 Elouise E. Kroon1*
Elouise E. Kroon1* Anna K. Coussens2,3,4
Anna K. Coussens2,3,4 Craig Kinnear1
Craig Kinnear1 Marianna Orlova5,6,7
Marianna Orlova5,6,7 Marlo Möller1
Marlo Möller1 Allison Seeger2
Allison Seeger2 Robert J. Wilkinson2,8,9
Robert J. Wilkinson2,8,9 Eileen G. Hoal1
Eileen G. Hoal1 Erwin Schurr5,6,7
Erwin Schurr5,6,7- 1DST-NRF Centre of Excellence for Biomedical Tuberculosis Research, South African Medical Research Council Centre for Tuberculosis Research, Division of Molecular Biology and Human Genetics, Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa
- 2Wellcome Centre for Infectious Diseases Research in Africa, Institute of Infectious Disease and Molecular Medicine, University of Cape Town, Cape Town, South Africa
- 3Infection and Immunity Division, Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, Australia
- 4Division of Medical Biology, Faculty of Medicine, Dentistry and Health Sciences, University of Melbourne, Melbourne, VIC, Australia
- 5Program in Infectious Diseases and Immunity in Global Health, The Research Institute of the McGill University Health Centre, Montreal, QC, Canada
- 6McGill International TB Centre, McGill University, Montreal, QC, Canada
- 7Departments of Medicine and Human Genetics, McGill University, Montreal, QC, Canada
- 8Department of Medicine, Imperial College London, London, United Kingdom
- 9The Francis Crick Institute, London, United Kingdom
Certain individuals are able to resist Mycobacterium tuberculosis infection despite persistent and intense exposure. These persons do not exhibit adaptive immune priming as measured by tuberculin skin test (TST) and interferon-γ (IFN-γ) release assay (IGRA) responses, nor do they develop active tuberculosis (TB). Genetic investigation of individuals who are able to resist M. tuberculosis infection shows there are likely a combination of genetic variants that contribute to the phenotype. The contribution of the innate immune system and the exact cells involved in this phenotype remain incompletely elucidated. Neutrophils are prominent candidates for possible involvement as primers for microbial clearance. Significant variability is observed in neutrophil gene expression and DNA methylation. Furthermore, inter-individual variability is seen between the mycobactericidal capacities of donor neutrophils. Clearance of M. tuberculosis infection is favored by the mycobactericidal activity of neutrophils, apoptosis, effective clearance of cells by macrophages, and resolution of inflammation. In this review we will discuss the different mechanisms neutrophils utilize to clear M. tuberculosis infection. We discuss the duality between neutrophils' ability to clear infection and how increasing numbers of neutrophils contribute to active TB severity and mortality. Further investigation into the potential role of neutrophils in innate immune-mediated M. tuberculosis infection resistance is warranted since it may reveal clinically important activities for prevention as well as vaccine and treatment development.
Introduction
Not all individuals exposed to Mycobacterium tuberculosis become infected as inferred by a lack of T cell memory response to M. tuberculosis antigens. Moreover, these individuals do not develop signs and symptoms suggestive of ‘active tuberculosis' (TB). The majority of M. tuberculosis infected individuals remain asymptomatic with what is known as latent tuberculosis infection (LTBI).
Only 5–15% of those infected will progress to active TB disease, given they have no underlying co-morbidity which would increase their risk further (1, 2). This resulted in an estimated 10.4 million new cases and 1,674 million TB deaths reported in 2016 (3). The remaining 85–95% of persons with LTBI who do not develop disease indicates that the majority of those infected have a natural immunity to prevent the progression from infection to disease. Similarly, certain individuals who are highly exposed, never develop evidence of infection. This suggests that they are naturally resistant to M. tuberculosis and can prevent infection via an innate immune response prior to adaptive immune cell priming, and are known as “innate resisters” (4). The mechanisms that underlie the resistance to infection in persons of the “innate resister” phenotype are not fully known. In the present article, we explore the possible contribution of neutrophils to innate infection resistance.
Evidence of M. tuberculosis Infection
LTBI is defined as the presence of M. tuberculosis-specific T-cell sensitization in the absence of clinical signs and symptoms of TB. Host sensitization is used as a proxy for this assumed latent M. tuberculosis infection in human hosts and is measured by reactivity to mycobacterial antigens using the tuberculin skin test (TST) or interferon-γ (IFN-γ) release assays (IGRAs). The TST is performed by injecting purified protein derivate (PPD) intradermally (5). A delayed-type hypersensitivity reaction occurs if the host is reactive to M. tuberculosis antigens. Due to the limited M. tuberculosis specificity of TST, more specific in vitro blood-based assays (T-SPOT.TB and QuantiFERON-TB Gold) were developed using early secretory antigen target-6 (ESAT-6), culture filtrate protein 10 (CFP-10), and TB-7.7 as M. tuberculosis antigens. These assays measure the ex-vivo IFN-γ release by T cells in response to the aforementioned M. tuberculosis peptide antigens (6). A disadvantage of TST and IGRA for the diagnosis of infection is that they are unable to distinguish between an amnestic response and persistent infection. It is therefore possible that an unknown proportion of persons who test positive in the immune assays are no longer infected with M. tuberculosis. Conversely, persons who test negative in the immune assays may be (i) not sufficiently exposed to M. tuberculosis, (ii) anergic to M. tuberculosis antigens used in the assays, or (iii) exposed but able to clear M. tuberculosis infection without triggering the onset of acquired anti-M. tuberculosis immunity.
Natural Immunity Against M. tuberculosis
While the lack of a direct assay for the determination of current infection complicates studying resistance to infection, multiple lines of evidence support human variability in resistance to infection with M. tuberculosis. Historical epidemiological studies have long supported the concept of infection resistance as a bona-fide biological phenotype. During an outbreak on a US naval ship, 66 sailors shared a cabin with 7 sailors who had active TB. Of the 66 sailors, 13 (20%) remained TST negative after 6 months (7). Fifty-seven (55%) of 104 elderly residents with a previously TST negative result remained uninfected after being exposed for at least 12 months to a fellow resident with sputum positive TB (8). An average of 50% of close contacts of TB patients develop positive TST or IGRA tests in overcrowded living conditions or household contact studies (9, 10). In Uganda only 4.1% of adults (age > 15 years old) with close household contacts remained PPD negative (<10 mm for HIV- adults, <5 mm for all HIV+) over a 2 year follow up period (11). Other studies done in individuals in environments with high exposure to M. tuberculosis, show that 10–20% do not become TST/IGRA positive (12–14). In South African goldminers who have a documented high exposure to M. tuberculosis and an estimated LTBI prevalence of 89% in 2006, 13% of the HIV-negative participants had a TST = 0 mm response (15). Together, these studies suggest that 5–20% of the population may possess resistance to M. tuberculosis infection.
Molecular genetics studies support the concept of resistance to M. tuberculosis infection. In a highly TB endemic area in South Africa 20% of the highly exposed population remained TST negative which was stringently defined as TST = 0 mm. This phenotype is linked to a major locus, TST1, which represents T cell-independent M. tuberculosis infection resistance (13). A genome-wide association study in HIV-infected persons identified a locus on chromosome region 5q31.1 in proximity of IL9 which significantly associates with TST positivity (16). In addition, the study replicated associations in the region of TST1 as well as on chromosome regions 2q21-2q24 and 5p13-5q22 that had been identified by genome-wide linkage analysis of Ugandan families (13, 16, 17). Current genetic evidence suggests that the resistance phenotype is likely due to a combination of genetic variants synergistically contributing to the phenotype rather than a single genetic variant.
The Heterogeneous Nature of Neutrophils
It is tempting to speculate that neutrophils of individuals who exhibit M. tuberculosis infection resistance are a unique subset of cells genetically or epigenetically programmed to control infection and inflammation. Epigenetic reprogramming of neutrophils offers an attractive avenue of investigation as neutrophils show increased variability in both gene expression and DNA methylation compared to phenotypically naïve T-cells and classic monocytes (18). This observation supports the concept of physiologically distinct inter-individual neutrophil populations.
Different intra-individual neutrophil subsets have also been defined in multiple studies investigating various diseases including cancer, systemic lupus erythematosus (SLE), TB, and HIV-1 (19–23). However, the heterogeneous nature of neutrophils with subsets displaying functional as well as phenotypic differences is still under debate and most subsets remain incompletely defined and phenotyped (20, 24–29).
Genetic variants, which underlie epigenetic and transcriptional variability, also contribute to differences in neutrophil activity. For example, 21 neutrophil genes showed significant differences in expression levels between males and females while a SNP in SELL, which encodes the CD62L receptor, strongly influenced expression levels of CD62L cell receptors on neutrophils (18, 30). Not surprisingly, genes of the inflammasome pathway are significantly enriched in neutrophils and play an important role in the regulation of interleukin 1 (IL-1)-dependent cytokine production (31). In murine studies, IL-1 deficiency predisposes to a lack of M. tuberculosis infection control and non-resolving inflammation (32). During persistent infections, such as active TB, inflammasome activation correlates with pathology (33, 34). Taken together, these data suggest that if neutrophils contribute to M. tuberculosis infection resistance the effector mechanisms involved are likely to be under both genetic and epigenetic regulation. However, at least some of the underlying variability may be ascribed to the inherent difficulties in working with these cells since they cannot be cryopreserved, are easily activated and are short-lived (35, 36). Possible genetic variability is further highlighted by the conflicting results published around the role of neutrophils in M. tuberculosis infection.
Neutrophils in M. tuberculosis Infection and Disease
M. tuberculosis is an airborne pathogen and is transmitted via the aerosol inhalation of transmitted droplets containing the bacteria from an infected individual. M. tuberculosis enters the airways and reaches the pulmonary alveolus where some of the first cells encountered are resident alveolar macrophages (AM) (37) which release pro-inflammatory cytokines tumor necrosis factor (TNF), IL-6, IL-1α, and IL-1β (38). If this first line of defense fails, M. tuberculosis enters the pulmonary interstitial tissue by either using the infected AM as a host vehicle to migrate or by infecting the epithelium or pneumocytes (2). Acute inflammatory signals are released and the other phagocytes are recruited to the site of infection. Local tissue macrophages recognize M. tuberculosis by Toll-like receptors (TLR) and are also activated to release pro-inflammatory cytokines including TNF, IL-6, and IL-1β (39, 40) (Figure 1A).
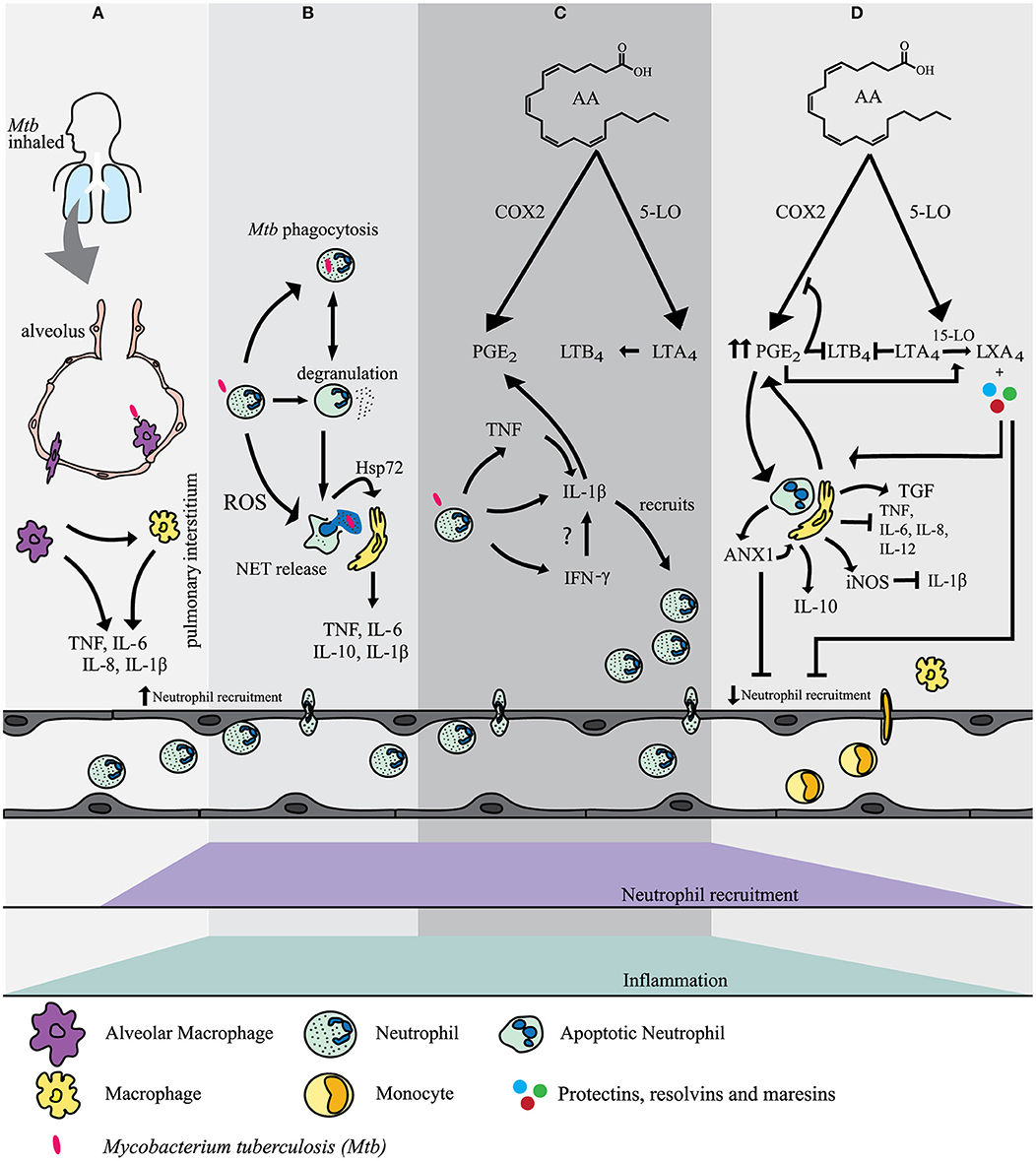
Figure 1. (A) Alveolar macrophages (AM) are the first cells to encounter M. tuberculosis after inhalation of the bacillus. Acute pro-inflammatory signals are released by AM and local tissue macrophages to recruit neutrophils to the site of infection.(B) Neutrophils use a variety of mechanisms to mediate M. tuberculosis infection. These included phagocytosis, degranulation, ROS formation and NET release. NETs transfer Hsp72 to adjacent macrophages inducing a pro-inflammatory response.(C) Interaction of recruited neutrophils with M. tuberculosis mediates the activation of several pathways which contribute to inflammation and clearance of M. tuberculosis infection. Interleukin-1β (IL-1β) release is mostly mediated in an inflammasome dependent manner. Tumour necrosis factor (TNF) induces NF-κB which mediates the induction of gene expression of IL-1β in neutrophils. Interferon-γ (IFN-γ) may also regulate the release of IL-1β. IL-1β is a key player in mediating the release of prostaglandin E2 (PGE2) and leukotriene B4 (LTB4) both of which contribute to inflammation and the recruitment of neutrophils. (D) PGE2 eventually becomes a stop signal and has a negative feedback on cyclo-oxygenase-2 (COX-2) and 5-lipoxygenase (5-LO). The production of lipoxin A4 (LXA4) is favoured. In addition, AnnexinA1 (ANX1) stimulates IL-10 release by macrophages. Neutrophils express inducible nitric oxide synthase (iNOS) which has a further negative feedback on IL-1β release. The net effect is an increase in neutrophil apoptosis and clearance by tissue macrophages. More macrophages are recruited and further neutrophil recruitment is inhibited and inflammation is resolved.
Neutrophils are some of the first phagocytes recruited from the pulmonary vasculature to the pulmonary interstitium (41). Multiple receptors (including TLRs and C-type lectins receptors (CLRs) and cytokine receptors) have been implicated in the interaction between neutrophils, M. tuberculosis and pro-inflammatory cytokines (42–46). Upon exposure to M. tuberculosis neutrophil blood counts in human pulmonary TB (PTB) contacts are initially higher than in unexposed control subjects and subside after 6 weeks (47). Interestingly, low neutrophil counts are associated with IGRA positivity in TB contacts (47). The initial neutrophil peak seen in TB contacts, implicates neutrophils in the acute inflammatory response to M. tuberculosis.
Individuals in contact with patients with pulmonary TB are less likely to be infected with M. tuberculosis if they have higher peripheral blood neutrophil counts (47). One hour after in vitro infection with virulent M. tuberculosis and stimulation with TNF, neutrophils suppressed the growth of the inoculum by 50–95% (48). Unstimulated neutrophils inhibit on average 40.6% of the growth of the M. tuberculosis inoculum. Interestingly, there was significant variability in this mycobactericidal capacity between donor neutrophils. Neutrophils from some donors were capable of inhibiting M. tuberculosis growth spontaneously while, despite the addition of TNF or IFN-γ, others were not. Neutrophil-depleted whole blood had a 3.1 fold decreased capacity to control M. tuberculosis infection ex vivo (47). This finding was recently confirmed and highlights the importance of neutrophils in M. tuberculosis infection (49). Granulocyte (CD15+) depleted blood does not control M. tuberculosis infection as efficiently as blood depleted of CD4+, CD8+, or CD14+ cells. Addition of viable CD15+ granulocytes significantly improved M. tuberculosis control (49).
However, infection in highly susceptible strains of mice shows the detrimental effect of uncontrolled neutrophil recruitment on TB infection and inflammation control and eventually an increase in TB disease severity (50). Most studies concur that neutrophils are final mediators of lung damage and disease (51–53). C57BL/6 mice with neutrophil and monocyte derived-cells lacking Atg5 succumb after 30–40 days post M. tuberculosis infection due to a massive influx of neutrophils, and increased lesion number and bacterial load, that is not observed in wild type mice (54, 55). Whilst ATG5 is normally associated with autophagy, the neutrophilic influx associated with premature death was independent of any autophagic response. Granulomas of various susceptible mouse strains contain a substantial number of necrotic neutrophils (53, 56–58) in comparison to more “resistant” mouse strains showing only scattered neutrophils and little or no necrosis (59).
In humans, as in the mouse model, necrotic neutrophils are unable to control M. tuberculosis infection (49). Phagocytosis of M. tuberculosis-induced necrotic neutrophils by macrophages promotes bacterial growth (60, 61). M. tuberculosis mostly remains encapsulated in apoptotic neutrophils (60). This enables fusion of neutrophil granular contents with macrophage lysosomes after efferocytosis of the apoptotic neutrophil by the macrophage (60). The neutrophil membranes surrounding M. tuberculosis prevent direct contact between the bacillus and the macrophage phagosomal membrane thus preventing M. tuberculosis inhibition of phagolysosome maturation (60). However, during neutrophil necrosis, M. tuberculosis is released from the disintegrated phagosome and enters the phagocytosing macrophage as extracellular bacteria (60). Once phagocytosed by a macrophage, the bacillus is able to evade phagolysosomal fusion in the macrophage and mycobacterial growth is promoted (60, 61).
Necrotic neutrophils added to whole blood increased the metabolism of M. tuberculosis, as measured by mycobacterial luminescence, and released IL-10 as well as growth factors, granulocyte- and granulocyte macrophage-colony-stimulating factors (G-CSF and GM-CSF), and the monocyte chemotactic protein chemokine ligand 2 (CCL2) (49). The predominant role of these molecules is to attract and prime more cells (49). G-CSF supports the growth and proliferation of neutrophils and their precursors (62). GM-CSF has the potential to act on earlier progenitor cells than G-CSF and therefore neutrophil progenitors as well as monocytes proliferate (63). G-CSF and GM-CSF not only drive the increased production of neutrophils and monocytes but also have the ability to indirectly affect neutrophil function and phenotype (64–68). Both of these growth factors delay neutrophil apoptosis and “prime” neutrophils for enhanced oxidative effects that can lead to tissue destruction (67, 69). In a setting where M. tuberculosis induces necrotic cell death, the newly released cells would undergo the same cycle of necrosis, release tissue damaging substances, recruit more cells, and contribute to continuous inflammation as seen in TB disease, with neutrophilia being an independent predictor of TB mortality (70).
Neutrophil Mechanisms to Clear M. tuberculosis Infection
Despite the involvement of neutrophils in tissue damage in late stage clinical TB, they display an inter-individual ability to control M. tuberculosis infection. Neutrophils can use oxidative and non-oxidative mechanisms to clear M. tuberculosis infection. Both mechanisms are involved in either the direct clearance of M. tuberculosis or in the mediation thereof.
Oxidative Mechanisms
Neutrophils are primed or activated by M. tuberculosis and pro-inflammatory cytokines, which in turn triggers degranulation and respiratory burst (71–79). Proteases (e.g., elastase, cathepsin G, and protease 3), hydrolyses, antimicrobial peptides and oxidants are released. The oxidants mediate tissue breakdown by activating matrix metalloproteinases (MMPs) (80, 81). These effectors do not discriminate between pathogen and host tissue and collateral damage is inevitable.
Neutrophil-produced reactive oxygen species (ROS) have been shown to drive M. tuberculosis-induced necrosis (60). Inhibiting myeloperoxidase (MPO) derived ROS prevents neutrophil necrosis and improves efferocytosis of these cells by macrophages and therein the control of M. tuberculosis growth (60). Similarly, chronic granulomatous disease (CGD) neutrophils are protected from necrosis after infection with M. tuberculosis (79). One would therefore expect an improved control of M. tuberculosis infection in CGD patients who are characterized by an inability to produce ROS but this does not always seem to be the case (82). Indeed, CGD patients are more susceptible to active TB supporting the possible role of neutrophils in mediating M. tuberculosis infection resistance (83–86). This view is supported by multiple studies that have shown neutrophils to be protective in control of early infection (47–49).
The NOX2 complex is an isoform of the large family of NAPDH oxidases (NOX) and is found in phagocytes including neutrophils (87, 88). It is an enzyme that is involved in infection and inflammation control and is activated by neutrophil chemotactic factors such as IL-8 and leukotriene B4 (LTB4) (88, 89). Hydrogen peroxide (H2O2) that is produced during respiratory burst contributes to neutrophil migration and subsequently retention at the site of infection (89). CGD patients have impaired neutrophil accumulation, in contrast to the increase in granuloma formation seen in CGD (89). Inflammatory leukotrienes are released by neutrophils in CGD patients but due to a lack of ROS there is a lack of degradation of these leukotrienes and delayed clearance of inflammation (44, 89, 90).
Reactive oxygen species have been shown to affect transcription factors such as NF-κβ (91, 92) which mediates the induction of IL-1β and IL-8 expression. However, CGD shows that NF-κβ activation is independent of ROS and is also mediated by TNF and IL-1 (93, 94) and so neutrophils in these individuals are still able to release these pro-inflammatory factors and uncontrolled chronic inflammation ensues (95, 96). Pro-inflammatory mediators alone, such as leukotrienes and IL-1β, are not enough to control infection and it is likely that the overproduction thereof augments the lack of M. tuberculosis infection control in CGD patients (44).
M. tuberculosis is relatively resistant to the bactericidal effects of H2O2 mediated by DNA damage (97). However, even if ROS does not have a direct bactericidal effect on M. tuberculosis, it still amplifies the neutrophil antimicrobial response. It does this by activating the formation of neutrophil extracellular traps (NET, discussed in 3.1.3), stimulating the release of pro-inflammatory cytokines such as TNF and macrophage inflammatory protein 2 (MIP-2), as well as decondensed DNA to which the contents of cytoplasmic granules adhere in a net-like structure (98–100). This is extensively reviewed by Deffert et al. (44).
Non Oxidative Mechanisms
Neutrophil granules can fuse with the phagolysosome, degranulate and release antimicrobial peptides (AMPs) (Figure 1B). Antimicrobial peptides (AMPs) are classified according to their amino acid motif and structure. Three classes are found in humans: defensins, cathelicidins, and histatins (101–103). Neutrophils contain α-defensins in azurophilic granules and cathelicidin LL-37 in specific granules, as well as other neutrophil specific AMPs as will be discussed below (101, 104). Macrophages can traffic phagocytosed apoptotic neutrophil debris, including neutrophil granules, to endosomes. The purified neutrophil granules in the endosomes fuse with the macrophage phagosome in which the M. tuberculosis bacillus resides. This mechanism of cell-cell cooperation provides an effective antimicrobial response to M. tuberculosis (105). Although this efferocytosis occurs between macrophages and apoptotic neutrophil debris, it is not known whether alveolar macrophages do the same. AMPs can also be associated extracellularly with NETs and facilitate in the clearing of microbial infection.
a. AMPs in azurophilic granules
Azurophilic granules are poorly mobilized in response to M. tuberculosis infection. Pathogenic mycobacteria block the fusion of azurophil granules with the phagosome and consequentially unlike specific granules they are unable to release their contents in to the phagosome for antimicrobial effect (106). However, azurophilic proteins obtained from apoptotic neutrophil debris, increase macrophage ability to restrict M. tuberculosis growth either by direct action or by lysosome fusion with the maturation-arrested mycobacterial phagosome in the macrophage (107).
-Defensins: Human neutrophil peptide 1 (HNP-1), one of four α-defensins found in the primary or azurophilic granules of neutrophils (101) has the ability in vitro to reduce the growth of M. tuberculosis in culture as well as within macrophages (105, 108, 109). Furthermore, HNP-1 also shows in vivo antimycobacterial activity in mice (110).
-Azurocidin: Defensin depleted azurophilic granules at 100 μg/ml were shown to restrict the growth of 55% of M. tuberculosis in culture after 24 h of incubation. However, the specific role of azurocidin in M. tuberculosis infection remains unclear (107).
-Cathepsins: M. tuberculosis infection decreases cathepsin gene expression in macrophages, with a parallel decrease in cathepsin protein levels (111). Genetic linkage and association studies have previously implicated cathepsin Z in susceptibility to TB (112, 113). A likely alternative source of cathepsin for macrophages is through the phagocytosis of apoptotic neutrophil material. Uptake of liposomal encapsulated cathepsin G and neutrophil elastase (NE) by alveolar macrophages in mice improves antimicrobial activity against Mycobacterium bovis bacillus Calmette-Guérin (BCG) (114).
b. Specific granules
-Cathelicidin: Neutrophils produce LL-37, the 37 amino acid biologically active C-terminal domain cleaved from the human cathelicidin propeptide (hCAP18) by proteinase 3, when infected with M. tuberculosis (115). LL-37 has been shown to restrict growth of M. tuberculosis in neutrophils (47). Similarly it restricts growth of M. tuberculosis in infected macrophages when hCAP18 is exogenously activated by neutrophil proteinase 3, which has only a low level of constitutive expression in macrophages (107, 116).
c. Gelatinase granules
-Lipocalin 2: Lipocalin 2 binds mycobacterial siderophores which scavenge iron for the bacillus in iron-limiting conditions (47). Lipocalin 2 has a greater mycobacterial suppressive effect (60%) in an iron-depleted broth (10 nM iron) compared to iron-replete broth (150 μM Fe) of 45%. It may be more effective in the phagolysosome where the molar ratio to siderophores would be higher.
d. Neutrophil cytoplasmic proteins
Calprotectin (S100A8/S110A9): Calprotectin is known as a damage-associated molecular pattern (DAMP) molecule and is a heterodimer of S100A8/A9. It sequesters free zinc and limits mycobacterial growth (107, 117). M. tuberculosis infection induces S100A8/A9 proteins. This is associated with neutrophil accumulation and exacerbated inflammation (52, 118).
NET Formation
During NETosis, neutrophils release their DNA contents coated in cytoplasmic and granular proteins to trap and possibly clear invading pathogens (119, 120). NETosis is an alternative form of cell death, different to apoptosis and necrosis, and mediated by phagocytosis and the generation of ROS by NADPH oxidase in M. tuberculosis infection (121, 122). Once activated, neutrophils lose their lobulated morphology (123). The nuclear membrane initially remains intact whilst the chromatin (histones and DNA) starts to decondense. Once the nuclear and granular membranes rupture, the decondensed chromatin comes into contact with the granular as well as cytoplasmic components of the cell. The NET components are released extracellularly when the cell membrane breaks (122). The most abundant non-histone protein in NETs is NE (124). In addition to this, NETs contain myeoloperoxidase (MPO) as well as other proteins from intracellular neutrophil organelles. These include substances from the primary neutrophil granule (cathepsin G, defensins, BPI-bactericidal substance), the secondary neutrophil granule (alcaic phosphatase, lactoferrins, lysozyme, cathelicidins, collagenase), tertiary granules [gelatinase, matrix metalloproteinase 9 (MMP-9)]; and catalase from peroxisomes (125–128). Other components include calprotectin, constituents of the neutrophil cytoskeleton and glycolytic enzymes (125, 128).
Although M. tuberculosis has been shown to induce NETosis, no experimental evidence exists that NET formation improves resolution of M. tuberculosis infection (129). However, the AMP NET components have been shown to restrict M. tuberculosis growth as discussed earlier. Also neutrophils can assist macrophages to clear M. tuberculosis infection. During infection, NET formation and M. tuberculosis-induced apoptosis occur independently. M. tuberculosis-induced NETs transfer the danger signal heat shock protein 72 (Hsp72) to adjacent macrophages (121). This interaction induces a pro-inflammatory response in macrophages leading to the release of IL-6, TNF, IL-1β, IL-10. In addition to these cytokines, calprotectin is released from the neutrophil cytoplasm into NETs (130). IL-10 is also released as part of the anti-inflammatory regulatory response via inhibiting IFN-γ and TNF production and downstream Th1 responses (121). It is possible that NETs play a role in trapping and localizing the infection. The sequestration of AMPs in the NET structures may also increase their effective concentrations. Furthermore; NETs contain the release of cellular contents to prevent distal tissue destruction (121, 123). Hence, NETs are potentially an effective defense mechanism that neutrophils could use to mediate M. tuberculosis infection resistance (Figure 1B).
Neutrophils and the Role of Cytokines and Chemokines in Inflammation in M. tuberculosis Infection Resistance
Initial Inflammation
M. tuberculosis infection triggers TLR signaling and induces NF-κB which mediates the induction of gene expression of pro-inflammatory cytokines such as IL-1β and TNF in neutrophils (42, 131). Inflammasomes are multimeric protein complexes and play a key role in the activation of IL-1α, and IL-1β (132). Neutrophils express components of the NOD-like receptor protein 3 (NLRP3) and absent in melanoma 2 (AIM2) inflammasomes (133). The latter are found in the cytoplasm as well as secretory and tertiary granule compartments (133). Neutrophils release IL-1β mostly in an inflammasome-dependent manner and do not release IL-1α (133). The inflammasome subunit caspase-1 activates pro-IL-1β to form IL-1β (132, 133). IL-1β activation can also occur in a caspase-1 independent manner via neutrophil proteases; NE, and proteinase 3 (PR3) (133). Furthermore, it is of interest that inflammasome components are found in neutrophil secretory vesicles. The components may play a role in phagosomal functionality or may be released into the extracellular environment and utilized by other phagocytes, but this remains to be proven in neutrophils (133).
One of the key roles of IL-1β is to mediate the release of prostaglandin E2 (PGE2), an eicosanoid. Eicosanoids are important lipid mediators derived from arachidonic acid (AA) and are rapidly synthesized by phagocytes after acute challenge with M. tuberculosis (134, 135). Cyclo-oxygenase-2 (COX-2) competes with 5-Lipoxygenase (5-LO) or 15-lipoxygenase (15-LO) for the generation of each of the different eicosanoids. During inflammation macrophages and other cells, including neutrophils, can produce COX-2, which converts AA to PGE2. 5-Lipoxygenase (5-LO) converts AA to LTB4 from leukotriene A4 (LTA4). PGE2 and LTB4 mainly have proinflammatory effects and mediate the rapid recruitment of neutrophils to the site of infection and inflammation (136, 137). LTB4 promotes phagocytosis and the bactericidal activity of neutrophils (136, 138, 139) (Figure 1C).
Furthermore, neutrophils are a possible source of IL-12 mediated IFN-γ release (140). However, whether this occurs through direct M. tuberculosis stimulation is unknown. Neutrophils release IFN-γ after stimulation by degranulating agents which is due to an available small storage of IFN-γ (140). In addition, neutrophil stimulation by IL-12 alone or in combinations with lipopolysaccharide (LPS), IL-2, IL-18, or IL-15, induces IFN-γ synthesis by neutrophils (140).
Neutrophils matured with IFN-γ have marked upregulation of multiple transcripts where Guanylate Binding Protein (GBP) showed the highest changes. GBPs are a subfamily of the IFN inducible GTPase superfamily (141, 142). GBP-5, in particular, is strongly upregulated in transcriptomes from an immature myeloid cell line (PLB-985) matured in the presence of IFN-γ (143). PLB-985 cells can differentiate into terminally mature neutrophils and have the ability to mimic the physiological conditions of stimulation (144). The exact role of GBP-5 has not been described in neutrophils yet, but it is possible that it enhances the NLRP3 inflammasome and IL-1β production, as in macrophages (143) (Figure 1C).
IFN-γ may increase the half-life of neutrophils in culture by being anti-apoptotic (143) and in this manner contributes to the pro-inflammatory state. Pathology in pulmonary tuberculosis is associated with neutrophils expressing IFN-γ and type I IFNs (145). This transcriptional signature is found in patients with active TB but infrequently in healthy individuals or those with latent TB (145). Type 1 IFNs may contribute to disease progression but the pro-inflammatory effect of IFN-γ from a neutrophil perspective may be effective for short bursts and in a setting where M. tuberculosis is effectively killed. The promotion of this initial pro-inflammatory state and release of TNF and IFN-γ by neutrophils is essential to effectively clear M. tuberculosis infection (48, 143).
The Resolution of Inflammation in M. tuberculosis Infection
Apoptosis represents a pivotal point in the control of inflammation as well as in the control of the cellular immune response (146). A delicate balance exists between apoptotic cell death, clearance of apoptotic cells and ongoing inflammatory responses (80, 147, 148). Not only does the efferocytosis of apoptotic neutrophils by tissue resident macrophages prevent spillage of neutrophil content into surrounding tissue (80, 147, 149, 150), but it also decreases pro-inflammatory mediators (148). Clearance of infection without a significant acquired immune response is favored by early killing of M. tuberculosis by neutrophils, followed by apoptotic neutrophil death, and an anti-inflammatory response in the phagocytosing macrophage (35, 105).
A hallmark of the anti-inflammatory response is the production of TGF-β and PGE2, and the inhibition of IL-6, IL-8, IL-12, and TNF release by the phagocytosing macrophages (151). Studies have shown that cAMP-elevating agents such as PGE2 result in increased levels of AnnexinA1 (ANXA1) (152). ANXA1, a protein found in neutrophils, stimulates release of the anti-inflammatory cytokine, IL-10, by macrophages, and inhibits neutrophil migration (153). In addition, ANXA1 promotes efferocytosis of apoptotic cells (154, 155) (Figure 1D).
In addition to the release of endogenous anti-inflammatory mediators, pro-resolution action is also required. Lipoxins, protectins, resolvins and macrophage mediator in resolving inflammation (maresins) are unique mediators fulfilling this duality (137, 156, 157). Rising PGE2 levels eventually act as a “lipid mediated class switch” by transcriptionally inducing 15-LO in neutrophils and shifting the production of PGE2 and LTB4 in favor of lipoxin A4 (LXA4) (158). LXA4 decreases neutrophil-mediated tissue damage, neutrophil proliferation, and adhesion, and increases efferocytosis of apoptotic neutrophils and IL-10 production by macrophages (159). Resolvins, protectins and maresins are oxygenated metabolites derived from eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) that is biosynthesized from omega-3 essential polyunsaturated fatty acids (137, 160). Collectively resolvins, protectins, and maresins regulate neutrophil apoptosis, efferocytosis by macrophages, inhibition of pro-inflammatory cytokines, release of IL-10 by local macrophages and tissue regeneration (159) (Figure 1D).
Finally, neutrophils express inducible nitric oxide synthase (iNOS) which converts the amino acid L-arginine to L-citrulline and nitric oxide (NO). iNOS/NO limits the production of IL-1β and therefore limits further recruitment of neutrophils (34, 161, 162). It is not known to what extent these neutrophil anti-inflammatory mechanisms are at play during early encounters of PMNs with M. tuberculosis in the lung (Figure 1D).
Conclusion
At first glance, the association of uncontrolled neutrophil recruitment and pathology in TB would argue against a role of these cells in M. tuberculosis infection resistance. However, neutrophils are multi-functional cells with variable roles in host defense. For example, there is documented inter-individual variability in the ability of neutrophils to kill M. tuberculosis suggesting that the role of neutrophils in an early encounter with M. tuberculosis may differ from the more integrated role in the presence of a strongly developed acquired immune response to the bacillus. As reviewed, the neutrophil has a large armamentarium of highly effective anti-microbial effector mechanisms that may come into play during the early stage of M. tuberculosis infection. Investigating the possible role of neutrophils in persons who remain free of M. tuberculosis infection despite documented high exposure to the bacillus offer an interesting opportunity. It may be that resisters possess a different ratio of neutrophil subpopulations, predominated by effective killers with a propensity to undergo apoptosis, compared to those who develop TB, predominated by inflammatory necrotising damage causing neutrophils. By comparing neutrophils and their anti-microbial responses from “innate resisters” with those from M. tuberculosis infection susceptible persons might illuminate if and how neutrophils play a protective role in the very stage of M. tuberculosis infection. Experiments along these lines will not only provide a better understanding of TB pathogenesis but also contribute to a better understanding of neutrophil biology in general.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was partially funded by the South African government through the South African Medical Research Council. The content is solely the responsibility of the authors and does not necessarily represent the official views of the South African Medical Research Council. This work was also supported by the National Research Foundation of South Africa and a grant from the National Institutes of Health (NIH) (1R01AI124349-01). Work in the laboratory of ES is supported by a Foundation grant from CIHR (FDN-143332). AC is supported by the Walter and Eliza Hall Institute of Medical Research, the Medical Research Council of South Africa (SHIP-02-2013), the National Institute of Health TB Research Unit (U19AI111276) and the South African National Research Foundation (UID109040). RW is supported by the Francis Crick Institute, which receives funding from the Cancer Research (UK), Wellcome (10218), and UKRI (10218). RW is also supported by Wellcome (104803, 203135). We thank Dr Elizna Maasdorp who assisted with the figure preparation.
References
1. Comstock GW, Livesay VT, Woolpert SF. The prognosis of a positive tuberculin reaction in childhood and adolescence. Am J Epidemiol. (1974) 99:131–8.
2. Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, et al. Tuberculosis. Nat Rev Dis Primers (2016) 2:16076. doi: 10.1038/nrdp.2016.76
3. WHO. Global Tuberculosis Report 2017. WHO. Available online at: http://www.who.int/tb/publications/global_report/en/
4. Simmons JD, Stein CM, Seshadri C, Campo M, Alter G, Fortune S, et al. Immunological mechanisms of human resistance to persistent Mycobacterium tuberculosis infection. Nat Rev Immunol. (2018) 18:575–89. doi: 10.1038/s41577-018-0025-3
5. Reichman LB. Tuberculin skin testing: the state of the art. Chest (1979) 76:764–70. doi: 10.1378/chest.76.6.764
6. Richeldi L. An update on the diagnosis of tuberculosis infection. Am J Respir Crit Care Med. (2006) 174:736–42. doi: 10.1164/rccm.200509-1516PP
7. Houk VN, Baker JH, Sorensen K, Kent DC. The epidemiology of tuberculosis infection in a closed environment. Arch Environ Health (1968) 16:26–35.
8. Stead WW. Tuberculosis among elderly persons: an outbreak in a nursing home. Ann Intern Med. (1981) 94:606–10.
9. Fox GJ, Barry SE, Britton WJ, Marks GB. Contact investigation for tuberculosis: a systematic review and meta-analysis. Eur Respir J. (2013) 41:140–56. doi: 10.1183/09031936.00070812
10. Morrison J, Pai M, Hopewell PC. Tuberculosis and latent tuberculosis infection in close contacts of people with pulmonary tuberculosis in low-income and middle-income countries: a systematic review and meta-analysis. Lancet Infect Dis. (2008) 8:359–68. doi: 10.1016/S1473-3099(08)70071-9
11. Stein CM, Zalwango S, Malone LL, Thiel B, Mupere E, Nsereko M, et al. Resistance and susceptibility to Mycobacterium tuberculosis infection and disease in tuberculosis households in Kampala, Uganda. Am J Epidemiol. (2018) 187:1477–89. doi: 10.1093/aje/kwx380
12. Abel L, El-Baghdadi J, Bousfiha AA, Casanova J-L, Schurr E. Human genetics of tuberculosis: a long and winding road. Philos Trans R Soc Lond B Biol Sci. (2014) 369: doi: 10.1098/rstb.2013.0428
13. Cobat A, Gallant CJ, Simkin L, Black GF, Stanley K, Hughes J, et al. Two loci control tuberculin skin test reactivity in an area hyperendemic for tuberculosis. J Exp Med. (2009) 206:2583–91. doi: 10.1084/jem.20090892
14. Cobat A, Gallant CJ, Simkin L, Black GF, Stanley K, Hughes J, et al. High heritability of antimycobacterial immunity in an area of hyperendemicity for tuberculosis disease. J Infect Dis. (2010) 201:15–9. doi: 10.1086/648611
15. Hanifa Y, Grant AD, Lewis J, Corbett EL, Fielding K, Churchyard G. Prevalence of latent tuberculosis infection among gold miners in South Africa. Int J Tuberc Lung Dis. (2009) 13:39–46.
16. Sobota RS, Stein CM, Kodaman N, Maro I, Wieland-Alter W Jr., Igo RP, et al. A chromosome 5q31.1 locus associates with tuberculin skin test reactivity in HIV-positive individuals from tuberculosis hyper-endemic regions in east Africa. PLoS Genet. (2017) 13:e1006710. doi: 10.1371/journal.pgen.1006710
17. Stein CM, Zalwango S, Malone LL, Won S, Mayanja-Kizza H, Mugerwa RD, et al. Genome scan of M. tuberculosis infection and disease in Ugandans. PLoS ONE (2008) 3:e4094. doi: 10.1371/journal.pone.0004094
18. Ecker S, Chen L, Pancaldi V, Bagger FO, Fernández JM, Carrillo de Santa Pau E, et al. Genome-wide analysis of differential transcriptional and epigenetic variability across human immune cell types. Genome Biol. (2017) 18:18. doi: 10.1186/s13059-017-1156-8
19. Carmona-Rivera C, Kaplan MJ. Low density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Semin Immunopathol. (2013) 35:455–63. doi: 10.1007/s00281-013-0375-7
20. Cloke T, Munder M, Taylor G, Müller I, Kropf P. Characterization of a novel population of low-density granulocytes associated with disease severity in HIV-1 infection. PLoS ONE (2012) 7:e48939. doi: 10.1371/journal.pone.0048939
21. Deng Y, Ye J, Luo Q, Huang Z, Peng Y, Xiong G, Guo Y, Jiang H, Li J. Low-density granulocytes are elevated in mycobacterial infection and associated with the severity of tuberculosis. PLoS ONE (2016) 11:e0153567. doi: 10.1371/journal.pone.0153567
22. Liu Y, Hu Y, Gu F, Liang J, Zeng Y, Hong X, et al. Phenotypic and clinical characterization of low density neutrophils in patients with advanced lung adenocarcinoma. Oncotarget (2017) 8:90969–78. doi: 10.18632/oncotarget.18771
23. Marini O, Costa S, Bevilacqua D, Calzetti F, Tamassia N, Spina C, et al. Mature CD10+ and immature CD10- neutrophils present in G-CSF-treated donors display opposite effects on T cells. Blood (2017) 129:1343–56. doi: 10.1182/blood-2016-04-713206
24. Mishalian I, Granot Z, Fridlender ZG. The diversity of circulating neutrophils in cancer. Immunobiology (2017) 222:82–88. doi: 10.1016/j.imbio.2016.02.001
25. Pillay J, Tak T, Kamp VM, Koenderman L. Immune suppression by neutrophils and granulocytic myeloid-derived suppressor cells: similarities and differences. Cell Mol Life Sci. (2013) 70:3813–27. doi: 10.1007/s00018-013-1286-4
26. Pokkali S, Rajavelu P, Sudhakar R, Das SD. Phenotypic modulation in Mycobacterium tuberculosis infected neutrophil during tuberculosis. Indian J Med Res. (2009) 130:185–92.
27. Scapini P, Cassatella MA. Social networking of human neutrophils within the immune system. Blood (2014) 124:710–9. doi: 10.1182/blood-2014-03-453217
28. Scapini P, Marini O, Tecchio C, Cassatella MA. Human neutrophils in the saga of cellular heterogeneity: insights and open questions. Immunol Rev. (2016) 273:48–60. doi: 10.1111/imr.12448
29. Silvestre-Roig C, Hidalgo A, Soehnlein O. Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood (2016) 127:2173–81. doi: 10.1182/blood-2016-01-688887
30. Patin E, Hasan M, Bergstedt J, Rouilly V, Libri V, Urrutia A, et al. Natural variation in the parameters of innate immune cells is preferentially driven by genetic factors. Nat Immunol. (2018) 19:302–14. doi: 10.1038/s41590-018-0049-7
31. Chen L, Ge B, Casale FP, Vasquez L, Kwan T, Garrido-Martín D, et al. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell (2016) 167:1398–414.e24. doi: 10.1016/j.cell.2016.10.026
32. Di Paolo NC, Shafiani S, Day T, Papayannoupoulou T, Russell DW, Iwakura Y, et al. Interdependence between interleukin-1 and tumor necrosis factor controls TNF-dependent effector functions during Mycobacterium tuberculosis infection. Immunity (2015) 43:1125–1136. doi: 10.1016/j.immuni.2015.11.016
33. Dorhoi A, Nouailles G, Jörg S, Hagens K, Heinemann E, Pradl L, et al. Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur J Immunol. (2012) 42:374–84. doi: 10.1002/eji.201141548
34. Mishra BB, Rathinam VAK, Martens GW, Martinot AJ, Kornfeld H, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat Immunol. (2013) 14:52–60. doi: 10.1038/ni.2474
35. Lowe DM, Redford PS, Wilkinson RJ, O'Garra A, Martineau AR. Neutrophils in tuberculosis: friend or foe? Trends Immunol. (2012) 33:14–25. doi: 10.1016/j.it.2011.10.003
36. Pillay J, Braber I den, Vrisekoop N, Kwast LM, Boer RJ de, Borghans JAM, et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood (2010) 116:625–7. doi: 10.1182/blood-2010-01-259028
37. Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol. (2001) 2:569–86. doi: 10.1038/35085034
38. Law K, Weiden M, Harkin T, Tchou-Wong K, Chi C, Rom WN. Increased release of interleukin-1 beta, interleukin-6, and tumor necrosis factor-alpha by bronchoalveolar cells lavaged from involved sites in pulmonary tuberculosis. Am J Respir Crit Care Med. (1996) 153:799–804. doi: 10.1164/ajrccm.153.2.8564135
39. Giacomini E, Iona E, Ferroni L, Miettinen M, Fattorini L, Orefici G, et al. Infection of human macrophages and dendritic cells with Mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T cell response. J Immunol. (2001) 166:7033–41.
40. Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature (2007) 449:819–26. doi: 10.1038/nature06246
41. Haschek WM, Rousseaux CG, Wallig MA. Chapter 6 - Respiratory System, In: Fundamentals of Toxicologic Pathology 2nd Edn San Diego: Academic Press. (2010). 93–133. doi: 10.1016/B978-0-12-370469-6.00006-4
42. Futosi K, Fodor S, Mócsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol. (2013) 17:638–50. doi: 10.1016/j.intimp.2013.06.034
43. Lyadova IV. Neutrophils in tuberculosis: heterogeneity shapes the way? Mediators Inflamm. (2017) 2017:8619307. doi: 10.1155/2017/8619307
44. Deffert C, Cachat J, Krause K-H. Phagocyte NADPH oxidase, chronic granulomatous disease and mycobacterial infections. Cell Microbiol. (2014) 16:1168–78. doi: 10.1111/cmi.12322
45. Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol. (2014) 9:181–218. doi: 10.1146/annurev-pathol-020712-164023
46. Stamm CE, Collins AC, Shiloh MU. Sensing of Mycobacterium tuberculosis and consequences to both host and bacillus. Immunol Rev. (2015) 264:204–19. doi: 10.1111/imr.12263
47. Martineau AR, Newton SM, Wilkinson KA, Kampmann B, Hall BM, Nawroly N, et al. Neutrophil-mediated innate immune resistance to mycobacteria. J Clin Invest. (2007) 117:1988–94. doi: 10.1172/JCI31097
48. Kisich KO, Higgins M, Diamond G, Heifets L. Tumor Necrosis Factor Alpha Stimulates Killing of Mycobacterium tuberculosis by Human Neutrophils. Infect Immun. (2002) 70:4591–99. doi: 10.1128/IAI.70.8.4591-4599.2002
49. Lowe DM, Demaret J, Bangani N, Nakiwala JK, Goliath R, Wilkinson KA, et al. Differential effect of viable versus necrotic neutrophils on Mycobacterium tuberculosis growth and cytokine induction in whole blood. Front Immunol. (2018) 9: doi: 10.3389/fimmu.2018.00903
50. Eruslanov EB, Lyadova IV, Kondratieva TK, Majorov KB, Scheglov IV, Orlova MO, et al. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun. (2005) 73:1744–53. doi: 10.1128/IAI.73.3.1744-1753.2005
51. Dorhoi A, Iannaccone M, Farinacci M, Faé KC, Schreiber J, Moura-Alves P, et al. MicroRNA-223 controls susceptibility to tuberculosis by regulating lung neutrophil recruitment. J Clin Invest. (2013) 123:4836–48. doi: 10.1172/JCI67604
52. Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med. (2013) 188:1137–46. doi: 10.1164/rccm.201304-0803OC
53. Nandi B, Behar SM. Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J Exp Med. (2011) 208:2251–62. doi: 10.1084/jem.20110919
54. Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature (2015) 528:565–9. doi: 10.1038/nature16451
55. Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci USA. (2012) 109:E3168–76. doi: 10.1073/pnas.1210500109
56. Kondratieva E, Logunova N, Majorov K, Jr MA, Apt A. Host genetics in granuloma formation: human-like lung pathology in mice with reciprocal genetic susceptibility to M. tuberculosis and M. avium. PLoS ONE (2010) 5:e10515. doi: 10.1371/journal.pone.0010515
57. Marzo E, Vilaplana C, Tapia G, Diaz J, Garcia V, Cardona P-J. Damaging role of neutrophilic infiltration in a mouse model of progressive tuberculosis. Tuberculosis (Edinb) (2014) 94:55–64. doi: 10.1016/j.tube.2013.09.004
58. Yeremeev V, Linge I, Kondratieva T, Apt A. Neutrophils exacerbate tuberculosis infection in genetically susceptible mice. Tuberculosis (Edinb) (2015) 95:447–51. doi: 10.1016/j.tube.2015.03.007
59. Vesosky B, Rottinghaus EK, Stromberg P, Turner J, Beamer G. CCL5 participates in early protection against Mycobacterium tuberculosis. J Leukoc Biol. (2010) 87:1153–65. doi: 10.1189/jlb.1109742
60. Dallenga T, Repnik U, Corleis B, Eich J, Reimer R, Griffiths GW, et al. M. tuberculosis-induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe. (2017) 22:519–30.e3. doi: 10.1016/j.chom.2017.09.003
61. Dallenga T, Schaible UE. Neutrophils in tuberculosis—first line of defence or booster of disease and targets for host-directed therapy? Pathog Dis. (2016) 74:ftw012. doi: 10.1093/femspd/ftw012
62. Nicola NA, Metcalf D, Matsumoto M, Johnson GR. Purification of a factor inducing differentiation in murine myelomonocytic leukemia cells. Identification as granulocyte colony-stimulating factor. J Biol Chem. (1983) 258:9017–23.
63. Metcalf D. The molecular biology and functions of the granulocyte-macrophage colony-stimulating factors. Blood (1986) 67:257–67.
64. Bober LA, Grace MJ, Pugliese-Sivo C, Rojas-Triana A, Waters T, Sullivan LM, et al. The effect of GM-CSF and G-CSF on human neutrophil function. Immunopharmacology (1995) 29:111–19. doi: 10.1016/0162-3109(94)00050-P
65. Höglund M, Håkansson L, Venge P. Effects of in vivo administration of G-CSF on neutrophil functions in healthy volunteers. Eur J Haematol. (1997) 58:195–202.
66. Lopez AF, Williamson DJ, Gamble JR, Begley CG, Harlan JM, Klebanoff SJ, et al. Recombinant human granulocyte-macrophage colony-stimulating factor stimulates in vitro mature human neutrophil and eosinophil function, surface receptor expression, and survival. J Clin Invest. (1986) 78:1220–8.
67. Spiekermann K, Roesler J, Emmendoerffer A, Elsner J, Welte K. Functional features of neutrophils induced by G-CSF and GM-CSF treatment: differential effects and clinical implications. Leukemia (1997) 11:466–78. doi: 10.1038/sj.leu.2400607
68. Weisbart RH, Golde DW, Clark SC, Wong GG, Gasson JC. Human granulocyte-macrophage colony-stimulating factor is a neutrophil activator. Nature (1985) 314:361–3.
69. Simon H-U. Neutrophil apoptosis pathways and their modifications in inflammation. Immunol Rev. (2003) 193:101–110.
70. Lowe DM, Bandara AK, Packe GE, Barker RD, Wilkinson RJ, Griffiths CJ, et al. Neutrophilia independently predicts death in tuberculosis. Eur Respir J. (2013) 42:1752–7. doi: 10.1183/09031936.00140913
71. Condliffe AM, Chilvers ER, Haslett C, Dransfield I. Priming differentially regulates neutrophil adhesion molecule expression/function. Immunology (1996) 89:105–111.
72. Dapino P, Dallegri F, Ottonello L, Sacchetti C. Induction of neutrophil respiratory burst by tumour necrosis factor-alpha; priming effect of solid-phase fibronectin and intervention of CD11b-CD18 integrins. Clin Exp Immunol. (1993) 94:533–8.
73. Detmers PA, Lo SK, Olsen-Egbert E, Walz A, Baggiolini M, Cohn ZA. Neutrophil-activating protein 1/interleukin 8 stimulates the binding activity of the leukocyte adhesion receptor CD11b/CD18 on human neutrophils. J Exp Med. (1990) 171:1155–62.
74. Liles W. Conrad, Ledbetter Jeffrey A., Waltersdorph Ann W., Klebanoff Seymour J. Cross-linking of CD18 primes human neutrophils for activation of the respiratory burst in response to specific stimuli: implications for adhesion-dependent physiological responses in neutrophils. J Leukoc Biol. (1995) 58:690–7. doi: 10.1002/jlb.58.6.690
75. Miralda I, Uriarte SM, McLeish KR. Multiple phenotypic changes define neutrophil priming. Front Cell Infect Microbiol. (2017) 7:217. doi: 10.3389/fcimb.2017.00217
76. Nathan CF. Neutrophil activation on biological surfaces. Massive secretion of hydrogen peroxide in response to products of macrophages and lymphocytes. J Clin Invest. (1987) 80:1550–60. doi: 10.1172/JCI113241
77. Neufert C, Pai RK, Noss EH, Berger M, Boom WH, Harding CV. Mycobacterium tuberculosis 19-kDa lipoprotein promotes neutrophil activation. J Immunol. (2001) 167:1542–9.
78. Wright HL, Thomas HB, Moots RJ, Edwards SW. RNA-Seq reveals activation of both common and cytokine-specific pathways following neutrophil priming. PLoS ONE (2013) 8:e58598. doi: 10.1371/journal.pone.0058598
79. Corleis B, Korbel D, Wilson R, Bylund J, Chee R, Schaible UE. Escape of Mycobacterium tuberculosis from oxidative killing by neutrophils. Cell Microbiol. (2012) 14:1109–21. doi: 10.1111/j.1462-5822.2012.01783.x
81. Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. (1989) 320:365–76. doi: 10.1056/NEJM198902093200606
82. Lau YL, Chan GCF, Ha SY, Hui YF, Yuen KY. The role of phagocytic respiratory burst in host defense against Mycobacterium tuberculosis. Clin Infect Dis. (1998) 26:226–7. doi: 10.1086/517036
83. Kulkarni M, Desai M, Gupta M, Dalvi A, Taur P, Terrance A, et al. Clinical, immunological, and molecular findings of patients with p47phox defect chronic granulomatous disease (CGD) in indian families. J Clin Immunol. (2016) 36:774–84. doi: 10.1007/s10875-016-0333-y
84. Movahedi M, Aghamohammadi A, Rezaei N, Shahnavaz N, Jandaghi AB, Farhoudi A, et al. Chronic granulomatous disease: a clinical survey of 41 patients from the iranian primary immunodeficiency registry. IAA (2004) 134:253–9. doi: 10.1159/000078774
85. van den Berg JM, van Koppen E, Åhlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the european experience. PLoS ONE (2009) 4:e5234. doi: 10.1371/journal.pone.0005234
86. Wolach Baruch, Gavrieli Ronit, de Boer Martin, van Leeuwen Karin, Berger-Achituv Sivan, Stauber Tal, et al. Chronic granulomatous disease: clinical, functional, molecular, and genetic studies. The Israeli experience with 84 patients. Am J Hematol. (2016) 92:28–36. doi: 10.1002/ajh.24573
87. Nauseef WM. Biological Roles for the NOX Family NADPH Oxidases. J Biol Chem. (2008) 283:16961–5. doi: 10.1074/jbc.R700045200
88. Singel KL, Segal BH. NOX2-dependent regulation of inflammation. Clin Sci (Lond) (2016) 130:479–90. doi: 10.1042/CS20150660
89. Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist's guide to reactive oxygen species. Nat Rev Immunol. (2013) 13:349–61. doi: 10.1038/nri3423
90. Henderson WR, Klebanoff SJ. Leukotriene production and inactivation by normal, chronic granulomatous disease and myeloperoxidase-deficient neutrophils. J Biol Chem. (1983) 258:13522–7.
91. Mitra S, Abraham E. Participation of superoxide in neutrophil activation and cytokine production. Biochim et Biophys Acta (2006) 1762:732–41. doi: 10.1016/j.bbadis.2006.06.011
92. Warnatsch A, Tsourouktsoglou T-D, Branzk N, Wang Q, Reincke S, Herbst S, et al. Reactive oxygen species localization programs inflammation to clear microbes of different size. Immunity (2017) 46:421–32. doi: 10.1016/j.immuni.2017.02.013
93. Bylund J, MacDonald KL, Brown KL, Mydel P, Collins LV, Hancock REW, et al. Enhanced inflammatory responses of chronic granulomatous disease leukocytes involve ROS-independent activation of NF-κB. Eur J Immunol. (2007) 37:1087–96. doi: 10.1002/eji.200636651
94. Fialkow L, Wang Y, Downey GP. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic Biol Med. (2007) 42:153–64. doi: 10.1016/j.freeradbiomed.2006.09.030
95. Roxo-Junior P, Simão HML. Chronic granulomatous disease: why an inflammatory disease? Braz J Med Biol Res. (2014) 47:924–8. doi: 10.1590/1414-431X20143735
96. Schäppi MG, Jaquet V, Belli DC, Krause K-H. Hyperinflammation in chronic granulomatous disease and anti-inflammatory role of the phagocyte NADPH oxidase. Semin Immunopathol. (2008) 30:255–271. doi: 10.1007/s00281-008-0119-2
97. Voskuil MI, Bartek IL, Visconti K, Schoolnik GK. The response of Mycobacterium Tuberculosis to reactive oxygen and nitrogen species. Front Microbiol. (2011) 2:105. doi: 10.3389/fmicb.2011.00105
98. Brinkmann V, Laube B, Abu Abed U, Goosmann C, Zychlinsky A. Neutrophil extracellular traps: how to generate and visualize them. J Vis Exp. (2010) 24: 1724. doi: 10.3791/1724
99. Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. (2011) 208:417–20. doi: 10.1084/jem.20110367
100. Sheshachalam A, Srivastava N, Mitchell T, Lacy P, Eitzen G. Granule protein processing and regulated secretion in neutrophils. Front Immunol. (2014) 5:448. doi: 10.3389/fimmu.2014.00448
101. Bals R. Epithelial antimicrobial peptides in host defense against infection. Respir Res. (2000) 1:141–50. doi: 10.1186/rr25
102. Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. (2003) 3:710–720. doi: 10.1038/nri1180
103. Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol. (2004) 75:39–48. doi: 10.1189/jlb.0403147
104. Wiesner J, Vilcinskas A. Antimicrobial peptides: the ancient arm of the human immune system. Virulence (2010) 1:440–64. doi: 10.4161/viru.1.5.12983
105. Tan BH, Meinken C, Bastian M, Bruns H, Legaspi A, Ochoa MT, et al. Macrophages acquire neutrophil granules for antimicrobial activity against intracellular pathogens. J Immunol. (2006) 177:1864–71.
106. N'Diaye EN, Darzacq X, Astarie-Dequeker C, Daffé M, Calafat J, Maridonneau-Parini I. Fusion of azurophil granules with phagosomes and activation of the tyrosine kinase Hck are specifically inhibited during phagocytosis of mycobacteria by human neutrophils. J Immunol. (1998) 161:4983–91.
107. Jena P, Mohanty S, Mohanty T, Kallert S, Morgelin M, Lindstrøm T, et al. Azurophil granule proteins constitute the major Mycobactericidal proteins in human neutrophils and enhance the killing of Mycobacteria in macrophages. PLoS ONE (2012) 7:e50345. doi: 10.1371/journal.pone.0050345
108. Sharma S, Verma I, Khuller GK. Biochemical interaction of human neutrophil peptide-1 with Mycobacterium tuberculosis H37Ra. Arch Microbiol. (1999) 171:338–42.
109. Sharma S, Verma I, Khuller GK. Antibacterial activity of human neutrophil peptide-1 against Mycobacterium tuberculosis H37Rv: in vitro and ex vivo study. Eur Respir J. (2000) 16:112–7.
110. Sharma S, Verma I, Khuller GK. Therapeutic potential of human neutrophil peptide 1 against experimental tuberculosis. Antimicrob Agents Chemother. (2001) 45:639–40. doi: 10.1128/AAC.45.2.639-640.2001
111. Pires D, Marques J, Pombo JP, Carmo N, Bettencourt P, Neyrolles O, et al. Role of Cathepsins in Mycobacterium tuberculosis survival in human macrophages. Sci Rep. (2016) 6:32247. doi: 10.1038/srep32247
112. Adams LA, Möller M, Nebel A, Schreiber S, van der Merwe L, van Helden PD, et al. Polymorphisms in MC3R promoter and CTSZ 3′UTR are associated with tuberculosis susceptibility. Eur J Hum Genet. (2011) 19:676–81. doi: 10.1038/ejhg.2011.1
113. Cooke GS, Campbell SJ, Bennett S, Lienhardt C, McAdam KPWJ, Sirugo G, et al. Mapping of a novel susceptibility locus suggests a role for MC3R and CTSZ in human tuberculosis. Am J Respir Crit Care Med. (2008) 178:203–7. doi: 10.1164/rccm.200710-1554OC
114. Steinwede K, Maus R, Bohling J, Voedisch S, Braun A, Ochs M, et al. Cathepsin G and neutrophil elastase contribute to lung-protective immunity against mycobacterial infections in mice. J Immunol. (2012) 188:4476–87. doi: 10.4049/jimmunol.1103346
115. Rivas-Santiago B, Hernandez-Pando R, Carranza C, Juarez E, Contreras JL, Aguilar-Leon D, et al. Expression of cathelicidin LL-37 during Mycobacterium tuberculosis infection in human alveolar macrophages, monocytes, neutrophils, and epithelial cells. Infect Immun. (2008) 76:935–41. doi: 10.1128/IAI.01218-07
116. Coussens AK, Wilkinson RJ, Martineau AR. Phenylbutyrate Is Bacteriostatic against Mycobacterium tuberculosis and regulates the macrophage response to infection, Synergistically with 25-Hydroxy-Vitamin D3. PLoS Pathog. (2015) 11:e1005007. doi: 10.1371/journal.ppat.1005007
117. Padhi A, Sengupta M, Sengupta S, Roehm KH, Sonawane A. Antimicrobial peptides and proteins in mycobacterial therapy: current status and future prospects. Tuberculosis (2014) 94:363–73. doi: 10.1016/j.tube.2014.03.011
118. Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, et al. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol. (2013) 191:773–84. doi: 10.4049/jimmunol.1300113
119. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
120. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
121. Braian C, Hogea V, Stendahl O. Mycobacterium tuberculosis-induced neutrophil extracellular traps activate human macrophages. J Innate Immun. (2013) 5:591–602. doi: 10.1159/000348676
122. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. (2007) 176:231–41. doi: 10.1083/jcb.200606027
123. Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol. (2007) 5:577–582. doi: 10.1038/nrmicro1710
124. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against candida albicans. PLoS Pathog. (2009) 5:e1000639. doi: 10.1371/journal.ppat.1000639
125. Dabrowska D, Jabłonska E, Garley M, Ratajczak-Wrona W, Iwaniuk A. New aspects of the biology of neutrophil extracellular traps. Scand J Immunol. (2016) 84:317–22. doi: 10.1111/sji.12494
127. Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, Wahn V, et al. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood (2011) 117:953–9. doi: 10.1182/blood-2010-06-290171
128. Vorobjeva NV, Pinegin BV. Neutrophil extracellular traps: mechanisms of formation and role in health and disease. Biochem Mosc. (2014) 79:1286–96. doi: 10.1134/S0006297914120025
129. Ramos-Kichik V, Mondragón-Flores R, Mondragón-Castelán M, Gonzalez-Pozos S, Muñiz-Hernandez S, Rojas-Espinosa O, et al. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (2009) 89:29–37. doi: 10.1016/j.tube.2008.09.009
130. Murthy AR, Lehrer RI, Harwig SS, Miyasaki KT. In vitro candidastatic properties of the human neutrophil calprotectin complex. J Immunol. (1993) 151:6291–301.
131. Hilda JN, Das SD. TLR stimulation of human neutrophils lead to increased release of MCP-1, MIP-1α, IL-1β, IL-8 and TNF during tuberculosis. Hum Immunol. (2016) 77:63–67. doi: 10.1016/j.humimm.2015.10.005
132. Wawrocki S, Druszczynska M. Inflammasomes in Mycobacterium tuberculosis-Driven Immunity. Can J Infect Dis Med Microbiol. (2017) 2017:2309478. doi: 10.1155/2017/2309478
133. Bakele M, Joos M, Burdi S, Allgaier N, Pöschel S, Fehrenbacher B, et al. Localization and functionality of the inflammasome in neutrophils. J Biol Chem. (2014) jbc.M113.505636. doi: 10.1074/jbc.M113.505636
134. Agard M, Asakrah S, Morici LA. PGE2 suppression of innate immunity during mucosal bacterial infection. Front Cell Infect Microbiol. (2013) 3:45. doi: 10.3389/fcimb.2013.00045
135. Funk CD. Prostaglandins and Leukotrienes: Advances in Eicosanoid Biology. Science (2001) 294:1871–5. doi: 10.1126/science.294.5548.1871
136. Dallenga T, Linnemann L, Paudyal B, Repnik U, Griffiths G, Schaible UE. Targeting neutrophils for host-directed therapy to treat tuberculosis. Int. J. Med. Microbiol. (2018) 308:142–7. doi: 10.1016/j.ijmm.2017.10.001
137. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. (2008) 8:349–61. doi: 10.1038/nri2294
138. Coffey MJ, Phare SM, Peters-Golden M. Role of leukotrienes in killing of Mycobacterium bovis by neutrophils. Prostaglandins Leukot Essent Fatty Acids (2004) 71:185–90. doi: 10.1016/j.plefa.2004.03.012
139. Toda A, Yokomizo T, Shimizu T. Leukotriene B4 receptors. Prostaglandins Other Lipid Mediat. (2002) 68–69:575–585.
140. Ethuin F, Gérard B, Benna JE, Boutten A, Gougereot-Pocidalo M-A, Jacob L, et al. Human neutrophils produce interferon gamma upon stimulation by interleukin-12. Lab Invest. (2004) 84:1363–71. doi: 10.1038/labinvest.3700148
141. Kim B-H, Shenoy AR, Kumar P, Bradfield CJ, MacMicking JD. IFN-inducible GTPases in host cell defense. Cell Host Microbe. (2012) 12:432–44. doi: 10.1016/j.chom.2012.09.007
142. Pilla-Moffett D, Barber MF, Taylor GA, Coers J. Interferon-inducible GTPases in host resistance, inflammation and disease. J Mol Biol. (2016) 428:3495–513. doi: 10.1016/j.jmb.2016.04.032
143. Ellison MA, Gearheart CM, Porter CC, Ambruso DR. IFN-γ alters the expression of diverse immunity related genes in a cell culture model designed to represent maturing neutrophils. PLoS ONE (2017) 12:1–31. doi: 10.1371/journal.pone.0185956
144. Pedruzzi E, Fay M, Elbim C, Gaudry M, Gougerot-Pocidalo M-A. Differentiation of PLB-985 myeloid cells into mature neutrophils, shown by degranulation of terminally differentiated compartments in response to N-formyl peptide and priming of superoxide anion production by granulocyte-macrophage colony-stimulating factor. Br J Haematol. (2002) 117:719–26.
145. Berry MPR, Graham CM, McNab FW, Xu Z, Bloch SAA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature (2010) 466:973–7. doi: 10.1038/nature09247
146. Kobayashi SD, Voyich JM, Buhl CL, Stahl RM, DeLeo FR. Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: cell fate is regulated at the level of gene expression. Proc Natl Acad Sci USA. (2002) 99:6901–6. doi: 10.1073/pnas.092148299
147. Greenlee-Wacker MC. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev. (2016) 273:357–70. doi: 10.1111/imr.12453
148. Kobayashi SD, Voyich JM, Braughton KR, DeLeo FR. Down-regulation of proinflammatory capacity during apoptosis in human polymorphonuclear leukocytes. J Immunol. (2003) 170:3357–68. doi: 10.4049/jimmunol.170.6.3357
149. Hart SP, Dransfield I, Rossi AG. Phagocytosis of apoptotic cells. Methods (2008) 44:280–5. doi: 10.1016/j.ymeth.2007.11.009
150. Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. (1989) 83:865–75. doi: 10.1172/JCI113970
151. Krysko DV, D'Herde K, Vandenabeele P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis (2006) 11:1709–26. doi: 10.1007/s10495-006-9527-8.
152. Lima KM, Vago JP, Caux TR, Negreiros-Lima GL, Sugimoto MA, Tavares LP, et al. The resolution of acute inflammation induced by cyclic AMP is dependent on annexin A1. J Biol Chem. (2017) 292:13758–73. doi: 10.1074/jbc.M117.800391
153. Ferlazzo V, D'Agostino P, Milano S, Caruso R, Feo S, Cillari E, et al. Anti-inflammatory effects of annexin-1: stimulation of IL-10 release and inhibition of nitric oxide synthesis. Int Immunopharmacol. (2003) 3:1363–69. doi: 10.1016/S1567-5769(03)00133-4
154. Arur S, Uche UE, Rezaul K, Fong M, Scranton V, Cowan AE, et al. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev Cell (2003) 4:587–598.
155. Parente L, Solito E. Annexin 1: more than an anti-phospholipase protein. Inflamm Res. (2004) 53:125–32. doi: 10.1007/s00011-003-1235-z
156. Ohira T, Arita M, Omori K, Recchiuti A, Van Dyke TE, Serhan CN. Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J Biol Chem. (2010) 285:3451–61. doi: 10.1074/jbc.M109.044131
157. Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. (2002) 196:1025–37.
158. Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. (2001) 2:612–9. doi: 10.1038/89759
159. Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity (2014) 40:315–27. doi: 10.1016/j.immuni.2014.02.009
160. Schwanke RC, Marcon R, Bento AF, Calixto JB. EPA- and DHA-derived resolvins' actions in inflammatory bowel disease. Eur J Pharmacol. (2016) 785:156–64. doi: 10.1016/j.ejphar.2015.08.050
161. Kaufmann SHE, Dorhoi A. Inflammation in tuberculosis: interactions, imbalances and interventions. Curr Opin Immunol. (2013) 25:441–9. doi: 10.1016/j.coi.2013.05.005
Keywords: Mycobacterium, tuberculosis, inflammation, NETs, antimicrobial, protection, necrosis
Citation: Kroon EE, Coussens AK, Kinnear C, Orlova M, Möller M, Seeger A, Wilkinson RJ, Hoal EG and Schurr E (2018) Neutrophils: Innate Effectors of TB Resistance? Front. Immunol. 9:2637. doi: 10.3389/fimmu.2018.02637
Received: 17 July 2018; Accepted: 26 October 2018;
Published: 14 November 2018.
Edited by:
Tamás Laskay, Universität zu Lübeck, GermanyReviewed by:
Nathalie Winter, Institut National de la Recherche Agronomique (INRA), FranceTobias Dallenga, Forschungszentrum Borstel (LG), Germany
Copyright © 2018 Kroon, Coussens, Kinnear, Orlova, Möller, Seeger, Wilkinson, Hoal and Schurr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elouise E. Kroon, ZWxvdWlzZV9rQHN1bi5hYy56YQ==