 Yehui Duan
Yehui Duan Liming Zeng2†
Liming Zeng2† Kang Xu
Kang Xu- 1Hunan Provincial Key Laboratory of Animal Nutritional Physiology and Metabolic Process, Key Laboratory of Agro-ecological Processes in Subtropical Region, Institute of Subtropical Agriculture, Chinese Academy of Sciences, Hunan Provincial Engineering Research Center for Healthy Livestock and Poultry Production, Scientific Observing and Experimental Station of Animal Nutrition and Feed Science in South-Central, Ministry of Agriculture, National Engineering Laboratory for Pollution Control and Waste Utilization in Livestock and Poultry Production, Changsha, China
- 2Science College of Jiangxi Agricultural University, Nanchang, China
- 3Guangdong Provincial Key Laboratory of Animal Nutrition Regulation, South China Agricultural University, Guangzhou, China
In recent years, chronic overnutrition, such as consumption of a high-fat diet (HFD), has been increasingly viewed as a significant modifiable risk factor for diseases such as diabetes and certain types of cancer. However, the mechanisms by which HFDs exert adverse effects on human health remains poorly understood. Here, this paper will review the recent scientific literature about HFD-induced inflammation and subsequent development of diseases and cancer, with an emphasis on mechanisms involved. Given the expanding global epidemic of excessive HFD intake, understanding the impacts of a HFD on these medical conditions, gaining great insights into possible underlying mechanisms, and developing effective therapeutic strategies are of great importance.
Introduction
At present, obesity has reached epidemic proportions. It develops from an imbalance of energy homeostasis and contributes dramatically to the global disease burden, predisposing individuals to chronic diseases such as type 2 diabetes mellitus (T2DM), cardiovascular disease (CVD), and certain types of cancer (1, 2). Excessive consumption of high fat diets (HFDs) has undoubtedly exacerbated the obesity epidemic and the development of obesity-related metabolic disorders (3, 4). Despite substantial work demonstrating that obesity develops from an imbalance between energy intake and energy expenditure, the underlying mechanisms for the detrimental effects of HFDs seem to be more complicated than the simple concept of energy imbalance (5). In this context, a better understanding of the pathogenesis of HFD-driven metabolic disorders may help to reduce the disease burden worldwide. This review will elaborate on the precise pathology and etiology of diseases induced by a HFD, and will evaluate the possibility of therapeutic targeting of free fatty acids (FFAs) and low-grade systemic inflammation to reduce HFDs-stimulated diseases.
HFDs Induce a Systemic Chronic Low-Grade Inflammation
Following the ingestion of HFDs, inflammation develops in the central nervous system (CNS) including the hypothalamus and in the peripheral tissues including the liver, adipose tissue, skeletal muscle, and intestine (6). Concerning the development of chronic systemic inflammation, alterations in the gut microbiota triggered by HFDs and direct effects of FFAs on intestinal cells may be the first step (7). Evidence to support this hypothesis is that germ-free mice exhibited neither obesity nor upregulation of intestinal TNF-α level compared to conventionalized mice when fed a HFD, whereas reconstitution of the gut microbiota from obese mice in the germ-free mice produced an increase in body fat (8). Furthermore, after comparing microbiota of obese vs. lean subjects, a recent study suggested that individuals with high bacterial richness were associated with less significant adiposity and inflammation than low bacterial richness subjects (9), which is line with HFD-induced inflammation and decrease of bacterial diversity. In particular, increased levels of Firmicutes and a reduced relative abundance of Bacteroidetes were observed in both humans and animals following HFD intake. Shifts in gut microbiota populations activate Toll-like receptor (TLR) signaling pathway, leading to increased intestinal permeability to endotoxins [such as lipopolysaccharides (LPS)] and thus promoting the translocation of LPS to the circulation (10–12). In addition, increased amounts of FFAs present in HFDs may directly act on intestinal cells. Therefore, elevated release of LPS and/or increased FFAs levels led to elevated production of pro-inflammatory cytokines [i.e., interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α] in the gut (13–17). The second step may consist in increased delivery of intestinal LPS, pro-inflammatory cytokines, and FFAs into the systemic circulation and portal circulation, thus leading to a systemic low-grade inflammation (15, 18). Elevated plasma FFAs and LPS can upregulate the expression of TLRs in circulating macrophages, enabling macrophages to be activated (M1 phenotype), which in turn produce proinflammmatory cytokines (11, 12).
Before the onset of obesity, these factors in the circulating system triggers inflammatory pathways in the brain. More especially, elevated FFAs and cytokines first activate hypothalamic IκB kinase β (IKKβ)/nuclear factor of kappa B (NF-κB) signaling directly or indirectly through the following two ways: (i) through activating TLR located at cellular surface; (ii) through inducing various cellular stresses including oxidative stress and endoplasmic reticulum stress (ERS) in the hypothalamus (19–21). Hence, activated IKKβ/NF-κB signaling blunts central leptin and insulin sensitivity and initiates gene expression of inflammatory response molecules in the hypothalamus (19, 22, 23). Meanwhile, activated inflammatory macrophages (M1) in plasma can reach the adipose and muscular tissues, pancreatic islets, and blood vessels, leading to peripheral inflammation (12, 24). Notably, accumulation of CD8+ T-cells in the adipose tissue contributes to M1 macrophage recruitment in the adipose tissue (25). In addition, under HFD feeding stress the adipose tissue fails to store the excess lipids, which are thereafter deposited into other tissues including the liver, pancreas, skeletal muscle, and blood vessels (24). Ectopic lipid accumulation contributes additionally to the expression of proinflammatory mediators and the recruitment of M1 macrophages, thus aggravating systemic inflammation (26, 27). Moreover, the liver is also exposed to relatively high concentrations of different mediators (i.e., LPS, proinflammatory cytokines, and FFAs) released by the gastrointestinal tract (15). These mediators lead to accumulation of Natural killer T (NKT) cells and activation of Kupffer cells in the liver, thereby contributing to hepatic and systemic inflammation (28).
Altogether, under HFD conditions, a complicated network of signals interconnecting several organs acts in synergy to elicit a low-grade systemic inflammation (Figure 1). HFD-related inflammation leads to the failure of adipocytes to effectively remove circulating FFAs, and is pivotal to disease progression and the development of complications, such as T2DM, CVD, liver disease, atherosclerosis, and certain types of cancer (Figure 2, discussed in detail below).
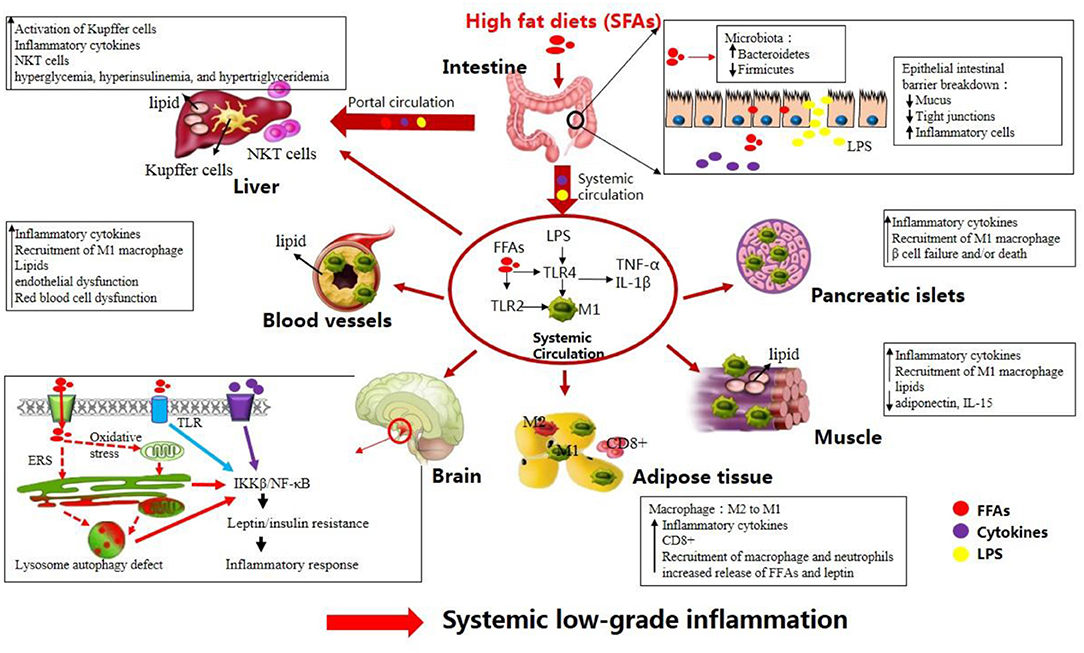
Figure 1. High-fat diets (HFDs) induce metabolic inflammation throughout the organism. The levels of endotoxins (e.g., LPS), circulating free fatty acids, and inflammatory mediators are increased in response to HFDs, resulting in low-grade systemic inflammation and altered homeostasis in many organs (see text).
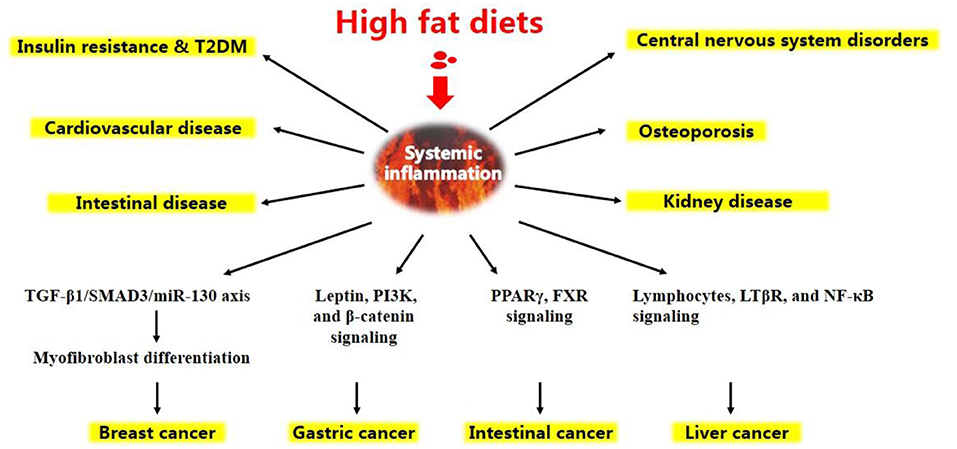
Figure 2. Chronic diseases and certain types of cancer are induced by high-fat diets.
HFD-Induced Diseases
Insulin Resistance and T2DM
Insulin resistance is the condition where the body fails to respond appropriately to circulating insulin, leading to the impaired systemic glucose clearance and uptake in several tissues including the adipose tissue, liver, and muscle (29). Exposure to a HFD often results in insulin resistance and pancreatic β cell dysfunction. For example, after only 3 days of HFD feeding, systemic insulin resistance, and glucose intolerance developed in mice (30). Other studies have also shown that mice fed a HFD for several weeks exhibited increased glucose intolerant and impaired insulin sensitive, reflecting insulin resistance and initiating β cell compensation (31, 32).
Although not fully explored, current evidence has established causative and correlative links between HFD-induced inflammation and the pathogenesis of insulin resistance (27, 33). Under HFD conditions, insulin secretion from β cells and peripheral insulin action can be impaired by hypothalamic inflammation (34). Proinflammatory cytokines produced by β cells themselves due to macrophages accumulation in islets may further block β cell function (35). In addition, HFDs can activate 12- lipoxygenase activity in β cells through cytokines, FFAs, and increased blood glucose, thus increasing the production of 12-hydroxyeicosatetraenoic acid (12-HETE). 12-HETE promotes oxidative stress and blunts Nrf2 function, eventually leading to β cell apoptosis and glucose intolerance (36). Therefore, HFDs lead to an increase in insulin release from β cells and thus hyperinsulinemia. In adipose tissue, hyperinsulinemia combined with an inflammatory response promotes lipolysis, thus enabling FFAs and glycerol to flow to the liver and to promote gluconeogenesis. At the same time, elevated insulin concentrations increases muscle glycolysis and lactate production, which is released into the circulation and can be used as a substrate for gluconeogenesis in the liver (37). Subsequently, increased gluconeogenesis enhances glucose production in the liver and thereby leads to systemic insulin resistance (38). As a consequence, T2DM ensues due to systemic insulin resistance and relatively diminished insulin secretion from pancreatic β cells (29). In addition, recent studies indicated that the A2b adenosine receptor (A2bAR), an established mediator of inflammation, control pancreatic dysfunction in HFD-induced obesity (39). In particular, macrophage A2bAR has been recently identified as an important mediator of HFD-induced hallmarks of T2DM (40, 41). Therefore, regulation of macrophage A2bAR, either by genetic manipulations of monocytes or pharmacologically, may be a promising therapeutic approach. Taken together, chronically overconsumption of HFDs is mechanistically associated with the development of insulin resistance and T2DM.
Cardiovascular Disease
Although some controversy still exists, an increasing number of studies have highlighted the crucial effects of HFDs on the development of CVD (42, 43). For instance, administration of HFDs (20 g of fat/100 g of diet) to adult male rats for 30 days led to simultaneous increase in nitric oxide (NO) production and oxidative stress, causing the augmentation of infarct size after myocardial infarction (43). Low-grade systemic inflammation related to a HFD resulted in cardiovascular complications partially through infiltration of adipose and vascular tissues by effector T cells (44). In support of this notion, a cohort of 1,172 subjects was selected and stratified in three groups (i.e., lean, overweight, and obese). Results showed that the blood of overweight and patients with obesity had elevated numbers of CXCR3+ effector memory T cells (a pro-inflammatory effector memory-like phenotype) compared to lean subjects. Similar phenotype was also found in mice. Upon further investigation, the same group found that this differentiation of CXCR3+ effector memory T cells was partially mediated by the PI3K p110δ-Akt signaling pathway (44). Apart from infiltration of adipose and vascular tissues by effector T cells, the vascular endothelial dysfunction may also exert a role in the onset and propagation of CVD induced by a HFD (45, 46). Evidence for this hypothesis is provided by observations that mice fed with a HFD rich in lard exhibited an inhibition of the L-arginine-NO pathway in red blood cells (RBCs) of mice (47). RBCs-produced NO can contribute to the intravascular NO pool and suppression of platelet aggregation, thus regulating vascular homeostasis (48). Hence, decreasing NO bioavailability contributes to CVD via impairing vascular endothelial function. The adverse effects of HFD on endothelial function may be mediated independently by hyperglycemia, insulin resistance, and FFAs consequent to a HFD (49). Analogous to endothelial dysfunction, RBCs dysfunction takes place early during HFD-induced obesity and may contribute to the development of CVD (50). More especially, mice fed a 60% HFD for 12 weeks displayed a marked increase in the level of chemokines, which were bound to RBC via Duffy antigen receptor for chemokines. Moreover, RBCs from HFD-fed mice showed elevated cholesterol content and membrane phosphatidylserine externalization, accompanied by increased phagocytosis of RBCs by macrophages in vitro and by the spleen in vivo, thus contributing to atherosclerosis (50). Altogether, a HFD may promote the development of CVD via infiltration of adipose and vascular tissues by effector T cells, endothelial dysfunction and RBCs dysfunction.
Intestinal Diseases
A HFD-induced intestinal inflammation contributes to not only a systemic low-grade inflammation but localized tissue dysfunction (51, 52). For example, HFD feeding can exacerbate chemically-induced dextran sodium sulfate colitis by increasing pro-inflammatory cytokines (53–55) and can increase mucosal tissue damage in mouse models of ileitis (Mdr1a−/− mice) (56, 57) and spontaneous colitis (Muc2−/−, TNFΔARE mice) (58). Consistently, human studies have involved HFD consumption with an increased risk of functional bowel symptoms (59), such as inflammatory bowel diseases (IBD) (60). The increased incidence of IBD such as Crohn's disease (CD) in developed countries parallels escalating consumption of fat (60, 61). A HFD promotes the development of CD partially via promoting gut inflammation and microbiome perturbations. Previous studies have shown that a HFD increased intestinal permeability, promoted intestinal mucosa dysbiosis, and altered microbiota composition with a profile characterized by the expansion of pro-inflammatory Firmicutes and Proteobacteria and the reduction of protective bacteria such as Bacteroidetes. These effects triggered by a HFD were accompanied by downregulated expression of PPARγ and GPR43. Thus, the activation of PPARγ signaling and GPR43 receptor pathway could be viewed as an effective strategy to treat CD patients (62, 63). Moreover, after intestinal inflammation, intestinal morphofunctional rearrangement of the enteric nervous system occurs in the murine model of HFD-induced obesity, contributing to the pathophysiology of digestive motor abnormalities (64, 65). Recent studies further report that excitatory tachykiniergic pathways, which can be regulated by A2bAR, are involved in the pathogenesis of colonic dysmotility induced by intestinal macrophages in HFD-induced obesity (16, 17).
Osteoporosis
Osteoblasts and adipocytes are derived from the same mesenchymal stem cell (MSC) source (66), leading to interactions between osteoclastogenesis and adipogenesis that can be affected by dietary fat intake (67). Epidemiological evidence has suggested that consuming HFDs promotes obesity, which is closely associated with lower bone mineral density (BMD) and a higher risk of osteoporotic fractures (68–70). Similar observations have also been observed in animal studies. Mice given HFDs (21.1% fat) for 13 weeks had lower bone mass, which was associated with oxidative stress (71). Likewise, HFDs (45% energy as fat) for 14 weeks led to decreased trabecular bone volume and trabecular number in the proximal tibia (72). These results suggest that HFDs affect adversely bone quantity. In contrast, other human and animal studies have shown that HFDs induced higher BMD because of elevated mechanical loading (73, 74). Recently, it has been reported that short-term consumption (8 weeks) of HFDs increased bone mass via elevated mechanical loading; however, long-term consumption (16 and 24 weeks) of HFDs exerted negative effects on bone mass (75). Therefore, it raises the possibility that HFDs initially augment bone deposition because of elevated mechanical demand but eventually impairs bone formation and turnover due to metabolic dysregulation (76, 77).
Mice received HFDs (60 kcal% fat) for 6 weeks before challenge with B16F10 melanoma cells exhibited elevated bone marrow (BM) adipocytes compared with mice received with normal diets. The increased presence of adipocytes largely changes the hematopoietic stem cell niche in the BM microenvironment and sets up a niche for tumor cells, increasing their proliferation by IL-6 and Janus Kinase 2 (JAK2). Moreover, cancer cells interact with fat in the BM facilitates the accumulation of osteoclasts in the BM by expressing osteopontin. Therefore, the IL-6-JAK2-osteopontin axis is a critical signaling pathway for building the metabolic tumor niche in the context of a HFD-induced obesity (78). In addition, HFD can also regulate bone deposition via the hormone leptin secreted by HFD-induced adiposity (79). Taken together, HFDs regulate bone metabolism and lead to osteoporosis partially through proinflammatory cytokines (IL-6) and adipokines (leptin).
Chronic Kidney Diseases
A strong association between intake of SFAs, hyperalbuminuria and kidney dysfunction was observed in a cross-sectional study including more than 19,000 adults > 45 years of age (80). Increasing studies using rodent models have address potential mechanisms for the development of kidney disease driven by HFDs. Rats fed HFDs (35% kcal from fat) for 10 weeks developed proteinuria, accompanied by increased inflammatory markers (TNF-α and NF-κB) and oxidative stress markers (NADPH oxidase) in the renal cortex (81). Another study using HFD-fed Sprague-Dawley rats (58% fat-derived calories, 6 weeks) has demonstrated similar molecular and metabolic effects (82). The effect of hyperlipidemia-driven inflammation on the development of kidney dysfunction was further demonstrated in Apo E−/− mice fed HFDs for 4 weeks (83). These mice displayed greatly increased levels of TNF-α and IL-6, inducing renal inflammation, mesangial cell proliferation, and the progression of proteinuria. However, these effects were strongly attenuated in mice after treatment with anti-IL-6 receptor antibody. Upon further investigation, studies found that the activity of proinflammatory cytokines and profibrotic growth factors may be enhanced by renal sterol-regulatory element binding protein (SREBP1). Mice fed HFDs (60% fat) became obese, hyperinsulinemic, and hyperglycemic, associated with increased levels of SREBP1/2 (84). More importantly, deletion of SREBP1 in the mice blocked renal inflammation and proteinuria (85). These data suggest that a SREBP1-enhanced renal inflammation may represent a mechanism for the development of HFD-induced chronic kidney diseases.
Central Nervous System Disorders
The neuroinflammation has been observed in brain structures such as the hypothalamus, hippocampus, amygdala, cortex, and brainstem, increasing the incidence of CNS pathologies such as cognitive impairments (86), behavioral abnormalities (such as depressive- and anxiety-like behaviors) (87), Alzheimer's disease (AD) (88). For instance, after 14 weeks feeding of HFDs (40% energy from fat), mice exhibited altered brain insulin signaling and cognitive functions (89). Without induction of obesity, HFDs (60% fat calories, 10 weeks) via alterations in gut microbiota can decrease memory and augment anxiety and stereotypical behaviors in mice (90). Long-term (16 months) exposition to HFDs has been shown to favor the deposition of amyloid beta (Aβ) peptide in the brain of mice, which is associated with the incidence of AD (91). These data underscore the strong effects of a HFD-induced changes on brain damage.
HFD-induced cognitive deficits may be associated with oxidative stress in the brain. Total levels of reactive oxygen species, superoxide, and peroxynitrite were significantly increased in mice fed HFDs (45% kcals from fat) compared to mice fed a control diet (10% kcals from fat) (92). Other studies also found elevated oxidative stress in HFD-fed mice which show clear cognitive impairment (86, 93). These studies highlight a role for oxidative stress in HFD-induced cognitive impairment. Unlike the underlying mechanisms for the induction of cognitive impairment, HFD promotes the development of anxiety-like behavior probably via upregulated expression of IL-1β in the amygdala (86) and changes in the GABAergic neurotransmission within the dorsomedial hypothalamus (94). In addition, HFD dramatically promotes the progression of AD-like pathology via elevation of cerebral amyloid deposition and oxidative stress, as demonstrated by previous studies (95). In particular, intake of HFDs results in peripheral insulin resistance and obesity, resulting in the disruption of the blood-brain barrier. Therefore, the transport of glucose and insulin to the CNS is limited, thus decreasing insulin signaling in the CNS. Hence, HFD-triggered peripheral hyperinsulinemia to some extent can initially provokes insulin deficiency in the brain, leading to lower expression of insulin degrading enzyme and less degradation (accumulation) of Aβ (96). At the same time, HFD-induced oxidative stress further contributes to impaired insulin signaling, thus increasing GSK-3β levels and Tau hyperphosphorylation. Increased Aβ levels and Tau hyperphosphorylation give rise to neurodegeneration and diminished synaptic plasticity, respectively, thus leading to the incidence of AD (89). Taken together, a HFD-induced damage to the brain is triggered by a number of mediators including inflammation, changes to integrity of blood-brain barrier, insulin resistance, and oxidative stress. These findings have important implications for better understanding how HFD may potentially induce brain dysfunction and the development of neurodegenerative disorders such as AD.
Liver Cancer
The current Western lifestyle will increase the risk of fatty liver disease. Non-alcoholic steatohepatitis (NASH) can be developed from fatty liver disease (steatosis) and can proceed to fibrosis, cirrhosis, and even hepatocellular carcinoma (HCC). Therefore, to better understand how dietary-induced metabolic changes contribute to the development of hepatic inflammation and NASH, a recent study used a HFD-induced obese murine model of NASH progression over 16 weeks (28). After 16 weeks, HFDs induced an obese and inflammatory phenotype in mice. More importantly, mice fed HFDs exhibited increased infiltration of NKT-cells and clusters of differentiation CD8+ T-cells in the liver. To further confirm a role for NKT cells and CD8+ T-cells, the same group used CD1d knockout (KO) and mAb mice experiments, respectively. Notably, CD1dKO mice have no functional NKT cells. After 16 weeks of HFDs, the researchers found that CD1dKO mice were protected against obesity and NASH progression, and CD8+ T-cell-depleted mice were also protected from NASH while continuing to result in weight gain. In addition, increased CD8+ T-cells were also observed in human liver sections from patients with NASH. Therefore, these data suggest that under HFD feeding stress, NKT cells and CD8+ T-cells synergistically contribute to the inflammation and fibrosis related to NASH progression (28). To further explore the underlying mechanisms for the development of NASH and HCC, a recent study used a rodent mole of long-term choline-deficient HFD to recapitulate NASH and NASH-induced HCC in humans. The analysis revealed that NKT cells primarily lead to steatosis through secreted LIGHT (97). Interestingly, unlike molecular mechanism determining NASH development, the underlying mechanisms for the HCC development may involve hepatocellular LTβR and canonical NF-κB signaling, which facilitates the transition from NASH to HCC (97). Overall, several hepatocellular signaling pathways including lymphocytes, LTβR, and NF-κB facilitate HFD-driven liver cancer.
Breast Cancer
Human breast adipose tissue, a heterogeneous cell population, is composed of multipotent MSCs, mature white adipocytes, committed progenitor cells, immune cells, endothelial cells, and fibroblasts. Under the effects of external stimulation, these MSCs can differentiate into various cell types, such as adipocytes, fibroblasts, and myofibroblasts (98). It has been recognized that obesity can promote myofibroblast differentiation within mammary adipose tissue, contributing to the microenvironment fibrotic remodeling and breast cancer progression (99). Upon further investigations, the researchers found that myofibroblast differentiation was increased by HFDs via downregulating microRNA (miR)-140 expression in mammary adipose tissue through a TGF-β1/SMAD3/miR-130 negative-feedback loop. In particular, HFD-increased TGF-β1 signaling leads to activation of SMAD3, which binds to and suppresses miR-140. This blocks miR-140 from targeting SMAD3 for degradation, causing amplified TGF-β1/SMAD3 signaling and the downregulation of miR-140, thus promoting myofibroblast differentiation (100). In vivo and in vitro studies have shown that myofibroblasts and its induced fibrotic microenvironment are related to increased initiation and growth of breast cancer (99, 100). Overall, HFDs may increase the initiation and growth of breast cancer through miR-140 downregulation-dependent myofibroblast differentiation in human breast adipose tissue.
Gastric Cancer
Epidemiological evidence has demonstrated that dietary fat is a critical risk factor for gastric cancer, which is a malignant tumor that originated from gastric mucosal epithelial cells (101–103). However, little is known about how dietary fat influences the function of gastric mucosal cells and contributes to precancerous lesions and hence gastric cancer. One recent study showed that in the precancerous gastric mucosa, fat deposition was obtained in the gastric pits in mice exposed to a HFD (60% fat calories) at 12 weeks. Ectopic fat accumulation then led to the disruption of organelle homeostasis in the gastric mucosa, as evidenced by remarkable increases in the expression of LAMP2A (the lysosomal marker), COX IV (the mitochondrial marker), Calnexin (the ER marker), and GM130 (the Golgi marker) in the gastric mucosa at 20 weeks (104). Therefore, HFD feeding first leads to excess lipid incorporation in the stomach, and then causes the disruption of organelle homeostasis in the precancerous gastric mucosa. In addition, HFD promoted stem-cell divisions and the development of gastric malignancies in the gastric mucosa, accompanied by activation of β-catenin pathway. However, β-catenin activation and stem cell marker-positive cells were not observed in mice with no leptin signaling. Meanwhile, the PI3K pathway, the downstream of leptin receptor, was also decreased (104). These data suggest that leptin signaling regulates β-catenin signaling. Taken together, HFD-driven lipid accumulation and organelle biosynthesis dysregulation confer cancer stem cell-like properties to the gastric mucosa via leptin, PI3K, and β-catenin signaling pathways (104).
Intestinal Cancer
Existing evidence has linked a HFD to intestinal cancer (2, 105–107). For instance, a recently published article showed that a long-term HFD (60% fat diet, 9–14 months) contributes to the early intestinal tumorigenesis (108). Similar findings were obtained in another study, showing that HFDs could promote intestinal tumor progression in genetically susceptible K-rasG12Dint mice in the absence of obesity but based on marked alterations in gut bacterial communities (109). Exposure to HFDs led to many intestinal adaptations: (i) intestinal villi became shorter, while intestinal crypt depth was increased (108, 110); (ii) the numbers of intestinal stem cells (ISCs) increased while those of Paneth niche cells decreased; (iii) ISCs could expand in the absence of signals from Paneth cells; (iv) progenitors became more stem-cell like, acquiring the capacity to form intestinal organoids. These adaptations may partially contribute to the development of early intestinal tumorigenesis and subsequently cancer (108). There are two well-defined mechanisms for increased cancer risk induced by HFDs. One is via enhancing stemness and tumorigenicity of intestinal progenitors through activation of PPARδ/β-catenin signaling cascade (108). Consistently, other studies also show that HFDs and sustained PPARδ activation are associated with colorectal cancer initiation and progression (107, 111, 112). However, inhibition of PPARδ signaling may exert modest anti-cancer effects (113). The other is through inactivation of nuclear bile acid (BA) receptor farnesoid X receptor (FXR), which resulted in BA deregulation and promoted colon cell proliferation (110). These data indicate that inhibition of PPARδ and activation of FXR after exposure to HFDs may blunt tumor initiation and progression. Future investigations are needed to address how HFDs induce activation of PPARδ signaling and inhibition of FXR signaling.
Potential Treatment Options
The demonstration of a link between HFDs and metabolic disorders and certain types of cancer has prompted research into strategies to reduce HFD-driven diseases and cancer. Undoubtedly, lifestyle changes, such as improving dietary regiments and enhancing physical activity, are the most successful interventions. In this regard, dietary interventions aimed at decreasing circulating FFAs and limiting inflammation originated from alterations in gut microbial communities are attracting increasing clinical interest. Although still in its infancy, promising findings have been reported by several studies. Circulating FFAs levels are strongly decreased without causing ectopic lipid accumulation in HFD-fed mice with dietary supplementation of Atglistatin, a selective inhibitor of adipose TG lipase, accompanied by decreased weight gain, insulin resistance, and liver diseases (114). Preclinical studies have also reported a metabolic benefit associated with the anti-inflammatory impacts of targeting PPARγ (115), ERS (116), IKKβ/NF-κB (117), and shifts in gut microbial communities (118). For instance, HFD-fed mice treated with Lactobacillus plantarum KY1032 and Lactobacillus curvatus HY7601 exhibited decreased body weight, fat mass, and levels of circulating leptin, insulin, total cholesterol and liver toxicity biomarkers, accompanied by alterations in gut bacterial composition and diversity (118). In addition, markers of inflammation and obesity are reduced with dietary supplementation with amino acid (especially leucine) and plant components (such as luteolin and flavonoids), alone or in combination, as were performed in HFD-fed mice (46, 119–123). Although there are additional undefined approaches, the above-mentioned findings give the field a critical boost and may be translated into human studies, thus improving interventional strategies to stem the negative effects of HFDs.
Conclusions and Perspectives for Future Studies
The literature reviewed here suggests that FFAs and LPS have been established as independent factors aggravating inflammation during HFD-induced chronic diseases and certain type of cancer. Targeting these factors may provide effective approaches for therapies against associated metabolic perturbations. Although great strides have been made in animal models to understand the signaling pathways implicated in HFDs-induced chronic diseases and certain type of cancer, more effects should be made to confirm whether these signaling pathways are physiologically valid in humans, and additional transcription factors and target genes need to be identified to reduce the overwhelming burden of chronic diseases and cancer.
Author Contributions
YD and LZ wrote the manuscript, KX designed the manuscirpt, CZ, BS, FL, and XK revised the manuscript. All authors read and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was jointly supported by the Nature Science Foundation of Hunan Province (S2014J504I), the National Natural Science Foundation of China (31601953), the Guangdong Province Key Laboratory of Animal Nutritional Regulation (DWYYTK-18KF002), the Youth Innovation Promotion Association CAS (2016326), the Science and Technology Projects of Hunan Province (2016SK3022, 2017RS3058), the Major Project of Hunan Province (2015NK1002), the Key Project of Research and Development Plan of Hunan Province (2016NK2170) and Youth Innovation Team Project of ISA, CAS (2017QNCXTD_ZCS), the Earmarked Fund for China Agriculture Research System (CARS-35), and the Plant Germplasm Resources Innovation Project of Strategic Biological Resources Service Network Plan From The Chinese Academy of Sciences (ZSZC-011).
References
1. Stevens GA, Singh GM, Lu Y, Danaei G, Lin JK, Finucane MM., et al. National, regional, and global trends in adult overweight and obesity prevalences. Popul Health Metr. (2012) 10:22. doi: 10.1186/1478-7954-10-22
2. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer (2004) 4:579–91. doi: 10.1038/nrc1408
3. Kennedy A, Martinez K, Chuang CC, LaPoint K, McIntosh M. Saturated fatty acid-mediated inflammation and insulin resistance in adipose tissue: mechanisms of action and implications. J Nutr. (2009) 139:1–4. doi: 10.3945/jn.108.098269
4. Tchernof A, Després JP. Pathophysiology of human visceral obesity: an update. Physiol Rev. (2013) 93:359–404. doi: 10.1152/physrev.00033.2011
5. Murphy EA, Velazquez KT, Herbert KM. Influence of high-fat diet on gut microbiota: a driving force for chronic disease risk. Curr Opin Clin Nutr. (2015) 18:515–20. doi: 10.1097/MCO.0000000000000209
6. Guillemot-Legris O, Masquelier J, Everard A, Cani PD, Alhouayek M, Muccioli GG. High-fat diet feeding differentially affects the development of inflammation in the central nervous system. J Neuroinflamm. (2016) 13:206. doi: 10.1186/s12974-016-0666-8
7. Sanmiguel C, Gupta A, Mayer EA. Gut microbiome and obesity: a plausible explanation for obesity. Curr Obes Rep. (2015) 4:250–61. doi: 10.1007/s13679-015-0152-0
8. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE., et al. A core gut microbiome in obese and lean twins. Nature (2009) 457:480–4. doi: 10.1038/nature07540
9. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G., et al. Richness of human gut microbiome correlates with metabolic markers. Nature (2013) 500:541–6. doi: 10.1038/nature12506
10. Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NM, Magness S., et al. High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE (2010) 5:e12191. doi: 10.1371/journal.pone.0012191
11. Kim KA, Gu W, Lee IA, Joh EH, Kim DH. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE (2012) 7:e47713. doi: 10.1371/journal.pone.0047713
12. Bleau C, Karelis AD, St-Pierre DH, Lamontagne L. Crosstalk between intestinal microbiota, adipose tissue and skeletal muscle as an early event in systemic low-grade inflammation and the development of obesity and diabetes. Diabetes Metab Res Rev. (2015) 31:545–61. doi: 10.1002/dmrr.2617
13. Fujiyama Y, Hokari R, Miura S, Watanabe C, Komoto S, Oyama T., et al. Butter feeding enhances TNF-alpha production from macrophages and lymphocyte adherence in murine small intestinal microvessels. J Gastroen Hepatol. (2007) 22:1838–45. doi: 10.1111/j.1440-1746.2007.04905.x
14. Yoshida H, Miura S, Kishikawa H, Hirokawa M, Nakamizo H, Nakatsumi RC., et al. Fatty acids enhance GRO/CINC-1 and interleukin-6 production in rat intestinal epithelial cells. J Nutr. (2001) 131:2943–50. doi: 10.1093/jn/131.11.2943
15. Konrad D, Wueest S. The gut-adipose-liver axis in the metabolic syndrome. Physiology (2014) 29:304–13. doi: 10.1152/physiol.00014.2014
16. Antonioli L, Caputi V, Fornai M, Pellegrini C, Gentile D, Giron MC., et al. Interplay between colonic inflammation and tachykininergic pathways in the onset of colonic dysmotility in a mouse model of diet-induced obesity. Int J Obes. (2018). doi: 10.1038/s41366-018-0166-2. [Epub ahead of print].
17. Antonioli L, Pellegrini C, Fornai M, Tirotta E, Gentile D, Benvenuti L., et al. Colonic motor dysfunctions in a mouse model of high-fat diet-induced obesity: an involvement of A2B adenosine receptors. Purinergic Signal. (2017) 13:497–510. doi: 10.1007/s11302-017-9577-0
18. Tsukumo DM, Carvalho BM, Carvalho Filho MA, Saad MJ. Translational research into gut microbiota: new horizons on obesity treatment: updated 2014. Arch Endocrinol Metab. (2015) 59:154–60. doi: 10.1590/2359-3997000000029
19. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKK beta/NF-kappa B and ER stress link overnutrition to energy imbalance and obesity. Cell (2008) 135:61–73. doi: 10.1016/j.cell.2008.07.043
20. De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC., et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology (2005) 146:4192–9. doi: 10.1210/en.2004-1520
21. Cai D, Liu T. Hypothalamic inflammation: a double-edged sword to nutritional diseases. Ann NY Acad Sci. (2011) 1243:E1–39. doi: 10.1111/j.1749-6632.2011.06388.x
22. Wang J, Obici S, Morgan K, Barzilai N, Feng Z, Rossetti L. Overfeeding rapidly induces leptin and insulin resistance. Diabetes (2001) 50:2786–91. doi: 10.2337/diabetes.50.12.2786
23. Woods SC, D'Alessio DA, Tso P, Rushing PA, Clegg DJ, Benoit SC., et al. Consumption of a high-fat diet alters the homeostatic regulation of energy balance. Physiol Behav. (2004) 83:573–8. doi: 10.1016/j.physbeh.2004.07.026
24. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. (2011) 121:2111–7. doi: 10.1172/JCI57132
25. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M., et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. (2009) 15:914–20. doi: 10.1038/nm.1964
26. Caesar R, Tremaroli V, Kovatcheva-Datchary P, Cani PD, Bäckhed F. Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metab. (2015) 22:658–68. doi: 10.1016/j.cmet.2015.07.026
27. Lee BC, Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. BBA Mol. Basis Dis. (2014) 1842:446–62. doi: 10.1016/j.bbadis.2013.05.017
28. Bhattacharjee J, Kirby M, Softic S, Miles L, Salazar-Gonzalez RM, Shivakumar P., et al. Hepatic natural killer T-cell and CD8+ T-cell signatures in mice with nonalcoholic steatohepatitis. Hepatol Commun. (2017) 1:299–310. doi: 10.1002/hep4.1041
29. Yao K, Duan Y, Li F, Tan B, Hou Y, Wu G., et al. Leucine in obesity: therapeutic prospects. Trends Pharmacol Sci. (2016) 37:714–27. doi: 10.1016/j.tips.2016.05.004
30. Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes (2011) 60:2474–83. doi: 10.2337/db11-0194
31. Wu Y, Wu T, Wu J, Zhao L, Li Q, Varghese Z., et al. Chronic inflammation exacerbates glucose metabolism disorders in C57BL/6J mice fed with high-fat diet. J Endocrinol. (2013) 219:195–204. doi: 10.1530/JOE-13-0160
32. Ellenbroek JH, Töns HA, de Graaf N, Loomans CJ, Engelse MA, Vrolijk H., et al. Topologically heterogeneous beta cell adaptation in response to high-fat diet in mice. PLoS ONE (2013) 8:e56922. doi: 10.1371/journal.pone.0056922
33. Heydemann A. An overview of murine high fat diet as a model for type 2 diabetes mellitus. J Diabetes Res. (2016) 2016:2902351. doi: 10.1155/2016/2902351
34. Calegari VC, Torsoni AS, Vanzela EC, Araújo EP, Morari J, Zoppi CC., et al. Inflammation of the hypothalamus leads to defective pancreatic islet function. J Biol Chem. (2011) 286:12870–80. doi: 10.1074/jbc.M110.173021
35. Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R., et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes (2007) 56:2356–70. doi: 10.2337/db06-1650
36. Tersey SA, Maier B, Nishiki Y, Maganti AV, Nadler JL, Mirmira RG. 12-lipoxygenase promotes obesity-induced oxidative stress in pancreatic islets. Mol Cell Biol. (2014) 34:3735–45. doi: 10.1128/MCB.00157-14
37. Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. (2017) 23:804–14. doi: 10.1038/nm.4350
38. Benard O, Lim J, Apontes P, Jing X, Angeletti RH, Chi Y. Impact of high-fat diet on the proteome of mouse liver. J Nutr Biochem. (2016) 31:10–9. doi: 10.1016/j.jnutbio.2015.12.012
39. Csóka B, Töro G, Vindeirinho J, Varga ZV, Koscsó B, Németh ZH., et al. A2A adenosine receptors control pancreatic dysfunction in high-fat-diet-induced obesity. FASEB J. (2017) 31:4985–97. doi: 10.1096/fj.201700398R
40. Johnston-Cox H, Koupenova M, Yang D, Corkey B, Gokce N, Farb MG., et al. The A2b adenosine receptor modulates glucose homeostasis and obesity. PLoS ONE (2012) 7:e40584. doi: 10.1371/journal.pone.0040584
41. Johnston-Cox H, Eisenstein AS, Koupenova M, Carroll S, Ravid K. The macrophage A2b adenosine receptor regulates tissue insulin sensitivity. PLoS ONE (2014) 9:e98775. doi: 10.1371/journal.pone.0098775
42. Martins F, Campos DH, Pagan LU, Martinez PF, Okoshi K, Okoshi MP., et al. High-fat diet promotes cardiac remodeling in an experimental model of obesity. Arq Bras Cardiol. (2015) 105:479–86. doi: 10.5935/abc.20150095
43. Aghajani M, Imani A, Faghihi M, Mahdavi MRV, Mahboubi S, et al. Does increased nitric oxide production and oxidative stress due to high fat diet affect cardiac function after myocardial infarction? J Cell Mol Anesth. (2017) 2:3–8. doi: 10.22037/jcma.v2i1.14288
44. Mauro C, Smith J, Cucchi D, Coe D, Fu H, Bonacina F., et al. Obesity-induced metabolic stress leads to biased effector memory CD4+ T cell differentiation via PI3K p110delta-Akt-mediated signals. Cell Metab. (2017) 25:593–609. doi: 10.1016/j.cmet.2017.01.008
45. Dunn SM, Hilgers RHP, Das KC. Endothelial dysfunction and outward remodeling in coronary and mesenteric arteries in response to high fat diet in mice. FASEB J. (2016) 30:1282– 6. doi: 10.1096/fasebj.30.1_supplement.1282.6
46. Gentile D, Fornai M, Pellegrini C, Colucci R, Benvenuti L, Duranti E., et al. Luteolin prevents cardiometabolic alterations and vascular dysfunction in mice with HFD-induced obesity. Front Pharmacol. (2018) 9:1094. doi: 10.3389/fphar.2018.01094
47. Martins MA, Catta-Preta M, Mandarim-de-Lacerda CA, Aguila MB, Brunini TC, Mendes-Ribeiro AC. High fat diets modulate nitric oxide biosynthesis and antioxidant defence in red blood cells from C57BL/6 mice. Arch Biochem Biophys. (2010) 499:56–61. doi: 10.1016/j.abb.2010.04.025
48. Ozüyaman B, Grau M, Kelm M, Merx MW, Kleinbongard P. RBC NOS: regulatory mechanisms and therapeutic aspects. Trends Mol Med. (2008) 14:314–22. doi: 10.1016/j.molmed.2008.05.002
49. Zhang H, Dellsperger KC, Zhang C. The link between metabolic abnormalities and endothelial dysfunction in type 2 diabetes: an update. Basic Res Cardiol. (2012) 107:237. doi: 10.1007/s00395-011-0237-1
50. Unruh D, Srinivasan R, Benson T, Haigh S, Coyle D, Batra N., et al. Red blood cell dysfunction induced by high-fat diet: potential implications for obesity-related atherosclerosis. Circulation (2015) 132:1898–908. doi: 10.1161/CIRCULATIONAHA.115.017313
51. Gulhane M, Murray L, Lourie R, Tong H, Sheng YH, Wang R., et al. High fat diets induce colonic epithelial cell stress and inflammation that is reversed by IL-22. Sci Rep. (2016) 6:28990. doi: 10.1038/srep28990
52. Ho W, Spiegel BM. The relationship between obesity and functional gastrointestinal disorders: causation, association, or neither? Gastroenterol Hepatol. (2008) 4:572–8.
53. Kim IW, Myung SJ, Do MY, Ryu YM, Kim MJ, Do EJ., et al. Western-style diets induce macrophage infiltration and contribute to colitis-associated carcinogenesis. J Gastroen Hepatol. (2010) 25:1785–94. doi: 10.1111/j.1440-1746.2010.06332.x
54. Laroui H, Ingersoll SA, Liu HC, Baker MT, Ayyadurai S, Charania MA., et al. Dextran sodium sulfate (DSS) induces colitis in mice by forming nano-lipocomplexes with medium-chain-length fatty acids in the colon. PLoS ONE (2012) 7:e32084. doi: 10.1371/journal.pone.0032084
55. Okada Y, Tsuzuki Y, Sato H, Narimatsu K, Hokari R, Kurihara C., et al. Trans fatty acids exacerbate dextran sodium sulphate-induced colitis by promoting the up-regulation of macrophage-derived proinflammatory cytokines involved in T helper 17 cell polarization. Clin Exp Immunol. (2013) 174:459–71. doi: 10.1111/cei.12200
56. Lu P, Bar-Yoseph F, Levi L, Lifshitz Y, Witte-Bouma J, de Bruijn AC., et al. High beta-palmitate fat controls the intestinal inflammatory response and limits intestinal damage in mucin muc2 deficient mice. PLoS ONE (2013) 8:e65878. doi: 10.1371/journal.pone.0065878
57. Paik J, Fierce Y, Treuting PM, Brabb T, Maggio-Price L. High-fat diet-induced obesity exacerbates inflammatory bowel disease in genetically susceptible Mdr1a(-/-) male mice. J Nutr. (2013) 143:1240–7. doi: 10.3945/jn.113.174615
58. Gruber L, Kisling S, Lichti P, Martin FP, May S, Klingenspor M., et al. High fat diet accelerates pathogenesis of murine crohn's disease-like ileitis independently of obesity. PLoS ONE (2013) 8:e71661. doi: 10.1371/journal.pone.0071661
59. Fysekidis M, Bouchoucha M, Bihan H, Reach G, Benamouzig R, Catheline JM. Prevalence and co-occurrence of upper and lower functional gastrointestinal symptoms in patients eligible for bariatric surgery. Obes Surg. (2011) 22:403–10. doi: 10.1007/s11695-011-0396-z
60. Hou JK, Abraham B, El-Serag H. Dietary intake and risk of developing inflammatory bowel disease: a systematic review of the literature. Am J Gastroenterol. (2011) 106:563–73. doi: 10.1038/ajg.2011.44
61. Chapman-Kiddell CA, Davies PS, Gillen L, Radford-Smith GL. Role of diet in the development of inflammatory bowel disease. Inflamm Bowel Dis. (2010) 16:137–51. doi: 10.1002/ibd.20968
62. Tomas J, Mulet C, Saffarian A, Cavin JB, Ducroc R, Regnault B., et al. High-fat diet modifies the PPAR-γ pathway leading to disruption of microbial and physiological ecosystem in murine small intestine. Proc Natl Acad Sci USA. (2016) 113:E5934–43. doi: 10.1073/pnas.1612559113
63. Agus A, Denizot J, Thévenot J, Martinez-Medina M, Massier S, Sauvanet P., et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci Rep. (2016) 6:19032. doi: 10.1038/srep19032
64. Bertrand RL, Senadheera S, Markus I, Liu L, Howitt L, Chen H., et al. A western diet increases serotonin availability in rat small intestine. Endocrinology (2011) 152:36–47. doi: 10.1210/en.2010-0377
65. Nezami BG, Mwangi SM, Lee JE, Jeppsson S, Anitha M, Yarandi SS., et al. MicroRNA 375 mediates palmitate-induced enteric neuronal damage and high-fat diet-induced delayed intestinal transit in mice. Gastroenterology (2014) 146:473–83. doi: 10.1053/j.gastro.2013.10.053
66. Chen Q, Shou P, Zheng C, Jiang M, Cao G, Yang Q., et al. Fate decision of mesenchymal stem cells: adipocytes or osteoblasts? Cell Death Differ. (2016) 23:1128–39. doi: 10.1038/cdd.2015.168
67. Styner M, Thompson WR, Galior K, Uzer G, Wu X, Kadari S., et al. Bone marrow fat accumulation accelerated by high fat diet is suppressed by exercise. Bone (2014) 64:39–46. doi: 10.1016/j.bone.2014.03.044
68. Corwin RL, Hartman TJ, Maczuga SA, Graubard BI. Dietary saturated fat intake is inversely associated with bone density in humans: analysis of NHANES III. J Nutr. (2006) 136:159–65. doi: 10.1093/jn/136.1.159
69. Zhao LJ, Liu YJ, Liu PY, Hamilton J, Recker RR, Deng HW. Relationship of obesity with osteoporosis. J Clin Endocr Metab. (2007) 92:1640–6. doi: 10.1210/jc.2006-0572
70. Shapses SA, Sukumar D. Bone metabolism in obesity and weight loss. Annu Rev Nutr. (2012) 32:287–309. doi: 10.1146/annurev.nutr.012809.104655
71. Xiao Y, Cui J, Li YX, Shi YH, Wang B, Le GW., et al. Dyslipidemic high-fat diet affects adversely bone metabolism in mice associated with impaired antioxidant capacity. Nutrition (2011) 27:214–20. doi: 10.1016/j.nut.2009.11.012
72. Cao JJ, Gregoire BR, Gao H. High-fat diet decreases cancellous bone mass but has no effect on cortical bone mass in the tibia in mice. Bone (2009) 44:1097–104. doi: 10.1016/j.bone.2009.02.017
74. Lecka-Czernik B, Stechschulte LA, Czernik PJ, Dowling AR. High bone mass in adult mice with diet-induced obesity results from a combination of initial increase in bone mass followed by attenuation in bone formation; implications for high bone mass and decreased bone quality in obesity. Mol Cell Endocrinol. (2015) 410:35–41. doi: 10.1016/j.mce.2015.01.001
75. Tian L, Wang C, Xie Y, Wan S, Zhang K, Yu X. High fructose and high fat exert different effects on changes in trabecular bone micro-structure. J Nutr Health Aging (2018) 22:361–70. doi: 10.1007/s12603-017-0933-0
76. Malvi P, Piprode V, Chaube B, Pote ST, Mittal M, Chattopadhyay N., et al. High fat diet promotes achievement of peak bone mass in young rats. Biochem. Biophys. Res. Commun. (2014) 455:133–8. doi: 10.1016/j.bbrc.2014.10.131
77. Núñez NP, Carpenter CL, Perkins SN, Berrigan D, Jaque SV, Ingles SA., et al. Extreme obesity reduces bone mineral density: complementary evidence from mice and women. Obesity (2007) 15:1980–7. doi: 10.1038/oby.2007.236
78. Chen GL, Luo Y, Eriksson D, Meng X, Qian C, Bäuerle T., et al. High fat diet increases melanoma cell growth in the bone marrow by inducing osteopontin and interleukin 6. Oncotarget (2016) 7:26653–69. doi: 10.18632/oncotarget.8474
79. Upadhyay J, Farr OM, Mantzoros CS. The role of leptin in regulating bone metabolism. Metabolism (2015) 64:105–13. doi: 10.1016/j.metabol.2014.10.021
80. Lin J, Judd S, Le A, Ard J, Newsome BB, Howard G., et al. Associations of dietary fat with albuminuria and kidney dysfunction. Am J Clin Nutr. (2010) 92:897–904. doi: 10.3945/ajcn.2010.29479
81. Ebenezer PJ, Mariappan N, Elks CM, Haque M, Francis J. Diet-induced renal changes in Zucker rats are ameliorated by the superoxide dismutase mimetic TEMPOL. Obesity (2009) 17:1994–2002. doi: 10.1038/oby.2009.137
82. Elmarakby AA, Imig JD. Obesity is the major contributor to vascular dysfunction and inflammation in high-fat diet hypertensive rats. Clin Sci. (2010) 118:291–301. doi: 10.1042/CS20090395
83. Tomiyama-Hanayama M, Rakugi H, Kohara M, Mima T, Adachi Y, Ohishi M, et al. Effect of interleukin-6 receptor blockage on renal injury in apolipoprotein E-deficient mice. Am J Physiol Renal (2009) 297:F679–84. doi: 10.1152/ajprenal.90680.2008
84. Jiang T, Wang Z, Proctor G, Moskowitz S, Liebman SE, Rogers T., et al. Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulosclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. J Biol Chem. (2005) 280:32317–25. doi: 10.1074/jbc.M500801200
85. Wang XX, Jiang T, Shen Y, Adorini L, Pruzanski M, Gonzalez FJ, et al. The farnesoid X receptor modulates renal lipid metabolism and diet-induced renal inflammation, fibrosis, and proteinuria. Am J Physiol Renal (2009) 297:F1587–96. doi: 10.1152/ajprenal.00404.2009
86. Almeida-Suhett CP, Graham A, Chen Y, Deuster P. Behavioral changes in male mice fed a high-fat diet are associated with IL-1beta expression in specific brain regions. Physiol Behav. (2017) 169:130–40. doi: 10.1016/j.physbeh.2016.11.016
87. Aslani S, Vieira N, Marques F, Costa PS, Sousa N, Palha JA. The effect of high-fat diet on rat's mood, feeding behavior and response to stress. Transl Psychiatry (2015) 5:e684. doi: 10.1038/tp.2015.178
88. Mayeux R, Stern Y. Epidemiology of Alzheimer disease. CSH. Perspect Med. (2012) 2:a006239. doi: 10.1101/cshperspect.a006239
89. Kothari V, Luo Y, Tornabene T, O'Neill AM, Greene MW, Geetha T., et al. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim Biophys Acta (2017) 1863:499–508. doi: 10.1016/j.bbadis.2016.10.006
90. Bruce-Keller AJ, Salbaum JM, Luo M, Blanchard E, Taylor CM, Welsh DA., et al. Obese-type gut microbiota induce neurobehavioral changes in the absence of obesity. Biol Psychiatry (2015) 77:607–15. doi: 10.1016/j.biopsych.2014.07.012
91. Busquets O, Ettcheto M, Pallàs M, Beas-Zarate C, Verdaguer E, Auladell C., et al. Long-term exposition to a high fat diet favors the appearance of beta-amyloid depositions in the brain of C57BL/6J mice. A potential model of sporadic Alzheimer's disease. Mech Ageing Dev. (2017) 162:38–45. doi: 10.1016/j.mad.2016.11.002
92. Freeman LR, Zhang L, Nair A, Dasuri K, Francis J, Fernandez-Kim SO., et al. Obesity increases cerebrocortical reactive oxygen species and impairs brain function. Free Radic Biol Med. (2013) 56:226–33. doi: 10.1016/j.freeradbiomed.2012.08.577
93. Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK., et al. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J Neuroimmunol. (2010) 219:25–32. doi: 10.1016/j.jneuroim.2009.11.010
94. de Noronha SR, Campos GV, Abreu AR, de Souza AA, Chianca DA, de Menezes RC. High fat diet induced-obesity facilitates anxiety-like behaviors due to GABAergic impairment within the dorsomedial hypothalamus in rats. Behav Brain Res. (2017) 316:38–46. doi: 10.1016/j.bbr.2016.08.042
95. Lin B, Hasegawa Y, Takane K, Koibuchi N, Cao C, Kim-Mitsuyama S. High-fat-diet intake enhances cerebral amyloid angiopathy and cognitive impairment in a mouse model of alzheimer's disease, independently of metabolic disorders. J Am Heart Assoc. (2016) 5:e003154. doi: 10.1161/JAHA.115.003154
96. Petrov D, Pedrós I, Artiach G, Sureda FX, Barroso E, Pallàs M., et al. High-fat diet-induced deregulation of hippocampal insulin signaling and mitochondrial homeostasis deficiences contribute to Alzheimer disease pathology in rodents. BBA Mol. Basis Dis. (2015) 1852:1687–99. doi: 10.1016/j.bbadis.2015.05.004
97. Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K., et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell (2014) 26:549–64. doi: 10.1016/j.ccell.2014.09.003
98. Gimble JM, Guilak F, Nuttall ME, Sathishkumar S, Vidal M, Bunnell BA. In vitro differentiation potential of mesenchymal stem cells. Transfus. Med. Hemother. (2008) 35:228–38. doi: 10.1159/000124281
99. Seo BR, Bhardwaj P, Choi S, Gonzalez J, Andresen Eguiluz RC, Wang K., et al. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci Transl Med. (2015) 7:301ra130. doi: 10.1126/scitranslmed.3010467
100. Wolfson B, Zhang Y, Gernapudi R, Duru N, Yao Y, Lo PK., et al. A High-fat diet promotes mammary gland myofibroblast differentiation through microRNA 140 downregulation. Mol Cell Biol. (2017) 37:e00461–00416. doi: 10.1128/MCB.00461-16
101. Graham S, Haughey B, Marshall J, Brasure J, Zielezny M, Freudenheim J, et al. Diet in the epidemiology of gastric-cancer. Nutr Cancer (1990) 13:19–34. doi: 10.1080/01635589009514042
102. Hu J, La Vecchia C, Negri E, de Groh M, Morrison H, Mery L., et al. Macronutrient intake and stomach cancer. Cancer Cause Control (2015) 26:839–47. doi: 10.1007/s10552-015-0557-9
103. Han J, Jiang Y, Liu X, Meng Q, Xi Q, Zhuang Q., et al. Dietary fat intake and risk of gastric cancer: a meta-analysis of observational studies. PLoS ONE (2015) 10:e0138580. doi: 10.1371/journal.pone.0138580
104. Arita S, Kinoshita Y, Ushida K, Enomoto A, Inagaki-Ohara K. High-fat diet feeding promotes stemness and precancerous changes in murine gastric mucosa mediated by leptin receptor signaling pathway. Arch Biochem Biophys. (2016) 610:16–24. doi: 10.1016/j.abb.2016.09.015
105. Mihaylova MM, Sabatini DM, Yilmaz ÖH. Dietary and metabolic control of stem cell function in physiology and cancer. Cell Stem Cell (2014) 14:292–305. doi: 10.1016/j.stem.2014.02.008
106. Finucane MM, Stevens GA, Cowan MJ, Danaei G, Lin JK, Paciorek CJ., et al. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet (2011) 377:557–67. doi: 10.1016/S0140-6736(10)62037-5
107. Baltgalvis KA, Berger FG, Peña MM, Davis JM, Carson JA. The interaction of a high-fat diet and regular moderate intensity exercise on intestinal polyp development in Apc(Min/+) mice. Cancer Prev Res. (2009) 2:641–9. doi: 10.1158/1940-6207.CAPR-09-0017
108. Beyaz S, Mana MD, Roper J, Kedrin D, Saadatpour A, Hong SJ., et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature (2016) 531:53–8. doi: 10.1038/nature17173
109. Schulz MD, Atay C, Heringer J, Romrig FK, Schwitalla S, Aydin B., et al. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature (2014) 514:508–12. doi: 10.1038/nature13398
110. Dermadi D, Valo S, Ollila S, Soliymani R, Sipari N, Pussila M., et al. Western diet deregulates bile acid homeostasis, cell proliferation and tumorigenesis in colon. Cancer Res. (2017) 77:3352–63. doi: 10.1158/0008-5472.CAN-16-2860
111. Wang D, Fu L, Ning W, Guo L, Sun X, Dey SK., et al. Peroxisome proliferator-activated receptor delta promotes colonic inflammation and tumor growth. Proc Natl Acad Sci USA. (2014) 111:7084–9. doi: 10.1073/pnas.1324233111
112. Zuo X, Peng Z, Moussalli MJ, Morris JS, Broaddus RR, Fischer SM., et al. Targeted genetic disruption of peroxisome proliferator-activated receptor-delta and colonic tumorigenesis. J Natl Cancer (2009) 101:762–67. doi: 10.1093/jnci/djp078
113. Neels JG, Grimaldi PA. Physiological functions of peroxisome proliferator-activated receptor beta. Physiol Rev. (2014) 94:795–858. doi: 10.1152/physrev.00027.2013
114. Schweiger M, Romauch M, Schreiber R, Grabner GF, Hütter S, Kotzbeck P, et al. Pharmacological inhibition of adipose triglyceride lipase corrects high-fat diet-induced insulin resistance and hepatosteatosis in mice. Nat Commun. (2017) 8:14859. doi: 10.1038/ncomms14859
115. Hevener AL, Olefsky JM, Reichart D, Nguyen MT, Bandyopadyhay G, Leung HY., et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest. (2007) 117:1658–69. doi: 10.1172/JCI31561
116. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO., et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science (2006) 313:1137–40. doi: 10.1126/science.1128294
117. Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J., et al. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci. (2008) 1:36–43. doi: 10.1111/j.1752-8062.2008.00026.x
118. Park DY, Ahn YT, Park SH, Huh CS, Yoo SR, Yu R., et al. Supplementation of Lactobacillus curvatus HY7601 and Lactobacillus plantarum KY1032 in diet-induced obese mice is associated with gut microbial changes and reduction in obesity. PLoS ONE (2013) 8:e59470. doi: 10.1371/journal.pone.0059470
119. Seo DB, Jeong HW, Cho D, Lee BJ, Lee JH, Choi JY., et al. Fermented green tea extract alleviates obesity and related complications and alters gut microbiota composition in diet-induced obese mice. J Med Food (2015) 18:549–56. doi: 10.1089/jmf.2014.3265
120. Zhang Y, Guo K, LeBlanc RE, Loh D, Schwartz GJ, Yu YH. Increasing dietary leucine intake reduces diet-induced obesity and improves glucose and cholesterol metabolism in mice via multimechanisms. Diabetes (2007) 56:1647–54. doi: 10.2337/db07-0123
121. Bruckbauer A, Zemel MB, Thorpe T, Akula MR, Stuckey AC, Osborne D., et al. Synergistic effects of leucine and resveratrol on insulin sensitivity and fat metabolism in adipocytes and mice. Nutr Metab. (2012) 9:77. doi: 10.1186/1743-7075-9-77
122. Gentile D, Fornai M, Pellegrini C, Colucci R, Blandizzi C, Antonioli L. Dietary flavonoids as a potential intervention to improve redox balance in obesity and related co-morbidities: a review. Nutr Res Rev. (2018) 31:239–47. doi: 10.1017/S0954422418000082
Keywords: high-fat diets, inflammation, disease, mechanisms, drug therapy
Citation: Duan Y, Zeng L, Zhang C, Song B, Li F, Kong X and Xu K (2018) Inflammatory Links Between High Fat Diets and Diseases. Front. Immunol. 9:2649. doi: 10.3389/fimmu.2018.02649
Received: 12 September 2018; Accepted: 26 October 2018;
Published: 13 November 2018.
Edited by:
Philippe Saas, INSERM U1098 Interactions Hôte-Greffon-Tumeur and Ingénierie Cellulaire et Génique, FranceReviewed by:
Luca Antonioli, Università degli Studi di Pisa, ItalyMatteo Fornai, Università degli Studi di Pisa, Italy
Kawaljit Kaur, University of California, Los Angeles, United States
Copyright © 2018 Duan, Zeng, Zheng, Song, Li, Kong and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kang Xu, eHVrYW5nQGlzYS5hYy5jbg==
†These authors have contributed equally to this work