases, such as COPD and severe asthma. Evidence for a pathogenic role for respiratory viruses (e.g., Rhinovirus), bacteria (e.g., Staphylococcus aureus) and fungi (e.g., Aspergillus fumigatus) in NET induction is emerging. Dysregulation of this process may exert localized NET burden and contribute to NETopathic lung inflammation. Disentangling the role of NETs in human health and disease offer unique opportunities for therapeutic modulation. The chemokine CXCR2 receptor regulates neutrophil activation and migration, and small molecule CXCR2 antagonists (e.g., AZD5069, danirixin) have been developed to selectively block neutrophilic inflammatory pathways. NET-stabilizing agents using CXCR2 antagonists are being investigated in proof-of-concept studies in patients with COPD to provide mechanistic insights. Clinical validation of this type could lead to novel therapeutics for multiple CXCR2-related NETopathologies. In this Review, we discuss the emerging role of NETs in the clinicopathobiology of COPD and severe asthma and provide an outlook on how novel NET-stabilizing therapies via CXCR2 blockade could be leveraged to disrupt NETopathic inflammation in disease-specific phenotypes.
Neutrophils
Optimal regulation of neutrophil-mediated immunity is essential for confining pathogens, resolving infection and inflammation (1). Neutrophils are the most abundant circulating leukocyte and are first responders of the host-protective innate immunity (2). These cells fulfill their sentinel role by safeguarding the host immune homeostasis through maintaining a strict equilibrium of the innate immune and acute inflammatory responses (3). While a robust innate immune response is vitally important for an effective host defense, dysfunction of neutrophils is associated with bystander immune responses in certain treatment-refractory lung diseases (4, 5).
Circulating neutrophils are short-lived innate immune cells and limited by an inherent pathway of apoptosis. Apoptosis is a form of programmed cell death and phagocytic clearance of dying neutrophils by macrophage facilitates the resolution of inflammation (6). Defective neutrophil apoptosis has been reported in the airways of patients with asthma with the degree of dysfunction corresponding to disease severity (7), suggesting derangements in inflammation resolution. Conflicting data exist on the potential role of impaired apoptosis in COPD (8, 9), whereas defects in efferocytosis have been identified in both neutrophilic asthma (10) and COPD (11). Targeting defective cell death pathways in these refractory airway diseases represents an attractive therapeutic strategy.
Neutrophil Extracellular Traps (NETs)
Neutrophils kill and clear invading pathogens via phagocytosis, degranulation and oxidative burst. There is an increasing recognition that neutrophils can extrude cytosolic and nuclear material via a conserved cell death process distinct to apoptosis and necrosis (12). This active process is commonly referred to as neutrophil extracellular traps (NETs) and was first coined in 2004 by Brinkmann and colleagues as a novel antimicrobial defense system (13). NETs are web-like scaffolds of extracellular DNA in complex with histones and antimicrobial neutrophil granular proteins, such as myeloperoxidase and neutrophil elastase (13). Entrapment of microorganisms by NETs restrict potential pathogen dissemination from the initial site of infection and it is hypothesized that electrostatic interactions govern the specificity of binding between the cationic nature of NETs and the anionic bacterial cell wall (14). Physicochemical perturbations are reported to modulate NET formation, including hypoxia, osmolarity, and extracellular pH shifts (15–19).
Though NETs are considered an essential part of neutrophil-mediated immunity, they have also been incriminated in NET-based immunopathology (20, 21). In a sense, NETs represent a “double-edged sword” in innate immunity (22), heavily dependent on maintaining a tight equilibrium between protective and detrimental immune responses. Aberrant NET production in the circulation and affected tissues have been reported in patients with cystic fibrosis and ARDS (23–25)—airway diseases typified by neutrophilic mucosal inflammation. Persistence of exaggerated NET formation may induce collateral damage to the airway tissues. For instance, NETs can directly trigger epithelial cell death (26) or when excessive, may impair lung epithelial barrier function during respiratory viral infection in vivo (27). Murine studies have demonstrated a role for NETs in inducing airway mucus hypersecretion (28). Furthermore, emerging translational studies lend further support to the assertion that NET burden could have a pathobiological role in treatment-refractory airways diseases. We (29, 30) and others (31–33), have recently provided the first evidence for induction of NET formation in neutrophils derived from COPD and severe asthma patients, suggesting that this process may also influence airway immunopathology (“lung NETopathy”) in these diseases (Table 1). A schematic representation of the proposed mechanism involving NETs in mediating airway NETopathic inflammation in NET-rich COPD and in the asthma smoking phenotype is depicted on Figure 1A.

Table 1. Overview of translational evidence of NET formation in patients with COPD and asthma.
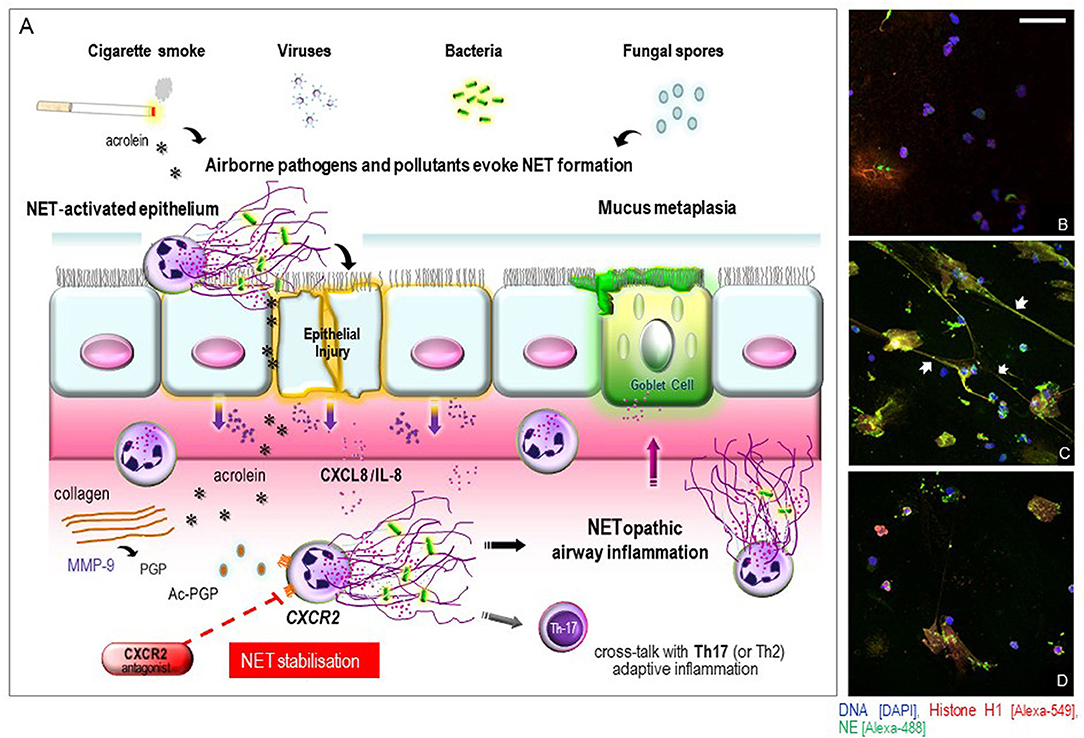
Figure 1. (A) schematic illustration of proposed mechanisms of airway epithelial and innate immune cell responses to NET induction in COPD or in the asthma smoking phenotype and the potential influence of CXCR2 signaling. Exposure to airborne pathogens and pollutant stimuli evoke a NET-permissive microenvironment leading to a cycle of airway mucosal inflammation. A dysregulated epithelium releases neutrophil-attracting mediators that can transduce their effects via CXCR2 signaling. The oxidants in cigarette smoke and other reactive components (e.g., acrolein) can also directly trigger NET formation or via acetylation of proteolytically cleaved collagen PGP to yield potent CXCR2-signaling matrikines, attracting neutrophils into the airways to further perpetuate NETopathic inflammation. NETs may also contribute to mucus-hypersecretion by submucosal glands via induction of respiratory mucins. Additionally, defective NET production can induce Th2 and Th17 adaptive immune responses which may further contribute to the disease pathomechanism. Our hypothesis is that selective CXCR2 antagonists [e.g., AZD5069] could essentially block CXCR2-stimulated NET induction, and may thereby represent a potential NET-stabilizing agent capable of disrupting NETopathic airway inflammation in NET-rich COPD or in the asthma smoking phenotype. (B–D) CXCR2 receptor antagonism stabilizes heightened NET formation in COPD ex vivo. Representative merged 3D confocal-fluorescence images of the NETs formed in COPD-derived peripheral blood neutrophils in response to stimulation with autologous COPD sputum supernatant from the same consenting patient. All specimens were fixed in phosphate buffered 4% paraformaldehyde. Immunofluorescence staining for detection of histone H1 (green), neutrophil elastase (red) and nuclear DNA with 4,6-diamidino-2-phenylindole (DAPI) (blue) was performed as described previously (30). Representative visual fields were acquired using a confocal laser scanning microscope (Leica microsystems TCS SP5) and captured by applying identical magnification, laser intensities, and detector settings. (B) unstimulated blood neutrophils with DAPI staining showing nuclear DNA; (C) blood neutrophils stimulated with autologous COPD sputum supernatant ex vivo with merges of the three colocalized images for histone H1, neutrophil elastase, and DAPI staining indicating projection of NET formation (arrowheads); (D) blood neutrophils stimulated with autologous COPD sputum supernatant after pretreatment with 100 μM AZD5069 ex vivo, with merges of the three color images indicating stabilized NET formation. Scale bar 50 μm.
NETopathic Inflammation in COPD
COPD is projected to become the third leading cause of death world-wide and is a major cause of mortality in adults (34). It is characterized by airflow limitation that is poorly reversible caused by narrowing of the small airways combined with emphysematous destruction (34). COPD is heterogeneous and encompasses a clinical spectrum of disease-specific phenotypes (35). Chronic exposure to cigarette smoke has been recognized to contribute to COPD pathogenesis (34), where it can elicit neutrophil retention within the airways (36). In general, the degree of neutrophilia correlates with COPD severity (37, 38), exacerbations (39), disease progression (40), and GC resistance (35).
Two independent translational studies in 2015, by Pedersen et al. (29) and Grabcanovic-Musija et al. (32) first observed NET formation in sputum from both stable and exacerbated COPD patients using confocal fluorescent and electron microscopy, respectively. These findings suggested that NET formation may be a feature of a NET-rich COPD phenotype. NET production has been shown to occur in peripheral blood-derived neutrophils from a small cohort of patients with stable COPD (41). Elevated sputum NETs are also associated with lung function and COPD symptoms and peptidylarginine deiminase 4 (PAD4) gene expression was found to be upregulated in neutrophilic COPD relative to non-neutrophilic patients (42). The increase in PAD4 expression at the protein level has previously been reported in the lungs of COPD patients (43), and it is believed that activation of PAD4 is an important regulator of NET formation (44). More recently, Dicker and colleagues reported increased NET formation in the airways of patients of COPD that was associated with disease severity (45). They went onto demonstrate a relationship between sputum-enriched NETs and non-eosinophilic COPD exacerbations and reduced bacterial diversity accompanied by an abundance of Haemophilus species. Functionally, the phagocytic capacity of neutrophils to engulf bacteria ex vivo was impaired in cells derived from patients with high sputum NET complexes, supporting their potential role in microbial dysbiosis in COPD (45).
CXCR2 Signaling in COPD
CXCR2 is a G-protein-coupled receptor that binds human CXC chemokine ligands, such as CXCL8/IL-8, CXCL1/GRO-α, and CXCL5/epithelial neutrophil-activating peptide-78 (ENA-78) with high affinity (46, 47). CXCL8/IL-8 is considered to be the predominant neutrophil-attracting chemokine in COPD airway secretions, accounting for the trafficking of approximately one-third of neutrophilic infiltrates in sputum (48). Additional CXCR2 ligands, such as CXCL1/GRO-α and CXCL5/ENA-78 are elevated in COPD sputa, airway fluids, and bronchial tissues (38, 49, 50). CXCR2 is upregulated in exacerbations of COPD where its expression co-localizes with accumulation of airway mucosal neutrophils (49). Recent clinical studies with the CXCR2 antagonist MK-7123 (SCH527123) in patients with COPD have shown a significant neutrophil-lowering effect leading to improvements in FEV1 and a reduction in exacerbations in active smokers compared to placebo (51). However, a dramatic fall in circulating neutrophils (neutropenia) was noted in a large proportion of COPD patients, raising concerns about undesirable immunosuppression. Avoidance of off-target effects are the major safety concerns to address, however, there has been scientific debate as to whether this could be mitigated by local delivery of inhalable CXCR2 antagonists directly to the lungs. From an efficacy perspective, it remains to be determined whether the observed anti-CXCR2 effects on improved lung function in that study were associated with NET stabilization within the COPD airways.
As further clinical trial data with CXCR2 antagonists emerge in NET-rich lung diseases, it will be important to dissect how well the steroid-resistant phenotypes respond to these neutrophil-trafficking CXCR2 inhibitors. Further mechanistic insights into CXCR2-mediated signaling is emerging from studies involving extracellular matrix (ECM)-derived fragments, such as the pro-neutrophilic matrikine, proline-glycine-proline (PGP). This tripeptide has structural homology to ELR+ chemokines and can chemotactically recruit neutrophils via CXCR2 receptors (52). Levels of PGP are markedly raised in the airways of patients with COPD (52, 53) which can occur in response to cigarette smoke (54). This airway irritant is also able to directly induce NET production (55), through its bioactive constituents, such as the reactive aldehyde and tussive agent acrolein (56). Acrolein can induce release of PGP fragments, including acetyl-proline-glycine-proline [AcPGP] (57) that function as potent CXCR2 signaling ligands. AcPGP has been utilized as a potential biomarker in a recent clinical study involving COPD patients with chronic bronchitis (58). Expanding the concept of PGP-CXCR2 crosstalk in COPD, we propose it is pharmacologically plausible that PGP could also trigger NET formation during COPD-related airway inflammation. Future studies need to establish whether PGP can directly contribute to lung NETopathologies in a CXCR2-dependent manner.
Interestingly, emerging evidence also implicates CXCR2 signaling in regulating NET production in COPD neutrophils. Selective anti-CXCR2 blockade using AZD5069 limited NET formation in autologous blood and sputum neutrophils derived from the same COPD patients ex vivo (30), representing the first reported evidence that a CXCR2 antagonist can stabilize COPD-derived NET formation (Figures 1B–D). This unique profile of CXCR2 dependence in regulating COPD-related NETopathic inflammation, and its consequent antagonism, differentiates it from the conventional neutrophil-trafficking functions. Applying this approach clinically, it is noteworthy that CXCR2 antagonists are now being trialed to evaluate their modulatory effects on sputum NET production in patients with COPD [ClinicalTrials.gov-Identifier: NCT03250689]. If successful, this could introduce an emerging field of therapeutic research for “NET stabilizers” in a range of CXCR2-related NETopathologies, including NET-rich COPD.
Pulmonary Pathogen-initiated NETopathic Inflammation
Respiratory viral infections, such as respiratory syncytial virus (RSV), rhinovirus and influenza virus A, are associated with exacerbations of COPD (59) and can induce NET formation (60–62). NETs have been ascribed an anti-viral immune role (63, 64), involving molecular interactions between NET-derived bactericidal/permeability-increasing protein and the fusion (F) protein on the viral surface (65). However, an aberrant NETopathic response can be damaging to the host when excessive. This is consistent with murine studies demonstrating that viral-induced NET perturbance can elicit acute lung injury and airway inflammatory responses following challenges with influenza virus A and parainfluenza virus type I (Sendai virus) in experimental models of pneumonitis and asthma in vivo, respectively (27, 66).
COPD patients are highly susceptible to recurrent bacterial infections which often occur after encountering respiratory viral infections (67). Chronic airway bacterial colonization with the pathogenic organisms, Haemophilus influenzae and Streptococcus pneumoniae are strongly associated with recurrent COPD exacerbations (68, 69). The pneumonia-causing pathogen, Staphylococcus aureus (S. aureus), has also been detected in the airways of COPD patients (70, 71), notably in much older populations (72). This is particularly important because S. aureus can directly trigger NET formation (73), and might thus influence the cycle of pathogen–NETopathic inflammation in COPD. Despite there being high levels of NETs in the COPD airways, there is a deficiency in effective pathogen clearance. Reports suggest that community-acquired pneumonia (CAP) is more common in patients with COPD relative to the adult general population and is higher in the winter period (67). Circulatory NETs have recently been shown to be associated with 30-day all-cause mortality in a cohort of 310 patients with CAP of whom 17.4% had COPD as a comorbidity (74). These findings are suggestive of an innate immunity conundrum we refer to as “the NET paradox”; if NET-forming neutrophils are committed to the eradication of invading microbes, then why do respiratory pathogens frequently colonize and induce pathogenicity in the CAP airways? Recent findings indicate that certain respiratory pathogens have developed effective mechanisms to evade NET-mediated entrapment and killing [reviewed in (75)]. This immune evasion strategy deployed by respiratory pathogens could explain the apparent NET paradox in pathogen-colonized airways in these patients, and might be an important factor in determining the outcome of CAP infections.
In asthma, respiratory viral infections represent acute pathobiological events that can precipitate exacerbations (76, 77). Rhinovirus-induced respiratory infections elicit rapid recruitment of neutrophils in the asthmatic airways with a concomitant increase in CXCL8/IL-8 levels (78, 79) and degree of airway responsiveness (80). It has been shown that CXCR2 signaling is essential for rhinovirus-induced neutrophilic airway inflammation and development of airway hyperresponsiveness in murine model in vivo (81). Recent work by Toussaint et al. have shown that NETs cross-talk with viral-provoked immune responses in the respiratory tract, thereby driving type-2 allergic airway inflammatory responses (82, 83). Elevated levels of host double stranded DNA (dsDNA) were observed in the upper asthmatic airways elicited by rhinovirus infection that correlated with subsequent type-2 immune-mediated asthma exacerbation severity. The study recapitulated the pathobiological features that mimic RV-induced asthma exacerbation using a house dust mite (HDM)-allergen-sensitized mouse model in vivo. Depletion of dsDNA production using DNase was found to constrain RV-induced exacerbation and hyper-responsiveness after HDM allergen challenge (82), in agreement with reports of lung function improvements in another pre-clinical model of asthma (84). Further interrogation of the role and regulation of viral-induced NETopathy with synergistic interaction following allergen exposure in triggering asthma exacerbations are warranted.
In addition to respiratory viral and bacterial infections, airborne transmission of fungi is associated with a subphenotype of asthma denoted as severe asthma with fungal sensitization (SAFS) (85). Sensitization to Aspergillus fumigatus and consequent airway neutrophilic inflammation correlate with asthma severity and reduced lung function (86). An essential role for NETs in the local confinement of A. fumigatus has been reported in vitro (87) and in vivo (88). It remains to be seen whether fungal-neutrophil interactions could uncover specific pathological roles in driving NETopathic airway inflammation in severe asthma.
NETopathic Inflammation in Asthma
Severe asthma is distinguished from milder forms of the disease by distinct underlying endotypes (89, 90). Neutrophilic predominant asthmatic inflammation differs from Th2-high eosinophilic inflammation, thus often being associated with Th2-low asthma (T2-low) (91–93). The presence of viable neutrophils in the airways often correlates with poorer clinical outcome, disease severity and response to GC therapies (7, 94–100). Translational evidence incriminating neutrophils in the pathobiology of severe asthma have provided a rationale for exploring therapeutic agents that selectively block inflammatory pathways (4, 101–103). However, the precise nature of neutrophilic asthmatic inflammation remains contentious (104, 105).
In asthma, both NETs and eosinophil extracellular traps (EETs) have been detected in bronchial biopsies from atopic patients, although levels were not different between pre- and post-allergen challenge (106). Increased NETs and their components, namely extracellular DNA and LL-37, as well as CXCL8/IL-8 in induced sputum were significantly higher in neutrophilic asthmatics relative to the non-neutrophilic cohort (42). This was inversely correlated with lung function and asthma control, suggestive of a potential link between airway NETopathy and worst asthma outcomes. Further, peripheral blood-derived neutrophils from severe asthmatics displayed greater NET production than did cells from non-severe patients after CXCL8/IL-8 stimulation ex vivo (33). Excessive NET load has been theorized as a potential driver for disordered mucosal epithelium in relation to asthma severity. Ex vivo stimulation of airway epithelial cells with NETs can markedly induce endogenous CXCL8/IL-8 production (33, 107), creating a possible self-amplifying cycle of airway inflammation, consistent with reports of a mucosal epithelial-neutrophil interplay in severe asthma (108). In addition, recent findings show that a subset of severe asthmatics has NETopathic inflammation mediated by neutrophil cytoplasts which may skew the immune responses toward Th17 adaptive inflammation (109, 110). Augmented NET formation has also been reported in eosinophilic granulomatosis with polyangiitis (EGPA; traditionally termed Churg–Strauss), an asthma plus syndrome characterized by eosinophilic inflammation and paranasal sinusitis (111). Circulatory NETs have been shown to positively correlate with anti-lactoferrin-specific antibody titers in patients with EGPA in another study (112). Together, these data indicate that defective NET formation might underpin some aspects of airway inflammation in more severe asthma.
CXCR2 Signaling in Severe Asthma
Elevated expression levels of CXC chemokines (CXCL1/GRO-α, CXCL5/ENA-78, or CXCL8/IL-8) associate with increased neutrophil infiltrates in the asthmatic bronchial mucosa, airway wall, and sputum (113–117). Anti-CXCR2 blockade can negate multiple features of pulmonary neutrophilic inflammation in vivo, supporting a pro-inflammatory role for CXCR2 signaling (118). AZD5069 is a selective CXCR2 antagonist developed by AstraZeneca that blocks neutrophil trafficking without hindering neutrophil effector responses of host defense (119, 120). Recently, AZD5069 has been shown to diminish CXCR2-mediated neutrophil infiltrates in spontaneous sputum from bronchiectasis patients (69% inhibition) (121), as well as pronounced reduction in induced sputum and bronchial biopsies from patients with more severe asthma (90% inhibition) (122). Treatment with another oral CXCR2 antagonist (MK-7123) was shown to attenuate sputum neutrophilia by 36% in patients with severe asthma (n = 34), yet had no clinical effect on asthma outcomes (123). Despite the observed neutrophil infiltrate lowering effects in severe asthma, results from trials of orally-active CXCR2 antagonists have not shown any clinical benefits to date.
More recently, a larger (6 months/n = 640) double-blind randomized phase 2b study has been conducted using three different doses of AZD5069 in patients with uncontrolled persistent asthma (102). AZD5069 induced a dose-related and sustained reduction in circulating neutrophils, whereas no impact was observed on the clinical outcomes in that study which had an overall low severe exacerbation rate, leaving uncertainties about the pathomechanism of CXCR2-mediated neutrophil recruitment in severe asthma. Targeting well-defined endotypes of neutrophilic non-eosinophilic/T2-low asthma using a biomarker-directed approach has recently been proposed (101, 103). In this context, the exclusion of patients who were current smokers or ex-smokers (smoking history of ≥ 20 pack years) in this clinical study (102), meant that it was not possible to determine the therapeutic potential of CXCR2 antagonists in smokers with asthma. Bearing in mind that nearly a quarter of adult asthma patients are current cigarette smokers in the “real world” (124), chronic exposure to tobacco smoke can modify the airway inflammatory profile (whilst reducing fractional exhaled nitric oxide [FeNO] levels) and worsen clinical outcomes in smokers with more severe asthma (125, 126). Data from the recent U-BIOPRED cluster analyses highlight that smoking pack year history differentially induces a pro-inflammatory signature profile (but not Th2-associated) in sputum of patients with severe asthma (127). As noted, cigarette smoke exposure leads to induction of NET formation (55) in association with CXCL8/IL-8 upregulation in the airway mucosa of smoking asthmatics (128), an inflammatory response that is insensitive to systemic GC therapy (129). This could lead to a feed-forward process of smoke-triggered CXCR2-dependent neutrophil accumulation, thereby maintaining the chronicity of NETopathic inflammation in the airways of the asthma smoking phenotype (Figure 1A).
Concluding Remarks
In just a decade, the identification of NETs has reshaped the paradigm of neutrophil-mediated innate immunity. Disentangling the function of NET biology in human health and disease is ongoing. It is now recognized that NET formation is an effective antimicrobial defense system, yet its dysregulation may impart bystander consequences and airway NETopathic inflammation. Despite considerable research efforts, important questions remain unanswered in the context of treatment-refractory lung diseases. For instance, how the upstream cues consequent to airborne insults (e.g., respiratory viruses, cigarette smoke, fungal exposure) are integrated during induction of NET perturbance (Figure 1A) and whether they operate concurrently and/or redundantly in COPD or the smoking asthma phenotype. Furthermore, there is an apparent NET paradox of heightened infective susceptibility to respiratory pathogens in the face of concomitant NETopathic airway inflammation, and whether there is a preferential deployment of NET-forming subsets of neutrophils? Whether and how NET formation contributes to the pathogenesis of COPD and in the smoking asthma phenotype, or stabilizing this process may be an efficacious therapeutic strategy is unknown. It is important to consider that NETs are vital to host defense and that therapeutic intervention could cautiously be aimed at stabilizing the aggressive potential of NETs to homeostatic control rather than completely neutralizing this process as an ideal outcome. In any case, in our opinion, it seems reasonable that stabilizing airway NET burden might provide a rationale for disrupting this process in NET-rich COPD or in the smoking asthma phenotype. These studies await clinical validation in phenotypically-defined patient populations. With no curative therapy currently available for these fatal lung diseases, we envisage that disentangling the pathways underpinning NETopathic inflammation could lead to the development of innovative therapeutics for disease-specific phenotypes in respiratory medicine.
Author Contributions
All authors were involved in the development of the manuscript. MU designed the outline of this manuscript and co-wrote it with HW, AM, and FP. All authors have proofread the drafts and edited/approved the final manuscript.
Funding
The publication fees of this Frontiers article was provided by AstraZeneca.
Conflict of Interest Statement
MU and AM are employees of AstraZeneca and hold shares in the company. AstraZeneca provided the AZD5069 compound for the exploratory research in this manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Torsten Goldmann and Dr. Thomas Scholzen of core facility Fluorescence Cytometry, Research Center Borstel for assistance with 3D confocal fluorescence microscopy imaging/photography, and also grateful to Dr. Catherine Betts (AstraZeneca, Cambridge, U.K) for carefully proofreading the manuscript.
References
1. Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol. (2014) 15:602–11. doi: 10.1038/ni.2921
2. Leliefeld PH, Koenderman L, Pillay J. How neutrophils shape adaptive immune responses. Front Immunol. (2015) 6:471. doi: 10.3389/fimmu.2015.00471
3. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. (2006) 6:173–82. doi: 10.1038/nri1785
4. Barnes PJ. New molecular targets for the treatment of neutrophilic diseases. J Allergy Clin Immunol. (2007) 119:1055–62. doi: 10.1016/j.jaci.2007.01.015
5. Mardh CK, Root J, Uddin M, Stenvall K, Malmgren A, Karabelas K, et al. Targets of neutrophil influx and weaponry: therapeutic opportunities for chronic obstructive airway disease. J Immunol Res. (2017) 2017:5273201. doi: 10.1155/2017/5273201
6. Haslett C, Savill JS, Whyte MK, Stern M, Dransfield I, Meagher LC. Granulocyte apoptosis and the control of inflammation. Philos Trans R Soc Lond B Biol Sci. (1994) 345:327–33. doi: 10.1098/rstb.1994.0113
7. Uddin M, Nong G, Ward J, Seumois G, Prince LR, Wilson SJ, et al. Prosurvival activity for airway neutrophils in severe asthma. Thorax (2010) 65:684–9. doi: 10.1136/thx.2009.120741
8. Rytila P, Plataki M, Bucchieri F, Uddin M, Nong G, Kinnula VL, et al. Airway neutrophilia in COPD is not associated with increased neutrophil survival. Eur Respir J. (2006) 28:1163–9. doi: 10.1183/09031936.00149005
9. Brown V, Elborn JS, Bradley J, Ennis M. Dysregulated apoptosis and NFkappaB expression in COPD subjects. Respir Res. (2009) 10:24. doi: 10.1186/1465-9921-10-24
10. Simpson JL, Gibson PG, Yang IA, Upham J, James A, Reynolds PN, et al. Impaired macrophage phagocytosis in non-eosinophilic asthma. Clin Exp Allergy (2013) 43:29–35. doi: 10.1111/j.1365-2222.2012.04075.x
11. Hodge S, Hodge G, Ahern J, Jersmann H, Holmes M, Reynolds PN. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. (2007) 37:748–55. doi: 10.1165/rcmb.2007-0025OC
12. Takei H, Araki A, Watanabe H, Ichinose A, Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leukoc Biol. (1996) 59:229–40. doi: 10.1002/jlb.59.2.229
13. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
14. Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol. (2007) 5:577–82. doi: 10.1038/nrmicro1710
15. McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW, Rondina MT, et al. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 alpha. Blood (2012) 120:3118–25. doi: 10.1182/blood-2012-01-405993
16. Behnen M, Moller S, Brozek A, Klinger M, Laskay T. Extracellular acidification inhibits the ROS-dependent formation of neutrophil extracellular traps. Front Immunol. (2017) 8:184. doi: 10.3389/fimmu.2017.00184
17. Naffah de Souza C, Breda LCD, Khan MA, de Almeida SR, Camara NOS, Sweezey N, et al. Alkaline pH promotes NADPH oxidase-independent neutrophil extracellular trap formation: a matter of mitochondrial reactive oxygen species generation and citrullination and cleavage of histone. Front Immunol. (2017) 8:1849. doi: 10.3389/fimmu.2017.01849
18. Nadesalingam A, Chen JHK, Farahvash A, Khan MA. Hypertonic saline suppresses NADPH oxidase-dependent neutrophil extracellular trap formation and promotes apoptosis. Front Immunol. (2018) 9:359. doi: 10.3389/fimmu.2018.00359
19. Khan MA, Philip LM, Cheung G, Vadakepeedika S, Grasemann H, Sweezey N, et al. Regulating NETosis: increasing pH promotes NADPH oxidase-dependent NETosis. Front Med. (2018) 5:19. doi: 10.3389/fmed.2018.00019
20. Cheng OZ, Palaniyar N. NET balancing: a problem in inflammatory lung diseases. Front Immunol. (2013) 4:1. doi: 10.3389/fimmu.2013.00001
21. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. (2017) 23:279–87. doi: 10.1038/nm.4294
22. Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol. (2012) 189:2689–95. doi: 10.4049/jimmunol.1201719
23. Dwyer M, Shan Q, D'Ortona S, Maurer R, Mitchell R, Olesen H, et al. Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J Innate Immun. (2014) 6:765–79. doi: 10.1159/000363242
24. Porto BN, Stein RT. Neutrophil extracellular traps in pulmonary diseases: too much of a good thing? Front Immunol. (2016) 7:311. doi: 10.3389/fimmu.2016.00311
25. Lefrancais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight (2018) 3. doi: 10.1172/jci.insight.98178
26. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE (2012) 7:e32366. doi: 10.1371/journal.pone.0032366
27. Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol. (2011) 179:199–210. doi: 10.1016/j.ajpath.2011.03.013
28. Zou Y, Chen X, Xiao J, Bo Zhou D, Xiao Lu X, Li W, et al. Neutrophil extracellular traps promote lipopolysaccharide-induced airway inflammation and mucus hypersecretion in mice. Oncotarget (2018) 9:13276–86. doi: 10.18632/oncotarget.24022
29. Pedersen F, Marwitz S, Holz O, Kirsten A, Bahmer T, Waschki B, et al. Neutrophil extracellular trap formation and extracellular DNA in sputum of stable COPD patients. Respir Med. (2015) 109:1360–2. doi: 10.1016/j.rmed.2015.08.008
30. Pedersen F, Waschki B, Marwitz S, Goldmann T, Kirsten A, Malmgren A, et al. Neutrophil extracellular trap formation is regulated by CXCR2 in COPD neutrophils. Eur Respir J. (2018) 51. doi: 10.1183/13993003.00970-2017
31. Obermayer A, Stoiber W, Krautgartner WD, Klappacher M, Kofler B, Steinbacher P, et al. New aspects on the structure of neutrophil extracellular traps from chronic obstructive pulmonary disease and in vitro generation. PLoS ONE (2014) 9:e97784. doi: 10.1371/journal.pone.0097784
32. Grabcanovic-Musija F, Obermayer A, Stoiber W, Krautgartner WD, Steinbacher P, Winterberg N, et al. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir Res. (2015) 16:59. doi: 10.1186/s12931-015-0221-7
33. Pham DL, Ban GY, Kim SH, Shin YS, Ye YM, Chwae YJ, et al. Neutrophil autophagy and extracellular DNA traps contribute to airway inflammation in severe asthma. Clin Exp Allergy (2017) 47:57–70. doi: 10.1111/cea.12859
34. Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet (2017) 389:1931–40. doi: 10.1016/S0140-6736(17)31222-9
35. Hansel TT, Barnes PJ. New drugs for exacerbations of chronic obstructive pulmonary disease. Lancet (2009) 374:744–55. doi: 10.1016/S0140-6736(09)61342-8
36. MacNee W, Wiggs B, Belzberg AS, Hogg JC. The effect of cigarette smoking on neutrophil kinetics in human lungs. N Engl J Med. (1989) 321:924–8. doi: 10.1056/NEJM198910053211402
37. Quint JK, Wedzicha JA. The neutrophil in chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2007) 119:1065–71. doi: 10.1016/j.jaci.2006.12.640
38. Di Stefano A, Caramori G, Gnemmi I, Contoli M, Bristot L, Capelli A, et al. Association of increased CCL5 and CXCL7 chemokine expression with neutrophil activation in severe stable COPD. Thorax (2009) 64:968–75. doi: 10.1136/thx.2009.113647
39. Aaron SD, Angel JB, Lunau M, Wright K, Fex C, Le Saux N, et al. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2001) 163:349–55. doi: 10.1164/ajrccm.163.2.2003122
40. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. (2004) 350:2645–53. doi: 10.1056/NEJMoa032158
41. Stockley JA, Walton GM, Lord JM, Sapey E. Aberrant neutrophil functions in stable chronic obstructive pulmonary disease: the neutrophil as an immunotherapeutic target. Int Immunopharmacol. (2013) 17:1211–7. doi: 10.1016/j.intimp.2013.05.035
42. Wright TK, Gibson PG, Simpson JL, McDonald VM, Wood LG, Baines KJ. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirology (2016) 21:467–75. doi: 10.1111/resp.12730
43. Lugli EB, Correia RE, Fischer R, Lundberg K, Bracke KR, Montgomery AB, et al. Expression of citrulline and homocitrulline residues in the lungs of non-smokers and smokers: implications for autoimmunity in rheumatoid arthritis. Arthritis Res Ther. (2015) 17:9. doi: 10.1186/s13075-015-0520-x
44. Leshner M, Wang S, Lewis C, Zheng H, Chen XA, Santy L, et al. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol. (2012) 3:307. doi: 10.3389/fimmu.2012.00307
45. Dicker AJ, Crichton ML, Pumphrey EG, Cassidy AJ, Suarez-Cuartin G, Sibila O, et al. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2018) 141:117–27. doi: 10.1016/j.jaci.2017.04.022
46. Baggiolini M. CXCL8 - the first chemokine. Front Immunol. (2015) 6:285. doi: 10.3389/fimmu.2015.00285
47. Stadtmann A, Zarbock A. CXCR2: from bench to bedside. Front Immunol. (2012) 3:263. doi: 10.3389/fimmu.2012.00263
48. Beeh KM, Kornmann O, Buhl R, Culpitt SV, Giembycz MA, Barnes PJ. Neutrophil chemotactic activity of sputum from patients with COPD: role of interleukin 8 and leukotriene B4. Chest (2003) 123:1240–7. doi: 10.1378/chest.123.4.1240
49. Qiu Y, Zhu J, Bandi V, Atmar RL, Hattotuwa K, Guntupalli KK, et al. Biopsy neutrophilia, neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2003) 168:968–75. doi: 10.1164/rccm.200208-794OC
50. Chung KF. Inflammatory mediators in chronic obstructive pulmonary disease. Curr Drug Targets Inflamm Allergy (2005) 4:619–25. doi: 10.2174/156801005774912806
51. Rennard SI, Dale DC, Donohue JF, Kanniess F, Magnussen H, Sutherland ER, et al. CXCR2 Antagonist MK-7123. A phase 2 proof-of-concept trial for chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2015) 191:1001–11. doi: 10.1164/rccm.201405-0992OC
52. Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, et al. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. (2006) 12:317–23. doi: 10.1038/nm1361
53. O'Reilly P, Jackson PL, Noerager B, Parker S, Dransfield M, Gaggar A, et al. N-alpha-PGP and PGP, potential biomarkers and therapeutic targets for COPD. Respir Res. (2009) 10:38. doi: 10.1186/1465-9921-10-38
54. Snelgrove RJ, Jackson PL, Hardison MT, Noerager BD, Kinloch A, Gaggar A, et al. A critical role for LTA4H in limiting chronic pulmonary neutrophilic inflammation. Science (2010) 330:90–4. doi: 10.1126/science.1190594
55. Sabbione F, Keitelman IA, Iula L, Ferrero M, Giordano MN, Baldi P, et al. Neutrophil extracellular traps stimulate proinflammatory responses in human airway epithelial cells. J Innate Immun. (2017) 9:387–402. doi: 10.1159/000460293
56. Arumugam S, Girish Subbiah K, Kemparaju K, Thirunavukkarasu C. Neutrophil extracellular traps in acrolein promoted hepatic ischemia reperfusion injury: therapeutic potential of NOX2 and p38MAPK inhibitors. J Cell Physiol. (2018) 233:3244–61. doi: 10.1002/jcp.26167
57. Hardison MT, Brown MD, Snelgrove RJ, Blalock JE, Jackson P. Cigarette smoke enhances chemotaxis via acetylation of proline-glycine-proline. Front Biosci. (2012) 4:2402–9. doi: 10.2741/E552
58. Wells JM, Jackson PL, Viera L, Bhatt SP, Gautney J, Handley G, et al. A randomized, placebo-controlled trial of roflumilast. effect on proline-glycine-proline and neutrophilic inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2015) 192:934–42. doi: 10.1164/rccm.201503-0543OC
59. Wilkinson TMA, Aris E, Bourne S, Clarke SC, Peeters M, Pascal TG, et al. A prospective, observational cohort study of the seasonal dynamics of airway pathogens in the aetiology of exacerbations in COPD. Thorax (2017) 72:919–27. doi: 10.1136/thoraxjnl-2016-209023
60. Cortjens B, de Boer OJ, de Jong R, Antonis AF, Sabogal Pineros YS, Lutter R, et al. Neutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease. J Pathol. (2016) 238:401–11. doi: 10.1002/path.4660
61. Schonrich G, Raftery MJ. Neutrophil extracellular traps go viral. Front Immunol. (2016) 7:366. doi: 10.3389/fimmu.2016.00366
62. Delgado-Rizo V, Martinez-Guzman MA, Iniguez-Gutierrez L, Garcia-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. (2017) 8:81. doi: 10.3389/fimmu.2017.00081
63. Galani IE, Andreakos E. Neutrophils in viral infections: current concepts and caveats. J Leukoc Biol. (2015) 98:557–64. doi: 10.1189/jlb.4VMR1114-555R
64. Camp JV, Jonsson CB. A role for neutrophils in viral respiratory disease. Front Immunol. (2017) 8:550. doi: 10.3389/fimmu.2017.00550
65. Souza PSS, Barbosa LV, Diniz LFA, da Silva GS, Lopes BRP, Souza PMR, et al. Neutrophil extracellular traps possess anti-human respiratory syncytial virus activity: possible interaction with the viral F protein. Virus Res. (2018) 251:68–77. doi: 10.1016/j.virusres.2018.04.001
66. Akk A, Springer LE, Pham CT. Neutrophil extracellular traps enhance early inflammatory response in sendai virus-induced asthma phenotype. Front Immunol. (2016) 7:325. doi: 10.3389/fimmu.2016.00325
67. Williams NP, Coombs NA, Johnson MJ, Josephs LK, Rigge LA, Staples KJ, et al. Seasonality, risk factors and burden of community-acquired pneumonia in COPD patients: a population database study using linked health care records. Int J Chron Obstruct Pulmon Dis. (2017) 12:313–22. doi: 10.2147/COPD.S121389
68. Hirschmann JV. Do bacteria cause exacerbations of COPD? Chest (2000) 118:193–203. doi: 10.1378/chest.118.1.193
69. Clancy RL, Dunkley M. Acute exacerbations in COPD and their control with oral immunization with non-typeable haemophilus influenzae. Front Immunol. (2011) 2:7. doi: 10.3389/fimmu.2011.00007
70. Sethi S, Evans N, Grant BJ, Murphy TF. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med. (2002) 347:465–71. doi: 10.1056/NEJMoa012561
71. Linge HM, Andersson C, Nordin SL, Olin AI, Petersson AC, Morgelin M, et al. Midkine is expressed and differentially processed during chronic obstructive pulmonary disease exacerbations and ventilator-associated pneumonia associated with Staphylococcus aureus infection. Mol Med. (2013) 19:314–23. doi: 10.2119/molmed.2013.00045
72. Albertson TE, Louie S, Chan AL. The diagnosis and treatment of elderly patients with acute exacerbation of chronic obstructive pulmonary disease and chronic bronchitis. J Am Geriatr Soc. (2010) 58:570–9. doi: 10.1111/j.1532-5415.2010.02741.x
73. Hoppenbrouwers T, Sultan AR, Abraham TE, Lemmens-den Toom NA, Hansenova Manaskova S, van Cappellen WA, et al. Staphylococcal protein A is a key factor in neutrophil extracellular traps formation. Front Immunol. (2018) 9:165. doi: 10.3389/fimmu.2018.00165
74. Ebrahimi F, Giaglis S, Hahn S, Blum CA, Baumgartner C, Kutz A, et al. Markers of neutrophil extracellular traps predict adverse outcome in community-acquired pneumonia: secondary analysis of a randomised controlled trial. Eur Respir J. (2018) 51. doi: 10.1183/13993003.01389-2017
75. Storisteanu DM, Pocock JM, Cowburn AS, Juss JK, Nadesalingam A, Nizet V, et al. Evasion of neutrophil extracellular traps by respiratory pathogens. Am J Respir Cell Mol Biol. (2017) 56:423–31. doi: 10.1165/rcmb.2016-0193PS
76. Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asthma in adults. BMJ (1993) 307:982–6. doi: 10.1136/bmj.307.6910.982
77. Mallia P, Johnston SL. How viral infections cause exacerbation of airway diseases. Chest (2006) 130:1203–10. doi: 10.1378/chest.130.4.1203
78. Jarjour NN, Gern JE, Kelly EA, Swenson CA, Dick CR, Busse WW. The effect of an experimental rhinovirus 16 infection on bronchial lavage neutrophils. J Allergy Clin Immunol. (2000) 105(6 Pt 1):1169–77. doi: 10.1067/mai.2000.106376
79. Rohde G, Message SD, Haas JJ, Kebadze T, Parker H, Laza-Stanca V, et al. CXC chemokines and antimicrobial peptides in rhinovirus-induced experimental asthma exacerbations. Clin Exp Allergy (2014) 44:930–9. doi: 10.1111/cea.12313
80. Grunberg K, Timmers MC, Smits HH, de Klerk EP, Dick EC, Spaan WJ, et al. Effect of experimental rhinovirus 16 colds on airway hyperresponsiveness to histamine and interleukin-8 in nasal lavage in asthmatic subjects in vivo. Clin Exp Allergy (1997) 27:36–45. doi: 10.1111/j.1365-2222.1997.tb00670.x
81. Nagarkar DR, Wang Q, Shim J, Zhao Y, Tsai WC, Lukacs NW, et al. CXCR2 is required for neutrophilic airway inflammation and hyperresponsiveness in a mouse model of human rhinovirus infection. J Immunol. (2009) 183:6698–707. doi: 10.4049/jimmunol.0900298
82. Toussaint M, Jackson DJ, Swieboda D, Guedan A, Tsourouktsoglou TD, Ching YM, et al. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat Med. (2017) 23:681–91. doi: 10.1038/nm.4332
83. Busse WW. A role for neutrophils in asthma exacerbations. Nat Med. (2017) 23:658–9. doi: 10.1038/nm.4351
84. da Cunha AA, Nunez NK, de Souza RG, Moraes Vargas MH, Silveira JS, Antunes GL, et al. Recombinant human deoxyribonuclease therapy improves airway resistance and reduces DNA extracellular traps in a murine acute asthma model. Exp Lung Res. (2016) 42:66–74. doi: 10.3109/01902148.2016.1143537
85. Denning DW, O'Driscoll BR, Hogaboam CM, Bowyer P, Niven RM. The link between fungi and severe asthma: a summary of the evidence. Eur Respir J. (2006) 27:615–26. doi: 10.1183/09031936.06.00074705
86. Fairs A, Agbetile J, Hargadon B, Bourne M, Monteiro WR, Brightling CE, et al. IgE sensitization to Aspergillus fumigatus is associated with reduced lung function in asthma. Am J Respir Crit Care Med. (2010) 182:1362–8. doi: 10.1164/rccm.201001-0087OC
87. McCormick A, Heesemann L, Wagener J, Marcos V, Hartl D, Loeffler J, et al. NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect. (2010) 12(12–13):928–36. doi: 10.1016/j.micinf.2010.06.009
88. Rohm M, Grimm MJ, D'Auria AC, Almyroudis NG, Segal BH, Urban CF. NADPH oxidase promotes neutrophil extracellular trap formation in pulmonary aspergillosis. Infect Immun. (2014) 82:1766–77. doi: 10.1128/IAI.00096-14
89. Haldar P, Pavord ID. Noneosinophilic asthma: a distinct clinical and pathologic phenotype. J Allergy Clin Immunol. (2007) 119:1043–52; quiz 53–4. doi: 10.1016/j.jaci.2007.02.042
90. Svenningsen S, Nair P. Asthma endotypes and an overview of targeted therapy for asthma. Front Med. (2017) 4:158. doi: 10.3389/fmed.2017.00158
91. Samitas K, Zervas E, Gaga M. T2-low asthma: current approach to diagnosis and therapy. Curr Opin Pulm Med. (2017) 23:48–55. doi: 10.1097/MCP.0000000000000342
92. Liu W, Liu S, Verma M, Zafar I, Good JT, Rollins D, et al. Mechanism of TH2/TH17-predominant and neutrophilic TH2/TH17-low subtypes of asthma. J Allergy Clin Immunol. (2017) 139:1548–58e4. doi: 10.1016/j.jaci.2016.08.032
93. Kuo CS, Pavlidis S, Loza M, Baribaud F, Rowe A, Pandis I, et al. T-helper cell type 2 (Th2) and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in U-BIOPRED. Eur Respir J. (2017) 49. doi: 10.1183/13993003.02135-2016
94. The ENFUMOSA cross-sectional European multicentre study of the clinical phenotype of chronic severe asthma. European network for understanding mechanisms of severe asthma. Eur Respir J. (2003) 22:470–7. doi: 10.1183/09031936.03.00261903
95. Fahy JV, Kim KW, Liu J, Boushey HA. Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J Allergy Clin Immunol. (1995) 95:843–52. doi: 10.1016/S0091-6749(95)70128-1
96. Wenzel SE, Szefler SJ, Leung DY, Sloan SI, Rex MD, Martin RJ. Bronchoscopic evaluation of severe asthma. Persistent inflammation associated with high dose glucocorticoids. Am J Respir Crit Care Med. (1997) 156(3 Pt 1):737–43. doi: 10.1164/ajrccm.156.3.9610046
97. Shaw DE, Berry MA, Hargadon B, McKenna S, Shelley MJ, Green RH, et al. Association between neutrophilic airway inflammation and airflow limitation in adults with asthma. Chest (2007) 132:1871–5. doi: 10.1378/chest.07-1047
98. Little SA, MacLeod KJ, Chalmers GW, Love JG, McSharry C, Thomson NC. Association of forced expiratory volume with disease duration and sputum neutrophils in chronic asthma. Am J Med. (2002) 112:446–52. doi: 10.1016/S0002-9343(02)01047-1
99. Moore WC, Hastie AT, Li X, Li H, Busse WW, Jarjour NN, et al. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J Allergy Clin Immunol. (2014) 133:1557–63.e5. doi: 10.1016/j.jaci.2013.10.011
100. Pavord ID, Brightling CE, Woltmann G, Wardlaw AJ. Non-eosinophilic corticosteroid unresponsive asthma. Lancet (1999) 353:2213–4. doi: 10.1016/S0140-6736(99)01813-9
101. Fricker M, Heaney LG, Upham JW. Can biomarkers help us hit targets in difficult-to-treat asthma? Respirology (2017) 22:430–42. doi: 10.1111/resp.13014
102. O'Byrne PM, Metev H, Puu M, Richter K, Keen C, Uddin M, et al. Efficacy and safety of a CXCR2 antagonist, AZD5069, in patients with uncontrolled persistent asthma: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. (2016) 4:797–806. doi: 10.1016/S2213-2600(16)30227-2
103. Chung KF. Neutrophilic asthma: a distinct target for treatment? Lancet Respir Med. (2016) 4:765–7. doi: 10.1016/S2213-2600(16)30232-6
104. Snelgrove RJ, Patel DF, Patel T, Lloyd CM. The enigmatic role of the neutrophil in asthma: friend, foe or indifferent? Clin Exp Allergy (2018) 48:1275–85. doi: 10.1111/cea.13191
105. Barnes PJ. Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. (2018) 18:454–66. doi: 10.1038/s41577-018-0006-6
106. Dworski R, Simon HU, Hoskins A, Yousefi S. Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J Allergy Clin Immunol. (2011) 127:1260–6. doi: 10.1016/j.jaci.2010.12.1103
107. Choi Y, Pham LD, Lee DH, Ban GY, Lee JH, Kim SH, et al. Neutrophil extracellular DNA traps induce autoantigen production by airway epithelial cells. Mediators Inflamm. (2017) 2017:5675029. doi: 10.1155/2017/5675029
108. Uddin M, Lau LC, Seumois G, Vijayanand P, Staples KJ, Bagmane D, et al. EGF-induced bronchial epithelial cells drive neutrophil chemotactic and anti-apoptotic activity in asthma. PLoS ONE (2013) 8:e72502. doi: 10.1371/journal.pone.0072502
109. Krishnamoorthy N, Douda DN, Bruggemann TR, Ricklefs I, Duvall MG, Abdulnour RE, et al. Neutrophil cytoplasts induce TH17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci Immunol. (2018) 3. doi: 10.1126/sciimmunol.aao4747
110. Wills-Karp M. Neutrophil ghosts worsen asthma. Sci Immunol. (2018) 3. doi: 10.1126/sciimmunol.aau0112
111. Natorska J, Zabczyk M, Siudut J, Krawiec P, Mastalerz L, Undas A. Neutrophil extracellular traps formation in patients with eosinophilic granulomatosis with polyangiitis: association with eosinophilic inflammation. Clin Exp Rheumatol. (2017) 35(Suppl. 103):27–32.
112. Shida H, Nakazawa D, Tateyama Y, Miyoshi A, Kusunoki Y, Hattanda F, et al. The presence of anti-lactoferrin antibodies in a subgroup of eosinophilic granulomatosis with polyangiitis patients and their possible contribution to enhancement of neutrophil extracellular trap formation. Front Immunol. (2016) 7:636. doi: 10.3389/fimmu.2016.00636
113. Ordonez CL, Shaughnessy TE, Matthay MA, Fahy JV. Increased neutrophil numbers and IL-8 levels in airway secretions in acute severe asthma: clinical and biologic significance. Am J Respir Crit Care Med. (2000) 161(4 Pt 1):1185–90. doi: 10.1164/ajrccm.161.4.9812061
114. Shannon J, Ernst P, Yamauchi Y, Olivenstein R, Lemiere C, Foley S, et al. Differences in airway cytokine profile in severe asthma compared to moderate asthma. Chest (2008) 133:420–6. doi: 10.1378/chest.07-1881
115. Keglowich L, Roth M, Philippova M, Resink T, Tjin G, Oliver B, et al. Bronchial smooth muscle cells of asthmatics promote angiogenesis through elevated secretion of CXC-chemokines (ENA-78, GRO-alpha, and IL-8). PLoS ONE (2013) 8:e81494. doi: 10.1371/journal.pone.0081494
116. Kikuchi S, Kikuchi I, Takaku Y, Kobayashi T, Hagiwara K, Kanazawa M, et al. Neutrophilic inflammation and CXC chemokines in patients with refractory asthma. Int Arch Allergy Immunol. (2009) 149(Suppl. 1):87–93. doi: 10.1159/000211379
117. Hastie AT, Steele C, Dunaway CW, Moore WC, Rector BM, Ampleford E, et al. Complex association patterns for inflammatory mediators in induced sputum from subjects with asthma. Clin Exp Allergy (2018) 48:787–97. doi: 10.1111/cea.13129
118. Nicholls DJ, Wiley K, Dainty I, MacIntosh F, Phillips C, Gaw A, et al. Pharmacological characterization of AZD5069, a slowly reversible CXC chemokine receptor 2 antagonist. J Pharmacol Exp Ther. (2015) 353:340–50. doi: 10.1124/jpet.114.221358
119. Jurcevic S, Humfrey C, Uddin M, Warrington S, Larsson B, Keen C. The effect of a selective CXCR2 antagonist (AZD5069) on human blood neutrophil count and innate immune functions. Br J Clin Pharmacol. (2015) 80:1324–36. doi: 10.1111/bcp.12724
120. Uddin M, Betts C, Robinson I, Malmgren A, Humfrey C. The chemokine CXCR2 antagonist (AZD5069) preserves neutrophil-mediated host immunity in non-human primates. Haematologica (2017) 102:e65–8. doi: 10.3324/haematol.2016.152371
121. De Soyza A, Pavord I, Elborn JS, Smith D, Wray H, Puu M, et al. A randomised, placebo-controlled study of the CXCR2 antagonist AZD5069 in bronchiectasis. Eur Respir J. (2015) 46:1021–32. doi: 10.1183/13993003.00148-2015
122. Watz H, Uddin M, Pedersen F, Kirsten A, Goldmann T, Stellmacher F, et al. Effects of the CXCR2 antagonist AZD5069 on lung neutrophil recruitment in asthma. Pulm Pharmacol Ther. (2017) 45:121–3. doi: 10.1016/j.pupt.2017.05.012
123. Nair P, Gaga M, Zervas E, Alagha K, Hargreave FE, O'Byrne PM, et al. Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: a randomized, placebo-controlled clinical trial. Clin Exp Allergy (2012) 42:1097–103. doi: 10.1111/j.1365-2222.2012.04014.x
124. Thomson NC, Chaudhuri R, Livingston E. Asthma and cigarette smoking. Eur Respir J. (2004) 24:822–33. doi: 10.1183/09031936.04.00039004
125. Thomson NC, Chaudhuri R, Heaney LG, Bucknall C, Niven RM, Brightling CE, et al. Clinical outcomes and inflammatory biomarkers in current smokers and exsmokers with severe asthma. J Allergy Clin Immunol. (2013) 131:1008–16. doi: 10.1016/j.jaci.2012.12.1574
126. Thomson NC. Novel approaches to the management of noneosinophilic asthma. Ther Adv Respir Dis. (2016) 10:211–34. doi: 10.1177/1753465816632638
127. Lefaudeux D, De Meulder B, Loza MJ, Peffer N, Rowe A, Baribaud F, et al. U-BIOPRED clinical adult asthma clusters linked to a subset of sputum omics. J Allergy Clin Immunol. (2017) 139:1797–807. doi: 10.1016/j.jaci.2016.08.048
128. St-Laurent J, Bergeron C, Page N, Couture C, Laviolette M, Boulet LP. Influence of smoking on airway inflammation and remodelling in asthma. Clin Exp Allergy (2008) 38:1582–9. doi: 10.1111/j.1365-2222.2008.03032.x
129. Fukakusa M, Bergeron C, Tulic MK, Fiset PO, Al Dewachi O, Laviolette M, et al. Oral corticosteroids decrease eosinophil and CC chemokine expression but increase neutrophil, IL-8, and IFN-gamma-inducible protein 10 expression in asthmatic airway mucosa. J Allergy Clin Immunol. (2005) 115:280–6. doi: 10.1016/j.jaci.2004.10.036
Keywords: airway, asthma, COPD, CXCR2, neutrophil extracellular traps (NETs), NETopathic inflammation
Citation: Uddin M, Watz H, Malmgren A and Pedersen F (2019) NETopathic Inflammation in Chronic Obstructive Pulmonary Disease and Severe Asthma. Front. Immunol. 10:47. doi: 10.3389/fimmu.2019.00047
Received: 11 November 2018; Accepted: 09 January 2019;
Published: 05 February 2019.
Edited by:
Cees Van Kooten, Leiden University, NetherlandsReviewed by:
Yi Zhao, Sichuan University, ChinaRostyslav Bilyy, Danylo Halytsky Lviv National Medical University, Ukraine
Copyright © 2019 Uddin, Watz, Malmgren and Pedersen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohib Uddin, mohib.uddin@astrazeneca.com