 Yao Chen
Yao Chen Ryan Zander2
Ryan Zander2 Weiguo Cui
Weiguo Cui
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 07 December 2018
Sec. Immunological Memory
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02826
This article is part of the Research Topic Differentiation, Tissue Adaptation and Function of Memory T Cells View all 12 articles
Immune protection and lasting memory are accomplished through the generation of phenotypically and functionally distinct CD8 T cell subsets. Understanding how these effector and memory T cells are formed is the first step in eventually manipulating the immune system for therapeutic benefit. In this review, we will summarize the current understanding of CD8 T cell differentiation upon acute infection, with a focus on the transcriptional and epigenetic regulation of cell fate decision and memory formation. Moreover, we will highlight the importance of high throughput sequencing approaches and single cell technologies in providing insight into genome-wide investigations and the heterogeneity of individual CD8 T cells.
During an acute viral or bacterial infection, pathogen-specific T cells robustly proliferate, acquire effector functions, and migrate to the site of infection to eliminate the pathogen. The majority (>90%) of antigen-specific CD8 T cells die via apoptosis upon pathogen clearance, leaving behind distinct memory subsets with unique phenotypic and functional properties. However, the molecular and genetic mechanisms that guide how these cell fate decisions are made remains incompletely understood. Additionally, although it is well-appreciated that antigen specific memory CD8 T cells can persist for extended periods of time in a functionally quiescent state, and that this is important for conferring long-term protective immunity against previously encountered pathogens, the underlying mechanisms that endow memory CD8 T cells with this longevity remain unclear. Moreover, the molecular pathways that help maintain the phenotypic and functional heterogeneity of memory subsets, and enable memory CD8 T cells to remain poised to quickly recall their effector function are still incompletely understood. Current evidence suggests that multiple signals, such as T-cell receptor (TCR), co-stimulation, inflammation, and metabolic signals can orchestrate CD8 T cell fate decisions, with some of these commitment choices occurring early in the immune response (1, 2). As the incorporation of multiple distinct signals received by individual T cells likely triggers diverse transcriptional programs, it is important to discuss the key transcription factors that have been known to orchestrate CD8 T cell fate decision. Moreover, we highlight the field's current understanding of CD8 T cell differentiation on the epigenetic and single-cell level, and provide a brief discussion on how modern technologies may help to refine the CD8 T cell differentiation paradigm.
The process of memory CD8 T cell selection is not entirely stochastic, as originally proposed (3), as effector cells can display inherently distinct memory cell potential, with some CD8 T cells being intrinsically better at persisting and populating the memory pool. It was previously identified that a small subset of effector T cells survive the contraction phase and serve as the precursors of the memory CD8 T cell compartment (4–8). This minor population of effector cells, termed memory precursor effector cells (MPECs), can be distinguished based on their high expression levels of CD127, the IL-7 receptor alpha (IL-7Rα), and their decreased expression of killer cell lectin-like receptor G1 (KLRG1) (5, 6). Other surface proteins that co-segregate with increased IL-7Rα expression on MPECs include CD27, CD28, CD62L, and CXCR3 (1). By contrast, a larger proportion of effector CD8 T cells display high expression of KLRG1 and low expression of IL-7Rα and are more terminally differentiated than their MPEC counterparts. This subset of KLRG1hi effector CD8 T cells is collectively referred to as short-lived effector cells (SLECs). Of note, although MPECs and SLECs were observed in various infectious settings in different species including humans, these phenotypic distinctions are not exclusive criteria for forming memory T cells nor do they represent universal markers for memory precursor cells across all types of immune response (9–11). Furthermore, several studies previously demonstrated that MPECs can give rise to both T central memory (TCM) and T effector memory (TEM) populations (1, 5–7) and recent evidence further indicates that the precursors of tissue resident memory cells (TRM) in the skin and small intestine are also derived from less differentiated KLRG1lo memory T cell precursor cells (12, 13). It is important to note however that these phenotypic distinctions are not exclusive criteria for memory T cell formation, as cell death may also occur among IL-7Rαhi effector T cells following infection, and many long-lived KLRG1hiIL-7Rαhi memory CD8 T cells have been observed following secondary infections (14–17). Moreover, the frequency of KLRG1hi cells can vary widely depending on the type of infection or vaccination. Indeed, a recent study further highlighted the limitations of these markers and elegantly demonstrated that some KLRG1hi cells can downregulate KLRG1 during the contraction phase and differentiate into all memory T cell lineages (18). Thus, a higher degree of developmental plasticity than previously appreciated may exist during the effector to memory CD8 T cell transition phase. Importantly, however, these cell surface markers do offer a useful framework of determining the relative memory cell potential of effector CD8 T cells in several circumstances, and they have become invaluable for identifying molecular pathways that regulate these effector-to-memory cell fate decisions.
As CD8 T cells transition from naïve to effector to memory cells, their overall gene expression profiles changed, resulting in phenotypic and functional variations among the different populations. As such, several fundamental studies have demonstrated that memory CD8 T cells can be compartmentalized into at least 3 distinct subsets on the basis of their effector function, proliferative potential, migration patterns and transcriptional program (19–23). For well over a decade, the population of circulating memory CD8 T cells has been broadly categorized into two distinct subsets, conventionally designated TCM and TEM (20, 24). These two subsets can be distinguished based on their differential expression of CCR7 and CD62L (L-selectin), with TCM cells expressing both of these lymph node homing receptor molecules which facilitates their trafficking to and retention within secondary lymphoid tissues (19, 20). By contrast, TEM cells lack expression of CCR7 and CD62L and are most commonly found in the blood and in non-lymphoid tissues (e.g., lung, liver, intestine) (20, 21, 25). Compared to TEM, TCM cells display an enhanced proliferative potential and an increased capacity to produce the cytokine IL-2, but are unable to immediately produce effector molecules until they undergo secondary proliferation and differentiate into effector cells (20, 26–28). Conversely, TEM cells constitutively display effector functions such as cytolytic activity and IFN-γ production (1, 21, 29, 30). Notably, within the past 10 years, TRM have emerged as the third major memory CD8 T cell subset and have been identified to permanently reside in peripheral tissues after pathogen clearance and provide site-specific protection upon re-infection (22, 23). The specific anatomical location of where TRM cells develop and are maintained can depend on the nature or route of the infection and the inflammatory signals experienced during the effector phase of the T cell response (31). TRM cells can generally be distinguished from TEM cells infiltrating non-lymphoid tissues based on their high expression of CD69 and the integrin CD103 (12, 32–35), although not all TRM cells constitutively express CD103 (31, 34). An important component of TRM differentiation is the migration of T effector cells to target sites (such as the skin or intestines) and their subsequent downregulation of tissue egress receptors, such as S1PR1 (35) and upregulation of adhesion molecules, such as CD103 (12). Other distinguishable features of TRM cells are their sustained expression of granzyme B (that may vary by location) and their maintained high levels of mRNAs encoding TNFα, IFNγ, and IL-2 (31), which allow them to eliminate any re-occurring microbial threat at portal entry sites. Whether TRM undergo homeostatic proliferation to maintain a stable population has not been clearly demonstrated. TRM cells from brain, skin and mucosal sites showed much lower homeostatic proliferation ability and turnover rate compare to their circulating counterparts (22, 31, 36, 37). Interestingly, TRM cells in the lung airway may require constant replenishment from recirculating memory cells (38). As TRM and TEM subsets display constitutive effector functions and occupy the frontline sites of pathogen entry, they are uniquely positioned to be among the first responders of the adaptive recall response. Conversely, TCM recall is critical for the rapid generation of a pool of secondary effector cells that may help contain pathogens that breach the initial containment. In humans and mice, there is a newly defined subset, called T memory stem (TSCM) cells. The characteristic of TSCM is stemness. TSCM cells represent increased proliferative, self-renewal and long-term persistence capacity (39, 40). Additionally, only naive T cells and TSCM cells were able to reconstitute the entire heterogeneity of memory T cell subsets, indicating that TSCM cells are multipotent (39). In patients undergoing haploidentical hematopoietic stem cell transplantation (HSCT), TSCM cells are preferentially generated from naïve cells and the dominant long-term clonotypes appeared to preferentially originate from infused TSCM rather than TCM clones (41, 42). Gene expression data also showed that there is a progressive change moving from naïve to TSCM to TCM and TEM cells (39). These evidences indicate that TSCM cells are at the apex of the hierarchical tree of T cell differentiation and at a hierarchically superior level over the TCM cells (40). Moreover, memory T cells can be further subdivided based on differential expression of additional phenotypic markers. As one example of such endeavor, CX3CR1 has been recently used to identify a peripheral memory (TPM) subset that possesses high cytotoxicity and provides global immune surveillance (43, 44). Collectively, the formation of these distinct memory CD8 T cell subsets and their division of labor likely ensures optimal protective immunity upon pathogen re-challenge. However, a key question that remains to be addressed is whether these distinct memory subsets are maintained by signals from the tissue microenvironment or preprogrammed by cell-intrinsic mechanisms, such as transcription profiles and the chromatin landscape.
During infection or vaccination, naïve CD8 T cells engage with antigen-presenting dendritic cells (DCs) and are presented cognate peptide in a major histocompatibility complex (MHC) class 1-restricted manner (45, 46). Upon TCR-mediated recognition of the MHC-peptide complex, antigen-specific CD8 T cells will start to rapidly proliferate and acquire effector functions and the ability to migrate to sites of infection. During this process of T cell priming, newly activated T cells will integrate multiple signals in the form of TCR signaling, co-stimulation, cytokine, chemokine, and metabolic signals, all of which can have a major impact on the accumulation, survival, and cell-fate decision of effector T cells (1, 2, 47) (Figure 1).
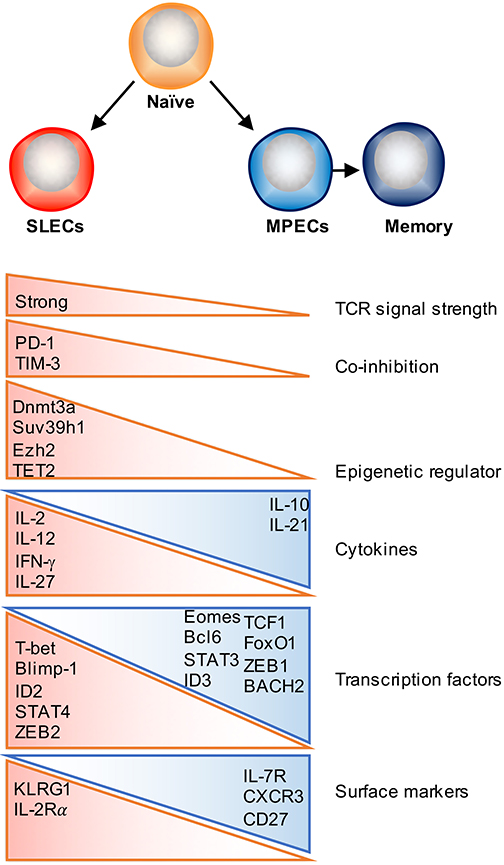
Figure 1. Factors that regulate effector and memory cell fate decision. Following activation, antigen-specific naïve CD8 T cells proliferate and differentiate into a heterogeneous pool of effector T cells that consist of two major subsets: SLECs and MPECs. Majority of SLECs die by apoptosis during contraction phase, whereas MPECs survive and become long-lived memory cells. Numerous factors as depicted can contribute to this cell fate decision process, which include TCR signal strength, co-stimulatory/co-inhibitory molecules, cytokines, transcription factors, and epigenetic regulators.
TCR signaling is one of the initiating signals that helps shape T cell memory. The strength and quality of TCR signaling, which is determined by the affinity of the TCR for peptide–MHC molecules (pMHC), the dose of antigen presented by APCs, the duration of the TCR–pMHC interaction, and the timing of TCR recognition (early or late during infection phase) have been shown to partially contribute to memory commitment, function and the diversity of the memory pool (48). The balance between co-stimulatory and co-inhibitory signaling is not only required for effector T cell activation and expansion, but also determines the size and quality of the memory T cell pool (49). Co-stimulatory molecules, such as CD28, 4-1BB, CD27 and OX40 have been shown to promote memory formation as well as contribute to secondary responses (50, 51). As for co-inhibitory signals, it has previously been reported that lower PD-1 expression may drive T cell differentiation away from a SLEC fate and skew toward TEM memory generation (52). TIM-3 is another inhibitory receptor and blockade of TIM-3 increases transcription of genes involved in T cell effector function and differentiation but decreases expression of genes associated with memory T cell formation (52, 53). Further studies are required to determine how co-stimulatory and co-inhibitory signaling pathways coordinately regulate memory T cell development.
Several studies have identified that exposure to certain inflammatory signals can play a major role in regulating the differentiation of effector and memory CD8 T cell subsets in a context dependent manner. For example, IL-12 or IL-27 enhances SLEC formation during acute bacterial or viral infection, whereas type I and type II interferons can either promote memory or enhance SLEC differentiation under different settings (17, 54–59). Other studies have further identified that exposure to IL-15 helps skew CD8 T cells along a memory pathway (60–62), whereas IL-2 signaling is implicated in promoting the differentiation of short-lived effector T cells (63, 64). However, the effects of IL-2 signaling on CD8 T cell memory formation may be regulated in a temporal manner, as administration of recombinant IL-2 (rIL-2) during the expansion phase diminishes T cell survival, whereas treatment with rIL-2 during the contraction phase promotes T cell proliferation, survival, and memory formation (65). By contrast, IL-10 and IL-21 signaling through a STAT3-SOCS3 pathway was found to promote memory formation, potentially by insulating T cells from excessive inflammatory stimuli (66). Recent studies have also begun to shed light on potential cytokine signaling pathways that contribute to TRM development, with recent findings elucidating an important role for the cytokine transforming growth factor β (TGF-β) and IL-15 in facilitating TRM differentiation by inducing CD103 expression on TRM precursor cells infiltrating the skin, lung, and small intestine (12, 13, 34, 67, 68).
During naïve to effector transition, dynamic changes occur at both the transcriptional and epigenetic level (Figures 2A, 3). Learning from CD4 differentiation (69), the fundamental identity of these heterogeneous effector CD8 T cells can generally be established by upstream “pioneer transcription factors” that regulate the entire transcriptional network to initiate early effector differentiation. Additionally, current evidence suggests that the majority gain-of-methylation and loss-of-methylation events, which represent a repressed and active transcription state respectively, happen within the first 4 days of activation, and more than half of these differentially methylated regions (DMRs) were similarly acquired in both SLECs and MPECs (70) (Figure 2B). Furthermore, effector and memory CD8 T cells have been found to share a more similar pattern of chromatin accessibility as compared to naïve CD8 T cells (71). Among these shared accessible regions, the binding motifs for bZIP, IRF, and T-box transcription factors are highly enriched (71–73) (Figure 2D). This then brings to a question, which transcription factors are initiating this early effector differentiation? Among naïve CD8 T cells, bivalency (H3K4me3+H3K27me3+) was observed at the promotors of transcription factors that are known to be crucial for initiating an effector program, such as T-bet, Eomes, Blimp1, and IRF4 (74) (Figure 2C). This finding indicates that these transcription factors may remain poised in naïve T cells but rapidly start transcription by acquiring a permissive histone methylation signature upon TCR stimulation within 24 h (74). Indeed, it has been demonstrated that IRF4 cooperates with BATF (belongs to AP-1 family) to serve as “pioneer transcription factors” that promote chromatin accessibility and gene expression associated with various aspects of effector CD8 T cell differentiation (75–78). In addition, Runx3 is another potential “pioneer transcription factor” that can initiate changes in chromatin accessibility after CD8 T cell activation, especially at the binding sites of IRF, bZIP transcription factors, and Blimp1 (73) (Figure 2D).
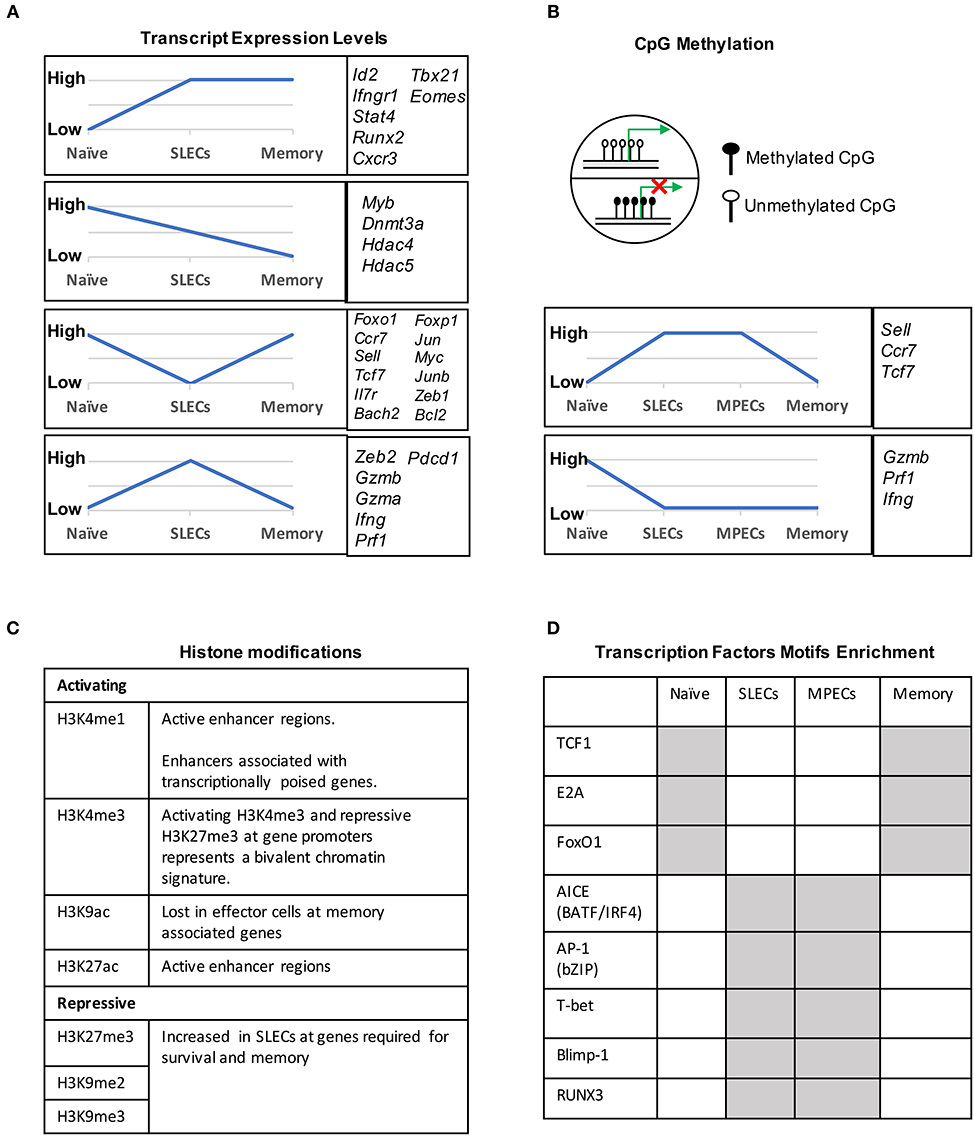
Figure 2. Transcriptional and epigenetic profiling during naïve to effector to memory transition. (A) Gene clusters defined by the mRNA expression levels in naïve, SLECs and memory cells. Four major expression patterns emerged: genes that were up- or downregulated during the effector stage and persisted into the memory phase, and genes that were up- or downregulated during the effector phase and then reverted to the naive state. (B) CpG methylation levels of different genes in naïve, SLECs, MPECs and memory cells. Naïve/memory genes are similarly acquired CpG methylation in both SLECs and MPECs. MPECs, not SLECs, have the capacity to erase their newly acquired methylation programs and re-express naïve/memory genes as they develop into memory CD8 T cells. SLECs and MPECs both show demethylation of several effector-associated genes which remain demethylated in memory cells for a long period of time. (C) Histones posttranslational modification (PTM) and their functions that are essential for CD8 T cell differentiation. For example, the epigenetic bivalency for H3K27me3 and H3K4me3 represent an epigenetic state from which a gene can be rapidly activated or repressed depending on the differentiation pathways. (D) Differentially enriched motifs of transcription factors in naïve, effector and memory cells. Motif analysis identified the cell-subsets specific transcription factors binding sites in enhancer or promoter regions. Gray depicts highly enriched motifs.
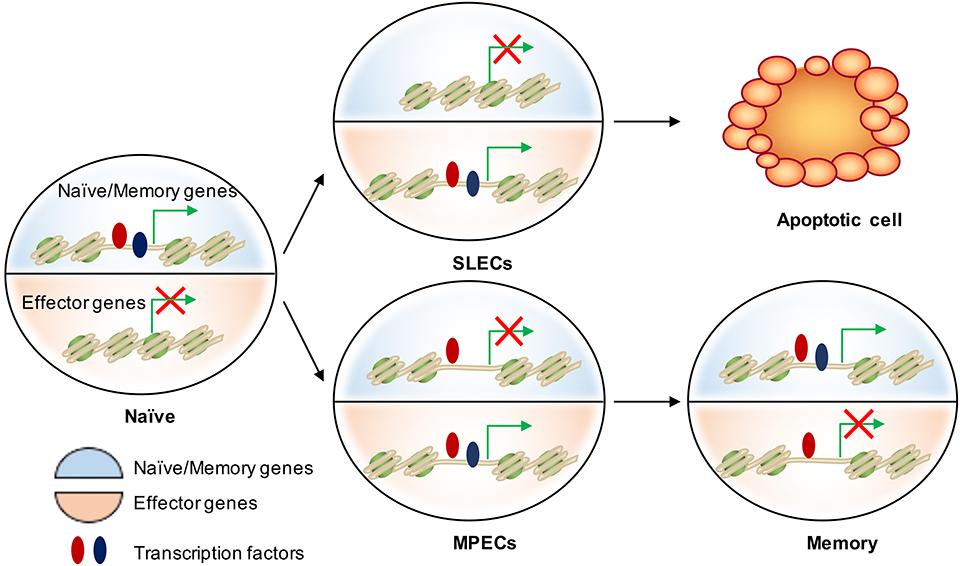
Figure 3. Epigenetic regulation of cell fate decision during acute infection. There are two major transcriptional circuits in regulating CD8 T cell differentiation: one of them associated with effector function and another one is essential in naïve/memory cells controlling T cell quiescence and homeostasis. In naïve CD8 T cells, naïve/memory genes are open (epigenetically by TFs in red) and on (transcriptionally by TFs in blue), while the effector genes are closed and off. When naïve cells are activated, effector genes are turned on mainly by “pioneer TFs” (in red) in both SLECs and MPECs. These genetic regions remain open but poised as MPECs develop into memory cells. Transcriptional repression of naïve/memory genes in MPECs cells can be reversed in memory CD8 T cells through recruiting additional TFs (in blue) to restart gene expression. In contrast, SLECs lose the accessibility at these TF-bound cis-regulatory elements and therefore permanently turn off the naïve/memory gene expression. This leads to their loss of memory potential and long-term survival.
After the initial expansion phase, effector T cells can be bifurcated into two distinct effector populations, SLECs and MPECs. Early on in the effector phase, the chromatin landscape has already been universally prepared by “pioneer transcription factors,” and now lineage-specifying transcription factors start to take effect. Considering that TCR signal strength is negatively associated with memory formation (2, 48), it is possible that TCR-induced transcription factors can influence the type of progeny derived from a single T cell. One such transcription factor is IRF4, expression of which is highly dependent on the signal strength of TCR signaling (79). Indeed, IRF4 has been found to be crucial for initial expansion and promoting SLEC formation (79). In addition, the expression level of memory associated transcription factors, Eomes and TCF1 appear to be highly sensitive to graded expression levels of IRF4 both in acute and chronic viral infection, indicating potential mechanisms by which IRF4 may regulate CD8 T cell fate decision (79, 80). Furthermore, Nuclear receptor subfamily 4 group A member 1 (NR4A1) supports formation of MPECs and TCM via inhibiting the expression of IRF4 by directly binding to its promoter region (81). Similarly, the transcription factor BACH2 represses genes associated with terminal differentiation by binding to their enhancer regions and attenuating the availability of AP-1 binding sites (82). In this manner, BACH2 suppresses the differentiation of SLECs and tips the balance in favor of generating memory cells (82). Collectively these findings indicate that TCR-responsive transcription factors, such as IRF4 and AP-1 family members establish effector differentiation while NR4A1 and BACH2 suppress effector-associated genes. Thus, these transcription factors may cooperatively or antagonistically regulate cell fate decisions in response to different TCR signal strength intensities.
Importantly, there is an ever-expanding list of transcription factors known to orchestrate various signals experienced during the effector phase to polarize terminal differentiation or memory formation. STATs are cytokine-induced lineage-specifying transcription factors. Inflammatory cytokines, such as IL-2, IL-12, IFN-γ, and type I IFNs, signal through STAT1, 2, 4, and 5 respectively, and direct effector CD8 T cell proliferation and differentiation by inducing T-bet and Blimp-1 expression, and downregulating Bcl6, TCF1, and IL-7Rα expression (6, 83, 84). Conversely, STAT3 activation, which is induced by IL-10 and IL-21, is necessary for memory formation by promoting the expression of memory related transcription factors, such as Bcl6, Eomes, the SOCS3 (66, 85, 86). Moreover, T-bet/Eomes, Id2/Id3 and Blimp-1/Bcl6 (1), and the newly defined ZEB1/ZEB2 axis (87, 88) have reciprocal expression patterns in SLECs and MPECs, and drive differentiation toward opposing cell fates (Figure 1). As cell identity determined by complicated gene regulatory networks, further studies should focus on how these networks cooperatively regulate downstream target genes and cell fate decisions.
Transcription factors that are critical for naïve T cell homeostasis have also been identified to promote memory CD8 T cell self-renewal and maintenance. For example, FoxO1, TCF1, and LEF1 are all highly expressed in naïve CD8 T cells, downregulated in effector cells, and re-acquired in memory cells (84, 89–93) (Figures 2A,D). Their expression is continuously required for the long-term survival and homeostatic proliferation but not the initial activation and clonal expansion, or effector function (91, 94–96). FoxO1 promotes the expression of pro-memory and pro-survival genes, such as Il7r, Bcl2, Sell, Ccr7, Eomes, Tcf7, Bach2, Zeb1, and Socs3, potentially by shielding these genes from deposition of repression associated histone 3 lysine 27 trimethyl (H3K27me3) chromatin modifications (91, 93). TCF1 and LEF1 are downstream factors of the Wnt-signaling pathway and their downregulation in effector cells is due to cell cycle and IL-12-dependent CpG methylation at the TCF1 promoter (84). Intriguingly, TCF1 and LEF1 can induce deacetylation at effector genes regions, such as Prdm1, to favor memory formation (97).
In parallel with circulating memory cell subset differentiation, TRM acquire a unique transcriptional program during differentiation and adaptation to a particular microenvironment (98–101). As early as 7 days after acute infection, a unique transcriptional signature and chromatin landscape is already established in intestinal intraepithelial lymphocytes (IELs) (102). The transcription factor Runx3 has been identified as a central regulator for TRM specification by controlling a core tissue-residency gene-expression program in barrier tissues (such as lung, skin, and small intestine) and non-barrier tissues (such as salivary glands and kidney), as well as in tumors (102). Blimp-1 and its homolog protein, Hobit, establish a universal transcriptional program of tissue-residency in lymphocytes, and they have been shown to be required for TRM retention in the gut, skin, liver, kidneys and lung by promoting CD103 expression while repressing Klf2, S1pr1, and Ccr7 expression (99). In addition, Notch controls TRM maintenance by promoting CD103 expression and regulating metabolic programs (98). Recently, NR4A1 was shown to be critical in regulating the tissue residence and function of human TRM (103), and AhR was also shown to be required for skin TRM (104). By contrast, the transcription factors ZEB2, T-bet (87), and KLF2 (100) have been demonstrated to inhibit TRM formation by promoting tissue egress. Although T-bet and Eomes can inhibit TRM formation, certain levels of T-bet expression are required for CD122 expression and IL-15 mediated TRM survival (105).
A critical feature of memory CD8 T cells is their ability to rapidly re-acquire effector functions upon secondary challenge with the same pathogen. We are now learning that changes in the epigenetic landscape of memory CD8 T cells, including DNA methylation, histone modifications, and chromatin accessibility, play a substantial role in this phenomenon. In this section, we will discuss how these epigenetic changes shape the effector and memory fate decision as well as memory T cell formation and function (Figure 3).
DNA methylation occurs primarily at CpG dinucleotides with the cytosine being methylated. Genomic regions with high frequencies of these CpG dinucleotide sequences are known as CpG islands and are often found in promoters. DNA methylation is commonly thought of as a repressive epigenetic mark, exerting its downstream effects by influencing transcription factor binding and acting as a docking site for various histone modifying enzymes (Figure 2B). In CD8 T cells, the DNA methyltransferase Dnmt3a has been shown to reduce MPECs formation by catalyzing DNA methylation at sites such as the promoter of Tcf7, a critical transcription factor for memory CD8 T cells (106). TET2 is methylcytosine dioxygenase and mediates active DNA demethylation. TET2 gene expression is rapidly and transiently induced by TCR signaling. TET2-deficient CD8 T cells rapidly acquired memory associated surface markers such as CD62L, CD27, and CXCR3 to promote memory formation (107). Interestingly, while naïve genes become methylated and effector genes become demethylated in both MPECs and SLECs, MPECs erase these DNA methylation marks at naïve genes as they develop into long-lived memory CD8 T cells, indicating that epigenetic repression in the form of DNA methylation can be reversed (70) (Figure 2B).
Genomic DNA is packaged in nucleosomes, comprised of DNA wrapped around histone octamers made up of two copies each of the histones H2A, H2B, H3, and H4. Each histone has a flexible N-terminal tail that is subject to post-translational modifications that subsequently influence transcription of nearby genes. These modifications can affect gene expression by recruiting other transcriptional regulators or, in the case of acetylation, by neutralizing the positively charged histone N-terminal tail and decreasing its interaction with negatively charged phosphates on DNA. Large-scale genomic studies have found patterns of histone modifications that can identify cis-regulatory elements such as promoters and enhancers, as well as provide information regarding their activity (108–111) (Figure 2C). Additionally, active promoters and enhancers tend to have a central region that is depleted of nucleosomes, where transcription factors can more easily access their binding sites. It is therefore reasonable to suspect that a combination of histone modifications and accessible regions also contribute to the enhanced function of memory CD8 T cells. From studies investigating chromatin accessibility using assay for transposase-accessible chromatin (ATAC)-seq (112) and the deposition of histone modifications (H3K4Me1, H3K27Ac, H3K27Me3) by chromatin immunoprecipitation (ChIP)-seq in CD8 T cells during acute infections with Listeria monocytogenes and lymphocytic choriomeningitis virus (LCMV), we now have a genome-wide overview of the epigenetic changes accompanying memory CD8 T cell differentiation (71, 72, 113). These studies provide important insights into the epigenetic differences between MPECs and SLECs and through which their differentiation is regulated. Regulatory regions that are more open in MPECs than SLECs are genetic loci regulate feature genes related to naïve and memory T cell properties. However, these regulatory regions are less open or permanently silenced in terminally differentiated SLECs or exhausted CD8 T cells, suggesting that MPECs keep their memory potential through maintaining accessibility at critical memory-related cis-regulatory elements (71). Terminally differentiated SLECs have increased levels of the repressive histone modification H3K27Me3 at genes required for survival and memory cell formation, and deposition of this mark is catalyzed by the polycomb repressive complex 2 (PRC2) (93). The histone methyltransferase Suv39h1 also promotes terminal differentiation by trimethylating histone H3 lysine 9 at memory-related genes, repressing their expression (114). These differences in the epigenetic landscape between the two subsets of effector CD8 T cells provides a potential mechanism for their divergent gene expression profiles and cell fate decisions.
The chromatin accessible regions of memory CD8 T cell are quite similar to effector cells, especially around effector gene regions (115). Moreover, their promoter regions remain demethylated from effector to memory transition (70, 115). Much work has been done investigating DNA methylation at the Ifng locus in CD8 T cells, which encodes the important cytokine IFNγ that is rapidly expressed by memory cells (116–120). Naïve CD8 T cells possess substantial DNA methylation at the Ifng promoter, at least in part due to the activity of the DNA methyltransferase Dnmt1 (117). After activation, effector CD8 T cells have this site demethylated and turn on the expression of Ifng. Despite no longer expressing Ifng, memory CD8 T cells maintain a demethylated state at the Ifng promoter, thereby decreasing the number of steps required before gene expression. Help from CD4 T cells during initial activation appears to play a role in this process (119). Similar patterns seem to exist at the sites of other critical CD8 T cell effector molecules, including Gzmb and Prf1, which were found to maintain their demethylated state for at least 12 years in humans who received the yellow fever virus (YFV) vaccine (115). Therefore, regulation of DNA methylation provides a mechanism for the ability of memory CD8 T cells to quickly respond to infection.
Levels of histone H3 acetylation (119, 121) and, more specifically, H3 lysine 9 acetylation (H3K9Ac) contributes to the rapid reactivation in memory CD8 T cells (122–124). Furthermore, several studies have characterized a number of different histone modifications and chromatin accessibility at a genome-wide level over the course of a CD8 T cell response to infection or vaccination (71, 74, 93, 113, 115, 125–128). In the same study mentioned earlier, YFV-specific CD8 T cells in vaccinated humans maintain open, accessible chromatin at the promoters of the effector molecules Ifng and Gzmb (115). Overall, the establishment of specific patterns of DNA methylation, histone modifications, and chromatin accessibility prime memory CD8 T cells to more rapidly produce effector molecules and clear the pathogen.
Individual transcription factors can affect the epigenetic landscape through the recruitment of chromatin modifying enzymes or their own intrinsic activity. Blimp-1, for example, directly binds to the genes Il2ra and Cd27, recruits the histone methyltransferase G9a and the histone deacetylase HDAC2, and leads to increased deposition of the repressive marks H3K9Me2, H3K9Me3, and H3K27Me3 and decreased levels of permissive marks H3Ac and H3K4Me3 (129). The AP-1 factor BATF has been proposed to act as a pioneer transcription factor, in cooperation with its binding partner IRF4, by directly binding to tightly packed chromatin and promoting its accessibility to other transcription factors (130). Runx3 was recently shown to drive memory CD8 T cell formation by regulating chromatin accessibility of memory cell cis-regulatory elements (73). While TCF7 has not yet been shown to affect the epigenetic landscape during the differentiation of activated mature CD8 T cells, it establishes critical regions of open chromatin during T cell development in the thymus (131). Additionally, studies performed in thymocytes have shown that TCF7 has intrinsic histone deacetylase activity (97). Given its importance in memory CD8 T cell formation (94, 95, 132), it is likely that TCF7 uses a combination of these two methods to regulate the memory differentiation process. Other transcription factors will likely continue to be identified that either directly or indirectly lead to epigenetic changes in activated CD8 T cells, and untangling this complex network of transcription factors and the epigenetic changes they induce will help decode the differentiation of memory CD8 T cells.
Previous studies show that there can be anywhere from ~80 to 1,200 naïve CD8 T cells or from ~20 to 200 CD4 T cells specific for a particular epitope in one mouse (133, 134). Following infection, each antigen specific CD8 T cell can interpret and integrate signals in a distinct way to create differential responses in the generation of terminally differentiated effector cells and self-renewing memory T cells (6, 20). However, when and how this fate decision is made following infection has been a topic of research for many years (127, 135–139).
In terms of fate specification from a single T cell, two obvious possibilities can happen: (1) one T cell can give rise to two daughters cells with each being capable of choosing multiple fates or (2) one T cell can give rise to daughter cells with only one fate (140). Different experimental approaches have been applied to understand the in vivo fate of single CD8 T cells following acute viral or bacterial infections. Using an OT-I TCR transgenic adoptive cell transfer model, it has been demonstrated that diverse cellular progeny, including both effector and memory T cells, could develop out of a single naïve T cell following infection with L. monocytogenes (135). Similar results have been found using tetramer enrichment to isolate antigen specific naïve CD4 T cells followed by a single cell adoptive transfer approach for the in vivo fate mapping for CD4 T cells (141). Surprisingly, in both cases single naïve T cells displayed diverse patterns of differentiation, yet when combined together, they resembled the endogenous T cell response in the same individual mouse. Although these studies were instrumental in developing our understanding of T cell fate decision at the single cell level, a limitation of these approaches is that they only allow for deciphering the fate of one T cell at a time per mouse. To overcome this hurdle and to facilitate the analysis of multiple T cell families at the same time, one elegant study performed adoptive transfer experiments using barcoded TCR transgenic CD8 T cells. Upon bulk transfer of single barcoded naïve CD8 T cells the authors demonstrated that individual naïve T cells have multiple fates and can differentiate into both effector and memory subsets during acute infection (136). This approach offers the opportunity to analyze large numbers of barcoded TCR transgenic single naïve CD8 T cells and their fates at the same time. However, this experimental strategy is limited by its dependency on using indirect approaches (microarray, sequencing) for barcode identification and by its inability to conduct a functional assessment of T cells at the protein level. Notably, other powerful tools have emerged that help alleviate some of these pitfalls. To serve the purpose of analyzing multiple T cell families simultaneously, adoptive transfer experiments have been accompanied with the use of a matrix co-expressing congenic markers, followed by their breeding to TCR transgenic mice (137). This innovative approach allowed for the transfer and assessment of eight naïve TCR transgenic CD8 T cells at a time and revealed differential subset diversification by each single cell resulting in broad and vigorous CD8 T cell immunity. To rule out any TCR-based influence, a limiting dilution strategy has been developed with the aim of transferring a single naïve antigen specific CD8 T cell into recipient mice, which is plausible mathematically but in vivo difficult to prove (138). With this approach, single naïve CD8 T cells have been found to exhibit differential cell fates as well as display some extreme bias toward a particular cell fate. Importantly, using the latest powerful technology- single-cell RNA sequencing (scRNA-seq), it has recently been demonstrated that virus-specific CD8 T cells display vast transcriptional heterogeneity and can give rise to multiple cell fates, which unexpectedly was found to occur as early as the first cell division (127). This study highlights the power of using scRNA-seq and computational analyses to elucidate cell-fate decisions at the singe-cell level.
With the knowledge of possible cellular fates (single fate vs. multiple fates) at the single cell level, the next question to ask is: how does this subset diversification occur following infection? There are two possibilities to support this: the model of asymmetric division driven differentiation vs. progressive differentiation model (140, 142) (Figure 4A). According to the asymmetric division model, the generation of long and short-lived progenies from a single precursor T cell occurs at the immediate onset of response, i.e., as early as the first cell division (143). Asymmetric segregation of cytokine receptors like IL-2Rα and IFN-γR and intracellular signaling pathways like PI3K and mTORC1 during mitosis (92, 139, 143–146) can cause proximal and distal daughter cells to have differential cytokine signaling that may lead them toward an effector or memory cell differentiation process, respectively. Supporting this, three groups (127, 138, 139) have found that at the single cell level, cellular bifurcation is possible during early rounds of cellular division in response to acute infection. On the other hand, the progressive differentiation model supports the subset diversification process from a single cell via a gradual differentiation process, from a memory-like stage to terminally differentiated cells, which is affected by the signaling strength of the signals that are received in the priming phase (115, 142). This model has been supported by a study (147) using unbiased mathematical model and probabilistic framework. It has been shown that a linear developmental pathway is responsible for cell fate diversification, that progresses from slowly proliferating memory precursors to the rapidly expanding effector population. However, none of these models alone can explain why during differentiation some cells take multiple fates while some show extreme bias toward a singular fate. On this note, it is important to consider that cellular differentiation is a dynamic process and can be accompanied by encountering stochastic initial priming events, which can make a difference in the fate of every single cell, depending on their reception and interpretation of various signals. In this respect, both T cell intrinsic factors like: signaling strength, co-stimulation, amount of cell intrinsic signaling molecules, the epigenetic landscape, and cellular metabolism and also cell extrinsic factors like: anatomical location to interact with APC, and inflammation can affect the fate of a single T cell undergoing differentiation (1, 148–150). Intriguingly, a recent finding that showed, depending on the developmental origin of naïve CD8 T cells: either fetal derived CD8 T cells or adult bone marrow derived CD8 T cells, can give rise to either memory-like CD8 T cells in adulthood or in the generation of naïve-like CD8 T cells, respectively (151). The diverse in vivo response generated at the single cell level during acute infections may potentially be a result of the recruitment of heterogeneous naïve CD8 T cells, a topic, which demands further research. In terms of technological advancement, it is now possible to do in vivo fate mapping of single naïve CD8 T cells in a way which was limited previously with the usage of cell number, while simultaneously accounting for the influence of TCR and more importantly to recapitulate an in vivo natural infection scenario without the reliance on adoptive transfer strategies.
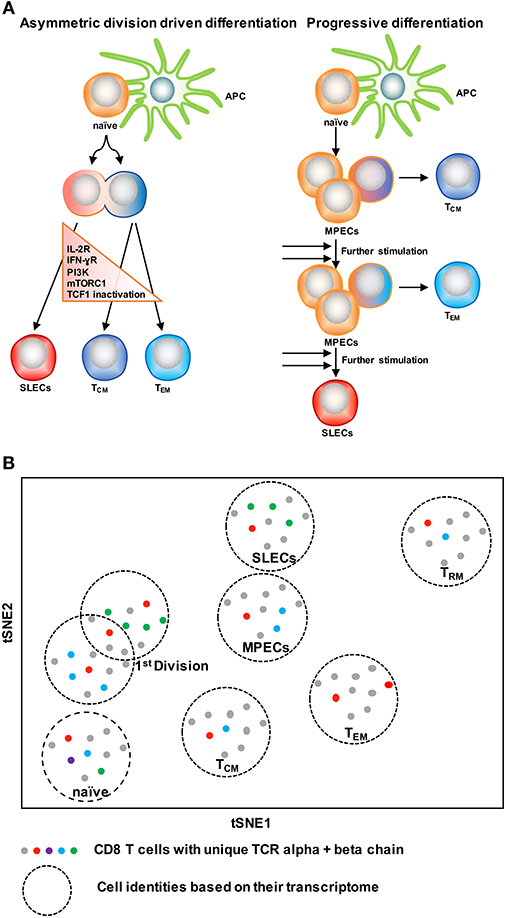
Figure 4. Two CD8 T cell differentiation models. (A) The asymmetric division model emphasizes the significance of asymmetric segregation of cytokine receptors and signals pathways as early as the first division in dictating the memory or effector potential of T cells. The proximal daughter cell (red) inherits molecules that make it more likely to become an effector cell, while the distal daughter (blue) inherits more memory-related molecules. The progressive differentiation model is a linear model in which the cumulative history of encounters with antigen and inflammation dictate the cell fate from a memory-like stage to terminally differentiated cells. (B) A method to depict “one cell, one fate” and “one cell, multiple fates” models. Single cell identity of a T cell can be profiled based on their transcriptome using scRNA-seq. TCRs are nature molecular tags to track T cells. Integration of TCR clonotypes to a gene expression profile on a single-cell level can monitor the dynamics of effect and memory CD8 T cell fate decision during infection.
ScRNA-seq has been emerged as an innovative platform to understand the cellular development and differentiation process (127, 152, 153). With the power of computational analysis, it also offers an assessment of the subset diversification and developmental trajectory in an unbiased manner without reliance on the preexisting knowledge of cellular types (153, 154). To understand single T cell fate and its kinship with subsequent progenies, it is ideal to trace the cell fate decision by using a natural T cell lineage barcode, the TCR sequence (155) (Figure 4B). Using TCR sequencing to uncover the identity of single T cells was limited with the determination of both TCR alpha and beta chain information in a single cell (156–158). With the use of more powerful algorithms, it is now possible to reconstruct TCR alpha-beta gene information from single cell RNA sequencing data and to couple the cellular identity of a T cell with its transcriptomic profile at the single cell level (159, 160). This approach can overcome the usage of TCR transgenic T cells and can allow for in vivo single cell fate mapping by observing and tracing thousands of single T cells simultaneously in a natural infection setting (152–154).
Current studies on genome-wide transcriptional and epigenetic changes during infection have revealed that DNA methylation, histone modifications and transcriptional signatures define CD8 T cell subsets and regulate CD8 T differentiation. Eventually, an identification of a core set of transcription factors or epigenetic regulatory molecules that can regulate memory formation could potentially be sufficient to help reprogram terminally differentiated CD8 T cells. Such findings will undoubtedly have an impact on T cell-based therapies and vaccine designs. Although the epigenetic patterns associated with distinct T cell subsets are starting to be unraveled, additional functional analyses are needed to further reveal the role of epigenetic modifying proteins and their relationship to key transcription factors that coordinately work together to determine cell-fate decisions. Moreover, as naïve CD8 T cells go through tremendous changes in their cell cycle, metabolism, cell signaling, and genetic landscape, it is starting to become well-appreciated that individual effector cells may acquire distinct cell fates, that as a whole results in the generation of a heterogeneous pool of memory T cells. While our current understanding of CD8 memory formation is derived from investigations using pooled cell populations to study cell fate decisions, recent technological advances in scRNA-seq and computational approaches hold great promise for deciphering the true transcriptional heterogeneity of individual CD8 T cells.
YC, RZ, DS, and AK wrote the manuscript. YC, RZ, and WC edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work is supported by NIH grants AI125741 (WC), DK108557 (DS), and by American Cancer Society (ACS) Research Scholar Grant and A Healthier Wisconsin (AHW) Grant (WC). RZ is supported by the Cancer Research Institute Irvington Fellowship. DS is a member of the Medical Scientist Training Program at MCW, which is partially supported by a training grant from NIGMS T32-GM080202.
1. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. (2012) 12:749–61. doi: 10.1038/nri3307
2. Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. (2014) 15:1104–15. doi: 10.1038/ni.3031
3. Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science (1996) 272:54–60. doi: 10.1126/science.272.5258.54
4. Schluns KS, Kieper WC, Jameson SC, Lefrancois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. (2000) 1:426–32. doi: 10.1038/80868
5. Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. (2003) 4:1191–8. doi: 10.1038/ni1009
6. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity (2007) 27:281–95. doi: 10.1016/j.immuni.2007.07.010
7. Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity (2007) 27:393–405. doi: 10.1016/j.immuni.2007.08.007
8. Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. (2008) 205:625–40. doi: 10.1084/jem.20071641
9. Ibegbu CC, Xu YX, Harris W, Maggio D, Miller JD, Kourtis AP. Expression of killer cell lectin-like receptor G1 on antigen-specific human CD8+ T lymphocytes during active, latent, and resolved infection and its relation with CD57. J Immunol. (2005) 174:6088–94. doi: 10.4049/jimmunol.174.10.6088
10. van Leeuwen EM, de Bree GJ, Remmerswaal EB, Yong SL, Tesselaar K, ten Berge IJ, et al. IL-7 receptor alpha chain expression distinguishes functional subsets of virus-specific human CD8+ T cells. Blood (2005) 106:2091–8. doi: 10.1182/blood-2005-02-0449
11. Bengsch B, Spangenberg HC, Kersting N, Neumann-Haefelin C, Panther E, von Weizsacker F, et al. Analysis of CD127 and KLRG1 expression on hepatitis C virus-specific CD8+ T cells reveals the existence of different memory T-cell subsets in the peripheral blood and liver. J Virol. (2007) 81:945–53. doi: 10.1128/JVI.01354-06
12. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol. (2013) 14:1294–301. doi: 10.1038/ni.2744
13. Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, Lefrancois L. Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity (2014) 40:747–57. doi: 10.1016/j.immuni.2014.03.007
14. Masopust D, Ha SJ, Vezys V, Ahmed R. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol. (2006) 177:831–9. doi: 10.4049/jimmunol.177.2.831
15. Croom HA, Denton AE, Valkenburg SA, Swan NG, Olson MR, Turner SJ, et al. Memory precursor phenotype of CD8+ T cells reflects early antigenic experience rather than memory numbers in a model of localized acute influenza infection. Eur J Immunol. (2011) 41:682–93. doi: 10.1002/eji.201040625
16. Nolz JC, Harty JT. Protective capacity of memory CD8+ T cells is dictated by antigen exposure history and nature of the infection. Immunity (2011) 34:781–93. doi: 10.1016/j.immuni.2011.03.020
17. Obar JJ, Jellison ER, Sheridan BS, Blair DA, Pham QM, Zickovich JM, et al. Pathogen-induced inflammatory environment controls effector and memory CD8+ T cell differentiation. J Immunol. (2011) 187:4967–78. doi: 10.4049/jimmunol.1102335
18. Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, et al. KLRG1(+) Effector CD8(+) T cells lose KLRG1, differentiate into all memory T cell lineages, and convey enhanced protective immunity. Immunity (2018) 48:716–29.e8. doi: 10.1016/j.immuni.2018.03.015
19. Hamann D, Baars PA, Rep MH, Hooibrink B, Kerkhof-Garde SR, Klein MR, et al. Phenotypic and functional separation of memory and effector human CD8+ T cells. J Exp Med. (1997) 186:1407–18. doi: 10.1084/jem.186.9.1407
20. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401:708–12. doi: 10.1038/44385
21. Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science (2001) 291:2413–7. doi: 10.1126/science.1058867
22. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. (2009) 10:524–30. doi: 10.1038/ni.1718
23. Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med. (2010) 207:553–64. doi: 10.1084/jem.20090858
24. von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med. (2000) 343:1020–34. doi: 10.1056/NEJM200010053431407
25. Reinhardt RL, Khoruts A, Merica R, Zell T, Jenkins MK. Visualizing the generation of memory CD4 T cells in the whole body. Nature (2001) 410:101–5. doi: 10.1038/35065111
26. Tussey L, Speller S, Gallimore A, Vessey R. Functionally distinct CD8+ memory T cell subsets in persistent EBV infection are differentiated by migratory receptor expression. Eur J Immunol. (2000) 30:1823–9. doi: 10.1002/1521-4141(200007)30:7<1823::AID-IMMU1823>3.0.CO;2-6
27. Jameson SC, Masopust D. Diversity in T cell memory: an embarrassment of riches. Immunity (2009) 31:859–71. doi: 10.1016/j.immuni.2009.11.007
28. Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol. (2013) 31:137–61. doi: 10.1146/annurev-immunol-032712-095954
29. Bouneaud C, Garcia Z, Kourilsky P, Pannetier C. Lineage relationships, homeostasis, and recall capacities of central- and effector-memory CD8 T cells in vivo. J Exp Med. (2005) 201:579–90. doi: 10.1084/jem.20040876
30. Sheridan BS, Lefrancois L. Regional and mucosal memory T cells. Nat Immunol. (2011) 12:485–91. doi: 10.1038/ni.2029
31. Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity (2014) 41:886–97. doi: 10.1016/j.immuni.2014.12.007
32. Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol. (2006) 176:2079–83. doi: 10.4049/jimmunol.176.4.2079
33. Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci USA. (2010) 107:17872–9. doi: 10.1073/pnas.1010201107
34. Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol. (2012) 188:4866–75. doi: 10.4049/jimmunol.1200402
35. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. (2013) 14:1285–93. doi: 10.1038/ni.2745
36. Steinbach K, Vincenti I, Kreutzfeldt M, Page N, Muschaweckh A, Wagner I, et al. Brain-resident memory T cells represent an autonomous cytotoxic barrier to viral infection. J Exp Med. (2016) 213:1571–87. doi: 10.1084/jem.20151916
37. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. (2017) 20:2921–34. doi: 10.1016/j.celrep.2017.08.078
38. Ely KH, Cookenham T, Roberts AD, Woodland DL. Memory T cell populations in the lung airways are maintained by continual recruitment. J Immunol. (2006) 176:537–43. doi: 10.4049/jimmunol.176.1.537
39. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. (2011) 17:1290–7. doi: 10.1038/nm.2446
40. Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med. (2017) 23:18–27. doi: 10.1038/nm.4241
41. Cieri N, Oliveira G, Greco R, Forcato M, Taccioli C, Cianciotti B, et al. Generation of human memory stem T cells after haploidentical T-replete hematopoietic stem cell transplantation. Blood (2015) 125:2865–74. doi: 10.1182/blood-2014-11-608539
42. Oliveira G, Ruggiero E, Stanghellini MT, Cieri N, D'Agostino M, Fronza R, et al. Tracking genetically engineered lymphocytes long-term reveals the dynamics of T cell immunological memory. Sci Transl Med. (2015) 7:317ra198. doi: 10.1126/scitranslmed.aac8265
43. Bottcher JP, Beyer M, Meissner F, Abdullah Z, Sander J, Hochst B, et al. Functional classification of memory CD8(+) T cells by CX3CR1 expression. Nat Commun. (2015) 6:8306. doi: 10.1038/ncomms9306
44. Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, et al. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity (2016) 45:1270–84. doi: 10.1016/j.immuni.2016.10.018
45. Zinkernagel RM, Doherty PC. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature (1974) 248:701–2. doi: 10.1038/248701a0
46. Doherty PC, Zinkernagel RM. H-2 compatibility is required for T-cell-mediated lysis of target cells infected with lymphocytic choriomeningitis virus. J Exp Med. (1975) 141:502–7. doi: 10.1084/jem.141.2.502
47. Obar JJ, Lefrancois L. Memory CD8+ T cell differentiation. Ann N Y Acad Sci. (2010) 1183:251–66. doi: 10.1111/j.1749-6632.2009.05126.x
48. Daniels MA, Teixeiro E. TCR signaling in T cell memory. Front Immunol. (2015) 6:617. doi: 10.3389/fimmu.2015.00617
49. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
50. Hendriks J, Xiao Y, Rossen JW, van der Sluijs KF, Sugamura K, Ishii N, et al. During viral infection of the respiratory tract, CD27, 4-1BB, and OX40 collectively determine formation of CD8+ memory T cells and their capacity for secondary expansion. J Immunol. (2005) 175:1665–76. doi: 10.4049/jimmunol.175.3.1665
51. Boesteanu AC, Katsikis PD. Memory T cells need CD28 costimulation to remember. Semin Immunol. (2009) 21:69–77. doi: 10.1016/j.smim.2009.02.005
52. Bally AP, Tang Y, Lee JT, Barwick BG, Martinez R, Evavold BD, et al. Conserved region C functions to regulate PD-1 expression and subsequent CD8 T cell memory. J Immunol. (2017) 198:205–17. doi: 10.4049/jimmunol.1601464
53. Sabins NC, Chornoguz O, Leander K, Kaplan F, Carter R, Kinder M, et al. TIM-3 engagement promotes effector memory T cell differentiation of human antigen-specific CD8 T cells by activating mTORC1. J Immunol. (2017) 199:4091–102. doi: 10.4049/jimmunol.1701030
54. Badovinac VP, Porter BB, Harty JT. CD8+ T cell contraction is controlled by early inflammation. Nat Immunol. (2004) 5:809–17. doi: 10.1038/ni1098
55. Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. (2005) 202:637–50. doi: 10.1084/jem.20050821
56. Pearce EL, Shen H. Generation of CD8 T cell memory is regulated by IL-12. J Immunol. (2007) 179:2074–81. doi: 10.4049/jimmunol.179.4.2074
57. Shaulov A, Murali-Krishna K. CD8 T cell expansion and memory differentiation are facilitated by simultaneous and sustained exposure to antigenic and inflammatory milieu. J Immunol. (2008) 180:1131–8. doi: 10.4049/jimmunol.180.2.1131
58. Cui W, Joshi NS, Jiang A, Kaech SM. Effects of Signal 3 during CD8 T cell priming: bystander production of IL-12 enhances effector T cell expansion but promotes terminal differentiation. Vaccine (2009) 27:2177–87. doi: 10.1016/j.vaccine.2009.01.088
59. Wiesel M, Crouse J, Bedenikovic G, Sutherland A, Joller N, Oxenius A. Type-I IFN drives the differentiation of short-lived effector CD8+ T cells in vivo. Eur J Immunol. (2012) 42:320–9. doi: 10.1002/eji.201142091
60. Weninger W, Crowley MA, Manjunath N, von Andrian UH. Migratory properties of naive, effector, and memory CD8(+) T cells. J Exp Med. (2001) 194:953–66. doi: 10.1084/jem.194.7.953
61. Berard M, Brandt K, Bulfone-Paus S, Tough DF. IL-15 promotes the survival of naive and memory phenotype CD8+ T cells. J Immunol. (2003) 170:5018–26. doi: 10.4049/jimmunol.170.10.5018
62. Richer MJ, Pewe LL, Hancox LS, Hartwig SM, Varga SM, Harty JT. Inflammatory IL-15 is required for optimal memory T cell responses. J Clin Invest. (2015) 125:3477–90. doi: 10.1172/JCI81261
63. Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity (2010) 32:91–103. doi: 10.1016/j.immuni.2009.11.010
64. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity (2010) 32:79–90. doi: 10.1016/j.immuni.2009.11.012
65. Blattman JN, Grayson JM, Wherry EJ, Kaech SM, Smith KA, Ahmed R. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat Med. (2003) 9:540–7. doi: 10.1038/nm866
66. Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity (2011) 35:792–805. doi: 10.1016/j.immuni.2011.09.017
67. El-Asady R, Yuan R, Liu K, Wang D, Gress RE, Lucas PJ, et al. TGF-{beta}-dependent CD103 expression by CD8(+) T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J Exp Med. (2005) 201:1647–57. doi: 10.1084/jem.20041044
68. Zhang N, Bevan MJ. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity (2013) 39:687–96. doi: 10.1016/j.immuni.2013.08.019
69. Evans CM, Jenner RG. Transcription factor interplay in T helper cell differentiation. Brief Funct Genomics (2013) 12:499–511. doi: 10.1093/bfgp/elt025
70. Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, et al. (2017). Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 552:404. doi: 10.1038/nature25144
71. Scott-Browne JP, Lopez-Moyado IF, Trifari S, Wong V, Chavez L, Rao A, et al. Dynamic changes in chromatin accessibility occur in CD8(+) T cells responding to viral infection. Immunity (2016) 45:1327–40. doi: 10.1016/j.immuni.2016.10.028
72. Yu B, Zhang K, Milner JJ, Toma C, Chen R, Scott-Browne JP, et al. Epigenetic landscapes reveal transcription factors that regulate CD8+ T cell differentiation. Nat Immunol. (2017) 18:573–82. doi: 10.1038/ni.3706
73. Wang D, Diao H, Getzler AJ, Rogal W, Frederick MA, Milner J, et al. The transcription factor Runx3 establishes chromatin accessibility of cis-regulatory landscapes that drive memory cytotoxic T lymphocyte formation. Immunity (2018) 48:659–74.e6. doi: 10.1016/j.immuni.2018.03.028
74. Russ BE, Olshanksy M, Smallwood HS Li J, Denton AE, Prier JE, et al. Distinct epigenetic signatures delineate transcriptional programs during virus-specific CD8(+) T cell differentiation. Immunity (2014) 41:853–65. doi: 10.1016/j.immuni.2014.11.001
75. Yao S, Buzo BF, Pham D, Jiang L, Taparowsky EJ, Kaplan MH, et al. Interferon regulatory factor 4 sustains CD8(+) T cell expansion and effector differentiation. Immunity (2013) 39:833–45. doi: 10.1016/j.immuni.2013.10.007
76. Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol. (2014) 15:373–83. doi: 10.1038/ni.2834
77. Godec J, Cowley GS, Barnitz RA, Alkan O, Root DE, Sharpe AH, et al. Inducible RNAi in vivo reveals that the transcription factor BATF is required to initiate but not maintain CD8+ T-cell effector differentiation. Proc Natl Acad Sci USA. (2015) 112:512–7. doi: 10.1073/pnas.1413291112
78. Xin G, Schauder DM, Lainez B, Weinstein JS Dai Z, Chen Y, et al. A critical role of IL-21-induced BATF in sustaining CD8-T-cell-mediated chronic viral control. Cell Rep. (2015) 13:1118–24. doi: 10.1016/j.celrep.2015.09.069
79. Nayar R, Schutten E, Bautista B, Daniels K, Prince AL, Enos M, et al. Graded levels of IRF4 regulate CD8+ T cell differentiation and expansion, but not attrition, in response to acute virus infection. J Immunol. (2014) 192:5881–93. doi: 10.4049/jimmunol.1303187
80. Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, et al. Transcription factor IRF4 promotes CD8(+) T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity (2017) 47:1129–41.e5. doi: 10.1016/j.immuni.2017.11.021
81. Nowyhed HN, Huynh TR, Thomas GD, Blatchley A, Hedrick CC. Cutting edge: the orphan nuclear receptor Nr4a1 regulates CD8+ T cell expansion and effector function through direct repression of Irf4. J Immunol. (2015) 195:3515–9. doi: 10.4049/jimmunol.1403027
82. Roychoudhuri R, Clever D, Li P, Wakabayashi Y, Quinn KM, Klebanoff CA, et al. BACH2 regulates CD8(+) T cell differentiation by controlling access of AP-1 factors to enhancers. Nat Immunol. (2016) 17:851–60. doi: 10.1038/ni.3441
83. Kim MT, Harty JT. Impact of inflammatory cytokines on effector and memory CD8+ T cells. Front Immunol. (2014) 5:295. doi: 10.3389/fimmu.2014.00295
84. Danilo M, Chennupati V, Silva JG, Siegert S, Held W. Suppression of Tcf1 by inflammatory cytokines facilitates effector CD8 T cell differentiation. Cell Rep. (2018) 22:2107–17. doi: 10.1016/j.celrep.2018.01.072
85. Siegel AM, Heimall J, Freeman AF, Hsu AP, Brittain E, Brenchley JM, et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity (2011) 35:806–18. doi: 10.1016/j.immuni.2011.09.016
86. Abdelsamed HA, Moustaki A, Fan Y, Dogra P, Ghoneim HE, Zebley CC, et al. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp Med. (2017) 214:1593–606. doi: 10.1084/jem.20161760
87. Dominguez CX, Amezquita RA, Guan T, Marshall HD, Joshi NS, Kleinstein SH, et al. The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J Exp Med. (2015) 212:2041–56. doi: 10.1084/jem.20150186
88. Guan T, Dominguez CX, Amezquita RA, Laidlaw BJ, Cheng J, Henao-Mejia J, et al. ZEB1, ZEB2, and the miR-200 family form a counterregulatory network to regulate CD8(+) T cell fates. J Exp Med. (2018) 215:1153–68. doi: 10.1084/jem.20171352
89. Kim EH, Sullivan JA, Plisch EH, Tejera MM, Jatzek A, Choi KY, et al. Signal integration by Akt regulates CD8 T cell effector and memory differentiation. J Immunol. (2012) 188:4305–14. doi: 10.4049/jimmunol.1103568
90. Rao RR Li Q, Gubbels Bupp MR, Shrikant PA. Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity (2012) 36:374–87. doi: 10.1016/j.immuni.2012.01.015
91. Kim MV, Ouyang W, Liao W, Zhang MQ Li MO. The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity (2013) 39:286–97. doi: 10.1016/j.immuni.2013.07.013
92. Lin WW, Nish SA, Yen B, Chen YH, Adams WC, Kratchmarov R, et al. CD8(+) T lymphocyte self-renewal during effector cell determination. Cell Rep. (2016) 17:1773–82. doi: 10.1016/j.celrep.2016.10.032
93. Gray SM, Amezquita RA, Guan T, Kleinstein SH, Kaech SM. Polycomb repressive complex 2-mediated chromatin repression guides effector CD8(+) T cell terminal differentiation and loss of multipotency. Immunity (2017) 46:596–608. doi: 10.1016/j.immuni.2017.03.012
94. Jeannet G, Boudousquie C, Gardiol N, Kang J, Huelsken J, Held W. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc Natl Acad Sci USA. (2010) 107:9777–82. doi: 10.1073/pnas.0914127107
95. Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity (2010) 33:229–40. doi: 10.1016/j.immuni.2010.08.002
96. Utzschneider DT, Delpoux A, Wieland D, Huang X, Lai CY, Hofmann M, et al. Active maintenance of T cell memory in acute and chronic viral infection depends on continuous expression of FOXO1. Cell Rep. (2018) 22:3454–67. doi: 10.1016/j.celrep.2018.03.020
97. Xing S, Li F, Zeng Z, Zhao Y, Yu S, Shan Q, et al. Tcf1 and Lef1 transcription factors establish CD8(+) T cell identity through intrinsic HDAC activity. Nat Immunol. (2016) 17:695–703. doi: 10.1038/ni.3456
98. Hombrink P, Helbig C, Backer RA, Piet B, Oja AE, Stark R, et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat Immunol. (2016) 17:1467–78. doi: 10.1038/ni.3589
99. Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science (2016) 352:459–63. doi: 10.1126/science.aad2035
100. Mackay LK, Kallies A. Transcriptional Regulation of Tissue-Resident Lymphocytes. Trends Immunol. (2017) 38:94–103. doi: 10.1016/j.it.2016.11.004
101. Milner JJ, Goldrath AW. Transcriptional programming of tissue-resident memory CD8(+) T cells. Curr Opin Immunol. (2018) 51:162–9. doi: 10.1016/j.coi.2018.03.017
102. Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature (2017) 552:253–7. doi: 10.1038/nature24993
103. Boddupalli CS, Nair S, Gray SM, Nowyhed HN, Verma R, Gibson JA, et al. ABC transporters and NR4A1 identify a quiescent subset of tissue-resident memory T cells. J Clin Invest. (2016) 126:3905–16. doi: 10.1172/JCI85329
104. Zaid A, Mackay LK, Rahimpour A, Braun A, Veldhoen M, Carbone FR, et al. Persistence of skin-resident memory T cells within an epidermal niche. Proc Natl Acad Sci USA (2014) 111:5307–12. doi: 10.1073/pnas.1322292111
105. Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, et al. T-box transcription factors combine with the cytokines TGF-beta and IL-15 to control tissue-resident memory T cell fate. Immunity (2015) 43:1101–11. doi: 10.1016/j.immuni.2015.11.008
106. Ladle BH Li K-P, Phillips MJ, Pucsek AB, Haile A, Powell JD, et al. De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8+ T-cell fate decisions following activation. Proc Natl Acad Sci USA. (2016) 113:10631–6. doi: 10.1073/pnas.1524490113
107. Carty SA, Gohil M, Banks LB, Cotton RM, Johnson ME, Stelekati E, et al. The loss of TET2 promotes CD8(+) T cell memory differentiation. J Immunol. (2018) 200:82–91. doi: 10.4049/jimmunol.1700559
108. ENCODE Project Consortium, Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature (2007) 447:799–816. doi: 10.1038/nature05874
109. Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. (2007) 39:311–8. doi: 10.1038/ng1966
110. Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature (2009) 459:108–12. doi: 10.1038/nature07829
111. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. (2010) 107:21931–6. doi: 10.1073/pnas.1016071107
112. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods (2013) 10:1213–8. doi: 10.1038/nmeth.2688
113. He B, Xing S, Chen C, Gao P, Teng L, Shan Q, et al. CD8+ T Cells Utilize Highly Dynamic Enhancer Repertoires and Regulatory Circuitry in Response to Infections. Immunity (2016) 45:1341–54. doi: 10.1016/j.immuni.2016.11.009
114. Pace L, Goudot C, Zueva E, Gueguen P, Burgdorf N, Waterfall JJ, et al. The epigenetic control of stemness in CD8+ T cell fate commitment. Science (2018) 359:177–86. doi: 10.1126/science.aah6499
115. Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT Li KW, et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature (2017) 552:362–7. doi: 10.1038/nature24633
116. Fitzpatrick DR, Shirley KM, Kelso A. Cutting edge: stable epigenetic inheritance of regional IFN-γ promoter demethylation in CD44high CD8+ T lymphocytes. J Immunol. (1999) 162:5053–7.
117. Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity (2001) 15:763–74. doi: 10.1016/S1074-7613(01)00227-8
118. Kersh EN, Fitzpatrick DR, Murali-Krishna K, Shires J, Speck SH, Boss JM, et al. Rapid demethylation of the IFN-γ gene occurs in memory but not naive CD8 T cells. J Immunol. (2006) 176:4083–93. doi: 10.4049/jimmunol.176.7.4083
119. Northrop JK, Thomas RM, Wells AD, Shen H. Epigenetic remodeling of the IL-2 and IFN-γ loci in memory CD8 T cells is influenced by CD4 T cells. J Immunol. (2006) 177:1062–9. doi: 10.4049/jimmunol.177.2.1062
120. Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA methylation remodeling accompanies CD8 T cell effector function. J Immunol. (2013) 191:3419–29. doi: 10.4049/jimmunol.1301395
121. Northrop JK, Wells AD, Shen H. Cutting edge: chromatin remodeling as a molecular basis for the enhanced functionality of memory CD8 T cells. J Immunol. (2008) 181:865–8. doi: 10.4049/jimmunol.181.2.865
122. Fann M, Godlove JM, Catalfamo M, Wood WH, Chrest FJ, Chun N, et al. Histone acetylation is associated with differential gene expression in the rapid and robust memory CD8+ T-cell response. Blood (2006) 108:3363–70. doi: 10.1182/blood-2006-02-005520
123. Araki Y, Fann M, Wersto R, Weng NP. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (Eomesodermin and its targets: Perforin and Granzyme B). J Immunol. (2008) 180:8102–8. doi: 10.4049/jimmunol.180.12.8102
124. Denton AE, Russ BE, Doherty PC, Rao S, Turner SJ. Differentiation-dependent functional and epigenetic landscapes for cytokine genes in virus-specific CD8+ T cells. Proc Natl Acad Sci USA. (2011) 108:15306–11. doi: 10.1073/pnas.1112520108
125. Araki Y, Wang Z, Zang C, Wood WH III, Schones D, Cui K, et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity (2009) 30:912–25. doi: 10.1016/j.immuni.2009.05.006
126. Crompton JG, Narayanan M, Cuddapah S, Roychoudhuri R, Ji Y, Yang W, et al. Lineage relationship of CD8+ T cell subsets is revealed by progressive changes in the epigenetic landscape. Cell Mol Immunol. (2016) 13:502. doi: 10.1038/cmi.2015.32
127. Kakaradov B, Arsenio J, Widjaja CE, He Z, Aigner S, Metz PJ, et al. Early transcriptional and epigenetic regulation of CD8(+) T cell differentiation revealed by single-cell RNA sequencing. Nat Immunol. (2017) 18:422–32. doi: 10.1038/ni.3688
128. Rodriguez RM, Suarez-Alvarez B, Lavín JL, Mosén-Ansorena D, Baragaño Raneros A, Márquez-Kisinousky L, et al. Epigenetic networks regulate the transcriptional program in memory and terminally differentiated CD8+ T cells. J Immunol. (2017) 198:937–49. doi: 10.4049/jimmunol.1601102
129. Shin HM, Kapoor V, Guan T, Kaech SM, Welsh RM, Berg LJ. Epigenetic modifications induced by Blimp-1 Regulate CD8(+) T cell memory progression during acute virus infection. Immunity (2013) 39:661–75. doi: 10.1016/j.immuni.2013.08.032
130. Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, et al. A validated regulatory network for Th17 cell specification. Cell (2012) 151:289–303. doi: 10.1016/j.cell.2012.09.016
131. Johnson JL, Georgakilas G, Petrovic J, Kurachi M, Cai S, Harly C, et al. Lineage-determining transcription factor TCF-1 initiates the epigenetic identity of T cells. Immunity (2018) 48:243–57.e10. doi: 10.1016/j.immuni.2018.01.012
132. Zhou X, Xue HH. Cutting edge: generation of memory precursors and functional memory CD8+ T cells depends on T cell factor-1 and lymphoid enhancer-binding factor-1. J Immunol. (2012) 189:2722. doi: 10.4049/jimmunol.1201150
133. Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, et al. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity (2007) 27:203–13. doi: 10.1016/j.immuni.2007.07.007
134. Jenkins MK, Chu HH, McLachlan JB, Moon JJ. On the composition of the preimmune repertoire of T cells specific for Peptide-major histocompatibility complex ligands. Annu Rev Immunol. (2010) 28:275–94. doi: 10.1146/annurev-immunol-030409-101253
135. Stemberger C, Huster KM, Koffler M, Anderl F, Schiemann M, Wagner H, et al. A single naive CD8+ T cell precursor can develop into diverse effector and memory subsets. Immunity (2007) 27:985–97. doi: 10.1016/j.immuni.2007.10.012
136. Gerlach C, van Heijst JW, Swart E, Sie D, Armstrong N, Kerkhoven RM, et al. One naive T cell, multiple fates in CD8+ T cell differentiation. J Exp Med. (2010) 207:1235–46. doi: 10.1084/jem.20091175
137. Buchholz VR, Flossdorf M, Hensel I, Kretschmer L, Weissbrich B, Graf P, et al. Disparate individual fates compose robust CD8+ T cell immunity. Science (2013) 340:630–5. doi: 10.1126/science.1235454
138. Plumlee CR, Sheridan BS, Cicek BB, Lefrancois L. Environmental cues dictate the fate of individual CD8+ T cells responding to infection. Immunity (2013) 39:347–56. doi: 10.1016/j.immuni.2013.07.014
139. Arsenio J, Kakaradov B, Metz PJ, Kim SH, Yeo GW, Chang JT. Early specification of CD8+ T lymphocyte fates during adaptive immunity revealed by single-cell gene-expression analyses. Nat Immunol. (2014) 15:365–72. doi: 10.1038/ni.2842
140. Reiner SL, Sallusto F, Lanzavecchia A. Division of labor with a workforce of one: challenges in specifying effector and memory T cell fate. Science (2007) 317:622–5. doi: 10.1126/science.1143775
141. Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, et al. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell (2013) 153:785–96. doi: 10.1016/j.cell.2013.04.007
142. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol (2004) 22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702
143. Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science (2007) 315:1687–91. doi: 10.1126/science.1139393
144. Chang JT, Ciocca ML, Kinjyo I, Palanivel VR, McClurkin CE, Dejong CS, et al. Asymmetric proteasome segregation as a mechanism for unequal partitioning of the transcription factor T-bet during T lymphocyte division. Immunity (2011) 34:492–504. doi: 10.1016/j.immuni.2011.03.017
145. Lin WH, Adams WC, Nish SA, Chen YH, Yen B, Rothman NJ, et al. Asymmetric PI3K signaling driving developmental and regenerative cell fate bifurcation. Cell Rep. (2015) 13:2203–18. doi: 10.1016/j.celrep.2015.10.072
146. Pollizzi KN, Sun IH, Patel CH, Lo YC, Oh MH, Waickman AT, et al. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8(+) T cell differentiation. Nat Immunol. (2016) 17:704–11. doi: 10.1038/ni.3438
147. Buchholz VR, Graf P, Busch DH. The smallest unit: effector and memory CD8(+) T cell differentiation on the single cell level. Front Immunol. (2013) 4:31. doi: 10.3389/fimmu.2013.00031
148. Rohr JC, Gerlach C, Kok L, Schumacher TN. Single cell behavior in T cell differentiation. Trends Immunol. (2014) 35:170–7. doi: 10.1016/j.it.2014.02.006
149. Buchholz VR, Schumacher TN, Busch DH. T cell fate at the single-cell level. Annu Rev Immunol. (2016) 34:65–92. doi: 10.1146/annurev-immunol-032414-112014
150. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol. (2018) 18:340–56. doi: 10.1038/nri.2017.146
151. Smith NL, Patel RK, Reynaldi A, Grenier JK, Wang J, Watson NB, et al. Developmental origin governs CD8(+) T cell fate decisions during infection. Cell (2018) 174:117–130.e14. doi: 10.1016/j.cell.2018.05.029
152. Lonnberg T, Svensson V, James KR, Fernandez-Ruiz D, Sebina I, Montandon R, et al. Single-cell RNA-seq and computational analysis using temporal mixture modelling resolves Th1/Tfh fate bifurcation in malaria. Sci Immunol. (2017) 2:eaal2192. doi: 10.1126/sciimmunol.aal2192
153. Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, Benoist C. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat Immunol. (2018) 19:291–301. doi: 10.1038/s41590-018-0051-0
154. Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. (2018) 24:978–85. doi: 10.1038/s41591-018-0045-3
155. Han A, Glanville J, Hansmann L, Davis MM. Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat Biotechnol. (2014) 32:684–92. doi: 10.1038/nbt.2938
156. Venturi V, Quigley MF, Greenaway HY, Ng PC, Ende ZS, McIntosh T, et al. A mechanism for TCR sharing between T cell subsets and individuals revealed by pyrosequencing. J Immunol. (2011) 186:4285–94. doi: 10.4049/jimmunol.1003898
157. Ji X, Lyu SC, Spindler M, Bacchetta R, Goncharov I, Han A, et al. Deep profiling of single T cell receptor repertoire and phenotype with targeted RNA-seq (TECH2P.927). J Immunol. (2015) 194(1 Suppl.):206.237–206.237.
158. Li B, Li T, Pignon JC, Wang B, Wang J, Shukla SA, et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat Genet. (2016) 48:725–32. doi: 10.1038/ng.3581
159. Stubbington MJT, Lonnberg T, Proserpio V, Clare S, Speak AO, Dougan G, et al. T cell fate and clonality inference from single-cell transcriptomes. Nat Methods (2016) 13:329–32. doi: 10.1038/nmeth.3800
Keywords: CD8 T cell, memory differentiation, cell fate decision, transcriptional, epigenetic, single cell sequencing
Citation: Chen Y, Zander R, Khatun A, Schauder DM and Cui W (2018) Transcriptional and Epigenetic Regulation of Effector and Memory CD8 T Cell Differentiation. Front. Immunol. 9:2826. doi: 10.3389/fimmu.2018.02826
Received: 31 August 2018; Accepted: 15 November 2018;
Published: 07 December 2018.
Edited by:
Surojit Sarkar, University of Washington, United StatesReviewed by:
Karl Kai McKinstry, University of Central Florida, United StatesCopyright © 2018 Chen, Zander, Khatun, Schauder and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiguo Cui, d2VpZ3VvLmN1aUBiY3cuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.