
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 December 2018
Sec. Microbial Immunology
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02943
This article is part of the Research Topic Secondary Respiratory Infections in the Context of Acute and Chronic Pulmonary Diseases View all 13 articles
 David J. Morgan†
David J. Morgan† Joshua Casulli†
Joshua Casulli† Christine Chew†Emma Connolly
Christine Chew†Emma Connolly Sylvia Lui
Sylvia Lui Oliver J. Brand
Oliver J. Brand Rizwana Rahman
Rizwana Rahman Christopher Jagger
Christopher Jagger Tracy Hussell*
Tracy Hussell*Secondary infections arise as a consequence of previous or concurrent conditions and occur in the community or in the hospital setting. The events allowing secondary infections to gain a foothold have been studied for many years and include poor nutrition, anxiety, mental health issues, underlying chronic diseases, resolution of acute inflammation, primary immune deficiencies, and immune suppression by infection or medication. Children, the elderly and the ill are particularly susceptible. This review is concerned with secondary bacterial infections of the lung that occur following viral infection. Using influenza virus infection as an example, with comparisons to rhinovirus and respiratory syncytial virus infection, we will update and review defective bacterial innate immunity and also highlight areas for potential new investigation. It is currently estimated that one in 16 National Health Service (NHS) hospital patients develop an infection, the most common being pneumonia, lower respiratory tract infections, urinary tract infections and infection of surgical sites. The continued drive to understand the mechanisms of why secondary infections arise is therefore of key importance.
It has been appreciated for a long time that infections following surgical cases are caused by a breach of skin barrier integrity. This breach of barrier tissue (e.g. the skin or epithelial surfaces lining the lung, gastro-intestinal or urogenital tract) however, is common during non-surgical infection and was one of the first causes identified to enhance bacterial outgrowth in the lung, by providing different substrates for adhesion and access to additional proteins for bacteria to metabolize. In this review we will discuss the changes in immunity that lead to dysregulation of responses and how prior viral infection in the lung suppresses cellular innate immunity facilitating bacterial outgrowth to occur. Though we assume that cellular innate immunity is adequate before viral infection, it is important to consider that patients most at risk of developing secondary bacterial complications may have a complex inflammatory history, medications, co-morbidities or mental-health history that has already influenced innate immunity. We will not cover the more soluble innate elements such as anti-microbial peptides or surfactant proteins, as these have been covered extensively elsewhere (1).
Innate immunity in the lung is important since it can facilitate the elimination of many pathogens in the absence of adaptive immunity and without immunopathological side effects. The actual location of some innate immune cell subsets is unclear due to the changing environment within the branching structure of the lung. A general rule however is that the density of immune cells gets lower the further down the respiratory tract you look, which facilitates optimal gaseous exchange.
As will be described for macrophages later, the immune components present in a healthy lung are specialized and sparse. Innate lymphoid cells exist in the naïve mouse lung at a low frequency of 0.4–1%. Their precise lung location in health however, has not been determined (2, 3), though they do expand during lung inflammation [for a review see (4)]. Gamma delta (γδ) T cells are also present and rare, accounting for ~1–5% of blood (5) and 8–10% of lung lymphocytes. They display a restricted profile of variable genes (Vγ4, Vδ1, and Vδ6) (6) in their T cell receptor, which changes with age to become predominantly Vγ4+ (7, 8). NK cells constitute 10% of resident lymphocytes in the lung (9) and it is thought their survival depends on IL-15 production by bronchial epithelial cells (10). NK cells detect an absence of MHC class I molecules using a variety of cell surface receptors and are induced to kill target cells by an activating receptor that binds stress ligands (11). In this way, NK cells present in the interstitial compartment are poised to recognize abnormality.
Dendritic cells (DCs) are present in the lung interstitial spaces (12) and the pulmonary epithelium (13), but are absent from the airspaces. In mice, DCs in the epithelium (CD103+ CD11blo) require Batf3, IRF8, and Flt3 ligand for development, whereas those in the lung parenchyma require M-CSF (14). Either population may derive from bone marrow or a local precursor cell population (15). In the steady state, the DCs present in the epithelium may be important for sampling luminal content and/or clearing apoptotic cell turnover (16). As with other innate immune cells, the density of dendritic cells will depend on the position in the respiratory tree with more being present in bronchi than alveoli. Dendritic cells and follicular dendritic cells are also located in sparse B cell follicles. Though typically absent in naïve mice and humans, aggregates of B and T cells may be located next to the major bronchi and include follicular dendritic, dendritic, and stromal, cells (17).
Generally, an absence in any of the innate immune cells described above has little affect in healthy lungs. However, lung macrophages have a unique role in health by performing general housekeeping duties, as exemplified by the build-up of proteinaceous material due to an absence of macrophages in mice lacking granulocyte macrophage-colony-stimulating factor (GM-CSF) (18). In rodents and humans, the lungs are home to two distinct macrophage subsets: airway macrophages and interstitial macrophages (19). We refer to airway, rather than alveolar macrophages since bronchoalveolar lavage (BAL) samples the whole airway. This procedure typically elutes 90–95% macrophages in health, the majority of which will be derived from the alveoli, in addition to a small number of lymphocytes (20, 21). Alveolar macrophages are remarkably long-lived and self-renewing and therefore do not require continuous replenishment from bone marrow-derived precursors in health (22–24). In contrast, interstitial macrophages have a higher turnover rate and are shorter lived in the steady state (25). Interstitial macrophages are located in the interstitial space between the alveoli and capillaries and are less abundant than alveolar macrophages (26).
Alveolar macrophages are initially derived from fetal monocytes and their development is reliant on GM-CSF, of which there is an abundance of in the airspaces shortly after birth (27, 28). GM-CSF drives production of alveolar macrophages through induction of peroxisome proliferator-activated receptor-γ (PPARγ) expression (27, 29, 30). Mice lacking GM-CSF (or its receptor), and patients with defects in GM-CSF signaling, develop pulmonary alveolar proteinosis due to a build-up of surfactant in the airways because of a lack of clearance by macrophages (31, 32). Following irradiation (27) or influenza infection (22) airway macrophages become depleted and are replenished from the periphery or the interstitial lung macrophage pool, respectively. On the other hand, interstitial macrophages originate from bone marrow derived-monocytes and are preferentially replenished by this population during inflammation (33). A recent study has identified 3 populations of interstitial macrophages based on phenotypic and transcriptomic studies, which are different to airway macrophages (34).
The mechanisms leading to bacterial outgrowth following lung viral infection are, to a large extent, driven by the attempt to return the lung to health. Understanding the role of innate immune cells in lung health therefore, may provide clues to why complications can occur. Due to their location, macrophages in the airways display phenotypic and functional differences to other macrophage populations. Alveolar macrophages reside in the alveolar lumen and are surrounded by surfactant, which contains proteins that dampen macrophage activity (35). This allows alveolar macrophages to be tolerant to cellular debris and innocuous antigens, thereby preventing excessive tissue damage, while setting an activation threshold that needs to be overcome to efficiently clear more pathogenic microorganisms (21). On the other hand, interstitial macrophages are in close contact with the extracellular matrix (ECM) and, as such, have a more prominent role in modulating tissue fibrosis, as well as being better equipped for antigen presentation (36, 37). Moreover, alveolar macrophages have reduced phagocytic activity and respiratory burst in comparison to interstitial macrophages (38, 39). Both subsets of macrophages inhibit T cell activation and subsequent onset of adaptive immunity via the suppression of DC activation; a process dependent on the anti-inflammatory cytokine interleukin-10 (IL-10), transforming growth factor-β (TGFβ) and prostaglandins (40, 41). Alveolar macrophages are poor at presenting antigen to T cells (42), although they are capable of transporting antigens to the lung-draining lymph nodes (43). Likewise, human alveolar macrophages induce T cell antigen-specific unresponsiveness as a result of poor antigen presentation and a lack of expression of co-stimulatory molecules, such as CD86 (44); which in itself promotes tolerance to innocuous antigens.
With respect to bacterial complications following viral infection, it is important to appreciate the role of the epithelium in regulating airway macrophage activity. Due to their direct exposure to environmental challenges in the alveolar lumen, strategies need to be in place for alveolar macrophages to discern a harmless antigen from a serious pathogenic threat. For this reason, alveolar macrophages are tightly regulated in order to prevent an inflammatory response against cellular debris and innocuous antigens, whilst still providing protection against harmful pathogens by propelling an inflammatory response (35). For example, alveolar macrophages are hypo-responsive to low levels of endotoxins, which are present in ambient air (21), thereby preventing an inappropriate innate immune response to innocuous antigens. A number of mechanisms are in place to suppress the activity of alveolar macrophages, including their interaction with the airway epithelium. The airway epithelium, through both direct contact and secreted products, negatively regulates alveolar macrophage activity. These factors include CD200, TGF-β, IL-10 and surfactant proteins (SP-A and SP-D), which act to suppress macrophage phagocytic ability and production of pro-inflammatory cytokines (45–47) (Figure 1). Though beneficial in some instances, these pathways can slow immediate immune activity. For example, knockout of IL-10 is beneficial as it allows immediate protection against acute influenza with better survival at lethal infection levels (48, 49). However, inhibiting IL-10 after acute influenza infection results in tissue inflammation and damage, with decreased survival (49, 50), similar to IL-10 knockout S.pneumoniae bacterial models (51). For an in-depth discussion on this see (48–51).
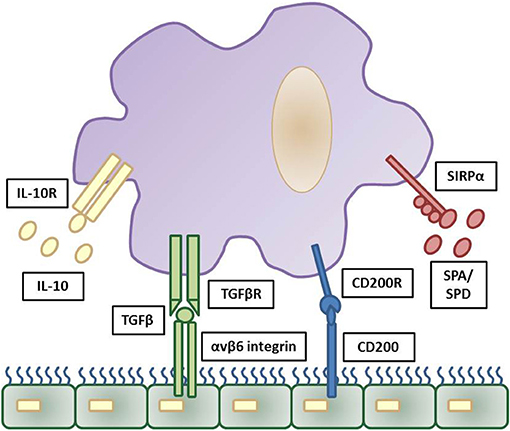
Figure 1. Inhibitory regulation of alveolar macrophages by the airway epithelium. Strict regulation of macrophage activation is required for homeostatic control of the general lung environment. As alveolar macrophages are under constant exposure to airborne endotoxins hypo-responsiveness is required for normal airway macrophage function. This is contributed through a number of downstream pathways triggered by airway epithelial cells production of IL-10, TGFβ, CD200, and surfactant proteins (SPA and SPD) and these reduce pro-inflammatory signaling and phagocytosis in airway macrophages via their respective cell surface receptors. The cascade of downstream inhibitory pathways to suppress macrophage activation are summarized elsewhere. Adapted from (35).
In addition, these mechanisms set a threshold of activation that needs to be overcome in order for an inflammatory response to be triggered. Activation of toll-like receptor (TLR) signaling, through recognition of an invading pathogen, elicits a strong enough immune response to exceed the inhibitory regulation of alveolar macrophages and causes up-regulation of TLR co-receptors including CD14 and triggering receptor expressed on myeloid cells 1 (TREM1) (52). Furthermore, loss of epithelial integrity during inflammation reduces the level of regulatory factors, releasing alveolar macrophages from epithelial-induced inhibition. This increases their phagocytic capabilities and initiates the production of pro-inflammatory cytokines (37, 53). The inhibitory factors that are important in maintaining airway homeostasis are also crucial in resolving inflammation after elimination of the microbial pathogen. Both CD200 and TGF-β assist in the suppression of inflammation, promote resolution and restore homeostasis (47).
Human respiratory syncytial virus (hRSV), human rhinovirus (hRV), human parainfluenza virus (hPIV) and human metapneumovirus (hMPV), are the major types of viruses responsible for acute infections of the upper and lower respiratory tract (54). These respiratory viruses represent a significant burden on global public health, with acute respiratory tract infections (ARTIs) being the fourth highest cause of global mortality (55).
Influenza virus is a member of the orthomyxovirus family and a negative sense, single stranded RNA virus (56). The viral envelope of influenza virus is composed of haemagglutinin (HA) and neuraminidase (NA) (57), which are used as identifiers of virus subtypes (58, 59). There are four genera of influenza virus; A, B, C, and D, with the influenza A subtypes H1N1 and H3N2 causing the largest proportion of influenza cases (60). Influenza virus infection is one of the leading causes of respiratory tract infections worldwide, with ~5–20% of the global population infected and a mortality rate of up to 650,000 patients annually1 (61). The influenza virus predominantly invades human upper airway epithelial cells by binding to α-2,6 or α-2,3-linked sialy glycans expressed on their surface (62–64). The influenza virus can effectively evade detection by the host immune system. Genetic changes due to the error-prone nature of the viral RNA polymerase, that result in antigenic drift or recombination events between influenza viruses, can give rise to new subtypes of influenza that can lead to epidemic or pandemic outbreaks (65–67). Currently, our best options to combat influenza are by prevention using vaccines and treatment with antiviral medications. However, the variable nature of the virus limits the efficacy of both approaches as they need to be updated annually to keep up with the evolution of new subtypes (68).
hRSV is the main cause of acute lower respiratory tract infection (ALTRI) in infants, young children and older adults (aged ≥65 years) (69). hRSV is an enveloped negative-sense single-stranded RNA virus belonging to the Pneumoviridae family, Orthopneumovirus genus (69, 70). There are 2 major antigenic groups of hRSV, A and B, which can be further subdivided into 10 A genotypes and 13 B genotypes (71). The highly contagious nature of the virus means nearly all children will have been infected with hRSV by the age of 2 years old (72). Bronchiolitis or pneumonia caused by hRSV infection is the major cause of hospitalisations in children under the age of 2 years old. Additionally, hRSV infection has been implicated in the development of childhood asthma and recurrent wheezing (72–75). The global public health burden of hRSV is significant, with ~10% of all hospital admissions for severe bronchiolitis or pneumonia due to the virus, representing an annual cost of about 394 million USD (76–78). The severity of hRSV infection and associated clinical symptoms can be controlled by the use of palivizumab, a neutralizing monoclonal antibody to the fusion glycoprotein (F protein), which is a transmembrane surface protein in the viral envelope of hRSV (79–82). However, an effective vaccine against hRSV has yet to be developed.
The development of childhood asthma and recurrent wheezing is not only closely linked with infant hRSV-induced bronchiolitis, but is also associated with wheezing illnesses due to hRV infection in infancy (83–86). A member of the Picornaviridae, genus Enterovirus, hRV is a non-enveloped positive single-stranded virus (87, 88). hRVs can be classified into three species, with RV-A and RV-C, causing more severe respiratory illness, when compared to RV-B (88, 89). The species can be further categorized into genotypes, of which there are over 100 (87, 90). hRVs circulate throughout the year, are transmitted through direct contact or aerosol particles and are capable of infecting both the lower and upper respiratory tracts (87, 91, 92). Symptoms following infection are generally that of the common cold, including sore throat, cough, nasal congestion, sneezing and rhinorrhoea. However, in infants, the elderly, immunocompromised adults or those suffering from chronic respiratory illnesses, infection with hRV can be more severe. For example, hRV is responsible for 20–40% of all hospitalisations due to wheezing in infants aged 12 months or less (93, 94). Development of a vaccine and antivirals against hRV has been hindered by the vast quantity of genetically distinct genotypes (90, 95).
hPIV is second most common cause of ALTRI in children, after hRSV (96). hPIV, like hRSV, is an enveloped negative-sense single-stranded RNA virus of the Paramyxoviridae family (97–99). hPIV consists of four major serotypes—hPIV-1 and hPIV-3, genus Respirovirus and hPIV-2 and hPIV-4, genus Rubulavirus (100). By the age of 2 years old 60% of children have been infected by hPIV-3 and at the age of 5 years the majority have been infected by hPIV-1, hPIV-2 and hPIV-3 (97, 101). Although hPIV has been predominantly viewed as a cause of respiratory illness in pediatric patients, both immunocompromised and older adults are also susceptible to infection (97, 100). Clinical manifestations of infection by hPIV include the common cold, croup (laryngotracheobronchitis), tracheobronchitis, bronchiolitis and pneumonia (100). However, as of yet there is no effective antiviral treatment or vaccine available for hPIV.
Since its discovery in 2001, hMPV has been identified as one of the major causes of upper and lower respiratory tract infection in children, immunocompromised patients and the elderly, being detected in 4–16% of patients with ARTIs (102–108). hMPV, a negative-sense single stranded RNA virus, is a member of the Paramyxoviridae family, genus Metapneumovirus, and is closely related to hRSV and parainfluenza (108). Most infections with hMPV elicit mild to moderate clinical symptoms, although 5–10% of cases result in admission to pediatric intensive care (102, 107, 109).
A significant contributor to morbidity and mortality in respiratory viral infections is bacterial invasion. Given the colonization of the upper respiratory tract with common pathogens including Streptococcus (S) pneumoniae, Haemophilus (H) influenzae and most of the Staphylococcus species, a shift in immunological balance and the airway environment can undoubtedly cause severe secondary bacterial infection in the host. The most famous reports of bacterial colonization after lung viral infection stem from the 1918 influenza pandemic where between 20 and 60 million deaths were due to bacterial co-infection (110). It is estimated that ~25% of all influenza-related deaths are associated with co-infections, particularly during seasonal outbreaks (111, 112). Viral respiratory infections elevate nasopharyngeal bacterial density (113, 114), which may promote their colonization in the lower airways, though the precise mechanisms are unclear.
Bacterial co-infection is not limited to influenza virus. A retrospective cohort study of 6,000 hospitalized neonates in China showed that 94% had RSV infection, with the remainder having parainfluenza, influenza virus or adenovirus. The dominant co-infections in RSV infected neonates were E. coli, Klebsiella (K) pneumoniae, S. aureus, and Enterobacter cloacae (115). The high frequency of RSV and pneumococci co-infection in hospitalized children is reduced by prior pneumococcal conjugate vaccination and has led to the suggestion that treatment for secondary bacterial infections should be considered for pneumonia cases even if a child tests positive for RSV (116). The choice of antibacterial strategy may be critical since RSV can increase S. pneumoniae virulence by binding to penicillin binding protein 1a (117) and so penicillin derivatives may be ineffective. Experimental studies on human Rhinovirus 16 infection enhances H. parainfluenzae, Neisseria subflava, and to a lesser extent S. aureus in throat swabs (118). One study in adults revealed that rhinovirus was the most common (23.6%), then parainfluenza virus (20.8%), hMPV (18.1%), influenza (16.7%), and RSV (13.9%). However, virus strain occurrence may also be influenced by co-infections as RSV was significantly more common in those that also had community-associated pneumonia (119).
Bacterial and viral infections co-exist, and the post-viral bacterial outgrowths are often co-infections made up of different species of bacteria. In a recent meta-analysis, 28–35% of patients demonstrated positive laboratory culture with the co-infective species, S. aureus and S. pneumoniae, respectively (120). S. pneumoniae is the most common pathogen that causes community-acquired pneumonia and potential overwhelming sepsis, and is associated with high mortality and morbidity during influenza epidemics and pandemics (121, 122).
S. aureus, a gram-positive cocci and a common commensal in the nose and skin, is a major cause of bacteraemia (123). It is unclear why S. aureus has become a major cause of concern particularly in the pediatric population, of which a study of the 2003–4 season in the USA found that this organism not only dominated influenza-associated childhood mortalities, but was also found to be the most common causative bacterial agent in 46% of isolates, whereby more than 50% were methicillin-resistant strains (111). A rare and severe complication of community-acquired pneumonia is necrotising pneumonia, characterized by pulmonary consolidation, inflammation, necrosis, and ultimately gangrene, which is caused by methicillin-resistant S. aureus, a major public health concern due to its resistance to antimicrobials. Prior or co-infection with influenza infection and the presence of Panton-Valentine leukocidin (PVL) are both significantly associated with the necrotising pneumonia (124).
Other than a breach of the epithelial barrier, there are a number of modifications to cellular innate immunity in the lung that contribute to secondary bacterial infection.
Cellular turnover by apoptosis features in health and inflammation. Airway macrophages play an important function in clearing apoptotic cells, a process known as efferocytosis, which is essential in maintaining airway homeostasis (125). Inefficient clearance of apoptotic cells leads to secondary necrosis and the release of damage associated molecular patterns (DAMPs) that subsequently promote an inflammatory response (126). Efferocytosis is mediated by a plethora of receptors that recognize externalized proteins on the cell surface of apoptotic cells. One of the most commonly studied proteins mediating efferocytosis is phosphatidylserine (PtdSer). PtdSer is present on the inner plasma membrane in living cells, but is externalized upon induction of apoptosis (127) by caspase inactivation of flippase (ATP11C) that is required to “flip” PtdSer back into the plasma membrane (128). Caspases also activate scramblases that “scramble” phospholipids in the plasma membrane; promoting exposure of PtdSer on apoptotic cells (129). Other proteins that flag up the presence of an apoptotic cell include oxidized low-density lipoprotein, calreticulin, annexin A1, ICAM-3, C1q, and thrombospondin (130). In parallel there are a number of receptors that recognize these proteins on apoptotic cells, including many that bind PtdSer: Triggering receptor expressed by myeloid cells-2 (TREM2) (131), CD300 (132), receptor for advanced glycation end products (RAGE) (133), Stabilin-2 (134), brain-specific angiogenesis inhibitor-1 (BAI1) (135) and TIM family members (T cell/transmembrane, immunoglobulin, and mucin) (136, 137) (Figure 2). For a review of other receptors recognizing externalized molecules on apoptotic cells see (130).
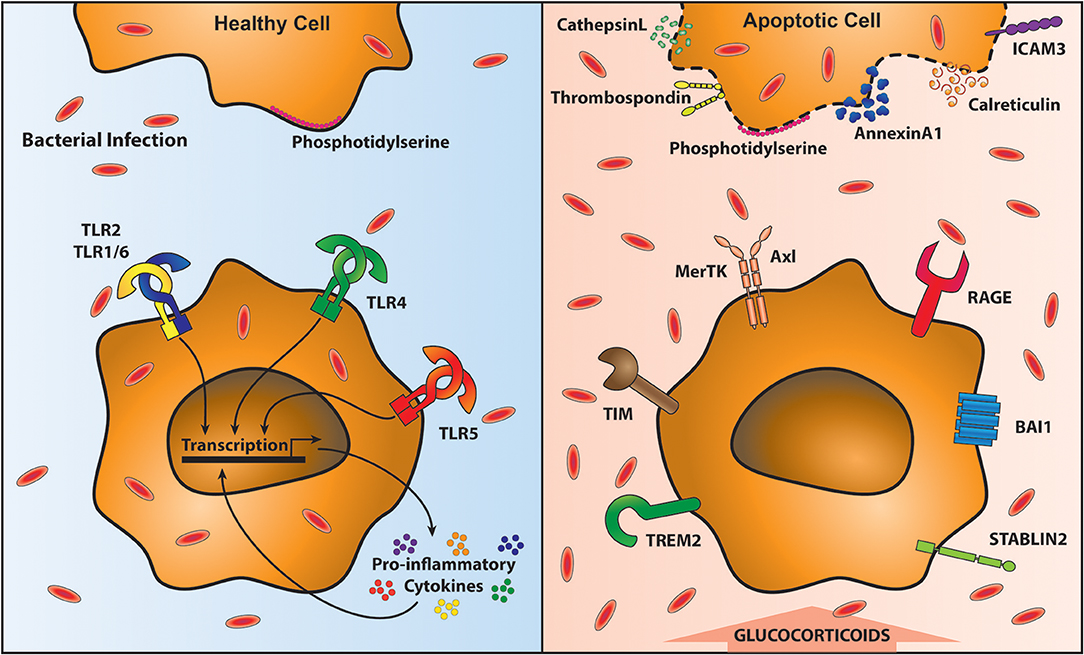
Figure 2. Clearance of apoptotic cells impairs anti-bacterial immunity. Removal of apoptotic cells requires their recognition by specialized receptors on phagocytic cells, including macrophages. In the presence of healthy cells (top left) Phosphatidylserine (PtdSer) is on the inner leaflet of the membrane. Local macrophages do not recognize them and therefore are able to signal through Toll-like receptors (TLR) unimpeded, resulting in the proinflammatory cytokine response. This optimal response is able to contain and clear bacterial infections (shown in red ovals). However, upon programmed cell death, PtdSer and a variety of other proteins are translocated to the outside of the cell membrane (top right). Macrophages recognize these exposed proteins via specific receptors (bottom right). These receptors facilitate apoptotic cell recognition and engulfment (known as efferocytosis) however, during efferocytosis macrophages are unable to respond to bacteria leading to their outgrowth (bottom right).
One PtdSer recognizing receptor family pertinent to the lung and its susceptibility to bacterial complications is the TAM receptor family (Tyro3, Axl, and Mertk receptors). These engulfment receptors require bridging molecules to link them to externalized PtdSer; protein S or growth arrest specific 6 (Gas6) (138). MerTK is ubiquitously expressed on macrophages, and even used as a defining marker for them. Axl, however, shows a more restricted distribution and is constitutively expressed on airway macrophages driven by GM-CSF and up-regulated during viral infection (139). Ligation of TAM receptors, in the presence of type 1 interferons (IFNs) enhances the expression of suppressor of cytokine signaling (SOCS) 1 and SOCS3, which reduce TLR and cytokine receptor signaling pathways (140–142). Furthermore, signaling via TAM receptors also induces TGFβ, IL-10 and prostaglandin production (143–146). This anti-inflammatory airway macrophage state is important to tolerate self-cells (125) but also reduces responses to subsequent coinfections (see Figure 2). Expression of IL-10 is raised following secondary bacterial coinfection after influenza virus exposure (147, 148). This is likely designed to prevent further tissue damage and to allow a return to homeostasis.
Lung viral infection enhances the apoptotic load due to cytopathology of infected cells and also the requirement to clear the large recruited immune cell infiltrate (149, 150). An absence of the TAM receptor Axl leads to excessive weight loss upon influenza infection in mice (139) that is likely linked to heightened secondary necrosis, which liberates DAMPS (151) recognized by pattern recognition receptors (PRRs), such as RAGE and ST2 (151–153). Axl knockout mice display increased nucleosome release in the airways corroborating the idea of enhanced secondary necrosis. This propagation of severe inflammation is likely to damage the lungs further and enhance the likelihood of secondary bacterial infections. Supporting this idea, prior exposure of mouse airway macrophages to apoptotic cells results in suppression of FcR-mediated phagocytosis and killing of bacteria. Furthermore, intrapulmonary administration of apoptotic cells impairs S. pneumoniae clearance from the infected lung (154). Also, suppression of antimicrobial responses of airway macrophages is enhanced by glucocorticoids, which promote efferocytosis, and treatment of mice with apoptotic cells in the presence of glucocorticoids is associated with elevated bacterial burden in the infected lungs (155).
Therefore, the normal process of clearing dying cells can have long term consequences and is particularly evident in chronic lung diseases (156) [for a review see: (157)]. However, further studies are required to determine the importance of this process, including analysis of the redundancy between apoptotic recognition receptors and the long term outcome of their manipulation. Efficient clearance of apoptotic cells may therefore provide an opportunity for therapeutic manipulation to lessen the severity of lung viral infections and prevent bacterial complications. In addition to the clearance of apoptotic cells by phagocytes, the phagocytes themselves (neutrophils and macrophages) may also undergo apoptosis.
Another natural process that occurs following viral infection is the cessation of inflammation. This is particularly important to allow efficient repair. Therefore, a prolonged inhibition of innate immunity is a common occurrence. However, a timely response to bacterial infection is critical to limit the pathogen load. Any delay in early immunity results in logarithmically higher bacterial loads that are difficult to clear and cause extensive bystander tissue damage. PRRs are important in this regard, but may be impaired by previous or concurrent inflammatory conditions. PRRs are widely expressed in the lungs on airway epithelial cells, alveolar macrophages and DCs and their ligation leads to the release of cytokines, chemokines, eicosanoids and type I IFNs into the airspaces (158, 159). The kinetics of this initial inflammatory wave limits early pathogen replication (159) by recruitment of monocytes, neutrophils and natural killer (NK) cells. NK cells target infected airway epithelial cells that have lost or reduced MHC class I expression (160), whereas monocytes and neutrophils aid alveolar macrophages in removing infected dead cells (161) and co-existing bacteria. Furthermore, type I IFNs stimulate the production of interferon-stimulated genes (ISGs), leading to cell-intrinsic and extrinsic antiviral activity (162). However, many studies have observed that subsequent stimulation via PRRs is defective following lung viral infection. This effect is not restricted to PRRs as defects in multiple processes employed by the mononuclear phagocyte system have been observed (69).
Following an acute viral infection, mouse airway macrophages display a similar phenotype to those in health (CD11c, CD11b, F4/80, and Siglec F). However, their responsiveness to TLR agonists is significantly dampened (163). We called this “innate imprinting” in 2004 (164), which is similar to the concept of “trained immunity” described by others in which monocytes acquire a tolerant phenotype after stimulation (165–167). This un-responsive state has recently been described as “immune paralysis” (168). In addition to influenza virus, human rhinovirus infection also predisposes to bacterial infection via degradation of IRAK-1 (interleukin 1 receptor associated kinase) leading to enhanced infection of respiratory epithelial cells by H. influenza (169). Defective TLR signaling would clearly lead to a reduction in many aspects of inflammation. With respect to subsequent bacterial infections, however, the most damaging consequences are the IFNγ induced impairment of macrophage phagocytosis (170, 171) and the reduction in neutrophil recruitment due to suppressed IL-8 production. In addition to reduced recruitment, neutrophil function also appears impaired following viral infection with reported reductions in myeloperoxidase, reactive oxygen species and the bactericidal properties of neutrophil extracellular traps (NETs) [for a review see (172)]. Reduced recruitment of neutrophils would also impact on airway macrophage NLRP3 inflammasome activation that is important for the production of IL-1β (173–176).
Reduced TLR signaling during viral infection may contribute to the impairment of the IL-17 response required for bacterial containment. Th17 cells produce IL-17 and IL-22 and are regulated by IL-23 (177, 178). These cytokines are crucial for lung epithelial production of neutrophil recruiting chemokines and anti-microbial peptides (179). Influenza virus induced type 1 IFNs reduce IL-17, IL-22, and IL-23 and impair the clearance of S. aureus; an outcome that can be rescued by adenoviral delivery of IL-23 (180). Type 1 IFNs also impair IL-17 production from γδ T cells (180).
The anti-inflammatory state that occurs following lung viral infection creates some confusion as patients and mice that succumb to secondary bacterial infection ultimately display enhanced inflammation (147, 181–183). However, a sluggish immune response will ultimately lead to enhanced inflammation due to an exponentially higher bacterial load.
In addition to viral induced modification of airway macrophages, other innate immune cells are also affected. Type-2 innate lymphoid cells (ILCs) increase during influenza virus infection and secrete IL-13 (3), which although important for wound repair, are not useful during bacterial infection. A similar population of Lin− CD127+ ST2+ CRTH2+ ILC2s have also been identified in human lung tissue and BAL and are known to produce IL-13. In mice, methicillin-resistant Staphylococcus aureus induces IL-13 up to 3 days after influenza virus infection and impairs viral clearance. Later infection of MRSA after influenza however, exacerbates bacterial replication due to inhibition of IL-13 and an upregulation of IFNγ (184). A detrimental impact of IL-13 is also evident following chlamydia (185) and tuberculosis (186). IL-13 also promotes Mycoplasma pneumoniae and non-typeable H. influenza adhesion in cultured bronchial epithelial cells by increasing MUC18 (187) and overcomes the enhanced bactericidal effects on epithelial cells of beta-2 agonists (188). Collectively, these studies suggest that ILC2s can be beneficial or harmful depending on their kinetics.
Viral infection induces the early recruitment of NK cells to the lungs where they promote anti-viral immune cells through the release of cytokines and limit viral replication by removing infected cells that have down-regulated MHC class I. If NK cells are depleted, adaptive immunity is not optimal, which could lead to prolonged viral infection (189, 190). NK cells also influence dendritic cells to support Th17 and Th1 cells that are important in anti-bacterial immunity (191) and NK cell production of IL-22 is protective against Klebsiella lung infection (192). However, NK cells appear early in the antiviral response to lung viral infection and so may not be present during secondary bacterial infection. Indeed influenza virus is reported to decrease NK cells, which reduces clearance of S. aureus in a process dependent on TNF-mediated enhancement of macrophage phagocytosis (193).
IL-10 is upregulated by viral infection and dampens the activation of invariant natural killer T (iNKT) cells by inhibiting the production of IL-12 by lung monocyte-derived dendritic cells, which contributes to S. pneumoniae outgrowth (194). IFNγ increases susceptibility to secondary bacterial infection by promoting inflammation and damage in the upper respiratory tract through both the ligand IFNγ and IFNγ receptor (245). Though IFNγ stimulates a pro-inflammatory phenotype in alveolar macrophages, it inhibits bacterial phagocytosis (49)
Neutrophils are critical components of anti-bacterial immunity. In addition to their reduced recruitment due to impaired chemokine production, influenza virus also inhibits their activity by inhibiting Th17 cell induction of anti-microbial peptides (195). Viral induction of Setdb2 (a protein lysine methyltransferase) also represses the expression of the CXCL1 gene that recruits neutrophils (196) and defective G-CSF production impairs neutrophil digestion and/or killing of phagocytized bacteria via myeloperoxidase (MPO) activity (197).
γδ T cells are also important in susceptibility to secondary bacterial infections. These rare T cells directly recognize pathogen-associated molecular patterns (PAMPs), express a range of cytokine receptors that modulate their function, mediate cell cytolysis via FAS and TRAIL and release anti-microbial peptides and cytotoxic molecules. They also produce IFN-γ, TNF-α, and IL-17. γδ T cell IL-17 production is impaired during influenza infection by type I IFNs causing susceptibility to S. pneumoniae infection (198). The role of γδ T cells in the extent of lung inflammation during viral infection depends, however, on whether other underlying conditions are present. For example, γδ T cell depletion in murine models of rhinovirus infection in asthmatic mice enhances airway hyper-reactivity (199).
Mucosal-associated invariant T (MAIT) cells (200, 201) are a recently studied population that are important in mucosal tissues for anti-bacterial immunity. They express cytotoxic markers such as CD107a and granzyme B via synergistic actions of IL-12 and IL-7 (202) and produce IFN-γ, TNF-α, and IL-17A (203). Their role in the lung is beginning to emerge. Lower numbers of peripheral blood CD161(+)Vα7.2(+) MAIT cells are associated with fatality in hospitalized patients with avian H7N9 influenza (204). However, it is not currently known whether defects in this population may predispose to bacteria following virus infections in the lung.
In addition to reduced neutrophil chemoattractants, the post-viral lung may be skewed toward wound repair that will not be conducive for bacterial recognition and clearance. The molecules mediating wound repair are often immune suppressive. IL-10 is enhanced following influenza infection and promotes bacterial replication in the post-influenza virus infected lung (148) by inhibiting multiple facets of immunity; a process that may be driven by the upregulation of indoleamine 2,3-dioxygenase (IDO) (205). Furthermore, regulatory T cells and TGFβ are raised post-viral infection to dampen inflammation and facilitate processes of wound repair; for example by inducing the synthesis of collagen (206). However, TGFβ is also anti-inflammatory and is required to limit the activity of dendritic cells (168). A recent study by the Schulz-Cherry group showed that knockout of the β6 integrin prevents the activation of latent TGFβ leading to the presence of constitutively activated airway macrophages (207). Wound repair therefore represents a double edged sword where anti-inflammatory components limit inflammation and promote repair, but at the same time leave hosts susceptible to bacterial infection.
A few studies have described that epithelial cell proliferation and the expression of lung repair genes are reduced following respiratory viral infection (208, 209). This implies that barrier repair is delayed, which may prolong the access to alternative adhesion and nutrition sources for bacteria.
The importance of the repair process in the outcome of viral and bacterial infection of the respiratory tract is elegantly illustrated by the administration of amphiregulin, which decreases inflammation and lung damage to influenza virus (3) and prevents mortality to a secondary bacterial infection in the absence of any discernible influence on bacterial load (208).
Extracellular matrix is a highly organized structure containing precise patterning of 43 different types of collagen, 200 glycoproteins and 40 proteoglycans (210). These components combine to form the interstitial matrix and the basement membrane. Alterations in both of these impacts on the cellular content of the lung and airways, and the adhesion, growth and location of bacterial species.
The basement membrane contributes to tissue architecture and is a highly organized structure made up of collagen IV, laminins, proteoglycans (decorin, biglycan, aggrecan and versican), heparan sulfate proteoglycans (perlecan and agrin), and nidogen (211). Some of these components can bind to other proteins that have immune modulatory properties. Decorin and biglycan for example, bind TGF-β1 (212) and so any alteration of their density or position will impact on lung inflammation and tissue repair. Similarly, fibrillar collagens type I and III of the interstitial matrix, in addition to binding other collagens and ECM components, also interact with inflammatory cell surface receptors particularly integrins. VLA-1, for example, is expressed on influenza-specific lung CD8+ T cells and binds α1β1 on interstitial matrix facilitating retention of memory CD8+ T cells in the lung (213). It is not hard to imagine that matrix re-modeling due to viral infection will have numerous consequences, such as the retention of a higher immune cell burden (214). Those retained immune cells, however, may not be optimal for subsequent bacterial infections and may even hinder the early migration of anti-bacterial immunity. For a recent review on immune cell:matrix interactions in the lung see (215).
The degradation of matrix can also liberate bioactive fragments now called matrikines which have immune modulatory properties. For example, the proteolytic processing by matrix metalloproteinases, MMP8 and MMP9, of interstitial collagens liberates the bioactive fragment, acetylated tripeptide Pro-Gly-Pro (acetyl-PGP) which promotes lung neutrophil recruitment (216, 217).
Accumulation of extracellular matrix components requires additional effort from interstitial and alveolar macrophages to clear them. This renders them hypo-responsive to subsequent bacteria. Recently we have found that excess hyaluronan induces adverse events in this way (218). Hyaluronan is a glycosaminoglycan that is abundant in the lung interstitial matrix. It is extruded from cells by hyaluronan synthases forming long cable-like polysaccharide structures. Degradation of hyaluronan is mediated by hyaluronidases. High- and -low molecular weight hyaluronan is reported to be anti-inflammatory and pro-inflammatory, respectively (219). Furthermore, hyaluronan can be sampled in sputum and by bronchoalveolar lavage, suggesting accumulation in the airways (220, 221). We have recently reported that hyauronan continues to accumulate in the lung and airway long after resolution of acute influenza virus infection in mice due to excess production via HA synthase 2. Furthermore, this excess hyaluronan is cross-linked with inter-α-inhibitor heavy chains due to elevated TNF-stimulated gene 6 expression. IαI is a proteoglycan containing two heavy chains of ~80 kDa, and a light chain (bikunin) of ~25 kDa that confers protease inhibitory properties (222, 223). Circulating IαI leaks into tissues during inflammation. Its synthesis has also been described in lung epithelia where it mediates repair after lung injury (224). In our study, administration of intranasal hyaluronidase completely restored lung function without any deleterious side effects (218).
There are other examples of matrix alterations contributing to the pathogenesis of lung viral infections (225). Influenza infection induces the recruitment of myeloid cells expressing membrane type I matrix metalloprotease (MT1-MMP/MMP-14) that is important in lung development and homeostasis (226). MT1-MMP inhibition rescues tissue damage and mortality in influenza-infected mice and combined with the anti-viral, oseltamivir, affords complete recovery. Furthermore, MT1-MMP inhibition also prevents outgrowth of S. pneumoniae following influenza infection (227). The modulation of extracellular matrix may depend on the viral strain. Analysis of RNA datasets from patients infected with pandemic associated influenza strains shows that H5N1 and H7N9 infection are enriched for genes involved with the extracellular matrix pathway (228). The importance of lung recovery and resilience is also demonstrated in mice lacking endophilin B2 that display improved mechanosensing and collagen and elastin ECM remodeling compared to wild-type mice (229). There are many other examples where matrix and associated components impact on lung immunity, which have been comprehensively reviewed elsewhere (230).
In addition to viruses directly promoting bacterial adherence (e.g., the neuraminidase in influenza virus exposes bacterial attachment sites by cleaving sialic acids, which are also metabolized by bacteria as a food source (231)), viral induced changes in extracellular matrix will change the lung microbiome. Dysbiosis of microbial commensalism can significantly impact on the overall health and progression of disease. Bacteria and bacterial products induce phenotypic and functional changes in immune pro-inflammatory gene expression, cellular adhesion and migration, and cell death (232). Binding to the ECM allows bacteria to adhere to, and colonize, host tissue. In addition, bacteria demonstrate affinity for different matrix substrates and changes in ECM components may increase host-pathogen accessibility and increase of bacterial virulence (233).
A number of microbes have elastase activity and/or express binding proteins for elastin that aid their pathogenicity (234). S aureus binds to elastin rich sites and expresses elastin binding proteins (EbpS) which bind to soluble, but not structurally intact chains of elastin (234). The expression of EbpS is also associated with greater bacterial cell growth, promoting cell proliferation and colonization (234, 235) and evasion of phagocytosis (234). In addition, elastin proteolytic products induce MMP activity and a number of bacteria express elastases (234, 235) further promoting elastin availability and consequently bacterial binding.
S. aureus encodes the fibronectin binding proteins (FnBPs), MSCRAMM (microbial surface component recognizing adhesive matrix molecule) that adhere to fibronectin and fibrinogen (236). Since components of fibronectin influence TLR4 receptor signaling, FnBPs may also promote immune regulation (237). Bacteria express collagen receptors and their binding appears to depend on collagen fiber tensile strength, conformation and structural dynamics. In an in vitro model, applying increasing high tensile forces to collagen peptides restricts receptor binding, suggesting that structurally normal collagen fibers decrease available sites for bacterial adhesion. Injured states, where collagen fibers are cleaved by high MMP activity, may increase susceptibility toward bacterial colonization with reduced structural strength and increased accessibility for more bacterial binding capacity (238).
Von Willebrand factor (vWF) is a large multimeric adhesion molecule and stimulates adhesion of bacteria. In bacteria such as S aureus, adherence to host can also be mediated via vWF and bacterial binding protein staphylococcal protein A (SPA). SPA binds to soluble and insoluble forms of vWF, promoting bacterial attachment and enhancing virulence in the absence of immune cell detection and clearance (239).
Glycosaminoglycan (GAG) interactions are ubiquitously used for cellular and extracellular signaling in all biological processes. Microbes utilize this universal process of the host for binding, and colonization of the host environment. Bacteria express GAG species and different binding domains across their entire surface. Studies blocking, removing or decreasing expression of these GAG binding domains decrease bacterial virulence (attachment, colonization and infection) in a number of bacterial strains (240). Bacterial communities have different affinities for GAG species (240). A large study manipulating GAG binding domains showed that the removal of heparin sulfate in S. aureus and S. pneumoniae decreases bacterial attachment to lung epithelial cells and fibroblasts and the inhibition of synthesis produced the same effect (240). The normal GAG interactions of the host are also used by microbes to prevent immune detection and clearance. Bacteria such as Streptococcus coat their surface with soluble high molecular weight hyaluronan, inhibiting detection and clearance by macrophages (241). Degradation of hyaluronan from the host tissues or bacteria into the low molecular weight protein stimulates phagocytosis, demonstrating bacterial colonization and infection can be influenced by the processing of GAGs from both the bacterium and host (241).
Interestingly, a time limited burst of inflammation from the outset is beneficial during influenza infection in mice, which results in faster clearance and less collateral damage (Figure 3). The evidence to support this comes from detailed studies on IL-10 knockout mice and the response to pathogen clearance discussed earlier, and our studies with CD200 or CD200R knockout mice. CD200R signaling on myeloid cells limits inflammatory activity (242). Mice lacking CD200 or CD200R show heightened weight loss during influenza infection due to raised levels of inflammation (47). However, when these mice are next exposed to S. pneumoniae, they do not show susceptibility, because the first inflammatory event to influenza was quicker, thus causing less collateral damage (243). The benefit of a short burst of inflammation has recently been supported by data from the Metzger group where mice lacking SOCS-1 or IFNγ cleared influenza virus faster than littermate controls due to a rapid induction of immunity. By contrast, in the presence of SOCS-1, inflammation was prolonged and collateral damage increased (171). It would be interesting to test the impact of subsequent respiratory bacterial infection in the SOCS1 and IFNγ deficient model. Such studies might suggest that patients experiencing severe disease do so because their immune system is too sluggish. However, upon presentation at care facilities it would be too late to consider boosting immunity. The speed of immunity could possibly be specifically tackled in patients with other underlying conditions that render their innate immune system suppressed, as in the case of chronic obstructive pulmonary disease, or of the wrong phenotype to limit viral replication, as in the case of asthma. These patient groups are known to be at risk of severe viral infections [for example see (244, 245)].
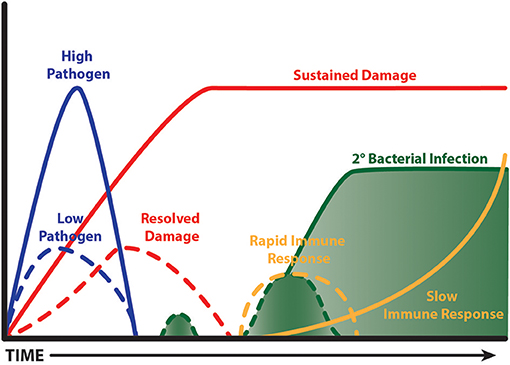
Figure 3. Fast and limited immunity is good. A time limited burst of inflammation limits bystander tissue damage, which in turn limits the extent of tissue repair. This leads to less impairment of anti-bacterial immunity and so a secondary bacterial infection is cleared. A virulent pathogen, or one that isn't cleared quickly, causes prolonged bystander tissue damage leading to a lengthy period of repair; the processes of which are anti-inflammatory. A subsequent bacterial infection is ignored and grows exponentially. Ultimately, innate immunity is activated when the bacterial load is excessive causing deleterious consequences.
The benefits of rapid induction of immunity to viral infection are also supported by research on IL-22. IL-22 is an interesting cytokine that is produced by innate immune cells and is critical for host protective immunity to lung K. pneumoniae (246), and S. aureus (183), but not to Mycobacterium tuberculosis or M. avium infection (164). IL-22 is upregulated during lung infection, but its neutralization has no effect on the kinetics of the disease or viral clearance. Rather it seems to function by promoting epithelial integrity and limiting lung damage (3, 247–249), which in turn prevents secondary lung infections by S. pneumoniae in mice (250). Interestingly, progesterone treatment of female mice also induces heightened IL-22 (and TGFβ and IL-6) and promotes faster recovery from influenza infection in female mice via epithelial production of amphiregulin. The resultant improvement of pulmonary function and reduced protein leakage is likely to diminish the risk of bacterial outgrowth, though this was not tested (251). In murine models of influenza infection, administration of GM-CSF promotes resistance to S. pneumoniae by promoting neutrophil recruitment and reactive oxygen species production from macrophages (252).
Another study that supports stimulation of immunity to prevent bacterial super-infections showed that the TLR-2 agonist, macrophage-activating lipopeptide 2 (MALP-2), reduces pneumococcal outgrowth in influenza virus infected mice (253). Also administration of nanoparticles containing the coat protein of a plant virus (papaya mosaic virus) and a single-stranded RNA causes the rapid recruitment of neutrophils, monocytes/macrophages and lymphocytes with beneficial effects on influenza virus and subsequent S. pneumoniae infection (254).
Matrix modulation research is a field with great potential in restoring immune function via alternative key mechanisms. Extracellular matrix production is elevated following severe acute viral infection, which could have consequences on cell retention, immune paralysis of phagocytic cells and the physical properties of the airspaces into which it leaks. Respiratory fluids from COPD patients for example, contain higher levels of hyaluronan (HA) than healthy controls (59, 218) and we have recently shown this is exacerbated further by viral infection in COPD patients. Hyaluronidase treatment of mice after resolution of influenza virus infection restores lung function suggesting that the consequences of increased airway and lung hyaluronan is an impaired lung physiology (218). Airway hyper-reactivity is also improved during ozone-induced airway disease in CD44 or IαI deficient mice (60, 255) that bind HA or cross-link it, respectively. TNF-stimulated gene 6 catalyzes the transfer of IαI heavy chains onto HA (256) and TSG-6 null mice are resistant to airway hyporesponsiveness (257). Also TSG-6 promotes anti-inflammatory macrophages, (258) and inhibits neutrophil recruitment (259–262) and NFκB nuclear translocation. Just considering one matrix protein such as hyaluronan, the method of its production and degradation and the proteins that cross link it, provides multiple avenues for modulation. Therapeutic development in this area, to our knowledge, is poor with most focus on neutralizing enzymes that degrade matrix to prevent the liberation of small chemotactic matrix products. However, recombinant human hyaluronidase is licensed for therapeutic use in humans to increase barrier permeability, and although it is currently approved to enhance delivery and absorption of subcutaneous anesthetics, increase uptake of fluids, and to improve resorption of radiopaque agents (263, 264), it has the potential to be used to improve inflammatory diseases by immune-matrix modulation.
Bacterial susceptibility following lung viral infection has been recognized for over a century and yet treatment options have not really altered since the introduction of antibiotics. It is now clear that long term suppression of innate immune mechanisms occurs following severe acute or chronic inflammation. In contrast to the clinical susceptibility toward bacterial infection that can occur in the 7 days following a viral infection, there are multiple long term modifications in immune mechanisms long after severe viral infections. These changes re-set the inflammatory tone of various immune cells by processes now known as trained immunity, innate imprinting or immune paralysis (164–166, 168). These molecular changes are evident during peak infection, but not in naïve un-infected lungs. This modified, tardy innate immunity immune response contributes to dysregulation of immune mechanisms to secondary bacterial exposure, rather than the clearance of the initial pathogen, and hence may explain the higher risk of long term bacterial outgrowth and chronic infection that cumulatively leads to excessive inflammatory disease. The majority of pathways leading to bacterial complications following viral infection have been discovered in single mouse strain studies. A recent report from the Metzger group shows that different mouse strains (BALB/C and C57BL/6) react differently to alveolar macrophage depletion following acute influenza infection. BALB/c mice respond to an acute influenza insult via IFNγ dependent alveolar macrophage depletion, whereas C57BL/6 mice do not. However, both are susceptible to post-viral bacterial coinfection (265). The precise combination of changes leading to bacterial super-infection may therefore be slightly different depending on genetic background.
Another area that requires development is that the known “at risk” patient groups currently identified for priority influenza vaccination (the elderly, asthmatic, pregnant etc.) do not account for the vast hospitalization numbers over the winter seasons. This suggests there may be other “at risk” groups.
This as exemplified by the rise of bacterial pneumonias in those experiencing low mood, stress, anxiety or mental health issues (266, 267). A mucosal barrier breach cannot explain all infectious complications. A population-based Danish study of 976,398 individuals, including 142,169 with a history of depression, onset of depression was associated with increased respiratory viral or bacterial complications (IRR = 1.58; CI = 1.36–1.85; p = 0.000) (268). Depression and stress are linked to suppression of multiple arms of innate and adaptive immunity [see (269) and references within], including a reduction of neutrophils (270) that are important for bacterial clearance. The link between mental health and infection is an area that will gain momentum in the next few years. Another area of concern that will likely garner research effort in the future is the influence of polypharmacy on respiratory infectious risk. In elderly patients hospitalized for pneumonia in Canada, 45% were taking 5 or more medications prior to hospital admission (271). A number of these medications may also modulate the immune system, though research in this area is sparse.
There is a window of opportunity between recovery from viral infection and the onset of bacterial outgrowth where innate immunity could be primed to react quicker. This may involve removal of immune suppressive pathways (CD200R, IL-10, and TGFβ), facilitation of apoptotic cell clearance (as apoptotic cell recognition receptors switch off innate immunity) or timely removal of high molecular weight matrix components from the airways. To identify these, studies are required that take into account other comorbidities, mental health status and the impact of polypharmacy on outcome.
EC, DM, CC, OB, SL, and TH drafted the manuscript. SL, OB, CJ, JC, and RR provided critical revisions and conceptual diagrams. All the authors made substantial contributions to the conception and design of the work, approved the submitted version of the manuscript, and agreed to be accountable for all aspects of the work.
This work was supported in part by grants from the Wellcome Trust (202865/Z/16/Z) (TH, DM, EC, OB, SL, and CJ) and Arthritis UK (CC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. ^https://www.who.int/influenza/en/
1. Seiler F, Lepper PM, Bals R, Beisswenger C. Regulation and function of antimicrobial peptides in immunity and diseases of the lung. Protein Pept Lett. (2014) 21 341–51. doi: 10.2174/09298665113206660100
2. Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. (2012) 88:1503–13. doi: 10.4049/jimmunol.1102832
3. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. (2011) 12:1045–54. doi: 10.1038/ni.2131
4. Walker JA, McKenzie A, Innate lymphoid cells in the airways. Eur J Immunol. (2012) 42:1368–74. doi: 10.1002/eji.201242425
5. Cheng M, Hu S. Lung-resident gammadelta T cells and their roles in lung diseases. Immunology (2017) 151:375–384. doi: 10.1111/imm.12764
6. Augustin A, Kubo RT, Sim G. K. Resident pulmonary lymphocytes expressing the gamma/delta T-cell receptor. Nature (1989) 340:239–41.
7. Sim G. K., Rajaserkar R, Dessing M, and Augustin A. Homing and in situ differentiation of resident pulmonary lymphocytes. Int Immunol. (1994) 6:1287–95.
8. Wands JM, Roark CL, Aydintug MK, Jin N, Hahn YS, Cook L, et al. Distribution and leukocyte contacts of gammadelta T cells in the lung. J Leukoc Biol. (2005) 78:1086–96. doi: 10.1189/jlb.0505244
9. Culley FJ. Natural killer cells in infection and inflammation of the lung. Immunology (2009) 128:151–63. doi: 10.1111/j.1365-2567.2009.03167.x
10. Ge N, Nishioka Y, Nakamura Y, Okano Y, Yoneda K, Ogawa H, et al. Synthesis and secretion of interleukin-15 by freshly isolated human bronchial epithelial cells. Int Arch Allergy Immunol. (2004) 135:235–42. doi: 10.1159/000081309
11. Curtis JL. Cell-mediated adaptive immune defense of the lungs. Proc Am Thorac Soc. (2005) 2:412–6. doi: 10.1513/pats.200507-070JS
12. Guilliams M, Lambrecht B. N., Hammad H. Division of labor between lung dendritic cells and macrophages in the defense against pulmonary infections. Mucosal Immunol. (2013) 6:464–73. doi: 10.1038/mi.2013.14
13. Sung SS, Fu SM, Rose CE Jr, Gaskin F, Ju ST, Beaty SR. A major lung CD103 (αE)-β7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J Immunol. (2006) 176:2161–72. doi: 10.4049/jimmunol.176.4.2161
14. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
15. von Garnier C, Blank F, Rothen-Rutishauser B, Goethert JR, Holt PG, Stumbles PA, et al. Identification and characterization of a dendritic cell precursor in parenchymal lung tissue. Am J Respir Cell Mol Biol. (2017) 56:353–361. doi: 10.1165/rcmb.2016-0058OC
16. Desch AN, Randolph GJ, Murphy K, Gautier EL, Kedl RM, Lahoud MH, et al. CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J Exp Med. (2011) 208:1789–97. doi: 10.1084/jem.20110538
17. Randall T. D. Bronchus-associated lymphoid tissue (BALT) structure and function. Adv Immunol. (2010) 107:187–241. doi: 10.1016/B978-0-12-381300-8.00007-1
18. Kumar A, Abdelmalak B, Inoue Y, Culver DA. Pulmonary alveolar proteinosis in adults: pathophysiology and clinical approach. Lancet Respir Med. (2018) 6:554–65. doi: 10.1016/S2213-2600(18)30043-2
19. Schyns J, Bureau F, and Marichal T. (2018). Lung Interstitial Macrophages: Past, Present, and Future. J Immunol Res 2018:5160794.
20. Ely KH, Cookenham T, Roberts AD, Woodland D. L. Memory T cell populations in the lung airways are maintained by continual recruitment. J Immunol. (2006) 176:537–43. doi: 10.4049/jimmunol.176.1.537
21. Snelgrove R. J., Godlee A, and Hussell T. Airway immune homeostasis and implications for influenza-induced inflammation. Trends Immunol. (2011) 32:328–34. doi: 10.1016/j.it.2011.04.006
22. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
23. Janssen WJ, Barthel L, Muldrow A, Oberley-Deegan RE, Kearns MT, Jakubzick C, et al. Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am J Respir Crit Care Med. (2011) 184:547–60. doi: 10.1164/rccm.201011-1891OC
24. Murphy J, Summer R, Wilson AA, Kotton D. N., Fine A. The prolonged life-span of alveolar macrophages. Am J Respir Cell Mol Biol. (2008) 38:380–5. doi: 10.1165/rcmb.2007-0224RC
25. Cai Y, Sugimoto C, Arainga M, Alvarez X, Didier ES, Kuroda M. J. In vivo characterization of alveolar and interstitial lung macrophages in rhesus macaques: implications for understanding lung disease in humans. J Immunol. (2014) 192:2821–9. doi: 10.4049/jimmunol.1302269
26. Duan M, Hibbs M. L., Chen W. The contributions of lung macrophage and monocyte heterogeneity to influenza pathogenesis. Immunol Cell Biol. (2017) 95:225–35. doi: 10.1038/icb.2016.97
27. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
28. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
29. Bonfield TL, Farver CF, Barna BP, Malur A, Abraham S, Raychaudhuri B, et al. Peroxisome proliferator-activated receptor-gamma is deficient in alveolar macrophages from patients with alveolar proteinosis. Am J Respir Cell Mol Biol. (2003) 29:677–82. doi: 10.1165/rcmb.2003-0148OC
30. Schneider C, Nobs S. P., Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol. (2014) 15:1026–37. doi: 10.1038/ni.3005
31. Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, et al., Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science (1994) 264:713–6. doi: 10.1126/science.8171324
32. Trapnell BC, Whitsett J. A. Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu Rev Physiol. (2002) 64:775–802. doi: 10.1146/annurev.physiol.64.090601.113847
33. Landsman L, Jung S. Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. J Immunol. (2007) 179:3488–94. doi: 10.4049/jimmunol.179.6.3488
34. Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, et al. Three unique interstitial macrophages in the murine lung at steady state. Am J Respir Cell Mol Biol. (2017) 57:66–76. doi: 10.1165/rcmb.2016-0361OC
35. Hussell T, Bell T. J. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. (2014) 14:81–93. doi: 10.1038/nri3600
36. Schneberger D, Aharonson-Raz K, Singh B. Monocyte and macrophage heterogeneity and Toll-like receptors in the lung. Cell Tissue Res. (2011) 343:97–106. doi: 10.1007/s00441-010-1032-2
37. Steinmuller C, Franke-Ullmann G, Lohmann-Matthes ML, Emmendorffer A. Local activation of nonspecific defense against a respiratory model infection by application of interferon-gamma: comparison between rat alveolar and interstitial lung macrophages. Am J Respir Cell Mol Biol. (2000) 22:481–90. doi: 10.1165/ajrcmb.22.4.3336
38. Hoidal JR, Schmeling D, Peterson PK. Phagocytosis, bacterial killing, and metabolism by purified human lung phagocytes. J Infect Dis. (1981) 144:61–71.
39. Holt PG, Inhibitory activity of unstimulated alveolar macrophages on T-lymphocyte blastogenic response. Am Rev Respir Dis. (1978) 118:791–3. doi: 10.1164/arrd.1978.118.4.791
40. Bedoret D, Wallemacq H, Marichal T, Desmet C, Quesada Calvo F, Henry E, et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J Clin Invest. (2009) 119:3723–38. doi: 10.1172/JCI39717
41. Roth MD, Golub SH. Human pulmonary macrophages utilize prostaglandins and transforming growth factor beta 1 to suppress lymphocyte activation. J Leukoc Biol. (1993) 53:366–71.
42. Lipscomb MF, Lyons CR, Nunez G, Ball EJ, Stastny P, Vial W, et al. Human alveolar macrophages: HLA-DR-positive macrophages that are poor stimulators of a primary mixed leukocyte reaction. J Immunol. (1986) 136:497–504.
43. Kirby AC, Coles MC, Kaye PM. Alveolar macrophages transport pathogens to lung draining lymph nodes. J Immunol. (2009) 183:1983–9. doi: 10.4049/jimmunol.0901089
44. Blumenthal RL, Campbell DE, Hwang P, DeKruyff RH, Frankel LR, Umetsu DT. Human alveolar macrophages induce functional inactivation in antigen-specific CD4 T cells. J Allergy Clin Immunol. (2001) 107:258–64. doi: 10.1067/mai.2001.112845
45. Bonfield TL, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, et al. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med. (1995) 152:2111–8. doi: 10.1164/ajrccm.152.6.8520783
46. Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell (1999) 96:319–28. doi: 10.1016/S0092-8674(00)80545-0
47. Snelgrove RJ, Goulding J, Didierlaurent AM, Lyonga D, Vekaria S, Edwards L, et al. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat Immunol. (2008) 9:1074–83. doi: 10.1038/ni.1637
48. Sun K, Torres L, Metzger DW. A detrimental effect of interleukin-10 on protective pulmonary humoral immunity during primary influenza A virus infection. J Virol. (2010) 84:5007–14. doi: 10.1128/JVI.02408-09
49. Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat Med. (2008) 14:558–64. doi: 10.1038/nm1765
50. Wainwright NW, Surtees PG, Gilks WR. Diagnostic boundaries, reasoning and depressive disorder, I Development of a probabilistic morbidity model for public health psychiatry Psychol Med. (1997) 27:835–45.
51. Penaloza HF, Nieto PA, Munoz-Durango N, Salazar-Echegarai FJ, Torres J, Parga MJ, et al. Interleukin-10 plays a key role in the modulation of neutrophils recruitment and lung inflammation during infection by Streptococcus pneumoniae. Immunology (2015) 146:100–12. doi: 10.1111/imm.12486
52. Klesney-Tait J, Turnbull I. R., and Colonna M. The TREM receptor family and signal integration. Nat Immunol. (2006) 7:1266–73. doi: 10.1038/ni1411
53. Lohmann-Matthes ML, Steinmuller C, Franke-Ullmann G. Pulmonary macrophages. Eur Respir J. (1994) 7:1678–89. doi: 10.1183/09031936.94.07091678
54. Lessler J, Reich NG, Brookmeyer R, Perl TM, Nelson KE, Cummings DA. Incubation periods of acute respiratory viral infections: a systematic review. Lancet Infect Dis. (2009) 9:291–300. doi: 10.1016/S1473-3099(09)70069-6
55. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al., Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet (2012) 380:2095–128. doi: 10.1016/S0140-6736(12)61728-0
56. Lamb RA, Choppin PW. The gene structure and replication of influenza virus. Annu Rev Biochem. (1983) 52:467–506.
57. Hsu AC. Influenza virus: a master tactician in innate immune evasion and novel therapeutic interventions. Front Immunol. (2018) 9:743. doi: 10.3389/fimmu.2018.00743
58. Souquette A, Thomas PG. Past life and future effects-how heterologous infections alter immunity to influenza viruses. Front Immunol. (2018) 9:1071. doi: 10.3389/fimmu.2018.01071
59. Zhou F, Trieu MC, Davies R, Cox RJ. Improving influenza vaccines: challenges to effective implementation. Curr Opin Immunol. (2018) 53:88–95. doi: 10.1016/j.coi.2018.04.010
60. Parrish CR, Murcia PR, Holmes EC. Influenza virus reservoirs and intermediate hosts: dogs, horses, and new possibilities for influenza virus exposure of humans. J Virol. (2015) 89:2990–4. doi: 10.1128/JVI.03146-14
61. Kwong JC, Stukel TA, Lim J, McGeer AJ, Upshur RE, Johansen H, et al. The effect of universal influenza immunization on mortality and health care use. PLoS Med. (2008) 5:e211. doi: 10.1371/journal.pmed.0050211
62. Ito T, Suzuki Y, Mitnaul L, Vines A, Kida H, Kawaoka Y. Receptor specificity of influenza A viruses correlates with the agglutination of erythrocytes from different animal species. Virology (1997) 227:493–9.
63. Ito T, Suzuki Y, Suzuki T, Takada A, Horimoto T, Wells K, et al. Recognition of N-glycolylneuraminic acid linked to galactose by the alpha2,3 linkage is associated with intestinal replication of influenza A virus in ducks. J Virol. (2000) 74:9300–5. doi: 10.1128/JVI.74.19.9300-9305.2000
64. Ryan-Poirier K, Suzuki Y, Bean WJ, Kobasa D, Takada A, Ito T, et al. Changes in H3 influenza A virus receptor specificity during replication in humans. Virus Res. (1998) 56:169–76. doi: 10.1016/S0168-1702(98)00067-7
65. Kim H, Webster RG, Webby RJ. Influenza Virus: Dealing with a Drifting and Shifting Pathogen. Viral Immunol. (2018) 31:174–83. doi: 10.1089/vim.2017.0141
66. Simonsen L, Spreeuwenberg P, Lustig R, Taylor RJ, Fleming DM, Kroneman M, et al. Global mortality estimates for the 2009 Influenza Pandemic from the GLaMOR project: a modeling study. PLoS Med. (2013) 10:e1001558. doi: 10.1371/journal.pmed.1001558
67. Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, et al. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA (2003) 289:179–86. doi: 10.1001/jama.289.2.179
68. Krammer F, Palese P. Advances in the development of influenza virus vaccines. Nat Rev Drug Discov. (2015) 14:167–82. doi: 10.1038/nrd4529
69. Bohmwald K, Espinoza JA, Pulgar RA, Jara EL, and Kalergis AM. Functional impairment of mononuclear phagocyte system by the human respiratory syncytial virus. Front Immunol. (2017) 8:1643. doi: 10.3389/fimmu.2017.01643
70. Afonso CL, Amarasinghe GK, Banyai K, Bao Y, Basler CF, Bavari S, et al. Taxonomy of the order Mononegavirales: update 2016. Arch Virol. (2016) 161:2351–60. doi: 10.1007/s00705-016-2880-1
71. Hotard AL, Laikhter E, Brooks K, Hartert TV, Moore ML. Functional analysis of the 60-nucleotide duplication in the respiratory syncytial virus buenos aires strain attachment glycoprotein. J Virol. (2015) 89:8258–66. doi: 10.1128/JVI.01045-15
72. Henderson J, Hilliard TN, Sherriff A, Stalker D, Al Shammari N, Thomas HM. Hospitalization for RSV bronchiolitis before 12 months of age and subsequent asthma, atopy and wheeze: a longitudinal birth cohort study. Pediatr Allergy Immunol. (2005) 16:386–92. doi: 10.1111/j.1399-3038.2005.00298.x
73. Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, et al. The burden of respiratory syncytial virus infection in young children. N Engl J Med. (2009) 360:588–98. doi: 10.1056/NEJMoa0804877
74. Sweetman LL, Ng YT, Butler IJ, Bodensteiner JB. Neurologic complications associated with respiratory syncytial virus. Pediatr Neurol. (2005) 32:307–10. doi: 10.1016/j.pediatrneurol.2005.01.010
75. Tripp RA. Pathogenesis of respiratory syncytial virus infection. Viral Immunol. (2004) 17:165–81. doi: 10.1089/0882824041310513
76. Bohmwald K, Espinoza JA, Rey-Jurado E, Gomez RS, Gonzalez PA, Bueno SM, et al. Human respiratory syncytial virus: infection and pathology. Semin Respir Crit Care Med. (2016) 37:522–37. doi: 10.1055/s-0036-1584799
77. Nair H, Simoes EA, Rudan I, Gessner BD, Azziz-Baumgartner E, Zhang JS, et al. Global and regional burden of hospital admissions for severe acute lower respiratory infections in young children in 2010: a systematic analysis. Lancet (2013) 381:1380–90. doi: 10.1016/S0140-6736(12)61901-1
78. Paramore LC, Ciuryla V, Ciesla G, Liu L. Economic impact of respiratory syncytial virus-related illness in the US: an analysis of national databases. Pharmacoeconomics (2004) 22:275–84. doi: 10.2165/00019053-200422050-00001
79. Fonseca W, Ozawa M, Hatta M, Orozco E, Martinez MB, Kawaoka Y. A recombinant influenza virus vaccine expressing the F protein of respiratory syncytial virus. Arch Virol. (2014) 159:1067–77. doi: 10.1007/s00705-013-1932-z
80. Graham BS. Vaccine development for respiratory syncytial virus. Curr Opin Virol. (2017) 23:107–12. doi: 10.1016/j.coviro.2017.03.012
81. Hu J, Robinson JL. Treatment of respiratory syncytial virus with palivizumab: a systematic review. World J Pediatr. (2010) 6:296–300. doi: 10.1007/s12519-010-0230-z
82. Mazur NI, Martinon-Torres F, Baraldi E, Fauroux B, Greenough A, Heikkinen T, et al. Lower respiratory tract infection caused by respiratory syncytial virus: current management and new therapeutics. Lancet Respir Med. (2015) 3:888–900. doi: 10.1016/S2213-2600(15)00255-6
83. Jackson DJ, Gangnon RE, Evans MD, Roberg KA, Anderson EL, Pappas TE, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med. (2008) 178:667–72. doi: 10.1164/rccm.200802-309OC
84. Kotaniemi-Syrjanen A, Vainionpaa R, Reijonen T. M., Waris M, Korhonen K, Korppi M. Rhinovirus-induced wheezing in infancy–the first sign of childhood asthma? J Allergy Clin Immunol. (2003) 111:66–71. doi: 10.1067/mai.2003.33
85. Lukkarinen M, Koistinen A, Turunen R, Lehtinen P, Vuorinen T, Jartti T. Rhinovirus-induced first wheezing episode predicts atopic but not nonatopic asthma at school age. J Allergy Clin Immunol. (2017) 140:988–995. doi: 10.1016/j.jaci.2016.12.991
86. Ruotsalainen M, Hyvarinen MK, Piippo-Savolainen E, Korppi M. Adolescent asthma after rhinovirus and respiratory syncytial virus bronchiolitis Pediatr Pulmonol. (2013) 48:633–9. doi: 10.1002/ppul.22692
87. Drysdale SB, Mejias A, Ramilo O. Rhinovirus - not just the common cold. J Infect. (2017) 74(Suppl. 1):S41–6. doi: 10.1016/S0163-4453(17)30190-1
88. McIntyre CL, Knowles NJ, Simmonds P. Proposals for the classification of human rhinovirus species A, B and C into genotypically assigned types. J Gen Virol. (2013) 94:1791–806. doi: 10.1099/vir.0.053686-0
89. Lee WM, Lemanske RF Jr, Evans MD, Vang F, Pappas T, Gangnon R, et al. Human rhinovirus species and season of infection determine illness severity. Am J Respir Crit Care Med. (2012) 186:886–91. doi: 10.1164/rccm.201202-0330OC
90. Jartti T, Gern JE. Role of viral infections in the development and exacerbation of asthma in children. J Allergy Clin Immunol. (2017) 140:895–906. doi: 10.1016/j.jaci.2017.08.003
91. Mosser AG, Vrtis R, Burchell L, Lee WM, Dick CR, Weisshaar E, et al. Quantitative and qualitative analysis of rhinovirus infection in bronchial tissues. Am J Respir Crit Care Med. (2005) 171:645–51. doi: 10.1164/rccm.200407-970OC
92. Stenberg-Hammar K, Hedlin G, Soderhall C. Rhinovirus and preschool wheeze. Pediatr Allergy Immunol. (2017) 28:513–520. doi: 10.1111/pai.12740
93. Marguet C, Lubrano M, Gueudin M, Le Roux P, Deschildre A, Forget C, et al. In very young infants severity of acute bronchiolitis depends on carried viruses. PLoS ONE (2009) 4:e4596. doi: 10.1371/journal.pone.0004596
94. Midulla F, Scagnolari C, Bonci E, Pierangeli A, Antonelli G, De Angelis D, et al. Respiratory syncytial virus, human bocavirus and rhinovirus bronchiolitis in infants. Arch Dis Child. (2010) 95:35–41. doi: 10.1136/adc.2008.153361
95. Lambkin-Williams R, Noulin N, Mann A, Catchpole A, Gilbert AS. The human viral challenge model: accelerating the evaluation of respiratory antivirals, vaccines and novel diagnostics. Respir Res. (2018) 19:123. doi: 10.1186/s12931-018-0784-1
96. Reed G, Jewett PH, Thompson J, Tollefson S, Wright PF. Epidemiology and clinical impact of parainfluenza virus infections in otherwise healthy infants and young children < 5 years old. J Infect Dis. (1997) 175:807–13.
97. Hall CB. Respiratory syncytial virus and parainfluenza virus. N Engl J Med. (2001) 344:1917–28. doi: 10.1056/NEJM200106213442507
98. Pawelczyk M, Kowalski ML. The role of human parainfluenza virus infections in the immunopathology of the respiratory tract. Curr Allergy Asthma Rep. (2017) 17:16. doi: 10.1007/s11882-017-0685-2
99. Russell E, Ison MG. Parainfluenza Virus in the Hospitalized Adult. Clin Infect Dis. (2017) 65:1570–6. doi: 10.1093/cid/cix528
100. Henrickson KJ. Parainfluenza viruses. Clin Microbiol Rev. (2003) 16:242–64. doi: 10.1128/CMR.16.2.242-264.2003
101. Tsukagoshi H, Ishioka T, Noda M, Kozawa K, Kimura H. Molecular epidemiology of respiratory viruses in virus-induced asthma. Front Microbiol. (2013) 4:278. doi: 10.3389/fmicb.2013.00278
102. Edwards KM, Zhu Y, Griffin MR, Weinberg GA, Hall CB, Szilagyi PG, et al. Burden of human metapneumovirus infection in young children. N Engl J Med. (2013) 368:633–43. doi: 10.1056/NEJMoa1204630
103. Englund JA, Boeckh M, Kuypers J, Nichols WG, Hackman RC, Morrow RA, et al. Brief communication: fatal human metapneumovirus infection in stem-cell transplant recipients. Ann Intern Med. (2006) 144:344–9. doi: 10.7326/0003-4819-144-5-200603070-00010
104. Esper F, Martinello RA, Boucher D, Weibel C, Ferguson D, Landry ML, et al. A 1-year experience with human metapneumovirus in children aged < 5 years. J Infect Dis. (2004) 189:1388–96. doi: 10.1086/382482
105. Falsey AR, Erdman D, Anderson LJ, Walsh EE. Human metapneumovirus infections in young and elderly adults. J Infect Dis. (2003) 187:785–90. doi: 10.1086/367901
106. Panda S, Mohakud NK, Pena L, Kumar S. Human metapneumovirus: review of an important respiratory pathogen. Int J Infect Dis. (2014) 25:45–52. doi: 10.1016/j.ijid.2014.03.1394
107. Principi N, Esposito S. Paediatric human metapneumovirus infection: epidemiology, prevention and therapy. J Clin Virol. (2014) 59:141–7. doi: 10.1016/j.jcv.2014.01.003
108. van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R, Fouchier RA, et al. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med. (2001) 7:719–24. doi: 10.1038/89098
109. Williams JV, Harris PA, Tollefson SJ, Halburnt-Rush LL, Pingsterhaus JM, Edwards KM, et al. Human metapneumovirus and lower respiratory tract disease in otherwise healthy infants and children. N Engl J Med. (2004) 350:443–50. doi: 10.1056/NEJMoa025472
110. Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. (2008) 198:962–70. doi: 10.1086/591708
111. Bhat N, Wright JG, Broder KR, Murray EL, Greenberg ME, Glover MJ, et al. Influenza-associated deaths among children in the United States, 2003-2004. N Engl J Med. (2005) 353:2559–67. doi: 10.1056/NEJMoa051721
112. Simonsen L. The global impact of influenza on morbidity and mortality. Vaccine (1999) 17(Suppl.) 1:S3–10.
113. Bisgaard H, Hermansen MN, Buchvald F, Loland L, Halkjaer LB, Bonnelykke K, et al. Childhood asthma after bacterial colonization of the airway in neonates. N Engl J Med. (2007) 357:1487–95. doi: 10.1056/NEJMoa052632
114. Wolter N, Tempia S, Cohen C, Madhi SA, Venter M, Moyes J, et al. High nasopharyngeal pneumococcal density, increased by viral coinfection, is associated with invasive pneumococcal pneumonia. J Infect Dis. (2014) 210:1649–57. doi: 10.1093/infdis/jiu326
115. Zhong Q, Feng H, Lu Q, Liu X, Zhao Q, Du Y, et al. Recurrent wheezing in neonatal pneumonia is associated with combined infection with Respiratory Syncytial Virus and Staphylococcus aureus or Klebsiella pneumoniae. Sci Rep. (2018) 8:995. doi: 10.1038/s41598-018-19386-y
116. Weinberger DM, Klugman KP, Steiner CA, Simonsen L, Viboud C. Association between respiratory syncytial virus activity and pneumococcal disease in infants: a time series analysis of US hospitalization data. PLoS Med. (2015) 12:e1001776. doi: 10.1371/journal.pmed.1001776
117. Smith CM, Sandrini S, Datta S, Freestone P, Shafeeq S, Radhakrishnan P, et al. Respiratory syncytial virus increases the virulence of Streptococcus pneumoniae by binding to penicillin binding protein 1a. A new paradigm in respiratory infection. Am J Respir Crit Care Med. (2014) 190:196–207. doi: 10.1164/rccm.201311-2110OC
118. Hofstra JJ, Matamoros S, van de Pol MA, de Wever B, Tanck MW, Wendt-Knol H, et al. Changes in microbiota during experimental human Rhinovirus infection. BMC Infect Dis. (2015) 15:336. doi: 10.1186/s12879-015-1081-y
119. Choi SH, Hong SB, Ko GB, Lee Y, Park HJ, Park SY, et al. Viral infection in patients with severe pneumonia requiring intensive care unit admission. Am J Respir Crit Care Med. (2012) 186:325–32. doi: 10.1164/rccm.201112-2240OC
120. Klein EY, Monteforte B, Gupta A, Jiang W, May L, Hsieh YH, et al. The frequency of influenza and bacterial coinfection: a systematic review and meta-analysis. Influenza Other Respir Viruses (2016) 10:394–403. doi: 10.1111/irv.12398
121. Brundage JF. Cases and deaths during influenza pandemics in the United States. Am J Prev Med. (2006) 31:252–6. doi: 10.1016/j.amepre.2006.04.005
122. Joseph C, Togawa Y, Shindo N. Bacterial and viral infections associated with influenza. Influenza Other Respir Viruses (2013) 7(Suppl. 2):105–13. doi: 10.1111/irv.12089
123. Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, et al. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis. (2005) 5:751–62. doi: 10.1016/S1473-3099(05)70295-4
124. DeLeo FR, Musser JM. Axis of coinfection evil. J Infect Dis. (2010) 201:488–90. doi: 10.1086/650304
125. Krysko DV, D'Herde K, Vandenabeele P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis (2006) 11:1709–26. doi: 10.1007/s10495-006-9527-8
126. Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptotic cells: getting rid of the corpses. Mol Cell (2004) 14:277–87. doi: 10.1016/S1097-2765(04)00237-0
127. Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. (2010) 39:407–27. doi: 10.1146/annurev.biophys.093008.131234
128. Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science (2014) 344:1164–8. doi: 10.1126/science.1252809
129. Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science (2013) 341:403–6. doi: 10.1126/science.1236758
130. Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. (2015) 16:907–17. doi: 10.1038/ni.3253
131. Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. (2005) 201:647–57. doi: 10.1084/jem.20041611
132. Murakami Y, Tian L, Voss OH, Margulies DH, Krzewski K, Coligan JE. CD300b regulates the phagocytosis of apoptotic cells via phosphatidylserine recognition. Cell Death Differ. (2014) 21:1746–57. doi: 10.1038/cdd.2014.86
133. Friggeri A, Banerjee S, Biswas S, de Freitas A, Liu G, Bierhaus A, et al. Participation of the receptor for advanced glycation end products in efferocytosis. J Immunol. (2011) 186:6191–8. doi: 10.4049/jimmunol.1004134
134. Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. (2008) 15:192–201. doi: 10.1038/sj.cdd.4402242
135. Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature (2007) 450:430–4. doi: 10.1038/nature06329
136. Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity (2007) 27:927–40. doi: 10.1016/j.immuni.2007.11.011
137. Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature (2007) 450:435–9. doi: 10.1038/nature06307
138. Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol. (2013) 5:a009076. doi: 10.1101/cshperspect.a009076
139. Fujimori T, Grabiec AM, Kaur M, Bell TJ, Fujino N, Cook PC, et al. The Axl receptor tyrosine kinase is a discriminator of macrophage function in the inflamed lung. Mucosal Immunol. (2015) 8:1021–30. doi: 10.1038/mi.2014.129
140. Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol. (2008) 8:327–36. doi: 10.1038/nri2303
141. Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell (2007) 131:1124–36. doi: 10.1016/j.cell.2007.10.034
142. Sharif MN, Sosic D, Rothlin CV, Kelly E, Lemke G, Olson EN, et al. Twist mediates suppression of inflammation by type I IFNs and Axl. J Exp Med. (2006) 203:1891–901. doi: 10.1084/jem.20051725
143. Chung EY, Liu J, Homma Y, Zhang Y, Brendolan A, Saggese M, et al. Interleukin-10 expression in macrophages during phagocytosis of apoptotic cells is mediated by homeodomain proteins Pbx1 and Prep-1. Immunity (2007) 27:952–64. doi: 10.1016/j.immuni.2007.11.014
144. Korns D, Frasch SC, Fernandez-Boyanapalli R, Henson PM, Bratton DL. Modulation of macrophage efferocytosis in inflammation. Front Immunol. (2011) 2:57. doi: 10.3389/fimmu.2011.00057
145. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature (1997) 390:350–1.
146. Xiao YQ, Freire-de-Lima CG, Schiemann WP, Bratton DL, Vandivier RW, Henson PM. Transcriptional and translational regulation of TGF-beta production in response to apoptotic cells. J Immunol. (2008) 181:3575–85. doi: 10.4049/jimmunol.181.5.3575
147. Smith MW, Schmidt JE, Rehg JE, Orihuela CJ, McCullers JA. Induction of pro- and anti-inflammatory molecules in a mouse model of pneumococcal pneumonia after influenza. Comp Med. (2007) 57:82–9.
148. van der Sluijs KF, van Elden LJ, Nijhuis M, Schuurman R, Pater JM, Florquin S, et al. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J Immunol. (2004) 172:7603–9. doi: 10.4049/jimmunol.172.12.7603
149. Kosai K, Seki M, Tanaka A, Morinaga Y, Imamura Y, Izumikawa K, et al. Increase of apoptosis in a murine model for severe pneumococcal pneumonia during influenza A virus infection. Jpn J Infect Dis. (2011) 64:451–7.
150. McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev. (2006) 19:571–82. doi: 10.1128/CMR.00058-05
151. Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. (2014) 14:166–80. doi: 10.1038/nri3607
152. Bonilla WV, Frohlich A, Senn K, Kallert S, Fernandez M, Johnson S, et al. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science (2012) 335:984–9. doi: 10.1126/science.1215418
153. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. (2008) 8:279–89. doi: 10.1038/nri2215
154. Medeiros AI, Serezani CH, Lee SP, Peters-Golden M. Efferocytosis impairs pulmonary macrophage and lung antibacterial function via PGE2/EP2 signaling. J Exp Med. (2009) 206:61–8. doi: 10.1084/jem.20082058
155. Stolberg VR, McCubbrey AL, Freeman CM, Brown JP, Crudgington SW, Taitano SH, et al. Glucocorticoid-augmented efferocytosis inhibits pulmonary pneumococcal clearance in mice by reducing alveolar macrophage bactericidal function. J Immunol. (2015) 195:174–84. doi: 10.4049/jimmunol.1402217
156. Grabiec AM, Denny N, Doherty JA, Happonen KE, Hankinson J, Connolly E, et al. Diminished airway macrophage expression of the Axl receptor tyrosine kinase is associated with defective efferocytosis in asthma. J Allergy Clin Immunol. (2017) 140:1144–6.e4. doi: 10.1016/j.jaci.2017.03.024
157. Grabiec AM, Hussell T. The role of airway macrophages in apoptotic cell clearance following acute and chronic lung inflammation. Semin Immunopathol. (2016) 38:409–23. doi: 10.1007/s00281-016-0555-3
158. Iwasaki A, Foxman EF, Molony RD. Early local immune defences in the respiratory tract. Nat Rev Immunol. (2017) 17:7–20. doi: 10.1038/nri.2016.117
159. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. (2014) 14:315–28. doi: 10.1038/nri3665
160. Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol. (2006) 7:517–23. doi: 10.1038/ni1322
161. Hashimoto S, Kawado M, Murakami Y, Izumida M, Ohta A, Tada Y, et al. Epidemics of vector-borne diseases observed in infectious disease surveillance in Japan, 2000-2005. J Epidemiol. (2007) 17(Suppl.) S48–55. doi: 10.2188/jea.17.S48
162. Chen K, Liu J, Cao X. Regulation of type I interferon signaling in immunity and inflammation: a comprehensive review. J Autoimmun. (2017) 83:1–11. doi: 10.1016/j.jaut.2017.03.008
163. Didierlaurent A, Goulding J, Patel S, Snelgrove R, Low L, Bebien M, et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med. (2008) 205:323–9. doi: 10.1084/jem.20070891
164. Williams AE, Edwards L, Humphreys IR, Snelgrove R, Rae A, Rappuoli R, et al. Innate imprinting by the modified heat-labile toxin of Escherichia coli (LTK63) provides generic protection against lung infectious disease. J Immunol. (2004) 173:7435–43. doi: 10.4049/jimmunol.173.12.7435
165. Bordon Y. Macrophages: innate memory training. Nat Rev Immunol. (2014) 14:713. doi: 10.1038/nri3759
166. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345:1250684. doi: 10.1126/science.1250684
167. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science (2014) 345:1251086. doi: 10.1126/science.1251086
168. Roquilly A, McWilliam HEG, Jacqueline C, Tian Z, Cinotti R, Rimbert M, et al. Local modulation of antigen-presenting cell development after resolution of pneumonia induces long-term susceptibility to secondary infections. Immunity (2017) 47:135–47.e5. doi: 10.1016/j.immuni.2017.06.021
169. Unger BL, Faris AN, Ganesan S, Comstock AT, Hershenson MB, Sajjan US. Rhinovirus attenuates non-typeable Hemophilus influenzae-stimulated IL-8 responses via TLR2-dependent degradation of IRAK-1. PLoS Pathog. (2012) 8:e1002969. doi: 10.1371/journal.ppat.1002969
170. Sun K, Metzger DW. Influenza infection suppresses NADPH oxidase-dependent phagocytic bacterial clearance and enhances susceptibility to secondary methicillin-resistant Staphylococcus aureus infection. J Immunol. (2014) 192:3301–7. doi: 10.4049/jimmunol.1303049
171. Sun K, Salmon S, Yajjala VK, Bauer C, Metzger DW. Expression of suppressor of cytokine signaling 1 (SOCS1) impairs viral clearance and exacerbates lung injury during influenza infection. PLoS Pathog. (2014) 10:e1004560. doi: 10.1371/journal.ppat.1004560
172. Bellinghausen C, Rohde GGU, Savelkoul PHM, Wouters EFM, Stassen FRM. Viral-bacterial interactions in the respiratory tract. J Gen Virol. (2016) 97:3089–102. doi: 10.1099/jgv.0.000627
173. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity (2009) 30:556–65. doi: 10.1016/j.immuni.2009.02.005
174. Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol. (2010) 11:404–10. doi: 10.1038/ni.1861
175. Peiro T, Patel DF, Akthar S, Gregory LG, Pyle CJ, Harker JA, et al. Neutrophils drive alveolar macrophage IL-1beta release during respiratory viral infection. Thorax (2018) 73:546–56. doi: 10.1136/thoraxjnl-2017-210010
176. Pirhonen J, Sareneva T, Kurimoto M, Julkunen I, Matikainen S. Virus infection activates IL-1 beta and IL-18 production in human macrophages by a caspase-1-dependent pathway. J Immunol. (1999) 162:7322–9.
177. McGeachy MJ, Cua DJ. Th17 cell differentiation: the long and winding road. Immunity (2008) 28:445–53. doi: 10.1016/j.immuni.2008.03.001
178. Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. (2007) 282:9358–63. doi: 10.1074/jbc.C600321200
179. Minegishi Y, Saito M, Nagasawa M, Takada H, Hara T, Tsuchiya S, et al. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J Exp Med. (2009) 206:1291–301. doi: 10.1084/jem.20082767
180. Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol. (2011) 186:1666–74. doi: 10.4049/jimmunol.1002194
181. Huber VC, McKeon RM, Brackin MN, Miller LA, Keating R, Brown SA, et al. Distinct contributions of vaccine-induced immunoglobulin G1 (IgG1) and IgG2a antibodies to protective immunity against influenza. Clin Vaccine Immunol. (2006) 13:981–90. doi: 10.1128/CVI.00156-06
182. Kuri T, Sorensen AS, Thomas S, Karlsson Hedestam GB, Normark S, Henriques-Normark B, et al. Influenza A virus-mediated priming enhances cytokine secretion by human dendritic cells infected with Streptococcus pneumoniae. Cell Microbiol. (2013) 15:1385–400. doi: 10.1111/cmi.12122
183. Wu Y, Mao H, Ling MT, Chow KH, Ho PL, Tu W, Lau YL. Successive influenza virus infection and Streptococcus pneumoniae stimulation alter human dendritic cell function. BMC Infect Dis. (2011) 11:201. doi: 10.1186/1471-2334-11-201
184. Rynda-Apple A, Harmsen A, Erickson AS, Larson K, Morton RV, Richert LE, et al. Regulation of IFN-gamma by IL-13 dictates susceptibility to secondary postinfluenza MRSA pneumonia. Eur J Immunol. (2014) 44:3263–72. doi: 10.1002/eji.201444582
185. Asquith KL, Horvat JC, Kaiko GE, Carey AJ, Beagley KW, Hansbro PM, et al. Interleukin-13 promotes susceptibility to chlamydial infection of the respiratory and genital tracts. PLoS Pathog. (2011) 7:e1001339. doi: 10.1371/journal.ppat.1001339
186. Heitmann L, Abad Dar M, Schreiber T, Erdmann H, Behrends J, McKenzie AN, et al. The IL-13/IL-4Ralpha axis is involved in tuberculosis-associated pathology. J Pathol. (2014) 234:338–50. doi: 10.1002/path.4399
187. Simon GC, Martin RJ, Smith S, Thaikoottathil J, Bowler RP, Barenkamp SJ, et al. Up-regulation of MUC18 in airway epithelial cells by IL-13: implications in bacterial adherence. Am J Respir Cell Mol Biol. (2011) 44:606–13. doi: 10.1165/rcmb.2010-0384OC
188. Gross CA, Bowler RP, Green RM, Weinberger AR, Schnell C, Chu HW. Beta2-agonists promote host defense against bacterial infection in primary human bronchial epithelial cells. BMC Pulm Med. (2010) 10:30. doi: 10.1186/1471-2466-10-30
189. Graham MB, Dalton DK, Giltinan D, Braciale VL, Stewart TA, Braciale TJ. Response to influenza infection in mice with a targeted disruption in the interferon gamma gene. J Exp Med. (1993) 178:1725–32. doi: 10.1084/jem.178.5.1725
190. Hussell T, Openshaw PJ. Intracellular IFN-gamma expression in natural killer cells precedes lung CD8+ T cell recruitment during respiratory syncytial virus infection. J Gen Virol. (1998) 79(Pt 11):2593–601.
191. Shekhar S, Peng Y, Gao X, Joyee AG, Wang S, Bai H, et al. NK cells modulate the lung dendritic cell-mediated Th1/Th17 immunity during intracellular bacterial infection. Eur J Immunol. (2015) 45:2810–20. doi: 10.1002/eji.201445390
192. Xu X, Weiss ID, Zhang HH, Singh SP, Wynn TA, Wilson MS, et al. Conventional NK cells can produce IL-22 and promote host defense in Klebsiella pneumoniae pneumonia. J Immunol. (2014) 192:1778–86. doi: 10.4049/jimmunol.1300039
193. Small CL, Shaler CR, McCormick S, Jeyanathan M, Damjanovic D, Brown EG, et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J Immunol. (2010) 184:2048–56. doi: 10.4049/jimmunol.0902772
194. Barthelemy A, Ivanov S, Fontaine J, Soulard D, Bouabe H, Paget C, et al. Influenza A virus-induced release of interleukin-10 inhibits the anti-microbial activities of invariant natural killer T cells during invasive pneumococcal superinfection. Mucosal Immunol. (2017) 10:460–9. doi: 10.1038/mi.2016.49
195. Robinson KM, McHugh KJ, Mandalapu S, Clay ME, Lee B, Scheller EV, et al. Influenza A virus exacerbates Staphylococcus aureus pneumonia in mice by attenuating antimicrobial peptide production. J Infect Dis. (2014) 209:865–75. doi: 10.1093/infdis/jit527
196. Schliehe C, Flynn EK, Vilagos B, Richson U, Swaminanthan S, Bosnjak B, et al. The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nat Immunol. (2015) 16:67–74. doi: 10.1038/ni.3046
197. Ishikawa H, Fukui T, Ino S, Sasaki H, Awano N, Kohda C, and Tanaka K, Influenza virus infection causes neutrophil dysfunction through reduced G-CSF production and an increased risk of secondary bacteria infection in the lung. Virology (2016) 499:23–9. doi: 10.1016/j.virol.2016.08.025
198. Li W, Moltedo B, Moran TM. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of gammadelta T cells. J Virol. (2012) 86:12304–12. doi: 10.1128/JVI.01269-12
199. Glanville N, Message SD, Walton RP, Pearson RM, Parker HL, Laza-Stanca V, et al. gammadeltaT cells suppress inflammation and disease during rhinovirus-induced asthma exacerbations. Mucosal Immunol. (2013) 6:1091–100. doi: 10.1038/mi.2013.3
200. Meermeier EW, Harriff MJ, Karamooz E, Lewinsohn DM. MAIT cells and microbial immunity. Immunol Cell Biol. (2018) 96:607–17. doi: 10.1111/imcb.12022
201. van Wilgenburg B, Scherwitzl I, Hutchinson EC, Leng T, Kurioka A, Kulicke C, et al. MAIT cells are activated during human viral infections. Nat Commun. (2016) 7:11653. doi: 10.1038/ncomms11653
202. Wallington JC, Williams AP, Staples KJ, T.Wilkinson MA. IL-12 and IL-7 synergize to control mucosal-associated invariant T-cell cytotoxic responses to bacterial infection. J Allergy Clin Immunol. (2018) 141:2182–95.e6. doi: 10.1016/j.jaci.2017.08.009
203. Meierovics A, Yankelevich WJ, Cowley SC. MAIT cells are critical for optimal mucosal immune responses during in vivo pulmonary bacterial infection. Proc Natl Acad Sci USA. (2013) 110:E3119–28. doi: 10.1073/pnas.1302799110
204. Loh L, Wang Z, Sant S, Koutsakos M, Jegaskanda S, Corbett AJ, et al. Human mucosal-associated invariant T cells contribute to antiviral influenza immunity via IL-18-dependent activation. Proc Natl Acad Sci USA. (2016) 113:10133–8. doi: 10.1073/pnas.1610750113
205. van der Sluijs KF, Nijhuis M, Levels JH, Florquin S, Mellor AL, Jansen HM, et al. Influenza-induced expression of indoleamine 2,3-dioxygenase enhances interleukin-10 production and bacterial outgrowth during secondary pneumococcal pneumonia. J Infect Dis. (2006) 193:214–22. doi: 10.1086/498911
206. Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, et al., Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci USA. (1986) 83:4167–71. doi: 10.1073/pnas.83.12.4167
207. Meliopoulos VA, Van de Velde LA, Van de Velde NC, Karlsson EA, Neale G, Vogel P, et al. An epithelial integrin regulates the amplitude of protective lung interferon responses against multiple respiratory pathogens. PLoS Pathog. (2016) 12:e1005804. doi: 10.1371/journal.ppat.1005804
208. Jamieson AM, Pasman L, Yu S, Gamradt P, Homer RJ, Decker T, Medzhitov R. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science (2013) 340:1230–4. doi: 10.1126/science.1233632
209. Kash JC, Walters KA, Davis AS, Sandouk A, Schwartzman LM, Jagger BW, et al. Lethal synergism of 2009 pandemic H1N1 influenza virus and Streptococcus pneumoniae coinfection is associated with loss of murine lung repair responses. MBio (2011) 2:e00172-11. doi: 10.1128/mBio.00172-11
210. Naba A, Hoersch S, Hynes RO. Towards definition of an ECM parts list: an advance on GO categories. Matrix Biol. (2012) 31:371–2. doi: 10.1016/j.matbio.2012.11.008
211. Burgstaller G, Oehrle B, Gerckens M, White ES, Schiller HB, Eickelberg O. The instructive extracellular matrix of the lung: basic composition and alterations in chronic lung disease. Eur Respir J. (2017) 50:1601805. doi: 10.1183/13993003.01805-2016
212. Kolb M, Margetts PJ, Sime PJ, Gauldie J. Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung. Am J Physiol Lung Cell Mol Physiol. (2001) 280:L1327–34. doi: 10.1152/ajplung.2001.280.6.L1327
213. Ray SJ, Franki SN, Pierce RH, Dimitrova S, Koteliansky V, Sprague AG, et al. The collagen binding alpha1beta1 integrin VLA-1 regulates CD8 T cell-mediated immune protection against heterologous influenza infection. Immunity (2004) 20:167–79. doi: 10.1016/S1074-7613(04)00021-4
214. Tighe RM, Garantziotis S. Hyaluronan interactions with innate immunity in lung biology. Matrix Biol. (2018). doi: 10.1016/j.matbio.2018.01.027. [Epub ahead of print].
215. O'Dwyer DN, Gurczynski SJ, Moore BB. Pulmonary immunity and extracellular matrix interactions. Matrix Biol. (2018). 73:122–34. doi: 10.1016/j.matbio.2018.04.003
216. Akthar S, Patel DF, Beale RC, Peiro T, Xu X, Gaggar A, et al. Matrikines are key regulators in modulating the amplitude of lung inflammation in acute pulmonary infection. Nat Commun. (2015) 6:8423. doi: 10.1038/ncomms9423
217. Snelgrove RJ, Jackson PL, Hardison MT, Noerager BD, Kinloch A, Gaggar A, et al. A critical role for LTA4H in limiting chronic pulmonary neutrophilic inflammation. Science (2010) 330:90–4. doi: 10.1126/science.1190594
218. Bell TJ, Brand OJ, Morgan DJ, Salek-Ardakani S, Jagger C, Fujimori T, et al. Defective lung function following influenza virus is due to prolonged, reversible hyaluronan synthesis. Matrix Biol. (2018). doi: 10.1016/j.matbio.2018.06.006. [Epub ahead of print].
219. Lauer ME, Dweik RA, Garantziotis S, Aronica MA. The Rise and fall of hyaluronan in respiratory diseases. Int J Cell Biol. (2015) 2015:712507. doi: 10.1155/2015/712507
220. Dentener MA, Vernooy JH, Hendriks S, Wouters EF. Enhanced levels of hyaluronan in lungs of patients with COPD: relationship with lung function and local inflammation. Thorax (2005) 60:114–9. doi: 10.1136/thx.2003.020842
221. Eurlings IM, Dentener MA, Cleutjens JP, Peutz CJ, Rohde GG, et al. Similar matrix alterations in alveolar and small airway walls of COPD patients. BMC Pulm Med. (2014) 14:90. doi: 10.1186/1471-2466-14-90
222. Forteza R, Casalino-Matsuda SM, Monzon ME, Fries E, Rugg MS, Milner CM, et al. TSG-6 potentiates the antitissue kallikrein activity of inter-alpha-inhibitor through bikunin release. Am J Respir Cell Mol Biol. (2007) 36:20–31. doi: 10.1165/rcmb.2006-0018OC
223. Ishiura S. Proteolytic cleavage of the Alzheimer's disease amyloid A4 precursor protein. J Neurochem. (1991) 56:363–9. doi: 10.1111/j.1471-4159.1991.tb08160.x
224. Adair JE, Stober V, Sobhany M, Zhuo L, Roberts JD, Negishi M, et al. Inter-alpha-trypsin inhibitor promotes bronchial epithelial repair after injury through vitronectin binding. J Biol Chem. (2009) 284:16922–30. doi: 10.1074/jbc.M808560200
225. Pociask DA, Robinson KM, Chen K, McHugh KJ, Clay ME, Huang GT, et al. Epigenetic and transcriptomic regulation of lung repair during recovery from influenza infection. Am J Pathol. (2017) 187:851–63. doi: 10.1016/j.ajpath.2016.12.012
226. Greenlee KJ, Werb Z, Kheradmand F. Matrix metalloproteinases in lung: multiple, multifarious, and multifaceted. Physiol Rev. (2007) 87:69–98. doi: 10.1152/physrev.00022.2006
227. Talmi-Frank D, Altboum Z, Solomonov I, Udi Y, Jaitin DA, Klepfish M, et al. Extracellular matrix proteolysis by MT1-MMP contributes to influenza-related tissue damage and mortality. Cell Host Microbe (2016) 20:458–470. doi: 10.1016/j.chom.2016.09.005
228. Wen F, Guo J, Tong G, Bi D, Wang Q, Liu X, et al. A meta-analysis of transcriptomic characterization revealed extracellular matrix pathway involved in the H5N1 and H7N9 infections. Oncotarget (2017) 8:62561–72. doi: 10.18632/oncotarget.19315
229. Fino KK, Yang L, Silveyra P, Hu S, Umstead TM, DiAngelo S, et al. SH3GLB2/endophilin B2 regulates lung homeostasis and recovery from severe influenza A virus infection. Sci Rep. (2017) 7:7262. doi: 10.1038/s41598-017-07724-5
230. Wight TN, Frevert CW, Debley JS, Reeves SR, Parks WC, Ziegler SF. Interplay of extracellular matrix and leukocytes in lung inflammation. Cell Immunol. (2017) 312:1–14. doi: 10.1016/j.cellimm.2016.12.003
231. Siegel SJ, Roche AM, Weiser JN. Influenza promotes pneumococcal growth during coinfection by providing host sialylated substrates as a nutrient source. Cell Host Microbe (2014) 16:55–67. doi: 10.1016/j.chom.2014.06.005
232. Dickson RP, Huffnagle GB. The lung microbiome: new principles for respiratory bacteriology in health and disease. PLoS Pathog. (2015) 11:e1004923. doi: 10.1371/journal.ppat.1004923
233. Singh B, Fleury C, Jalalvand F, Riesbeck K. Human pathogens utilize host extracellular matrix proteins laminin and collagen for adhesion and invasion of the host. FEMS Microbiol Rev. (2012) 36:1122–80. doi: 10.1111/j.1574-6976.2012.00340.x
234. Downer R, Roche F, Park PW, Mecham RP, Foster TJ. The elastin-binding protein of Staphylococcus aureus (EbpS) is expressed at the cell surface as an integral membrane protein and not as a cell wall-associated protein. J Biol Chem. (2002) 277:243–50. doi: 10.1074/jbc.M107621200
235. Nakakido M, Aikawa C, Nakagawa I, Tsumoto K. The staphylococcal elastin-binding protein regulates zinc-dependent growth/biofilm formation. J Biochem (2014) 156:155–62. doi: 10.1093/jb/mvu027
236. Westerlund B, Korhonen TK. Bacterial proteins binding to the mammalian extracellular matrix. Mol Microbiol. (1993) 9:687–94. doi: 10.1111/j.1365-2958.1993.tb01729.x
237. Sandig H, McDonald J, Gilmour J, Arno M, Lee TH, Cousins DJ. Fibronectin is a TH1-specific molecule in human subjects. J Allergy Clin Immunol. (2009) 124:528–35:535.e1-5. doi: 10.1016/j.jaci.2009.04.036
238. Madani A, Garakani K M.Mofrad RK. Molecular mechanics of Staphylococcus aureus adhesin, CNA, and the inhibition of bacterial adhesion by stretching collagen. PLoS ONE (2017) 12:e0179601. doi: 10.1371/journal.pone.0179601
239. Hartleib J, Kohler N, Dickinson RB, Chhatwal GS, Sixma JJ, Hartford OM, et al. Protein A is the von Willebrand factor binding protein on Staphylococcus aureus. Blood (2000) 96:2149–56.
240. Rajas O, Quiros LM, Ortega M, Vazquez-Espinosa E, Merayo-Lloves J, Vazquez F, et al. Glycosaminoglycans are involved in bacterial adherence to lung cells. BMC Infect Dis. (2017) 17:319. doi: 10.1186/s12879-017-2418-5
241. Schommer NN, Muto J, Nizet V, Gallo RL. Hyaluronan breakdown contributes to immune defense against group A Streptococcus. J Biol Chem. (2014) 289:26914–21. doi: 10.1074/jbc.M114.575621
242. Holmannova D, Kolackova M, Kondelkova K, Kunes P, Krejsek J, Andrys C. CD200/CD200R paired potent inhibitory molecules regulating immune and inflammatory responses; Part I: CD200/CD200R structure, activation, and function. Acta Medica (Hradec Kralove) (2012) 55:12–7. doi: 10.14712/18059694.2015.68
243. Goulding J, Godlee A, Vekaria S, Hilty M, Snelgrove R, Hussell T. Lowering the threshold of lung innate immune cell activation alters susceptibility to secondary bacterial superinfection. J Infect Dis. (2011) 204:1086–94. doi: 10.1093/infdis/jir467
244. Kopsaftis Z, Wood-Baker R, Poole P. Influenza vaccine for chronic obstructive pulmonary disease (COPD). Cochrane Database Syst Rev. (2018) 6:Cd002733. doi: 10.1002/14651858.CD002733.pub3
245. Matsumoto K, Inoue H. Viral infections in asthma and COPD. Respir Investig. (2014) 52:92–100. doi: 10.1016/j.resinv.2013.08.005
246. Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. (2008) 14:275–81. doi: 10.1038/nm1710
247. Guo H, Topham DJ, Interleukin-22 (IL-22) production by pulmonary Natural Killer cells and the potential role of IL-22 during primary influenza virus infection. J Virol. (2010) 84:7750–9. doi: 10.1128/JVI.00187-10
248. Paget C, Ivanov S, Fontaine J, Renneson J, Blanc F, Pichavant M, et al. Interleukin-22 is produced by invariant natural killer T lymphocytes during influenza A virus infection: potential role in protection against lung epithelial damages. J Biol Chem. (2012) 287:8816–29. doi: 10.1074/jbc.M111.304758
249. Pociask DA, Scheller EV, Mandalapu S, McHugh KJ, Enelow RI, Fattman CL, et al. IL-22 is essential for lung epithelial repair following influenza infection. Am J Pathol. (2013) 182:1286–96. doi: 10.1016/j.ajpath.2012.12.007
250. Ivanov S, Renneson J, Fontaine J, Barthelemy A, Paget C, Fernandez EM, et al. Interleukin-22 reduces lung inflammation during influenza A virus infection and protects against secondary bacterial infection. J Virol. (2013) 87:6911–24. doi: 10.1128/JVI.02943-12
251. Hall OJ, Limjunyawong N, Vermillion MS, Robinson DP, Wohlgemuth N, Pekosz A, et al. Progesterone-based therapy protects against influenza by promoting lung repair and recovery in females. PLoS Pathog. (2016) 12:e1005840. doi: 10.1371/journal.ppat.1005840
252. Subramaniam R, Barnes PF, Fletcher K, Boggaram V, Hillberry Z, Neuenschwander P, et al. Protecting against post-influenza bacterial pneumonia by increasing phagocyte recruitment and ROS production. J Infect Dis. (2014) 209:1827–36. doi: 10.1093/infdis/jit830
253. Reppe K, Radunzel P, Dietert K, Tschernig T, Wolff T, Hammerschmidt S, et al. Pulmonary immunostimulation with MALP-2 in influenza virus-infected mice increases survival after pneumococcal superinfection. Infect Immun. (2015) 83:4617–29. doi: 10.1128/IAI.00948-15
254. Mathieu C, Rioux G, Dumas MC, Leclerc D. Induction of innate immunity in lungs with virus-like nanoparticles leads to protection against influenza and Streptococcus pneumoniae challenge. Nanomedicine (2013) 9:839–48. doi: 10.1016/j.nano.2013.02.009
255. Li Z, Potts-Kant EN, Garantziotis S, Foster WM, Hollingsworth JW. Hyaluronan signaling during ozone-induced lung injury requires TLR4, MyD88, and TIRAP. PLoS ONE (2011) 6:e27137. doi: 10.1371/journal.pone.0027137
256. Stober VP, Johnson CG, Majors A, Lauer ME, Cali V, Midura RJ, et al. TNF-stimulated gene 6 promotes formation of hyaluronan-inter-alpha-inhibitor heavy chain complexes necessary for ozone-induced airway hyperresponsiveness. J Biol Chem. (2017) 292:20845–58. doi: 10.1074/jbc.M116.756627
257. Swaidani S, Cheng G, Lauer ME, Sharma M, Mikecz K, Hascall VC, et al. TSG-6 protein is crucial for the development of pulmonary hyaluronan deposition, eosinophilia, and airway hyperresponsiveness in a murine model of asthma. J Biol Chem. (2013) 288:412–22. doi: 10.1074/jbc.M112.389874
258. Mittal M, Tiruppathi C, Nepal S, Zhao YY, Grzych D, Soni D, et al. TNFalpha-stimulated gene-6 (TSG6) activates macrophage phenotype transition to prevent inflammatory lung injury. Proc Natl Acad Sci USA. (2016) 113:E8151–8. doi: 10.1073/pnas.1614935113
259. Cao TV, La M, Getting SJ, Day AJ, Perretti M, Inhibitory effects of TSG-6 Link module on leukocyte-endothelial cell interactions in vitro and in vivo. Microcirculation (2004) 11:615–24. doi: 10.1080/10739680490503438
260. Dyer DP, Thomson JM, Hermant A, Jowitt TA, Handel TM, Proudfoot AE, et al. TSG-6 inhibits neutrophil migration via direct interaction with the chemokine CXCL8. J Immunol. (2014) 192:2177–85. doi: 10.4049/jimmunol.1300194
261. Getting SJ, Mahoney DJ, Cao T, Rugg MS, Fries E, Milner CM, et al. The link module from human TSG-6 inhibits neutrophil migration in a hyaluronan- and inter-alpha -inhibitor-independent manner. J Biol Chem. (2002) 277:51068–76. doi: 10.1074/jbc.M205121200
262. Wisniewski HG, Hua JC, Poppers DM, Naime D, Vilcek J, Cronstein BN. TNF/IL-1-inducible protein TSG-6 potentiates plasmin inhibition by inter-alpha-inhibitor and exerts a strong anti-inflammatory effect in vivo. J Immunol. (1996) 156:1609–15.
263. Frost GI, Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration. Expert Opin Drug Deliv. (2007) 4:427–40. doi: 10.1517/17425247.4.4.427
264. van Bon AC, Bode BW, Sert-Langeron C, DeVries JH, Charpentier G. Insulin glulisine compared to insulin aspart and to insulin lispro administered by continuous subcutaneous insulin infusion in patients with type 1 diabetes: a randomized controlled trial. Diabetes Technol Ther. (2011) 13:607–14. doi: 10.1089/dia.2010.0224
265. Califano D, Furuya Y, Metzger DW. Effects of influenza on alveolar macrophage viability are dependent on mouse genetic strain. J Immunol. (2018) 201:134–44. doi: 10.4049/jimmunol.1701406
266. Baran S, Teul-Swiniarska I, Dzieciolowska-Baran E, Lorkowski J, Gawlikowska-Sroka A. Mental health of Polish students and the occurrence of respiratory tract infections. Adv Exp Med Biol. (2013) 755:275–81. doi: 10.1007/978-94-007-4546-9_35
267. Maxwell L, Barrett B, Chase J, Brown R, Ewers T. Self-reported mental health predicts acute respiratory infection. WMJ (2015) 114:100–4.
268. Andersson NW, Goodwin RD, Okkels N, Gustafsson LN, Taha F, Cole SW, et al. Depression and the risk of severe infections: prospective analyses on a nationwide representative sample. Int J Epidemiol. (2016) 45:131–9. doi: 10.1093/ije/dyv333
269. Rudzki L, Pawlak D, Pawlak K, Waszkiewicz N, Malus A, Konarzewska B, et al. Immune suppression of IgG response against dairy proteins in major depression. BMC Psychiatry (2017) 17:268. doi: 10.1186/s12888-017-1431-y
270. Duggal NA, Upton J, Phillips AC, Hampson P, Lord JM. Depressive symptoms are associated with reduced neutrophil function in hip fracture patients. Brain Behav Immun. (2013) 33:173–82. doi: 10.1016/j.bbi.2013.07.004
Keywords: lung, macrophage, innate immunity, bacteria, virus, matrix, apoptotic cells, training
Citation: Morgan DJ, Casulli J, Chew C, Connolly E, Lui S, Brand OJ, Rahman R, Jagger C and Hussell T (2018) Innate Immune Cell Suppression and the Link With Secondary Lung Bacterial Pneumonia. Front. Immunol. 9:2943. doi: 10.3389/fimmu.2018.02943
Received: 02 August 2018; Accepted: 30 November 2018;
Published: 14 December 2018.
Edited by:
François Trottein, Centre National de la Recherché Scientifique (CNRS), FranceReviewed by:
Dennis Metzger, Albany Medical College, United StatesCopyright © 2018 Morgan, Casulli, Chew, Connolly, Lui, Brand, Rahman, Jagger and Hussell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tracy Hussell, dHJhY3kuaHVzc2VsbEBtYW5jaGVzdGVyLmFjLnVr
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.