 Louise M. Ince
Louise M. Ince Jasmin Weber
Jasmin Weber Christoph Scheiermann
Christoph Scheiermann- 1Department of Pathology and Immunology, Faculty of Medicine, University of Geneva, Geneva, Switzerland
- 2Walter-Brendel-Centre of Experimental Medicine, University Hospital, Ludwig-Maximilians-University Munich, BioMedical Centre, Planegg-Martinsried, Germany
- 3DZHK (German Centre for Cardiovascular Research), Partner Site Munich Heart Alliance, Munich, Germany
Leukocyte migration is a crucial process in both homeostatic and inflammatory conditions. The spatiotemporal distribution of immune cells is balanced between processes of cellular mobilization into the bloodstream, their adhesion to vascular beds and trafficking into tissues. Systemic regulation of leukocyte mobility is achieved by different signals including neuronal and hormonal cues, of which the catecholamines and glucocorticoids have been most extensively studied. These hormones are often associated with a stress response, however they regulate immune cell trafficking also in steady state, with effects dependent upon cell type, location, time-of-day, concentration, and duration of signal. Systemic administration of catecholamines, such as the sympathetic neurotransmitters adrenaline and noradrenaline, increases neutrophil numbers in the bloodstream but has different effects on other leukocyte populations. In contrast, local, endogenous sympathetic tone has been shown to be crucial for dynamic daily changes in adhesion molecule expression in the bone marrow and skeletal muscle, acting as a key signal to the endothelium and stromal cells to regulate immune cell trafficking. Conversely, glucocorticoids are often reported as anti-inflammatory, although recent data shows a more complex role, particularly under steady-state conditions. Endogenous changes in circulating glucocorticoid concentration induce redistribution of cells and potentiate inflammatory responses, and in many paradigms glucocorticoid action is strongly influenced by time of day. In this review, we discuss the current knowledge of catecholamine and glucocorticoid regulation of leukocyte migration under homeostatic and stimulated conditions.
Introduction
Leukocytes migrate through the body by shuttling between the vascular system and tissues. Within the vasculature, immune cells freely circulate or are firmly attached to the vessel wall, effectively removing them from the circulation in what is known as the marginal pool. Adherent cells may be in the process of exiting the circulation to immigrate into organs [reviewed in (1)]. However, still vasculature-bound, marginated cells can also detach and be remobilized into the bloodstream—a process which is called demargination (Figure 1). Leukocytes adhere to the vasculature in a sequence of events known as the leukocyte adhesion cascade. This cascade is crucial for a functioning immune system, allowing immune cells to infiltrate tissues that are in need of pathogen clearance or regeneration. Leukocytes initially roll along the vessel wall with the help of cell adhesion molecules where they can be activated by chemokines on the vascular endothelium, leading to their arrest, and transmigration through the endothelial barrier to exit the bloodstream and enter underlying tissues [reviewed in (18) and illustrated in Figure 1]. These different stages of the adhesion cascade can be modulated by various factors, including circulating hormones such as catecholamines and glucocorticoids. It has been known for decades that the sympathetic nervous system, a key source of catecholamines, regulates the maturation and function of leukocytes via adrenoceptors on their surface [see (19) for an in-depth overview, also on the expression profile of adrenoreceptors]. However, the regulation of leukocyte trafficking by catecholamines and glucocorticoids (typically classed as stress hormones) and their interplay in steady state and stress conditions is multifaceted and therefore incompletely understood. In this review we focus on the recent findings in this field, which we have summarized in Table 1.
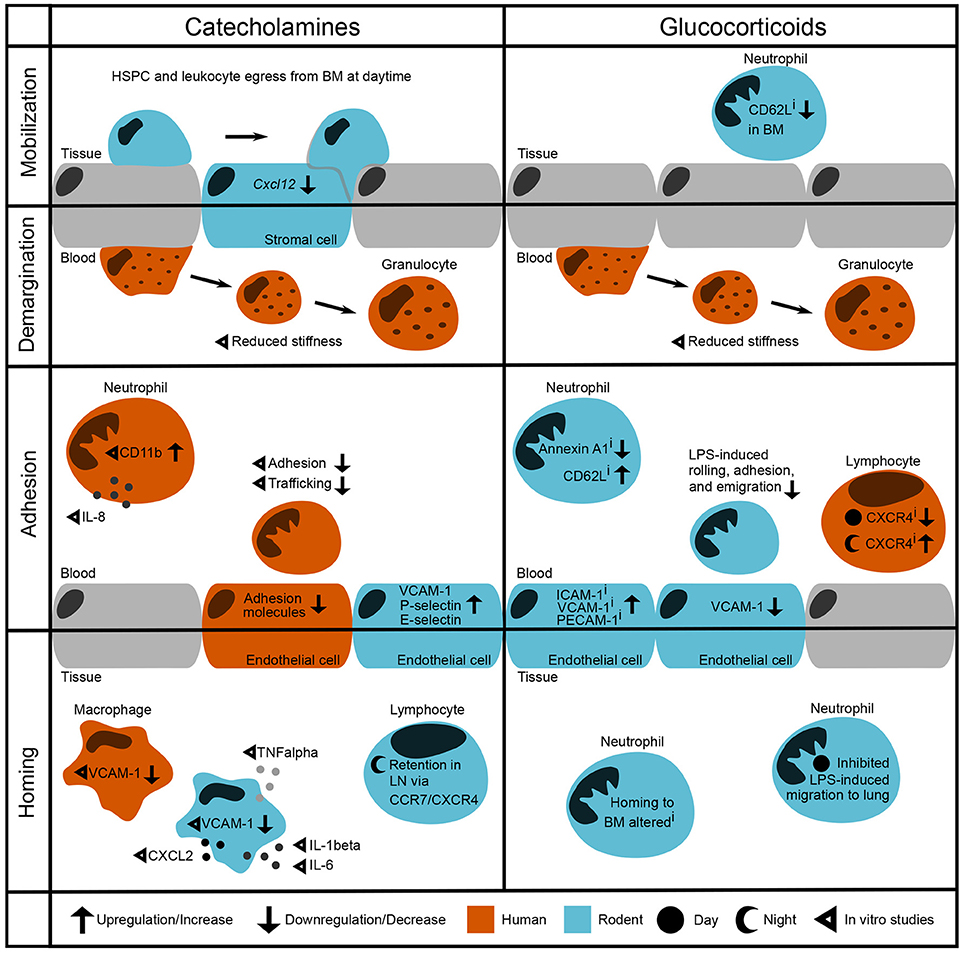
Figure 1. Modulation of leukocyte trafficking by stress-associated hormones. Leukocyte migration can be broadly broken down into mobilization and homing (entering/leaving the vasculature, respectively) as well as adhesion and demargination (attachment to/detachment from the vessel wall, respectively). Catecholamines control hematopoietic stem and progenitor cell (HSPC) and leukocyte egress from the bone marrow during daytime under steady-state conditions by downregulation of the retention factor CXCL12 in stromal cells (2). in vitro studies showed that after incubation with catecholamines and glucocorticoids, human granulocytes detach more easily by reducing their stiffness (3). In the bloodstream, human neutrophils show increased levels of CD11b as well as IL-8 after stimulation with adrenaline (4). However, their adhesion and trafficking in vitro are reduced due to downregulation of endothelial adhesion molecules (5, 6). In contrast, mouse endothelial cells upregulate VCAM-1, P-selectin, and E-selectin after catecholamine stimulation (7). In both humans and rodent macrophages, VCAM-1 levels are regulated through β2-adrenoceptor signaling (8). In addition, catecholamines induce cytokine release by murine macrophages (9). In mice, sympathetic stimulation leads to a retention of T cells in the lymph node via upregulation of CCR7 and CXCR4 (10, 11). Inhibition of glucocorticoid receptors downregulates Annexin A1 levels (12) and upregulates CD62L expression on circulating murine neutrophils whilst downregulating its expression in the bone marrow (13). Furthermore, murine neutrophils show increased LPS-induced adhesion when treated with a GR antagonist—although endothelial VCAM-1 is downregulated (14). Human naïve T cells show upregulated CXCR4 levels when treated with a GR antagonist during the night, whereas CXCR4 is downregulated when treated during the day (15). Similarly, GR agonism with dexamethasone inhibits LPS-induced neutrophil migration to the lung in the behavioral resting phase (16, 17). i denotes effects of inhibition.
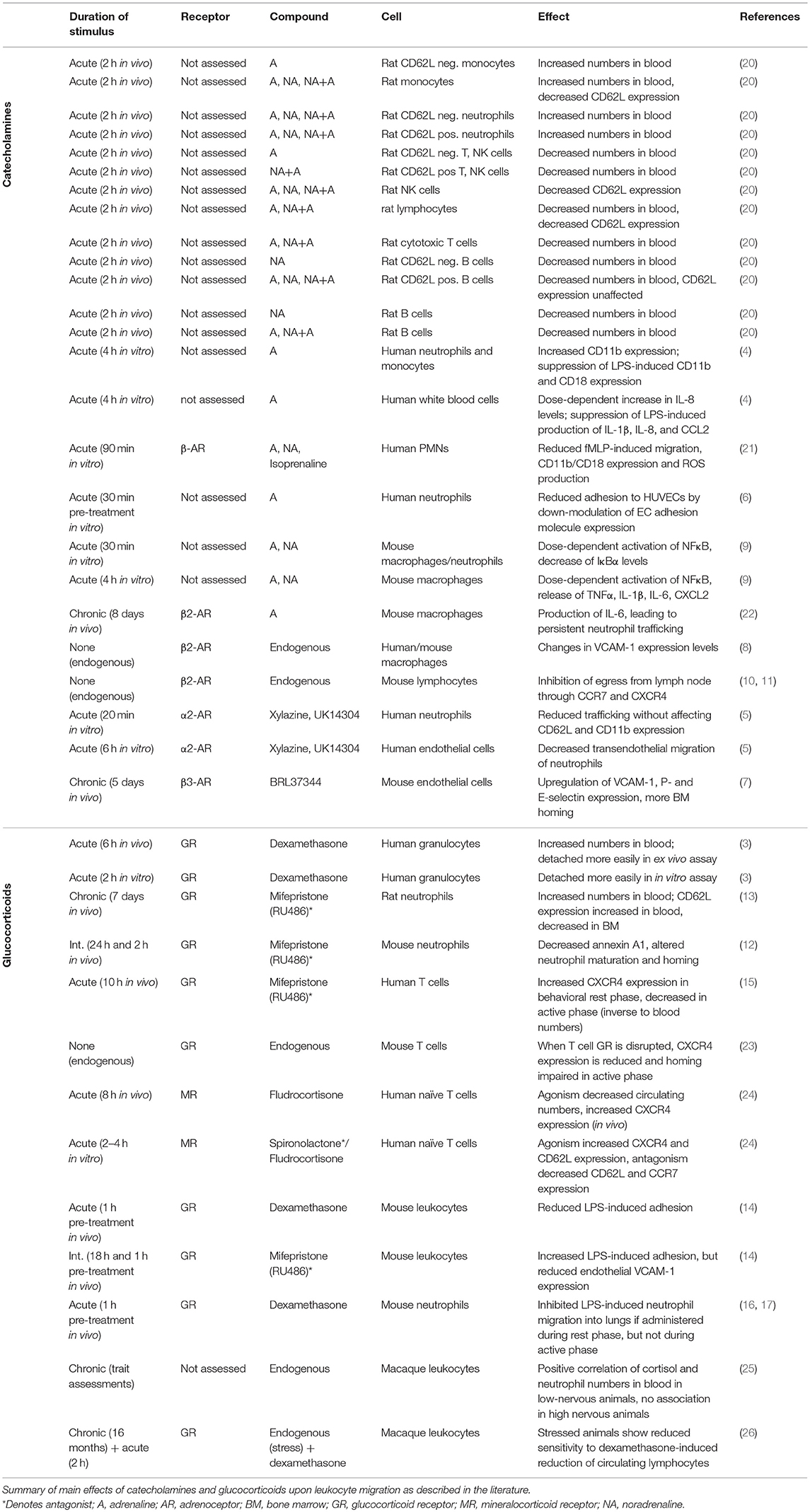
Table 1. Effects of hormonal signals on leukocyte trafficking.
Catecholamines
Catecholamines, such as adrenaline and noradrenaline, are an important class of systemic immune-modulators, released systemically by the adrenal gland and locally mainly by sympathetic nerves. These hormones have immune-enhancing or immune-suppressing effects, depending on the duration of the signal (acute vs. chronic), the microenvironment, and the timing of their release (27). In mice it was demonstrated that under steady-state conditions, the release of hematopoietic stem and progenitor cells requires local delivery of noradrenergic signals to the bone marrow by sympathetic nerves, where they are transmitted to stromal cells via β3-adrenoceptors, leading to a downregulation of the key retention factor CXCL12 (2). A similar phenomenon may contribute to the release of leukocytes into the bloodstream in acute stress, as in rats administration of adrenaline and noradrenaline has been shown to increase circulating myeloid and lymphoid cell numbers within a few minutes. In this scenario, most subpopulations have left the blood after 2 h, except for neutrophils, whose numbers continue to increase (20). Differences in the effect on subpopulation specificity are evidenced by the fact that noradrenaline increases numbers of circulating neutrophils and B cells, whereas adrenaline increases the number of neutrophils and monocytes but decreases lymphocyte numbers in blood. (20) (Table 1). The underlying signaling pathways and receptors responsible for these distinct outcomes are, however, ambiguous. For example, it is currently not clear how much of the increase of blood leukocyte numbers is caused by a stress-induced mobilization from hematopoietic tissues into blood, or by demargination from the vessel wall.
Stress hormones can affect leukocyte migratory properties via diverse mechanisms. A recent publication provided the first evidence that catecholamines can induce the rearrangement of cellular cortical actin in human granulocytes, thereby decreasing cell stiffness and leading to leukocyte demargination (3). This could explain the very fast increase in circulating leukocyte numbers by these hormones without the need of mobilization from tissues, allowing the organism to respond quickly to acute signals. Additionally, catecholamines can alter cytokine levels and expression of adhesion molecules. Exposure to adrenaline in vitro increases interleukin-8 (IL-8) expression and CD11b (alpha-M-integrin) levels in human neutrophils (4). Under LPS-induced inflammatory conditions the production of IL-1, IL-8, and CCL2 is reduced, indicating that regulation of cytokines and chemokines by adrenaline is highly dependent on the inflammatory milieu (4). In contrast to this study, in vitro stimulation with the adrenergic agents adrenaline, noradrenaline, or the agonist isoproterenol reduced N-formyl-methionyl-leucyl-phenylalanine (fMLP)-induced human polymorphonuclear cell (PMN) migration, CD11b/CD18 (Mac-1) integrin expression, as well as production of reactive oxygen species, without affecting IL-8 levels (21). Furthermore, adrenaline and dopamine, a structurally-related catecholaminergic neurotransmitter, facilitated the down-modulation of adhesion molecule expression in human umbilical cord vein endothelial cells (HUVECs), reducing neutrophil adhesion (6). Thus, experiments using catecholamines or their agonists have thus far provided different outcomes, which is most likely dependent on the dosage used and the microenvironmental context. What is clear, however, is that they exert effects on both the immune cell and the endothelial aspects of the adhesion cascade, by modulating expression of adhesion molecules, cytokine levels and leukocyte stiffness.
In addition to their direct influence on the leukocyte adhesion cascade, catecholamines also modulate functions of macrophages, a resident leukocyte subset. As major producers of cytokines, these phagocytic cells are likely largely responsible for the effects of catecholamines on cytokine levels. Adrenaline and noradrenaline can directly activate NF-κB in isolated peritoneal mouse macrophages, resulting in the release of pro-inflammatory cytokines including TNFα, CXCL2, IL-1β, and IL-6 (9). In murine skin wounds, tissue-resident macrophages produce IL-6 in response to chronic β2-adrenergic receptor activation, which in turn leads to a persistent trafficking of neutrophils to the site of injury (22). This is one potential mechanism by which long-term stress may be associated with a delayed wound healing. However, phagocytes themselves can also produce catecholamines and in a rat model of acute lung injury, elevated levels of macrophage-derived catecholamines were associated with increased expression of pulmonary intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) via α2-adrenoceptors. Work using knockout models of adrenoceptors could show that in mice and humans the expression of VCAM-1 in macrophages is sensitive to stimulation of β2-adrenoceptors, which plays an important role in the cardiac infiltration of leukocytes to facilitate an early inflammatory repair response to an acute myocardial injury (8). Taken together, these findings demonstrate that catecholamines act on resident macrophages but can also be released by these cells, providing an additional, indirect mechanism in regulating the behavior of migratory cells.
β2-adrenoceptors are the most common adrenergic receptor type expressed on leukocytes [reviewed in (28)]. However, mRNA for other adrenoceptor subtypes is also present in human immune cells (21). Pharmacological agonists for the α2-adrenoceptor reduced trafficking of IL-8 activated human neutrophils by inhibition of CD62L shedding with simultaneous prevention of increased CD11b expression (5). In vitro flow chamber assays revealed that targeting the α2-adrenoceptor in HUVECs, but not the neutrophils, decreased transendothelial migration of neutrophils (5). These data indicate that both leukocytes and the endothelium are important targets for catecholaminergic signaling in the regulation of leukocyte trafficking. However, the exact mechanisms in different cell types and the interplay of systemic and local factors remain to be identified.
Whereas most studies have investigated the effects of systemic administration of catecholamines and thereby mimicking a stress response, other reports focused on the ablation of catecholaminergic signaling and thus the endogenous role these hormones play in steady state. One study examined the consequence of unilateral surgical ablation of local nerves in mice upon leukocyte adhesion to innervated tissues such as bone marrow and skeletal muscle. Whilst leukocyte adhesion in nerve-intact organs showed a diurnal rhythm (high at night onset, lower during the day), this was abolished in denervated tissues. This pattern corresponded to a rhythmic expression pattern of ICAM-1 in mouse vascular endothelial cells, which was flattened after denervation (7). Rhythms in adherent leukocyte cell numbers were equally lost in mice lacking β2- or β3-adrenoceptors, indicating that rhythmic adhesion requires local delivery of adrenergic signals by nerves and that the microenvironment is an important regulator of leukocyte trafficking and target site of stress hormones. In murine lymph nodes, activation of β2-adrenoceptors leads to the retention of lymphocytes and therefore affects the extent of adaptive immune responses (11). Under steady state conditions, lymphocyte numbers in lymph nodes peak at night (11, 23, 29), which coincides with peak levels of noradrenaline in these tissues (11). After functional depletion of adrenergic nerves using a sympathetic neurotoxin (6-OHDA), restricted lymphocyte egress from the lymph node in the active phase of the animals was observed. The same group had previously demonstrated the physical interaction of β2-adrenoceptors with the chemokine receptors CCR7 and CXCR4, which are critically involved in lymphocyte homing to and their retention in murine lymph nodes (10). These data therefore provide evidence for an important time-of-day-dependent regulation of migratory factors on leukocytes and non-hematopoietic cells by β2-adrenergic signaling under homeostatic conditions.
Lack-of-function assays are also suited to tease apart the complex interplay of signaling pathways involved in the hormonal regulation of leukocyte migration. Previous data reported that adrenergic signaling through β3-adrenoceptors promotes rhythmic egress of hematopoietic stem cells from the mouse bone marrow via downregulation of the retention factor CXCL12 (2). Activation of β3-adrenoceptors during the day promotes egress from bone marrow, yet activation of β2- or β3-adrenoceptors at night promotes homing of murine leukocytes to tissues (2, 7). This apparent paradox was recently investigated in the context of cholinergic signaling, which is a potent inhibitor of endothelial activation in inflammatory scenarios (30) and part of the inflammatory reflex pathway (31, 32). Using mice with decreased cholinergic tone, García-García et al. found that during the day, acetylcholine inhibits vascular adhesion while noradrenergic signals promote egress via β3-adrenoceptors, providing complementary effects which increase leukocyte content in blood. At night, the higher circulating adrenaline levels preferentially stimulate β2-adrenoceptors while at the same time sympathetic cholinergic signals downregulate β3-adrenoceptor expression, promoting nocturnal homing (33). This series of studies highlights the complex interactions between different signaling pathways in vivo and the importance of considering neuroendocrine regulation of leukocyte trafficking in an integrative manner.
Glucocorticoids
The adrenal-derived steroid hormones (glucocorticoids and mineralocorticoids) are another significant class of stress hormones which influence leukocyte migration. Produced in the adrenal cortex, these hormones bind to their cognate receptors [glucocorticoid receptor (GR) and mineralocorticoid receptor (MR)] but with significant overlap. Whilst mineralocorticoids such as aldosterone can only bind MR, endogenous glucocorticoids such as cortisol (humans) and corticosterone (rodents) can bind both receptors. However, due to the higher affinity of endogenous glucocorticoids for MR, this receptor is favored at lower glucocorticoid concentrations and signaling via GR emerges at higher concentrations (34, 35). Appropriate balance between the MR/GR pathways is regulated by the 11β-hydroxysteroid dehydrogenase (11β-HSD) enzymes. 11β-HSD2 converts cortisol and corticosterone into inactive forms, effectively restricting MR signaling to mineralocorticoids in tissues where it is highly expressed, such as the kidney. On the other hand, 11β-HSD1, highly expressed in the liver, can “reactivate” these inactive compounds and locally increase glucocorticoid signaling [see (36) for a review of 11β-HSD functions]. With the DNA-binding domains of human GR and MR showing 94% identity (37), there is also a degree of commonality in their target genes and effects. Innate and adaptive immune cell populations express both MR [reviewed in (38)] and GR, although some sex-specific differences in GR expression levels and isoform distribution are reported in human cells (39). The use of specific, synthetic compounds is therefore more commonly employed to allow more refined investigations into the relative contributions of these pathways, as synthetic glucocorticoids such as dexamethasone show much higher affinity for GR than endogenous ligands (approx. 5-fold) and remain a major class of anti-inflammatory agents in clinical use.
Recently, Fay et al. investigated the influence of glucocorticoid administration on leukocyte demargination and found similar effects to that of catecholamines. Dexamethasone led to increased leukocyte numbers in the bloodstream of patients. In vitro experiments showed that dexamethasone increased granulocyte demargination independently of changes in vascular adhesion molecule expression. Although not to the same extent as adrenaline, in vitro dexamethasone treatment also induced changes to the actin cytoskeleton, leading to softening of granulocytes and enabling their detachment (3). In addition to effects on biophysical properties of leukocytes, glucocorticoids modulate expression of key receptors on leukocytes to influence maturation, homing, and egress. Neutrophil maturation is accelerated in rats treated with a GR antagonist (mifepristone/RU486) (13), an effect which may be attributable to reduced expression of Annexin A1. Annexin A1 is up-regulated by glucocorticoids, and circulating neutrophils from Annexin A1-deficient mice express higher levels of CXCR4, representing an ‘aged’ phenotype (12). Annexin A1−/− neutrophils did not migrate as efficiently as wild-type cells to CXCL12 in vitro, and stromal cells from Annexin A1−/− mice also produced less CXCL12 in vivo. The accelerated maturation and inability to home leads to persistent neutrophilia in these mice, and may be a route through which GR antagonism exerts its effects (12). Recent work has also shown this pathway to be involved in the redistribution of T cells (15, 23). In humans, GR antagonism using mifepristone affected T cell CXCR4 expression in a manner dependent on circulating cortisol levels. Using timed administration of mifepristone it was revealed that when endogenous cortisol was low, the GR antagonist increases CXCR4 expression on CD4+ and CD8+ subsets through a partial agonist effect, whereas administration when cortisol was high led to reduced CXCR4 expression by traditional antagonism (15). This axis has been more extensively investigated in mice, where GR agonism was shown to increase expression of the IL-7 receptor, which then drove increased CXCR4 expression when circulating glucocorticoids were high. Significantly fewer memory CD4+ T cells were observed in spleen, lymph node, and lungs of mice lacking GR in T cells than in wild type controls, suggesting that cell-intrinsic GR signaling enhances survival of this population and promotes migration to peripheral lymphoid tissues (23). Furthermore, MR signaling also increases CXCR4 expression on naïve human T cells but does so along with CD62L and CCR7, suggesting that MR activation facilitates homing to lymph nodes whereas GR activation preferentially drives cells toward the bone marrow (24). In an inflammatory scenario, glucocorticoid administration generally inhibits the immune response, as GR activation decreases expression of many pro-inflammatory cytokines. In a mouse model of LPS-induced inflammation, dexamethasone treatment also resulted in reduced leukocyte rolling flux, adhesion and emigration, along with reduced circulating leukocyte counts, whereas mifepristone treatment increased adhesion and emigration (14). These data show that GR agonism attenuates interactions between leukocytes and the endothelium in this model, consistent with dexamethasone-induced inhibition of ICAM-1 and VCAM-1 expression on the inflamed endothelium. Interestingly, the blockade of endogenous GR signaling by mifepristone resulted in a counter-intuitive decrease in VCAM-1 expression, suggesting that there may be a difference between endogenous and exogenous glucocorticoids and their effects on leukocyte-endothelium interactions (14). It will be interesting to see whether further studies can dissect the relative contributions of endogenous or exogenous glucocorticoids and their signaling through GR and/or MR.
In addition to sensitivity of adrenergic and glucocorticoid signaling to acute environmental signals and stressors, these signals are also regulated on a longer time scale by the circadian rhythm [see (40) for a review of circadian regulation of immune function]. Circulating glucocorticoids and adrenergic tone both increase at the start of an organism's behavioral active phase, providing a rhythmic signal promoting redistribution of leukocytes across the body. The influence of such rhythmic signal is seen in the results of Besedovsky et al. (15), where the diurnal oscillation in endogenous cortisol significantly influenced the ability of GR antagonism to elicit changes in human T cell CXCR4 expression. Shimba et al. (23) also addressed the role of GR in a rhythmic manner, supporting data by other groups (11, 29) and providing an additional mechanism to regulate leukocyte trafficking in a daily cycle. In inflammatory scenarios a rhythmic glucocorticoid signal is known to modulate chemokine signaling and neutrophil trafficking to the mouse lung via time-of-day dependent inhibition of epithelial CXCL5 production (16, 17), providing an additional layer of fine-tuning the inflammatory response. Under chronic stress conditions, however, elevated glucocorticoid levels are associated with a reduction in cellular sensitivity to these hormones. Experiments using rhesus macaques have illustrated a link between both nervous temperament and social stress and impaired leukocyte trafficking patterns (25, 26). Whilst control animals showed glucocorticoid-induced redistribution of circulating leukocytes, those exposed to social stress showed a reduced correlation between cortisol concentration and blood lymphocyte content (26). In further experiments without social manipulation but with analysis of behavior and temperament, the expected correlation between cortisol and blood neutrophil counts was found at a population level, but this was significantly attenuated in nervous macaques (25). These results have interesting implications for human scenarios of disrupted neuroendocrine functions, stress, and anxiety. In these situations, a disconnection appears between circulating hormone levels and inflammatory cell responsiveness, which may explain the lack of efficacy of glucocorticoid treatment in some patients. Furthermore, there may even be a cycle of inflammatory exacerbation due to the effects of stress upon monocyte trafficking and microglial activation, whereby reactive endothelium and enhanced trafficking of cells to the brain releases cytokines and reinforces stress- and anxiety-like signaling [reviewed in (41, 42)]. Similar to catecholamines, glucocorticoids are key systemic orchestrators of immune cell migration. Yet, due to the complexity of the interlocking signaling cascades in different leukocyte subsets and tissues, the precise effects in different sites of the body remain elusive.
Conclusion
In summary, the hormones adrenaline, noradrenaline and glucocorticoids, typically associated with a stress response, exert diverse effects on leukocyte migration under both steady-state and stimulated conditions. These effects are dependent not only on the responding cell type but also on location, duration and source of the stress/hormone signal, inflammatory context, and even time of day. Whereas adrenaline increases circulating neutrophil numbers, it reduces lymphocyte numbers in blood. Noradrenaline, on the other hand, increases both neutrophil and B cell numbers with distinct temporal profiles. Glucocorticoids can act to redistribute T cells from the bloodstream into organs at their endogenous peak levels, but synthetic agonists are widely used in inflammatory scenarios to inhibit chemokine production and disrupt excessive inflammatory responses. This potential divergence between the function of endogenous hormones and their clinical counterparts should be explored further, particularly with respect to cell-specific differences in receptor expression and diurnal rhythms in endogenous hormone concentrations. To achieve this, analyses using lineage specific ablation of hormone receptors will be needed in combination with well-controlled in vitro and in vivo studies to dissect their complex and highly interwoven signaling pathways and functions.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the German Research Foundation (DFG) (Emmy-Noether grant (SCHE 1645/2-1) and SFB914 project B09) and the DZHK (German Center for Cardiovascular Research) and BMBF (German Ministry of Education and Research), in addition to a European Research Council (ERC) starting grant (635872, CIRCODE) and IMPRS funding.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. (2010) 31:318–24. doi: 10.1016/j.it.2010.05.006
2. Méndez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature (2008) 452:442–7. doi: 10.1038/nature06685
3. Fay ME, Myers DR, Kumar A, Turbyfield CT, Byler R, Crawford K, et al. Cellular softening mediates leukocyte demargination and trafficking, thereby increasing clinical blood counts. Proc Natl Acad Sci USA. (2016) 113:1987–92. doi: 10.1073/pnas.1508920113
4. Margaryan S, Hyusyan A, Martirosyan A, Sargsian S, Manukyan G. Differential modulation of innate immune response by epinephrine and estradiol. Horm Mol Biol Clin Investig. (2017) 30:20160046. doi: 10.1515/hmbci-2016-0046
5. Herrera-García AM, Domínguez-Luis MJ, Arce-Franco M, Armas-González E, Álvarez de La Rosa D, Machado JD, et al. Prevention of neutrophil extravasation by α2-adrenoceptor–mediated endothelial stabilization. J Immunol. (2014) 193:3023–35. doi: 10.4049/jimmunol.1400255
6. Trabold B, Lunz D, Gruber M, Fröhlich D, Graf B. Immunomodulation of neutrophil–endothelial interaction by inotropes. Injury (2010) 41:1079–83. doi: 10.1016/j.injury.2010.05.034
7. Scheiermann C, Kunisaki Y, Lucas D, Chow A, Jang J, Zhang D, et al. Adrenergic nerves govern circadian leukocyte recruitment to tissues. Immunity (2012) 37:290–301. doi: 10.1016/j.immuni.2012.05.021
8. Grisanti LA, Gumpert AM, Traynham CJ, Gorsky JE, Repas AA, Gao E, et al. Leukocyte-expressed β2-adrenergic receptors are essential for survival after acute myocardial injury. Circulation (2016) 134:153–67. doi: 10.1161/CIRCULATIONAHA.116.022304
9. Flierl MA, Rittirsch D, Nadeau BA, Sarma JV, Day DE, Lentsch AB, et al. Upregulation of phagocyte-derived catecholamines augments the acute inflammatory response. PLoS ONE (2009) 4:e4414. doi: 10.1371/journal.pone.0004414
10. Nakai A, Hayano Y, Furuta F, Noda M, Suzuki K. Control of lymphocyte egress from lymph nodes through β2-adrenergic receptors. J Exp Med. (2014) 211:2583–98. doi: 10.1084/jem.20141132
11. Suzuki K, Hayano Y, Nakai A, Furuta F, Noda M. Adrenergic control of the adaptive immune response by diurnal lymphocyte recirculation through lymph nodes. J Exp Med. (2016) 213:2567–74. doi: 10.1084/jem.20160723
12. Machado ID, Spatti M, Hastreiter A, Santin JR, Fock RA, Gil CD, et al. Annexin A1 is a physiological modulator of neutrophil maturation and recirculation acting on the CXCR4/CXCL12 pathway. J Cell Physiol. (2016) 231:2418–27. doi: 10.1002/jcp.25346
13. Cavalcanti DMH, Lotufo CMC, Borelli P, Ferreira ZS, Markus RP, Farsky SHP. Endogenous glucocorticoids control neutrophil mobilization from bone marrow to blood and tissues in non-inflammatory conditions. Br J Pharmacol. (2007) 152:1291–300. doi: 10.1038/sj.bjp.0707512
14. Gregory JL, Hall P, Leech M, Morand EF, Hickey MJ. Independent roles of macrophage migration inhibitory factor and endogenous, but not exogenous glucocorticoids in regulating leukocyte trafficking. Microcirculation (2009) 16:735–48. doi: 10.3109/10739680903210421
15. Besedovsky L, Born J, Lange T. Endogenous glucocorticoid receptor signaling drives rhythmic changes in human T-cell subset numbers and the expression of the chemokine receptor CXCR4. FASEB J. (2014) 28:67–75. doi: 10.1096/fj.13-237958
16. Gibbs JE, Ince L, Matthews L, Mei J, Bell T, Yang N, et al. An epithelial circadian clock controls pulmonary inflammation and glucocorticoid action. Nat Med. (2014) 20:919–26. doi: 10.1038/nm.3599
17. Ince LM, Zhang Z, Beesley S, Vonslow RM, Saer BR, Matthews LC, et al. Circadian variation in pulmonary inflammatory responses is independent of rhythmic glucocorticoid signaling in airway epithelial cells. FASEB J. (2019) 33:126–39. doi: 10.1096/fj.201800026RR
18. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) 7:678–89. doi: 10.1038/nri2156
19. Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve - an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. (2000) 52:595–638.
20. Dhabhar FS, Malarkey WB, Neri E, McEwen BS. Stress-induced redistribution of immune cells–from barracks to boulevards to battlefields: a tale of three hormones. Psychoneuroendocrinology (2012) 37:1345–68. doi: 10.1016/j.psyneuen.2012.05.008
21. Scanzano A, Schembri L, Rasini E, Luini A, Dallatorre J, Legnaro M, et al. Adrenergic modulation of migration, CD11b and CD18 expression, ROS and interleukin-8 production by human polymorphonuclear leukocytes. Inflamm Res. (2015) 64:127–35. doi: 10.1007/s00011-014-0791-8
22. Kim M-H, Gorouhi F, Ramirez S, Granick JL, Byrne BA, Soulika AM, et al. Catecholamine stress alters neutrophil trafficking and impairs wound healing by β2-adrenergic receptor-mediated upregulation of IL-6. J Invest Dermatol. (2014) 134:809–17. doi: 10.1038/jid.2013.415
23. Shimba A, Cui G, Tani-ichi S, Ogawa M, Abe S, Okazaki F, et al. Glucocorticoids drive diurnal oscillations in T cell distribution and responses by inducing interleukin-7 receptor and CXCR4. Immunity (2018) 48:286–98.e6. doi: 10.1016/j.immuni.2018.01.004
24. Besedovsky L, Linz B, Born J, Lange T. Mineralocorticoid receptor signaling reduces numbers of circulating human naïve T cells and increases their CD62L, CCR7, and CXCR4 expression. Eur J Immunol. (2014) 44:1759–69. doi: 10.1002/eji.201344265
25. Capitanio JP, Mendoza SP, Cole SW. Nervous temperament in infant monkeys is associated with reduced sensitivity of leukocytes to cortisol's influence on trafficking. Brain Behav Immun. (2011) 25:151–9. doi: 10.1016/j.bbi.2010.09.008
26. Cole SW, Mendoza SP, Capitanio JP. Social stress desensitizes lymphocytes to regulation by endogenous glucocorticoids: insights from in vivo cell trafficking dynamics in rhesus macaques. Psychosom Med. (2009) 71:591–7. doi: 10.1097/PSY.0b013e3181aa95a9
27. Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology. Neuroimmunomodulation (2009) 16:300–17. doi: 10.1159/000216188
28. Scanzano A, Cosentino M. Adrenergic regulation of innate immunity: a review. Front Pharmacol. (2015) 6:171. doi: 10.3389/fphar.2015.00171
29. Druzd D, Matveeva O, Ince L, Harrison U, He W, Schmal C, et al. Lymphocyte circadian clocks control lymph node trafficking and adaptive immune responses. Immunity (2017) 46:120–32. doi: 10.1016/j.immuni.2016.12.011
30. Saeed RW, Varma S, Peng-Nemeroff T, Sherry B, Balakhaneh D, Huston J, et al. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J Exp Med. (2005) 201:1113–23. doi: 10.1084/jem.20040463
31. Rosas-Ballina M, Olofsson PS, Ochani M, Valdés-Ferrer SI, Levine YA, Reardon C, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science (2011) 334:98–101. doi: 10.1126/science.1209985
32. Tracey KJ. Understanding immunity requires more than immunology. Nat Immunol. (2010) 11:561–4. doi: 10.1038/ni0710-561
33. García-García A, Korn C, García-Fernández M, Domingues O, Villadiego J, Martín-Perez D, et al. Dual cholinergic signals regulate daily migration of hematopoietic stem cells and leukocytes. Blood (2018). doi: 10.1182/blood-2018-08-867648. [Epub ahead of print].
34. Fuller PJ, Lim-Tio SS, Brennan FE. Specificity in mineralocorticoid versus glucocorticoid action. Kidney Int. (2000) 57:1256–64. doi: 10.1046/J.1523-1755.2000.00959.X
35. Reul JMHM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology (1985) 117:2505–11. doi: 10.1210/endo-117-6-2505
36. Chapman K, Holmes M, Seckl J. 11β-Hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev. (2013) 93:1139. doi: 10.1152/PHYSREV.00020.2012
37. Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, et al. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science (1987) 237:268–75. doi: 10.1126/science.3037703
38. Bene NC, Alcaide P, Wortis HH, Jaffe IZ. Mineralocorticoid receptors in immune cells: emerging role in cardiovascular disease. Steroids (2014) 91:38–45. doi: 10.1016/j.steroids.2014.04.005
39. Lu KD, Radom-Aizik S, Haddad F, Zaldivar F, Kraft M, Cooper DM. Glucocorticoid receptor expression on circulating leukocytes differs between healthy male and female adults. J Clin Transl Sci. (2017) 1:108–14. doi: 10.1017/cts.2016.20
40. Scheiermann C, Gibbs J, Ince L, Loudon A. Clocking in to immunity. Nat Rev Immunol. (2018) 18:423–37. doi: 10.1038/s41577-018-0008-4
41. Weber MD, Godbout JP, Sheridan JF. Repeated social defeat, neuroinflammation and behavior: monocytes carry the signal. Neuropsychopharmacology (2017) 42:46–61. doi: 10.1038/npp.2016.102
Keywords: catecholamine, glucocorticoid, adrenergic signaling, neutrophil, lymphocyte, circadian rhythm
Citation: Ince LM, Weber J and Scheiermann C (2019) Control of Leukocyte Trafficking by Stress-Associated Hormones. Front. Immunol. 9:3143. doi: 10.3389/fimmu.2018.03143
Received: 04 October 2018; Accepted: 19 December 2018;
Published: 11 January 2019.
Edited by:
Andres Hidalgo, Centro Nacional de Investigaciones Cardiovasculares (CNIC), SpainReviewed by:
Simon Mendez-Ferrer, University of Cambridge, United KingdomKrisztina Káldi, Semmelweis University, Hungary
Copyright © 2019 Ince, Weber and Scheiermann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Louise M. Ince, bG91aXNlLmluY2VAdW5pZ2UuY2g=
Christoph Scheiermann, Y2hyaXN0b3BoLnNjaGVpZXJtYW5uQG1lZC51bmktbXVlbmNoZW4uZGU=
†These authors have contributed equally to this work