 Santiago Sánchez-Alonso1†
Santiago Sánchez-Alonso1† Francisco Sánchez-Madrid
Francisco Sánchez-Madrid
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 November 2018
Sec. Vaccines and Molecular Therapeutics
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02799
This article is part of the Research Topic Extracellular Vesicle-Based Therapy for Regenerative Medicine View all 6 articles
Myocardial ischemia-related disorders constitute a major health problem, being a leading cause of death in the world. Upon ischemia, tissue remodeling processes come into play, comprising a series of inter-dependent stages, including inflammation, cell proliferation and repair. Neovessel formation during late phases of remodeling provides oxygen supply, together with cellular and soluble components necessary for an efficient myocardial reconstruction. Immune system plays a central role in processes aimed at repairing ischemic myocardium, mainly in inflammatory and angiogenesis phases. In addition to cellular components and soluble mediators as chemokines and cytokines, the immune system acts in a paracrine fashion through small extracellular vesicles (EVs) release. These vesicular structures participate in multiple biological processes, and transmit information through bioactive cargoes from one cell to another. Cell therapy has been employed in an attempt to improve the outcome of these patients, through the promotion of tissue regeneration and angiogenesis. However, clinical trials have shown variable results, which put into question the actual applicability of cell-based therapies. Paracrine factors secreted by engrafted cells partially mediate tissue repair, and this knowledge has led to the hypothesis that small EVs may become a useful tool for cell-free myocardial infarction therapy. Current small EVs engineering strategies allow delivery of specific content to selected cell types, thus revealing the singular properties of these vesicles for myocardial ischemia treatment.
Regenerative medicine is focused on repairing damaged or malfunctioning tissue through both cellular and non-cellular therapies, such as tissue engineering or treatments based on biomaterials. This biological strategy is applicable to a wide spectrum of processes, including wound healing (1), neurological conditions (2), bone disorders (3), and ischemic cardiovascular diseases (4). One feature common to all these conditions is the involvement of tissue regeneration and remodeling in their termination phase.
Myocardial infarction (MI) is the most common cause of early death in adults worldwide (5). It results from an imbalance between oxygen supply and demand, usually due to the occlusion of a coronary artery. Despite success in reducing early mortality, late morbidity and mortality in the form of heart failure (HF) are still an unmet clinical problem. HF after MI is the consequence of a non-optimal myocardial repair. After ischemia, a step-by-step myocardial remodeling takes place involving two main phases: an inflammatory phase, where recruited inflammatory cells clear the wound of tissue debris and prepare the tissue for the next phase, and the proliferative and reparative phase, where inflammation resolves, angiogenesis is induced, fibroblasts activated, and fibrosis takes place with scar formation (6). These highly orchestrated phases may seem to occur separately, but they actually overlap with a coordinated interplay between immune cells, fibroplasia and angiogenesis in order to provide an efficient microvascular perfusion of the remodeled heart, thus ensuring its subsequent repair and functionality (6) (Figure 1). Throughout the last decades, researchers have made efforts to promote myocardial repair by acting at different steps of the process, as is the case of angiogenesis (7). Indeed, the promotion of angiogenesis in MI patients seeks to improve the microcirculation by promoting new capillary and collateral arterial vessel formation, thus rescuing the myocardium at early stages post-MI and preventing long-term left ventricular remodeling and transition to HF (8–10).
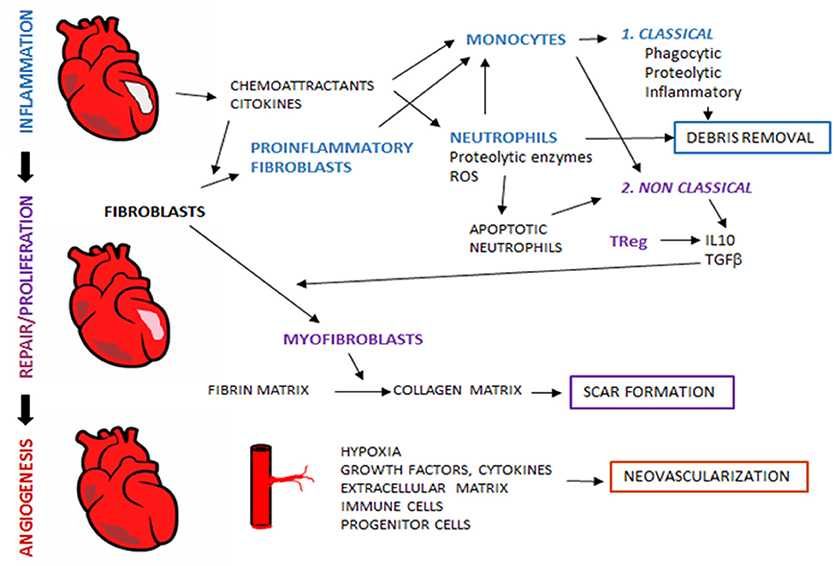
Figure 1. Tissue regeneration after myocardial infarction. After myocardial ischemia, remodeling process take place in order to reestablish tissue properties and myocardial function. During remodeling, sequential interdependent phases occur, which include inflammation, proliferation and repair. Al later stages, angiogenesis restores blood flow to ensure adequate tissue regeneration. Immune system is an essential player in tissue remodeling, and acts through the coordinated action with other non-immune cellular components and released mediators.
Extracellular vesicles (EVs) are membranous vesicles involved in intercellular communication, released to the extracellular environment by different types of living cells under normal or pathological conditions. They can be classified into three main groups based on their biological origin, size and composition: microvesicles, apoptotic bodies and exosomes. Exosomes, here referred to as small EVs, are nanosized (50–150 nm) EVs of endocytic origin (11), which may act as a vehicle to transfer bioactive cargoes (proteins, lipids or nucleic acids) from one cell to another, either allowing or enhancing functional communication between cells (12, 13). Loading of these molecules into small EVs is not a random process; on the contrary, these vesicles are enriched with specific contents, thus revealing the existence of sorting mechanisms that selectively regulate small EVs load (14). Uptake of small EVs is mediated by direct interaction. Binding of small EVs to the plasma membrane of recipient cells is mainly driven by adhesion molecules involved in cell-to-cell interaction, but there are other small EVs molecules that specifically act as receptors for their selective binding and engulfment. To deliver their cargo, small EVs may either fuse with plasma membrane of recipient cells or be internalized by endocytosis, pinocytosis or phagocytosis (15).
Small EVs participate in a number of biological processes, which include organogenesis (16), vascular regulation (17) and immune responses (18). In these processes, cells from different origin release small EVs to generate an intricate signaling network, which reciprocally regulates cellular functions. In this review, we offer a perspective on the interaction between immune system and angiogenesis, and examine the functional role and potential application of immune system-derived small EVs in the promotion of neovascularization after MI.
During inflammatory phase, the first cells recruited to the injured site are neutrophils, which migrate in response to signals as damage-associated molecular patterns (DAMPs), cytokines, chemokines, endogenous lipid mediators (prostaglandin E2, leukotriene B4), histamine and complement components (19–21). Emigrant neutrophils release proteolytic enzymes and reactive oxygen species at their arrival to the infarcted site and damage local cells (22). Another key role of neutrophils in cardiac repair is the release of chemotactic factors such as azurocidin, IL-37 and cathepsin G that recruit and accumulate splenic monocytes in the infarcted myocardium (21, 23, 24). Two monocyte populations have been described in ischemic myocardium of both mice and human: an inflammatory subset Gr1+CCR2+ CX3CX1lo(Gr1high), and the resident monocyte subset with a less inflammatory function Gr1− CCR2− CX3CR1hiGr1low, corresponding to classical human monocytes CD14hiCD16− and nonclassical monocytes CD14loCD16+, respectively (25). Classical monocyte subset is recruited in the early phase of MI through the activation of the MCP-1/CCR2axis. These cells display a phagocytic, proteolytic and inflammatory function, by digesting the damaged tissue and clearing cellular debris at the injury site (26) (Figure 1).
After this phase of degradation and digestion of injured cells, new subsets of immune cells intervene, such as non-classical monocytes (Gr1low) with anti-inflammatory properties, T-cell subpopulations as regulatory T cells (TRegs), and macrophages with anti-inflammatory phenotype which secrete transforming growth factor (TGF)-β and IL-10. These cellular effectors and molecular signals repress inflammation and enhance heart healing through myofibroblasts accumulation, angiogenesis and collagen deposition (27) (Figure 1).
As soon as necrotic cells and debris are removed by the inflammatory cells, cells that play a crucial role in the first phase of regeneration are then replaced by resident or newly recruited Gr1low monocytes, lymphocytes and mast cells that will coordinate the remodeling of the myocardium. In fact, neutrophils have no longer an important role in the proliferation phase and despite DAMPs, proinflammatory cytokines, hypoxia and acidosis survival signals, neutrophils are short-lived cells that rapidly undergo cell death (19, 28). Indeed, late-stage and apoptotic neutrophils are essential for the end of the inflammatory phase, as their transmigration is slowed down by the release of pro-resolving mediators (such as lipid mediators, annexin A1 and lactoferrin) and their phagocytosis by macrophages is enhanced both by these mediators and by the expression of eat-me signals (19, 21). Phagocytosis of apoptotic neutrophils induces a pro-resolving M2 phenotype in macrophages and the subsequent secretion of anti-inflammatory and profibrotic cytokines such as IL-10 and TGF-β, which suppress inflammation and enhance tissue repair (29) (Figure 1).
Despite the well-established time frame of events during cardiac remodeling after MI, a subset of cases shows a late progressive ventricular dilatation and HF mainly due to a chronic inflammation state of the myocardium (30). Accordingly, it has been recently described that in chronic ischemic HF, there is local and systemic expansion of proinflammatory macrophages, dendritic cells (DC) and T cells with an impaired left ventricular remodeling (31).
Angiogenesis is a highly orchestrated process that starts early after inflammatory phase of MI. The main players in hypoxia-driven angiogenesis are hypoxia-inducible factors (HIF). They are expressed in cardiomyocytes, endothelial and inflammatory cells early after MI, and may remain stable up to several weeks after MI (32). Both HIF-1α and endothelial-specific HIF-2α target proangiogenic genes such as VEGF (33), cyclooxygenase-2 (COX2) (34), the progenitor cell mobilizing chemokine Stromal Cell-Derived Factor 1-alpha (SDF1-alpha or CXCL12) (35), angiopoietins and others. Notably, several of these HIF-inducible genes also play an important role in inflammation, indicating a parallelism between inflammation and angiogenesis in ischemic-tissue remodeling.
A number of growth factors are implicated in angiogenesis promotion in the ischemic heart. Hence, an important increase of VEGF and its receptor VEGFR2 takes place in the viable border zone of myocardiocytes and in the area of forming vessels, respectively (36, 37). VEGF-B may also play a role in the induction of prosurvival signals that ensure the presence and maturation of new vessels in the ischemic myocardium, by providing anti-apoptotic signals in endothelial and mural cells (38). Similarly, PDGF-A and -D expression increases in the periphery of infarcted area, in parallel with the development of angiogenic process (39).
Different types of progenitor cells may act as a substrate for generation of new cells in angiogenesis in MI. Several studies have demonstrated proangiogenic and therapeutic effect of endothelial progenitor cells (EPCs) in experimental models of MI; however, the mechanisms involved are not well understood yet (40). Thus, although EPCs differentiate into mature ECs, there is growing evidence that their paracrine activity could play an important role in the injured endothelium repair (41). In a mouse model of MI, ischemic hearts injected with c-kit+ cardiac stem cells (CSCs) display higher angiopoietin-like 2 expression and staining for EC marker PECAM-1 in peri-infarct areas, suggesting that c-kit+ cells can differentiate into ECs and promote angiogenesis in vivo in these hypoxic sites (42). Other studies support this function of CSCs on vascularization recovery after MI, not only through differentiation into ECs, but also in a paracrine manner by promoting the secretion of multiple pro-angiogenic growth factors as VEGF, PDGF and HGF. (43). Mesenchymal stem cells (MSC) and adipose tissue-derived stem cells have also shown a proangiogenic potential after MI in experimental studies (44). MSC enhance several key processes in angiogenesis by releasing paracrine factors that stimulate vessel formation, differentiating into endothelial of vascular smooth muscle cell lineage, and acting as perivascular cells (45). The proangiogenic function of MSC has been related to both the release of soluble factors and miRNA, notably miR-146a, which increases VEGF secretion in MSCs leading to a reduction in fibrosis and enhancement of left ventricular ejection fraction (45, 46).
As mentioned above, inflammation and angiogenesis are closely inter-related processes. The immune system handles, along with other cells and mediators, the correct and efficient repair of the damaged heart, and more specifically an adequate angiogenesis development. This is achieved by the concerted action of cellular and molecular components (summarized in Table 1), which display pleiotropic functions that modulate their respective activities (67) (Figure 2).
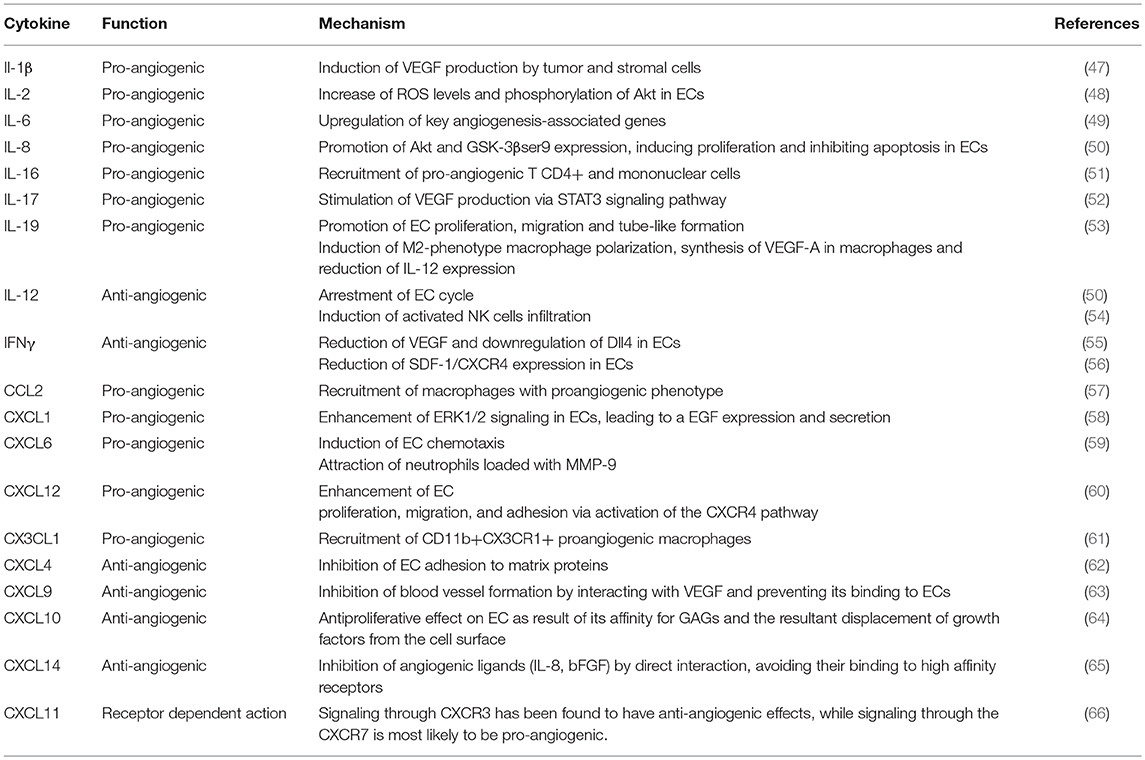
Table 1. Function of interleukins and chemokines in angiogenesis regulation.
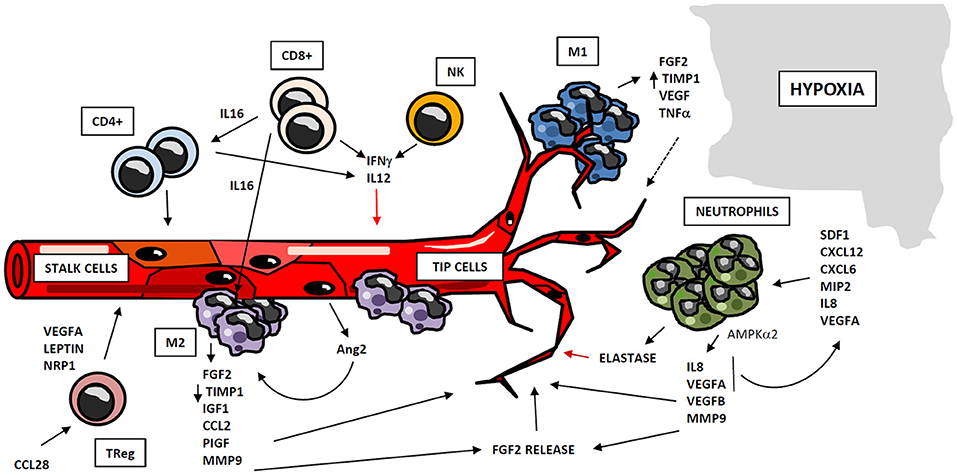
Figure 2. Regulation of angiogenesis by cellular compartment of immune system. Immune system participates in angiogenesis development after myocardial ischemia, through cellular and soluble components. Myeloid and lymphoid cells may operate as positive or negative regulators of angiogenesis, through the secretion of cytokines and soluble factors which act at different stages of the process. Monocytes act in two subsequent waves of infiltration, composed respectively by M1and M2-like cells. M1 macrophages co-localize with endothelial tip cells and display a mild pro-angiogenic phenotype, whereas M2 cells are located close to EC anastomosis, and possess potent pro-angiogenic properties. Neutrophils are recruited to hypoxic areas by chemoattractant cytokines, and accumulate in so-called “angiogenic hotspots” at vascular tips. Neutrophils mainly exert proangiogenic actions by the production of soluble factors and metalloproteinases, although these cells may act as negative regulators of this process, trough elastase release. Lymphocytes also play an important role in angiogenesis, either directly by the secretion of pro- and anti-angiogenic mediators, or by regulating the activity of other cell types as macrophages and different lymphocyte subsets.
Cells of myeloid lineage are the best described immune regulators of angiogenesis. Specifically, monocytes/macrophages exhibit well-know functions in vessel formation (68). A number of studies suggest that recruitment of monocytes to ischemic tissues takes place in two waves of infiltration: the first wave attracts monocytes with M1 inflammatory phenotype, which are subsequently replaced by a second wave, composed of pro-angiogenic and regenerative M2-like macrophages (69, 70). Macrophage-secreted Semaphorin 4A has been involved in angiogenesis occurring after ischemic injury in the heart (71). M2 macrophages promote neovascularization through the release of proangiogenic factors as FGF-2, insulin-like growth factor-1 (IGF-1), monocyte chemotactic protein-1 (CCL2), placental growth factor (PIGF) (72), and proteases as MMP-9 (73). M1 macrophages may have a positive role in angiogenesis as well, although to a different extent: both M1 and M2 macrophages produce MMP-9, but M2 macrophages display a reduced expression of tissue inhibitor of metalloproteinase 1 (TIMP-1), so their angiogenesis-inducing capacity is higher than that of the M1 macrophages (74). Additionally, M1 macrophages co-localize with endothelial tip cells and M2 macrophages with cell anastomosis sites, thus revealing a potential interaction of M1 phenotype with tip cells and a participation of M2 macrophages in cell fusion (75, 76). Therefore, macrophage-EC contact seems to be important in vascular morphogenesis. Furthermore, paracrine signaling may participate in macrophage pro-angiogenic functions. Activated ECs secrete angiopoietin-2 (Ang2), which binds neighbor Tie2-expressing monocytes/macrophages, enhancing their angiogenic potential (77). All these findings point to monocytes/macrophages as key regulators of angiogenesis, suggesting that there is a coordinated participation of both subsets of macrophages in vessel formation. Hence, M1 macrophages sustain the initial tissue inflammation, remove damaged tissue and finally begin angiogenic process and vessel growth. They are subsequently replaced by M2 macrophages, which display more pro-angiogenic capacities (Figure 2).
Neutrophils play an important role in angiogenesis through different mechanisms. One is regulation of vascular repair through AMP-activated protein kinase α2 (AMPKα2), a subunit of AMPK protein, which promotes survival of neutrophils in ischemic tissues as well as generation of pro-angiogenic factors (such as VEGFA and VEGFB) (78). Aside, ischemic-infiltrating neutrophils produce TIMP-1-free proMMP-9 that proteolytically induces functional angiogenic activity (74) through bioactive FGF-2 release (79). Furthermore, it has been demonstrated that VEGFA released under ischemic conditions promotes the recruitment of a specific subset of circulating CD49d+ VEGFR1high CXCR4high neutrophils with high MMP-9 expression levels, to facilitate rapid angiogenesis at hypoxic areas (80, 81). Other stimuli lead to neutrophil recruitment. Thus, CXCL1/macrophage inflammatory protein-2 (MIP-2) induces neutrophil recruitment and release of active VEGFA, which in turn activates the angiogenic cascade in tissues (82). Besides, IL-8/CXCL8 induces recruitment of neutrophils, which secrete VEGF and more IL-8, acting as a paracrine feedforward signal that amplifies the angiogenic effect of this cell type. Infiltrating neutrophils accumulate in “angiogenic hotspots,” specifically localized at the tips of the sprouting vessels; and this recruitment is directed by SDF-1 (CXCL12), a chemokine produced in the proangiogenic niche (83). However, some evidences suggest that neutrophils could secrete factors that restrain the angiogenic process. For instance, neutrophils release neutrophil elastase, a serine proteinase that cleaves plasminogen into angiostatin (fragments one to three), which in turn inhibits VEGF and FGF-2-mediated EC proliferation (84) (Figure 2). Despite the above mentioned proangiogenic characteristics of neutrophils, they are one of the major mediators of microvascular dysfunction after MI, and promote ischemia reperfusion injury and no-reflow phenomenon (10, 85).
Lymphocytes have been shown to mediate neovascularization as well. B lymphocytes infiltrating tumors contribute to angiogenesis through STAT3-mediated production of pro-angiogenic factors such as MMP-9 or CCL2 (86). Likewise, both CD4+ and CD8+ T lymphocytes play an important role in this process, together with other subpopulations as TReg and Tγδ cells. CD4+ T lymphocyte-deficient mice demonstrate impaired post-ischemic neovascularization, which is rescued by CD4 reconstitution (87). Similarly, CD8+ T lymphocyte-deficient mice also display defective blood flow recovery after hindlimb ischemia, which recovers upon CD8+ infusion through IL-16-mediated CD4+ and mononuclear cell recruitment (51). However, lymphocytes may participate also in restraining angiogenesis. It has been shown that lymphocytes (mostly CD4+)-EC crosstalk mediates IL-12-dependent inhibition of angiogenesis by reducing expression and activity of MMP-9 in vitro (88, 89); and in a mouse model of lung ischemia, neovascularization is limited by both CD8+ and CD4+ effector T cells through interferon gamma (IFNγ) secretion (90) (Figure 2). Likewise, Tγδ cells are able to promote angiogenesis in tumors by differentiating into IL-17 producing cells (91). Additionally, in a model of oxygen-induced retinopathy, acute retinal ischemia induces TReg recruitment, although this accumulation is not enough to completely prevent vascular damage by itself; but expanding TReg population by administrating IL-2/anti-IL2 monoclonal antibody complexes to mice, TRegs are able to reduce retinopathy (92). Furthermore, Natural Killer (NK) cells are involved in angiogenesis regulation as well. In a model of murine hindlimb ischemia, depletion of NK1.1-expressing cells led to defective perfusion recovery and collateral blood vessels growth (93). Moreover, IFNγ synthesis is increased in tumor-infiltrating NK and CD4+ T cells, and this may be responsible for production of tumor necrosis factor superfamily-15 (TNFSF15) by ECs, a cytokine that inhibits their proliferation and differentiation (94). In conclusion, lymphocytes may act as positive or negative regulators of neovascularization, frequently through the secretion of cytokines and soluble factors that operate in different steps of the angiogenic process.
Cells belonging to either non-immune or immune compartments contribute to angiogenesis development through the delivery of small EVs, which act in a paracrine manner and trigger functional responses in neighboring cells (Figure 3).
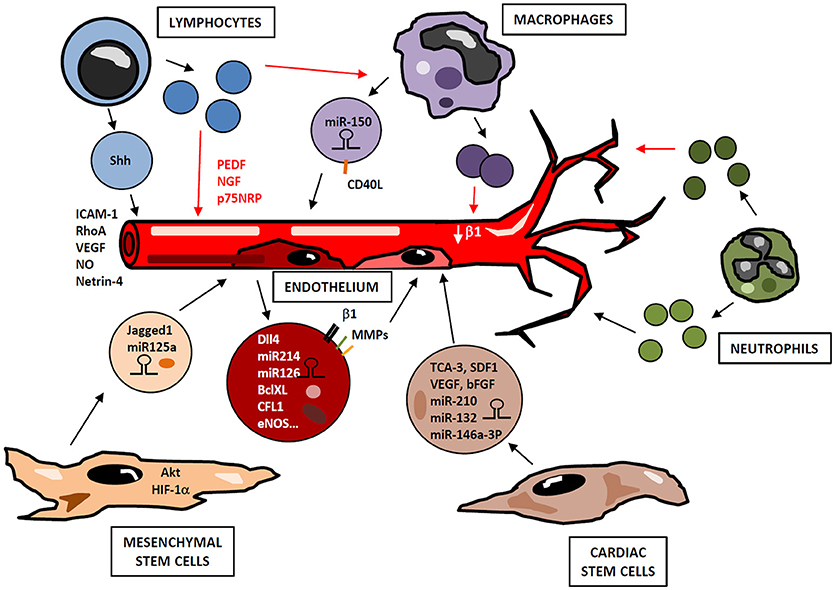
Figure 3. Small EVs in angiogenesis regulation. Cells from immune (lymphocytes, macrophages, neutrophils) and non-immune (cardiac and mesenchymal stem cells, endothelial cells) compartments release small EVs containing bioactive cargos (miRNAs, mRNAs, cytokines, signaling molecules), which modulate endothelial functions in a paracrine manner, positively or negatively regulating neovascularization. Depending on their activation state, lymphocytes may produce small EVs with either pro- or antiangiogenic effect. Similarly, small EVs derived from activated macrophages and neutrophils display different functional properties. Thus, a highly complex network of vesicles with potential opposite functions may be released during angiogenic process in response to immune cell activation and microenvironmental conditions.
ECs-derived microvesicles contain β1-integrin, MMP-2 and MMP-9; and their addition to human umbilical vein endothelial cells (HUVEC) stimulates formation of capillary-like structures in vitro (95). Interestingly, Dll4 is incorporated into EC-derived small EVs and can be transferred from one cell to another in a paracrine manner, thus inhibiting Notch receptor signaling and enhancing vessel growth and branching (96). Likewise, EC-derived small EVs contain miRNAs such as miR-214, which promotes angiogenesis both in vitro and in vivo by suppressing cell cycle arrest (97). miR-126-3p has also been identified as an important player in endothelial repair cell migration, proliferation and regeneration by inhibiting EVH1 domain-containing protein1 (SPRED1) and promoting Ras/MAPK signaling (98). Furthermore, small EVs derived from human EPCs induce angiogenesis by the horizontal mRNA transfer of genes such as BCL-XL, CFL1, CTNNB1, EDF1, MAPKAPK2, and eNOS (99) (Figure 3).
MSCs are another source of small EVs with pro-angiogenic potential (Figure 3). MSC-derived small EVs induce the formation of new vessels in the infarcted myocardium, inhibit cardiac remodeling and preserve the ejection function of the damaged heart (100, 101). In addition to this, a comparative study of small EVs and MSCs therapy revealed that small EVs injection was more effective and improved cardiac fibrosis, inflammation and cardiac performance (102). Regarding angiogenesis, in vivo studies have shown that small EVs from MSCs overexpressing Akt enhance neovascularization with a subsequent improvement of LVEF (103). Similar results were obtained with MSCs overexpressing HIF-1α. Indeed, these small EVs contain Jagged1, and induce angiogenesis in ECs both in vitro and in vivo by activating Notch signaling (104). In a renal ischemia-reperfusion model, treatment with umbilical cord MSC-derived small EVs improved capillary density by inducing VEGF elevation in a HIF1α independent manner (105). Small EVs derived from MSCs also transfer miRNAs as miR-125a to ECs, thus promoting angiogenesis (106, 107).
Other adult stem cells, as CSC, secrete small EVs enriched with cytokines and chemokines (TCA-3, SDF1), vascular growth factors (VEGF, erythropoietin, bFGF, osteopontin, SCF) and cardiac differentiation factors (Activin A, Dkk homolog-1, TGF-β) (108). In addition, CSCs derived small EVs analysis revealed high levels of miR-210, miR132 and miR-146a-3p that enhance anti-apoptotic and pro-angiogenic activity (Figure 3). This seems to be mediated by activation of their downstream targets such as ephrin A3, PTP1b and RasGaP-p120, that eventually leads to augmented cardiac function (109). Similarly, a study showed that bone marrow-derived CD34+ cells secreted small EVs with pro-angiogenic activity in ischemic cardiac tissue, mainly due to the delivery of pro-angiogenic microRNAs such as 130a and 126 (110).
Immune system-derived EVs originate from lymphocytes, monocytes/macrophages or neutrophils, and their selective cargo and surface biomolecules modulate neovascularization by different mechanisms. Several studies have been conducted to assess the effect of immune system-derived EVs in angiogenesis, however the specific role of small EVs in this process has not been exhaustively addressed. Lymphocytes constitute an important source of EVs that may exert pro- or anti-angiogenic effects depending on stimuli involved in their production (Figure 3). When lymphocytes undergo activation before apoptosis, they release proangiogenic microvesicles. Thus, in vitro experiments show that treatment of T cells with phytohemagglutinin, phorbol ester and actinomycin D induces production of microvesicles, so-called lymphocyte microparticles (LMPs), which lead to the formation of capillary-like structures. This pro-angiogenic effect is mediated by increasing cell adhesion and promoting expression of ICAM-1, Rho A and VEGF, through activation of Sonic Hedgehog (Shh) pathway (111). In a mouse model of ischemia-reperfusion, microvesicles from T lymphocytes carrying Shh are involved in restoration of endothelial function by NO production (112). Similarly, in a model of hindlimb ischemia, EVs harboring Shh promote post ischemic neovascularization by enhancing vascular density (113). HUVECs can internalize EVs from T lymphocytes, a process that modifies HUVEC gene expression profile, including modulation of some angiogenic-related genes as netrin-4, and increases tube length (114). Conversely, lymphocytes treated with actinomycin D without previous activation produce anti-angiogenic LMPs, through a mechanism not well-defined yet. Thus, these microvesicles suppress neovessel formation in rat choroidal explants by increasing mRNA levels of pigment epithelium-derived factor (PEDF), nerve growth factor (NGF) and p75 neurotropin receptor (p75NRP) (115). Likewise, in a model of corneal neovascularization, LMPs reduce angiogenesis and inhibit human ECs proliferation, survival and migration by inducing reactive ROS and interfering with VEGFR-2 pathway (116). Moreover, LMPs produced after treatment with actinomycin D alter the expression of pro-angiogenic factors in macrophages through CD36-mediated signal transduction pathways (117).
Microvesicles derived from monocytes and macrophages participate in angiogenesis regulation as well (Figure 3). Transfer of miR-150 to ECs by monocyte-derived microvesicles enhances neovascularization in in vivo and in vitro models by downregulating c-Myb (118) and enhancing ECs migration (119). In addition, CD40 Ligand+ (CD40L) microvesicles originated from atherosclerotic lesions stimulate EC proliferation and neovessel formation in vivo. Most of these microparticles express CD14 on their surface, suggesting that they are mainly of monocytic origin (120). However, the role of myeloid-derived small EVs in neovascularization is controversial. In an in vitro assay with HUVECs, small EVs derived from monocytes adhere to these ECs stimulating them and enhancing tube formation, but also inducing apoptosis (121). In another study, small EVs derived from macrophages modulate integrin-β1 trafficking by enhancing its internalization and degradation in ECs, thus impairing their migration (122). Furthermore, similar to LMPs after actinomycin D treatment, monocyte-derived EVs inhibit tube formation and migration in microvascular ECs through CD36 signaling. However, this inhibitory effect decreases when small EVs are isolated by differential centrifugation from other EVs, since small EVs-containing fraction inhibits EC migration to a lesser extent than small EVs-depleted EVs (123). In addition to monocyte/macrophage-derived EVs, EVs from DCs can also mediate neovascularization. DC-derived small EVs harbor active MMP-9 (124), a pro-angiogenic factor that acts in ECM remodeling to allow neovessel formation.
Finally, neutrophils also secrete small EVs in a stimulus-dependent manner. When stimulated with N-Formylmethionyl-leucyl-phenylalanine (fMLP), they secrete small EVs with anti-inflammatory potential, which up-regulate EC-protective factors as ANGPTL4 or CD55. On the contrary, neutrophils pre-incubated with a HUVEC monolayer before addition of fMLP produce a population of small EVs that display a pro-inflammatory, anti-apoptotic phenotype (125).
This experimental evidence highlights that leukocyte-derived EVs can generate both pro- and anti-angiogenic effects, which may be due to different cellular origin or microenvironment. Moreover, diverse EVs subpopulations, such as small EVs and other microvesicles, seem to play distinct roles in promoting or inhibiting angiogenesis. Such differences may rely either on diverse cargoes or on specific expression patterns of membrane molecules in these EVs subsets. A detailed study of mechanisms that regulate these functional differences may help to design therapies that profit from small EVs-mediated paracrine effect to foster tissue repair and angiogenesis in ischemic cardiac tissue.
Current therapies of MI mainly focus on macrovascular reperfusion of the infarcted myocardium with thrombolytic therapy, primary percutaneous coronary intervention and coronary artery bypass grafting. Early reperfusion of the culprit artery has proven to be the most successful therapy in reducing mortality, being the gold standard (126). Indeed, early mortality after MI has decreased over the last decades, from 20% in the late 80's to approximately 5–7% in contemporary series (127, 128). However, delayed HF secondary to inadequate cardiac remodeling is still a challenge in therapeutic management of MI. Angiogenesis plays an essential role in cardiac repair and regeneration after ischemic heart disease; therefore, therapeutic angiogenesis has been considered as a promising therapy for patients suffering MI. Over the past decades, a variety of strategies to promote therapeutic angiogenesis has been evaluated. However, there is still a need to come up with a balanced and targeted therapy that ensures an optimal restoration of blood supply into the damaged myocardium in order to enhance a correct tissue healing and prevent HF.
Both preclinical models (129, 130) and early human clinical trials (131) demonstrated promising outcomes with strategies consisting of administration of proangiogenic factors as VEGF; however larger clinical trials using individual proangiogenic factors showed mixed results (132, 133), thus indicating that delivery of a single growth factor was not sufficient to support a complete and mature angiogenesis in these patients.
After major failure of proangiogenic growth factors to show any benefit in clinical trials, cell transplantation quickly became the next frontier therapy for promoting angiogenesis and cardiac regeneration (134, 135). Several stem cells have been used as cellular sources to produce new cardiac cells such as endothelial mesenchymal stem cells (EMCs), MSCs, and cardiac progenitor cells (CPCs), showing safety and promising results (136, 137). Several clinical trials have been launched to probe therapeutic utility of progenitor cells from different sources in MI treatment, with different outcomes (138–143). However, these results could not be fully assigned to the exogenous cells, as it seems that in mouse models nearly 90% of newly formed cardiomyocytes and ECs are of endogenous origin and do not come from the exogenous cells (144). Thus, paracrine factors secreted by cell transplantation appear to mediate endogenous repair, through modification of functions of neighboring cells. In this line of evidence, a recent study demonstrated that intracoronary injection of cardiac-derived stem cells (CDC)-secreted small EVs reduced the number and modified the polarization state of CD68+ macrophages in the infarcted myocardium, leading to increased expression of anti-inflammatory genes such as Arg1, IL4ra, Tgfb1, and Vegfa. In this study, RNA-Seq analysis of macrophages primed with CDC small EVs showed enhanced levels of miR-181b, which targets protein kinase C δ (145). Another study showed that the most abundant RNA species in CDC small EVs was a Y RNA fragment (EV-YF1), which was transferred to macrophages. This transfer induced transcription and secretion of the anti-inflammatory IL-10, thus promoting protection of cardiomyocytes from oxidative stress and reducing infarct size (146).
These findings led to the idea that small EVs secreted by different cell populations could become a potential therapeutic tool to improve cardiac function after MI through tissue remodeling and more precisely by promoting angiogenesis in the damaged heart.
Small EVs have a biological signature that mimics the phenotype of the cells that produced them (15). Hence, there is growing interest in small EVs and their application in the clinical context. Indeed, they play an important role as transporters of paracrine factors in angiogenesis, immune regulation, and tissue regeneration (147). This has led to the notion that small EVs could become a delivering system for regenerative therapeutic factors. Thus, administration of CDC small EVs has shown the same benefits in infarcted hearts of animal models with enhancement of angiogenesis and promotion of cardiomyocytes survival and proliferation (148). Moreover, despite concerns regarding standarization of isolation procedures, small EVs offer clear advantages over cell-based therapies (summarized in Table 2). Hence, these vesicles have small size and low immunogenicity, are nontoxic, permeable to physiological barriers and stable in blood (149–151). Indeed, they can be preloaded with specific therapeutic agents as well as protected from degradation and deactivation. Protein changes on their surface ensure affinity to specific cell types, thus enhancing efficient and selected target cell communication (152). Cell modification is generally achieved either by hijacking biosynthesis to favor the production of specific endogenous material or by delivering exogenous species to the cytoplasmic membrane (158). Main current small EVs engineering strategies are summarized in Table 3.

Table 2. Main advantages of Small EVs over cell-based therapies.
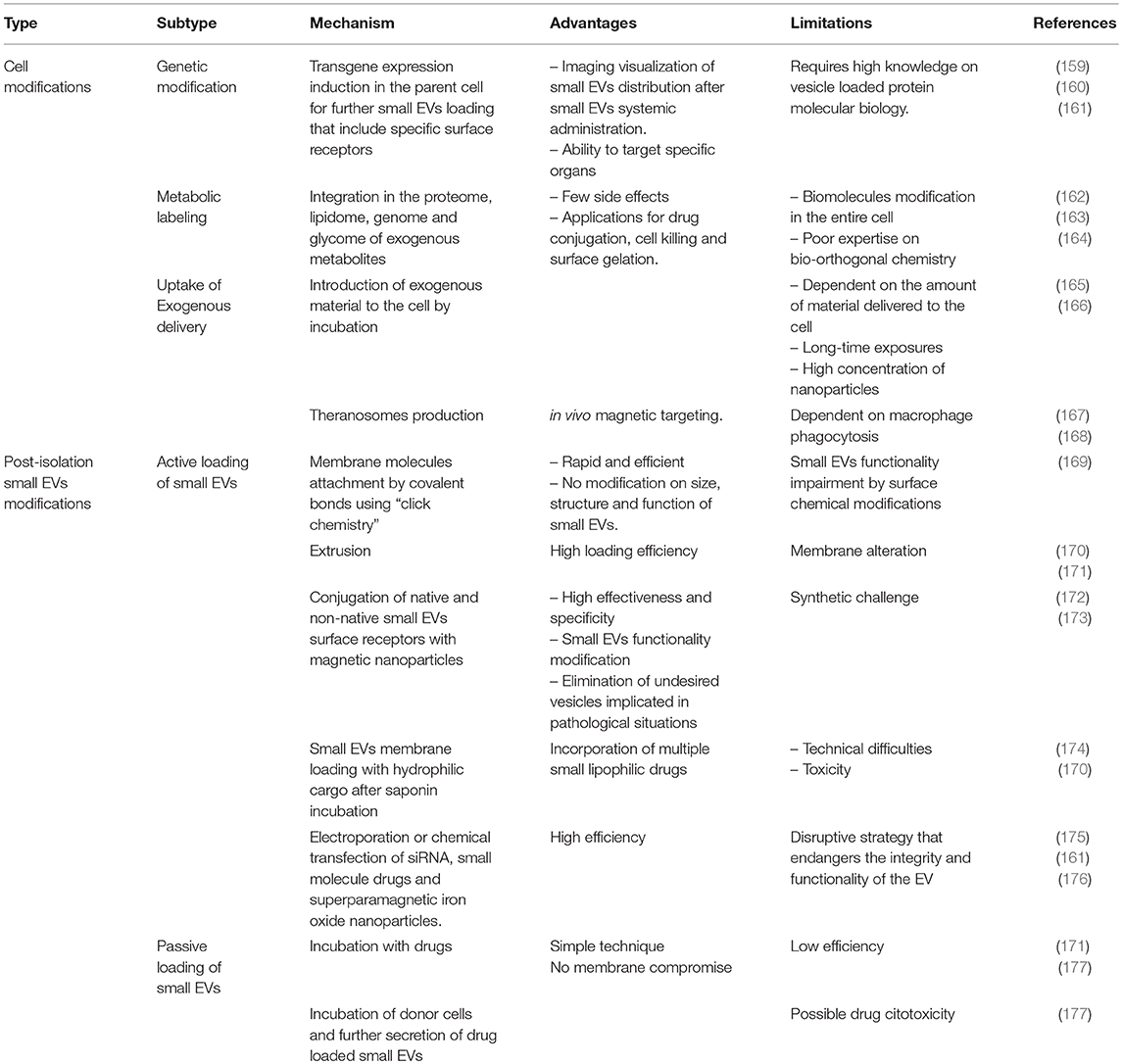
Table 3. Overview of main strategies for small EVs engineering.
Different strategies have been developed to enhance cardiac capture and retention of small EVs: Systemic administration of small EVs conjugated with a synthetic cardiac homing peptide using a DOPE-NHS linker, shows reduced fibrosis and scar size as well as increased cellular proliferation and angiogenesis (178). Another attempt to target small EVs to the heart has been performed using a fusion protein comprising synthetic cardiac-targeting peptide (CTP)-Lamp2b, which is expressed in small EVs membrane. This construct is expressed in small EVs-producing cells through transfection, and stabilized in small EVs membrane by the attachment of glycosylation sequences. Delivery of these modified small EVs to the heart has shown significant but mild results (179).
Small EVs derived from immune system have been also engineered both endogenously and exogenously through modifications aimed at improving their therapeutic potential, usually modifying their cargo. Small EVs derived from EL-4 cells (mouse lymphoma cell line) were passively loaded with anti-inflammatory curcumin, which led to increased drug stability and delivery in a lipopolysaccharide (LPS)-induced septic shock mouse model (174) and a LPS-induced brain inflammation model (180). Additionally, siRNA against α-synuclein gene, which generates pathological aggregates in Parkinson's disease, was loaded by electroporation into murine DCs small EVs; these small EVs specifically target brain by expressing rabies virus glycoprotein peptide (RVG)-Lamp2, where a downregulation of α-synuclein was achieved (181, 182). Likewise, small EVs derived from M12.4 murine B cell line were loaded with miR-155 by electroporation and delivered to macrophages, thus suppressing LPS-induced TNF-α production (183).
Both endogenous small EVs function in tissue repair and angiogenesis, and the feasibility of small EVs modification to optimize specific cargo loading and tissue targeting, highlight the potential of these EVs in proangiogenic MI therapy. However, further definition of the appropriate cellular source and characterization of specific functional characteristics of these vesicles is needed before their clinical application. Thus, pleiotropic effects on angiogenesis of small EVs derived from different types of immune cells with diverse activation states, may represent a challenge for optimizing the production of a suitable subset of vesicles. Similarly, most preclinical studies have been conducted using cell lines; and a clear advantage in terms of therapeutic efficacy, biosafety, and clinical application of small EVs derived from cell lines over those isolated from more physiological sources, has not been established yet.
When myocardial ischemia occurs, a tissue remodeling process takes place that involves sequential interdependent stages. These include an inflammatory phase and subsequent proliferative and reparative phases, where inflammation terminates and angiogenesis is induced. Immune system components are major regulators of cardiac tissue remodeling, which is especially remarkable in the case of angiogenesis. Some of these regulatory functions may be carried out through the release of small EVs loaded with bioactive molecules that act in a paracrine manner in adjacent cells. Likewise, benefit associated to cell therapy is in part mediated by paracrine factors including small EVs. Indeed, small EVs features demonstrate a significant advantage over cell-based therapies due to their stable physiology, their low immunogenicity and safety profile, their storage and application characteristics, and their ability to deliver therapeutic cargoes selectively to the damaged cardiac cells. These characteristics unveil the unique properties of small EVs as tools for the development of selective cell-free therapies to promote angiogenesis during tissue regeneration in the infarcted myocardium. Further investigation is needed to identify the optimum immune cell source and isolation conditions required to obtain small EVs adequate for successful clinical application in myocardial remodeling.
SS-A, AA-S, FS-M, and AA had scientific discussion for this work and wrote the manuscript.
This work was supported by grants to AA-S (FIS PI15/01491) and to FS-M (grants SAF2014-55579-R and SAF2017-82886-R to FS-M), BIOIMID PIE13/041 and CIBER CARDIOVASCULAR from the Instituto de Salud Carlos III (Fondo de Investigación Sanitaria del Instituto de Salud Carlos III with co-funding from the Fondo Europeo de Desarrollo Regional; FEDER), Programa de Actividades en Biomedicina de la Comunidad de Madrid-B2017/BMD-3671-INFLAMUNE to FS-M, and ERC-2011-AdG294340-GENTRIS to FS-M, and Fundació La Marató TV3 (20152330 31). The Centro Nacional de Investigaciones Cardiovasculares (CNIC) is supported by the Spanish Ministry of Economy and Competitiveness (MINECO) and the Pro-CNIC Foundation and is a Severo Ochoa Center of Excellence (MINECO award SEV-2015-0505).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Reinke JM, Sorg H. Wound repair and regeneration. Eur Surg Res. (2012) 49:35–43. doi: 10.1159/000339613
2. Egawa N, Lok J, Washida K, Arai K. Mechanisms of axonal damage and repair after central nervous system injury. Transl Stroke Res. (2017) 8:14–21. doi: 10.1007/s12975-016-0495-1
3. Majidinia M, Sadeghpour A, Yousefi B. The roles of signaling pathways in bone repair and regeneration. J Cell Physiol. (2018) 233:2937–48. doi: 10.1002/jcp.26042
4. Frangogiannis NG. Inflammation in cardiac injury, repair and regeneration. Curr Opin Cardiol. (2015) 30:240–5. doi: 10.1097/HCO.0000000000000158
5. Moran AE, Tzong KY, Forouzanfar MH, Rothy GA, Mensah GA, Ezzati M, et al. Variations in ischemic heart disease burden by age, country, and income: the Global Burden of Diseases, Injuries, and Risk Factors 2010 study. Global Heart (2014) 9:91–9. doi: 10.1016/j.gheart.2013.12.007
6. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circul Res. (2016) 119:91–112. doi: 10.1161/CIRCRESAHA.116.303577
7. Cochain C, Channon KM, Silvestre JS. Angiogenesis in the infarcted myocardium. Antioxidants Redox Signal. (2013) 18:1100–13. doi: 10.1089/ars.2012.4849
8. van der Laan AM, Piek JJ, van Royen N. Targeting angiogenesis to restore the microcirculation after reperfused MI. Nat Rev Cardiol. (2009) 6:515–23. doi: 10.1038/nrcardio.2009.103
9. Shah AM, Mann DL. In search of new therapeutic targets and strategies for heart failure: recent advances in basic science. Lancet (2011) 378:704–12. doi: 10.1016/S0140-6736(11)60894-5
10. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. (2007) 357:1121–35. doi: 10.1056/NEJMra071667
11. van der Pol E, Boing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. (2012) 64:676–705. doi: 10.1124/pr.112.005983
12. Mittelbrunn M, Sanchez-Madrid F. Intercellular communication: diverse structures for exchange of genetic information. Nat Rev Mol Cell Biol. (2012) 13:328–35. doi: 10.1038/nrm3335
13. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. (2018) 19:213–28. doi: 10.1038/nrm.2017.125
14. Villarroya-Beltri C, Baixauli F, Gutierrez-Vazquez C, Sanchez-Madrid F, Mittelbrunn M. Sorting it out: regulation of exosome loading. Semin Cancer Biol. (2014) 28:3–13. doi: 10.1016/j.semcancer.2014.04.009
15. Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles (2015) 4:27066. doi: 10.3402/jev.v4.27066
16. Hayashi T, Lombaert IM, Hauser BR, Patel VN, Hoffman MP. Exosomal MicroRNA transport from salivary mesenchyme regulates epithelial progenitor expansion during organogenesis. Dev Cell (2017) 40:95–103. doi: 10.1016/j.devcel.2016.12.001
17. Xu B, Zhang Y, Du XF, Li J, Zi HX, Bu JW, et al. Neurons secrete miR−132-containing exosomes to regulate brain vascular integrity. Cell Res. (2017) 27:882–97. doi: 10.1038/cr.2017.62
18. Olson JK, Luong N. Exosomes secreted from virus- infected microglia can activate an inflammatory response in the central nervous system. J Immunol. (2017) 198(1 Supplement):158.5.
19. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. (2011) 11:519–31. doi: 10.1038/nri3024
20. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. (2008) 8:349–61. doi: 10.1038/nri2294
21. Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. (2010) 10:427–39. doi: 10.1038/nri2779
22. Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. (2002) 53:31–47. doi: 10.1016/S0008-6363(01)00434-5
23. Soehnlein O, Zernecke A, Weber C. Neutrophils launch monocyte extravasation by release of granule proteins. Thromb Haemost. (2009) 102:198–205. doi: 10.1160/TH08-11-0720
24. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science (2009) 325:612–6. doi: 10.1126/science.1175202
25. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. (2007) 204:3037–47. doi: 10.1084/jem.20070885
26. Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circul Res. (2005) 96:881–9. doi: 10.1161/01.RES.0000163017.13772.3a
27. Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation (2010) 121:2437–45. doi: 10.1161/CIRCULATIONAHA.109.916346
28. Geering B, Stoeckle C, Conus S, Simon HU. Living and dying for inflammation: neutrophils, eosinophils, basophils. Trends Immunol. (2013) 34:398–409. doi: 10.1016/j.it.2013.04.002
29. Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J. (2017) 38:187–97. doi: 10.1093/eurheartj/ehw002
30. Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circul Res. (2002) 91:988–98. doi: 10.1161/01.RES.0000043825.01705.1B
31. Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circul Res. (2014) 114:266–82. doi: 10.1161/CIRCRESAHA.113.301720
32. Jurgensen JS, Rosenberger C, Wiesener MS, Warnecke C, Horstrup JH, Grafe M, et al. Persistent induction of HIF-1alpha and−2alpha in cardiomyocytes and stromal cells of ischemic myocardium. FASEB J. (2004) 18:1415–7. doi: 10.1096/fj.04-1605fje
33. Dai Y, Xu M, Wang Y, Pasha Z, Li T, Ashraf M. HIF-1α induced-VEGF overexpression in bone marrow stem cells protects cardiomyocytes against ischemia. J Mol Cell Cardiol. (2007) 42:1036–44. doi: 10.1016/j.yjmcc.2007.04.001
34. Kaidi A, Qualtrough D, Williams AC, Paraskeva C. Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Res. (2006) 66:6683–91. doi: 10.1158/0008-5472.CAN-06-0425
35. Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. (2004) 10:858. doi: 10.1038/nm1075
36. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circul Res. (2012) 110:159–73. doi: 10.1161/CIRCRESAHA.111.243162
37. Li J, Brown LF, Hibberd MG, Grossman JD, Morgan JP, Simons M. VEGF, flk-1, and flt-1 expression in a rat myocardial infarction model of angiogenesis. Am J Physiol. (1996) 270(5 Pt 2):H1803–11. doi: 10.1152/ajpheart.1996.270.5.H1803
38. Zhang F, Tang Z, Hou X, Lennartsson J, Li Y, Koch AW, et al. VEGF-B is dispensable for blood vessel growth but critical for their survival, and VEGF-B targeting inhibits pathological angiogenesis. Proc Natl Acad Sci USA. (2009) 106:6152–7. doi: 10.1073/pnas.0813061106
39. Zhao W, Zhao T, Huang V, Chen Y, Ahokas RA, Sun Y. Platelet-derived growth factor involvement in myocardial remodeling following infarction. J Mol Cell Cardiol. (2011) 51:830–8. doi: 10.1016/j.yjmcc.2011.06.023
40. Fadini GP, Losordo D, Dimmeler S. Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circul Res. (2012) 110:624–37. doi: 10.1161/CIRCRESAHA.111.243386
41. Zhang M, Malik AB, Rehman J. Endothelial progenitor cells and vascular repair. Curr Opin Hematol. (2014) 21:224–8. doi: 10.1097/MOH.0000000000000041
42. Li C, Matsushita S, Li Z, Guan J, Amano A. c-kit positive cardiac outgrowth cells demonstrate better ability for cardiac recovery against ischemic myopathy. J Stem Cell Res Ther. (2017) 7. doi: 10.4172/2157-7633.1000402
43. Bao L, Meng Q, Li Y, Deng S, Yu Z, Liu Z, et al. C-Kit positive cardiac stem cells and bone marrow-derived mesenchymal stem cells synergistically enhance angiogenesis and improve cardiac function after myocardial infarction in a paracrine manner. J Cardiac Fail. (2017) 23:403–15. doi: 10.1016/j.cardfail.2017.03.002
44. Kumar AH, Caplice NM. Clinical potential of adult vascular progenitor cells. Arterioscl Thromb Vasc Biol. (2010) 30:1080–7. doi: 10.1161/ATVBAHA.109.198895
45. Lemcke H, Voronina N, Steinhoff G, David R. Recent progress in stem cell modification for cardiac regeneration. Stem Cell Int. (2018) 2018:1909346. doi: 10.1155/2018/1909346
46. Seo HH, Lee SY, Lee CY, Kim R, Kim P, Oh S, et al. Exogenous miRNA−146a enhances the therapeutic efficacy of human mesenchymal stem cells by increasing vascular endothelial growth factor secretion in the ischemia/reperfusion-injured heart. J Vasc Res. (2017) 54:100–8. doi: 10.1159/000461596
47. Saijo Y, Tanaka M, Miki M, Usui K, Suzuki T, Maemondo M, et al. Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: in vivo analysis of tumor-stromal interaction. J Immunol. (2002) 169:469–75. doi: 10.4049/jimmunol.169.1.469
48. Bae J, Park D, Lee YS, Jeoung D. Interleukin-2 promotes angiogenesis by activation of Akt and increase of ROS. J Microbiol Biotechnol. (2008) 18:377–82.
49. Gertz K, Kronenberg G, Kalin RE, Baldinger T, Werner C, Balkaya M, et al. Essential role of interleukin-6 in post-stroke angiogenesis. Brain (2012) 135(Pt 6):1964–80. doi: 10.1093/brain/aws075
50. Xie Q, Sun Z, Chen M, Zhong Q, Yang T, Yi J. IL-8 up-regulates proliferative angiogenesis in ischemic myocardium in rabbits through phosphorylation of Akt/GSK-3beta(ser9) dependent pathways. Int J Clin Exp Med. (2015) 8:12498–508.
51. Stabile E, Kinnaird T, la Sala A, Hanson SK, Watkins C, Campia U, et al. CD8+ T lymphocytes regulate the arteriogenic response to ischemia by infiltrating the site of collateral vessel development and recruiting CD4+ mononuclear cells through the expression of interleukin−16. Circulation (2006) 113:118–24. doi: 10.1161/CIRCULATIONAHA.105.576702
52. Pan B, Shen J, Cao J, Zhou Y, Shang L, Jin S, et al. Interleukin−17 promotes angiogenesis by stimulating VEGF production of cancer cells via the STAT3/GIV signaling pathway in non-small-cell lung cancer. Sci Rep. (2015) 5:16053. doi: 10.1038/srep16053
53. Richards J, Gabunia K, Kelemen SE, Kako F, Choi ET, Autieri MV. Interleukin−19 increases angiogenesis in ischemic hind limbs by direct effects on both endothelial cells and macrophage polarization. J Mol Cell Cardiol. (2015) 79:21–31. doi: 10.1016/j.yjmcc.2014.11.002
54. Yao L, Sgadari C, Furuke K, Bloom ET, Teruya-Feldstein J, Tosato G. Contribution of natural killer cells to inhibition of angiogenesis by interleukin−12. Blood (1999) 93:1612–21.
55. Deng J, Liu X, Rong L, Ni C, Li X, Yang W, et al. IFNgamma-responsiveness of endothelial cells leads to efficient angiostasis in tumours involving down-regulation of Dll4. J Pathol. (2014) 233:170–82. doi: 10.1002/path.4340
56. Salvucci O, Basik M, Yao L, Bianchi R, Tosato G. Evidence for the involvement of SDF-1 and CXCR4 in the disruption of endothelial cell-branching morphogenesis and angiogenesis by TNF-alpha and IFN-gamma. J Leukocyte Biol. (2004) 76:217–26. doi: 10.1189/jlb.1203609
57. Low-Marchelli JM, Ardi VC, Vizcarra EA, van Rooijen N, Quigley JP, Yang J. Twist1 induces CCL2 and recruits macrophages to promote angiogenesis. Cancer Res. (2013) 73:662–71. doi: 10.1158/0008-5472.CAN-12-0653
58. Miyake M, Goodison S, Urquidi V, Gomes Giacoia E, Rosser CJ. Expression of CXCL1 in human endothelial cells induces angiogenesis through the CXCR2 receptor and the ERK1/2 and EGF pathways. Lab Invest. (2013) 93:768–78. doi: 10.1038/labinvest.2013.71
59. Gijsbers K, Gouwy M, Struyf S, Wuyts A, Proost P, Opdenakker G, et al. GCP-2/CXCL6 synergizes with other endothelial cell-derived chemokines in neutrophil mobilization and is associated with angiogenesis in gastrointestinal tumors.Exp Cell Res. (2005) 303:331–42. doi: 10.1016/j.yexcr.2004.09.027
60. Li B, Bai W, Sun P, Zhou B, Hu B, Ying J. The effect of CXCL12 on endothelial progenitor cells: potential target for angiogenesis in intracerebral hemorrhage. J Interferon Cytokine Res. (2015) 35:23–31. doi: 10.1089/jir.2014.0004
61. Park Y, Lee J, Kwak JY, Noh K, Yim E, Kim HK, et al. Fractalkine induces angiogenic potential in CX3CR1-expressing monocytes. J Leukocyte Biol. (2018) 103:53–66. doi: 10.1189/jlb.1A0117-002RR
62. Aidoudi S, Bujakowska K, Kieffer N, Bikfalvi A. The CXC-chemokine CXCL4 interacts with integrins implicated in angiogenesis. PLoS ONE (2008) 3:e2657. doi: 10.1371/journal.pone.0002657
63. Huang B, Wang W, Li Q, Wang Z, Yan B, Zhang Z, et al. Osteoblasts secrete Cxcl9 to regulate angiogenesis in bone. Nat Commun. (2016) 7:13885. doi: 10.1038/ncomms13885
64. Campanella GS, Colvin RA, Luster AD. CXCL10 can inhibit endothelial cell proliferation independently of CXCR3. PLoS ONE (2010) 5:e12700. doi: 10.1371/journal.pone.0012700
65. Shellenberger TD, Wang M, Gujrati M, Jayakumar A, Strieter RM, Burdick MD, et al. BRAK/CXCL14 is a potent inhibitor of angiogenesis and a chemotactic factor for immature dendritic cells. Cancer Res. (2004) 64:8262–70. doi: 10.1158/0008-5472.CAN-04-2056
66. Rupertus K, Sinistra J, Scheuer C, Nickels RM, Schilling MK, Menger MD, et al. Interaction of the chemokines I-TAC (CXCL11) and SDF-1 (CXCL12) in the regulation of tumor angiogenesis of colorectal cancer. Clin Exp Met. (2014) 31:447–59. doi: 10.1007/s10585-014-9639-4
67. Szade A, Grochot-Przeczek A, Florczyk U, Jozkowicz A, Dulak J. Cellular and molecular mechanisms of inflammation-induced angiogenesis. IUBMB Life (2015) 67:145–59. doi: 10.1002/iub.1358
68. David Dong ZM, Aplin AC, Nicosia RF. Regulation of angiogenesis by macrophages, dendritic cells, and circulating myelomonocytic cells. Curr Pharmaceut Design (2009) 15:365–79. doi: 10.2174/138161209787315783
69. Capoccia BJ, Gregory AD, Link DC. Recruitment of the inflammatory subset of monocytes to sites of ischemia induces angiogenesis in a monocyte chemoattractant protein-1-dependent fashion. J Leukocyte Biol. (2008) 84:760–8. doi: 10.1189/jlb.1107756
70. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science (2007) 317:666–70. doi: 10.1126/science.1142883
71. Meda C, Molla F, De Pizzol M, Regano D, Maione F, Capano S, et al. Semaphorin 4A exerts a proangiogenic effect by enhancing vascular endothelial growth factor-A expression in macrophages. J Immunol. (2012) 188:4081–92. doi: 10.4049/jimmunol.1101435
72. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis. (2014) 17:109–18. doi: 10.1007/s10456-013-9381-6
73. Johnson C, Sung HJ, Lessner SM, Fini ME, Galis ZS. Matrix metalloproteinase-9 is required for adequate angiogenic revascularization of ischemic tissues: potential role in capillary branching. Circul Res. (2004) 94:262–8. doi: 10.1161/01.RES.0000111527.42357.62
74. Deryugina EI, Zajac E, Juncker-Jensen A, Kupriyanova TA, Welter L, Quigley JP. Tissue-infiltrating neutrophils constitute the major in vivo source of angiogenesis-inducing MMP-9 in the tumor microenvironment. Neoplasia (2014) 16:771–88. doi: 10.1016/j.neo.2014.08.013
75. Zhu Y, Zhang L, Lu Q, Gao Y, Cai Y, Sui A, et al. Identification of different macrophage subpopulations with distinct activities in a mouse model of oxygen-induced retinopathy. Int J Mol Med. (2017) 40:281–92. doi: 10.3892/ijmm.2017.3022
76. Fantin A, Vieira JM, Gestri G, Denti L, Schwarz Q, Prykhozhij S, et al. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood (2010) 116:829–40. doi: 10.1182/blood-2009-12-257832
77. Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. (2010) 70:5270–80. doi: 10.1158/0008-5472.CAN-10-0012
78. Abdel Malik R, Zippel N, Fromel T, Heidler J, Zukunft S, Walzog B, et al. AMP-activated protein kinase alpha2 in neutrophils regulates vascular repair via hypoxia-inducible factor-1alpha and a network of proteins affecting metabolism and apoptosis. Circul Res. (2017) 120:99–109. doi: 10.1161/CIRCRESAHA.116.309937
79. Ardi VC, Van den Steen PE, Opdenakker G, Schweighofer B, Deryugina EI, Quigley JP. Neutrophil MMP-9 proenzyme, unencumbered by TIMP-1, undergoes efficient activation in vivo and catalytically induces angiogenesis via a basic fibroblast growth factor (FGF-2)/FGFR-2 pathway. J Biol Chem. (2009) 284:25854–66. doi: 10.1074/jbc.M109.033472
80. Christoffersson G, Vagesjo E, Vandooren J, Liden M, Massena S, Reinert RB, et al. VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood (2012) 120:4653–62. doi: 10.1182/blood-2012-04-421040
81. Massena S, Christoffersson G, Vagesjo E, Seignez C, Gustafsson K, Binet F, et al. Identification and characterization of VEGF-A-responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood (2015) 126:2016–26. doi: 10.1182/blood-2015-03-631572
82. Scapini P, Morini M, Tecchio C, Minghelli S, Di Carlo E, Tanghetti E, et al. CXCL1/macrophage inflammatory protein-2-induced angiogenesis in vivo is mediated by neutrophil-derived vascular endothelial growth factor-A. J Immunol. (2004) 172:5034–40. doi: 10.4049/jimmunol.172.8.5034
83. Christoffersson G, Lomei J, O'Callaghan P, Kreuger J, Engblom S, Phillipson M. Vascular sprouts induce local attraction of proangiogenic neutrophils. J Leukocyte Biol. (2017) 102:741–51. doi: 10.1189/jlb.1MA0117-018R
84. Scapini P, Nesi L, Morini M, Tanghetti E, Belleri M, Noonan D, et al. Generation of biologically active angiostatin kringle 1-3 by activated human neutrophils. J Immunol. (2002) 168:5798–804. doi: 10.4049/jimmunol.168.11.5798
85. Bekkers SC, Yazdani SK, Virmani R, Waltenberger J. Microvascular obstruction: underlying pathophysiology and clinical diagnosis. J Am College Cardiol. (2010) 55:1649–60. doi: 10.1016/j.jacc.2009.12.037
86. Yang C, Lee H, Pal S, Jove V, Deng J, Zhang W, et al. B cells promote tumor progression via STAT3 regulated-angiogenesis. PLoS ONE (2013) 8:e64159. doi: 10.1371/journal.pone.0064159
87. Stabile E, Burnett MS, Watkins C, Kinnaird T, Bachis A, la Sala A, et al. Impaired arteriogenic response to acute hindlimb ischemia in CD4-knockout mice. Circulation (2003) 108:205–10. doi: 10.1161/01.CIR.0000079225.50817.71
88. Strasly M, Cavallo F, Geuna M, Mitola S, Colombo MP, Forni G, et al. IL−12 inhibition of endothelial cell functions and angiogenesis depends on lymphocyte-endothelial cell cross-talk. J Immunol. (2001) 166:3890–9. doi: 10.4049/jimmunol.166.6.3890
89. Mitola S, Strasly M, Prato M, Ghia P, Bussolino F. IL−12 regulates an endothelial cell-lymphocyte network: effect on metalloproteinase-9 production. J Immunol. (2003) 171:3725–33. doi: 10.4049/jimmunol.171.7.3725
90. Zhong Q, Jenkins J, Moldobaeva A, D'Alessio F, Wagner EM. Effector T Cells and Ischemia-Induced Systemic Angiogenesis in the Lung. Am J Respir Cell Mol Biol. (2016) 54:394–401. doi: 10.1165/rcmb.2015-0087OC
91. Wakita D, Sumida K, Iwakura Y, Nishikawa H, Ohkuri T, Chamoto K, et al. Tumor-infiltrating IL−17-producing gammadelta T cells support the progression of tumor by promoting angiogenesis. Eur J Immunol. (2010) 40:1927–37. doi: 10.1002/eji.200940157
92. Deliyanti D, Talia DM, Zhu T, Maxwell MJ, Agrotis A, Jerome JR, et al. Foxp3(+) Tregs are recruited to the retina to repair pathological angiogenesis. Nat Commun. (2017) 8:748. doi: 10.1038/s41467-017-00751-w
93. van Weel V, Toes RE, Seghers L, Deckers MM, de Vries MR, Eilers PH, et al. Natural killer cells and CD4+ T-cells modulate collateral artery development. Arterioscl Thrombosis Vasc Biol. (2007) 27:2310–8. doi: 10.1161/ATVBAHA.107.151407
94. Lu Y, Gu X, Chen L, Yao Z, Song J, Niu X, et al. Interferon-gamma produced by tumor-infiltrating NK cells and CD4+ T cells downregulates TNFSF15 expression in vascular endothelial cells. Angiogenesis (2014) 17:529–40. doi: 10.1007/s10456-013-9397-y
95. Taraboletti G, D'Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am J Pathol. (2002) 160:673–80. doi: 10.1016/S0002-9440(10)64887-0
96. Sheldon H, Heikamp E, Turley H, Dragovic R, Thomas P, Oon CE, et al. New mechanism for Notch signaling to endothelium at a distance by Delta-like 4 incorporation into exosomes. Blood (2010) 116:2385–94. doi: 10.1182/blood-2009-08-239228
97. van Balkom BW, de Jong OG, Smits M, Brummelman J, den Ouden K, de Bree PM, et al. Endothelial cells require miR−214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood (2013) 121:3997–4006, S1–15. doi: 10.1182/blood-2013-02-478925
98. Jansen F, Yang X, Hoelscher M, Cattelan A, Schmitz T, Proebsting S, et al. Endothelial microparticle-mediated transfer of MicroRNA−126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation (2013) 128:2026–38. doi: 10.1161/CIRCULATIONAHA.113.001720
99. Deregibus MC, Cantaluppi V, Calogero R, Lo Iacono M, Tetta C, Biancone L, et al. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood (2007) 110:2440–8. doi: 10.1182/blood-2007-03-078709
100. Zhang Z, Yang J, Yan W, Li Y, Shen Z, Asahara T. Pretreatment of cardiac stem cells with exosomes derived from mesenchymal stem cells enhances myocardial repair. J Am Heart Assoc. (2016) 5:e002856. doi: 10.1161/JAHA.115.002856
101. Borger V, Bremer M, Ferrer-Tur R, Gockeln L, Stambouli O, Becic A, et al. Mesenchymal stem/stromal cell-derived extracellular vesicles and their potential as novel immunomodulatory therapeutic agents. Int J Mol Sci. (2017) 18:E1450. doi: 10.3390/ijms18071450
102. Shao L, Zhang Y, Lan B, Wang J, Zhang Z, Zhang L, et al. MiRNA-sequence indicates that mesenchymal stem cells and exosomes have similar mechanism to enhance cardiac repair. BioMed Res Int. (2017) 2017:4150705. doi: 10.1155/2017/4150705
103. Ma J, Zhao Y, Sun L, Sun X, Zhao X, Sun X, et al. Exosomes derived from akt-modified human umbilical cord mesenchymal stem cells improve cardiac regeneration and promote angiogenesis via activating platelet-derived growth factor D. Stem Cell Transl Med. (2017) 6:51–9. doi: 10.5966/sctm.2016-0038
104. Gonzalez-King H, Garcia NA, Ontoria-Oviedo I, Ciria M, Montero JA, Sepulveda P. Hypoxia inducible factor-1alpha potentiates jagged 1-mediated angiogenesis by mesenchymal stem cell-derived exosomes. Stem Cell. (2017) 35:1747–59. doi: 10.1002/stem.2618
105. Zou X, Gu D, Xing X, Cheng Z, Gong D, Zhang G, et al. Human mesenchymal stromal cell-derived extracellular vesicles alleviate renal ischemic reperfusion injury and enhance angiogenesis in rats. Am J Transl Res. (2016) 8:4289–99.
106. Gong M, Yu B, Wang J, Wang Y, Liu M, Paul C, et al. Mesenchymal stem cells release exosomes that transfer miRNAs to endothelial cells and promote angiogenesis. Oncotarget (2017) 8:45200–12. doi: 10.18632/oncotarget.16778
107. Liang X, Zhang L, Wang S, Han Q, Zhao RC. Exosomes secreted by mesenchymal stem cells promote endothelial cell angiogenesis by transferring miR−125a. J Cell Sci. (2016) 129:2182–9. doi: 10.1242/jcs.170373
108. Tang YL, Zhu W, Cheng M, Chen L, Zhang J, Sun T, et al. Hypoxic preconditioning enhances the benefit of cardiac progenitor cell therapy for treatment of myocardial infarction by inducing CXCR4 expression. Circul Res. (2009) 104:1209–16. doi: 10.1161/CIRCRESAHA.109.197723
109. Kishore R, Khan M. More than tiny sacks: stem cell exosomes as cell-free modality for cardiac repair. Circul Res. (2016) 118:330–43. doi: 10.1161/CIRCRESAHA.115.307654
110. Sahoo S, Klychko E, Thorne T, Misener S, Schultz KM, Millay M, et al. Exosomes from human CD34(+) stem cells mediate their proangiogenic paracrine activity. Circul Res. (2011) 109:724–8. doi: 10.1161/CIRCRESAHA.111.253286
111. Soleti R, Benameur T, Porro C, Panaro MA, Andriantsitohaina R, Martinez MC. Microparticles harboring Sonic Hedgehog promote angiogenesis through the upregulation of adhesion proteins and proangiogenic factors. Carcinogenesis (2009) 30:580–8. doi: 10.1093/carcin/bgp030
112. Agouni A, Mostefai HA, Porro C, Carusio N, Favre J, Richard V, et al. Sonic hedgehog carried by microparticles corrects endothelial injury through nitric oxide release. FASEB J. (2007) 21:2735–41. doi: 10.1096/fj.07-8079com
113. Benameur T, Soleti R, Porro C, Andriantsitohaina R, Martinez MC. Microparticles carrying Sonic hedgehog favor neovascularization through the activation of nitric oxide pathway in mice. PLoS ONE (2010) 5:e12688. doi: 10.1371/journal.pone.0012688
114. Kaur S, Singh SP, Elkahloun AG, Wu W, Abu-Asab MS, Roberts DD. CD47-dependent immunomodulatory and angiogenic activities of extracellular vesicles produced by T cells. Matrix Biol. (2014) 37:49–59. doi: 10.1016/j.matbio.2014.05.007
115. Tahiri H, Yang C, Duhamel F, Omri S, Picard E, Chemtob S, et al. p75 neurotrophin receptor participates in the choroidal antiangiogenic and apoptotic effects of T-lymphocyte-derived microparticles. Invest Ophthalmol Visual Sci. (2013) 54:6084–92. doi: 10.1167/iovs.13-11896
116. Yang C, Mwaikambo BR, Zhu T, Gagnon C, Lafleur J, Seshadri S, et al. Lymphocytic microparticles inhibit angiogenesis by stimulating oxidative stress and negatively regulating VEGF-induced pathways. Am J Physiol Regul Integr Compar Physiol. (2008) 294:R467–76. doi: 10.1152/ajpregu.00432.2007
117. Tahiri H, Omri S, Yang C, Duhamel F, Samarani S, Ahmad A, et al. Lymphocytic microparticles modulate angiogenic properties of macrophages in laser-induced choroidal neovascularization. Sci Rep. (2016) 6:37391. doi: 10.1038/srep37391
118. Li J, Zhang Y, Liu Y, Dai X, Li W, Cai X, et al. Microvesicle-mediated transfer of microRNA−150 from monocytes to endothelial cells promotes angiogenesis. J Biol Chem. (2013) 288:23586–96. doi: 10.1074/jbc.M113.489302
119. Zhang Y, Liu D, Chen X, Li J, Li L, Bian Z, et al. Secreted monocytic miR−150 enhances targeted endothelial cell migration. Mol Cell. (2010) 39:133–44. doi: 10.1016/j.molcel.2010.06.010
120. Leroyer AS, Rautou PE, Silvestre JS, Castier Y, Leseche G, Devue C, et al. CD40 ligand+ microparticles from human atherosclerotic plaques stimulate endothelial proliferation and angiogenesis a potential mechanism for intraplaque neovascularization. J Am College Cardiol. (2008) 52:1302–11. doi: 10.1016/j.jacc.2008.07.032
121. Aharon A, Tamari T, Brenner B. Monocyte-derived microparticles and exosomes induce procoagulant and apoptotic effects on endothelial cells. Thromb Haemost. (2008) 100:878–85. doi: 10.1160/TH07-11-0691
122. Lee HD, Kim YH, Kim DS. Exosomes derived from human macrophages suppress endothelial cell migration by controlling integrin trafficking. Eur J Immunol. (2014) 44:1156–69. doi: 10.1002/eji.201343660
123. Ramakrishnan DP, Hajj-Ali RA, Chen Y, Silverstein RL. Extracellular vesicles activate a CD36-dependent signaling pathway to inhibit microvascular endothelial cell migration and tube formation. Arterioscler Thromb Vasc Biol. (2016) 36:534–44. doi: 10.1161/ATVBAHA.115.307085
124. Silva AM, Almeida MI, Teixeira JH, Maia AF, Calin GA, Barbosa MA, et al. Dendritic Cell-derived extracellular vesicles mediate mesenchymal stem/stromal cell recruitment. Sci Rep. (2017) 7:1667. doi: 10.1038/s41598-017-01809-x
125. Dalli J, Montero-Melendez T, Norling LV, Yin X, Hinds C, Haskard D, et al. Heterogeneity in neutrophil microparticles reveals distinct proteome and functional properties. Mol Cell Proteom. (2013) 12:2205–19. doi: 10.1074/mcp.M113.028589
126. Ibanez B, James S, Agewall S, Antunes MJ, Bucciarelli-Ducci C, Bueno H, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the task force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. (2018) 39:119–77. doi: 10.1093/eurheartj/ehx393
127. Puymirat E, Simon T, Steg PG, Schiele F, Gueret P, Blanchard D, et al. Association of changes in clinical characteristics and management with improvement in survival among patients with ST-elevation myocardial infarction. J Am Med Assoc. (2012) 308:998–1006. doi: 10.1001/2012.jama.11348
128. Schmidt M, Jacobsen JB, Lash TL, Botker HE, Sorensen HT. 25 year trends in first time hospitalisation for acute myocardial infarction, subsequent short and long term mortality, and the prognostic impact of sex and comorbidity: a Danish nationwide cohort study. Br Med J. (2012) 344:e356. doi: 10.1136/bmj.e356
129. Schwarz ER, Speakman MT, Patterson M, Hale SS, Isner JM, Kedes LH, et al. Evaluation of the effects of intramyocardial injection of DNA expressing vascular endothelial growth factor (VEGF) in a myocardial infarction model in the rat–angiogenesis and angioma formation. J Am College Cardiol. (2000) 35:1323–30. doi: 10.1016/S0735-1097(00)00522-2
130. Pearlman JD, Hibberd MG, Chuang ML, Harada K, Lopez JJ, Gladstone SR, et al. Magnetic resonance mapping demonstrates benefits of VEGF-induced myocardial angiogenesis. Nat Med. (1995) 1:1085–9. doi: 10.1038/nm1095-1085
131. Henry TD, Annex BH, McKendall GR, Azrin MA, Lopez JJ, Giordano FJ, et al. The VIVA trial: vascular endothelial growth factor in Ischemia for Vascular Angiogenesis. Circulation (2003) 107:1359–65. doi: 10.1161/01.CIR.0000061911.47710.8A
132. Stewart DJ, Kutryk MJ, Fitchett D, Freeman M, Camack N, Su Y, et al. VEGF gene therapy fails to improve perfusion of ischemic myocardium in patients with advanced coronary disease: results of the NORTHERN trial. Mol Ther. (2009) 17:1109–15. doi: 10.1038/mt.2009.70
133. Simons M, Annex BH, Laham RJ, Kleiman N, Henry T, Dauerman H, et al. Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor-2: double-blind, randomized, controlled clinical trial. Circulation (2002) 105:788–93. doi: 10.1161/hc0802.104407
134. Gerbin KA, Murry CE. The winding road to regenerating the human heart. Cardiovasc Pathol. (2015) 24:133–40. doi: 10.1016/j.carpath.2015.02.004
135. Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, et al. Bone marrow cells regenerate infarcted myocardium. Nature (2001) 410:701–5. doi: 10.1038/35070587
136. Messina E, De Angelis L, Frati G, Morrone S, Chimenti S, Fiordaliso F, et al. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circul Res. (2004) 95:911–21. doi: 10.1161/01.RES.0000147315.71699.51
137. Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, et al. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet (2012) 379:895–904. doi: 10.1016/S0140-6736(12)60195-0
138. Malliaras K, Makkar RR, Smith RR, Cheng K, Wu E, Bonow RO, et al. Intracoronary cardiosphere-derived cells after myocardial infarction: evidence of therapeutic regeneration in the final 1-year results of the CADUCEUS trial (CArdiosphere-Derived aUtologous stem CElls to reverse ventricUlar dySfunction). J Am College Cardiol. (2014) 63:110–22. doi: 10.1016/j.jacc.2013.08.724
139. Chugh AR, Beache GM, Loughran JH, Mewton N, Elmore JB, Kajstura J, et al. Administration of cardiac stem cells in patients with ischemic cardiomyopathy: the SCIPIO trial: surgical aspects and interim analysis of myocardial function and viability by magnetic resonance. Circulation (2012) 126(11 Suppl. 1):S54–64. doi: 10.1161/CIRCULATIONAHA.112.092627
140. Bartunek J, Behfar A, Dolatabadi D, Vanderheyden M, Ostojic M, Dens J, et al. Cardiopoietic stem cell therapy in heart failure: the C-CURE (Cardiopoietic stem Cell therapy in heart failURE) multicenter randomized trial with lineage-specified biologics. J Am College Cardiol. (2013) 61:2329–38. doi: 10.1016/j.jacc.2013.02.071
141. Steinhoff G, Nesteruk J, Wolfien M, Kundt G, Group PTI, Borgermann J, et al. Cardiac function improvement and bone marrow response -: outcome analysis of the randomized perfect phase III clinical trial of intramyocardial CD133(+) application after myocardial infarction. EBio Med. (2017) 22:208–24. doi: 10.1016/j.ebiom.2017.07.022
142. Henry TD, Schaer GL, Traverse JH, Povsic TJ, Davidson C, Lee JS, et al. Autologous CD34(+) Cell therapy for refractory angina: 2-year outcomes from the ACT34-CMI study. Cell Transpl. (2016) 25:1701–11. doi: 10.3727/096368916X691484
143. Wollert KC, Meyer GP, Lotz J, Ringes-Lichtenberg S, Lippolt P, Breidenbach C, et al. Intracoronary autologous bone-marrow cell transfer after myocardial infarction: the BOOST randomised controlled clinical trial. Lancet (2004) 364:141–8. doi: 10.1016/S0140-6736(04)16626-9
144. Chimenti I, Smith RR, Li TS, Gerstenblith G, Messina E, Giacomello A, et al. Relative roles of direct regeneration versus paracrine effects of human cardiosphere-derived cells transplanted into infarcted mice. Circul Res. (2010) 106:971–80. doi: 10.1161/CIRCRESAHA.109.210682
145. de Couto G, Gallet R, Cambier L, Jaghatspanyan E, Makkar N, Dawkins JF, et al. Exosomal MicroRNA transfer into macrophages mediates cellular postconditioning. Circulation (2017) 136:200–14. doi: 10.1161/CIRCULATIONAHA.116.024590
146. Cambier L, de Couto G, Ibrahim A, Echavez AK, Valle J, Liu W, et al. Y RNA fragment in extracellular vesicles confers cardioprotection via modulation of IL−10 expression and secretion. EMBO Mol Med. (2017) 9:337–52. doi: 10.15252/emmm.201606924
147. Lai RC, Arslan F, Lee MM, Sze NS, Choo A, Chen TS, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. (2010) 4:214–22. doi: 10.1016/j.scr.2009.12.003
148. Ibrahim AG, Cheng K, Marban E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep. (2014) 2:606–19. doi: 10.1016/j.stemcr.2014.04.006
149. Mikamori M, Yamada D, Eguchi H, Hasegawa S, Kishimoto T, Tomimaru Y, et al. MicroRNA−155 controls exosome synthesis and promotes gemcitabine resistance in pancreatic ductal adenocarcinoma. Sci Rep. (2017) 7:42339. doi: 10.1038/srep42339
150. Rayner S, Bruhn S, Vallhov H, Andersson A, Billmyre RB, Scheynius A. Identification of small RNAs in extracellular vesicles from the commensal yeast Malassezia sympodialis. Sci Rep. (2017) 7:39742. doi: 10.1038/srep39742
151. Jiang J, Kao CY, Papoutsakis ET. How do megakaryocytic microparticles target and deliver cargo to alter the fate of hematopoietic stem cells? J Control Release (2017) 247:1–18. doi: 10.1016/j.jconrel.2016.12.021
152. Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. (2013) 21:185–91. doi: 10.1038/mt.2012.180
153. Amosse J, Martinez MC, Le Lay S. Extracellular vesicles and cardiovascular disease therapy. Stem Cell Invest. (2017) 4:102. doi: 10.21037/sci.2017.11.07
154. Van Buskirk RG, Baust JM, Snyder KK, Mathew AJ, Baust JG. Hypothermic storage and cryopreservation. BioProcess Int. (2004) 2:42–9.
155. Lamparski HG, Metha-Damani A, Yao JY, Patel S, Hsu DH, Ruegg C, et al. Production and characterization of clinical grade exosomes derived from dendritic cells. J Immunol Methods (2002) 270:211–26. doi: 10.1016/S0022-1759(02)00330-7
156. O'Cearbhaill ED, Ng KS, Karp JM. Emerging medical devices for minimally invasive cell therapy. Mayo Clinic Proc. (2014) 89:259–73. doi: 10.1016/j.mayocp.2013.10.020
157. Brindley D, Moorthy K, Lee JH, Mason C, Kim HW, Wall I. Bioprocess forces and their impact on cell behavior: implications for bone regeneration therapy. J Tissue Eng. (2011) 2011:620247. doi: 10.4061/2011/620247
158. Armstrong JP, Perriman AW. Strategies for cell membrane functionalization. Exp Biol Med. (2016) 241:1098–106. doi: 10.1177/1535370216650291
159. Stickney Z, Losacco J, McDevitt S, Zhang Z, Lu B. Development of exosome surface display technology in living human cells. Biochem Biophys Res Commun. (2016) 472:53–9. doi: 10.1016/j.bbrc.2016.02.058
160. Takahashi Y, Nishikawa M, Shinotsuka H, Matsui Y, Ohara S, Imai T, et al. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J Biotechnol. (2013) 165:77–84. doi: 10.1016/j.jbiotec.2013.03.013
161. Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ, et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials (2014) 35:2383–90. doi: 10.1016/j.biomaterials.2013.11.083
162. Wang M, Altinoglu S, Takeda YS, Xu Q. Integrating protein engineering and bioorthogonal click conjugation for extracellular vesicle modulation and intracellular delivery. PLoS ONE (2015) 10:e0141860. doi: 10.1371/journal.pone.0141860
163. Okeley NM, Toki BE, Zhang X, Jeffrey SC, Burke PJ, Alley SC, et al. Metabolic engineering of monoclonal antibody carbohydrates for antibody-drug conjugation. Bioconjugate Chem. (2013) 24:1650–5. doi: 10.1021/bc4002695
164. Iwasaki Y, Matsunaga A, Fujii S. Preparation of biointeractive glycoprotein-conjugated hydrogels through metabolic oligosacchalide engineering. Bioconjug Chem. (2014) 25:1626–31. doi: 10.1021/bc5003295
165. Neubert J, Glumm J. Promoting neuronal regeneration using extracellular vesicles loaded with superparamagnetic iron oxide nanoparticles. Neural Regen Res. (2016) 11:61–3. doi: 10.4103/1673-5374.175043
166. Correia Carreira S, Armstrong JP, Seddon AM, Perriman AW, Hartley-Davies R, Schwarzacher W. Ultra-fast stem cell labelling using cationised magnetoferritin. Nanoscale (2016) 8:7474–83. doi: 10.1039/C5NR07144E
167. Silva AK, Kolosnjaj-Tabi J, Bonneau S, Marangon I, Boggetto N, Aubertin K, et al. Magnetic and photoresponsive theranosomes: translating cell-released vesicles into smart nanovectors for cancer therapy. ACS Nano (2013) 7:4954–66. doi: 10.1021/nn400269x
168. Silva AK, Luciani N, Gazeau F, Aubertin K, Bonneau S, Chauvierre C, et al. Combining magnetic nanoparticles with cell derived microvesicles for drug loading and targeting. Nanomedicine (2015) 11:645–55. doi: 10.1016/j.nano.2014.11.009
169. Smyth T, Petrova K, Payton NM, Persaud I, Redzic JS, Graner MW, et al. Surface functionalization of exosomes using click chemistry. Bioconjug Chem. (2014) 25:1777–84. doi: 10.1021/bc500291r
170. Fuhrmann G, Serio A, Mazo M, Nair R, Stevens MM. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J Control Release (2015) 205:35–44. doi: 10.1016/j.jconrel.2014.11.029
171. Haney MJ, Klyachko NL, Zhao Y, Gupta R, Plotnikova EG, He Z, et al. Exosomes as drug delivery vehicles for Parkinson's disease therapy. J Control Release (2015) 207:18–30. doi: 10.1016/j.jconrel.2015.03.033
172. Qi H, Liu C, Long L, Ren Y, Zhang S, Chang X, et al. Blood exosomes endowed with magnetic and targeting properties for cancer therapy. ACS Nano (2016) 10:3323–33. doi: 10.1021/acsnano.5b06939
173. Maguire CA, Balaj L, Sivaraman S, Crommentuijn MH, Ericsson M, Mincheva-Nilsson L, et al. Microvesicle-associated AAV vector as a novel gene delivery system. Mol Ther. (2012) 20:960–71. doi: 10.1038/mt.2011.303
174. Sun D, Zhuang X, Xiang X, Liu Y, Zhang S, Liu C, et al. A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol Ther. (2010) 18:1606–14. doi: 10.1038/mt.2010.105
175. El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C, et al. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc. (2012) 7:2112–26. doi: 10.1038/nprot.2012.131
176. Wahlgren J, De LKT, Brisslert M, Vaziri Sani F, Telemo E, Sunnerhagen P, et al. Plasma exosomes can deliver exogenous short interfering RNA to monocytes and lymphocytes. Nucleic Acids Res. (2012) 40:e130. doi: 10.1093/nar/gks463
177. Pascucci L, Cocce V, Bonomi A, Ami D, Ceccarelli P, Ciusani E, et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: a new approach for drug delivery. J Control Release (2014) 192:262–70. doi: 10.1016/j.jconrel.2014.07.042
178. Vandergriff A, Huang K, Shen D, Hu S, Hensley MT, Caranasos TG, et al. Targeting regenerative exosomes to myocardial infarction using cardiac homing peptide. Theranostics (2018) 8:1869–78. doi: 10.7150/thno.20524
179. Kim H, Yun N, Mun D, Kang JY, Lee SH, Park H, et al. Cardiac-specific delivery by cardiac tissue-targeting peptide-expressing exosomes. Biochem Biophys Res Commun. (2018) 499:803–8. doi: 10.1016/j.bbrc.2018.03.227
180. Zhuang X, Xiang X, Grizzle W, Sun D, Zhang S, Axtell RC, et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol Ther. (2011) 19:1769–79. doi: 10.1038/mt.2011.164
181. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. (2011) 29:341–5. doi: 10.1038/nbt.1807
182. Cooper JM, Wiklander PB, Nordin JZ, Al-Shawi R, Wood MJ, Vithlani M, et al. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Move Disor. (2014) 29:1476–85. doi: 10.1002/mds.25978
Keywords: small EVs, tissue remodeling, myocardial infarction, immune system, angiogenesis
Citation: Sánchez-Alonso S, Alcaraz-Serna A, Sánchez-Madrid F and Alfranca A (2018) Extracellular Vesicle-Mediated Immune Regulation of Tissue Remodeling and Angiogenesis After Myocardial Infarction. Front. Immunol. 9:2799. doi: 10.3389/fimmu.2018.02799
Received: 28 May 2018; Accepted: 13 November 2018;
Published: 29 November 2018.
Edited by:
Denise Doolan, James Cook University, AustraliaReviewed by:
Sai Kiang Lim, Institute of Medical Biology (A*STAR), SingaporeCopyright © 2018 Sánchez-Alonso, Alcaraz-Serna, Sánchez-Madrid and Alfranca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francisco Sánchez-Madrid, ZnNtYWRyaWRAc2FsdWQubWFkcmlkLm9yZw==
Arantzazu Alfranca, bWFyaWFhcmFuemF6dS5hbGZyYW5jYUBzYWx1ZC5tYWRyaWQub3Jn
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.