 Jahnavi Aluri1
Jahnavi Aluri1 Mukesh Desai2
Mukesh Desai2 Maya Gupta1Aparna Dalvi1Antony Terance3
Maya Gupta1Aparna Dalvi1Antony Terance3 Sergio D. Rosenzweig4
Sergio D. Rosenzweig4 Jennifer L. Stoddard4
Jennifer L. Stoddard4 Julie E. Niemela4Vasundhara Tamankar5
Julie E. Niemela4Vasundhara Tamankar5 Snehal Mhatre1Umair Bargir1
Snehal Mhatre1Umair Bargir1 Manasi Kulkarni1Nitin Shah6
Manasi Kulkarni1Nitin Shah6 Amita Aggarwal7Harsha Prasada Lashkari8Vidya Krishna9
Amita Aggarwal7Harsha Prasada Lashkari8Vidya Krishna9 Geeta Govindaraj10Manas Kalra11
Geeta Govindaraj10Manas Kalra11 Manisha Madkaikar1*
Manisha Madkaikar1*- 1Department of Pediatric Immunology and Leukocyte Biology, National Institute of Immunohaematology (ICMR), Mumbai, India
- 2Division of Immunology, Bai Jerbai Wadia Children's Hospital, Mumbai, India
- 3Department of Pediatric Pulmonology, G. Kuppuswamy Naidu Memorial Hospital, Coimbatore, India
- 4Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, United States
- 5Centre for Medical Genetics, Mumbai, India
- 6Pediatric Hematology-Oncology, P. D. Hinduja National Hospital & Research Center, Mumbai, India
- 7Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, India
- 8Department of Pediatrics, Kasturba Medical College, Mangalore, India
- 9Department of Pediatrics, Sri Ramachandra Medical College and Research Institute, Chennai, India
- 10Department of Pediatrics, Institute of Maternal and Child Health, Government Medical College, Kozhikode, India
- 11Department of Pediatrics Hematology and Oncology, Indraprastha Apollo Hospital, New Delhi, India
Severe combined immunodeficiency (SCID) represents one of the most severe forms of primary immunodeficiency (PID) disorders characterized by impaired cellular and humoral immune responses. Here, we report the clinical, immunological, and molecular findings in 57 patients diagnosed with SCID from India. Majority of our patients (89%) presented within 6 months of age. The most common clinical manifestations observed were recurrent pneumonia (66%), failure to thrive (60%), chronic diarrhea (35%), gastrointestinal infection (21%), and oral candidiasis (21%). Hematopoietic Stem Cell Transplantation (HSCT) is the only curative therapy available for treating these patients. Four patients underwent HSCT in our cohort but had a poor survival outcome. Lymphopenia (absolute lymphocyte counts/μL <2,500) was noted in 63% of the patients. Based on immunophenotypic pattern, majority of the cases were T−B− SCID (39%) followed by T−B+ SCID (28%). MHC class II deficiency accounted for 10.5% of our patient group. A total of 49 patients were molecularly characterized in this study and 32 novel variants were identified in our cohort. The spectrum of genetic defects in our cohort revealed a wide genetic heterogeneity with the major genetic cause being RAG1/2 gene defect (n = 12) followed by IL2RG (n = 9) and JAK3 defects (n = 9). Rare forms of SCID like Purine nucleoside phosphorylase (PNP) deficiency, reticular dysgenesis, DNA-Protein Kinase (DNA-PKcs) deficiency, six cases of MHC class II deficiency and two ZAP70 deficiency were also identified in our cohort. Fourteen percent of the defects still remained uncharacterized despite the application of next generation sequencing. With the exception of MHC class II deficiency and ZAP70 deficiency, all SCID patients had extremely low T cell receptor excision (TRECs) (<18 copies/μL).
Introduction
Severe combined immunodeficiency (SCID) refers to a heterogeneous group of primary immunodeficiency disorders characterized by impaired T lymphocyte development with an effect on the B cell and NK cell number and/or function. SCID pathogenesis involves multiple genes whose defect which leads to abnormal cellular and humoral immune responses. Affected children suffer from recurrent infections, notably infections with opportunistic organisms such as Pneumocystis jiroveci, chronic diarrhea, failure to thrive and persistent mucocutaneous candidiasis (1). SCID is not apparent at birth and the presence of maternally derived antibodies provide some protection in the initial few months which further delays the diagnosis. As a consequence, these children also get the routine administration of live vaccines which is known to be contraindicated in SCID patients. Diagnosis of SCID is supported by a low absolute lymphocyte counts, abnormalities in lymphocyte subpopulations, absent/reduced naïve T cell population and recent thymic emigrants, absent T cell receptor excision circles (TRECs) and a low or absent T cell response to mitogens.
The Primary Immune Deficiency Treatment Consortium (PIDTC) classifies patients into Typical SCID with total T cell (CD3) count of <300 cells/microL (2). Depending on the B and NK cell status, patients are further classified as T−B+NK−, T−B+NK−, T−B−NK+, T−B−NK−. Some patients can have T cells (leaky/atypical SCID) which are mostly oligoclonal and are classified as T+ or T++ SCID. These modify the counts from T−B−NK− to T+B−NK−,T−B+NK− to T+B+NK−, from T−B−NK+ to T+B−NK+, or from T−B+NK+ to T+B+NK+ (3).
More than 30 genes are involved in SCID pathogenesis (3). Despite a wide genetic heterogeneity, patients are clinically indistinguishable. Importantly, the clinical features in SCID infants can also be found in patients with human immunodeficiency virus (HIV) infection/acquired immunodeficiency syndrome (AIDS). Hence, it is essential to rule out such secondary causes of lymphopenia by determining the presence of maternal HIV antibodies and measuring the levels of HIV by polymerase chain reaction (PCR).
With the exception of common gamma chain cytokine receptor (IL2RG) deficiency which follows X-linked pattern of inheritance, all the other causes are inherited in an autosomal recessive pattern. There are many SCID cases where the genetic defect is still unknown. In countries with a low rate of consanguinity, approximately 50% of all SCID cases are X-linked (4). Of the AR forms of SCID, 20–30% of all SCID patients are T−, B−, NK+, and approximately half of these patients have mutations in the RAG1 or RAG2 genes.
The incidence of SCID was previously reported at approximately 1 in 100,000 but the implementation of TREC assay for Newborn screening of SCID revealed the true incidence of SCID to be 1 in 58,000 live births (95% CI, 1 in 46,000–1 in 80,000) for typical SCID, leaky/atypical SCID, and Omenn syndrome (5). SCID is a fatal disorder and without treatment, death from infection usually occurs within the first 2 years of life. Diagnosis must be made before severe life-threatening infections occur so that the immunity can be restored with enzyme replacement or Hematopoietic Stem Cell Transplantation (HSCT); early transplantation (before 3.5 months of age) can lead to long-term survival (6). Gene therapy is an alternative option available especially for patients with ADA-SCID and X-SCID.
Here, we report the first largest series on the clinical, immunological, and molecular findings in SCID patients (n = 57) from India.
Materials and Methods
Patients and Samples
Patients (n = 57) suspected of Severe combined immunodeficiency (SCID) at National Institute of Immunohaematology (NIIH) between 2013 and 2018 were included in the study. Informed consent for participating in the study was procured from the family members in accordance with the declaration of Helsinki and 3 mL peripheral blood was collected in EDTA, Plain and Heparin vacutainers each. The study was approved by the Institutional Ethics Committee of NIIH.
A clinical proforma was filled for all patients which included the age, consanguinity, family history, clinical parameters like number of infections, site of infections, age of presentation, failure to thrive, diarrhea, presence of any skin rashes, administration of vaccines and post live vaccine complications, presence of dysmorphic features, hepatosplenomegaly, lymphadenopathy.
Prenatal diagnosis (PND) was provided to a total of four affected families. Two families were provided a molecular confirmation of the genetic defect on the chorionic villus sample. Maternal contamination was ruled out by Kleihauer-Betke (KB) staining and analysis of the variable number of tandem repeats (VNTR) using the apolipoprotein B (ApoB), ACTB2, D1S80, and IgJH genes. Phenotypic prenatal diagnosis was provided to 2 families on the Fetal cord blood (FB) sample (1–2 mL, <0.5% of expected weight in all cases) as molecular diagnosis was not available at the time of PND. The FB sample was collected at 18 weeks of gestation by ultrasound-guided cordocentesis after procuring informed consent from the parents. The FB sample accepted for analysis had a high MCV value (>110 fL) with narrow and single red cell distribution curve. The testing was performed within 3 h of sampling.
Immunological Workup
Initial investigations involved a complete blood cell count (CBC) on a Sysmex XS-800i (Sysmex Co., Cobe, Japan) 5-part automated hematological analyzer, lymphocyte subset analysis by flow cytometry using BD Multitest 6-color TBNK reagent followed by acquisition of cells on FACS Aria I; analysis was performed on FACS Diva and FlowJo software (BD Biosciences, San Jose, CA, USA). Serum immunoglobulin levels were estimated by nephelometry (BNProspec, Siemens).
The percentage of naïve and memory T cell subsets on CD4+ and CD8+ cells was measured by flow cytometry using anti-CD45RA phycoerythrin (PE), anti-CD45RO Phycoerythrin/Cy7 (PE-Cy7) and anti-CD62L allophycocyanin (APC) procured from BD Biosciences, San Jose, CA, USA.
T cell receptor excision circles (TRECs) were measured by an in-house modification of a previously described method (7).
Flow cytometric evaluation of Human Leukocyte antigen- D related (HLA-DR) expression on lymphocytes and monocytes using cell surface markers specific for T cells (anti-CD3 Peridinin-chlorophyll-protein Complex: CY5.5 Conjugate, PerCP-Cy5), B cells (anti-CD19 allophycocyanin [APC]), monocytes (anti-CD14 Phycoerythrin [PE]) and HLA-DR (anti-HLA-DR fluorescein isothiocyanate [FITC]) was performed.
T cell proliferation assay was performed using CellTrace Violet dye (Thermo Fisher Scientific). The PBMCs separated from the heparinized blood samples of the patients was suspended in complete RPMI medium (GIBCO, USA) containing 10% fetal calf serum (GIBCO, USA) and was stained at a density of 106 cells with Cell trace violet (1 μM) for 20 min at 37°C. The cells were aliquoted into 96 well tissue culture plates at a density of 105 cells per well and stimulated with phytohemagglutinin (PHA) (1 μg/ml) and cultured for 72 h. Flow cytometric analysis of T cell functionality was assessed on CD3+ and CD69+ (activation marker) T cells.
Clonality of the T cell receptor (TCR) was assessed by flow cytometric evaluation of TCR-Vβ repertoire by using the IOTest® Beta Mark.
Flow cytometric evaluation of CD132 expression on B cells was done using cell surface markers specific for B cells (anti-CD19 allophycocyanin [APC]) and CD132 (Phycoerythrin [PE]) procured from Biolegend. Flow cytometric evaluation of CD127 on T cells was done using cell surface markers specific for T cells (anti-CD3 fluorescein isothiocyanate [FITC]) and anti-CD127 allophycocyanin [APC]) procured from BD biosciences.
Phospho-STAT5 analysis was performed on whole blood after IL-2 (10 μg/ml) (Peprotech, NJ, USA) stimulation for 15 min at 37°C. The cells were fixed with BD Lyse Fix and permeabilized with Perm III Buffer. The cells were stained with anti-phospho-STAT5 (p-STAT5) Alexa 488 (Y694, clone 47, BD Biosciences) according to the manufacturer's instructions.
Determination of Adenosine deaminase (ADA) activity on RBC lysate was performed by the Giusti and Galant calorimetry method using a commercially available kit (ADA-MTB kit) from Tulip Diagnostics, India.
Data was presented in terms of median and percentages. One-way analysis of variance (ANOVA) test was used for comparison of >2 groups. Mann-Whitney U-test was used for comparing groups with non-parametric data. The p-values less than 0.05 were considered statistically significant. All statistical calculations were done using GraphPad prism (Chicago, IL, USA) version 15 for Microsoft Windows.
Molecular Investigations
Molecular investigations were done by Sanger sequencing of IL2RG, ADA, RAG1, RAG2, IL7RA, ZAP70 genes using the standard protocol. Targeted Next Generation sequencing was performed using a custom capture kit by Medgenome Labs Pvt Ltd. India, in samples where molecular diagnosis was not identified by Sanger Sequencing. The libraries were sequenced on Illumina sequencing platform (mean coverage >80 to 100X). The identified mutations were confirmed by Sanger sequencing.
Results
Patient Characteristics and Clinical Findings
In this study, a total of 57 SCID patients were diagnosed and followed up. Forty patients (70%) were male. Data on the status of consanguinity was available for 53 cases, from which 19 patients (36%) belonged to consanguineous parents. A positive family history of SCID was recorded in 32 families (56%). The median age at onset and diagnosis of all patients referred with a clinical suspicion of SCID was 60 days (range, 12–304) and 152 days (range, 12–730), respectively. Two patients received a pre-symptomatic diagnosis in view of strong family history.
The most common clinical manifestations were pneumonia (66%), failure to thrive (60%), chronic diarrhea (35%), gastrointestinal infection (21%), oral candidiasis (21%) and BCGiosis (12%). In our cohort, organisms isolated included both gram- negative and gram-positive bacterium: Staphylococcus aureus (n = 2), Klebsiella pneumonia (n = 8), Pseudomonas aeruginosa (n = 4), Burkholderia (n = 1), Chryseobacterium (n = 1). Viral organisms isolated included rotavirus (n = 1), cytomegalovirus (CMV) (n = 2), Rubella (n = 1), RSV (n = 1), Varicella (n = 1). Fungal infections included PCP (n = 1). Other features such as Erythematous skin rash was observed in 29% of the cases, Dysmorphism was seen in 8% cases, abscess in 8% and Hepatosplenomegaly in 3% cases. Table 1 presents the clinical findings in our patient cohort.
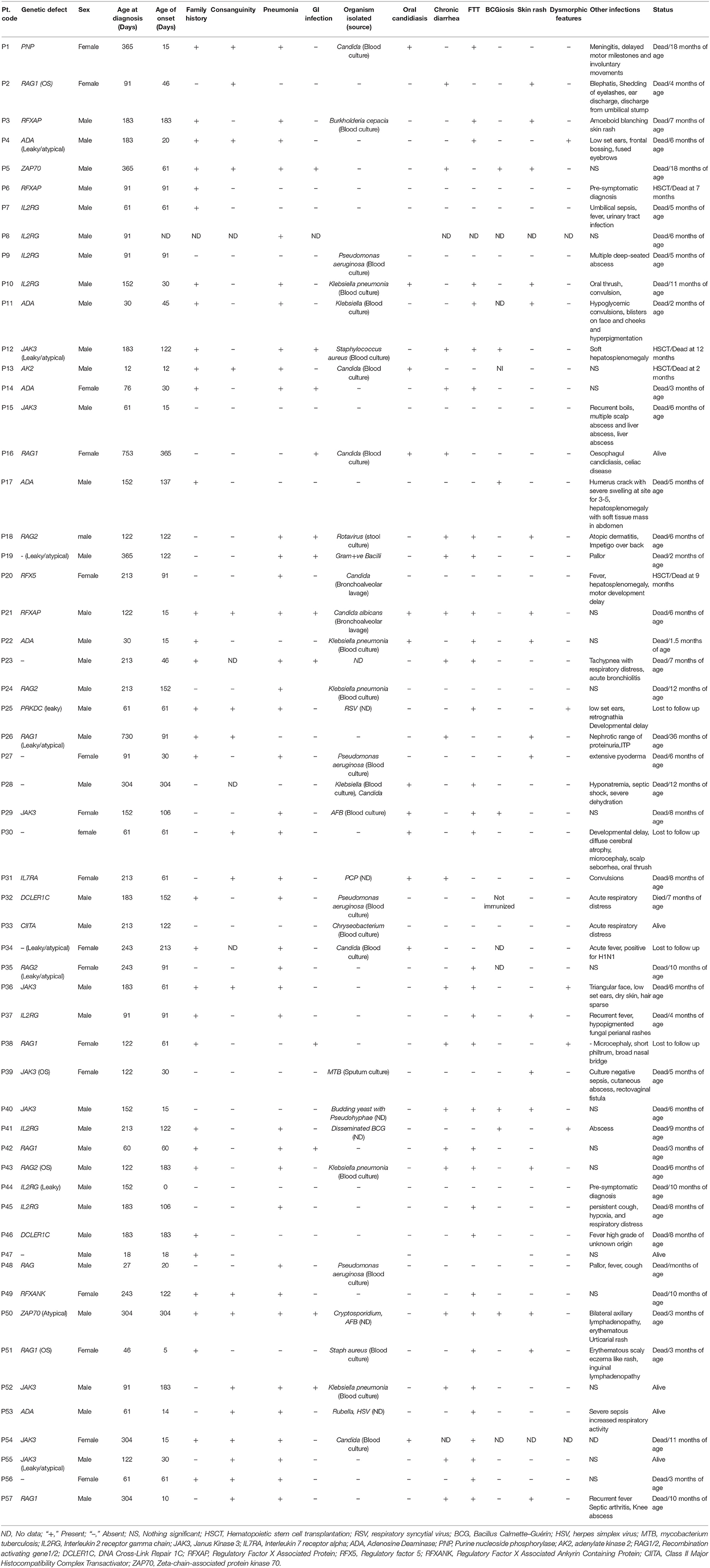
Table 1. Clinical findings in SCID patients.
Immunological Findings
Based on the Primary Immune Deficiency Treatment Consortium (PIDTC) case definition for SCID, 38 of 57 patients (67%) with absent or severely low T cell counts (<300 cells/μL) were classified as typical SCIDs (2). The remaining patients were classified as Leaky/Atypical SCIDs. Lymphopenia (<2,500 lymphocyte counts/μL) was noted in 36 of 57 (63%) patients including both typical and atypical SCID cases. Of these, the median lymphocyte count/μL was 680 (11–2,403) counts/μL. Classification of patients based on comparison of absolute counts/μL of B and NK cells with age matched reference ranges (8) revealed 14 patients with T−B+NK− phenotype (25%), 2 patients as T−B+NK+ (3.5%), 10 as T−B−NK− (17.5%), 12 patients with T−B−NK+ SCID (21%).
7 of 57 (12%) cases were identified with isolated T cell lymphopenia (selective deficiency of CD4+ T cells (n = 6) or CD8+ T cells (n = 1) and were eventually grouped under the category “Combined Immunodeficiency (CID) generally less profound than Severe combined Immunodeficiency” according to the IUIS classification (9).
The remaining cases with detectable T cells were classified as leaky/atypical SCID (12 cases, 21%). Of these cases, 6 patients had T+B+NK− phenotype (1 Omenn phenotype), 2 patients as T+B+NK+, 1 as T+B−NK−, 3 patients with T+B−NK+ SCID (1 Omenn phenotype).
The median T cell counts/μL in typical SCIDs was significantly lower than leaky SCID (1[range, 0–388] vs. 1,165 [range, 493–8,288]; p < 0.0001) and CID (1,565 [range, 927–13,900]; p < 0.0001).
Though the median ALC/μL was significantly lower in the SCID group (including typical and leaky SCID) than CID (771 [11–9,570] counts/μL vs. 4,408 [2,860–1,9041] counts/μL; p = 0.0003), the median age of onset in the SCID group (46 [range, 0–730] days) was not statistically significant from the median age of onset within CID group (106 days [range, 15–304], p = 0.08).
Apart from lymphocyte subset analysis, serum immunoglobulin levels were measured in a total of 41 available patient serum samples. The median serum IgG level in all SCID subtypes was lower than the age matched ranges. However, 14 patients had normal Sr. IgG levels in our cohort. These included 6 cases of B+ SCID, 7 cases of B− SCID, and 1 case of ZAP70 deficiency. Eight of these Fourteen patients were less than 6 months of age, suggesting the presence of maternal immunoglobulins in these children. Normal IgE was observed in 7 cases (1 case of B+ SCID, 2 cases of B−SCID, 4 patients with T−B−NK+) and elevated IgE was noted in 2 cases (T−B+ SCIDs).
Table 2 presents the basic immunological findings in our patient cohort.
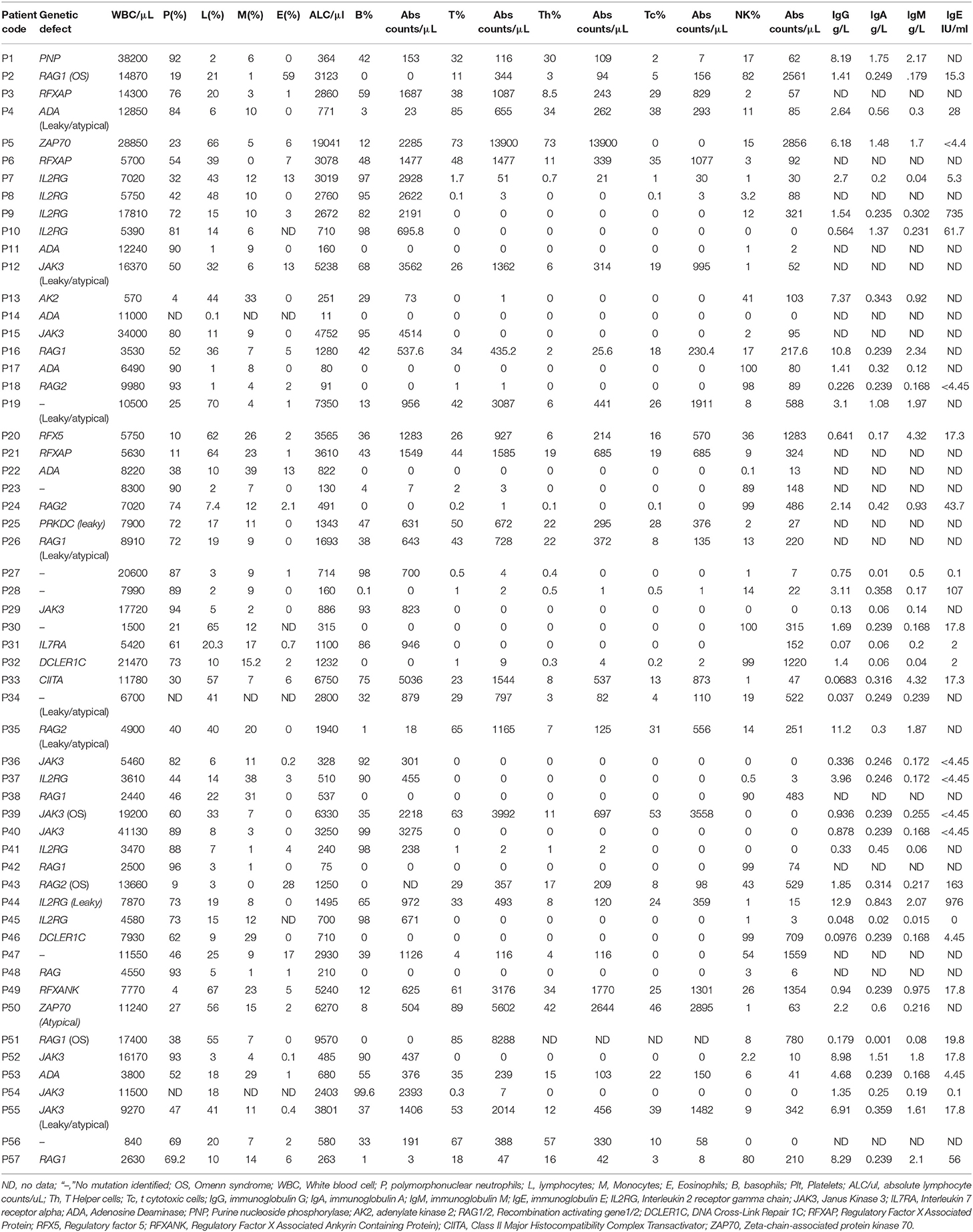
Table 2. Immunological findings in SCID patients.
Absent HLA-DR expression was noted in 6 of 57 patients and they were classified as MHC class II deficient cases. Elevated HLA-DR expression with other features of Omenn SCID such as skin rash, hepatosplenomegaly, elevated eosinophil counts was identified in 4 patients. T cell proliferation assay was performed in 3 leaky SCID patients and all had a poor T cell response to PHA.
The median percentage of naïve Th cells(0.3 [range, 0–8]%) within the typical and atypical SCID patient group was significantly lower than the median percentage of healthy age matched control group 73% ([range, 66–89]%; p < 0.0001).The median percentage of naïve Tc cells was significantly lower in SCID group than control group (7 [range, 0–29] vs. 64[range, 41–74]; p < 0.001). In MHC class II deficiency cases, a selective deficiency of naïve cells on CD4 cells was noted in 3 of 4 cases. The median percentage of naïve Th in MHC class II deficient cases was 39.50 (range, 29–48) which was significantly lower than the median percentage of healthy control group 77 ([range, 66–99]; p < 0.003). The naïve Tc percentage was within the normal ranges in these 3 cases. Measurement of naïve T cell percentages in one case of ZAP70 deficiency revealed reduction of naïve T cell subsets on both CD4 and CD8.
Four patients with T−B−NK− and 1 patient with T+B−NK− had RBC-ADA activity (≤0.5 U/g Hb) lower than the healthy controls (1.1–2.5 U/g Hb) and were sequenced for ADA gene defects.
Absent CD132 expression on B cells was observed in 6 patients and they were classified as X-SCID. Seven patients with B+ phenotype had detectable T cells and could be tested for JAK3-pSTAT5 signaling studies and CD127 expression. Of these, 2 patients (P12,39) had absent pSTAT5 expression on T cells after IL-2 stimulation and two patients (P12, P44) had reduced expression of CD127 on T cells.
Analysis of T cell receptor excision circles (TRECs) was done in all the SCID and CID patients and compared with TREC copies in age matched healthy controls samples (n = 55). The median TREC copies in T−B+ SCID (2.3[0.0175–16]) and T−B− SCID (3[0–11]) was significantly lower than the control group (139 copies [range, 62–348]; p < 0.0001). The median TREC copies in MHC class II deficiency 81.5(13–154) and ZAP70 deficiency 40.5(17–64) were significantly higher than SCID patients (p < 0.01).
Treatment and Outcome
Intravenous immunoglobulin was administered to 75% of the patients and 54% of patients were on prophylaxis (antibacterial, antiviral, and antifungal). During the study period, 4 patients (P6, P12, P13, P20) underwent HSCT however, had a poor survival outcome. The median age at transplant was 8 months (range, 2–12). P6 underwent umbilical cord blood transplant and died due to post-transplant complications like diarrhea and Gram-negative sepsis. P12 and P13 underwent haploidentical HSCT, however expired from Graft vs. Host disease and adenovirus infection, respectively. P20 underwent HSCT from HLA identical sibling, however, expired in the period immediately followed by HSCT due to lung damage and systemic candidiasis. P6 and P20 underwent HSCT using myeloablative conditioning (Treosulfan, Cyclophosphamide, anti-thymocyte globulin [ATG]). For the other two patients, no details were available on the conditioning regimen.
Of the remaining patients, 49 patients could be followed up. Presently, only 6 patients are surviving. These patients were recently diagnosed as SCID and the median age of these children is 5.5 months (range, 2–30). The patient aged 2.5 years (30 months) had a late onset of presentation (2 years). At the time of last available report, 2 of these 6 patients were awaiting a transplant.
The median age of death in patients who did not undergo HSCT (n = 43) was 6 months (range, 1.5 months−3 years). Majority of these children expired before 12 months of age (n = 38). Three patients survived beyond 1 year of age (Patient P1 with PNP deficiency, P5 with ZAP70 deficiency and P26 with hypomorphic RAG gene mutation). While P1 and P5 expired within 2 years of age, P26 expired at 3 years. The main cause of death in all the patients was respiratory failure, chronic diarrhea, sepsis and disseminated BCG in 1 patient.
Molecular Findings
As a first-line approach, molecular investigations were done in the patients by sanger sequencing of the common genes like IL2RG, IL7RA, ADA, RAG1, RAG2, ZAP70 gene depending on the immunophenotypic pattern. Twenty-five patients could be molecularly characterized using this approach. In a quest to identify the underlying genetic defect in the remaining cases, Targeted Next Generation sequencing (T-NGS)- Primary immunodeficiency (PID) panel was done (n = 32). Of the 32 cases referred for T-NGS, 24 cases could be molecularly characterized, however, 8 cases still remained uncharacterized.
Overall, a total of 49 patients could be molecularly characterized in this study. Of the 25 cases characterized by sanger sequencing, we identified 7 patients with mutations in IL2RG, 1 patient with IL7Ra deficiency, 5 with ADA deficiency, 1 with PNP defect, 1 with AK2 defect,9 patients with RAG1/2 deficiency and 1 case of ZAP70 defect. With the help of T-NGS, genetic cause could be identified in 24 cases with 9 cases of JAK3 deficiency (*1 case was identified with whole exome sequencing), 6 MHC class II deficient cases, 3 RAG 1/2mutations, 2 IL2RG defects, 1 PRKDC defect, 2 cases of DCLER1C, an atypical case of ZAP70 deficiency. The nature of novel missense mutations identified in our cohort was determined by in silico tools like Mutation Taster (10), SIFT (11) and Polyphen-2 (12). Depending on the availability of parent's sample, familial segregation analysis was performed.
The molecular findings are presented in Tables 3–7.
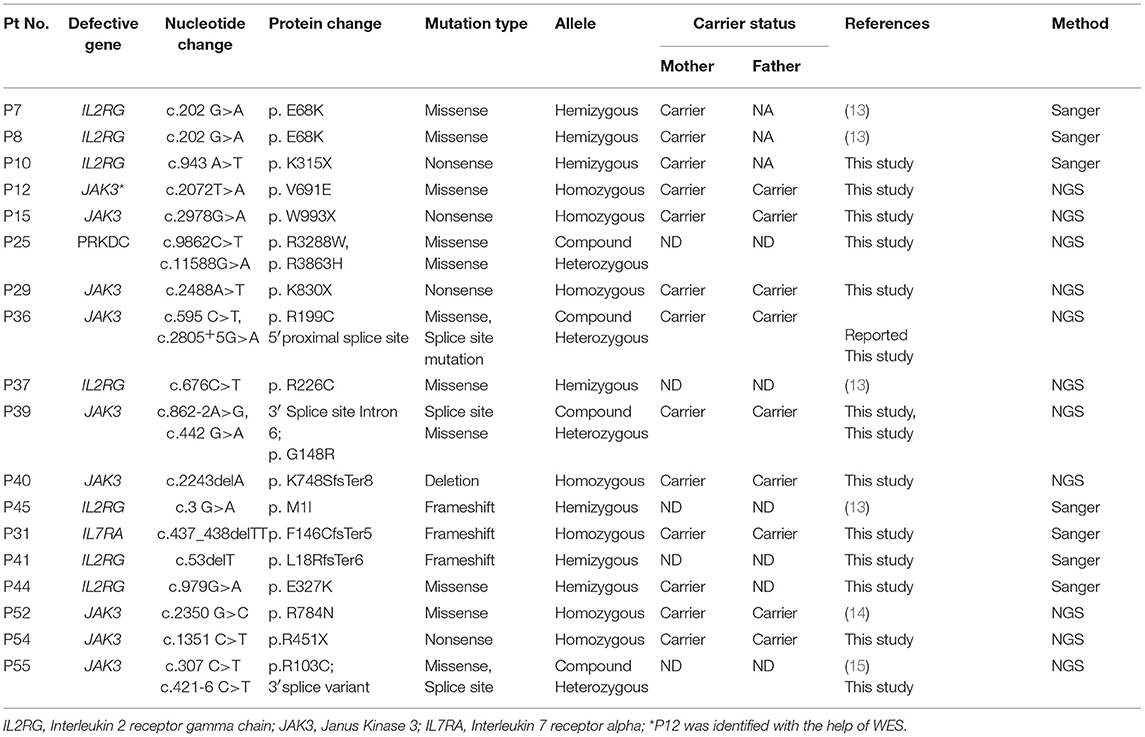
Table 3. Molecular findings in T−/+B+NK−SCID.
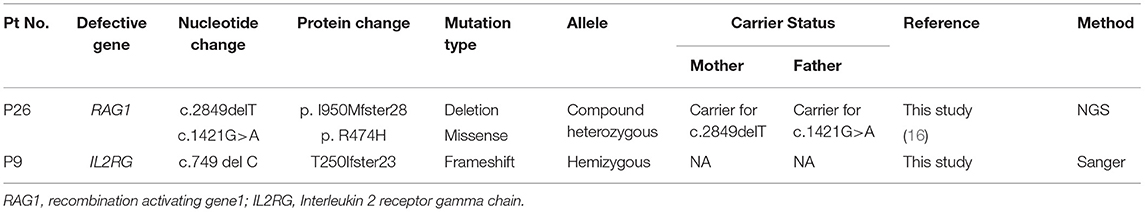
Table 4. Molecular findings in T−/+B+NK+SCID.
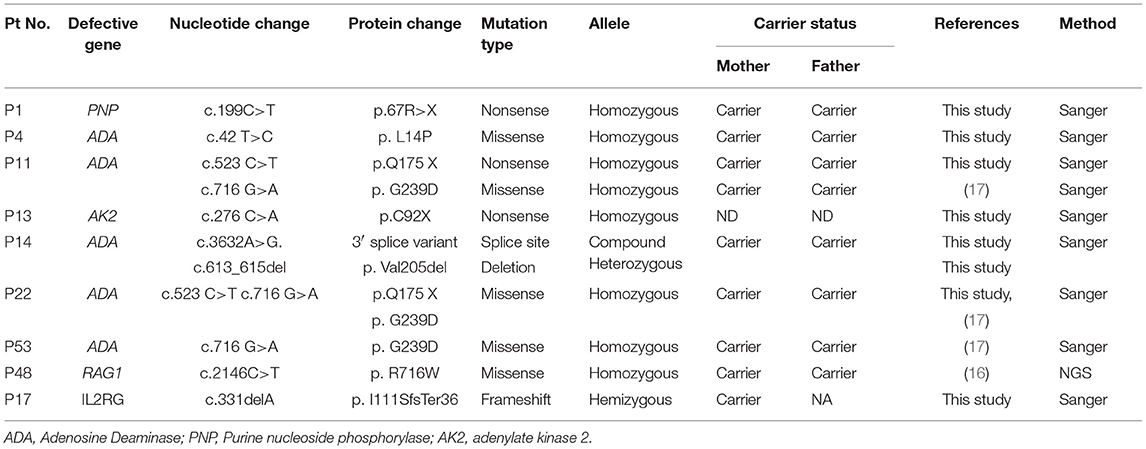
Table 5. Molecular findings in T−/+B−NK−SCID.
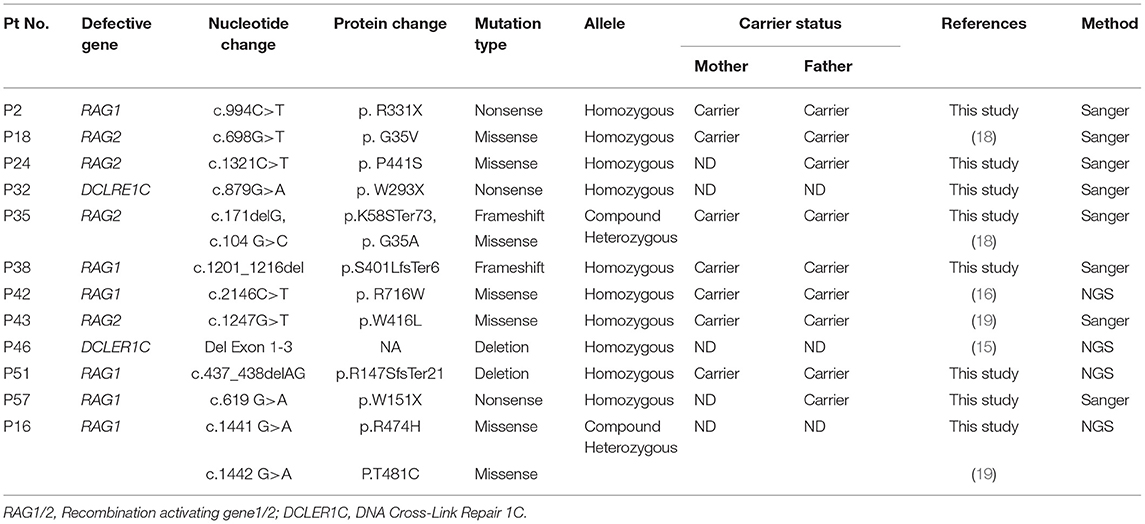
Table 6. Molecular findings in T−/+B−NK+SCID.
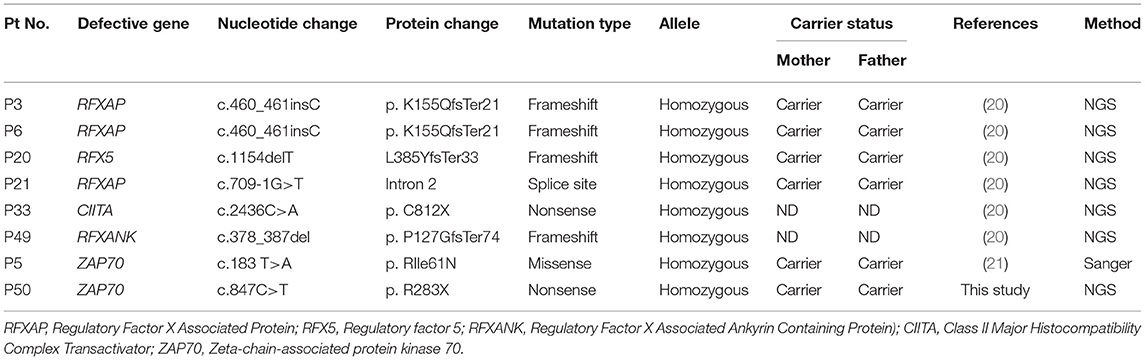
Table 7. Molecular findings in CID.
Discussion
SCID is a genetically heterogenous group of disorders that affects both the cellular and humoral immunity. The incidence and prevalence of SCID varies in different parts of the world and is reported to be higher in countries with a high rate of consanguinity. To the best of our knowledge, this is the first comprehensive report on clinical, immunological, and molecular studies in 57 SCID patients from India.
Majority of our patients (89%) presented within 6 months of age with a median age of onset of 2 months. The median age of diagnosis in our cohort (152 days; 5 months) was consistent with other studies such as Canada (4.2 months), China (5 months), Greece (6 months), and United States (6.59 months) (22–24). Patients with MHC class II deficiency and ZAP70 deficiency were also referred to us within 1 year of age with a clinical suspicion of SCID. Though, these groups are categorized under “Combined immunodeficiency less profound than Severe combined immunodeficiency” our patient data highlights the clinical severity of these disorders to be like SCID.
We observed 36% rate of consanguinity in our cohort which is intermediate to countries like Iran, Kuwait (18) where a very high rate of consanguinity (77%) exists and UK which has 3% rate of consanguineous marriages (23).
Lymphopenia, which is considered as a hallmark of SCID was observed in 67% of our patients (cut off <2,500 counts/μL). The patients with normal ALC/μl included B+ SCIDs (n = 10) and Omenn SCID (n = 4). MHC class II deficient patients and ZAP70 deficient patients also had normal ALC/μl. This is consistent with a study from china where 86% of the patients had a low lymphocyte count (25). Pilot series from United States identified most newborns with SCID based on ALC, but, 10% of SCID samples had normal lymphocyte counts.
The median TREC copies/μL were significantly lower in SCID patients as compared to healthy controls. Importantly, we found TREC copies to be higher than typical SCIDs in children with either CD4 or CD8 lymphopenia. Both TREC copies and ALC/μL are higher in MHC class II deficiency and ZAP70 deficiency than typical SCIDs. Hence, in a scenario of severe infections with normal ALC/μL and normal TRECs, extensive immunological evaluations should still be performed to rule out these forms of SCID. An important clue to underlying immunodeficiency in both these cases were reduced percentages of naïve Th/Tc which was evaluated in 4 cases of MHC class II deficiency and 1 case of ZAP70 deficiency. Notably, 3 of 4 MHC class II deficiency cases showed a selective reduction of Naïve Th population with normal percentages of naive Tc cells. Hence, a suspicion of MHC class II deficiency can be made in a scenario of selective reduction of naïve Th subset.
Traditionally SCID patients have been classified based on absolute counts of T, B, NK cells. Majority of our cases belonged to the category of T−B− SCID (39%) followed by T−B+ SCID (28%). In Greece, 40% SCID patients belonged to T−B−NK+ category and in Serbia and Montenegro, 57% patients with SCID and Omenn syndrome presented with T−B−NK+ SCID phenotype (26). In the registry of Saudi Arabia for combined immunodeficiencies, T−B− was the most common type; 17% followed by T−B+ found in 5% and Omenn syndrome in 3.6% (26). China's SCID registry has reported 66.7% of patients with B+ SCID and 7.1% of the cohort with B− SCID (25). Defective expression of major histocompatibility complex class II (MHC) molecules accounted for 5% of severe combined immunodeficiency (SCID) in Canadian Survey and almost 20–30% of SCID cases in Kuwait and North Africa (27). MHC class II deficiency accounted for 10.5%and ZAP70 deficiency constituted 3.5% of our patient group.
The pattern of lymphocyte subsets serves as a useful guide to perform genetic studies, however, molecular diagnosis of SCID is highly challenging due to the involvement of multiple genes whose defect can result in same immunophenotypic pattern. Hence, in such cases DNA sequencing of individual genes becomes tedious, time consuming and expensive. As there exists a significant clinical and immunophenotypic overlap between different genetic subtypes of SCID, we performed assays like measurement of RBC-ADA levels, flow cytometric evaluation of HLA-DR, CD132, CD127, and pSTAT5 expression to narrow down the list of possible genetic defects.
Spectrophotometric estimation of RBC-ADA levels was found to be a simple, cost- effective and accurate method for identification of ADA deficient patients. Five patients had low RBC-ADA levels and were also detected with a molecular defect in the ADA gene. Of the remaining patients with T−B−NK− phenotype (n = 6), a PNP and AK2 defect were identified. No pathogenic variant was identified in 2 patients despite T-NGS. In the remaining two patients, mutations were identified in IL2RG and RAG1 gene. These findings expand the phenotypic spectrum of typical X-SCID and RAG SCID.
Study of HLA-DR expression on immune cells helped in identification of MHC class II deficient patients (absent HLA-DR expression) and Omenn SCID (elevated HLA-DR expression suggesting activated T lymphocytes).
Absent/reduced expression of CD132 on B cells helped identify 6 patients (67%) with IL2RG gene defect. One patient with c.676 C>T in Exon 5 of IL2RG had a normal expression of CD132 on B cells (89%) suggesting this mutation did not affect the protein expression. Functional screening of STAT3 phosphorylation after IL-21 stimulation to assess the functionality of γchain could have helped us in this scenario, but this assay was not performed in our study.
Two patients with T+B+ phenotype were identified with RAG1/2 and PRKDC gene defect suggesting these mutations to be hypomorphic thereby, producing residual number of T and B cells. The oligoclonality of TCR-Vβ repertoire was tested in the RAG deficient patient and was found to be clonally restricted (Vβ13.1 on CD4+T cells [20.5%] and Vβ11[5%], Vβ16 [4.1%] on CD8+T cells). A case of IL7RA gene defect was detected in a patient with T−B+NK− phenotype. CD127 expression studies could not be performed in the patient due to lack of T cells.
We observed reduced CD127 expression in 2 patients (P12 and P44) who were later identified with a mutation in JAK3 gene and IL2RG gene, respectively. The possible explanation for this observation lies in the mechanism of IL7Ra receptor downregulation/ internalization due to high circulating levels of IL7 in some lymphopenic patients. However, studies to look for internalized CD127 or western blot from separated T cells to look for CD127 expression could not be performed in our study. Hence, a reduced CD127 expression on flow cytometry needs to be interpreted carefully.
A hemizygous deletion (c.749 del C) in Exon 5 of IL2RG gene leading to frameshift mutation was identified in a T−B+NK+ patient (male). This patient had a normal CD132 expression on B cells, but its functionality was not tested. Normal NK cell numbers in γchain deficient patients have been reported earlier (28) and they are predicted to be of maternal origin. In patients with mutations involving the γc portion of the IL2RG, normal NK cell numbers have been identified and these observations have raised the possibility of a potential downstream activation mechanism in NK cell differentiation (28).
Flow cytometric evaluation of proteins specifically expressed on T cells (for e.g.,: CD127) or assays that required TCR engagement with specific stimulants (JAK3-pSTAT5) could not be assessed in patients who lacked T cells. Almost 65% of our B+ SCID cohort had absent T cells hence, both CD127 and phospho-STAT5 assay had a limited utility in our study and we had to rely on genetic analysis to identify the defect.
The frequency of mutations in RAG1/2 (21 %) in the current study is like United States (21%) and the Netherlands (32%), but much less compared to Greece (41%) and Serbia (61%) where a common founder gene defect in RAG1 is likely (18). A patient with microcephaly, flat nasal bridge, short philtrum was suspected of a defect in DNA Ligase IV (1) but was identified with a RAG1 defect. Three patients within this group were classified as Omenn SCID (2 RAG1 mutations and 1 RAG2 mutation).
The molecular findings in five cases of MHC class II deficiency from our cohort has been recently described (20). Additionally, we have identified another patient with a defect in RFXANK gene. There have been only 2 patients with confirmed RFXANK mutations reported in Asia. RFXANK mutation causes bare lymphocyte syndrome type 2B (15) commonly observed in North Africa (15) and in other places such as France and Spain.
An atypical case of ZAP70 deficiency with a novel nonsense mutation in ZAP70 gene was identified in 1 patient with elevated CD8+T cells. Poor proliferative responses was the only clue to an underlying immunodeficiency in this patient. Unfortunately, we could not assess the intracellular ZAP70 expression or assess the TCR-Vβ repertoire on CD4 and CD8 T cells as the child expired by the time of receiving a molecular diagnosis. This mutation further extends the phenotypic spectrum of ZAP70 deficiency.
The spectrum of genetic defects in our cohort revealed a wide genetic heterogeneity with 21% RAG1/2 defects,15.8% IL2RG defects, 15.8% JAK3 defect, 1.7% IL7RA defect, 8.8% ADA defect, 1.8% AK2 defect, 1.8% PNP defect, 3.5% DCLER1C defect, 1.8% PRKDC defect, 10.5% with MHC class II deficiency and 3.5% ZAP70 defects. Thirty-two novel variants were identified in our study and no founder mutation was detected in our cohort. 14% of the defects still remained uncharacterized despite application of T-NGS (list of genes covered in the T-NGS panel is presented in Table 8) and it would be interesting to perform whole exome/genome sequencing in these cases as that may lead to discovery of novel genetic defects causing SCID (no pathogenic variant explaining the cause of immunodeficiency could be identified in one patient [P47] even after WES). Our data also shows a lack of demonstrable correlation between genotype and phenotype in few cases (7%). Certain genetic and environmental factors, concurrent mutations in other SCID genes, modifier gene(s), and mutations, that lead to sparing or disrupting developments of other lineages of lymphocytes may be the cause for lack of correlation (15).
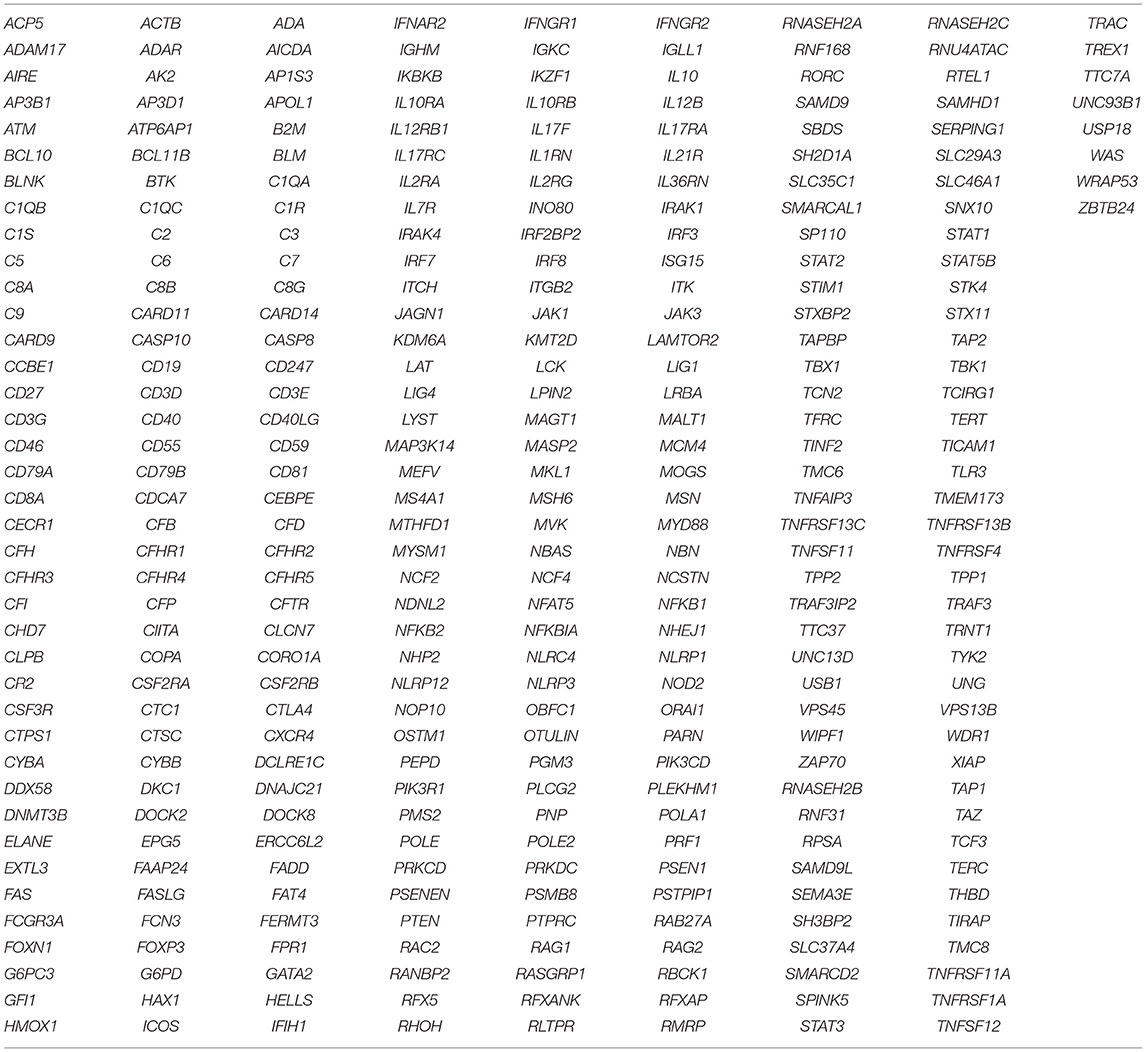
Table 8. Genes covered in Targeted NGS panel.
The autosomal recessive form of SCID (86%) was more common than X-SCID (14%) in our cohort. This is in contrast with several studies that reported a predominance of X-linked SCID which accounts for approximately half of SCID cases (24). This finding supports that genetic defects in SCID patients probably differ depending on diverse genetic backgrounds. Our data is similar to countries where consanguinity is practiced like in Turkey where the AR form accounts for 80% SCID cases (29) as compared to data from USA with AR form 20% and a low rate of consanguinity (23).
The overall outcome in our cohort is extremely poor with death occurring in 92% of the patients. Only 4 patients underwent HSCT, and none of them could survive the transplant. The median age at HSCT in our cohort was 7.5 months. It is well known that HSCT for (S)CID before the age of 3.5 months results in a superior outcome (6). One of our patients with MHC class II deficiency (P7) had a pre-symptomatic diagnosis and received an early transplant (at 3 months), however, he expired within 8 days of transplant due to severe diarrhea and gram-negative sepsis. As stated in a recent report, the reason for low survival rate in MHC class II deficiency patients may be due to presentation of donor antigens by donor antigen-presenting cells to recipient T cells leading to graft rejection (30).
We consider delayed diagnosis as the major cause for a poor survival outcome in our cohort. Our data is consistent with countries where newborn screening (NBS) for SCID has not yet been implemented (18). Without NBS, asymptomatic diagnosis for SCID is possible only in a scenario of strong family history. In several other cases, even before the child is diagnosed as SCID and the crucial decision for transplantation is taken, the child has significant number of infections which affects the survival outcome. Maintenance on IVIg therapy is expensive and a major constraint in the management of children with PIDs in India, as also in other developing countries (31). Only two state governments (Punjab and Karnataka) in India have taken a major policy initiative to provide IVIg freely to the patients. In the other states, several children are not privileged to get the recommended dose of IVIg (31). Many centers in India are now routinely performing HSCT for a variety of malignant diseases in both children and adults whose results are comparable to many international published reports (32), however, there is only a minority that have the requisite expertise for carrying out transplant for PIDs (31). A recent report by a tertiary referral center in India shared their 15-year experience of HSCT in children with PIDs with encouraging results of an overall survival rate of 62 and 55% survival rate for SCIDs (33). However, the number of such centers that specialize in transplantation for PIDs in India are very few and specific arrangements are needed to transfer the patients to centers in other states. Financial constraint is one of the major reasons for families to refuse transplantation. Overall, delayed diagnosis due to lack of awareness about SCID among the pediatricians, lack of expertise in HSCT for SCID, financial constraints and lack of a suitable donor are the reasons for such a poor survival rate observed in our cohort.
Prenatal diagnosis (PND) is hence, highly useful in families affected with SCID. The preferred procedure for prenatal diagnosis is genetic confirmation in the index case and parents and then performing PND by chorionic villus sampling or amniocentesis. This was helpful in two of our affected families (index case P5 and P6). Two families (index case P6 and P42) benefited from the facility of phenotypic prenatal diagnosis performed on cordocentesis sample at 18 weeks of gestation due to lack of genetic diagnosis at the time of PND.
Early detection of SCID and HSCT at a pre-symptomatic stage has proven to result in a better outcome in countries with NBS program for SCID and advanced health care systems. As NBS for SCID is not yet performed in our country, there exists a high chance that we are missing out many cases as these children may have expired before receiving a diagnosis. Pilot studies for NBS in our country could provide data on the true incidence of SCID in our population. Overall, our data reveals a wide genetic heterogeneity of SCID in the Indian population, confirms the poor prognosis of SCID due to delayed diagnosis and highlights the need for implementing NBS for SCID in India.
Author Contributions
JA analyzed the data and wrote the manuscript. MG, AD, SM, and MKu were involved in performing laboratory investigations of the different cases. MD, AT, NS, AA, HL, VK, GG, and MKa supervised the clinical care of the various patients. UB helped in procuring the clinical details and follow up of the patients. SR, JN, and JS provided the WES support. VT provided genetic diagnosis in AK2 deficient patient. MM supervised the study and reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge Medgenome Pvt. Ltd, India for Targeted NGS support and Dr. Daniel Douek, Vaccine Research Center, National Institute of Allergy and Infectious Diseases, USA, for kindly providing the hTREC plasmid for performing the TREC assay.
References
1. Sponzilli I, Notarangelo LD. Severe Combined Immunodeficiency (SCID): from molecular basis to clinical management. Acta BioMed. (2011) 82:5–13.
2. Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for Severe Combined Immunodeficiency Disease (SCID), leaky SCID, and omenn syndrome: the primary immune deficiency treatment consortium experience. J Allergy Clin Immunol. (2014) 133:1092–8. doi: 10.1016/j.jaci.2013.09.044
4. Tasher D, Dalal I. The genetic basis of severe combined immunodeficiency and its variants. Appl Clin Genet. (2012) 5:67–80. doi: 10.2147/TACG.S18693
5. Antonia K, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA (2015) 312:729–38. doi: 10.1001/jama.2014.9132
6. Dvorak CC, Cowan MJ, Logan BR, Notarangelo LD, Griffith LM, Puck JM, et al. The natural history of children with severe combined immunodeficiency: baseline features of the first fifty patients of the primary immune deficiency treatment consortium prospective study 6901. J Clin Immunol. (2013) 33:1156–64. doi: 10.1007/s10875-013-9917-y
7. Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature (1998) 396:690–5. doi: 10.1038/25374
8. Shearer WT, Rosenblatt HM, Gelman RS, Oyomopito R, Plaeger S, Stiehm ER, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the pediatric AIDS clinical trials group P1009 study. J Allergy Clin Immunol. (2003) 112:973–80. doi: 10.1016/j.jaci.2003.07.003
9. Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. (2018) 38:129–143. doi: 10.1007/s10875-017-0465-8
10. Schwarz JM, Cooper DN, Schuelke M, Seelow D. Mutationtaster2: mutation prediction for the deep-sequencing age. Nat Methods (2014) 11:361–2. doi: 10.1038/nmeth.2890
11. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. (2003) 31:3812–4. doi: 10.1093/nar/gkg509
12. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. (2013) Chapter 7, Unit7.20. doi: 10.1002/0471142905.hg0720s76
14. Barreiros LA, Segundo GRS, Grumach AS, Torgerson TR, Ochs HD, Condino-Neto A. A novel homozygous JAK3 mutation leading to T-B+NK– SCID in two Brazilian patients. Front Pediatr. (2018) 6:230. doi: 10.3389/fped.2018.00230
15. Luk ADW, Lee PP, Mao H, Chan KW, Chen XY, Chen TX, et al. Family history of early infant death correlates with earlier age at diagnosis but not shorter time to diagnosis for severe combined immunodeficiency. Front Immunol. (2017) 8:808. doi: 10.3389/fimmu.2017.00808
16. Sobacchi C, Marrella V, Rucci F, Vezzoni P, Villa A. RAG-dependent primary immunodeficiencies. Hum Mutat. (2006) 27:1174–84. doi: 10.1002/humu.20408
17. Adams SP, Wilson M, Harb E, Fairbanks L, Xu-Bayford J, Brown L, et al. Spectrum of mutations in a cohort of UK patients with ADA deficient SCID: segregation of genotypes with specific ethnicities. Clin Immunol. (2015) 161:174–79. doi: 10.1016/j.clim.2015.08.001
18. Al-Herz W, Massaad MJ, Chou J, Notarangelo LD, Geha RS. DNA recombination defects in Kuwait: clinical, immunologic and genetic profile. Clin Immunol. (2018) 187:68–75. doi: 10.1016/j.clim.2017.10.006
19. Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol. (2016) 16:234–46. doi: 10.1038/nri.2016.28
20. Aluri J, Gupta M, Dalvi A, Mhatre S, Kulkarni M, Hule G, et al. Clinical, immunological, and molecular findings in five patients with major histocompatibility complex class II deficiency from India. Front Immunol. (2018) 9:188. doi: 10.3389/fimmu.2018.00188
21. Aluri J, Italia K, Gupta M, Dalvi A, Bavdekar A, Madkaikar M. Low T cell receptor excision circles (TRECs) in a case of ZAP 70 deficient severe combined immunodeficiency (SCID) with a novel mutation from India. Blood Cells Mol Dis. (2017). 65:95–6. doi: 10.1016/j.bcmd.2016.10.022
22. Lee PP, Chan KW, Chen TX, Jiang LP, Wang XC, Zeng HS, et al. Molecular diagnosis of severe combined immunodeficiency - identification of IL2RG, JAK3, IL7R, DCLRE1C, RAG1, and RAG2 mutations in a cohort of Chinese and Southeast Asian children. J Clini Immunol. (2011) 31:281–96. doi: 10.1007/s10875-010-9489-z
23. Edgar JD, Buckland M, Guzman D, Conlon NP, Knerr V, Bangs C, et al. The United Kingdom Primary Immune Deficiency (UKPID) registry: report of the first 4 years' activity 2008-2012. Clin Exp Immunol. (2014) 175:68–78. doi: 10.1111/cei.12172
24. Michos A, Tzanoudaki M, Villa A, Giliani S, Chrousos G, Kanariou M. Severe combined immunodeficiency in Greek children over a 20-year period: rarity of γ c-chain deficiency (X-Linked) type. J Clin Immunol. (2011) 31:778–83. doi: 10.1007/s10875-011-9564-0
25. Yao CM, Han XH, Zhang YD, Zhang H, Jin YY, Cao RM, et al. Clinical characteristics and genetic profiles of 44 patients with Severe Combined Immunodeficiency (SCID): report from Shanghai, China (2004-2011). J Clin Immunol. (2013) 33:526–39. doi: 10.1007/s10875-012-9854-1
26. Fazlollahi MR, Pourpak Z, Hamidieh AA, Movahedi M, Houshmand M, Badalzadeh M, et al. Clinical, laboratory and molecular findings of 63 patients with severe combined immunodeficiency: a decade's experience. J Invest Allergol Clin Immunol. (2017) 27:299–304. doi: 10.18176/jiaci.0147
27. Saleem MA. Clinical course of patients with major histocompatibility complex class II deficiency. Arch Dis Childhood (2000) 83:356–9. doi: 10.1136/adc.83.4.356
28. Jamal A, Upton JEM. IL2RG : a series of three novel mutations with clinical manifestations. LymphoSign J. (2016) 3:111–8. doi: 10.14785/lymphosign-2016-0003
29. Abolhassani H, Chou J, Bainter W, Platt CD, Tavassoli M, Momen T, et al. Clinical, immunologic, and genetic spectrum of 696 patients with combined immunodeficiency. J Allergy Clin Immunol. (2018) 141:1450–8. doi: 10.1016/j.jaci.2017.06.049
30. Kallen ME, Pullarkat ST. Type II bare lymphocyte syndrome: role of peripheral blood flow cytometry and utility of stem cell transplant in treatment. J Pediatr Hematol Oncol. (2015) 37:e245–9. doi: 10.1097/MPH.0000000000000278
31. Jindal AK, Pilania RK, Rawat A, Singh S. Primary immunodeficiency disorders in India-a situational review. Front Immunol. (2017) 8:714. doi: 10.3389/fimmu.2017.00714
32. Shah CA, Karanwal A, Desai M, Pandya M, Shah R, Shah R. Hematopoietic stem-cell transplantation in the developing world: experience from a center in Western India. J Oncol. (2015) 2015:710543. doi: 10.1155/2015/710543
Keywords: PID, flow cytometry, TREC, sanger sequencing, targeted next generation sequencing
Citation: Aluri J, Desai M, Gupta M, Dalvi A, Terance A, Rosenzweig SD, Stoddard JL, Niemela JE, Tamankar V, Mhatre S, Bargir U, Kulkarni M, Shah N, Aggarwal A, Lashkari HP, Krishna V, Govindaraj G, Kalra M and Madkaikar M (2019) Clinical, Immunological, and Molecular Findings in 57 Patients With Severe Combined Immunodeficiency (SCID) From India. Front. Immunol. 10:23. doi: 10.3389/fimmu.2019.00023
Received: 19 October 2018; Accepted: 07 January 2019;
Published: 04 February 2019.
Edited by:
Waleed Al-Herz, Kuwait University, KuwaitReviewed by:
Kimberly Gilmour, Great Ormond Street Hospital for Children NHS Foundation Trust, United KingdomMonica Thakar, Fred Hutchinson Cancer Research Center, United States
Copyright © 2019 Aluri, Desai, Gupta, Dalvi, Terance, Rosenzweig, Stoddard, Niemela, Tamankar, Mhatre, Bargir, Kulkarni, Shah, Aggarwal, Lashkari, Krishna, Govindaraj, Kalra and Madkaikar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manisha Madkaikar, bWFka2Fpa2FybWFuaXNoYUBnbWFpbC5jb20=