 Sandra Jeremias Catarino1
Sandra Jeremias Catarino1 Fabiana Antunes Andrade1
Fabiana Antunes Andrade1 Angelica Beate Winter Boldt1,2
Angelica Beate Winter Boldt1,2 Luiza Guilherme3
Luiza Guilherme3 Iara Jose Messias-Reason1*
Iara Jose Messias-Reason1*- 1Molecular Immunopathology Laboratory, Department of Medical Pathology, Clinical Hospital, Federal University of Paraná, Curitiba, Brazil
- 2Human Molecular Genetics Laboratory, Department of Genetics, Federal University of Paraná, Curitiba, Brazil
- 3Heart Institute (InCor), School of Medicine, University of São Paulo, São Paulo, Brazil
Rheumatic fever (RF) and its subsequent progression to rheumatic heart disease (RHD) are chronic inflammatory disorders prevalent in children and adolescents in underdeveloped countries, and a contributing factor for high morbidity and mortality rates worldwide. Their primary cause is oropharynx infection by Streptococcus pyogenes, whose acetylated residues are recognized by ficolin-1. This is the only membrane-bound, as well as soluble activator molecule of the complement lectin pathway (LP). Although LP genetic polymorphisms are associated with RF, FCN1 gene's role remains unknown. To understand this role, we haplotyped five FCN1 promoter polymorphisms by sequence-specific amplification in 193 patients (138 with RHD and 55, RF only) and 193 controls, measuring ficolin-1 serum concentrations in 78 patients and 86 controls, using enzyme-linked immunosorbent assay (ELISA). Patients presented lower ficolin-1 serum levels (p < 0.0001), but did not differ according to cardiac commitment. Control's genotype distribution was in the Hardy-Weinberg equilibrium. Four alleles (rs2989727: c.−1981A, rs10120023: c.−542A, rs10117466: c.−144A, and rs10858293: c.33T), all associated with increased FCN1 gene expression in whole blood or adipose subcutaneous tissue (p = 0.000001), were also associated with increased protection against the disease. They occur within the *3C2 haplotype, associated with an increased protection against RF (OR = 0.41, p < 0.0001) and with higher ficolin-1 levels in patient serum (p = 0.03). In addition, major alleles of these same polymorphisms comprehend the most primitive *1 haplotype, associated with increased susceptibility to RF (OR = 1.76, p < 0.0001). Nevertheless, instead of having a clear-cut protective role, the minor c.−1981A and c.−144A alleles were also associated with additive susceptibility to valvar stenosis and mitral insufficiency (OR = 3.75, p = 0.009 and OR = 3.37, p = 0.027, respectively). All associations were independent of age, sex or ethnicity. Thus, minor FCN1 promoter variants may play a protective role against RF, by encouraging bacteria elimination as well as increasing gene expression and protein levels. On the other hand, they may also predispose the patients to RHD symptoms, by probably contributing to chronic inflammation and tissue injury, thus emphasizing the dual importance of ficolin-1 in both conditions.
Introduction
Rheumatic fever (RF) is an immune-mediated disease occurring in genetically susceptible individuals, as a sequelae of group A (GAS) Streptococcus pyogenes pharyngitis. It affects predominantly children and adolescents in low-income and developing countries, where it remains a considerable public health problem (1, 2).
Five major manifestations reflect target tissue involvement in RF, including synovium (inflammatory arthritis), heart valves (endocarditis), brain (Sydenham's chorea), skin (erythema marginatum), and subcutaneous tissue (nodules). Repeated or severe RF episodes can result in permanent damage to the heart valves, leading to rheumatic heart disease (RHD), the most common acquired cardiovascular disease in young adults (3, 4). Rheumatic heart disease (RHD) is associated with high morbidity and mortality, causing 9 million disability-adjusted life years lost, 33 million cases (5) and 275,000 deaths each year (6). This multifactorial disorder involves multiple genetic and environmental factors, not yet fully elucidated. Well-designed case-control studies strive to unravel the genetic susceptibility to this disease, given that the only genome-wide association study done with RHD has rendered no relevant results (7).
Autoimmunity is known to play a role in the pathogenesis of RF and RHD, with tissue damage being mediated by autoantibodies resulting from molecular mimicry between GAS and heart tissue proteins. It has been shown that GAS molecules such as N-acetyl-β-D-glucosamine (GlcNAc) and M protein display cross reactivity with valve and myocellular contractile proteins of the host. GlcNAc is the immunodominant cell wall antigen of GAS, recognized by ficolins, molecules that comprise important pattern-recognition receptors (PRRs) of the complement (2, 5, 8, 9).
The complement system plays an important role, both in the defense against GAS infection, as well as in the development of autoimmunity in RHD. This system promptly responds to any pathogen, bridging innate and adaptive immune responses (10–14). Ficolins initiate one of the three complement activation pathways, known as the lectin pathway, along with collectins, as mannose-binding lectin (MBL). They occur in oligomeric structures of a basic homotrimer, where each chain is formed by a collagenous strand and a C-terminal fibrinogen-like recognition domain. The oligomers are complexed with homodimers of serine proteases (MASP-1 or MASP-2). Ficolin oligomers bind specific patterns of acetylated residues on the surface of pathogens or altered cells (8, 15). MASP-1 then autoactivates, transactivating MASP-2 leading to subsequent cleavage of downstream complement components [reviewed by Boldt et al. (16)]. The lectin pathway, along with the classical and alternative pathways, converge at the cleavage of C3 and C5, with subsequent C3b opsonization and pathogen phagocytosis or its destruction by membrane attack complex (MAC) pores on the cell membrane (17).
Three human ficolins have been described: ficolin-1 (M-ficolin), ficolin-2 (L-ficolin), and ficolin-3 (H-ficolin, Hakata antigen) (15). Unlike any other PRRs, ficolin-1 is expressed by myeloid cells, being found in monocytes, neutrophils and macrophages of the lung and spleen (8). It is also the only one that occurs in both soluble form (0.05–1.0 μg/mL in serum) and on the cell membrane (18–20). Apart from other acetylated residues, ficolin-1 has the unique ability to bind sialic acid to capsular polysaccharides of pathogens including Streptococcus agalactiae, as well as to the surface of immune cells (21, 22).
Ficolin-1 is encoded by the FCN1 gene on chromosome 9q34 and contains nine exons. Among the several SNPs described for the FCN1 gene, at least eight are associated with Ficolin-1 levels, four of them are located in the promoter and one in the first exon (23). Polymorphisms of collectin and ficolin genes have been repeatedly associated with infectious and autoimmune diseases (23–28). FCN1 polymorphisms were associated with increased fatal outcome in patients with systemic inflammation (29), susceptibility to rheumatoid arthritis (28) and leprosy (26). Gene polymorphisms of the lectin pathway have already been associated with RF in case-control association studies with MBL (30–32), Ficolin-2 (33), and MASP-2 (34). Within this context, we are the first to investigate a membrane-bound molecule of the lectin pathway, able to activate the complement. More specifically, we evaluated the association of FCN1 polymorphisms and haplotypes, as well as Ficolin-1 serum levels, with the susceptibility to RF and RHD.
Materials and Methods
Subjects and Samples
This study was approved by the local medical ethics committee (CEP/HC 2658.265/2011-11). All patients and control subjects provided written informed consent in accordance with the Declaration of Helsinki. We investigated a total of 193 patients with a history of RF, all of them with ASO (anti streptolysin O) titers higher than 250 units, characterizing a precedent streptococcal infection and diagnosed according to Jones' modified criteria; 55 (28.5%) males and 138 (71.5%) females; with a mean age of 37 years (range = 7–76 years). Among them, 138 had RHD, confirmed by the transthoracic echocardiogram showing rheumatic involvement of the mitral or aortic valves (Table 1), and 55 did not present RHD, but had RF history and were designated as “rheumatic fever only” (RFo) patients. None of the patients presented other inflammatory disease, neoplasia, infective endocarditis, or other infections at the time their blood was collected. Values of high-sensitivity C-reactive protein (hs-CRP) levels, C3 levels and C4 levels, previously published by our group, are shown in Table 1 (30). The control group included 193 blood donors from The Clinical Hospital of the University Federal of Paraná, with a mean age of 37 years (range = 18–64 years), 68 (35%) males, and 125 (65%) females.
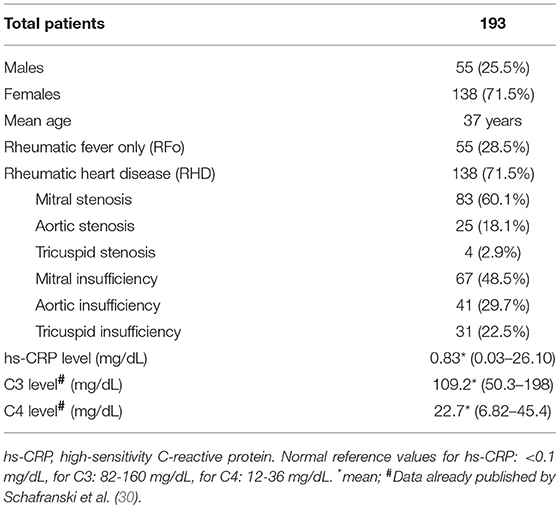
Table 1. Clinical characteristics of patients.
FCN1 Genotyping
DNA extraction from peripheral blood was performed using The QIAamp DNA Blood Mini Kit, QIAGEN (Hilden, Germany), following the manufacturer's instructions. The genotype method was adapted from a previously described multiplex PCR-SSP (sequence-specific amplification) method (30). Five FCN1 single nucleotide polymorphisms (SNPs) were genotyped: rs2989727 SNP (c.–1981G > A) in the distal FCN1 promoter, rs10120023 (c.–542G > A), rs17039495 (c.–399G > A), rs10117466 (c.–144C > A), and rs10858293 (c.+33T > G) SNPs in the proximal FCN1 promoter, with primers listed in Table S1.
FCN1_Prom-1981Af or FCN1_Prom-1981Gf were conjugated with the FCN1_Prom_r reverse primer to generate a fragment of 729 bp. PCR conditions were as follows: 0.2 μM of SSP primers, 1X Coral Load PCR buffer (Qiagen, Hilden, Germany), 1.6 mM MgCl2 (Qiagen, Hilden, Germany), 0.5% glycerol, 0.2 mM deoxyribonucleoside triphosphate (dNTP) (Invitrogen, São Paulo, Brazil), 0.03 U/uL of Taq polymerase (Invitrogen, São Paulo, Brazil), 0.1 μg/mL DNA, ultrapure water for 15 μL. The amplification protocol starts with a 5 min denaturation step at 94°C, followed by 10 cycles of 20 s at 94°C, 30 s at 60°C, and 30 s at 72°C; 10 cycles of 20 s at 94°C, 30 s at 56°C, and 30 s at 72°C; 10 cycles of 20 s at 94°C, 30 s at 52°C, and 30 s at 72°C, concluding with 5 min at 72°C in the final DNA extension step.
FCN1_Prom-542Af or FCN1_Prom-542Gf were conjugated with the FCN1_Prom-144Ar or FCN1_Prom-144Cr to generate a fragment of 434 bp. PCR conditions differed from those previously mentioned as follows: 0.6 μM of SSP primers, 1.7 mM MgCl2, and 1.5% glycerol. The amplification protocol differed from the previous one only by the annealing primer temperatures, which were 57°C in the first 10, 55°C in the next 10, and 53°C in the last 10 cycles.
FCN1_Prom-399Af or FCN1_Prom-399Gf were conjugated with the FCN1_Prom+33Gr or FCN1_Prom+33Tr to generate a fragment of 470 bp. PCR conditions differed from those previously noticed, as follows: 0.4 μM of SSP primers, 1.75 mM MgCl2, 0.5% glycerol. The amplification protocol differed from the first one only by the annealing primer temperatures, which were 58°C in the first 10, 56°C in the next 10 and 54°C in the final 15 cycles.
Interpretation was based on the electrophoretic pattern of the amplified fragments, on agarose gels 1.5% stained with Sybrsafe (Invitrogen, São Paulo, Brazil). This bispecific PCR-SSP approach allows the identification of 8 haplotypes: *1 (GGGCG), *3A (AGGCG), *3A.3C2.A (AGGCT), *3A.3C2.B (AGACT), *3B2 (AAGCG), *3C1 (AAGCT), *3C2 (AAGAT), and *3C2.3A (AGGCG), as previously described (26).
Ficolin-1 Measurement
Ficolin-1 serum concentrations were measured in 78 patients and 86 controls using enzyme-linked immunosorbent assay (ELISA) SEA786Hu Cloud-Clone Corp. (Texas, USA).
Statistics
Allele, haplotype and genotype frequencies were obtained by direct counting (the phase between distantly situated SNPs could be deduced due to the strong LD between the variants, and were verified with the Expectation-Maximization algorithm implemented in the PLINK software. Exact tests of Guo and Thompson for testing the hypothesis of Hardy-Weinberg equilibrium were accomplished using ARLEQUIN v.3.5.2.2 (http://cmpg.unibe.ch/software/arlequin35/). The investigated polymorphisms were evaluated for regulatory effects on gene expression, using information from The Genotype-Tissue Expression (GTEx) Project. Associations with alleles were tested by the Exact Fisher test using the SISA quantitative skill tables and including genotypes and haplotypes, by multivariate binary logistic regression with the software package STATA v. 9.2. Correction for associated demographic factors (sex, age) and clinical factors were applied in the reduced logistic regression model where sample size was adequate, and p-values ≤5% were considered significant. Distributions of ficolin-1 levels were tested for normality with the Shapiro-Wilk test. Since ficolin-1 levels presented a non-normal distribution, the two-tailed Mann-Whitney test was used to compare ficolin-1 levels between groups (GraphPad Prism v.7.03).
Results
FCN1 Alleles and Haplotypes Association With RF
Genotype distribution was in equilibrium with the Hardy-Weinberg model in both controls and patients, except for SNPs rs10120023 (c.−542G > A) and rs10117466 (c.−144C > A) in patients (p = 0.017 and p = 0.024, respectively). A total of eight haplotypes were found in patients and seven, in controls. The phase between the variants in the proximal promoter was determined with bispecific PCR-SSP and the phase of these variants with the SNP c.−1981G > A could be deduced due to a strong linkage disequilibrium (LD). Evidence for recombination was found between the SNP rs10858293 (c.33G > T) in exon 1 and promoter variants in patients (Figure 1).
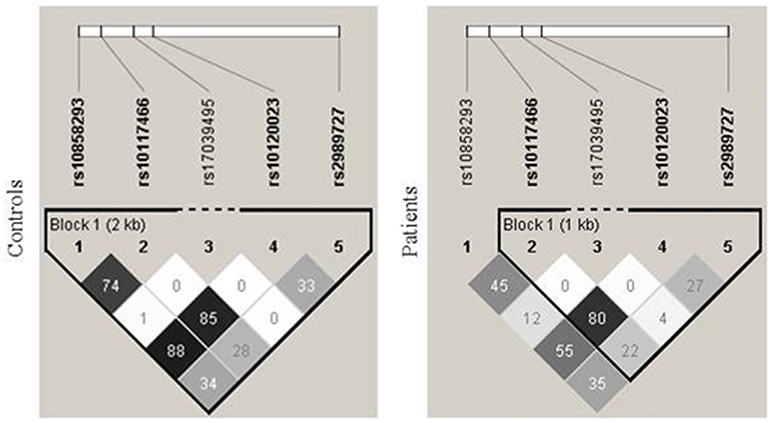
Figure 1. Linkage disequilibrium between FCN1 single nucleotide polymorphisms. There was strong linkage disequilibrium (LD) between the SNPs rs10120023 (−542G > A) and rs10117466 (−144C > A), as indicated by the correlation coefficient values (r2). An evidence of recombination was found between the SNP rs10858293 (+33T > G) in exon 1 and the promoter variants in patients.
The c.−1981A, c.−542A, c.−144A, and c.33T alleles were associated with an increased level of protection against RF, presenting an additive effect with homozygotes protecting more than heterozygotes. All four are also known to be associated with higher FCN1 expression in adipose subcutaneous tissue. Three of them present the same effect in peripheral blood cells (GTex portal) (35). All the protective alleles occur within the *3C2 haplotype (AAGAT). As expected, this haplotype was also associated with increased protection against RF under the additive model (OR = 0.41, p < 0.0001). On the other hand, the common alleles of these SNPs compose the phylogenetically ancestral *1 (GGGCG) haplotype, which was associated with increased susceptibility to RF (OR = 1.76, p < 0.0001, Table 2).
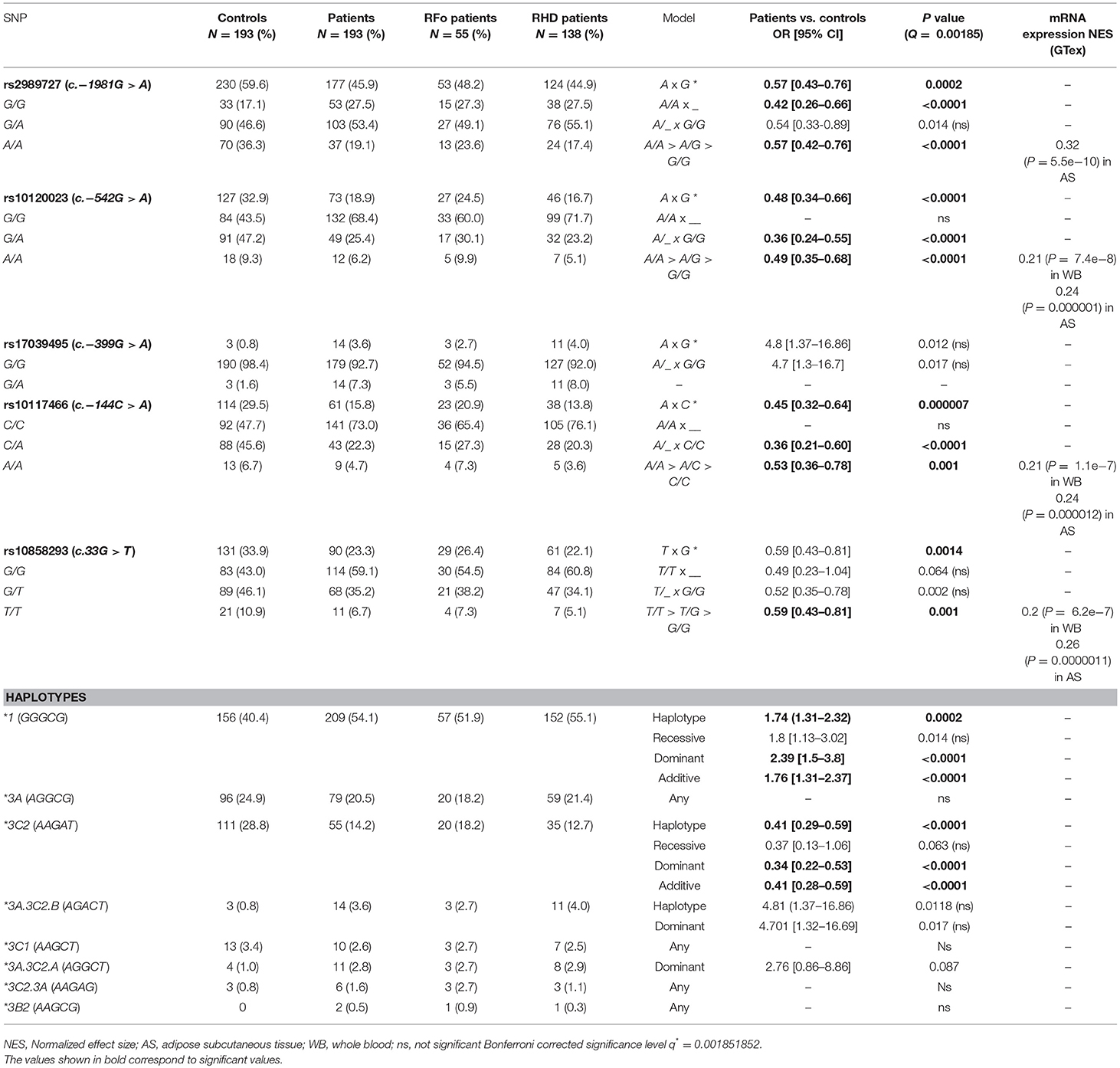
Table 2. FCN1 allele, genotype and haplotype frequencies and associations with RF.
In contrast, the c.−1981A allele was associated with increased susceptibility to valvar stenosis (5/19 or 26.3% vs. 6/89 or 6.7% A/A homozygotes and 13/19 or 68.4% vs. 39/89 or 43.8% A/G heterozygotes in patients with moderate to severe, vs. light or no valvar stenosis, respectively: OR = 3.75 [95%CI = 1.39–10.15], p = 0.009). Similarly, the c.−144A allele presented an increased susceptibility effect for mitral insufficiency (2/12 or 16.7% vs. 1/75 or 1.3% A/A homozygotes and 1/12 or 8.3% vs. 4/75 or 5.3% A/C heterozygotes in patients with moderate to severe vs. light or no mitral insufficiency, respectively: OR = 3.37 [95%CI = 1.15–9.92], p = 0.027). Although no other allele or haplotype were associated either with RHD or other clinical manifestation, this may be due to the relatively small sample size of RHD patients in our setting.
Ficolin-1 Levels
Ficolin-1 serum levels were lower in patients (median: 800.5 ng/mL [324.6–1,715 ng/mL]), compared to controls (1,208 ng/mL [488–2,852 ng/mL, p < 0.0001), but did not differ between RHD and RFo patient groups (Figure 2).
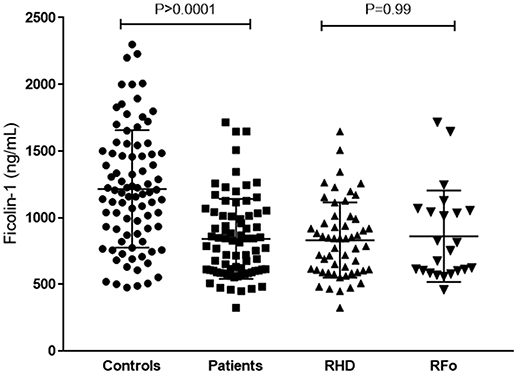
Figure 2. Ficolin-1 serum levels in controls and RF patients. RHD, Rheumatic Heart Disease; RFo, Rheumatic Fever only (not present RHD).
Patients with the *1 (GGGCG) “susceptibility” haplotype presented lower ficolin-1 levels, than those with the *3C2 (AAGAT) “protective” haplotype (p = 0.03, medians 770.8 and 975.9 ng/mL, respectively) (Figure 3).
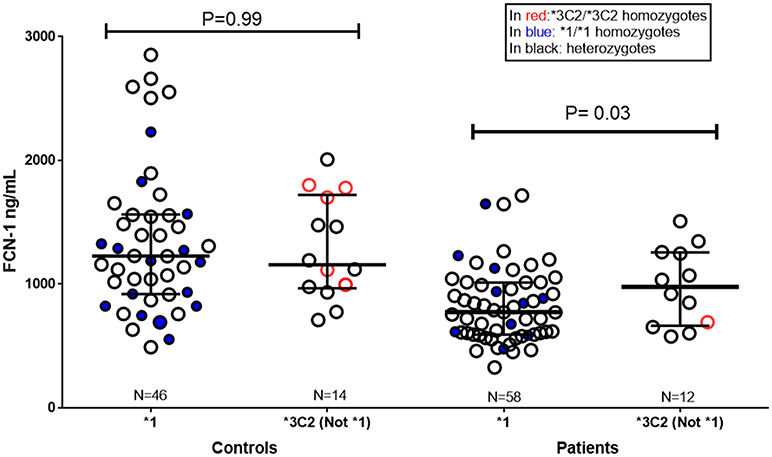
Figure 3. Ficolin-1 serum levels in controls and RF patients, according to FCN1 genotype. *1: carriers of the *1 “risk” haplotype (GGGCG). Not *1/*3C2: carriers of *3C2 “protective” haplotype (AAGAT), but not of the *1 “risk” haplotype. In red: *3C2/*3C2 homozygotes. In blue: *1/*1 homozygotes. Subjects carrying the other FCN1 haplotypes are not represented in the figure (Controls N = 26, Patients N = 8).
Discussion
The complement system plays an important role both in the defense against GAS infection, as well as in the development of RHD (28, 36, 37). Among studies focusing on complement genetic polymorphisms (30–34), this is the first considering the role of a complement membrane-bound molecule of the lectin pathway in the development of RF and RHD. Our results indicate that FCN1 polymorphisms may play a dual role in the physiopathology of RF. On one hand, they increase resistance to GAS infection and on the other hand, predispose the patient to RHD symptoms, once the infection is established.
A protective role for FCN1 promoter variants against RF has been observed (Figure 4). Among the investigated FCN1 alleles, those four occurring within the *3C2 haplotype that were associated with RF protection and ficolin-1 levels have also been associated with higher FCN1 gene expression (GTEx Portal) and ficolin-1 serum levels in other studies (23, 26, 29). Indeed, it has been suggested that the minor alleles −542A and −144A may facilitate the binding of transcription factors, causing amplified gene expression (29). Thus, it is conceivable that higher FCN1 gene and protein expression could increase resistance against GAS infection due to ficolin-1 anti-bacterial properties. In fact ficolin-1 is able to bind sialic acid on Group B Streptococcus bacteria (20, 38) as well as GAS's carbohydrate A (GlcNAc) which is a preferable ficolin ligand. GAS recognition by ficolins may culminate in complement activation, despite the described complement evasion mechanisms of the bacteria, e.g., through C5a, as well as C2–C9 cleavage (39). Moreover, it is known that GAS infection occurs through fibronectin binding in the extracellular matrix (39). Fibronectin is also a ligand for ficolin-1 (40), thus competition for fibronectin binding sites may occur. Additionally, ficolin-1 anchors to GPCR43, a G-protein coupled receptor on monocytes. Ficolin-1/GPCR43 activation results in signal transduction through NFkB and interleukin-8 (IL8) gene expression. IL8 is a chemokine that attracts phagocytes to the infection site, enhancing bacterial elimination (20). Taken together, these events corroborate the eventual protective effect of ficolin-1 in the development of RF.
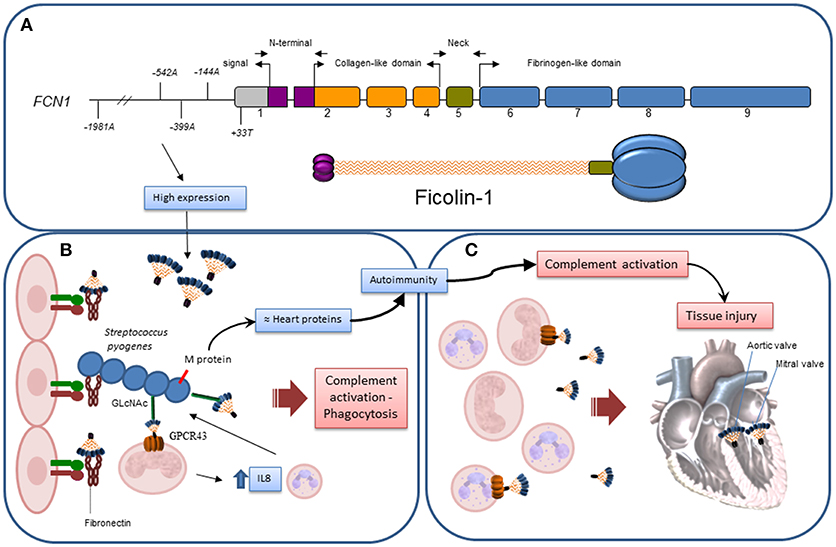
Figure 4. Hypothetical actions of Ficolin-1 in RF/RHD. (A) Exon structure and correspondence to domains in the structural trimeric ficolin-1 subunit: gray – untranslated region, purple – N-terminal trimerization domain, orange – collagenous stalk, green – neck region, blue – fibrinogen C-domain. Approximate location of the investigated polymorphisms is given according to number of base pairs, counting from the first transcribed nucleotide. (B) Higher ficolin-1 levels, associated with certain FCN1 alleles in the promoter region, improved resistance against GAS infection, slowing down the process by which autoantibodies are produced due to exposure of neoepitopes in damaged tissue and structural homology of GAS's M protein with myosin/tropomyosin. This may occur through competition for fibronectin binding in the extracellular matrix; recognition of GlcNAc in GAS carbohydrate A by phagocyte membrane-bound ficolin-1 and consequent bacteria internalization; complement activation, despite GAS's abilities to cleave C5a, as well as C2-C9; ficolin-1/GPCR43 activation on monocytes, transducing signals through NFkB and activating interleukin-8 (IL8) gene expression, leading to chemoattraction of more phagocytes to the infection site. (C) After established infection, structures on damaged/altered cells recognized by membrane-bound or soluble ficolin-1 mediate complement activation and increases tissue injury, which may become irreversible in the cardiac valves.
On the other hand, these FCN1 variants associated with high ficolin-1 expression may contribute to excessive complement activation, leading to chronic inflammation and tissue injury, thereby predisposing the patient to RHD symptoms such as valvar stenosis and mitral insufficiency in the advanced phase of the disease. In addition, exposure to neoepitopes on bacteria-damaged valvar tissue would not only induce autoantibody production, but also promote complement activation with MAC deposition, thereby increasing tissue injury. C9, the last MAC complement, was indeed exclusively found in RHD patients with mitral stenosis, compared to control subjects (13). Thus, in the advanced phase of the disease, high ficolin-1 levels would probably not be beneficial. Even though ficolin-1 levels did not differ between RHD and RFo patients, it is possible that the quantification of ficolin-1 in serum does not reflect the levels of membrane-bound ficolin-1, and that the valvar damage could be instead related to this last ficolin-1 form. Keeping this balance—pathogen elimination vs. host preservation, has proven to be very difficult in circumstances where ineffective pathogen elimination induces chronic persistence of the infection with autoimmune features, as is the case of RF/RHD. This would explain other apparently opposite associations of ficolin-1, reported formerly in leprosy for −542A, −144C, and +33T (26), −1981A in rheumatoid arthritis (28) and earlier chronic Pseudomonas aeruginosa colonization in cystic fibrosis patients (27), as well as ficolin-1 deficiency in a mouse model of collagen Ab-induced arthritis (41).
Lower ficolin-1 levels were found in RF patients, an effect that might purely indicate considerable ficolin-1 consumption in RF. In contrast to our study, ficolin-1 serum levels were higher in patients with vasculitis syndrome or rheumatoid arthritis, than in those with myositis, whereas no difference was observed among patients with systemic lupus erythematosus and Behcet's disease (42). Ficolin-1 levels further correlated with several inflammatory markers, including C-reactive protein (CRP), serum amyloid protein (SAP) and complement factor C3 (42) and strongly associated with the severity of ischemic stroke, in another group (43). Interestingly, part of the benefit of intravenous immunoglobulin (IVIG) therapy relies on ficolin-1 pulldown (reported for Kawasaki disease, the most common form of acquired heart disease in childhood) (44). IVIG therapy has also been proven beneficial for Sydenham's Chorea associated with rheumatic fever (45). Moreover, anti-ficolin-1 mAb ameliorated symptoms of collagen antibody-induced arthritis (CAIA) in animal model (42). Additionally, acute injury leads to higher FCN1 gene expression, due to specific regulatory proteins such as hypoxia factor HIF-1a (46). This collection of evidence is in line with our results associating high-FCN1 producing promoter variants with heart damage, in later stages of the disease.
This study has some limitations, especially regarding sample size of individuals with measured ficolin-1 levels which probably affected some of the results. Increased ficolin-1 levels were observed in patients carrying the *3C2 haplotype, however this effect was not evident among controls possibly due to low sample size. In addition we cannot dismiss the possibility that there may be other causal variants responsible for modulating FCN1 expression, not investigated in this study, such as rs12377780 (in intron 1), rs7857015 (5′ upstream) and rs7858307 (3′UTR) (http://raggr.usc.edu/). Well-designed case control studies including higher number of individuals and different FCN1 gene polymorphisms are necessary to better define the action of ficolin-1in RF. Concluding, we suggest a role for ficolin-1 in fighting GAS infection, with a possible damaging effect when infection succeeds, due to excessive complement activation. Inhibiting the final steps of complement activation may be a therapeutic clue for preventing valvar damage in patients with RHD.
Author Contributions
FA, AB, LG, and IM-R designed the study and analyzed the data. SC performed all the assays, analyzed the data, and performed statistical tests. All of the authors contributed toward manuscript preparation and revision, and provided final approval of the version to be published.
Funding
This work was supported by CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico), Fundação Araucária and BNDES (The National Bank for Economic and Social Development).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge all the patients for their participation in this study, and the staff of the Molecular Immunology Laboratory of HC/UFPR for their assistance. We are thankful to CAPES and CNPq for research grants to IM-R, and to BNDES (The National Bank for Economic and Social Development) for its research grants to LG.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.03009/full#supplementary-material
Abbreviations
RF, Rheumatic Fever; RHD, Rheumatic Heart Disease; RFo, Rheumatic Fever only; FCN1, ficolin-1 gene; LP, Lectin Pathway; CRP, C-Reactive Protein; MAC, Membrane Attack Complex; GAS, Group A Streptococcus; LD, Linkage Disequilibrium; GlcNAc, N-acetyl-β-D-glucosamine; MBL, Mannose-binding lectin.
References
1. Steer AC. Historical aspects of rheumatic fever. J Paediatr Child Health (2015) 51:21–27. doi: 10.1111/jpc.12808
2. Guilherme L, Kalil J. Rheumatic heart disease: molecules involved in valve tissue inflammation leading to the autoimmune process and Anti-S. pyogenes vaccine. Front Immunol. (2013) 4:352. doi: 10.3389/fimmu.2013.00352
3. Bono-Neri F. Acute rheumatic fever: global persistence of a preventable disease. J Pediatr Heal Care (2017) 31:275–84. doi: 10.1016/j.pedhc.2016.09.001
4. Martin WJ, Steer AC, Smeesters PR, Keeble J, Inouye M, Carapetis J, et al. Post-infectious group A streptococcal autoimmune syndromes and the heart. Autoimmun Rev. (2015) 14:710–25. doi: 10.1016/j.autrev.2015.04.005
5. Zühlke LJ, Beaton A, Engel ME, Hugo-Hamman CT, Karthikeyan G, Katzenellenbogen JM, et al. Group A streptococcus, acute rheumatic fever and rheumatic heart disease: epidemiology and clinical considerations. Curr Treat Options Cardiovasc Med. (2017) 19:15. doi: 10.1007/s11936-017-0513-y
6. Roberts K, Colquhoun S, Steer A, Reményi B, Carapetis J. Screening for rheumatic heart disease: current approaches and controversies. Nat Rev Cardiol. (2013) 10:49–58. doi: 10.1038/nrcardio.2012.157
7. Tian C, Hromatka BS, Kiefer AK, Eriksson N, Noble SM, Tung JY, et al. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat Commun. (2017) 8:599. doi: 10.1038/s41467-017-00257-5
8. Matsushita M. Ficolins in complement activation. Mol Immunol. (2013) 55:22–6. doi: 10.1016/j.molimm.2012.08.017
9. Garred P, Honoré C, Ma YJ, Rørvig S, Cowland J, Borregaard N, et al. The genetics of ficolins. J Innate Immun. (2009) 2:3–16. doi: 10.1159/000242419
10. Ermert D, Weckel A, Agarwal V, Frick IM, Björck L, Blom AM. Binding of complement inhibitor C4b-binding protein to a highly virulent Streptococcus pyogenes M1 strain is mediated by protein H and enhances adhesion to and invasion of endothelial cells. J Biol Chem. (2013) 288:32172–83. doi: 10.1074/jbc.M113.502955
11. Ramasawmy R, Spina GS, Fae KC, Pereira AC, Nisihara R, Messias Reason IJ, et al. Association of mannose-binding lectin gene polymorphism but not of mannose-binding serine protease 2 with chronic severe aortic regurgitation of rheumatic etiology. Clin Vaccine Immunol. (2008) 15:932–6. doi: 10.1128/CVI.00324-07
12. Beltrame MH, Catarino SJ, Goeldner I, Boldt ABW, de Messias-Reason IJ. The lectin pathway of complement and rheumatic heart disease. Front Pediatr. (2015) 2:148. doi: 10.3389/fped.2014.00148
13. Mukherjee S, Jagadeeshaprasad MG, Banerjee T, Ghosh SK, Biswas M, Dutta S, et al. Proteomic analysis of human plasma in chronic rheumatic mitral stenosis reveals proteins involved in the complement and coagulation cascade. Clin Proteomics (2014) 11:1–18. doi: 10.1186/1559-0275-11-35
14. Wu X, Yue Q, Jia W, Zhang J, Ouyang H, Xin D, et al. A novel approach for characterizing variations in serum peptides in rheumatic heart disease. Indian J Med Res. (2017) 145:365–72. doi: 10.4103/ijmr.IJMR_1581_14
16. Boldt ABW, Boschmann SE, Catarino SJ, Andrade FA, de Messias-Reason IJ. MASP1 and MASP2. In Choi S, editor. Encyclopedia of Signaling Molecules. New York, NY: Springer New York (2016). p.1–18.
17. Vignesh P, Rawat A, Sharma M, Singh S. Complement in autoimmune diseases. Clin Chim Acta (2017) 465:123–30. doi: 10.1016/j.cca.2016.12.017
18. Honoré C, Hummelshoj T, Hansen BE, Madsen HO, Eggleton P, Garred P. The innate immune component ficolin 3 (Hakata antigen) mediates the clearance of late apoptotic cells. Arthritis Rheum. (2007) 56:1598–607. doi: 10.1002/art.22564
19. Rørvig S, Honore C, Larsson L-I, Ohlsson S, Pedersen CC, Jacobsen LC, et al. Ficolin-1 is present in a highly mobilizable subset of human neutrophil granules and associates with the cell surface after stimulation with fMLP. J Leukoc Biol. (2009) 86:1439–49. doi: 10.1189/jlb.1008606
20. Zhang J, Yang L, Ang Z, Yoong SL, Tran TTT, Anand GS, et al. Secreted M-Ficolin anchors onto monocyte transmembrane G protein-coupled receptor 43 and cross talks with plasma C-reactive protein to mediate immune signaling and regulate host defense. J Immunol. (2010) 185:6899–910. doi: 10.4049/jimmunol.1001225
21. Kjaer TR, Hansen AG, Sørensen UBS, Nielsen O, Thiel S, Jensenius JC. Investigations on the pattern recognition molecule M-ficolin: quantitative aspects of bacterial binding and leukocyte association. J Leukoc Biol. (2011) 90:425–37. doi: 10.1189/jlb.0411201
22. Ren Y, Ding Q, Zhang X. Ficolins and infectious diseases. Virol Sin. (2014) 29:25–32. doi: 10.1007/s12250-014-3421-2
23. Ammitzbøll CG, Kjær TR, Steffensen R, Stengaard-Pedersen K, Nielsen HJ, Thiel S, et al. Non-synonymous polymorphisms in the FCN1 gene determine ligand-binding ability and serum levels of M-ficolin. PLoS ONE (2012) 7:e50585. doi: 10.1371/journal.pone.0050585
24. Kasperkiewicz K, Eppa Ł, Swierzko AS, Bartłomiejczyk MA, Zuber ZM, Siniewicz-Luzenczyk K, et al. Lectin pathway factors in patients suffering from juvenile idiopathic arthritis. Immunol Cell Biol. (2017) 95:666–75. doi: 10.1038/icb.2017.31
25. Addobbati C, de Azevêdo Silva J, Tavares NAC, Monticielo O, Xavier RM, Brenol JCT, et al. Ficolin gene polymorphisms in Systemic Lupus Erythematosus and rheumatoid arthritis. Ann Hum Genet. (2016) 80:1–6. doi: 10.1111/ahg.12129
26. Boldt ABW, Sanchez MIN, Stahlke ERS, Steffensen R, Thiel S, Jensenius JC, et al. Susceptibility to leprosy is associated with M-ficolin polymorphisms. J Clin Immunol. (2013) 33:210–9. doi: 10.1007/s10875-012-9770-4
27. Haerynck F, Van Steen K, Cattaert T, Loeys B, Van Daele S, Schelstraete P, et al. Polymorphisms in the lectin pathway genes as a possible cause of early chronic Pseudomonas aeruginosa colonization in cystic fibrosis patients. Hum Immunol. (2012) 73:1175–83. doi: 10.1016/j.humimm.2012.08.010
28. Vander Cruyssen B, Nuytinck L, Boullart L, Elewaut D, Waegeman W, Van thielen M, et al. Polymorphisms in the ficolin 1 gene (FCN1) are associated with susceptibility to the development of rheumatoid arthritis. Rheumatology (2007) 46:1792–5. doi: 10.1093/rheumatology/kem266
29. Munthe-Fog L, Hummelshoj T, Honoré C, Moller ME, Skjoedt MO, Palsgaard I, et al. Variation in FCN1 affects biosynthesis of ficolin-1 and is associated with outcome of systemic inflammation. Genes Immun. (2012) 13:515–22. doi: 10.1038/gene.2012.27
30. Schafranski MD, Stier A, Nisihara R, Messias-Reason IJT. Significantly increased levels of mannose-binding lectin (MBL) in rheumatic heart disease: a beneficial role for MBL deficiency. Clin Exp Immunol. (2004) 138:521–5. doi: 10.1111/j.1365-2249.2004.02645.x
31. Schafranski MD, Pereira Ferrari L, Scherner D, Torres R, Jensenius JC, de Messias-Reason IJ. High-producing MBL2 genotypes increase the risk of acute and chronic carditis in patients with history of rheumatic fever. Mol Immunol. (2008) 45:3827–31. doi: 10.1016/j.molimm.2008.05.013
32. Messias Reason IJ, Schafranski MD, Jensenius JC, Steffensen R. The association between mannose-binding lectin gene polymorphism and rheumatic heart disease. Hum Immunol. (2006) 67:991–8. doi: 10.1016/j.humimm.2006.08.296
33. Messias-Reason IJ, Schafranski MD, Kremsner PG, Kun JFJ. Ficolin 2 (FCN2) functional polymorphisms and the risk of rheumatic fever and rheumatic heart disease. Clin Exp Immunol. (2009) 157:395–9. doi: 10.1111/j.1365-2249.2009.03975.x
34. Catarino SJDS, Boldt ABW, Beltrame MH, Nisihara RM, Schafranski MD, de Messias-Reason IJ. Association of MASP2 polymorphisms and protein levels with rheumatic fever and rheumatic heart disease. Hum Immunol. (2014) 75:1197–202. doi: 10.1016/j.humimm.2014.10.003
35. Carithers LJ, Moore HM. The Genotype-Tissue Expression (GTEx) project. Biopreserv Biobank (2015) 13:307–8. doi: 10.1089/bio.2015.29031.hmm
36. Degn SE, Jensenius JC, Thiel S. Disease-causing mutations in genes of the complement system. Am J Hum Genet. (2011) 88:689–705. doi: 10.1016/j.ajhg.2011.05.011
37. Thiel S, Kolev M, Degn S, Steffensen R, Hansen AG, Ruseva M, et al. Polymorphisms in mannan-binding lectin (MBL)-associated serine protease 2 affect stability, binding to MBL, and enzymatic activity. J Immunol. (2009) 182:2939–47. doi: 10.4049/jimmunol.0802053
38. Ma YJ, Skjoedt M-O, Garred P. Collectin-11/MASP complex formation triggers activation of the lectin complement pathway–the fifth lectin pathway initiation complex. J Innate Immun. (2013) 5:242–50. doi: 10.1159/000345356
39. Hamada S, Kawabata S, Nakagawa I. Molecular and genomic characterization of pathogenic traits of group A Streptococcus pyogenes. Proc Jpn Acad Ser B Phys Biol Sci. (2015) 91:539–59. doi: 10.2183/pjab.91.539
40. Swierzko AS, Bartlomiejczyk MA, Brzostek A, Lukasiewicz J, Michalski M, Dziadek J, et al. Mycobacyerial antigen 85 complex (Ag85) as a target for ficolins and mannose-binding lectin. Int J Med Microbiol. (2016) 306:212–21. doi: 10.1016/j.ijmm.2016.04.004
41. Banda NK, Acharya S, Scheinman RI, Mehta G, Takahashi M, Endo Y, et al. Deconstructing the lectin pathway in the pathogenesis of experimental inflammatory arthritis: essential role of the lectin ficolin B and mannose-binding protein–associated serine protease 2. J Immunol. (2017) 199:1835–45. doi: 10.4049/jimmunol.1700119
42. Katayama M, Ota K, Nagi-Miura N, Ohno N, Yabuta N, Nojima H, et al. Ficolin-1 is a promising therapeutic target for autoimmune diseases. Int Immunol. (2018)1–10. doi: 10.1093/intimm/dxy056
43. Kouchaki E, Babamohammadi A, Nikoueinejad H, Sehat M. Association of serum levels of Pentraxin-3, M-ficolin, and surfactant protein A with the severity of ischemic stroke complement activators in ischemic stroke. Iran J Allergy Asthma Immunol. (2017) 16:140–6.
44. Okuzaki D, Ota K, Takatsuki SI, Akiyoshi Y, Naoi K, Yabuta N, et al. FCN1 (M-ficolin), which directly associates with immunoglobulin G1, is a molecular target of intravenous immunoglobulin therapy for Kawasaki disease. Sci Rep. (2017) 7:6–11. doi: 10.1038/s41598-017-11108-0
45. Dean SL, Singer HS. Treatment of sydenham's chorea: a review of the current evidence. Tremor Other Hyperkinet Mov. (2017) 7:456. doi: 10.7916/D8W95GJ2
Keywords: FCN1, ficolin-1, polymorphism, haplotype, rheumatic fever, rheumatic heart disease
Citation: Catarino SJ, Andrade FA, Boldt ABW, Guilherme L and Messias-Reason IJ (2018) Sickening or Healing the Heart? The Association of Ficolin-1 and Rheumatic Fever. Front. Immunol. 9:3009. doi: 10.3389/fimmu.2018.03009
Received: 15 June 2018; Accepted: 05 December 2018;
Published: 18 December 2018.
Edited by:
Joao P. B. Viola, Instituto Nacional de Câncer (INCA), BrazilReviewed by:
Thomas Vorup-Jensen, Aarhus University, DenmarkBryce Binstadt, University of Minnesota Twin Cities, United States
Copyright © 2018 Catarino, Andrade, Boldt, Guilherme and Messias-Reason. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iara Jose Messias-Reason, aWFyYXJlYXNvbkBoYy51ZnByLmJy