 Oskar A. Haas
Oskar A. Haas- Department of Clinical Genetics, Children's Cancer Research Institute, Vienna, Austria
Common understanding suggests that the normal function of a “healthy” immune system safe-guards and protects against the development of malignancies, whereas a genetically impaired one might increase the likelihood of their manifestation. This view is primarily based on and apparently supported by an increased incidence of such diseases in patients with specific forms of immunodeficiencies that are caused by high penetrant gene defects. As I will review and discuss herein, such constellations merely represent the tip of an iceberg. The overall situation is by far more varied and complex, especially if one takes into account the growing difficulties to define what actually constitutes an immunodeficiency and what defines a cancer predisposition. The enormous advances in genome sequencing, in bioinformatic analyses and in the functional in vitro and in vivo assessment of novel findings together with the availability of large databases provide us with a wealth of information that steadily increases the number of sequence variants that concur with clinically more or less recognizable immunological problems and their consequences. Since many of the newly identified hard-core defects are exceedingly rare, their tumor predisposing effect is difficult to ascertain. The analyses of large data sets, on the other hand, continuously supply us with low penetrant variants that, at least in statistical terms, are clearly tumor predisposing, although their specific relevance for the respective carriers still needs to be carefully assessed on an individual basis. Finally, defects and variants that affect the same gene families and pathways in both a constitutional and somatic setting underscore the fact that immunodeficiencies and cancer predisposition can be viewed as two closely related errors of development. Depending on the particular genetic and/or environmental context as well as the respective stage of development, the same changes can have either a neutral, predisposing and, in some instances, even a protective effect. To understand the interaction between the immune system, be it “normal” or “deficient” and tumor predisposition and development on a systemic level, one therefore needs to focus on the structure and dynamic functional organization of the entire immune system rather than on its isolated individual components alone.
Introduction
The neoplastic transformation of cells and their subsequent successful clonal expansion and progression into clinically apparent hematologic malignancies and solid tumors is a complex multifactorial process. On the one hand, this process requires changes in the respective cells' genetic program that modify their metabolism and performance and consequently alter their normal differentiation, replicative, and survival capacity. On the other hand, these cells have to learn to adapt themselves and to exploit external deterministic physiological stimuli as well as to flexibly react to a plethora of stochastic environmental challenges (1, 2). This, in turn, defines their capability to achieve successful interactions with and survival strategies within their normal surrounding tissue. With its interactive network of cells, humoral factors, and cytokines, the immune system plays a fundamental role in the recognition of and protection against any internal or external threads, be it abnormal cells, foreign tissues or infections agents. Inborn genetic defects or dysfunctions of the one or the other immune system components may thus unsettle the intricate physiological balance and maintenance of a body's functional homeostasis and thereby diminish its preventive capability or even promote the formation of neoplastic diseases in a proactive manner.
The recent methodological advances in deciphering the composition and structure of the human genome allow us now to identify virtually any DNA sequence alterations in a hitherto unimaginable fast and detailed manner. Various such technologies have in the meantime become invaluable diagnostic mutation screening tools that help to identify clear-cut disease-associated genetic defects in inborn errors of the immune system but also more elusive variants that may participate in the predisposition to malignant diseases in children. These developments are addressed in a large number of original publications as well as in many excellent reviews of these subjects (3–16). Rather than reiterating what has already extensively been written about, I intend to provide a more conceptional framework of this subject and focus my attention on often neglected and less well-appreciated fundamental facts and phenomena, which I consider particular relevant for an in-depth appreciation and understanding of this topic and which I will supplement with some specific examples that illustrate the developments and progress in this field.
To begin with, we first need to (re)define the current view and understanding of “primary immunodeficiency” as well “genetic predisposition and susceptibility.”
Primary Immunodeficiency Syndromes (PID)
The immune system is composed of highly specialized cells, tissues, organs and soluble factors that interact in a complex way to ensure an organism's immune defense. According to the current definition, PID are thus a group of diseases, which are caused by heritable DNA sequence alterations that impair the quantitative or qualitative function of cellular or humoral components of the adaptive or innate immune system (17). The spectrum of their clinical, often intimately interrelated symptoms includes developmental disorders, autoinflammation, chronic inflammation, autoimmunity, neoplasms as well as serious, recurrent, or unusual infections (18, 19). Initially, the diagnosis of these conditions was based on abnormal laboratory parameters and clinical problems, in particular recurrent, severe or unusual infections that in certain groups of patients occasionally concurred with familial clustering, syndromic features, radiation sensitivity and also a certain propensity to develop particular types of malignancies. With the advent of in vitro testing and immunophenotyping technologies, it became possible to better define and differentiate certain categories as well as to characterize even subtle cellular and humoral functional deviances already to a certain extent. In the early days of the molecular genetic era, the respective responsible genes were then identified in cases with highly penetrant genetic traits, which instigated a first, albeit restricted diagnostic mutation screening. With the introduction of more sophisticated sequencing technologies, the discovery of causative genetic defects increased steadily in parallel with the refined dissection, delineation, and definition of such immunodeficiency syndromes. The recent 2017 update of the “Primary Immunodeficiency Committee” of the “International Union of Immunological Societies” thus recognizes 344 genetic defects that define 354 distinct disorders of immunity in nine categories (20, 21). Some of these monogenetic disorders are extremely rare and were so far identified in single families only.
This compilation together with the commonly unconsidered use of the term PID leaves the impression that one indeed knows what the term PID stands for. It is therefore intriguing to note and especially important to point out that there is actually no clear consensus about its definition (22). The reason for this now newly flaring-up debate is the recognition that the perception of immunodeficiency has so far clearly focused only on the most obvious and clinically striking disorders in both adaptive and innate immunity that affect the lympho- and hematopoietic system. With the increasing appreciation that also non-hematopoietic cells and tissues participate in a significant manner in the immune defense this view is currently changing and necessitates an expansion of this concept. For instance, keratinocytes, endothelial cells, and fibroblast secrete as much and as many cytokines as hematopoietic cells do and can thus use their intrinsic pathways for protection against infectious agents also in a similar fashion. Another example are neurons and oligodendrocytes, which are similar essential and sufficient guardians against herpes simplex virus I and probably also other infection agents (22).
Another development that one has to consider in this context are the results that derive from the increasingly sophisticated diagnostic work-up of suspicious cases with technologies that enable nowadays the recognition of even clinically not readily apparent quantitative and qualitative deviations of particular cellular and humoral immune system components. As can be appreciated already in a normal setting, such differences are commonly due to and thus correlate with variations on the sequence level, either in form of single nucleotide polymorphisms/(SNP) alone or in form of definable haplotypes, which can make it more and more difficult to define a physiological norm and, under particular settings, a clear disease-relevant pathological state (23–32). One of the best documented and therefore most instructive example is the context-dependent implications of the highly variable serum levels of the mannan-binding lectin (MBL), the apparently most common deficiency of a humoral component of the innate immune system (33). The respective gene contains 87 different polymorphic sites with a multitude of possible combinations, of which seven common haplotypes stand out. These haplotypes determine the serum levels as well as the configuration and function of the encoded proteins in a predictable manner, although the possible effects are co-determined by the sex and age of the respective carrier, hormonal changes and immune system activation. Moreover, the frequency of the diverse haplotypes varies world-wide in an ethnicity specific manner. Thus, although the magnitude of particular MBL protein levels are clearly recognizable and determined by genetic factors, the ensuing effects, whether a low or high level becomes detrimental or beneficial or whether it remains irrelevant, are strictly context-dependent and therefore difficult to predict or interpret in a given individual.
Based on an estimate that ~5% of all genes participate in one or the other way in host defense and immune tolerance, it was predicted that with the new sequencing technologies up to 3,000 PIDs will be identified by the year 2021. Even if one considers only a still monogenic scenario with only two types of alleles per locus (i.e., heterozygous vs. homozygous, loss-of function vs. gain-of-function, hypomorphic vs. amorphic as well as various other variations), it is hardly imaginable that one will be able to functionally evaluate and analyze the magnitude of all the possible outcomes in a reasonable manner to make some sense of the ensuing (patho)physiological effects even in an perhaps otherwise well-defined setting (22, 34).
In the end, these reflections leave us with the question how one actually will define primary immunodeficiencies in the future. When, to which extent and in which form do they need to manifest themselves clinically? Will it be sufficient to just view them as pure monogenic disorders or does one eventually also need to consider the contribution of modifying gene, signaling pathway, and cellular networks in a much stronger way?
Genetic Predisposition and Susceptibility
The concept of genetic predisposition and susceptibility, which so far was also based primarily on the clinical perception of disease and inheritance patterns, experiences nowadays a similar reinterpretation and paradigm shift as the one of immunodeficiency. The emergence and continuous improvement of fine-scale and cost-efficient targeted, whole exome and whole genome, methylation as well as RNA sequencing approaches, increase the possibilities to investigate the genetic background of heritable and acquired diseases in a previously unprecedented manner (6, 10). Not only has it become much easier to screen all the eligible genes of already well-recognized conditions for causative mutations, it has also become much simpler to identify novel sequence abnormalities in rare, unusual, or merely suspected cases of immunodeficiencies and cancer predisposition. Thus, the special choice of the appropriate mode to search for and ascertain such genetic factors remains nowadays a matter of intention, clinical opportunities and individual demands that, in particular, is based on patient/family-relevant, gene-related, disease-associated, or population-based aspects (30). In a clinical setting, the direct patient-orientated approach is definitely the most important one. The ascertainment of an inborn genetic cause of a particular disease requires not only its appropriate work-up with the best-fitted mutation screening method but also the careful justification of its significance through the assessment of medical records and family history as well as the clinical and laboratory data of an affected patient (7, 13, 14, 34–47). Especially in those cases in which a cancer-prone condition is recognized already before the onset of a malignant disease, securing the specific genetic cause is essential to guide the necessary clinical measures, such as an appropriate treatment and surveillance program together with a suitable genetic counseling (7, 13, 14, 34–47). However, in many instances a potential predisposing germ-line alteration may only be suspected and searched for at the time a malignant disease is diagnosed. Especially if one screens the neoplastic tissue for disease-specific diagnostic alterations, one cannot avoid coming across inborn genetic errors, not only those in already known genes but occasionally also in novel ones. Distinguishing somatic from inherited defects in tumor tissue alone may turn out quite difficult because both types often affect the same genes, a fact that necessitates the verification of the inborn nature of any such changes by analyzing germ line material in addition. Based on such experiences, it is therefore becoming practice to screen for underlying germ-line defects in a more systematic fashion in form of so called “trio analyses,” which in addition to a patient's tumor and germ line DNA also requires the parental ones for comparison (4, 13, 34, 35). An increasing number of publications confirm that this approach is particular rewarding, not only for the sake of the patient and her family but also for scientific reasons. In case a particular gene defect is not already clearly indicative of a specific type of predisposing condition, it may be difficult or virtually impossible to decide whether concomitant immune system derangements at diagnosis are actually the cause or the effect of the respective disease. As I will point out later, more subtle predisposing gene alterations that merely modify the function of a gene, such as single nucleotid polymorphisms (SNPs), may not even exert any easily recognizable effects prior to onset of the malignant disease. Predisposing SNPs were originally discovered by large-scale genome wide association studies (GWAS) in regions of the genome, which are linked with particular disease traits. The biological relevance and functional consequences of some of these variants has in the meantime already been established and confirmed with appropriate experiments and test systems (23, 26, 48, 49).
Our current knowledge of the genetic basis of immunodeficiency and tumor predisposition is primarily based on monogenic disorders. We learned to appreciate the genetic heterogeneity of these conditions, meaning that single or similar phenotypes can be generated by different genetic mechanisms. Polygenic diseases, on the other hand, are caused by the joint contribution of several independent acting or interacting genes, whose individual contribution might be small or even unnoticeable. GWAS together with WGS studies have now allowed us to extend such analyses to the entire genome in a kind of omnigenic approach, which means that we will need to learn to cope with the combinatorial effects of a large number of genetic variants, whose individual contribution is not readily apparent (50). In contrast to Mendelian diseases, which are primarily caused by mutations in the protein-coding part of the genome, complex traits are mainly driven by non-coding variants that presumably affect regulatory elements of genes, such as promoters and enhancers. For instance, risk variants for autoimmune diseases show particular enrichment in active chromatin regions of immune cells (51–53).
The “omnigenic” model still accepts that only a modest number of “core genes” or pathways are etiologically important for a specific disease and their dysfunction will still have the strongest impact on the disease process (54). However, in this situation the particular risk will be driven by an accumulation of weak and heterogeneous effects of many modifying gene variants, whose specific configuration might even only become relevant in certain cell types and tissues, whereas in others they might remain completely inconsequential (51). The ultimate and most provocative conclusion and interpretation of this “omnigenic” model is of course that virtually any variant with regulatory effects in a given tissue is likely to have some (weak) effects on all diseases that are modulated through this particular tissue (51).
Whereas the identification of risk factors in monogenic diseases requires sequencing of specific genes and the careful functional assessment of any unusual sequence variant that pops up, polygenic risk scores of common diseases are statistically determined likelihoods that are calculated from genome-wide SNP patterns. Given the countless possibilities how defective and normal but functionally dissimilar allele variants of one or multiple genes can be combined and co-inherited, it is therefore astonishing that, as reported recently by Khera et al., the risk scores of such common diseases may under particular circumstances nevertheless reach at least the same magnitude as the ones achieved in monogenic diseases (55). Together with the cell- and tissue-specific utilization of the ensuing gene products, these findings provide a ready explanation for the highly variable penetrance of genuine gene defects and, even more so, for functionally modifying variants and, not least, why it is so difficult to foresee their biological consequences even in monogenic disorders (34). In addition, one has of course also to keep in mind that even in instances with a strong predisposing genetic component, the development of malignancies is always a multifactorial process that not only requires a liable genetic architecture but also some probabilistic elements as well as the participation and interaction of a multitude of other intrinsic and extrinsic factors and mechanisms. In case of hematologic malignancies, such cell-intrinsic defects and abnormalities consist of those that affect (I) (lympho- and hematopoietic stem) cell development, differentiation and apoptosis; (II) lymphocyte signaling, cytoskeleton, cytotoxicity and metabolism and (III) chromosome stability as well as DNA repair (3). Cell-extrinsic factors, on the other hand, comprise chronic inflammation; autoimmune- and autoinflammatory diseases, chronic (viral) infections and an impaired tumor surveillance (Figure 1).
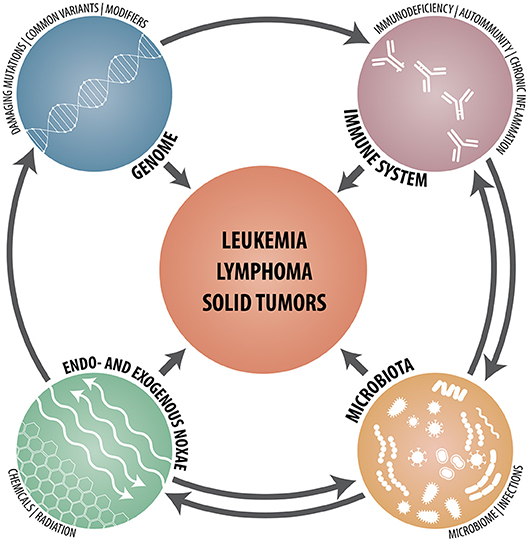
Figure 1. Schematic synopsis of the various genetic, immunological, microbial, and environmental constituents that contribute to and participate in the development of hematologic neoplasms, lymphomas and solid tumors.
Somatic Mutation (SMT) vs. Tissue Organization Field (TOFT) Theory
SMT and TOFT are two apparently competing theories of cancer development. The SMT postulates that cancer is a molecular, gene-based disease that derives from single cells whose autonomous and unrestrained proliferation is driven by the progressive accumulation of accidental and essentially unrelated events (2, 56–58). The TOFT, on the other hand, posits that cancer develops as an adaptive, emergent phenomenon whose fundamental determinants act at the level of tissue and organ homeostasis. In this scenario, inherent genetic constituents as well as a variety of physical, chemical and biological agents, such as cytokines, viruses, chemicals and/or radiation, perturb the functional interaction of diverse cellular modules and subsequently also the organizational state of tissues themselves (58). To a certain extent this process resembles morphogenesis and as such also replicates a tumor's capability to continuously balance novelty with stability and to combine plasticity with robustness (57, 59). The reductionist, bottom-up approach of SMT and the emergentist, top-down approach of TOFT are often considered incompatible because they view the problem from two different levels of biological complexity. The probably smallest and already generally agreed-upon common centerpiece where these two opinions meet is the tissue micro-environment (57).
Compared to the possibilities of compact tissues, the various closely interconnected humoral and cellular components of the immune system are in a unique situation, because they can exert their action not only in a local microenvironment, but they can also act over and cover the macroenvironment of tissues, organs and even an entire organism in a systemic fashion. Moreover, within a particular context and a respective cellular milieu, components of the immune system can either foster or suppress tumor development. It is thus not surprising that the highly flexible and adaptable immune system, be it normal, impaired, or defective, is one of the major players in the game of tumor predisposition and development (60–64).
The three prevalent and often closely connected complications of PIDs are thus infections, autoimmunity, and malignancies. Nevertheless, it is intriguing to note that except for a few distinct disorders, such as Nijmegen breakage syndrome (NBS), Ataxia telangectasia (AT), and autoimmune lymphoproliferative syndrome (ALPS)-related autoimmune diseases, PIDs do not cluster with malignancies in the human diseaseome network (60, 65). Most of the available information regarding cancer risk derives from specific subtypes that result from defects in genes that regulate DNA repair, cell cycle, apoptosis, bone marrow maturation as well as those that help to protect against virus infections (17, 60). As might be expected, the most common overall hitherto documented forms of malignancies in all these conditions are lymphomas, whereas other neoplasms occur predominantly in a more disorder-typical and -constrained manner (7, 14, 17, 38, 60, 66–72).
“Environment”
Multicellular organisms are organized in a modular fashion with distinct functional units and compartments. Based on the particular level of organization one can thus distinguish various internal as well as external forms of environment. From the perspective of cells, for instance, such environmental shells may constitute specific niches, tissues, organs and the entire organism. The environment of a developing fetus, on the other hand, is provided by the mother. Following birth, the organism becomes embedded in a milieu of beneficial as well as latent pathogenic microorganism and is then openly exposed to the potentially damaging biological, physical, and chemical agents of the outer world, with which it has then to interact in various ways.
Fetal-Maternal Immune System Interactions
With regard to immunology, pregnancy is a particular interesting condition because it requires the constructive co-existence of two genetically and immunological distinct individuals within a single body (73, 74). To succeed, this endeavor requires the beneficial cooperation of a fully developed, but during this period dampened, immune system with a just evolving one that still has to mature and achieve its independence. This interaction requires the temporary reorganization and adaptation of the maternal immune system as well as the acceptance of assistance and cooperation of the fetal one. Maternal immune cells therefore help to “teach” those of the fetus to balance the need of self-defense against that of immune tolerance: too much restraint would lead to lethal infections, whereas too little would lead to autoimmunity (75–77). During this especially vulnerable phase this intricate balance can easily be disturbed, in particular by both cell intrinsic (genetic) as well as cell extrinsic biological factors.
Although a fetus expresses genetically foreign paternal antigens, it coexists in harmony inside the mother because it resides in an immune-privileged cocoon (78). Nevertheless, maternal and fetal cells still traffic through the placenta during the entire gestation period. After birth, surviving cells become then the source of a lifelong micro-chimerism in the corresponding opposite bodies, a phenomenon that under particular circumstances may significantly impact the future life of the mother as well as the child in various positive or negative ways (78, 79). Changes in the number, phenotype or distribution of microchimeric cells, for instance, can have an effect on immune surveillance, tissue repair, autoimmune diseases and tumorigenesis. It has thus been suggested that microchimeric cells may modulate health and disease in a similar way as commensal microorganisms control the susceptibility to various immunological and non-immunological disorders (78, 80).
One of the striking pathological consequences of the bidirectional cell trafficking are particular forms of SCID, in which the accumulation of a significant number of maternal T cells can cause a kind of graft-versus-host disease (GVHD) in the immune incompetent child (81, 82). However, even asymptomatic infiltrations of maternal cells are still an independent predictor for the development of GVHD later in life in case of transplantations (82). Conversely, maternal micro-chimerism in cord blood can mediate a graft-versus-leukemia effect in cord blood transplantation (83). Finally, there are also rare instances of maternal-fetal transmissions of malignancies, such as has been reported for lymphoma, leukemia and melanoma (84).
Within the setting of such pre- and postnatal fetal/maternal immune system interactions, the human leukocyte antigen (HLA) system and, in particular, its paternal component plays a particular important role (78). Exposure to inherited paternal antigens as well as non-inherited maternal antigens during pregnancy can lead to either immunization or tolerization, the sequelae of which can have even consequences decades later, not least in form of an alloimmune response in case of transplantation (85).
HLA System
The HLA system is part of the human major histocompatibility complex (MHC), a region on the short arm of chromosome 6 with some 260 genes that are involved in the immune response. Unsurprisingly, this is also the region of the genome that is associated with the greatest number of diseases with an immune system component, including those “bare lymphocyte” SCID disorders that are caused by deleterious mutations in certain MHC genes (86–88). As one of the main players in immune system interactions, the HLA system orchestrates the induction, regulation and fine-tuning of immune reactions and, in particular, the selection of the T cell repertoire (89). It is highly polymorphic and comprises more than 15,000 allelic variants (86).
Although specific HLA genotypes do not per se predispose to any particular disease in the strict direct sense, they are still highly enriched in and closely associated with distinct forms of inflammatory, autoimmune, and malignant disorders, a fact that not only underlines the central position of this regulatory system in their pathogenesis but also somehow links these otherwise disparate diseases.
HLA genotype patterns are not only associated with distinct sub-types of leukemias and lymphomas, but they can even correlate to some extent with the prognosis and survival of the respective diseases (90, 91). This evidence derived originally from the atypical HLA segregation patterns in leukemic families. They revealed an increase in HLA-identical non-affected sibs, HLA homozygosity, and identical disease-related maternal class II DRB1 haplotypes (90). Together this data is also taken as an indication that ALL is a problem that results from a population level response of HLA to infectious disease. In other words, overrepresented HLA haplotypes can provide some valuable insights into gene environment interactions as well as why and how particular clones are selected that will eventually produce the leukemic cell mass (88, 92–94). According to Greaves, B cell precursor (BCP) ALL evolves in two discrete steps, the in utero formation of a preleukemic clone, which is then triggered by a delayed abnormal immune response to common infections, followed by its postnatal conversion to overt leukemia (95–97). In the absence of any direct evidence for a specific causative infectious agent, the respective HLA pattern can thus be used as an indirect proxy measures for a genetically directed immune response and can thereby deliver valuable clues for the involved mechanisms. Based on such investigations, Taylor et al. concluded that BCP ALL is an indirect outcome of a transient auto-immune induced inflammatory molecular mimicry reaction that in turn may also explain why this subtype appears to be associated with delayed infection (88, 92, 93). Other factors that can help tumor cells to escape immune surveillance include somatic mutations that result in structural and functional changes in HLA system components, loss of expression of tumor antigens, lack of co-stimulatory molecules, and production of immunosuppressive cytokines (86). However, even more intriguing is the recent discovery that the HLA class I genotype is also participating in sculpting the entire oncogenic mutation landscape of a neoplasm (98). This is achieved through the continuous elimination of tumor cells with mutations that primarily produce strong antigens, which leads to a selection of cells with mutations that avoid producing such neoantigens (98).
Since the first hit that initiates the formation of the majority of pediatric cancers and leukemias occurs already very early in fetal life, the largest part of their further development still takes place in utero (95–97). It is therefore conceivable that fetal/maternal interactions and, in particular, the maternal immune system can influence and modify also the course of the disease in the one or the other way. Because one cannot study these processes directly, one has to rely on the above discussed traces and patterns that remain imprinted especially in the child's immune system after birth and which at least can provide some indirect clues about what had happened prenatally. The essential role of the in utero environment is further underlined by the fact that in individuals with a preexistent germline predisposition, secondary leukemia-promoting mutations can only evolve in special niches during distinct stages of organ development. In Down syndrome, for instance, GATA1 mutations, which emerge exclusively during fetal liver hematopoiesis, cause an accumulation of immature erythro-megakaryocytic precursor cells (99–102). After birth, this pseudo- or preleukemic cell population is usually uncapable to maintain itself any longer and usually collapses within a short period. Although such spontaneous postnatal regressions of embryonic malignancies are quite common, it is not yet clear, whether at all or to which extent this phenomenon can also be attributed, in the strictest or in a broader sense, to the loss of the fetal/maternal interaction or altered activity of the newborns immune system (68, 103).
Disorders of the DNA Repair System
Embryonic malignancies as well as those associated with PIDs are similar unfortunate byproducts of the complex processes that control normal development (102, 104–106). Environmental triggers such as carcinogenic pollutants and radiation play in general only a subordinate pathogenetic role in the development of childhood malignancies (105, 106). Such factors become primarily relevant only in those PID forms that are due to some types of DNA repair deficiencies (60, 71, 107–112). One of the physiological tasks of this system is to orchestrate processes, such as V(D)J recombination, class switch recombination, and somatic hypermutation, which together generate those lymphocyte-specific reorganizations that provide the basis for the adaptive immune system's genetic diversity (71, 108). Therefore, immune deficient patients with a dysfunctional DNA repair, such as those with an AT, NBS, and Bloom syndrome, are prone to develop lymphomas, whereas those with a dysfunctional DNA repair but without immune deficiency, such as xeroderma pigmentosum, Fanconi anemia, Werner syndrome, and Rothmund–Thomson syndrome, will primarily develop other forms of cancers (71). These occur especially in organs with rapidly dividing cells and/or an increased metabolic activity, including the brain, skin, breast, and the gastrointestinal tract. Since particular DNA repair defects produce characteristic mutation patterns and predispose to specific tumor forms in PID patients, it is in turn even possible to infer already from such indicators which DNA recombination processes are impaired (71).
Patients with constitutional mismatch repair deficiency (CMMRD) are prone to develop gastrointestinal, genitourinary and brain tumors, lymphomas, and leukemias (38, 39, 46, 47, 109, 113–116). They may also develop antibody deficiencies of variable severity, although they are neither a constant nor obligatory feature and usually also have no clinical correlate (107).
Genetic defects that disrupt the normal function of the DNA nucleotide excision repair (NER) complex cause at least eight overlapping phenotypes, such as xeroderma pigmentosum (XP), Cockayne syndrome (CS), and trichothiodystrophy (TTD) (117, 118). NER is responsible for fixing UV-induced lesions, bulky chemical adducts and some forms of oxidative damage (118). The respective complex comprises at least 30 proteins, three of which (XPB, XPD, and TTDA) are also part of the basal transcription factor TFIIH. The most interesting and intriguing of these proteins is XPD, because, as Lehman pointed out so suitably, it is one gene with two functions (DNA repair and transcription) and three diseases (XP, CS, and TTD) (117). Some of the clinical features of these syndromes are quite similar but some are also markedly different (119). Although all three syndromes have an exceptionally sun-sensitive skin, cancers develop only in patients with XP but not in those with CS and TTD (118). CS and TTD, on the other hand, have neurodevelopmental abnormalities and the latter also ichthyosis and brittle hair. To explain this conflicting and somehow mysterious genotype-phenotype discrepancies, Bootsma and Hoeijmakers proposed already quite some time ago that XP would result, if the defect would concern the DNA repair but not the transcriptional function of the complex, whereas, vice versa, the developmental TTD-associated problems would arise, if only the transcriptional part would be affected (120).
DNA repair defects may also impair the formation and production of antibodies, which is the defining feature of CVID (19, 121–127). Patients with such a dysfunctional humoral immunity have an increased probability to develop extra-nodal non-Hodgkin B cell and mucosa-associated lymphomas as well as epithelial tumors of the stomach, breast, bladder, and cervix (70, 128–130). In contrast to most PIDs, CVID-associated lymphomas are more common in older people and usually EBV-negative (70, 128–130). Selective IgA deficiency, in particular, goes along with a 7- to 10-fold increase in gastric adenocarcinomas. This risk is most likely related to an inability to clear Helicobacter pylori infections and appears to decrease when these bacteria are eradicated (70, 129, 130).
Immunoediting
The immune system is in many, apparently paradoxical ways involved in the manifestation and evolution of malignancies. It can facilitate cellular transformation as well as prevent, promote, control and thus shape their development, phenomena that eventually were summarized under the term “cancer immunoediting” (62, 131–137). The concept of immunoediting evolved from the older and more controversial “cancer immune surveillance” one, which was based on the notion that, analogous to the “non-self” of pathogens, our immune system is also able to discriminate between the “malignant self” of pre-cancerous and cancerous cells and the “self” of normal cells (61, 62, 68). To discriminate cancer cells from normal cells, the immune system pursues two main strategies: T and B cells, which belong to the adaptive immune system, recognize altered self-proteins, whereas natural killer (NK) cells, gamma/delta T cells and macrophages, which are part of the innate immune system, take care of stress-induced self-molecules on transformed cells (61). Still, the necessity to establish an effective antitumor response, goes always hand in hand with the formidable challenge to circumvent the destruction of normal cells and to avoid autoimmunity.
Given the tight interaction between the immune system and neoplastic tissues, one naturally expects, and as Corthay put forward in eight arguments, that individuals with PIDs are more prone to develop tumors than the general population (61). At first sight this notion is well-supported by both animal models and clinical observations (61, 68). The best evidence that this is indeed the case derives from experiments with mice that lack key components of the immune system. They have not only an overall higher tumor incidence, but they are also more susceptible to transplanted or chemical carcinogen-induced tumors (64, 138). At second sight, however, all hitherto available data argues against the long-hold notion that potentially dysfunctional immune surveillance mechanisms indeed increase the general tumor risk in all PID patients. If at all, such processes can only play a subordinate and ancillary role.
Reliable information regarding the general and specific tumor risk of individuals with PIDs derives primarily from three large epidemiological studies from the USA, Australia, and the Netherlands (67, 128, 139). Together these studies comprise more than 5,000 patients with around 300 different forms of PIDs. Compared to the general population and previous estimates, these analyses revealed a surprisingly low, only 2-fold increased, overall tumor risk. However, since the respective risk is primarily confined to and therefore significantly higher in the nine most frequent high-penetrant PIDs, it is conversely also much lower or even absent in most of the other PIDs. Distinct genetic PID defects predispose to and concur with special and often unique types of malignancies, the most common of which are non-Hodgkin lymphomas, leukemias, digestive tract as well as virus-induced cancers (67, 68, 70, 128, 129, 139). This particular distribution patterns can already provide some important clues about the underlying defective, disrupted or impaired immune processes that trigger such disease developments. The two major driving forces that are responsible for the 8- to 10-fold excess lymphoma risk in subjects with PIDs, for instance, are a deficient DNA repair and an inadequate response to viral infections (61, 67, 68, 70). The intriguing part of this story is, however, that the incidence of the most frequent cancers, such as breast, lung, and colon, is in the remaining group of PIDs much lower than that in the general population.
Inadequate Activation and Response of the Immune System
Chronic inflammation, autoinflammation, autoimmunity, and infection-associated overstimulation are closely intertwined derangements of a deficient or compromised immune system (140).
Chronic Inflammation
Inflammation is a physiological response to tissue stressors such as tissue damage and infectious as well as non-infectious agents that ensures the maintenance of tissue homeostasis (141). Under normal circumstances it mitigates infections, clears damaged cells, and initiates tissue repair (142). If this process is not properly terminated, it can cause substantial collateral damage and contribute to tumor development (143–145). Chronic inflammation plays not only a pivotal role in different stages of tumorigenesis, but it may also impede the response to therapy (63, 145). Along the route to tumorigenesis, intrinsic genetic factors interact with extrinsic immune system and stromal humoral and cellular components to generate a mixed microenvironment that is composed of tumor-promoting and tumor-suppressive factors, including innate NK and adaptive T cells (62, 63, 145, 146). The fact that, despite their dissimilar etiology and physiology, autoimmune and infectious diseases take advantage of the same immunosuppressive pathways and logistics underlines the global relevance and central position of these particular activities (146).
Infection-provoked chronic inflammatory conditions predispose especially but not exclusively to the development of Hodgkin's, Burkitt's, and mucosa-associated lymphoid tissue (MALT) lymphomas (147–149). The best-known associations are those between Helicobacter pylori and gastric lymhomas, Chlamydophila psittaci and ocular adnexal lymphomas as well as Borrelia burgdorferi and cutaneous MALT lymphomas, while chronic infections with Epstein-Barr virus (EBV) usually predispose to Burkitt- & Hodgkin lymphomas, those with Hepatitis C virus (HCV) to marginal zone lymphomas and those with Hepatitis B virus (HBV) to hepatocellular carcinoma (147, 148). The pathogenetic role of chronic inflammation remains less clear in case of human papillomavirus-, herpes simplex virus 2-, and cytomegalovirus-triggered malignancies (150).
Inflammatory bowel diseases, such as Crohn's disease and ulcerative colitis, provoke especially the development of colorectal cancer (151, 152). Crohn's disease, in particular, is a multifactorial disease whose genetic underpinning encompasses 71 so far recognized susceptibility loci (31). Amongst these are the first recognized monogenic causes of such diseases, namely interleukin-10 (IL-10) and IL-10 receptor (IL-10R) loss of function mutations that are the specific cause of a severe, very early onset type of inflammatory bowel disease (153–156). Affected children have an extremely high probability to develop a unique form of monoclonal EBV-negative diffuse large B cell lymphoma, which is characterized by a constitutive activation of the NF-kappaB pathway and a defective local T cell immune response (153). These findings prompted Neven et al. to postulate that not gut inflammation itself but that the defective IL-10 pathway alone was the responsible pathogenetic trigger. Referring to the fact that all these children received azathioprine, which is a well-known lymphoma risk-increasing factor in adults with inflammatory bowel diseases, they suggested that this immunosuppressive treatment was also the final spark that ignited lymphoma development in these children (153).
Autoimmunity
Autoimmunity is a prominent element in many PIDs, especially in CVIDs, and not least in those, which predispose to malignancies (19, 125, 157–159). Despite their close clinical and genetic interrelationship, autoimmunity, and PIDs were up to now interpreted as two mutually exclusive conditions rather than as two sides of the same coin (157). In consideration of the fact that autoimmunity is the leading symptom in a variety of monogenic disorders that affect T cell development, tolerance, and interferon signaling, complement pathways as well as the resolution of inflammation, this view is changing nowadays (157). The two prototypic examples to illustrate their close interrelationship are the autoimmune lymphoproliferative syndrome (ALPS) and the IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy, X-linked). In case of ALPS, the respective lymphoproliferation and propensity to develop lymphomas results from apoptosis-impairing germline as well as somatic mutations in the FAS, FASL, and caspase 10 genes (160–162). Still, mutation carriers have only a <60% chance for disease manifestation (161). The IPEX syndrome, on the other hand, is caused by activating mutations in the FOXP3 gene (163–165). The encoded transcription factor controls the function of regulatory T cells that are essential for maintaining self-tolerance and immune homeostasis by suppressing aberrant responses such as autoimmunity and allergy (166). Deficiency of cytotoxic T lymphocyte antigen 4 (CTLA-4), which is a crucial inhibitor of T cell response that is also present on regulatory T cells, can therefore generate autoinflammation and autoimmunity in a corresponding fashion (166–169). Moreover, CTLA-4-deficient individuals are at risk to primarily develop EBV-related malignancies (170). Another recently recognized albeit rare cause of autoimmunity are leukemia-predisposing germline mutations in the IKZF1 gene, which encodes the hematopoietic transcription factor Ikaros (171–175). Finally, one should not forget that autoimmune diseases are also a common problem in one of the most remarkable forms of leukemia-predisposing immunodeficiencies, the Down syndrome (176–178).
Hyperactivation
The inability of cytotoxic lymphocytes to fend off and kill virus-infected or transformed cells leads to an often uncontrollable hyperactivation of the immune system in form of hemophagocytic lymphohistiocytosis (HLH) (179–181). This distinctive clinical feature is the common denominator of a related group of perforin-deficient hyperinflammatory disorders, so called “perforinopathies,” that may either be due to rare congenital gene-disrupting mono- or biallelic mutations or, in less severe form, due to functionally impairing hypomorphic alleles (182–186). Familial hemophagocytic lymphohistiocytosis type 2 (FHL2) is caused by biallelic mutations of the perforin gene (PRF1) (179). It shares some of its clinical characteristics with those of anaplastic large cell lymphoma (ALCL), which accounts for ~10 to 15% of all pediatric non-Hodgkin lymphomas (187–190). About a quarter of these lymphoma patients carry only monoallelic PRF1 mutations but, remarkably, virtually none in SH2D1A or UNC13D, genes that are implicated in two other forms of FHL (181, 187, 191–193). Moreover, an otherwise common activity-diminishing PRF1 gene variant (SNP A91V; rs35947132), is also postulated to predispose to the nasal form of NK/T cell lymphoma in adults, which is the most frequent EBV-related NK/T cell malignancy (188).
Microbiome
Human beings are holobiontic meta-organisms (194–197). They are composed of host as well as trillions of viral, fungal, bacterial, and eukaryotic microbes that are collectively referred to as the microbiota or microbiome (194–197). This microbiome is acquired and shaped during the first 2 years of life. It co-evolves with its respective host genome and, under physiological conditions, becomes part of a stable, life-long synergistic homoeostasis (194, 195, 197–200). Because of its tight functional link with and profound effects on the host's immune system in health and disease, the microbiome is therefore already regarded as a complex, polygenic trait (200–202). Environmental, and host-related perturbations of this microbial ecosystem reduce almost invariably its diversity. This is a common finding in many multifactorial inflammatory, autoimmune, metabolic, neoplastic, and neurodegenerative diseases, although it is rarely known whether such a dysbiosis is indeed the cause or the effect of the underlying ailment (197, 203). Nevertheless, the host's immune system is certainly the most important force that shapes the configuration of the normal and dysbiotic microbiome, which, in turn, may of course be a significant cofounding factor in immune-mediated and immune-associated diseases (197). A healthy or dysbiotic microbiota can influence the host innate immune system and it is therefore no surprise that microorganisms are also implicated in the pathogenesis of at least 20% of all human malignancies (204). In a dysbiotic state, alterations in the signature of microbial molecules that are sensed by the host can lead to a different activation state of the immune system. These changes may alter the balance of host cell proliferation and death, guide immune system function and influence metabolism of host-produced factors, ingested food and pharmaceuticals (205). Moreover, they may also drive transformation by affecting genomic stability, resistance to cell death and proliferative signaling (205). Both chronic high-grade as well as lower-grade smoldering inflammatory disorders drive a tumor-permissive milieu, a problem that was extensively studied and confirmed in mice that were deficient in various immunologically relevant genes (199, 205, 206). It is worth noting that such a cancer susceptibility can even be transferred to healthy mice by cohousing, fostering or fecal transplants (195, 199, 207). Since polymorphisms in immunologically relevant genes affect human microbiota composition and cancer predisposition, this observation is therefore also highly relevant for such human diseases (201, 202, 208, 209).
Based on these results, Dzutsev et al. therefore suggested that malignancies can be viewed as systemic diseases that alter the physiological homeostatic interaction of the entire meta-organism (143, 195). Mostly because of its effects on metabolism, cellular proliferation, inflammation, and immunity, the microbiome would interact with their development at virtually every level, including predisposing conditions, initiation, genetic instability, susceptibility to host immune response, progression, comorbidity and, not least, response to therapy (143, 195, 205). In support of this notion, Yamamoto et al reported, for instance, that variation in intestinal microbes between different animal facilities or as a consequence of experimental perturbations profoundly affected the incidence of lymphoma and survival of Atm (ataxia telangiectasia mutated)-deficient mice (210). Another very intriguing and instructive example of how we will one day perhaps be able to exploit particular constituents of the microbiome for medical and therapeutic purposes was recently provided by Bromberg et al. (211). They showed that with the delivery of stool samples from pregnant mice or gavage with isolated B. pseudolongum species to those with cardiac allografts they were able to improve their long-term survival as well as to prevent inflammation and fibrosis in this respective organ (211).
Bona Fide Infections
The IARC classifies 10 microbial agents (7 viruses, 2 parasites, and 1 bacterium) as group 1 human carcinogens (195, 204). Over 90% of all infection-attributed cancers are attributed to Helicobacter pylori, HBV and HCV, and human papillomaviruses (HPV) (204). Except for HCV, all human oncogenic viruses encode at least one oncogene and may therefore directly induce neoplastic transformation, although, as alluded to above, infection-associated inflammation and dysbiosis most likely play a likewise significant role in this context. Thus, Plottel et al. distinguish three classes of microbe-induced human malignancies, the first is defined as involving immunologic tissues, the second requires direct microbial interactions with parenchymal cells and the third involves distant effects from local interactions (212).
The likelihood to be either protected or to become infected as well as the infection outcome depends primarily on the diverse conditions that guide the interactions between the respective pathogen and its potential host. These include their specific genetic set-up and functional fitness to invade or defend themselves, as well as on a variety of other factors, such as concomitant infections as well as the age and microbial constitution of the respective host (213–220). To invade host cells, pathogens exploit and hijack particular cell surface proteins. Amongst the docking receptors that were identified so far in case of the Malaria parasite plasmodium falciparum, for instance, are CD55 and structural variants of the GYPA and GYPB genes (220–224). CD55-null erythrocytes, in particular, are refractory to invasion by all isolates of plasmodium falciparum (222). The second example is CCR5, which encodes a coreceptor for HIV entry. Consequently, carriers of an otherwise phenotypically and functionally completely inconsequential homozygous 32-bp deletion are resistant to HIV infection (225). Although HIV infections are indisputably the cause of the “acquired” immune deficiency, one can still argue that all these infection-related problems are nevertheless due to a genetically primed primary albeit clinically inapparent susceptibility. The point I want to stress here is that what one currently perceives as a “normal” wild-type or a “defective” susceptible gene variant is merely a matter of choice, frequency, common habit, and/or subjective interpretation. Since the functional consequences of any such variant will always remain context-dependent, one therefore needs to keep in mind that the distinction between “protective” and “defective” can never be an absolute dogma, but always lies in the eyes of the beholder.
Epstein-Barr virus (EBV) is a ubiquitous virus that infects virtually all humans and obligatory leads to a usually asymptomatic symbiotic lifelong latent persistence (214, 226). Although this EBV latency may provide an evolutionary mutualistic benefit to its hosts as an immune adjuvant that apparently protects against lethal Listeria monocytogenes and Yersinia pestis infections (227, 228), the many EBV-associated problems have nowadays become clinically far more relevant and interesting. At present, the literature distinguishes more than 25 EBV-related disease entities, including those that are associated with various forms of immunodeficiencies and those that concur with a high propensity to develop diverse hematopoietic, epithelial, and mesenchymal malignancies (66, 226, 229–234). These disease forms can be roughly divided into reactive EBV-associated lymphoid and histiocytic/dendritic proliferations (including reactive lesions with or without diverse malignant potential), B cell proliferations (including Hodgkin lymphoma and plasma cell neoplasms), T/NK cell proliferations, immunodeficiency-related lymphoid proliferations and histiocytic/dendritic proliferations (66, 226, 229, 231–236). Taken together, EBV contributes to about 1.5% of all cases of human cancer worldwide (229, 237).
The type and incidence of EBV associated diseases varies significantly in different parts of the world, an observation that can be attributed to the different distribution of genetic susceptibility factors, including individual-, HLA-, and ethnicity-specific ones, to environmental- and geographic-specific co-founding influences but also to the existence of particular EBV strains that may produce different disease patterns (27, 66, 153, 213, 214, 217, 218, 229, 230, 232–234, 236, 238–240).
In contrast to the above-mentioned HIV infection, which significantly increases the risk for malignant diseases and especially lymphomas, such a risk is, if at all, by far not as pronounced in case of Malaria (241–243). The only notable exception concerns the concomitant early and sustained infection of Plasmodium falciparum and EBV, which together are the essential pathogenetic ingredients in the endemic form of Burkitt lymphoma in Africa (217–219, 236, 238, 244–246). In this particular combination, the Plasmodium infection destabilizes the genome of rapidly dividing EBV-infested germinal center B cells by eliciting the protracted expression of the activation-induced cytidine deaminase, a mutation-aggravating enzyme (238, 245).
At one point in their life, virtually all humans become infected with EBV, most of them without any acute, severe or lasting health problems. However, in those who do, one can often identify an underlying disease-associated, more or less pronounced genetic susceptibility, which begs for the question whether EBV-associated disease processes indeed afflict also “immunocompetent” individuals, or put the other way around, what in the end will define such an “immune (in)competence” (235).
Predisposition to Hematologic Neoplasms in Children
The role of predisposing germline mutations and sequence variants in children and adults with various types of hematologic malignancies was hitherto largely underappreciated, because not all of them concur with nor create any easily recognizable clinical stigmata or suspicious family history. Especially if one screens neoplastic tissues for disease- and/or therapy relevant somatic mutations, it becomes of critical importance to distinguish those from germline ones, because the latter may also have a profound clinical impact as regards choice of therapy, donor selection in case of transplantations, evaluation of comorbidities as well as surveillance strategies (8, 173).
A recent paper by Duan et al. provides an excellent and very comprehensive overview about all the primary immunodeficiencies that in particular predispose to the development of various types of lymphomas and hematologic malignancies (3). The authors compiled more than 60 conditions, which comprised all subgroups of syndromic and non-syndromic cellular and humoral PID as well as defects in phagocytes and innate immunity.
To pack some of the underlying principles and problems into a practical and newsworthy perspective, I will now briefly turn to recent findings in three categories of childhood leukemia.
Constitutional Trisomy 21
Trisomy 21 is not only the most common chromosome abnormality in liveborns but in many aspects also one of the most outstanding and fascinating examples of an immune system disorder, although for a long time the precise nature of the respective immunological derangements remained elusive (178). Since it is not a monogenic ailment, it is also hardly ever viewed as a primary immunodeficiency, although it clearly concurs with multiple distinct immunological and developmental defects that affect the myeloid but also the early and committed B-lymphoid progenitor compartments in second trimester fetal liver (247). I chose this most intriguing and highly instructive example to explain in which way the predisposing risk score of a kind of “polygenic” disposition can easily reach at least the same magnitude as the one that is otherwise only achievable in a monogenic setting (55).The ensuing variable and clinically often inapparent immunological alterations in Down individuals comprise a mild to moderate decrease in T and B cells, impaired mitogen-induced T cell proliferation, reduced specific antibody responses to immunizations as well as defects of neutrophil chemotaxis (248). Affected individuals suffer from various types of autoimmune and autoinflammation diseases, whereas it is still a matter of debate whether they are also more prone to experience more or severer infections than non-Down individuals (176, 178, 248, 249). The first clues that helped to resolve the functional consequences of this immunological conundrum derived from recent transcriptome and proteome analyses. They revealed that the presence of an extra chromosome 21 leads, amongst others, to an overexpression of the four chromosome 21-encoded interferon receptors and, therefore, place this syndrome into the class of interferonopathies. Interferons are normally produced by cells in response to viral or bacterial infections, regulate genes in neighboring cells and shut down the production of proteins, which activate the immune system and thereby prevent the spread of the infection (250, 251). In line with interferonopathies and autoinflammatory conditions, individuals with Down syndrome display higher levels of many pro-inflammatory cytokines (including IL-6, IL-22, TNF-α, and MCP-1) as well as complement consumption, a state that indicates that the immune system is constantly fighting viral infections that are in fact not there (250). Whether and to which extent such a faulty overreaction may also participate in promoting the development of hematologic neoplasms perhaps in a similar fashion as in genuine virus infection-triggered malignancies, remains to be elucidated.
Individuals with an inborn trisomy 21 have also an extraordinary risk to acquire special forms of hematologic neoplasms in early life, whereas they are otherwise exquisitely protected against the development of any other malignancies (252, 253). Compared to normal age-matched children, the self-limiting transient myeloproliferative disorder (TMD) together with the acute megakaryoblastic leukemia (AML-M7), is ~150 times and the B cell precursor ALL ~33 times more common (99, 101, 102, 253, 254). Of particular note are also the absence of infant ALL, the rarity of T-ALL and, compared to normal children, the different distribution pattern of genetic B-ALL subtypes (255).
The occurrence of specific mutations in the receptive precursor cells determine which form of leukemia will eventually develop. In case of TMD, the perturbation of megakaryocyte-erythroid precursor cell differentiation fosters the appearance of a highly specific truncating mutation in exon 2 of the hematopoietic transcription factor GATA1. This in turn provides the receptive cellular and molecular environment for the occurrence of further mutations, primarily in the JAK and RAS signaling pathways as well as in epigenetic regulators and multiple cohesion components, which then facilitate the further progression into AML (99, 101, 104, 254). A reduced lymphoid gene expression in fetal liver hematopoietic precursor cells impairs B-lymphoid development in a similar fashion. The ensuing maturation arrest leads to an ~10-fold reduction in B cells. The concomitant accumulation of pro-B progenitors (247), on the other hand, increases the likelihood for illegitimate V(D)J recombination-mediated chromosomal rearrangements, in particular CRLF2 gene fusions, that can be found in approximately half of all Down syndrome BCP ALL cases (99, 104). To explore the potential contribution of chromosome 21-encoded and overexpressed genes, a set of 31 triplicated orthologous human genes were tested in germline mouse models (256). Their presence induced progenitor B cell self-renewal in vitro, maturation defects in vivo and the development of especially CRLF2-rearranged and JAK2 pathway-activated BCP ALL. Out of these 31 genes, the nucleosome-remodeling protein high mobility group nucleosome-binding domain- containing protein 1 (HMGN1), whose protein product suppresses H3K27me3, turned out to be the most relevant candidate. Together with secondary alterations in the CRLF2, JAK2, NRAS, or KRAS genes, it promotes both B cell proliferation in vitro and the development of B ALL in mice in vivo (256).
Given the extraordinary susceptibility and the high incidence of leukemias, one would intuitively expect that both myeloid and lymphoid forms should occasionally occur together by pure chance alone. However, such a coincidence has so far never been reported. On the one hand, this lack of co-occurrence might indicate that the development of a specific type of leukemia requires and is subject to very individual-specific predisposing conditions. On the other hand, it is also in keeping with the fact that these patients virtually never suffer from secondary neoplasms and that they are in a unique and matchless way also protected against the development of any other types of neoplasms (252, 253).
This protective effect also has been put down to a copy number-dependent gene dosage but also context-dependent effect of specific chromosome 21-encoded genes. The presence of three ETS2 copies, for instance, act as tumor repressor in the ApcMin intestinal cancer mouse model, whereas in the PyMT breast cancer mouse model it functions as tumor promoter, albeit within the non-cancerous stromal cells, where it regulates the expression of genes that produce the extracellular matrix, an essential component for tumor growth and metastasis (257–260). Two other relevant genes are the Down's syndrome candidate region-1 (DSCR1), which encodes a suppressor of the vascular endothelial growth factor (VEGF)-mediated angiogenic signaling by the calcineurin pathway, and DYRK1A, which encodes a regulator of cell proliferation (261). In mice, the presence of a single extra copy of Dscr1 is sufficient to diminish tumor growth by suppressing the calcineurin pathway and therefore also angiogenesis, an effect that is significantly enhanced by an extra copy of Dyrk1a. For the sake of completeness, one needs to take at least also note of several other trisomic chromosome21-encoded genes whose presence in the stromal compartment helps to reduce tumor angiogenesis, namely the angiogenic inhibitor ADAMTS1, the transcription regulator ERG and, finally, the endothelial cell-specific genes JAMB and PTTG1IP (257).
Bone Marrow Failure (BMF), Myelodysplastic Syndromes (MDS), and Myeloid Leukemias
Table 1 provides a comprehensive summary of the various disease entities together with their causative genetic background. For a more in-depth overview I refer the interested reader to recent publications that deal with these individual subjects in detail (8, 60, 158, 264, 265, 268, 269, 272, 275, 283, 299, 300). Herein, I merely select a few instructive examples to highlight some of the intriguing phenomena that are particularly pertinent for the topic discussed herein.
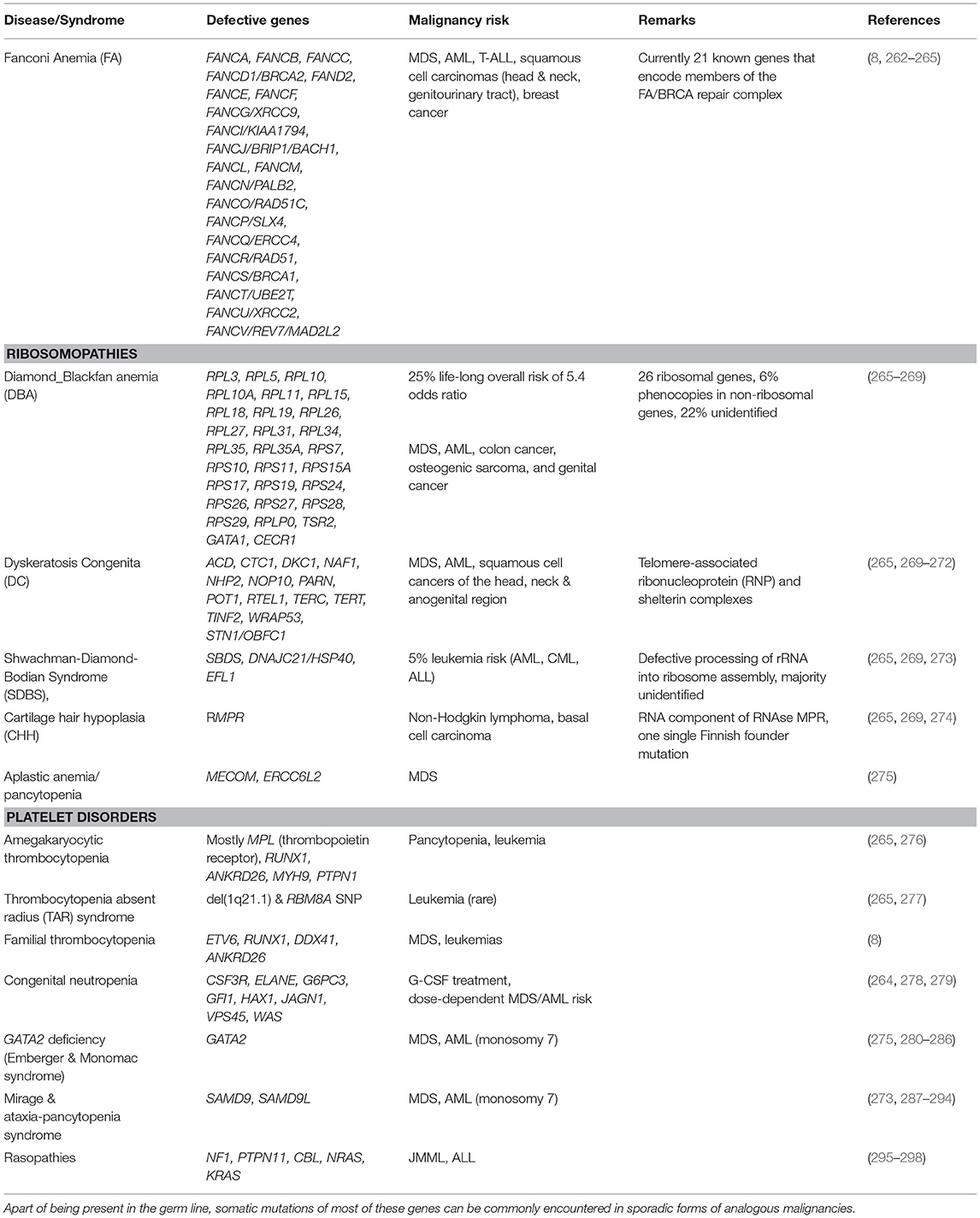
Table 1. Immunodeficiency syndromes that predispose to the development of bone marrow failure, myelodysplasia and myeloid leukemias.
Fanconi Anemia (FA) is not only the most common inherited BMF disorder but, with a 500- to 700-fold higher incidence of head and neck squamous cell carcinomas in older patients, also a highly penetrant cancer susceptibility syndrome (301). Except for the X-linked FANCB gene, it is due to bi-allelic mutations that can affect one of 21 genes, which encode various components of the evolutionarily conserved FA/BRCA repair complex. Five of these (BRCA2, PALB2, RAD51C, SLX4, and BACH1) specifically predispose also to breast cancer. The encoded proteins participate in biochemical pathways that safeguard not only against the effects of alkylating agents and radiation but, probably even more relevant, also against those of endogenous aldehydes, oxidative stress, inflammation, mitophagy, and virophagy (263, 265, 302, 303). All these factors damage DNA in form of distinct inter-strand DNA crosslinks. The inability to repair these damages is the primary driver of the various biological and clinical problems that define this disease category.
Small aldehydes, such as acetaldehyde and formaldehyde, are not only ubiquitously present in the environment, but also potentially toxic byproducts of the normal cellular metabolism and especially de-methylation reactions (304–306). Given that they provide already a rich source for endogenous inter-strand DNA and protein cross links, one can envisage that the pathogenic manifestations and consequences of FA mutations may also be strongly influenced and modified by the functional capability of aldehyde detoxifying enzymes, such as aldehyde dehydrogenase 2 (ALDH2) and alcohol dehydrogenase 5 (ADH5) (306, 307). In line with this notion, Japanese FA children that carried a functionally deficient ALDH2E504K allele were shown to progress more rapidly to aplastic anemia but not to MDS or AML (307–309). Moreover, malformations were only more severe in two of three homozygous carriers, which indicates that a deficient maternal genetic background might contribute to this outcome (310). Maternal-produced aldehydes diffuse indeed across the placenta and can thus damage the developing embryo's DNA, whereas embryo-derived ones can in turn be detoxified by the maternal organism. An inappropriate in utero exposure, such as an excessive maternal ethanol consumption during gestation, would therefore aggravate not only the formation of congenital abnormalities in FA but it provides also an intriguing etiological link to analogous phenotypic changes that define the alcohol embryopathy (308). Whether a disturbed aldehyde detoxifying system might also be causatively involved in the in utero initiation of childhood leukemias remains currently a matter of speculation (307).
In addition to stalling and destabilizing DNA replication forks directly, formaldehyde also selectively depletes BRCA2 via proteasomal degradation, a circumstance that poses a special risk for heterozygous BRCA2 mutation carriers. In these, formaldehyde-induced degradation can decrease the respective protein levels below the otherwise protective one of normal wild-type individuals and thereby potentiate their mutagenic vulnerability (311).
Taken together, these observations have significant implications for risk awareness and avoidance as well as the clinical management of FA patients. On the positive side, they offer new treatment opportunities, for instance in form of ALDH2 agonists and the widely used diabetes drug metformin, which acts as aldehyde scavenger. At least in mouse models, both of them are able to delay the onset of BMF and malignancies as well as improve hematopoiesis (263, 312, 313).
One remarkable feature of many heritable diseases is that somatic mutations can occasionally autocorrect the particular inherited gene defect in the respective cells (314). Such reverting mutations transform a homozygous or combined heterozygous state again back into a heterozygous functionally compensated state, either through somatic recombination, gene conversion or a compensatory mutation (314, 315). Although this phenomenon is well-known in immunodeficiencies, its effects are most probably still underappreciated. In BMF syndromes such an autocorrection can spontaneously improve or even resolve the specific underlying hematopoietic problem. Amongst others, such spontaneous remissions have repeatedly been documented in FA, dyskeratosis congenita, Diamond-Blackfan anemia, Shwachman-Diamond syndrome and, more recently, in the SAMD9- and SAMD9L-associated Mirage and ataxia-pancytopenia syndromes, respectively (273, 287–293, 301, 316). Heterozygous SAMD9L gain-of-function mutations decrease cell proliferation. The loss of the mutation-carrying chromosome 7 in bone marrow cells therefore leads to the development of MDS and acute myeloid leukemias, whereas a compensating duplication of the normal allele in form of an uniparental disomy (UPD) 7 or 7q is able to resolve the cytopenias (287–293).
The somatic appearance of a complete or partial UPD always draws attention to regions that are usually highly relevant for specific disease processes (317, 318). Such UPDs may either contain duplicated gain of function mutations or, as alluded to in the example above, eliminate them (317, 318). A similar important consequence is the mere transformation of a heterozygous to a homozygous state. In case of the HLA-containing region on the short arm of chromosome 6, for instance, it is clearly exerted through a selective pressure, since the loss of one HLA haplotype is an important immune-escape mechanism. It protects neoplastic cells from the immune surveillance machinery and therefore also plays a crucial role for disease recurrence after haploidentical stem cell transplantations (319, 320). Contrariwise, such a haplotype loss is able to shield hematopoietic cells from the destructive effects of autoimmunity, as has been demonstrated in case of aplastic anemia (321). The practical problems that arise from such a hematopoietic revertant mosaicism is that it may lead to an ascertainment bias and cause difficulties in identifying underlying disease-relevant mutations (322).
The formation of such well-adapted clones might suggest that such compensatory mechanisms are rare events. However, there is ample evidence that this is definitely not the case. Rather than being a life-long stable system the genome is a highly dynamic one that is continuously modified and shaped by ongoing mutational processes, which eventually promote the appearance of cell clones and populations with an increased survival fitness. The formation of somatic mosaicism is therefore the rule rather than the exception, as exemplified by Davis et al., who observed a remarkable clonal heterogeneity and diversity of lymphocytes in a patient with a Wiskott-Aldrich syndrome (323). The continuous generation of such cellular variants and the selective pressures they are exposed to is thus not only a characteristic of the exceptional dynamics of neoplastic but also of normal cells populations (324).
B-Cell Precursor Acute Lymphoblastic Leukemia (BCP ALL)
Germline lesions that predispose to BCP ALL in children comprise not only those which cause various cancer prone and chromosomal syndromes but also other genuine gene disrupting defects as well as high and low risk variants. The majority of these affect genes that encode B-cell development and transcription factors as well as components of various signal transduction pathways (Table 2) (42, 297, 342).
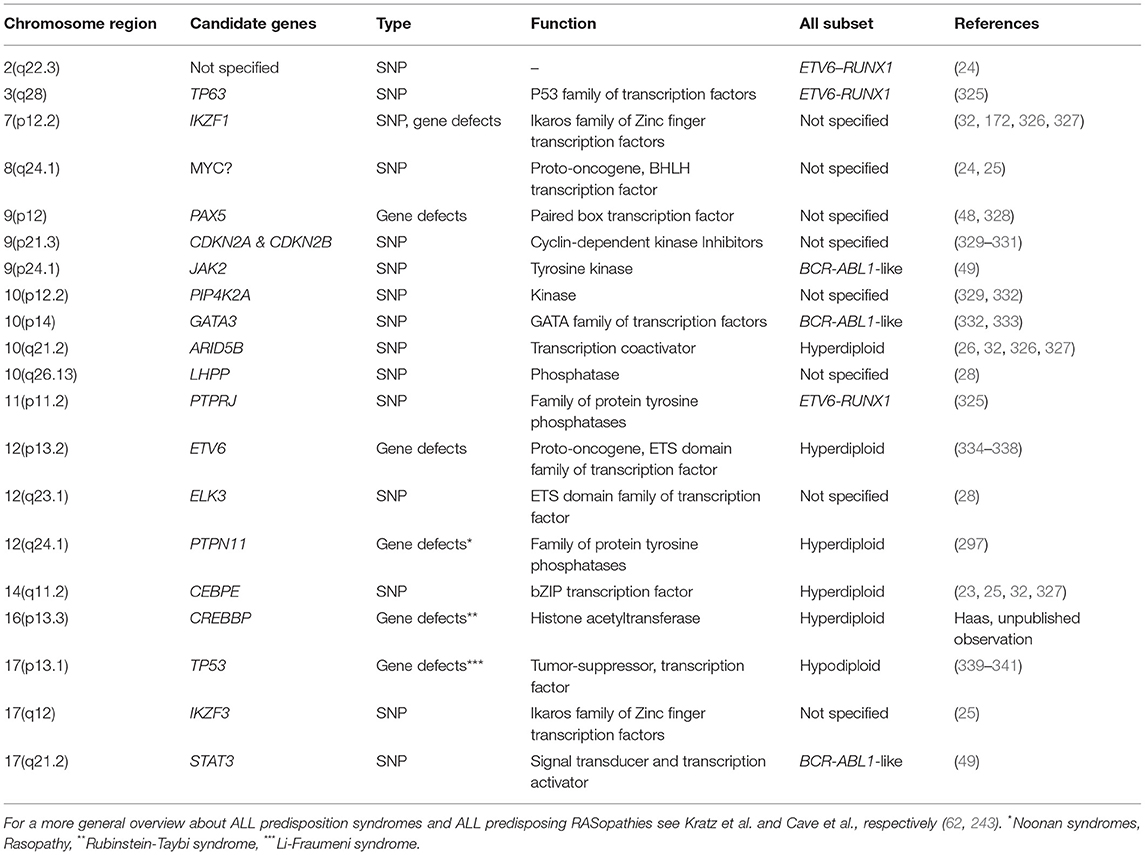
Table 2. Chromosomal locations of GWAS-verified SNPs or genuine germline gene defects that predispose to the development of particular types of childhood ALL.
The development of B lymphocytes, in particular, is coordinated by specific regulatory transcription networks that activate the respective B-lymphoid program and at the same time oppress alternate cell fates (343). Somatic mutations in several of these key regulators, such as IKZF1, TCF3, EBF1, and PAX5, induce leukemic transformation by blocking normal B cell differentiation, which then leads to the accumulation of leukemic B-cell precursors. Although their role in leukemogenesis has been known and explored already for quite some time, the awareness that otherwise apparently inconsequential germline variants may also exert a predisposing effect is quite new. The biological relevance and functional consequences of some of these variants has been confirmed in the meantime with appropriate in vitro and in vivo experiments and test systems (23–26, 28, 332, 333).
Take for instance PAX5, a member of the “paired box” family of transcription factors, which encodes the B cell lineage specific activator protein that is expressed at early but not late stages of B-cell differentiation (195). Out of the three up to now reported families with highly penetrant germline variants, 13 carriers developed ALL (48, 328). In the two families, in which the respective information was provided, all unaffected carriers as well as those with ALL had normal immunoglobulin levels and no evidence of an impaired B cell function at diagnosis (328). Moreover, in line with Greaves two step model, leukemia developed in mice only after exposure to common pathogens and the acquisition of second hits in the IL7R/JAK3/STAT5 signaling axis (344).
Another revealing example is IKZF1, which encodes IKAROS, a member of a hematopoietic zinc-finger transcription factor family (345–347). The mutational spectrum of human IKZF1-associated diseases ranges from somatic to germline and from haploinsufficient to dominant negative forms (171). Somatic mutations occur in overall 15% of BCP-ALL and especially in prognostic adverse genetic subtypes (348, 349). Heterozygous germline mutations cause two different forms of immunodeficiency. The haploinsufficient, autosomal dominant late onset form of common variable immunodeficiency (CVID) has an incomplete penetrance, is clinically mild and concurs with a marked decrease in B-cell numbers and immunoglobulin levels as well as autoimmunity (175, 350, 351). The early-onset dominant negative CVID, on the other hand, is characterized by innate and adaptive immune defects of the B, T and myeloid cell lineage (171). Notably, 2/29 of patients with the late onset form developed B-ALL and 1/7 patients as well as another independently reported one with the early onset form developed T-ALL (171, 346, 351). Based on these observations, Churchman and colleagues screened remission samples from 4,963 childhood ALL cases, identified a total of 28 unique IKZF1 variants in 43 and succeeded to prove a functional consequence in 22 of them (172, 173). Evans et al. even attempted to elucidate the influence of a parental environmental exposure on such leukemia-predisposing risk alleles (352). They found some preliminary albeit hitherto unexplainable evidence that the IKZF1 risk genotype might have a stronger effect if the mother took folic acid or if the father did not smoke prior to pregnancy (352).
Finally, Auer et al reported a first intriguing example of a “double-hit one-pathway” scenario, in which the biparental inherited combination of two rare germline variants, JAK2 (G571S) and STAT3 (K370R), whose products synergistically interact in the same disease-relevant JAK/STAT signal transduction pathway, is obviously sufficient to induce a Ph-like BCP-ALL (49).
Concluding Remarks
During their entire development and ongoing existence, both the immune system as well as malignant diseases need to adapt themselves to highly variable and continuously fluctuating environmental conditions, which requires a high flexibility that is largely driven by a combination of interacting antagonistic as well as synergistic deterministic events and regulatory probabilities. “Immunodeficiency” and “tumor susceptibility” are thus two closely intertwined concepts, whose original understanding was based on easily explicable clinical symptoms as well as certain genetic norms. As long as these were based on such more or less simple phenotypic and genotypic features, one did not require any further explanatory definitions. However, the switch from phenotypic to genetic ascertainment programs include now much less obvious disease categories, healthy carriers as well as only vaguely defined potentially predisposed individuals. Although this approach enables of course unprecedented insights into the fine-scale structural and regulatory organization of biological system, the boundaries of classification standards get increasingly blurred, which goes hand in hand with the awareness that it becomes increasingly difficult and virtually impossible to define either of these terms in an unambiguous manner anymore. The more closely one looks, the harder it gets to find genes that are not in one or the other way part of this game.
In his highly recommendable and readable book “Tending Adam's garden,” Irun Cohen portrays the immune system as a cognitive system (353). Like its prototypic equivalent, the brain, it learns through individual experience and thereby forms a functionally highly efficient, flexible, and to some extent also redundant interactive structure. As Cohen pointed out, a particular gene may only become essential once the system has organized itself around it so that thereafter the system becomes dependent on it. In case this particular gene is already defective at the beginning, the system often can compensate for this loss and organize itself around an alternative gene, which then becomes the essential one.
Thus, organizational entities depend not only on distinct features of particular sub-units but even more so on their functional interactions. In case of the immune system, such multi-component, self-emergent networks comprise a variety of distinct cellular as well as humoral host constituents, but also a manifold of environmental factors, such as the maternal immune system, the microbiome, infectious agents, as well as physical and chemical agents, that co-govern and modulate, but often also interfere and disrupt its normal development during different stages.
Thus, organizational entities depend perhaps less on the appropriate function of particular genetic sub-units alone but much more on the successful interaction of their cellular and humoral products, an observation that is readily evident in case of genetically determined developmental disorders that can cause both immune system deficiencies as well as malignant diseases.
The essential implication of Cohens' model is that we only may become more successful in curing such disease when we begin to understand the decision-making processes of the immune system rather than that of the effects of individual components alone.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This paper is dedicated to my late friend and colleague Andreas Heitger, a devoted immunologist, with whom it was always fun to discuss the philosophical part of the activity and behavioral pro and cons of the immune system.
I acknowledge the networking support by the COST Action CA16223 LEukaemia GENe Discovery by data sharing, mining and collaboration (LEGEND) as well as by the IBFM Leukemia & Lymphoma Genetic Predisposition Committee, the Österreichische Kinderkrebsforschung for the continuous funding of my research work and Fikret Rifatbegovic's invaluable help in designing and drawing the Figure.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:64674. doi: 10.1016/j.cell.2011.02.013
2. Sonnenschein C, Soto AM. Theories of carcinogenesis: an emerging perspective. Semin Cancer Biol. (2008) 18:372–77. doi: 10.1016/j.semcancer.2008.03.012
3. Duan L, Grunebaum E. Hematological malignancies associated with primary immunodeficiency disorders. Clin Immunol. (2018) 194:46–59. doi: 10.1016/j.clim.2018.06.011
4. Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature (2018) 555:321–7. doi: 10.1038/nature25480
5. Kager L, Jimenez Heredia R, Hirschmugl T, Dmytrus J, Krolo A, Müller H, et al. Targeted mutation screening of 292 candidate genes in 38 children with inborn haematological cytopenias efficiently identifies novel disease-causing mutations. Br J Haematol. (2018) 182:251–8. doi: 10.1111/bjh.15389
6. Ochs HD, Petroni D. From clinical observations and molecular dissection to novel therapeutic strategies for primary immunodeficiency disorders. Am J Med Genet A (2018) 176:784–803. doi: 10.1002/ajmg.a.38480
7. Desai AV, Perpich M, Godley LA. Clinical assessment and diagnosis of germline predisposition to hematopoietic malignancies: the university of chicago experience. Front Pediatry (2017) 5:252. doi: 10.3389/fped.2017.00252
8. Godley LA, Shimamura A. Genetic predisposition to hematologic malignancies: management and surveillance. Blood (2017) 130:424–32. doi: 10.1182/blood-2017-02-735290
9. Seleman M, Hoyos-Bachiloglu R, Geha RS, Chou J. Uses of next-generation sequencing technologies for the diagnosis of primary immunodeficiencies. Front Immunol. (2017) 8:847. doi: 10.3389/fimmu.2017.00847
10. Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, Coban Akdemir ZH, et al. Primary immunodeficiency diseases: genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. (2017) 139:232–45. doi: 10.1016/j.jaci.2016.05.042
11. Al-Mousa H, Abouelhoda M, Monies DM, Al-Tassan N, Al-Ghonaium A, Al-Saud B, et al. Unbiased targeted next-generation sequencing molecular approach for primary immunodeficiency diseases. J Allergy Clin Immunol. (2016) 137:1780–7. doi: 10.1016/j.jaci.2015.12.1310
12. Gallo V, Dotta L, Giardino G, Cirillo E, Lougaris V, D'Assante R, et al. Diagnostics of primary immunodeficiencies through next-generation sequencing. Front Immunol. (2016) 7:466. doi: 10.3389/fimmu.2016.00466
13. Kuhlen M, Borkhardt A. Cancer susceptibility syndromes in children in the area of broad clinical use of massive parallel sequencing. Eur J Pediatry (2015) 174:987–97. doi: 10.1007/s00431-015-2565-x
14. Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. (2015) 373:2336–46. doi: 10.1056/NEJMoa1508054
15. Nijman IJ, van Montfrans JM, Hoogstraat M, Boes ML, van de Corput L, Renner ED, et al. Targeted next-generation sequencing: a novel diagnostic tool for primary immunodeficiencies. J Allergy Clin Immunol. (2014) 133:529–34. doi: 10.1016/j.jaci.2013.08.032
16. Raje N, Soden S, Swanson D, Ciaccio CE, Kingsmore SF, Dinwiddie DL. Utility of next generation sequencing in clinical primary immunodeficiencies. Curr Allergy Asthma Rep. (2014) 14:468. doi: 10.1007/s11882-014-0468-y
17. Raje N, Soden S, Swanson D, Ciaccio CE, Kingsmore SF, Dinwiddie DL. Primary Immunodeficiency Diseases. Berlin Heidelberg: Springer (2016).
18. Arkwright PD, Gennery AR. Ten warning signs of primary immunodeficiency: a new paradigm is needed for the 21st century. Ann NY Acad Sci. (2011) 1238:7–14. doi: 10.1111/j.1749-6632.2011.06206.x
19. Cunningham-Rundles C. The many faces of common variable immunodeficiency. Hematol Am Soc Hematol Educ Program (2012) 2012:301–5. doi: 10.1182/asheducation-2012.1.301
20. Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. (2018) 38:129–43. doi: 10.1007/s10875-017-0465-8
21. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
22. Conley ME, Notarangelo LD, Casanova JL. Definition of primary immunodeficiency in 2011: a “trialogue” among friends. Ann N Y Acad Sci. (2011) 1238:1–6. doi: 10.1111/j.1749-6632.2011.06212.x
23. Studd JB, Yang M, Li Z, Vijayakrishnan J, Vijayakrishnan J, Lu Y, et al. Genetic predisposition to B-cell acute lymphoblastic leukemia at 14q11.2 is mediated by a CEBPE promoter polymorphism. Leukemia (2018) 100:347. doi: 10.1038/s41375-018-0184-z
24. Vijayakrishnan J, Studd J, Broderick P, Kinnersley B, Holroyd A, Law PJ, et al. Genome-wide association study identifies susceptibility loci for B-cell childhood acute lymphoblastic leukemia. Nat Commun. (2018) 9:1340. doi: 10.1038/s41467-018-03178-z
25. Wiemels JL, Walsh KM, de Smith AJ, Metayer C, Gonseth S, Hansen HM, et al. GWAS in childhood acute lymphoblastic leukemia reveals novel genetic associations at chromosomes 17q12 and 8q24.21. Nat Commun. (2018) 9:286. doi: 10.1038/s41467-017-02596-9
26. Studd JB, Vijayakrishnan J, Yang M, Migliorini G, Paulsson K, Houlston RS. Genetic and regulatory mechanism of susceptibility to high-hyperdiploid acute lymphoblastic leukaemia at 10p21.2. Nat Commun. (2017) 8:14616. doi: 10.1038/ncomms14616
27. Sud A, Thomsen H, Law PJ, Försti A, Filho MIDS, Holroyd A, et al. Genome-wide association study of classical Hodgkin lymphoma identifies key regulators of disease susceptibility. Nat Commun. (2017) 8:1892. doi: 10.1038/s41467-017-00320-1
28. Vijayakrishnan J, Kumar R, Henrion MY, Moorman AV, Rachakonda PS, Hosen I, et al. A genome-wide association study identifies risk loci for childhood acute lymphoblastic leukemia at 10q26.13 and 12q23.1. Leukemia (2017) 31:573–9. doi: 10.1038/leu.2016.271
29. odil N, Langlais D, Gros P. Primary immunodeficiencies and inflammatory disease: a growing genetic intersection. Trends Immunol. (2016) 37:126–40. doi: 10.1016/j.it.2015.12.006
30. Cerhan JR, Slager SL. Familial predisposition and genetic risk factors for lymphoma. Blood (2015) 126:2265–73. doi: 10.1182/blood-2015-04-537498
31. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. (2010) 42:1118–25. doi: 10.1038/ng.717
32. Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. (2009) 41:1006–10. doi: 10.1038/ng.430
33. Heitzeneder S, Seidel M, Forster-Waldl E, Heitger A. Mannan-binding lectin deficiency - Good news, bad news, doesn't matter? Clin Immunol. (2012) 143:22–38. doi: 10.1016/j.clim.2011.11.002
34. Taeubner J, Wieczorek D, Yasin L, Brozou T, Borkhardt A, Kuhlen M. Penetrance and expressivity in inherited cancer predisposing syndromes. Trends Cancer (2018) 4:718–28. doi: 10.1016/j.trecan.2018.09.002
35. Kuhlen M, Borkhardt A. Trio sequencing in pediatric cancer and clinical implications. EMBO Mol Med. (2018) 10:e8641–7. doi: 10.15252/emmm.201708641
36. Kratz CP, Achatz MI, Brugières L, Frebourg T, Garber JE, Greer MC, et al. Cancer screening recommendations for individuals with Li-Fraumeni syndrome. Clin Cancer Res. (2017) 23:e38–45. doi: 10.1158/1078-0432.CCR-17-0408
37. Porter CC, Druley TE, Erez A, Kuiper RP, Onel K, Schiffman JD, et al. Recommendations for surveillance for children with leukemia-predisposing conditions. Clin Cancer Res. (2017) 23:e14–22. doi: 10.1158/1078-0432.CCR-17-0428
38. Ripperger T, Bielack SS, Borkhardt A, Brecht IB, Burkhardt B, Calaminus G, et al. Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am J Med Genet A (2017) 173:1017–37. doi: 10.1002/ajmg.a.38142
39. Tabori U, Hansford JR, Achatz MI, Kratz CP, Plon SE, Frebourg T, et al. Clinical management and tumor surveillance recommendations of inherited mismatch repair deficiency in childhood. Clin Cancer Res. (2017) 23:e32–7. doi: 10.1158/1078-0432.CCR-17-0574
40. Walsh MF, Chang VY, Kohlmann WK, Scott HS, Cunniff C, Bourdeaut F, et al. Recommendations for childhood cancer screening and surveillance in DNA repair disorders. Clin Cancer Res. (2017) 23:e23–31. doi: 10.1158/1078-0432.CCR-17-0465
41. Jongmans MC, Loeffen JL, Waanders E, Hoogerbrugge PM, Ligtenberg MJ, Kuiper RP, et al. Recognition of genetic predisposition in pediatric cancer patients: an easy-to-use selection tool. Eur J Med Genet. (2016) 59:116–25. doi: 10.1016/j.ejmg.2016.01.008
42. Kratz CP, Stanulla M, Cave H. Genetic predisposition to acute lymphoblastic leukemia: overview on behalf of the I-BFM ALL Host Genetic Variation Working Group. Eur J Med Genet. (2016) 59:111–5. doi: 10.1016/j.ejmg.2015.10.003
43. Similuk M, Rao VK, Churpek J, Lenardo M. Predispositions to lymphoma: a practical review for genetic counselors. J Genet Couns. (2016) 25:1157–70. doi: 10.1007/s10897-016-9979-0
44. Hampel H, Bennett RL, Buchanan A, Pearlman R, Wiesner GL, Guideline Development Group, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. (2015) 17:70–87. doi: 10.1038/gim.2014.147
45. Schneider M, Chandler K, Tischkowitz M, Meyer S. Fanconi anaemia: genetics, molecular biology, and cancer - implications for clinical management in children and adults. Clin Genet. (2015) 88:13–24. doi: 10.1111/cge.12517
46. Vasen HF, Ghorbanoghli Z, Bourdeaut F, Cabaret O, Caron O, Duval A, et al. Guidelines for surveillance of individuals with constitutional mismatch repair-deficiency proposed by the European Consortium “Care for CMMR-D” (C4CMMR-D). J Med Genet. (2014) 51:283–93. doi: 10.1136/jmedgenet-2013-102238
47. Wimmer K, Kratz CP, Vasen HF, Caron O, Colas C, Entz-Werle N, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'care for CMMRD' (C4CMMRD). J Med Genet. (2014) 51:355–65. doi: 10.1136/jmedgenet-2014-102284
48. Auer F, Rüschendorf F, Gombert M, Husemann P, Ginzel S, Izraeli S, et al. Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G> A. Leukemia (2014) 28:1136–8. doi: 10.1038/leu.2013.363
49. Auer F, Lin M, Nebral K, Gertzen CGW, Haas OA, Kuhlen M, et al. Novel recurrent germline JAK2 G571S variant in childhood acute B-lymphoblastic leukemia: a double hit one pathway scenario. Blood (2018) 132:387. doi: 10.1182/blood-2018-99-115293
50. Mitchell KJ. What is complex about complex disorders? Genome Biol. (2012) 13:237. doi: 10.1186/gb-2012-13-1-237
51. Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: from polygenic to omnigenic. Cell (2017) 169:1177–86. doi: 10.1016/j.cell.2017.05.038
52. Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature (2015) 518:337–43. doi: 10.1038/nature13835
53. Maurano MT, Haugen E, Sandstrom R, Vierstra J, Shafer A, Kaul R, et al. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat Genet. (2015) 47:1393–401. doi: 10.1038/ng.3432
54. Wray NR, Wijmenga C, Sullivan PF, Yang J, Visscher PM. Common disease is more complex than implied by the core gene omnigenic model. Cell (2018) 173:1573–80. doi: 10.1016/j.cell.2018.05.051
55. Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. (2018) 50:1219–24. doi: 10.1038/s41588-018-0183-z
56. Sonnenschein C, Soto AM. Carcinogenesis explained within the context of a theory of organisms. Prog Biophys Mol Biol. (2016) 122:70–6. doi: 10.1016/j.pbiomolbio.2016.07.004
57. Soto AM, Sonnenschein C. The somatic mutation theory of cancer: growing problems with the paradigm? Bioessays (2004) 26:1097–107. doi: 10.1002/bies.20087
58. Laconi E, Sonnenschein C. Cancer development at tissue level. Semin Cancer Biol. (2008) 18:303–4. doi: 10.1016/j.semcancer.2008.03.002
59. Longo G, Montevil M, Sonnenschein C, Soto AM. In search of principles for a theory of organisms. J Biosci. (2015) 40:955–68. doi: 10.1007/s12038-015-9574-9
60. Hauck F, Voss R, Urban C, Seidel MG. Intrinsic and extrinsic causes of malignancies in patients with primary immunodeficiency disorders. J Allergy Clin Immunol. (2018) 141:59–68.e4. doi: 10.1016/j.jaci.2017.06.009
61. Corthay A. Does the immune system naturally protect against cancer? Front Immunol. (2014) 5:197. doi: 10.3389/fimmu.2014.00197
62. Chow MT, Moller A, Smyth MJ. Inflammation and immune surveillance in cancer. Semin Cancer Biol. (2012) 22:23–32. doi: 10.1016/j.semcancer.2011.12.004
63. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
64. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. (2007) 117:1137–46. doi: 10.1172/JCI31405
65. Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabási AL. The human disease network. Proc Natl Acad Sci USA. (2007) 104:8685–90. doi: 10.1073/pnas.0701361104
66. Latour S, Winter S. Inherited immunodeficiencies with high predisposition to epstein-barr virus-driven lymphoproliferative diseases. Front Immunol. (2018) 9:1103. doi: 10.3389/fimmu.2018.01103
67. Mayor PC, Eng KH, Singel KL, Abrams SI, Odunsi K, Moysich KB, et al. Cancer in primary immunodeficiency diseases: cancer incidence in the United States Immune Deficiency Network Registry. J Allergy Clin Immunol. (2018) 141:1028–35. doi: 10.1016/j.jaci.2017.05.024
68. Satge D. A tumor profile in primary immune deficiencies challenges the cancer immune surveillance concept. Front Immunol. (2018) 9:1149. doi: 10.3389/fimmu.2018.01149
69. Verhoeven D, Stoppelenburg AJ, Meyer-Wentrup F, Boes M. Increased risk of hematologic malignancies in primary immunodeficiency disorders: opportunities for immunotherapy. Clin Immunol. (2018) 190:22–31. doi: 10.1016/j.clim.2018.02.007
70. Mortaz E, Tabarsi P, Mansouri D, Khosravi A, Garssen J, Velayati A, et al. Cancers related to immunodeficiencies: update and perspectives. Front Immunol. (2016) 7:365. doi: 10.3389/fimmu.2016.00365
71. de Miranda NF, Björkman A, Pan-Hammarström Q. DNA repair: the link between primary immunodeficiency and cancer. Ann N Y Acad Sci. (2011) 1246:50–63. doi: 10.1111/j.1749-6632.2011.06322.x
72. Shapiro RS. Malignancies in the setting of primary immunodeficiency: implications for hematologists/oncologists. Am J Hematol. (2011) 86:48–55. doi: 10.1002/ajh.21903
73. Racicot K, Kwon JY, Aldo P, Silasi M, Mor G. Understanding the complexity of the immune system during pregnancy. Am J Reprod Immunol. (2014) 72:107–116. doi: 10.1111/aji.12289
74. Mor G, Cardenas I. The immune system in pregnancy: a unique complexity. Am J Reprod Immunol. (2010) 63:425–33. doi: 10.1111/j.1600-0897.2010.00836.x
75. Burlingham WJ. A lesson in tolerance–maternal instruction to fetal cells. N Engl J Med. (2009) 360:1355–7. doi: 10.1056/NEJMcibr0810752
76. Mold JE, Michaëlsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science (2008) 322:1562–5. doi: 10.1126/science.1164511
77. Trowsdale J, Betz AG. Mother's little helpers: mechanisms of maternal-fetal tolerance. Nat Immunol. (2006) 7:241–6. doi: 10.1038/ni1317
78. Kinder JM, Stelzer IA, Arck PC, Way SS. Immunological implications of pregnancy-induced microchimerism. Nat Rev Immunol. (2017) 17:483–94. doi: 10.1038/nri.2017.38
79. Cismaru CA, Pop L, Berindan-Neagoe I. Incognito: are microchimeric fetal stem cells that cross placental barrier real emissaries of peace? Stem Cell Rev. (2018) 14:632–41. doi: 10.1007/s12015-018-9834-9
80. Jeanty C, Derderian SC, Mackenzie TC. Maternal-fetal cellular trafficking: clinical implications and consequences. Curr Opin Pediatr. (2014) 26:377–82. doi: 10.1097/MOP.0000000000000087
81. Müller SM, Ege M, Pottharst A, Schulz AS, Schwarz K, Friedrich W. Transplacentally acquired maternal T lymphocytes in severe combined immunodeficiency: a study of 121 patients. Blood (2001) 98:1847–851.
82. Wahlstrom J, Patel K, Eckhert E, Kong D, Horn B, Cowan MJ, et al. Transplacental maternal engraftment and posttransplantation graft-versus-host disease in children with severe combined immunodeficiency. J Allergy Clin Immunol. (2017) 139:628–33.e10. doi: 10.1016/j.jaci.2016.04.049
83. van Rood JJ, Scaradavou A, Stevens CE. Indirect evidence that maternal microchimerism in cord blood mediates a graft-versus-leukemia effect in cord blood transplantation. Proc Natl Acad Sci USA. (2012) 109:2509–14. doi: 10.1073/pnas.1119541109
84. Isoda T, Ford AM, Tomizawa D, van Delft FW, De Castro DG, Mitsuiki N, et al. Immunologically silent cancer clone transmission from mother to offspring. Proc Natl Acad Sci USA. (2009) 106:17882–5. doi: 10.1073/pnas.0904658106
85. van den Boogaardt DE, van Rood JJ, Roelen DL, Claas FH. The influence of inherited and noninherited parental antigens on outcome after transplantation. Transpl Int. (2006) 19:360–71. doi: 10.1111/j.1432-2277.2006.00304.x
86. Dendrou CA, Petersen J, Rossjohn J, Fugger L. HLA variation and disease. Nat Rev Immunol. (2018) 18:325–39. doi: 10.1038/nri.2017.143
87. Trowsdale J, Knight JC. Major histocompatibility complex genomics and human disease. Annu Rev Genomics Hum Genet. (2013) 14:301–23. doi: 10.1146/annurev-genom-091212-153455
88. Taylor M, Hussain A, Urayama K, Chokkalingam A, Thompson P, Trachtenberg E, et al. The human major histocompatibility complex and childhood leukemia: an etiological hypothesis based on molecular mimicry. Blood Cells Mol Dis. (2009) 42:129–35. doi: 10.1016/j.bcmd.2008.10.009
89. Mosaad YM. Clinical role of human leukocyte antigen in health and disease. Scand J Immunol. (2015) 82:283–306. doi: 10.1111/sji.12329
90. Klitz W, Gragert L, Trachtenberg E. Spectrum of HLA associations: the case of medically refractory pediatric acute lymphoblastic leukemia. Immunogenetics (2012) 64:409–19. doi: 10.1007/s00251-012-0605-5
91. Masucci GV, Andersson E, Villabona L, Helgadottir H. Survival of the fittest or best adapted: HLA-dependent tumor development. J Nucleic Acids Investig. (2010) doi: 10.4081/jnai.2010.1654
92. Taylor GM, Dearden S, Ravetto P, Ayres M, Watson P, Hussain A, et al. Genetic susceptibility to childhood common acute lymphoblastic leukaemia is associated with polymorphic peptide-binding pocket profiles in HLA-DPB1*0201. Hum Mol Genet. (2002) 11:1585–97.
93. Taylor GM, Hussain A, Lightfoot TJ, Birch JM, Eden TO, Greaves MF. HLA-associated susceptibility to childhood B-cell precursor ALL: definition and role of HLA-DPB1 supertypes. Br J Cancer (2008) 98:1125–31. doi: 10.1038/sj.bjc.6604257
94. Taylor M, Harrison C, Eden T, Birch J, Greaves M, Lightfoot T, et al. HLA-DPB1 supertype-associated protection from childhood leukaemia: relationship to leukaemia karyotype and implications for prevention. Cancer Immunol Immunother. (2008) 57:53–61. doi: 10.1007/s00262-007-0349-5
95. Greaves M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat Rev Cancer (2018) 18:471–84. doi: 10.1038/s41568-018-0015-6
96. Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer (2006) 6:193–203. doi: 10.1038/nrc1816
97. Greaves M. In utero origins of childhood leukaemia. Early Hum Dev. (2005) 81:123–9. doi: 10.1016/j.earlhumdev.2004.10.004
98. Marty R, Kaabinejadian S, Rossell D, Slifker MJ, van de Haar J, Engin HB, et al. MHC-I genotype restricts the oncogenic mutational landscape. Cell (2017) 171:1272–1283.e15. doi: 10.1016/j.cell.2017.09.050
99. Roberts I, Izraeli S. Haematopoietic development and leukaemia in Down syndrome. Br J Haematol. (2014) 167:587–99. doi: 10.1111/bjh.13096
100. Yoshida K, Toki T, Okuno Y, Kanezaki R, Shiraishi Y, Sato-Otsubo A, et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat Genet (2013) 45:1293–1299. doi: 10.1038/ng.2759
101. Malinge S, Izraeli S, Crispino JD. Insights into the manifestations, outcomes, and mechanisms of leukemogenesis in Down syndrome. Blood (2009) 113:2619–28. doi: 10.1182/blood-2008-11-163501
102. Izraeli S. Down's syndrome as a model of a pre-leukemic condition. Haematologica (2006) 91:1448–52.
103. Raciborska A, Bilska K, Wecławek-Tompol J, Ussowicz M, Pogorzała M, Janowska J, et al. Solid cancers in the premature and the newborn: report of three national referral centers. Pediatr Neonatol. (2016) 57:295–301. doi: 10.1016/j.pedneo.2015.08.007
104. Roberts I, Fordham NJ, Rao A, Bain BJ. Neonatal leukaemia. Br J Haematol. (2018) 182:170–84. doi: 10.1111/bjh.15246
106. Perera FP. Environment and cancer: who are susceptible? Science (1997) 278:1068–73. doi: 10.1126/science.278.5340.1068
107. Tesch VK, IJspeert H, Raicht A, Rueda D, Dominguez-Pinilla N, Allende LM, et al. No overt clinical immunodeficiency despite immune biological abnormalities in patients with constitutional mismatch repair deficiency. Front Immunol. (2018) 9:1506. doi: 10.3389/fimmu.2018.01506
108. Prochazkova J, Loizou JI. Programmed DNA breaks in lymphoid cells: repair mechanisms and consequences in human disease. Immunology (2016) 147:11–20. doi: 10.1111/imm.12547
109. Lavoine N, Colas C, Muleris M, Bodo S, Duval A, Entz-Werle N, et al. Constitutional mismatch repair deficiency syndrome: clinical description in a French cohort. J Med Genet. (2015) 52:770–8. doi: 10.1136/jmedgenet-2015-103299
110. Chrzanowska KH, Gregorek H, Dembowska-Baginska B, Kalina MA, Digweed M. Nijmegen breakage syndrome (NBS). Orphanet J Rare Dis. (2012) 7:13. doi: 10.1186/1750-1172-7-13
111. Moraes MC, Neto JB, Menck CF. DNA repair mechanisms protect our genome from carcinogenesis. Front Biosci. (2012) 17:1362–88.
112. Slatter MA, Gennery AR. Primary immunodeficiencies associated with DNA-repair disorders. Expert Rev Mol Med. (2010) 12:e9. doi: 10.1017/S1462399410001419
113. Durno CA, Sherman PM, Aronson M, Malkin D, Hawkins C, Bakry D, et al. Phenotypic and genotypic characterisation of biallelic mismatch repair deficiency (BMMR-D) syndrome. Eur J Cancer (2015) 51:977–83. doi: 10.1016/j.ejca.2015.02.008
114. Bakry D, Aronson M, Durno C, Rimawi H, Farah R, Alharbi QK, et al. Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: report from the constitutional mismatch repair deficiency consortium. Eur J Cancer (2014) 50:987–96. doi: 10.1016/j.ejca.2013.12.005
115. Wimmer K, Kratz CP. Constitutional mismatch repair-deficiency syndrome. Haematologica (2010) 95:699–701. doi: 10.3324/haematol.2009.021626
116. Thoms KM, Kuschal C, Emmert S. Lessons learned from DNA repair defective syndromes. Exp Dermatol. (2007) 16:532–44. doi: 10.1111/j.1600-0625.2007.00559.x
117. Lehmann AR. The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev. (2001) 15:15–23.
118. de Boer J, Hoeijmakers JH. Nucleotide excision repair and human syndromes. Carcinogenesis (2000) 21:453–60.
119. Kraemer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience (2007) 145:1388–96. doi: 10.1016/j.neuroscience.2006.12.020
120. Bootsma D, Hoeijmakers JH. Repair DNA. Engagement with transcription. Nature (1993) 363:114–5. doi: 10.1038/363114a0
121. Bogaert DJ, Dullaers M, Lambrecht BN, Vermaelen KY, et al. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet. (2016) 53:575–90. doi: 10.1136/jmedgenet-2015-103690
122. Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova JL, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol. (2016) 7:220. doi: 10.3389/fimmu.2016.00220
123. Abbott JK, Gelfand EW. Common variable immunodeficiency: diagnosis, management, and treatment. Immunol Allergy Clin North Am. (2015) 35:637–58. doi: 10.1016/j.iac.2015.07.009
124. Gangemi S, Allegra A, Musolino C. Lymphoproliferative disease and cancer among patients with common variable immunodeficiency. Leuk Res. (2015) 39:389–96. doi: 10.1016/j.leukres.2015.02.002
125. Ameratunga R, Brewerton M, Slade C, Jordan A, Gillis D, Steele R, et al. Comparison of diagnostic criteria for common variable immunodeficiency disorder. Front Immunol. (2014) 5:415. doi: 10.3389/fimmu.2014.00415
126. Gathmann B, Mahlaoui N, Gérard L, Oksenhendler E, Warnatz K, Schulze I, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. (2014) 134:116–26. doi: 10.1016/j.jaci.2013.12.1077
127. Park JH, Resnick ES, Cunningham-Rundles C. Perspectives on common variable immune deficiency. Ann N Y Acad Sci. (2011) 1246:41–9. doi: 10.1111/j.1749-6632.2011.06338.x
128. Vajdic CM, Mao L, van Leeuwen MT, Kirkpatrick P, Grulich AE, Riminton S. Are antibody deficiency disorders associated with a narrower range of cancers than other forms of immunodeficiency? Blood (2010) 116:1228–34. doi: 10.1182/blood-2010-03-272351
129. Salavoura K, Kolialexi A, Tsangaris G, Mavrou A. Development of cancer in patients with primary immunodeficiencies. Anticancer Res. (2008) 28:1263–9.
130. Mellemkjaer L, Hammarstrom L, Andersen V, Yuen J, Heilmann C, Barington T, et al. Cancer risk among patients with IgA deficiency or common variable immunodeficiency and their relatives: a combined Danish and Swedish study. Clin Exp Immunol. (2002) 130:495–500.
131. Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Investig. (2015) 125:3338–46. doi: 10.1172/JCI80004
132. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol. (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
133. Gross E, Sunwoo JB, Bui JD. Cancer immunosurveillance and immunoediting by natural killer cells. Cancer J. (2013) 19:483–9. doi: 10.1097/PPO.0000000000000005
134. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science (2011) 331:1565–70. doi: 10.1126/science.1203486
135. Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. Int J Biol Sci. (2011) 7:651–8.
136. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology (2007) 121:1–14. doi: 10.1111/j.1365-2567.2007.02587.x
137. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity (2004) 21:137–48. doi: 10.1016/j.immuni.2004.07.017
138. Ribatti D. The concept of immune surveillance against tumors. The first theories. Oncotarget (2017) 8:7175–80. doi: 10.18632/oncotarget.12739
139. Jonkman-Berk BM, van den Berg JM, Ten Berge IJ, Bredius RG, Driessen GJ, Dalm VA, et al. Primary immunodeficiencies in the Netherlands: national patient data demonstrate the increased risk of malignancy. Clinical Immunol. (2015) 156:154–62. doi: 10.1016/j.clim.2014.10.003
140. McGonagle D, Aziz A, Dickie LJ, McDermott MF. An integrated classification of pediatric inflammatory diseases, based on the concepts of autoinflammation and the immunological disease continuum. Pediatr Res. (2009) 65:38R–45R. doi: 10.1203/PDR.0b013e31819dbd0a
141. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454:428–35. doi: 10.1038/nature07201
142. Karin M, Clevers H. Reparative inflammation takes charge of tissue regeneration. Nature (2016) 529:307–15. doi: 10.1038/nature17039
143. Dzutsev A, Goldszmid RS, Viaud S, Zitvogel L, Trinchieri G. The role of the microbiota in inflammation, carcinogenesis, and cancer therapy. Eur J Immunol. (2015) 45:17–31. doi: 10.1002/eji.201444972
144. Shalapour S, Karin M. Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest. (2015) 125:3347–55. doi: 10.1172/JCI80007
145. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454:436–44. doi: 10.1038/nature07205
146. Baniyash M, Sade-Feldman M, Kanterman J. Chronic inflammation and cancer: suppressing the suppressors. Cancer Immunol Immunother (2014) 63:11–20. doi: 10.1007/s00262-013-1468-9
147. Ferreri AJ, Govi S, Ponzoni M. Marginal zone lymphomas and infectious agents. Semin Cancer Biol. (2013) 23:431–40. doi: 10.1016/j.semcancer.2013.09.004
148. Thieblemont C, Bertoni F, Copie-Bergman C, Ferreri AJ, Ponzoni M. Chronic inflammation and extra-nodal marginal-zone lymphomas of MALT-type. Semin Cancer Biol. (2014) 24:33–42. doi: 10.1016/j.semcancer.2013.11.005
149. Valente AL, Schroeder B, Shriver CD, Henning JD, et al. Chronic inflammation in cancer: the role of human viruses. Adv Tumor Virol. (2015) 5:1–11. doi: 10.4137/atv.S19779
151. Boyapati R, Satsangi J, Ho GT. Pathogenesis of Crohn's disease. F1000Prime Rep. (2015) 7:44. doi: 10.12703/P7-44
152. Schreiber S, Rosenstiel P, Albrecht M, Hampe J, Krawczak M. Genetics of Crohn disease, an archetypal inflammatory barrier disease. Nat Rev Genet. (2005) 6:376–88. doi: 10.1038/nrg1607
153. Neven B, Mamessier E, Bruneau J, Kaltenbach S, Kotlarz D, Suarez F, et al. A Mendelian predisposition to B-cell lymphoma caused by IL-10deficiency R. Blood (2013) 122:3713–3722. doi: 10.1182/blood-2013-06-508267
154. Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology (2012) 143:347–55. doi: 10.1053/j.gastro.2012.04.045
155. Glocker EO, Kotlarz D, Klein C, Shah N, Grimbacher B. IL-10 and IL-10 receptor defects in humans. Ann N Y Acad Sci. (2011) 1246:102–7. doi: 10.1111/j.1749-6632.2011.06339.x
156. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. (2009) 361:2033–45. doi: 10.1056/NEJMoa0907206
157. Schmidt RE, Grimbacher B, Witte T. Autoimmunity and primary immunodeficiency: two sides of the same coin? Nat Rev Rheumatol. (2017) 14:7–18. doi: 10.1038/nrrheum.2017.198
158. Seidel MG. Autoimmune and other cytopenias in primary immunodeficiencies: pathomechanisms, novel differential diagnoses, and treatment. Blood (2014) 124:2337–44. doi: 10.1182/blood-2014-06-583260
159. Arkwright PD, Abinun M, Cant AJ. Autoimmunity in human primary immunodeficiency diseases. Blood (2002) 99:2694–702.
160. Rieux-Laucat F, Magerus-Chatinet A, Neven B. The autoimmune lymphoproliferative syndrome with defective FAS or FAS-ligand functions. J Clin Immunol. (2018) 38:558–68. doi: 10.1007/s10875-018-0523-x
161. Price S, Shaw PA, Seitz A, Joshi G, Davis J, Niemela JE, et al. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood (2014) 123:1989–99. doi: 10.1182/blood-2013-10-535393
162. Straus SE, Jaffe ES, Puck JM, Dale JK, Elkon KB, Rösen-Wolff A, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood (2001) 98:194–200.
163. Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, et al. Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: an international multicenter retrospective study. J Allergy Clin Immunol. (2018) 141:1036–49 e1035. doi: 10.1016/j.jaci.2017.10.041
164. Gambineri E, Perroni L, Passerini L, Bianchi L, Doglioni C, Meschi F, et al. Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: inconsistent correlation between forkhead box protein 3 expression and disease severity. J Allergy Clin Immunol. (2008) 122:1105–1112.e1. doi: 10.1016/j.jaci.2008.09.027
165. Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest. (2006) 116:1713–22. doi: 10.1172/JCI25112
166. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322:271–5. doi: 10.1126/science.1160062
167. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. (2014) 20:1410–6. doi: 10.1038/nm.3746
168. Rudd CE. The reverse stop-signal model for CTLA4 function. Nat Rev Immunol. (2008) 8:153–60. doi: 10.1038/nri2253
169. Gough SC, Walker LS, Sansom DM. CTLA4 gene polymorphism and autoimmunity. Immunol Rev. (2005) 204:102–15. doi: 10.1111/j.0105-2896.2005.00249.x
170. Egg D, Schwab C, Gabrysch A, Arkwright PD, Cheesman E, Giulino-Roth L, et al. Increased risk for malignancies in 131 affected CTLA4 mutation carriers. Front Immunol. (2018) 9:2012. doi: 10.3389/fimmu.2018.02012
171. Boutboul D, Kuehn HS, Van de Wyngaert Z, Niemela JE, Callebaut I, Stoddard J, et al. Dominant-negative IKZF1 mutations cause a T, B, and myeloid cell combined immunodeficiency. J Clin Invest. (2018) 128:3071–87. doi: 10.1172/JCI98164
172. Churchman ML, Qian M, Te Kronnie G, Zhang R, Yang W, Zhang H, et al. Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell (2018) 33:937–948.e8. doi: 10.1016/j.ccell.2018.03.021
173. Kamihara J, Shimamura A. It's ALL in the family: IKZF1 and hereditary leukemia. Cancer Cell (2018) 33:798–800. doi: 10.1016/j.ccell.2018.04.008
174. Van Nieuwenhove E, Garcia-Perez JE, Helsen C, Rodriguez PD, van Schouwenburg PA, Dooley J, et al. A kindred with mutant IKAROS, autoimmunity. J Allergy Clin Immunol. (2018) 142:699–702.e2. doi: 10.1016/j.jaci.2018.04.008
175. Hoshino A, Okada S, Yoshida K, Nishida N, Okuno Y, Ueno H, et al. Abnormal hematopoiesis and autoimmunity in human subjects with germline IKZF1 mutations. J Allergy Clin Immunol. (2017) 140:223–31. doi: 10.1016/j.jaci.2016.09.029
176. Pellegrini FP, Marinoni M, Frangione V, Tedeschi A, Gandini V, Ciglia F, et al. Down syndrome, autoimmunity and T regulatory cells. Clin Exp Immunol. (2012) 169:238–43. doi: 10.1111/j.1365-2249.2012.04610.x
178. Cuadrado E, Barrena MJ. Immune dysfunction in Down's syndrome: primary immune deficiency or early senescence of the immune system? Clin Immunol Immunopathol. (1996) 78:209–14.
179. Cetica V, Sieni E, Pende D, Danesino C, De Fusco C, Locatelli F, et al. Genetic predisposition to hemophagocytic lymphohistiocytosis: report on 500 patients from the Italian registry. J Allergy Clin Immunol. (2016) 137:188–96.e4. doi: 10.1016/j.jaci.2015.06.048
180. Otrock ZK, Eby CS. Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol. (2015) 90:220–24. doi: 10.1002/ajh.23911
181. Arico M, Imashuku S, Clementi R, Hibi S, Teramura T, Danesino C, et al. Hemophagocytic lymphohistiocytosis due to germline mutations in SH2D1A, the X-linked lymphoproliferative disease gene. Blood (2001) 97:1131–3.
182. Chaudhry MS, Gilmour KC, House IG, Layton M, Panoskaltsis N, Sohal M, et al. Missense mutations in the perforin (PRF1) gene as a cause of hereditary cancer predisposition. Oncoimmunology (2016) 5:e1179415. doi: 10.1080/2162402X.2016.1179415
183. Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. (2015) 15:388–400. doi: 10.1038/nri3839
184. Osinska I, Popko K, Demkow U. Perforin: an important player in immune response. Cent Eur J Immunol. (2014) 39:109–15. doi: 10.5114/ceji.2014.42135
185. Trapani JA, Thia KY, Andrews M, Davis ID, Gedye C, Parente P, et al. Human perforin mutations and susceptibility to multiple primary cancers. Oncoimmunology (2013) 2:e24185. doi: 10.4161/onci.24185
186. Voskoboinik I, Trapani JA. Perforinopathy: a spectrum of human immune disease caused by defective perforin delivery or function. Front Immunol. (2013) 4:441. doi: 10.3389/fimmu.2013.00441
187. Ciambotti B, Mussolin L, d'Amore ES, Pillon M, Sieni E, Coniglio ML, et al. Monoallelic mutations of the perforin gene may represent a predisposing factor to childhood anaplastic large cell lymphoma. J Pediatr Hematol Oncol. (2014) 36:e359–65. doi: 10.1097/MPH.0000000000000073
188. Manso R, Rodríguez-Pinilla SM, Lombardia L, Ruiz de Garibay G, Del Mar López M, Requena L, et al. An A91V SNP in the perforin gene is frequently found in NK/T-cell lymphomas. PLoS ONE (2014) 9:e91521. doi: 10.1371/journal.pone.0091521
189. Cannella S, Santoro A, Bruno G, Pillon M, Mussolin L, Mangili G, et al. Germline mutations of the perforin gene are a frequent occurrence in childhood anaplastic large cell lymphoma. Cancer (2007) 109:2566–71. doi: 10.1002/cncr.22718
190. Clementi R, Locatelli F, Dupré L, Garaventa A, Emmi L, Bregni M, et al. A proportion of patients with lymphoma may harbor mutations of the perforin gene. Blood (2005) 105:4424–8. doi: 10.1182/blood-2004-04-1477
191. Aricò M, Mussolin L, Carraro E, Buffardi S, Santoro N, D'Angelo P, et al. Non-Hodgkin lymphoma in children with an associated inherited condition: a retrospective analysis of the Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP). Pediatr Blood Cancer (2015) 62:1782–9. doi: 10.1002/pbc.25565
192. Booth C, Gilmour KC, Veys P, Gennery AR, Slatter MA, Chapel H, et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood (2011) 117:53–62. doi: 10.1182/blood-2010-06-284935
193. Filipovich AH, Zhang K, Snow AL, Marsh RA. X-linked lymphoproliferative syndromes: brothers or distant cousins? Blood (2010) 116:3398–408. doi: 10.1182/blood-2010-03-275909
194. Round JL, Palm NW. Causal effects of the microbiota on immune-mediated diseases. Sci Immunol. (2018) 3:eaao1603. doi: 10.1126/sciimmunol.aao1603
195. Dzutsev A, Badger JH, Perez-Chanona E, Roy S, Salcedo R, Smith CK, et al. Microbes and cancer. Annu Rev Immunol. (2017) 35:199–228. doi: 10.1146/annurev-immunol-051116-052133
196. Kundu P, Blacher E, Elinav E, Pettersson S. Our gut microbiome: the evolving inner self. Cell (2017) 171:1481–93. doi: 10.1016/j.cell.2017.11.024
197. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol. (2017) 17:219–32. doi: 10.1038/nri.2017.7
198. Tamburini S, Shen N, Wu HC, Clemente JC. The microbiome in early life: implications for health outcomes. Nat Med. (2016) 22:713–22. doi: 10.1038/nm.4142
199. Garrett WS, Gordon JI, Glimcher LH. Homeostasis and inflammation in the intestine. Cell (2010) 140:859–70. doi: 10.1016/j.cell.2010.01.023
200. Khachatryan ZA, Ktsoyan ZA, Manukyan GP, Kelly D, Ghazaryan KA, Aminov RI. Predominant role of host genetics in controlling the composition of gut microbiota. PLoS ONE (2008) 3:e3064. doi: 10.1371/journal.pone.0003064
201. Benson AK. Host genetic architecture and the landscape of microbiome composition: humans weigh in. Genome Biol. (2015) 16:203. doi: 10.1186/s13059-015-0775-1
202. Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA. (2010) 107:18933–8. doi: 10.1073/pnas.1007028107
203. Fulde M, Sommer F, Chassaing B, van Vorst K, Dupont A, Hensel M, et al. Neonatal selection by Toll-like receptor 5 influences long-term gut microbiota composition. Nature (2018) 560:489–93. doi: 10.1038/s41586-018-0395-5
204. de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. (2012) 13:607–15. doi: 10.1016/S1470-2045(12)70137-7
206. Gagliani N, Hu B, Huber S, Elinav E, Flavell RA. The fire within: microbes inflame tumors. Cell (2014) 157:776–83. doi: 10.1016/j.cell.2014.03.006
207. Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell (2007) 131:33–45. doi: 10.1016/j.cell.2007.08.017
208. Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature (2018) 555:210–5. doi: 10.1038/nature25973
209. Blekhman R, Goodrich JK, Huang K, Sun Q, Bukowski R, Bell JT, et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. (2015) 16:191. doi: 10.1186/s13059-015-0759-1
210. Yamamoto ML, Maier I, Dang AT, Berry D, Liu J, Ruegger PM, et al. Intestinal bacteria modify lymphoma incidence and latency by affecting systemic inflammatory state, oxidative stress, and leukocyte genotoxicity. Cancer Res. (2013) 73:4222–4232. doi: 10.1158/0008-5472.CAN-13-0022
211. Bromberg JS, Hittle L, Xiong Y, Saxena V, Smyth EM, Li L, et al. Gut microbiota-dependent modulation of innate immunity and lymph node remodeling affects cardiac allograft outcomes. JCI Insight (2018) 3:121045. doi: 10.1172/jci.insight.121045
212. Plottel CS, Blaser MJ. Microbiome and malignancy. Cell Host Microbe (2011) 10:324–35. doi: 10.1016/j.chom.2011.10.003
213. Correia S, Bridges R, Wegner F, Venturini C, Palser A, Middeldorp JM, et al. Sequence variation of Epstein-Barr virus: viral types, geography, codon usage and diseases. J Virol. (2018) 92:e01132–18. doi: 10.1128/JVI.01132-18
214. Young LS, Yap LF, Murray PG. Epstein-Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer (2016) 16:789–802. doi: 10.1038/nrc.2016.92
215. Yilmaz B, Portugal S, Tran TM, Gozzelino R, Ramos S, Gomes J, et al. Gut microbiota elicits a protective immune response against malaria transmission. Cell (2014) 159:1277–89. doi: 10.1016/j.cell.2014.10.053
216. Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M, et al. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe (2012) 12:233–45. doi: 10.1016/j.chom.2012.06.008
217. Magrath I. Epidemiology: clues to the pathogenesis of Burkitt lymphoma. Br J Haematol. (2012) 156:744–56. doi: 10.1111/j.1365-2141.2011.09013.x
218. Mutalima N, Molyneux E, Jaffe H, Kamiza S, Borgstein E, Mkandawire N, et al. Associations between Burkitt Lymphoma among Children in Malawi and Infection with HIV, EBV and Malaria: results from a case-control study. PLoS ONE (2008) 3:e2505. doi: 10.1371/journal.pone.0002505.t005
219. Thorley-Lawson DA, Allday MJ. The curious case of the tumour virus: 50 years of Burkitt's lymphoma. Nat Rev Microbiol. (2008) 6:913–24. doi: 10.1038/nrmicro2015
220. Kwiatkowski DP. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. (2005) 77:171–92. doi: 10.1086/432519
221. Leffler EM, Band G, Busby GBJ, Kivinen K, Le QS, Clarke GM, et al. Resistance to malaria through structural variation of red blood cell invasion receptors. Science (2017) 356: eaam6393. doi: 10.1126/science.aam6393
222. Egan ES, Jiang RH, Moechtar MA, Barteneva NS, Weekes MP, Nobre LV, et al. Malaria. A forward genetic screen identifies erythrocyte CD55 as essential for Plasmodium falciparum invasion. Science (2015) 348:711–4. doi: 10.1126/science.aaa3526
223. Williams TN. Human red blood cell polymorphisms and malaria. Curr Opin Microbiol. (2006) 9:388–94. doi: 10.1016/j.mib.2006.06.009
224. Hadley TJ, Peiper SC. From malaria to chemokine receptor: the emerging physiologic role of the Duffy blood group antigen. Blood (1997) 89:3077–91.
225. Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell (1996) 86:367–77.
226. Rezk SA, Zhao X, Weiss LM. Epstein-Barr virus (EBV)-associated lymphoid proliferations, a 2018 update. Hum Pathol. (2018) 79:18–41. doi: 10.1016/j.humpath.2018.05.020
227. Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, et al. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature (2007) 447:326–9. doi: 10.1038/nature05762
228. White DW, Keppel CR, Schneider SE, Reese TA, Coder J, Payton JE, et al. Latent herpesvirus infection arms cells NK. Blood (2010) 115:4377–83. doi: 10.1182/blood-2009-09-245464
229. Farrell PJ. Epstein-Barr virus and cancer. Annu Rev Pathol. (2018) doi: 10.1146/annurev-pathmechdis-012418-013023
230. Iwatsuki K, Xu Z, Ohtsuka M, Kaneko F. Overview of EBV-associated T/NK-cell lymphoproliferative diseases. (2018) 1–17.
231. Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. (2015) 42:291–303. doi: 10.1053/j.seminoncol.2014.12.030
232. Palendira U, Rickinson AB. Primary immunodeficiencies and the control of Epstein-Barr virus infection. Ann N Y Acad Sci. (2015) 1356:22–44. doi: 10.1111/nyas.12937
233. Kushekhar K, van den Berg A, Nolte I, Hepkema B, Visser L, Diepstra A. Genetic associations in classical hodgkin lymphoma: a systematic review and insights into susceptibility mechanisms. Cancer Epidemiol Biomarkers Prev. (2014) 23:2737–47. doi: 10.1158/1055-9965.EPI-14-0683
234. Parvaneh N, Filipovich AH, Borkhardt A. Primary immunodeficiencies predisposed to Epstein-Barr virus-driven haematological diseases. Br J Haematol. (2013) 162:573–86. doi: 10.1111/bjh.12422
235. Worth AJ, Houldcroft CJ, Booth C. Severe Epstein-Barr virus infection in primary immunodeficiency and the normal host. Br J Haematol. (2016) 175:559–76. doi: 10.1111/bjh.14339
236. Vockerodt M, Yap LF, Shannon-Lowe C, Curley H, Wei W, Vrzalikova K, et al. The Epstein-Barr virus and the pathogenesis of lymphoma. J Pathol. (2015) 235:312–22. doi: 10.1002/path.4459
237. Khan G, Hashim MJ. Global burden of deaths from Epstein-Barr virus attributable malignancies 1990-2010. Infect Agent Cancer (2014) 9:38. doi: 10.1186/1750-9378-9-38
238. Robbiani DF, Deroubaix S, Feldhahn N, Oliveira TY, Callen E, Wang Q, et al. Plasmodium infection promotes genomic instability and AID-dependent B Cell lymphoma. Cell (2015) 162:727–37. doi: 10.1016/j.cell.2015.07.019
239. Quintanilla-Martinez L, Ridaura C, Nagl F, Sáez-de-Ocariz M, Durán-McKinster C, Ruiz-Maldonado R, et al. Hydroa vacciniforme-like lymphoma: a chronic EBV+ lymphoproliferative disorder with risk to develop a systemic lymphoma. Blood (2013) 122:3101–10. doi: 10.1182/blood-2013-05-502203
240. Tang M, Zeng Y, Poisson A, Marti D, Guan L, Zheng Y, et al. Haplotype-dependent HLA susceptibility to nasopharyngeal carcinoma in a Southern Chinese population. Genes Immun. (2010) 11:334–42. doi: 10.1038/gene.2009.109
241. Qin L, Chen C, Chen L, Xue R, Ou-Yang M, Zhou C, et al. Worldwide malaria incidence and cancer mortality are inversely associated. Infect Agent Cancer (2017) 12:14. doi: 10.1186/s13027-017-0117-x
242. Murray CJ, Ortblad KF, Guinovart C, Lim SS, Wolock TM, Roberts DA, et al. Global, regional, and national incidence and mortality for HIV, tuberculosis, and malaria during 1990-2013: a systematic analysis for the global burden of disease study 2013. Lancet (2014) 384:1005–70. doi: 10.1016/S0140-6736(14)60844-8
243. Lehrer S. Association between malaria incidence and all cancer mortality in fifty U.S. States, and the District of Columbia. Anticancer Res. (2010) 30:1371–3.
244. Chene A, Donati D, Orem J, Mbidde ER, Kironde F, Wahlgren M, et al. Endemic Burkitt's lymphoma as a polymicrobial disease: new insights on the interaction between Plasmodium falciparum and Epstein-Barr virus. Semin Cancer Biol. (2009) 19:411–20. doi: 10.1016/j.semcancer.2009.10.002
245. Robbiani DF, Bothmer A, Callen E, B Reina-San-Martin, Dorsett Y, Difilippantonio S, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell (2008) 135:1028–38. doi: 10.1016/j.cell.2008.09.062
246. Rochford R, Cannon MJ, Moormann AM. Endemic Burkitt's lymphoma: a polymicrobial disease? Nat Rev Microbiol. (2005) 3:182–7. doi: 10.1038/nrmicro1089
247. Roy A, Cowan G, Mead AJ, Filippi S, Bohn G, Chaidos A, et al. Perturbation of fetal liver hematopoietic stem and progenitor cell development by trisomy 21. Proc Natl Acad Sci USA. (2012) 109:17579–84. doi: 10.1073/pnas.1211405109
248. Ram G, Chinen J. Infections and immunodeficiency in Down syndrome. Clin Exp Immunol. (2011) 164:9–16. doi: 10.1111/j.1365-2249.2011.04335.x
249. Giménez-Barcons M, Casteràs A, Armengol Mdel P, Porta E, Correa PA, Marín A, et al. Autoimmune predisposition in Down syndrome may result from a partial central tolerance failure due to insufficient intrathymic expression of AIRE, peripheral antigens. J Immunol. (2014) 193:3872–9. doi: 10.4049/jimmunol.1400223
250. Sullivan KD, Evans D, Pandey A, Hraha TH, Smith KP, Markham N, et al. Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci Rep. (2017) 7:14818. doi: 10.1038/s41598-017-13858-3
251. Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, et al. Trisomy 21 consistently activates the interferon response. Elife (2016) 5:1709. doi: 10.7554/eLife.16220
252. Hasle H. Pattern of malignant disorders in individuals with Down's syndrome. Lancet Oncol. (2001) 2:429–36. doi: 10.1016/S1470-2045(00)00435-6
253. Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down's syndrome. Lancet (2000) 355:165–9. doi: 10.1016/S0140-6736(99)05264-2
254. Izraeli S. Leukaemia – a developmental perspective. Br J Haematol. (2004) 126:3–10. doi: 10.1111/j.1365-2141.2004.04986.x
256. Lane AA, Chapuy B, Lin CY, Tivey T, Li H, Townsend EC, et al. Triplication of a 21q22 region contributes to B cell transformation through HMGN1 overexpression and loss of histone H3 Lys27 trimethylation. Nat Genet. (2014) 46:618–23. doi: 10.1038/ng.2949
257. Reynolds LE, Watson AR, Baker M, Jones TA, D'Amico G, Robinson SD, et al. Tumour angiogenesis is reduced in the Tc1 mouse model of Down's syndrome. Nature (2010) 465:813–7. doi: 10.1038/nature09106
258. Rabin KR, Whitlock JA. Malignancy in children with trisomy 21. Oncologist (2009) 14:164–73. doi: 10.1634/theoncologist.2008-0217
259. Sussan TE, Yang A, Li F, Ostrowski MC, Reeves RH. Trisomy represses Apc(Min)-mediated tumours in mouse models of Down's syndrome. Nature (2008) 451:73–5. doi: 10.1038/nature06446
260. Threadgill DW. Down's syndrome: paradox of a tumour repressor. Nature (2008) 451:21–2. doi: 10.1038/451021a
261. Baek KH, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, et al. Down's syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature (2009) 459:1126–30. doi: 10.1038/nature08062
262. Suliman A, Smith FO, Kupfer GM. Fanconi anemia: a pathway of hematopoiesis and cancer predisposition. In: Kupfer G, Reaman G, Smith F, editors. Bone Marrow Failure. Cham: Springer International Publishing (2018).p. 81–97. doi: 10.1007/978-3-319-61421-2_4
263. Cheung RS, Taniguchi T. Recent insights into the molecular basis of Fanconi anemia: genes, modifiers, and drivers. Int J Hematol. (2017) 106:335–44. doi: 10.1007/s12185-017-2283-4
264. West AH, Churpek JE. Old and new tools in the clinical diagnosis of inherited bone marrow failure syndromes. Hematol Am Soc Hematol Educ Program (2017) 2017:79–87. doi: 10.1182/asheducation-2017.1.79
265. Chirnomas SD, Kupfer GM. The inherited bone marrow failure syndromes. Pediatr Clin North Am. (2013) 60:1291–310. doi: 10.1016/j.pcl.2013.09.007
266. Aspesi A, Ellis SR. Ribosomopathies through a diamond lens. In: Kupfer G, Reaman G, Smith F, editors. Bone Marrow Failure. Pediatric Oncology. Cham: Springer International Publishing (2018).
267. Hattangadi SM, Lipton JM. Diamond-blackfan anemia. In: Kupfer G, Reaman G, Smith F, editors. Bone Marrow Failure. Cham: Springer International Publishing (2018) doi: 10.1007/978-3-319-61421-2_7
268. Ulirsch JC, Verboon JM, Kazerounian S, Guo MH, Yuan D, Ludwig LS, et al. The genetic landscape of diamond-blackfan anemia. Am J Hum Genet. (2018) 103:930–47. doi: 10.1016/j.ajhg.2018.10.027
269. Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood (2010) 115:3196–205. doi: 10.1182/blood-2009-10-178129
270. Dodson L, Bertuch AA. Dyskeratosis Congenita and the Telomere Biology Disorders. In: Kupfer G, Reaman G, Smith F, editors. Bone Marrow Failure, Cham: Springer International Publishing (2018) doi: 10.1007/978-3-319-61421-2_6
271. Savage SA. Beginning at the ends: telomeres and human disease. F1000Res (2018) 7: F1000 Faculty Rev-524. doi: 10.12688/f1000research.14068.1
272. Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood (2014) 124:2775–83. doi: 10.1182/blood-2014-05-526285
273. Myers KC, Shimamura A. Shwachman-Diamond Syndrome. In: Kupfer G, Reaman G, Smith F, editors. Bone Marrow Failure, Cham: Springer International Publishing (2018) doi: 10.1007/978-3-319-61421-2_8
274. Kostjukovits S, Klemetti P, Valta H, Martelius T, Notarangelo LD, Seppänen M, et al. Analysis of clinical and immunologic phenotype in a large cohort of children and adults with cartilage-hair hypoplasia. J Allergy Clin Immunol. (2017) 140:612–4 e615. doi: 10.1016/j.jaci.2017.02.016
275. Bluteau O, Sebert M, Leblanc T, Peffault de Latour R, Quentin S, Lainey E, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood (2018) 131:717–32. doi: 10.1182/blood-2017-09-806489
276. Nakano T, Di Paola J. Inherited Thrombocytopenias. In: Kupfer G, Reaman G, Smith F, editors. Bone Marrow Failure, Cham: Springer International Publishing (2018) doi: 10.1007/978-3-319-61421-2_11
277. Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, et al. Compound inheritance of a low-frequency regulatory SNP, a rare null mutation in exon-junction complex subunit RBM8A causes syndrome TAR. Nat Genet. (2012) 44:435–439, S431–432. doi: 10.1038/ng.1083
278. Mehta HM, Corey SJ. Inherited Neutropenias and Their Insights into Cellular and Developmental Biology. In: Kupfer G, Reaman G, Smith F, editors. Bone Marrow Failure, Cham: Springer International Publishing, (2018) doi: 10.1007/978-3-319-61421-2_10
279. Skokowa J, Dale DC, Touw IP, Zeidler C, Welte K. Severe congenital neutropenias. Nat Rev Dis Prim. (2017) 3:17032. doi: 10.1038/nrdp.2017.32
280. Hirabayashi S, Wlodarski MW, Kozyra E, Niemeyer CM. Heterogeneity of GATA2-related myeloid neoplasms. Int J Hematol. (2017) 106:175–82. doi: 10.1007/s12185-017-2285-2
281. Wlodarski MW, Collin M, Horwitz MS. GATA2 deficiency and related myeloid neoplasms. Semin Hematol. (2017) 54:81–6. doi: 10.1053/j.seminhematol.2017.05.002
282. Wlodarski MW, Hirabayashi S, Pastor V, Starý J, Hasle H, Masetti R, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood (2016) 127:1387–97; quiz 1518. doi: 10.1182/blood-2015-09-669937
283. Churpek JE, Pyrtel K, Kanchi KL, Shao J, Koboldt D, Miller CA, et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood (2015) 126:2484–90. doi: 10.1182/blood-2015-04-641100
284. Collin M, Dickinson R, Bigley V. Haematopoietic and immune defects associated with GATA2 mutation. Br J Haematol. (2015) 169:173–87. doi: 10.1111/bjh.13317
285. Ganapathi KA, Townsley DM, Hsu AP, Arthur DC, Zerbe CS, Cuellar-Rodriguez J, et al. GATA2 deficiency-associated bone marrow disorder differs from idiopathic aplastic anemia. Blood (2015) 125:56–70. doi: 10.1182/blood-2014-06-580340
286. Mir MA, Kochuparambil ST, Abraham RS, Rodriguez V, Howard M, Hsu AP, et al. Spectrum of myeloid neoplasms and immune deficiency associated with germline GATA2 mutations. Cancer Med. (2015) 4:490–9. doi: 10.1002/cam4.384
287. Davidsson J, Puschmann A, Tedgård U, Bryder D, Nilsson L, Cammenga J. SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia (2018) 32:1106–15. doi: 10.1038/s41375-018-0074-4
288. Pastor VB, Sahoo SS, Boklan J, Schwabe GC, Saribeyoglu E, Strahm B, et al. Constitutional SAMD9L mutations cause familial myelodysplastic syndrome and transient monosomy 7. Haematologica (2018) 103:427–37. doi: 10.3324/haematol.2017.180778
289. Shima H, Koehler K, Nomura Y, Sugimoto K, Satoh A, Ogata T, et al. Two patients with MIRAGE syndrome lacking haematological features: role of somatic second-site reversion SAMD9 mutations. J Med Genet. (2018) 55:81–5. doi: 10.1136/jmedgenet-2017-105020
290. Wong JC, Bryant V, Lamprecht T, Ma J, Walsh M, Schwartz J, et al. Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. JCI Insight (2018) 3: 121086. doi: 10.1172/jci.insight.121086
291. Buonocore F, Kühnen P, Suntharalingham JP, Del Valle I, Digweed M, Stachelscheid H, et al. Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans. J Clin Invest. (2017) 127:1700–13. doi: 10.1172/JCI91913
292. Collin M. I am SAMD9L: 7q regulator am I. Blood (2017) 129:2210–2. doi: 10.1182/blood-2017-03-770198
293. Tesi B, Davidsson J, Voss M, Rahikkala E, Holmes TD, Chiang SCC, et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood (2017) 129:2266–79. doi: 10.1182/blood-2016-10-743302
294. Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, et al. SAMD9 mutations cause a novel multisystem disorder, syndrome MIRAGE, and are associated with loss of chromosome 7. Nat Genet. (2016) 48:792–7. doi: 10.1038/ng.3569
295. Murakami N, Okuno Y, Yoshida K, Shiraishi Y, Nagae G, Suzuki K, et al. Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood (2018) 131:1576–86. doi: 10.1182/blood-2017-07-798157
296. Niemeyer CM. JMML genomics and decisions. Hematology Am Soc Hematol Educ Program. (2018) 2018:307–12. doi: 10.1182/asheducation-2018.1.307
297. Cavé H, Caye A, Strullu M, Aladjidi N, Vignal C, Ferster A, et al. Acute lymphoblastic leukemia in the context of RASopathies. Eur J Med Genet. (2016) 59:173–8. doi: 10.1016/j.ejmg.2016.01.003
298. Stieglitz E, Taylor-Weiner AN, Chang TY, Gelston LC, Wang YD, Mazor T, et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet. (2015) 47:1326–33. doi: 10.1038/ng.3400
299. Hofmann I. Pediatric Myelodysplastic Syndromes. In: Kupfer G, Reaman G, Smith F, editors. Bone Marrow Failure, Cham: Springer International Publishing (2018) doi: 10.1007/978-3-319-61421-2_3
300. Wegman-Ostrosky T, Savage SA. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br J Haematol. (2017) 177:526–42. doi: 10.1111/bjh.14535
301. Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. (2009) 668:4–10. doi: 10.1016/j.mrfmmm.2009.01.013
302. Bagby G. Recent advances in understanding hematopoiesis in Fanconi Anemia. F1000Res (2018) 7:105. doi: 10.12688/f1000research.13213.1
303. Garaycoechea JI, Patel KJ. Why does the bone marrow fail in Fanconi anemia? Blood (2014) 123:26–34. doi: 10.1182/blood-2013-09-427740
304. Pontel LB, Rosado IV, Burgos-Barragan G, Garaycoechea JI, Yu R, Arends MJ, et al. Endogenous formaldehyde is a hematopoietic stem cell genotoxin and metabolic carcinogen. Mol Cell (2015) 60:177–88. doi: 10.1016/j.molcel.2015.08.020
305. Parmar K, D'Andrea AD. Stressed out: endogenous aldehydes damage hematopoietic stem cells. Cell Stem Cell (2012) 11:583–4. doi: 10.1016/j.stem.2012.10.007
306. Rosado IV, Langevin F, Crossan GP, Takata M, Patel KJ. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat Struct Mol Biol. (2011) 18:1432–4. doi: 10.1038/nsmb.2173
307. Oberbeck N, Langevin F, King G, de Wind N, Crossan GP, Patel KJ. Maternal aldehyde elimination during pregnancy preserves the fetal genome. Mol Cell (2014) 55:807–17. doi: 10.1016/j.molcel.2014.07.010
308. Van Wassenhove LD, Mochly-Rosen D, Weinberg KI. Aldehyde dehydrogenase 2 in aplastic anemia, Fanconi anemia and hematopoietic stem cells. Mol Genet Metab. (2016) 119:28–36. doi: 10.1016/j.ymgme.2016.07.004
309. Hira A, Yabe H, Yoshida K, Okuno Y, Shiraishi Y, Chiba K, et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood (2013) 122:3206–9. doi: 10.1182/blood-2013-06-507962
310. Ruiz S, Fernandez-Capetillo O. The maternal side of Fanconi Anemia. Mol Cell (2014) 55:803–4. doi: 10.1016/j.molcel.2014.08.029
311. Tan SLW, Chadha S, Liu Y, Gabasova E, Perera D, Ahmed K, et al. A class of environmental and endogenous toxins induces BRCA2 haploinsufficiency and genome instability. Cell (2017) 169:1105–18.e5. doi: 10.1016/j.cell.2017.05.010
312. Zhang QS, Tang W, Deater M, Phan N, Marcogliese AN, Li H, et al. Metformin improves defective hematopoiesis and delays tumor formation in Fanconi anemia mice. Blood (2016) 128:2774–84. doi: 10.1182/blood-2015-11-683490
313. Chen CH, Ferreira JC, Gross ER, Mochly-Rosen D. Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol Rev. (2014) 94:1–34. doi: 10.1152/physrev.00017.2013
315. Youssoufian H, Pyeritz RE. Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet. (2002) 3:748–58. doi: 10.1038/nrg906
316. Venugopal P, Moore S, Lawrence DM, George AJ, Hannan RD, Bray SC, et al. Self-reverting mutations partially correct the blood phenotype in a Diamond Blackfan anemia patient. Haematologica (2017) 102:e506–9. doi: 10.3324/haematol.2017.166678
317. Makishima H, Maciejewski JP. Pathogenesis and consequences of uniparental disomy in cancer. Clin Cancer Res. (2011) 17:3913–23. doi: 10.1158/1078-0432.CCR-10-2900
318. O'Keefe C, McDevitt MA, Maciejewski JP. Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood (2010) 115:2731–9. doi: 10.1182/blood-2009-10-201848
319. Vago L, Perna SK, Zanussi M, Mazzi B, Barlassina C, Stanghellini MT, et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med. (2009) 361:478–88. doi: 10.1056/NEJMoa0811036
320. Villalobos IB, Takahashi Y, Akatsuka Y, Muramatsu H, Nishio N, Hama A, et al. Relapse of leukemia with loss of mismatched HLA resulting from uniparental disomy after haploidentical hematopoietic stem cell transplantation. Blood (2010) 115:3158–61. doi: 10.1182/blood-2009-11-254284
321. Katagiri T, Sato-Otsubo A, Kashiwase K, Morishima S, Sato Y, Mori Y, et al. Frequent loss of HLA alleles associated with copy number-neutral 6pLOH in acquired aplastic anemia. Blood (2011) 118:6601–9. doi: 10.1182/blood-2011-07-365189
322. Asur RS, Kimble DC, Lach FP, Jung M, Donovan FX, Kamat A, et al. Somatic mosaicism of an intragenic FANCB duplication in both fibroblast and peripheral blood cells observed in a Fanconi anemia patient leads to milder phenotype. Mol Genet Genomic Med. (2018) 6:77–91. doi: 10.1002/mgg3.350
323. Davis BR, Dicola MJ, Prokopishyn NL, Rosenberg JB, Moratto D, Muul LM, et al. Unprecedented diversity of genotypic revertants in lymphocytes of a patient with Wiskott-Aldrich syndrome. Blood (2008) 111:5064–7. doi: 10.1182/blood-2007-06-095299
324. Fernández LC, Torres M, Real FX. Somatic mosaicism: on the road to cancer. Nat Rev Cancer (2016) 16:43–55. doi: 10.1038/nrc.2015.1
325. Ellinghaus E, Stanulla M, Richter G, Ellinghaus D, te Kronnie G, Cario G, et al. Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia (2012) 26:902–9. doi: 10.1038/leu.2011.302
326. Treviño LR, Yang W, French D, Hunger SP, Carroll WL, Devidas M, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. (2009) 41:1001–5. doi: 10.1038/ng.432
327. Prasad RB, Hosking FJ, Vijayakrishnan J, Papaemmanuil E, Koehler R, Greaves M, et al. Verification of the susceptibility loci on 7p12.2, 10q21.2, and 14q11.2 in precursor B-cell acute lymphoblastic leukemia of childhood. Blood (2010) 115:1765–7. doi: 10.1182/blood-2009-09-241513
328. Shah S, Schrader KA, Waanders E, Timms AE, Vijai J, Miething C, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. (2013) 45:1226–31. doi: 10.1038/ng.2754
329. Xu H, Zhang H, Yang W, Yadav R, Morrison AC, Qian M, et al. Inherited coding variants at the CDKN2A locus influence susceptibility to acute lymphoblastic leukaemia in children. Nat Commun. (2015) 6:7553. doi: 10.1038/ncomms8553
330. Hungate EA, Vora SR, Gamazon ER, Moriyama T, Best T, Hulur I, et al. A variant at 9p21.3 functionally implicates CDKN2B in paediatric B-cell precursor acute lymphoblastic leukaemia aetiology. Nat Commun. (2016) 7:10635. doi: 10.1038/ncomms10635
331. Vijayakrishnan J, Henrion M, Moorman AV, Fiege B, Kumar R, da Silva Filho MI, et al. The 9p21.3 risk of childhood acute lymphoblastic leukaemia is explained by a rare high-impact variant in CDKN2. Sci Rep A (2015) 5:15065. doi: 10.1038/srep15065
332. Migliorini G, Fiege B, Hosking FJ, Ma Y, Kumar R, Sherborne AL, et al. Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood (2013) 122:3298–307. doi: 10.1182/blood-2013-03-491316
333. Perez-Andreu V, Roberts KG, Xu H, Smith C, Zhang H, Yang W, et al. A genome-wide association study of susceptibility to acute lymphoblastic leukemia in adolescents and young adults. Blood (2015) 125:680–6. doi: 10.1182/blood-2014-09-595744
334. Zhang MY, Churpek JE, Keel SB, Walsh T, Lee MK, Loeb KR, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet. (2015) 47:180–5. doi: 10.1038/ng.3177
335. Noetzli L, Lo RW, Lee-Sherick AB, Callaghan M, Noris P, Savoia A, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet. (2015) 47:535–8. doi: 10.1038/ng.3253
336. Topka S, Vijai J, Walsh MF, Jacobs L, Maria A, Villano D, et al. Germline ETV6 mutations confer susceptibility to acute lymphoblastic leukemia and thrombocytopenia. PLoS Genet. (2015) 11:e1005262. doi: 10.1371/journal.pgen.1005262
337. Moriyama T, Metzger ML, Wu G, Nishii R, Qian M, Devidas M, et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol. (2015) 16:1659–66. doi: 10.1016/s1470-2045(15)00369-1
338. Duployez N, Abou Chahla W, Lejeune S, Marceau-Renaut A, Letizia G, Boyer T, et al. Detection of a new heterozygous germline ETV6 mutation in a case with hyperdiploid acute lymphoblastic leukemia. Eur J Haematol. (2018) 100:104–7. doi: 10.1111/ejh.12981
339. Qian M, Cao X, Devidas M, Yang W, Cheng C, Dai Y, et al. TP53 germline variations influence the predisposition and prognosis of B-Cell acute lymphoblastic leukemia in children. J Clin Oncol. (2018) 36:591–9. doi: 10.1200/JCO.2017.75.5215
340. Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. (2013) 45:242–52. doi: 10.1038/ng.2532
341. Safavi S, Olsson L, Biloglav A, Veerla S, Blendberg M, Tayebwa J, et al. Genetic and epigenetic characterization of hypodiploid acute lymphoblastic leukemia. Oncotarget (2015) 6:42793–802. doi: 10.18632/oncotarget.6000
342. Furutani E, Shimamura A. Germline genetic predisposition to hematologic malignancy. J Clin Oncol. (2017) 35:1018–28. doi: 10.1200/JCO.2016.70.8644
343. Somasundaram R, Prasad MA, Ungerbäck J, Sigvardsson M. Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood (2015) 126:144–52. doi: 10.1182/blood-2014-12-575688
344. Martín-Lorenzo A, Hauer J, Vicente-Dueñas C, Auer F, González-Herrero I, García-Ramírez I, et al. Infection exposure is a causal factor in B-cell precursor acute lymphoblastic leukemia as a result of Pax5-inherited susceptibility. Cancer Discov. (2015) 5:1328–43. doi: 10.1158/2159-8290.CD-15-0892
345. John LB, Ward AC. The Ikaros gene family: transcriptional regulators of hematopoiesis and immunity. Mol Immunol. (2011) 48:1272–8. doi: 10.1016/j.molimm.2011.03.006
346. Heizmann B, Kastner P, Chan S. The Ikaros family in lymphocyte development. Curr Opin Immunol. (2018) 51:14–23. doi: 10.1016/j.coi.2017.11.005
347. Fan Y, Lu D. The Ikaros family of zinc-finger proteins. Acta Pharm Sin B (2016) 6:513–21. doi: 10.1016/j.apsb.2016.06.002
348. Marke R, van Leeuwen FN, Scheijen B. The many faces of IKZF1 in B-cell precursor acute lymphoblastic leukemia. Haematologica (2018) 103:565–74. doi: 10.3324/haematol.2017.185603
349. Stanulla M, Dagdan E, Zaliova M, Moricke A, Palmi C, Cazzaniga G, et al. IKZF1(plus) defines a new minimal residual disease-dependent very-poor prognostic profile in pediatric B-Cell precursor acute lymphoblastic leukemia. J Clin Oncol. (2018) 36:1240–9. doi: 10.1200/JCO.2017.74.3617
350. Bogaert DJ, Kuehn HS, Bonroy C, Calvo KR, Dehoorne J, Vanlander AV, et al. A novel IKAROS haploinsufficiency kindred with unexpectedly late and variable B-cell maturation defects. J Allergy Clin Immunol. (2018) 141:432–5.e437. doi: 10.1016/j.jaci.2017.08.019
351. Kuehn HS, Boisson B, Cunningham-Rundles C, Reichenbach J, Stray-Pedersen A, Gelfand EW, et al. Loss of B cells in patients with heterozygous mutations in IKAROS. N Engl J Med. (2016) 374:1032–43. doi: 10.1056/NEJMoa1512234
352. Evans TJ, Milne E, Anderson D, de Klerk NH, Jamieson SE, Talseth-Palmer BA, et al. Confirmation of childhood acute lymphoblastic leukemia variants, ARID5B and IKZF1, and interaction with parental environmental exposures. PLoS ONE (2014) 9:e110255. doi: 10.1371/journal.pone.0110255
Keywords: primary immunodeficiency, cancer predisposition, down syndrome, childhood leukemia, immune editing, immune activation, inflammation, microbiome
Citation: Haas OA (2019) Primary Immunodeficiency and Cancer Predisposition Revisited: Embedding Two Closely Related Concepts Into an Integrative Conceptual Framework. Front. Immunol. 9:3136. doi: 10.3389/fimmu.2018.03136
Received: 31 October 2018; Accepted: 19 December 2018;
Published: 12 February 2019.
Edited by:
Markus G. Seidel, Medical University of Graz, AustriaReviewed by:
Irun R. Cohen, Weizmann Institute of Science, IsraelKarl Welte, University of Tubingen, Germany
Copyright © 2019 Haas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oskar A. Haas, b3NrYXIuaGFhc0BjY3JpLmF0