 Nikos E. Papaioannou
Nikos E. Papaioannou Maria Pasztoi
Maria Pasztoi Barbara U. Schraml
Barbara U. Schraml- 1Biomedical Center, Institute for Cardiovascular Physiology and Pathophysiology, Ludwig-Maximilians-University Munich, Munich, Germany
- 2Walter-Brendel-Centre of Experimental Medicine, University Hospital, Ludwig-Maximilians-University Munich, Munich, Germany
Increased susceptibility to infectious diseases is a hallmark of the neonatal period of life that is generally attributed to a relative immaturity of the immune system. Dendritic cells (DCs) are innate immune sentinels with vital roles in the initiation and orchestration of immune responses, thus, constituting a promising target for promoting neonatal immunity. However, as is the case for other immune cells, neonatal DCs have been suggested to be functionally immature compared to their adult counterparts. Here we review some of the unique aspects of neonatal DCs that shape immune responses in early life and speculate whether the functional properties of neonatal DCs could be exploited or manipulated to promote more effective vaccination in early life.
Introduction
Early life immune balance is essential for survival and establishment of healthy immunity in later life. The neonatal period in mammals represents a critical window, in which the immune system has to keep a fine balance between efficient pathogen defense and maintenance of tolerance against a continuous flood of commensal microbes and environmental antigens (1–4). Reduced inflammatory capacity is an inherent feature of early life immunity that has been attributed to an altered repertoire of immune cells, as well as a relative functional immaturity of immune cells in early compared to later life (2–5). For example, the T and B cell pools are not fully expanded at birth and are biased to generate T helper (Th) 2 type responses compared to a more Th1 type response in adults (1). Neonatal, but not adult, monocytes and neutrophils potently suppress T cell activation in vitro and therefore strongly resemble myeloid derived suppressor cells (6). Although neutrophil-like myeloid derived suppressor cells show microbicidal activity (6), the inflammation-induced trafficking of neutrophils, as well as their ability to form extracellular traps, are reduced in fetal and early life compared to adult (7–9). Fetal monocytes are transcriptionally distinct from their adult counterparts and fetal, as well as, neonatal monocytes show distinct responsiveness to inflammatory stimuli than adult monocytes (10–14). Macrophages first develop before birth and are thought to aid in tissue remodeling during development, whereas they acquire their full-blown immune functions with increasing age (11, 14). Microglia of the brain for instance gain an immune-related gene signature over time and in response to microbial signals (15). Additionally, the existence of specific immune regulatory cells of erythroid origin in early life has been suggested to dampen inflammatory responses (4, 16).
As a result of these immune alterations, neonates exhibit an increased susceptibility to infections (2, 5). In humans, pathogens that are often asymptomatic in adults, such as Haemophilus influenzae type B, Bordetella pertussis and Streptococcus pneumoniae, account for the death of more than two million infants per year world-wide (17). Dendritic cells (DCs) have been implicated to promote immune responses to these pathogens in adults (18–21). It is possible to immunize infants under one year against these pathogens, but a single immunization does not necessarily provide immediate protection, or as in the case of H. influenzae type B, antibody titers may not persist (17). For other pathogens, such as rotavirus, immunization is first possible few weeks after birth leaving infants at risk of infection, when the disease is most severe (17, 22). The efficacy, success and challenges of vaccines in early life, as well as existing efforts to improve their effectiveness have recently been reviewed elsewhere (17). Here we focus on the functional differences between DCs in early and adult life. DCs sense the presence of pathogens or damage via so called pattern recognition receptors (PRRs) and initiate innate, as well as adaptive immune responses through cytokine production and antigen presentation (23–25). In their function as immune sentinels DCs have been extensively targeted to increase vaccine effectiveness and DC based vaccines hold promise in adults (26). However, as other immune cells, DCs in early life differ from their adult counterparts in phenotype and function, raising the question, whether targeting DCs could be used to elicit protective immunity in infants and increase vaccine effectiveness. Although most of the data discussed here derive from mouse studies, parallels likely exist in humans, as DC subsets and functions appear highly conserved across species (23, 25).
Dendritic Cells Develop as Functionally Distinct Subsets
Among DCs, we distinguish two main functionally and developmentally distinct cell lineages. Conventional or classical DCs (cDCs) are remarkable activators of adaptive immune responses with a remarkable capacity to capture, process, and present antigens to T cells (23–25, 27). Plasmacytoid DCs (pDCs) on the other hand are critical for defense against viruses, because of their capacity to respond to viral antigens and secrete type I interferons (IFN) (28, 29). Most of our knowledge about the development of these cells is based on studies in adult mice. In adults, DCs have a short lifespan and rely on constant replenishment from bone marrow-derived hematopoietic stem cells (30, 31). cDCs and pDCs were long thought to derive from a common myeloid precursor, the so-called common dendritic cell progenitor (CDP) (32, 33). Within this progenitor fraction, expression of the C type lectin receptor CLEC9A/DNGR-1 distinguishes cells with cDC-restricted developmental potential (34). These cDC restricted CDPs further differentiate into pre-cDCs, which leave the bone marrow and seed lymphoid and non-lymphoid tissues (31, 35) where they differentiate into the two main cDC1 and cDC2 subsets in response to environmental cues (23–25, 27). Of note, the signals that regulate cDC differentiation in tissues remain poorly defined and recent studies indicate that the commitment of pre-cDCs toward cDC1 or cDC2 may already be imprinted in the bone marrow (36, 37). In contrast to cDCs, pDCs exit the bone marrow as fully differentiated cells (38) and only a fraction of pDCs appears to belong to the myeloid lineage, whereas the majority of pDCs arises from lymphoid progenitors (39).
In adults cDC1 and cDC2 are developmentally and functionally distinct cell subsets that can be distinguished based on their differential dependence on transcription factors (23–25, 27). While cDC1 rely on BATF3, ID2 and IRF8 for their development, cDC2 require the transcription factors IRF4, ZEB2, and RELB and are additionally influenced by Notch2 signaling and retinoic acid (23–25, 27). Although not yet investigated in detail, at least some of these developmental pathways are conserved with age, as cDC1-like cells are missing in spleen, mesenteric lymph node and intestinal lamina propria of neonatal BATF3-deficient mice (40, 41). Additionally, DCs in early and late life require FMS-like tyrosine kinase 3 ligand (FLT3L) for their development (42, 43). In adults cDC1 can be reliably identified across tissues by expression of XCR-1, DNGR-1, and CD205 (23–25, 27). In addition, CD8α and CD24 mark cDC1 in lymphoid tissues (23–25, 27). The integrin CD103 marks cDC1 in non-lymphoid tissues, although it is also expressed on a subset of intestinal cDC2 (23–25, 27). cDC2 on the other hand are marked by expression of CD11b, CD172a, and CLEC4A4 (23–25, 27). Functionally, cDC1 are exceptional activators of CD8+ T cells, in part for their superior activity to cross-present cell associated antigens (23–25, 27). cDC1 are additionally dominant inducers of Th1 polarized immune responses due to their strong capacity to produce IL-12 (23–25, 27). In contrast, cDC2 are generally thought to be more efficient at activating CD4+ T cell and inducing Th2- or Th17-biased effector responses (23, 24, 44, 45). Since cDC1 and cDC2 have unique functions in immunity and can be distinguished by expression of select surface markers, they are attractive targets for the manipulation of immune responses in adults (26).
Altered Subset Distribution of Dendritic Cells in Early Life
Although DCs reportedly can be found in mice as early as embryonic day 17, the DC compartment of newborn mice is not fully developed and subject to dynamic age-dependent changes during development (46, 47). In mice the neonatal period includes the first 10 days after birth, which correlates to the first 28 days of life in humans (1, 17). However, it is important to note that in terms of immune development there is substantial temporal variation between mice and humans in early life (48). In murine neonates the DC compartment in lymphoid and non-lymphoid organs is much smaller than that of adults and hallmarked by distinct DC subset distribution (46, 47). The frequency of splenic cDCs at birth is about ten-fold lower than that of adult spleen and, similarly, neonatal splenic pDCs are seven-fold lower in terms of frequency compared to their adult counterparts (46, 47). This is also reflected in lower numbers of DCs in neonatal spleen, which is not fully developed at birth in terms of organ architecture and size (46, 47). By about 5 weeks of age, when the total splenic cellularity reaches adult levels, both cDC and pDC numbers also reach adult levels (46, 47). A relative scarcity of DCs in newborn mice is also evident in other organs, such as thymus, lymph nodes, lung and intestine (47, 49–53). Altered immune responses and infection susceptibility in early life could therefore simply be a by-product of low DC numbers. Administration of the DC growth factor FLT3L leads to a strong increase in DC numbers in neonatal mice and results in increased resistance to Listeria monocytogenes (L. monocytogenes) and herpes simplex virus 1 (HSV-1) (43). Similarly, administration of FLT3L significantly enhances resistance of neonates to the intestinal parasite Cryptosporidium parvum by increasing the number of intestinal CD103+ cDCs, which include both cDC1 and a fraction of cDC2 (54).
In adults, the cDC compartment is dominated by cDC2, while in early life cDC1 appear to be the dominant cDC subtype in spleen and lymph nodes (46, 47, 51). In thymus, cDC1 remain the dominant cDC population also in adults, possibly owing to a unique requirement of this cell type in ensuring central T cell tolerance (55). A systematic analysis of DC subset distribution with age across non-lymphoid organs has not been performed, with one exception being the lung, where cDC1 outnumber cDC2 in neonates, but this relationship is inversed in adults (50). It is important to note that most of these studies relied on the use of surface markers to identify cDC subsets. Notably, within the first 6 days of life, cDC1 from spleen and mesenteric lymph node lack CD8α, although they do express CD24, CLEC9A/DNGR-1, and CD205 (40, 46, 47). Expression of XCR-1 on neonatal cDC1 has not been investigated. These data have led to the suggestion that CD8α− cDC1 may represent a progenitor of bona fide cDC1 (40) and indicate that the use of surface markers to define cDC subsets in early life needs to be approached with caution. A summary of surface markers expressed on neonatal and adult DCs can be found in Figure 1.
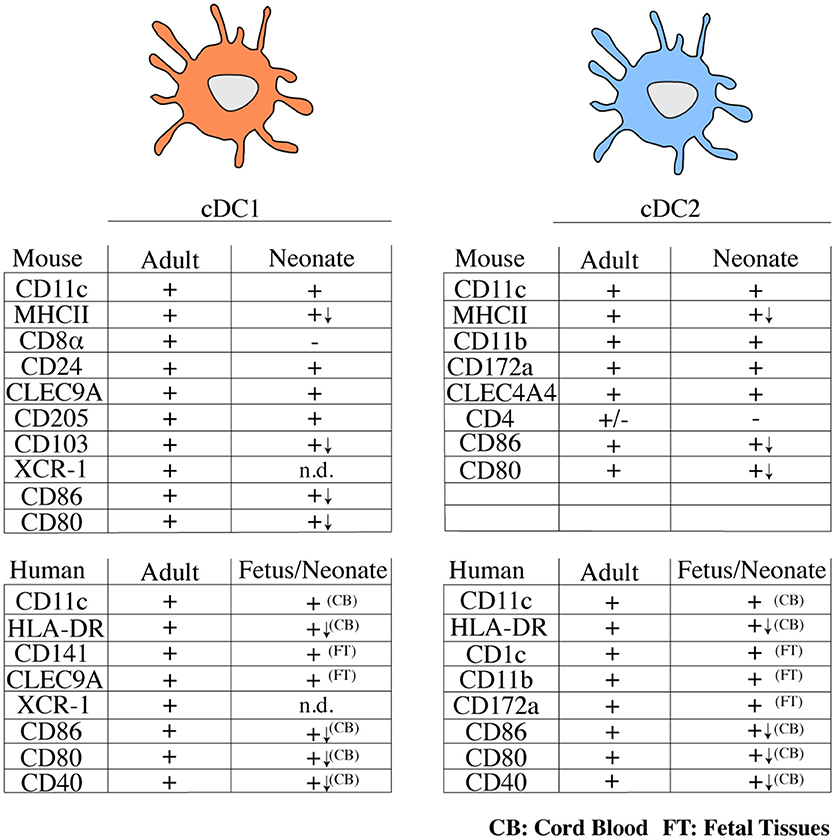
Figure 1. Overview of typical surface markers expressed on cDC subsets from mice and humans in early and adult life. (+) marker is expressed, (–) marker is not expressed or is expressed by a small fraction of cells, n.d., not determined; (↓) lower expression; CB, cord blood; FT, fetal tissues; cDC, conventional dendritic cell; MHCII, major histocompatibility complex class II; CLEC, C-type lectin domain family; HLA, human leukocyte antigen.
Why DC subset distribution differs between neonates and adults is unclear. It is possible that DC differentiation may be intrinsically programmed to generate a functionally adapted DC repertoire that meets the needs of immune responses in early life. However, age-specific changes in specific organ environments could alter DC subset development. cDC1 in neonatal mediastinal lymph nodes express lower levels of CD103 than cDC1 from adult mediastinal lymph nodes (49). This difference in CD103 expression has been attributed to the unique cytokine environment of the lung in early life, such as low expression of GM-CSF (49). Thus, the DC compartment of neonates differs from that of adults in terms of cell number, subset distribution and marker expression.
Dendritic Cell Function in Neonates vs. Adults
It is well established that neonatal DCs in both mice and men are functionally distinct from DCs in adults, which has been suggested to represent a level of functional immaturity. In mice, early life cDCs produce lower levels of pro-inflammatory cytokines than their adult counterparts. In the first week of life, splenic cDC1 produce lower amounts of IL-12 in response to CpG or after in vivo poly I:C treatment (40, 47). Similarly, splenic cDC2 produce less IFN-γ after stimulation with IL-12 and IL-18 compared to their adult counterparts and pDCs produce less IFN-α after combined treatment with CpG, GM-CSF, IL-4 and IFN-γ than pDCs from 6-week old mice (47). Interestingly, when cDC1 from Balb/C rather than C57BL/6 mice were analyzed, CpG-induced IL-12 production did not differ between cDC1 from 7-day old and adult mice (46). In cDC1 from C57BL/6 mice CpG-induced IL-12 production in early life can be augmented by addition of GM-CSF, IFN-γ, and IL-4 to culture conditions, however, the level of IL-12 produced still does not reach that of adult cDC1 (40, 47). These data indicate that some pathogen sensing pathways are fully functional in early life cDCs and that cytokine production of neonatal cDCs may at least in part be augmented through the use of additional costimulatory signals, which in turn could be used to boost immune responses in early life.
A key property of DCs is their ability to activate naïve T cells. Early life cDCs express lower basal levels of major histocompatibility complex class II (MHCII) and costimulatory molecules compared to adult cDCs (43, 47, 49) but expression of these molecules can be induced, for instance upon CpG stimulation in vivo (46). Despite these differences, splenic cDC1 and cDC2 from 1-week-old mice are able to induce allogeneic CD4+ T cell proliferation in vitro to a similar extent as the same subsets from 6-week old mice (47). In the first week of life, CD8α− cDC1 phagocytose L. monocytogenes and cross-present L. monocytogenes-derived antigens to CD8+ T cells as efficiently as adult cDC1 (40). However, while cDC1 from adults respond to this pathogen in a predominantly pro-inflammatory manner, neonatal cDC1 additionally produce the anti-inflammatory cytokine IL-10, which suppresses CD8+ T cell activation (40). Accordingly, IL-10 blockade augments the antigen-specific CD8+ T cell proliferation induced by neonatal CD8α− cDC1 in vitro (40). Whether this IL-10 production has an impact on pathogen burden in vivo has not been examined, but these results indicate that the response to L. monocytogenes is intrinsically different between cDC1 from neonatal and adult mice. Following infection with respiratory syncytial virus (RSV) 7-day-old mice show an altered CD8+ T cell response to that is hallmarked by an epitope shift toward DbM187−195, rather than the KdM282−90 epitope that is immunodominant in adults (49). This epitope shift can be partially rescued by administration of costimulatory signals in vivo (51), suggesting that lower levels of costimulatory molecules on cDCs contribute to the observed epitope bias. However, it is noteworthy, that cDC1 from 7-day-old RSV infected mice preferentially present DbM187−195 epitopes (49, 51), indicating that early life cDCs may exhibit intrinsic differences in antigen processing. Notably, epitope bias may also be found in humans, as infants infected with RSV show age-dependent differences in antibody specificities (56). cDC1 are also required for generating antigen-specific CD8+ T cell responses to rotavirus, a major cause of childhood gastroenteritis (41). Interestingly, neonatal Batf3-deficient mice have a stronger impairment in the antigen-specific CD8+ T cell response to rotavirus than adults, yet, Batf3 deficiency delays viral clearance only minimally in neonates (41). These data suggest, that in vivo other immune mechanisms are put in place that compensate for a lack of cDC1 in this case.
Antigen exposure in early life can elicit both Th1 and Th2 responses (1, 57, 58), however generates a strong bias for Th2 recall responses later in life. This has been partially attributed to a T cell intrinsic Th2 bias in early life but also to the altered cytokine production of cDCs in the first weeks of life, such as low-level IL-12 production (59, 60). Immunization with OVA before postnatal day 6 leads to an upregulation of IL-13 receptor α (IL-13Rα) in antigen specific Th1 cells (59). During secondary exposure to OVA, IL-13Rα expression renders antigen-specific Th1 cells sensitive to IL-4-induced apoptosis, thus leading to Th2-biased recall responses (58, 59). Exogenous administration of IL-12 or adoptive transfer of IL-12 competent cDCs from adult mice reverses the Th2-biased recall response (59), indicating that the low level IL-12 production by cDCs in early life exerts lasting effects on immunity. Why functional differences between neonatal and adult DCs exist, is unclear but several studies suggest that the neonatal environment functionally imprints DCs. As an example, in the developing lung IL-33 produced by epithelial cells during alveolarization on postnatal day 14 suppresses the ability of pulmonary cDC2 to produce the Th1 cytokine IL-12 (50). IL-33 instead promotes OX40L expression, which in turn leads to a stronger ability of cDC2 to promote Th2-biased responses and allergy (50). Th17 responses can be mounted in neonatal mice, for instance after infection with Yersinia enterolytica (61), but early life Th17 responses are damped by T cell derived IL-4 (62). The role of neonatal DCs in this process has not been investigated in detail.
Functional differences between early life and adult DCs have also been observed in humans. cDC2 from fetal spleen respond differently to various PAMPs than adult splenic cDC2 and as in mice, the response in the fetus is marked by higher production of anti-inflammatory cytokines (63). Although human fetal splenic cDC2s can induce allogeneic T cell proliferation, they limit T-cell-derived TNF-α production in an arginase 2-dependent manner and promote differentiation of Foxp3+ regulatory T cells (63). pDCs from cord blood exhibit a severe defect in IFN-I secretion upon TLR9 stimulation with CpG when compared to pDCs from peripheral blood in adults, whereas the cytokine response to influenza A or human immunodeficiency virus is similar (64, 65). Collectively, these data show that the response of neonatal DCs to pathogens differs from that of adults in many ways, however, some signaling pathways induce immune responses that are comparable to those in adults.
Neonatal Dendritic Cells as Targets for Vaccination?
The unique properties of neonatal cDCs discussed above likely contribute to the relative ineffectiveness of vaccines in early life (Figure 2A) and it will be important to determine to what extent tailoring vaccines to the properties of early life DCs can be used to boost immunity. Similarities in surface receptor expression exist between neonatal and adult cDC subsets in both mice and humans (Figure 1) and specific targeting of cDC subsets holds promise for immunization in adults (26). Subset specific targeting using selectively expressed surface receptors can induce protective immunity in murine neonates. Targeting OVA to cDC1 via antibodies directed against CLEC9A with poly I:C as adjuvant on postnatal day 3 efficiently protects murine neonates against lethal challenge with OVA-expressing L. monocytogenes in adulthood (40). Whether targeting cDC2 in early life can induce protective immunity remains to be investigated. The prominent capacity of cDC2 to migrate to draining lymph nodes from the lung (50) indicates that they may be potent targets for initiating T cell responses. But several functions of neonatal cDC2 have not been studied in detail and it is unclear whether they induce effector T cell responses and promote T follicular helper cell differentiation and concomitant antibody production as efficiently as their adult counterparts.
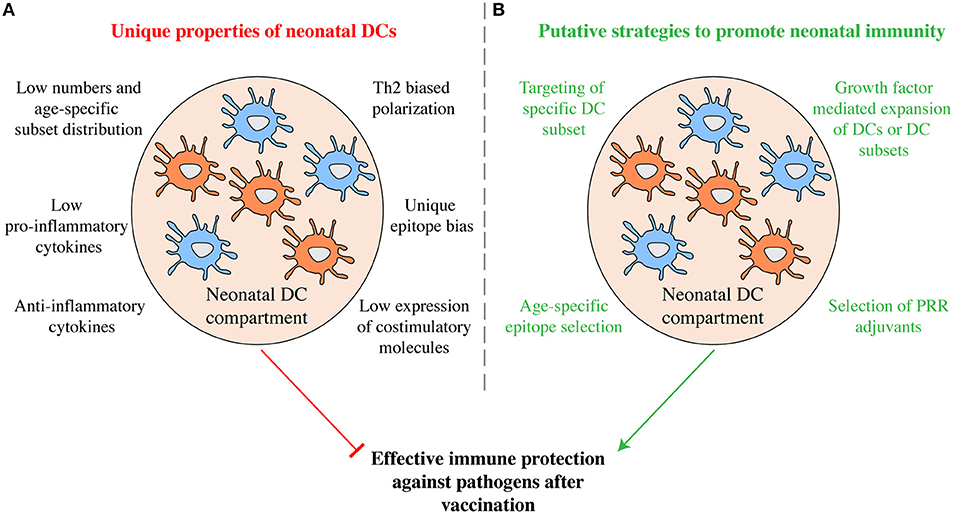
Figure 2. Manipulation of the neonatal DC compartment as putative strategy to promote immunity. (A) Some of the functional properties that characterize neonatal DCs are shown. Taken together they may inhibit efficient immunity in currently used vaccination protocols. (B) Exploiting the unique functions of neonatal DCs may prove promising in developing more efficient vaccination protocols tailored to early life. Through age-specific epitope selection, DC subset specific epitope delivery, the combinatorial use of select PRR agonists as adjuvants, as well as via the manipulation of the DC compartment using growth factors, increased protective humoral and cellular immunity may be promoted.
Synergistic use of TLR agonists greatly increases the Th1 polarizing capacity of human adult DCs (66) and thus the use of defined PRR agonists may be used to promote immunity in early life. A recent study showed that stimulation with multiple TLR agonists elicits stronger secretion of Th1 polarizing cytokines from total human cord blood mononuclear cells than stimulation with a single TLR agonist, however, in cord blood cDCs a single TLR agonist induced stronger pro-inflammatory cytokine production than a combinatorial treatment (67). Agonists of the stimulator of interferon genes (STING) induce expression of costimulatory molecules on neonatal bone marrow derived DCs in vitro and promote secretion of IFN-β (68). In vivo administration of STING agonists in alum in neonatal mice promotes germinal center formation, IFN-γ production by antigen specific T cells, as well as increased antibody titers (68). Taken together these data highlight that the correct choice of adjuvant, either alone or in combination, is important for the design of effective vaccine strategies (17).
Immunization critically depends on the pathogen epitope selected to be vaccinated against. In neonatal mice, T cell responses are directed against a distinct set of antigenic epitopes compared to adults (49, 51). Similarly, in human infants neutralizing antibodies against RSV are preferentially generated against a distinct array of viral epitopes than in adults (56). Thus, selection of age-specific epitopes may also serve as a strategy to foster early life immunity and may help to circumvent deprivation of antigen by pre-existing maternal antibodies (17). At the same time, expanding the DC compartment or specific DC subsets using growth factors may ultimately shift epitope bias and cytokine production. Understanding the functional and developmental properties of neonatal cDCs and how to manipulate them may potentially be used to increase the effectiveness of neonatal immunization beyond what is possible today (Figure 2B). However, immune responses in early life are complex and vaccine effectiveness is influenced by a wide array of factors, including preexisting maternal antibodies (17). Therefore, further studies, especially in humans, are required to better understand the unique aspects of DC development and function in neonates and in early life, as well as the interplay of DCs with other components of the immune system, in order to fully capture whether DCs could be exploited to alter early life vaccination.
Author Contributions
NP and BS wrote the manuscript with assistance from MP. NP designed the figures. All authors provided critical feedback throughout the writing process.
Funding
Work in the Schraml lab is funded by an ERC Starting Grant awarded to BS (ERC-2016-STG-715182) and by the German Research Foundation: Emmy Noether Grant: Schr 1444/1-1 and SFB914 project A11 and SFB1335 project 08.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nat Rev Immunol. (2004) 4:553–64. doi: 10.1038/nri1394
2. Kollmann TR, Kampmann B, Mazmanian SK, Marchant A, Levy O. Protecting the newborn and Young infant from infectious diseases: lessons from immune ontogeny. Immunity (2017) 46:350–63. doi: 10.1016/j.immuni.2017.03.009
3. Zhang X, Zhivaki D, Lo-Man R. Unique aspects of the perinatal immune system. Nat Rev Immunol. (2017) 17:495–507. doi: 10.1038/nri.2017.54
4. Dowling DJ, Levy O. Ontogeny of early life immunity. Trends Immunol. (2014) 35:299–310. doi: 10.1016/j.it.2014.04.007
5. PrabhuDas M, Adkins B, Gans H, King C, Levy O, Ramilo O, et al. Challenges in infant immunity: implications for responses to infection and vaccines. Nat Immunol. (2011) 12:189–94. doi: 10.1038/ni0311-189
6. He YM, Li X, Perego M, Nefedova Y, Kossenkov AV, Jensen EA, et al. Transitory presence of myeloid-derived suppressor cells in neonates is critical for control of inflammation. Nat Med. (2018) 24:224–31. doi: 10.1038/nm.4467
7. Nussbaum C, Gloning A, Pruenster M, Frommhold D, Bierschenk S, Genzel-Boroviczeny O, et al. Neutrophil and endothelial adhesive function during human fetal ontogeny. J Leukoc Biol. (2013) 93:175–84. doi: 10.1189/jlb.0912468.
8. Sperandio M, Quackenbush EJ, Sushkova N, Altstätter J, Nussbaum C, Schmid S, et al. Ontogenetic regulation of leukocyte recruitment in mouse yolk sac vessels. Blood (2013) 121:e118–28. doi: 10.1182/blood-2012-07-447144
9. Yost CC, Cody MJ, Harris ES, Thornton NL, McInturff AM, Martinez ML, et al. Impaired neutrophil extracellular trap (NET) formation: a novel innate immune deficiency of human neonates. Blood (2009) 113:6419–27. doi: 10.1182/blood-2008-07-171629
10. Ulas T, Pirr S, Fehlhaber B, Bickes MS, Loof TG, Vogl T, et al. S100-alarmin-induced innate immune programming protects newborn infants from sepsis. Nat Immunol. (2017) 18:622-632. doi: 10.1038/ni.3745
11. De Kleer, Stanislas Goriely I. Ontogeny of myeloid cells. Front Immunol. (2014) 9:432. doi: 10.3389/fimmu.2014.00423
12. Krow-Lucal ER, Kim CC, Burt TD, McCune JM. Distinct functional programming of human fetal and adult monocytes. Blood (2014) 123:1897–904. doi: 10.1182/blood-2013-11-536094
13. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb+ erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity (2015) 42:665–78. doi: 10.1016/j.immuni.2015.03.011
14. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity (2016) 44:439–49. doi: 10.1016/j.immuni.2016.02.024
15. Thion MS, Low D, Silvin A, Chen J, Grisel P, Schulte-Schrepping J, et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell (2018) 172:500–16.e16. doi: 10.1016/j.cell.2017.11.042
16. Elahi S, Ertelt JM, Kinder JM, Jiang TT, Zhang X, Xin L, et al. Immunosuppressive CD71+ erythroid cells compromise neonatal host defence against infection. Nature (2013) 504:158–62. doi: 10.1038/nature12675
17. Whittaker E, Goldblatt D, McIntyre P, Levy O. Neonatal immunization: rationale, current state, and future prospects. Front Immunol. (2018) 9:532. doi: 10.3389/fimmu.2018.00532
18. Dunne PJ, Moran B, Cummins RC, Mills KHG. CD11c+CD8alpha+ dendritic cells promote protective immunity to respiratory infection with Bordetella pertussis. J Immunol. (2009) 183:400–10. doi: 10.4049/jimmunol.0900169
19. Latz E, Franko J, Golenbock DT, Schreiber JR. Haemophilus influenzae type b-outer membrane protein complex glycoconjugate vaccine induces cytokine production by engaging human toll-like receptor 2 (TLR2) and requires the presence of TLR2 for optimal immunogenicity. J Immunol. (2004) 172:2431–38. doi: 10.4049/jimmunol.172.4.2431
20. Heesters BA, Carroll MC. The role of dendritic cells in S. pneumoniae transport to follicular dendritic cells. Cell Rep. (2016) 16:3130–37. doi: 10.1016/j.celrep.2016.08.049
21. Colino J, Shen Y, Snapper CM. Dendritic cells pulsed with intact Streptococcus pneumoniae elicit both protein- and polysaccharide-specific immunoglobulin isotype responses in vivo through distinct mechanisms. J Exp Med. (2002) 195:1–13. doi: 10.1084/jem.20011432
22. Levy O, Goriely S, Kollmann TR. Immune response to vaccine adjuvants during the first year of life. Vaccine (2013) 31:2500–05. doi: 10.1016/j.vaccine.2012.10.016
23. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
24. Durai V, Murphy KM. Functions of murine dendritic cells. Immunity (2016) 45:719–36. doi: 10.1016/j.immuni.2016.10.010
25. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. (2014) 14:571–8. doi: 10.1038/nri3712
26. Kastenmuller W, Kastenmüller K, Kurts C, Seder RA. Dendritic cell-targeted vaccines — hope or hype? Nat Rev Immunol. (2014) 14:705–11. doi: 10.1038/nri3727
27. Pakalniškyte D, Schraml BU. Tissue-specific diversity and functions of conventional dendritic cells. Adv Immunol. (2017) 134:89–135. doi: 10.1016/bs.ai.2017.01.003
28. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. (2015) 15:471–85. doi: 10.1038/nri3865
29. Reizis B. Regulation of plasmacytoid dendritic cell development. Curr Opin Immunol. (2010) 22:206–11. doi: 10.1016/j.coi.2010.01.005
30. Liu K, Waskow C, Liu X, Yao K, Hoh J, Nussenzweig M. Origin of dendritic cells in peripheral lymphoid organs of mice. Nat Immunol. (2007) 8:578–83. doi: 10.1038/ni1462
31. Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, et al. In vivo analysis of dendritic cell development and homeostasis. Science (2009) 324:392–7. doi: 10.1126/science.1170540
32. Naik SH, Sathe P, Park H-Y, Metcalf D, Proietto AI, Dakic A, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol. (2007) 8:1217–26. doi: 10.1038/ni1522
33. Onai N, Obata-Onai A, Schmid MA, Ohteki T, Jarrossay D, Manz MG. Identification of clonogenic common Flt3+M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol. (2007) 8:1207–16. doi: 10.1038/ni1518
34. Schraml BU, van Blijswijk J, Zelenay S, Whitney PG, Filby A, Acton SE, et al. Genetic tracing via DNGR-1 expression history defines dendritic cells as a hematopoietic lineage. Cell (2013) 154:843–58. doi: 10.1016/j.cell.2013.07.014
35. Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, et al. The origin and development of nonlymphoid tissue CD103 +DCs. J Exp Med. (2009) 206:3115–30. doi: 10.1084/jem.20091756
36. Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, KC W, et al. Batf3 maintains autoactivation of Irf8 for commitment of a CD8α+ conventional DC clonogenic progenitor. Nat Immunol. (2015) 16:708–17. doi: 10.1038/ni.3197
37. Schlitzer A, Sivakamasundari V, Chen J, Bin Sumatoh HR, Schreuder J, Lum J, et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol. (2015) 16:718–28. doi: 10.1038/ni.3200
38. Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell (2008) 135:37–48. doi: 10.1016/j.cell.2008.09.016
39. Rodrigues PF, Alberti-Servera L, Eremin A, Grajales-Reyes GE, Ivanek R, Tussiwand R. Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat Immunol. (2018) 19:711–22. doi: 10.1038/s41590-018-0136-9
40. Torres D, Köhler A, Delbauve S, Caminschi I, Lahoud MH, Shortman K, et al. IL-12p40/IL-10 producing preCD8α/Clec9A+ dendritic cells are induced in neonates upon listeria monocytogenes infection. PLoS Pathogen. (2016) 12:e1005561. doi: 10.1371/journal.ppat.1005561
41. Sun T, Rojas OL, Li C, Ward LA, Philpott DJ, Gommerman JL. Intestinal Batf3-dependent dendritic cells are required for optimal antiviral T-cell responses in adult and neonatal mice. Mucosal Immunol. (2016) 10:775–88. doi: 10.1038/mi.2016.79
42. Durai V, Bagadia P, Briseño CG, Theisen DJ, Iwata A, Davidson JT IV, et al. Altered compensatory cytokine signaling underlies the discrepancy between Flt3 –/–and Flt3l –/–mice. J Exp Med. (2018) 215:1417–35. doi: 10.1084/jem.20171784
43. Vollstedt S, Franchini M, Hefti HP, Odermatt B, O'Keeffe M, Alber G, et al. Flt3 ligand–treated neonatal mice have increased innate immunity against intracellular pathogens and efficiently control virus infections. J Exp Med. (2003) 197:575–84. doi: 10.1084/jem.20021900
44. Satpathy AT, Briseño CG, Lee JS, Ng D, Manieri NA, KC W, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol. (2013) 14:937–48. doi: 10.1038/ni.2679
45. Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hägerbrand K, Marsal J, et al. IRF4 transcription-factor-dependent CD103+CD11b+ dendritic cells drive mucosal T helper 17 cell differentiation. Immunity (2013) 38:958–69. doi: 10.1016/j.immuni.2013.03.009
46. Sun CM. Ontogeny and innate properties of neonatal dendritic cells. Blood (2003) 102:585–91. doi: 10.1182/blood-2002-09-2966
47. Dakic A, Shao QX, D'Amico A, O'Keeffe M, Chen WF, Shortman K, et al. Development of the dendritic cell system during mouse ontogeny. J Immunol. (2004) 172:1018–27. doi: 10.4049/jimmunol.172.2.1018
48. Mold JE, McCune JM. Immunological tolerance during fetal development: from mouse to man. Adv Immunol. (2012) 115:73–111. doi: 10.1016/B978-0-12-394299-9.00003-5
49. Ruckwardt TJ, Morabito KM, Bar-Haim E, Nair D, Graham BS. Neonatal mice possess two phenotypically and functionally distinct lung-migratory CD103+ dendritic cell populations following respiratory infection. Mucosal Immunol. (2018) 11:186–98. doi: 10.1038/mi.2017.28
50. de Kleer IM, Kool M, de Bruijn MJW, Willart M, van Moorleghem J, Schuijs MJ, et al. Perinatal activation of the interleukin-33 pathway promotes type 2 immunity in the developing lung. Immunity (2016) 45:1285–98. doi: 10.1016/j.immuni.2016.10.031
51. Ruckwardt TJ, Malloy AMW, Morabito KM, Graham BS. Quantitative and qualitative deficits in neonatal lung-migratory dendritic cells impact the generation of the CD8+ T cell response. PLOS Pathog. (2014) 10:e1003934. doi: 10.1371/journal.ppat.1003934
52. Torow N, Yu K, Hassani K, Freitag J, Schulz O, Basic M, et al. Active suppression of intestinal CD4+TCRαβ+ T-lymphocyte maturation during the postnatal period. Nat Commun. (2015) 6:7725. doi: 10.1038/ncomms8725
53. Bain CC, Bravo-Blas A, Scott CL, Gomez-Perdiguero E, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. (2014) 15:929–37. doi: 10.1038/ni.2967
54. Lantier L, Lacroix-Lamandé S, Potiron L, Metton C, Drouet F, Guesdon W, et al. Intestinal CD103+ dendritic cells are key players in the innate immune control of Cryptosporidium parvum infection in neonatal mice. PLOS Pathog. (2013) 9:e1003801. doi: 10.1371/journal.ppat.1003801
55. Ardouin L, Luche H, Chelbi R, Carpentier S, Shawket A, Montanana Sanchis F, et al. Broad and largely concordant molecular changes characterize tolerogenic and immunogenic dendritic cell maturation in thymus and periphery. Immunity (2016) 45:305–18. doi: 10.1016/j.immuni.2016.07.019
56. Goodwin E, Gilman MSA, Wrapp D, Chen M, Ngwuta JO, Moin SM, et al. Infants infected with respiratory syncytial virus generate potent neutralizing antibodies that lack somatic hypermutation. Immunity (2018) 48:339–49.e5. doi: 10.1016/j.immuni.2018.01.005
57. Adkins B, Du R-Q. Newborn mice develop balanced Th1/Th2 primary effector responses in vivo but are biased to Th2 secondary responses. J Immunol. (1998) 160:4217–24.
58. Li L, Lee H-H, Bell JJ, Gregg RK, Ellis JS, Gessner A, et al. IL-4 utilizes an alternative receptor to drive apoptosis of Th1 cells and skews neonatal immunity toward Th2. Immunity (2004) 20:429–40. doi: 10.1016/S1074-7613(04)00072-X
59. Lee H-H, Hoeman CM, Hardaway JC, Guloglu FB, Ellis JS, Jain R, et al. Delayed maturation of an IL-12–producing dendritic cell subset explains the early Th2 bias in neonatal immunity. J Exp Med. (2008) 205:2269–80. doi: 10.1084/jem.20071371
60. Ridge JP, Fuchs EJ, Matzinger P. Neonatal tolerance revisited: turning on newborn T cells with dendritic cells. Science (1996) 271:1723–6. doi: 10.1126/science.271.5256.1723
61. Echeverry A, Saijo S, Schesser K, Adkins B. Yersinia enterocolitica promotes robust mucosal inflammatory T-cell immunity in murine neonates. Infect Immun. (2010) 78:3595–608. doi: 10.1128/IAI.01272-09
62. Debock I, Delbauve S, Dubois A, Pétein M, Leo O, Goldman M, et al. Th17 alloimmunity prevents neonatal establishment of lymphoid chimerism in IL-4-deprived mice. Am J Transplant. (2012) 12:81–9. doi: 10.1111/j.1600-6143.2011.03778.x
63. McGovern N, Shin A, Low G, Low D, Duan K, Yao LJ, et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature (2017) 546:662–6. doi: 10.1038/nature22795
64. Danis B, George TC, Goriely S, Dutta B, Renneson J, Gatto L, et al. Interferon regulatory factor 7-mediated responses are defective in cord blood plasmacytoid dendritic cells. Eur J Immunol. (2008) 38:507–17. doi: 10.1002/eji.200737760
65. Zhang X, Lepelley A, Azria E, Lebon P, Roguet G, Schwartz O, et al. Neonatal Plasmacytoid Dendritic Cells (pDCs) display subset variation but can elicit potent anti-viral innate responses. PLoS ONE (2013) 8:e52003. doi: 10.1371/journal.pone.0052003
66. Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1–polarizing program in dendritic cells. Nat Immunol. (2005) 6:769–76. doi: 10.1038/ni1223
67. Surendran N, Simmons A, Pichichero ME. TLR agonist combinations that stimulate Th type I polarizing responses from human neonates. Innate Immun. (2018) 24:240–51. doi: 10.1177/1753425918771178
Keywords: dendritic cell (DC), DC subsets, early life immunity, vaccination, immune system development, T cell activation, innate immunity, DC targeting
Citation: Papaioannou NE, Pasztoi M and Schraml BU (2019) Understanding the Functional Properties of Neonatal Dendritic Cells: A Doorway to Enhance Vaccine Effectiveness? Front. Immunol. 9:3123. doi: 10.3389/fimmu.2018.03123
Received: 29 August 2018; Accepted: 18 December 2018;
Published: 10 January 2019.
Edited by:
Diana Dudziak, Universitätsklinikum Erlangen, GermanyReviewed by:
Joke M. M. Den Haan, VU University Medical Center, NetherlandsDaniela Verthelyi, State Food and Drug Administration, China
Copyright © 2019 Papaioannou, Pasztoi and Schraml. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara U. Schraml, YmFyYmFyYS5zY2hyYW1sQG1lZC51bmktbXVlbmNoZW4uZGU=