
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol., 16 November 2018
Sec. Alloimmunity and Transplantation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02602
This article is part of the Research TopicImmune Profile after Autologous Hematopoietic Stem Cell Transplantation for Autoimmune Diseases: Where Do We Stand?View all 12 articles
 Kelen Cristina Ribeiro Malmegrim1,2*
Kelen Cristina Ribeiro Malmegrim1,2* João Rodrigues Lima-Júnior2,3
João Rodrigues Lima-Júnior2,3 Lucas Coelho Marlière Arruda4,5Júlia Teixeira Cottas de Azevedo4,6
Lucas Coelho Marlière Arruda4,5Júlia Teixeira Cottas de Azevedo4,6 Gislane Lelis Vilela de Oliveira7
Gislane Lelis Vilela de Oliveira7 Maria Carolina Oliveira2,4
Maria Carolina Oliveira2,4Phase I/II clinical trials of autologous hematopoietic stem cell transplantation (AHSCT) have led to increased safety and efficacy of this therapy for severe and refractory autoimmune diseases (AD). Recent phase III randomized studies have demonstrated that AHSCT induces long-term disease remission in most patients without any further immunosuppression, with superior efficacy when compared to conventional treatments. Immune monitoring studies have revealed the regeneration of a self-tolerant T and B cell repertoire, enhancement of immune regulatory mechanisms, and changes toward an anti-inflammatory milieu in patients that are responsive to AHSCT. However, some patients reactivate the disease after transplantation due to reasons not yet completely understood. This scenario emphasizes that additional specific immunological interventions are still required to improve or sustain therapeutic efficacy of AHSCT in patients with AD. Here, we critically review the current knowledge about the operating immune mechanisms or established mechanistic biomarkers of AHSCT for AD. In addition, we suggest recommendations for future immune monitoring studies and biobanking to allow discovery and development of biomarkers. In our view, AHSCT for AD has entered a new era and researchers of this field should work to identify robust predictive, prognostic, treatment-response biomarkers and to establish new guidelines for immune monitoring studies and combined therapeutic interventions to further improve the AHSCT protocols and their therapeutic efficacy.
More than 20 years ago, autologous hematopoietic stem cell transplantation (AHSCT) was proposed as an alternative and innovative treatment for severe and refractory autoimmune diseases (AD) (1, 2). This therapeutic approach has been successfully used to treat several AD and over the years phase I/II clinical studies have led to increased safety and efficacy of the procedure (1, 2). More recently, phase III randomized trials have proven greater therapeutic efficacy of AHSCT than conventional therapies for some AD, such as multiple and systemic sclerosis. In addition, these trials have demonstrated that AHSCT can induce long-term disease remission without further use of immunosuppression (3–11).
Despite the overall positive outcomes of the procedure, subgroups of patients fail to remain in remission after AHSCT and reactivate the autoimmune disease (1, 2). The factors associated with disease reactivation, however, remain to be investigated. They may range from patient-specific (e.g., disease physiopathology, age, co-morbidities) to more general factors (e.g., infections, resurgence of autoreactive T, and B cells) (1, 12).
In the last decade, immune monitoring analyses have shown that AHSCT is able to regenerate a new auto-tolerant immune T and B cell repertoire, increase immune regulatory mechanisms, and induce changes to an anti-inflammatory milieu in patients with different AD (13–44). Most of the established immune mechanisms of AHSCT are common to AD in general, while others may be disease-specific (1, 2, 12). Few studies have analyzed the mechanistic results by comparing data of autoimmune patients with different outcomes after AHSCT (17, 18, 26, 31). Table 1 lists all 39 mechanistic studies of AHSCT for AD already published since 2004 and the most common immune mechanisms reported.
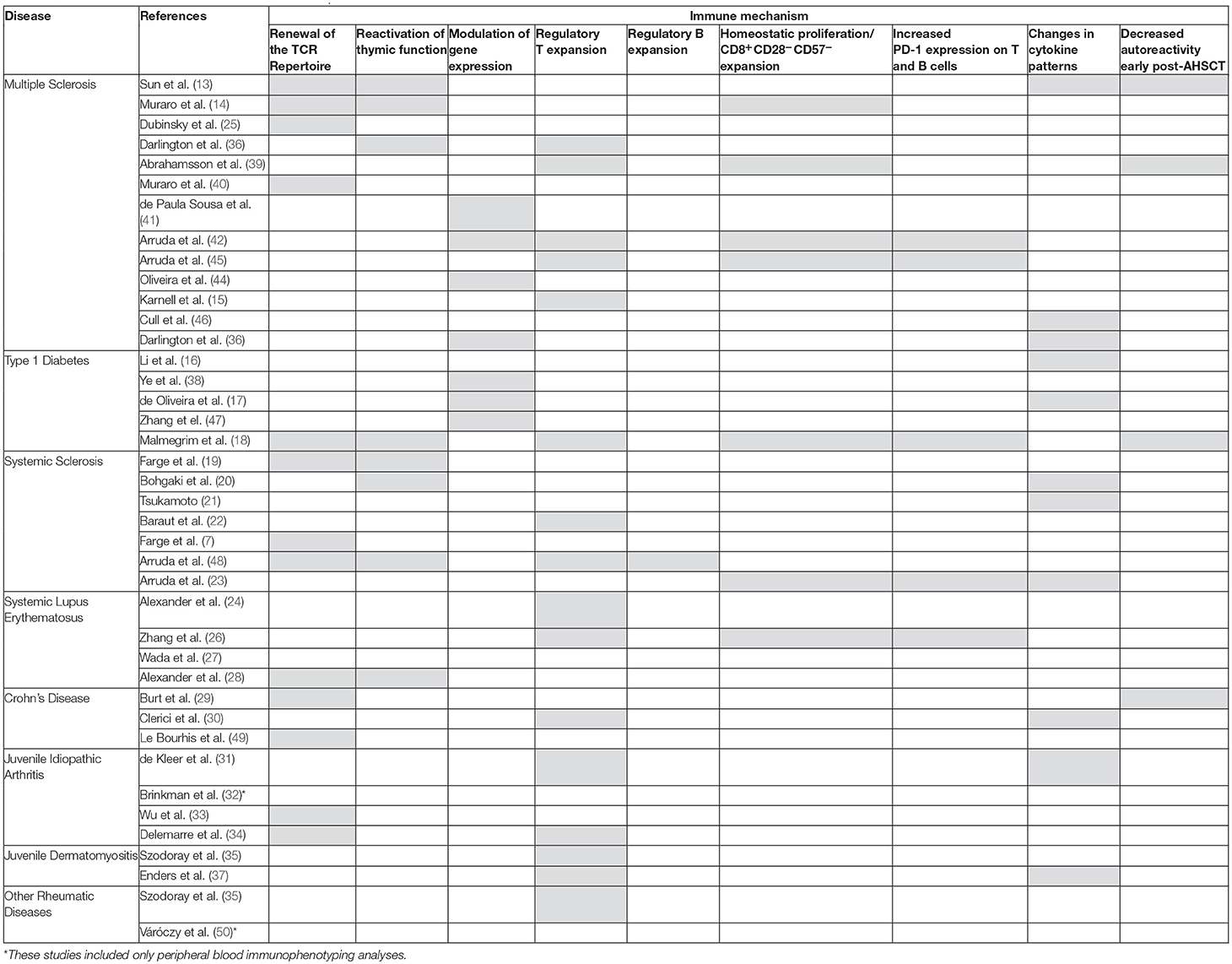
Table 1. Described immune mechanisms of AHSCT in patients with autoimmune diseases.
In this context, understanding of the immune mechanisms that lead to different clinical outcomes is essential to refine AHSCT protocols or to propose adjuvant/combined immunotherapy. Figure 1 indicates future directions to establish biomarkers of AHSCT for AD, including sample biobanking of multicenter randomized clinical studies and development of adjuvant/combined immunotherapy for patients that did not respond to AHSCT as single therapy.
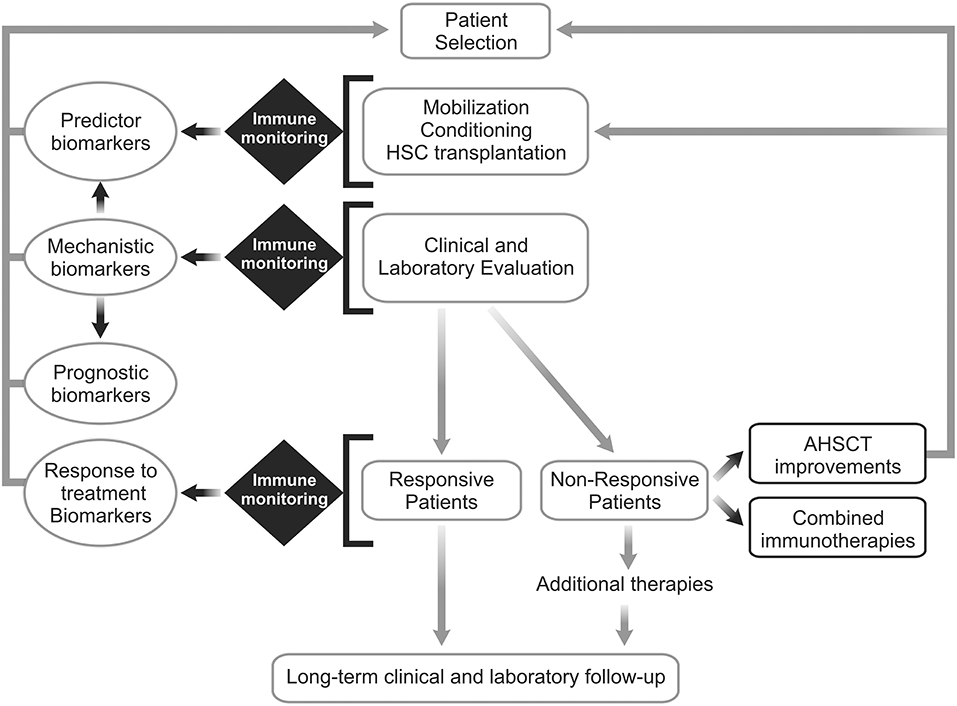
Figure 1. Future directions for establishment of biomarkers of autologous hematopoietic stem cell transplantation for autoimmune diseases. Patients with autoimmune disease (AD) must be are selected to undergo autologous hematopoietic stem cell transplantation (AHSCT). Based on the clinical, laboratory and immune monitoring results, patients will be classified in “responsive” or “non-responsive” to AHSCT. Well-performed immune monitoring evaluations are essential to settle mechanistic biomarkers and reveal new prognostic, predictor and/or response to treatment biomarkers. Further, additional therapeutic interventions can be proposed to treat patients who do not respond sufficiently to transplantation as a single treatment or reactivate the disease after some period of remission. New approaches for improving AHSCT efficacy in AD patients and/or combined immunotherapies are warranted.
In this perspective, we discuss the current knowledge about the immune mechanisms involved in AHSCT for AD, suggest recommendations for further immune monitoring studies and propose future directions in the field.
The rationale of AHSCT is the eradication of the autoreactive immunological memory and regeneration of the immune system. Ablation of the immune system, including depletion of autoreactive memory T and B cells, is followed by the reestablishment of immune tolerance (51). However, exactly how AHSCT corrects a deregulated immune system is not yet completely understood (1, 2).
Since 2004, and over the past 14 years, several immune mechanisms after AHSCT for AD have been described (Table 1). Many groups have investigated how the immunological renewal after AHSCT for ADs may reset a deregulated immune system into a self-tolerant status, inducing long-term remission (1, 2). Patients have been prospectively evaluated in studies that elucidate some immune mechanisms (Table 1). Currently, we may consider that these results, already reproduced on different patient cohorts, diseases, conditioning regimens, and laboratories around the world, are quite robust and consistent. Nevertheless, we still need to extend and worldwide standardize the immune monitoring evaluations in AD patients to enable the discovery of biomarkers, which will ultimately help to improve AHSCT protocols and their therapeutic efficacy (Table 2).
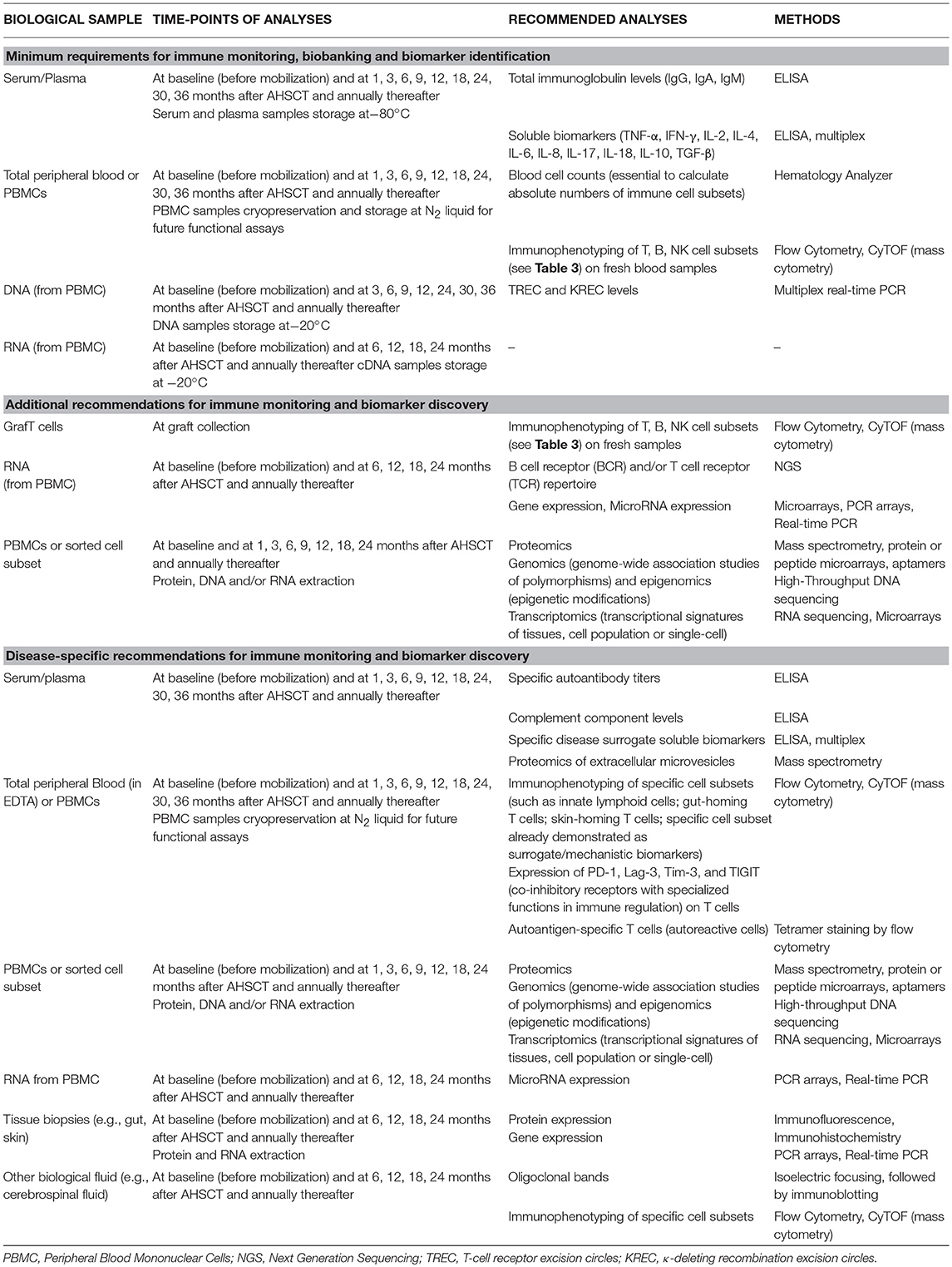
Table 2. Requirements for immune monitoring analyses in patients with AD undergoing AHSCT.
Thymus function translates into production of naive T cell populations and their exportation to the periphery (52). Generation of a diverse T cell repertoire throughout life is essential for immunity against pathogens. During development, T cells undergo central tolerance mechanisms, where positive selection ensures that T cells recognize self-MHC molecules and negative selection eliminates most T cells specific to autoantigens (52–54).
Thymic production of early naive T cells, named recent-thymic emigrants (RTE), can be determined through analysis of signal-joint T cell receptor excision circles (sjTRECs) (51). TRECs are DNA byproducts of T cell receptor (TCR) gene rearrangements that take place during T cell development in the thymus, and are reliable markers of RTE production and thymic function (55).
In the context of AHSCT, thymic rebound, which is defined by volumetric enlargement and functional reactivation of the thymus following lymphoid ablation, has been reported (14, 32, 55). Lymphoid and myeloid cells may be partially or completely depleted, depending on the intensity of the AHSCT conditioning regimen. Shortly after AHSCT, immune reconstitution predominantly relies on peripheral expansion of T and B cells that survived the highly immunosuppressive regimen or that were re-infused with the stem cell graft (15, 56).
Muraro et al. (14) were the first to demonstrate the so-called “immune resetting” mechanism in multiple sclerosis (MS) patients treated with AHSCT. Increased TREC levels indicated that peripheral T cells underwent thymic maturation after transplantation (14, 51). In addition, deep sequencing analysis demonstrated extensive replacement of a pre-existing TCR repertoire with new T cell clones that have emerged after transplantation. In fact, greater TCR repertoire diversity was found in patients with complete clinical response, indicating an interesting clinical correlation (18). In the following years, other researchers have also demonstrated, in different AD, that T cells could regenerate in adults with inactive thymuses (13–15, 18, 27, 31, 34, 35, 40, 41) and that the TCR repertoire after AHSCT was indeed renewed (13, 14, 16, 18, 26–28, 31, 34) (Table 1).
In summary, thymus reactivation and renewal of the TCR repertoire following AHSCT are the most significant mechanisms of action of this therapy so far described. Therefore, we should strongly recommend routine analyses of TREC levels in patients with AD undergoing AHSCT (Table 2). In addition, whenever possible, TCR repertoire evaluation by next generation sequencing should also be performed (Table 2).
Transcriptional analyses are currently used to describe disease signatures, evaluate response to treatments and to define patient subgroups, among other applications. However, to date few studies have evaluated modulation of gene expression of immune cells in the context of AHSCT for AD. Our group has evaluated the gene and microRNA expression profiles of reconstituted immune cells after AHSCT in MS patients. de Paula Sousa et al. (41) analyzed the global gene expression profiling of peripheral CD4+ or CD8+ T cells from MS patients at pre-transplantation and periodically after AHSCT. Hierarchical gene clustering analysis revealed that at 2 years after AHSCT, CD8+ T cells from MS patients were more similar to samples from healthy controls (19).
Other studies demonstrated normalization of deregulated gene expression following AHSCT for multiple sclerosis (20, 22, 25). Arruda et al. (23) demonstrated post-transplant normalization of the expression of mir-16, mir-155 and mir-142-3P, which have immmunoregulatory functions and are abnormally expressed in MS patients (20). As expected, expressions of their putative target genes, FOXP3, FOXO1, PDCD1, and IRF2BP2, were increased at 2 years post-transplantation (20) (Table 1).
In transplanted type 1 diabetes patients, Ye et al. (38) observed increased IL10, TGFβ, and FOXP3 mRNA expression, despite no significant regulatory T cell expansion (44).
In conclusion, few studies have so far explored transcriptomic analyses to understand the immune mechanisms of AHSCT for AD. One important challenge is to perform global transcriptome or single gene expression analysis for specific genes in purified T and B cell subsets from patients that undergo AHSCT, before and after disease reactivation. Lack of proper study design for appropriate cell separation/sorting and RNA isolation and/or technology cost are the most probable reasons for researchers not having so far explored this approach. In our opinion, this is a powerful method that should be explored in future studies. Transcriptional signatures of diseases for diagnosis, mechanisms and response to treatment are currently needed. Besides transcriptomics, other advanced new tools and technologies may be also useful to identify therapeutic biomarkers, such as proteomics, cytomics, lipidomics, and metabolomics (57–61).
Many mechanistic studies have demonstrated that AHSCT decreases the inflammatory status of patients with different AD (13, 24, 28, 37, 38, 43, 51–53) (Table 1). Sun et al. (13) reported the first study that deeply evaluated the immune reconstitution in MS patients treated with AHSCT (13). Serum levels of IL-12, IFN-γ, TNF-α, IL-10, and IL-4 were modulated after transplantation, however they decreased to baseline levels at 12 months (13). The authors also reported significant transient increase of IFN-γ, TNF-α, and IL-10 serum levels between 3 and 6 months after AHSCT, compared to the pre-transplant period. On the other hand, IL-12 serum levels consistently decreased in all patients following AHSCT (13).
Bohgaki et al. (20) evaluated IFN-γ and IL-4 production in T cells of SSc patients by intracellular staining. At inclusion, the cytokine production profile between transplanted SSc patients and healthy individuals was not significantly different (28). Frequencies of CD3+CD8− and CD3+CD8+ cytokine-producing T cells were not different between poor or good response groups. Notably, IFN-γ producing CD8+ T cells increased after transplantation in both groups (28).
Tsukamoto et al. (21) showed that while AHSCT was effective in controlling disease activity of systemic sclerosis (SSc) patients, Th1/Th2 ratio was significantly increased for at least 3 years after transplantation. The authors showed that the IFN-γ+/ CD4+ T cells to IL-4+/CD4+ T cell ratio increased early after transplantation, reaching a plateau 6 months after AHSCT (29).
In addition, Crohn's disease patients who fully responded to treatment with AHSCT had higher numbers of regulatory T cells (Treg) and lower IFN- γ and IL-12 serum levels than non-responders (37). In juvenile arthritis patients, de Kleer et al. (31) demonstrated increased expression of mRNA IL-10 and decreased expression of mRNA IFN-γ in hsp60-specific T cells after transplantation (38). Therefore, authors suggest that AHSCT promotes a shift in autoreactive cells from pro-inflammatory to a more tolerant phenotype (38).
Type 1 diabetes (T1D) patients showed decreased levels of serum autoantibodies and of pro-inflammatory cytokines IL-1, IL-17, and TNF-α after AHSCT (24). Enders et al. (37) reported that patients with active juvenile dermatomyositis had elevated levels of three pro-inflammatory biomarkers (CXCL10, TNFR2, and Galectin-9) that highly correlated with disease activity. Notably, levels of these biomarkers decreased in two patients after transplantation (43).
In summary, the above-described reports showed interesting modulation of cytokine/chemokine/other soluble markers (13, 28, 29, 38, 43, 46, 50) after AHSCT. However, these post-transplantation changes only make sense if related to disease pathogenesis and/or activity. In our opinion, serum evaluation of the main classical cytokines (TNF-α, IFN-γ, IL-2, IL-4, IL-6, IL-8, IL-17, IL-18, IL-10, TGF-β) is interesting, but not essential for most AD post-AHSCT follow-up (Table 2). In organ-specific AD, serum concentration of most general cytokines is low and heterogeneous. However, in systemic AD serum levels of important soluble factors (such as cytokines chemokines, growth factors, and surrogate disease-specific markers) may be evaluated at pre- and post-transplantation periods to search for predictive biomarkers and/or treatment-response biomarkers of AHSCT for systemic AD (Table 2).
Autoreactive T cells that escape negative selection in the thymus are found in the peripheral blood of healthy individuals (53). However, peripheral tolerance mechanisms control autoimmunity and prevent development of autoimmune disease (62, 63). T cell-intrinsic (anergy, clonal deletion, or immunological ignorance) and extrinsic mechanisms (mediated by suppressor/regulatory cells) are essential to peripheral tolerance. The CD4+CD25+Foxp3+ Treg, the main natural regulatory T cell subset, are very important for extrinsic control of peripheral tolerance, once they modulate autoreactive T and B cell responses.
Almeida et al. (64) demonstrated that in lymphopenic environments CD8+ T-cell subsets impact on each other during expansion (64). CD4+CD25+ Treg suppress and control repopulation of CD8+ T cells, leading to balanced repopulation of central-memory and effector-memory T cell subsets (64). Therefore, CD4+CD25+ Treg may have a major role in regulating the expansion of CD8+ T cell subsets during repopulation of the lymphopenic environment after AHSCT in AD patients (55, 64).
In the context of AHSCT, de Kleer et al. (31) were first to demonstrate that AHSCT for AD induces immunologic self-tolerance by restoring the CD4+CD25hi immune regulatory network (Table 1). AHSCT was able to normalize the frequency of CD4+CD25hi Treg in patients with idiopathic juvenile arthritis (IJA). Recovery of normal Treg levels after transplantation was due to both, homeostatic expansion during the lymphopenic phase of immune reconstitution and thymic generation of naive Treg CD4+CD25hi expressing FOXP3 mRNA (38). Since then, many groups have shown increased Treg numbers as an important and common immune mechanism of AHSCT for AD (16, 20, 23, 26, 31–33, 36–38, 41, 63) (Table 1). Delemarre et al. (34) demonstrated that remission of AD after transplantation involves renewal of the Treg TCR repertoire during thymic reactivation (41).
During immune reconstitution, the lack of Foxp3+ Treg results in development of “gaps” in the TCR repertoire and inappropriate responses to foreign antigens. Conversely, the presence of Foxp3+ Treg optimizes TCR repertoire diversity and foreign antigen responsiveness (64, 65). These studies provide an example of Treg activity that actually enhances immunity, which is very important in transplanted AD patients, contrasting with their generally accepted immunosuppressive function (64, 65).
The peripheral expansion of CD4+CD25hiFoxp3+ Treg and other regulatory populations during the lymphopenic phase post-transplantation is a very intriguing mechanism of AHSCT for AD. During this early phase of immune reconstitution, the total number of CD4+ T cells is very low. Therefore, expanded CD4+CD25hiFoxp3+ Treg cells correspond to the majority of CD4+ T cells in this phase. The expansion of cells with regulatory phenotype and function may allow the achievement of a “fine immune balance” in the patient until new naive T cells are produced by the reactivated thymus. Therefore, all future immune monitoring studies should assess the absolute number of phenotypically well-characterized Treg subsets and their immunosuppressive potential in well-designed assays (Tables 2, 3).
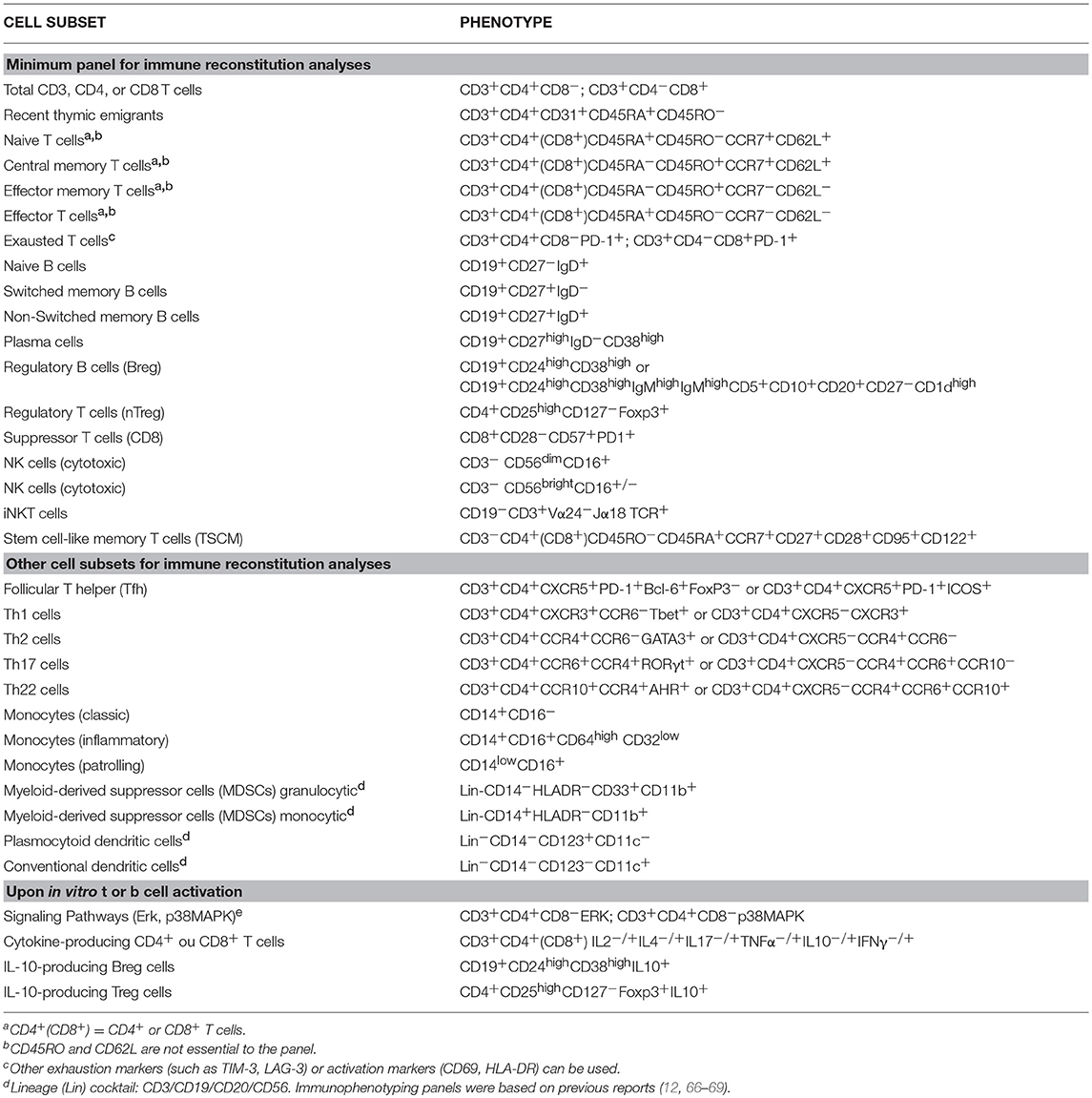
Table 3. Immunophenotyping of peripheral blood cell subsets in patients with AD undergoing AHSCT.
Regulatory B cells (Breg) are immunosuppressive cells that support immunological tolerance (70, 71). They modulate the immune responses mainly via IL-10 secretion (70, 71). Abnormalities in the Breg number or function have been described in many immune-mediated disorders. Therefore, Breg are considered essential to maintain immune homeostasis (70, 71). CD19+CD24hiCD38hi B-cell populations are well-characterized Breg in humans, playing an important role in the control of autoreactivity (72).
Recently, our group (31) showed that Breg frequencies transiently increased after AHSCT, tending to remain higher than pre-transplant values for at least 2 years (Table 1). We believe that Breg may be involved in the reestablishment of auto-tolerance after AHSCT, as suggested by persistently increased Breg/memory B cell ratio, as well as higher IL-10 production in SSc patients after transplantation (31). Notably, we found that Breg expansion occurs early post-transplantation [(31) and unpublished observation of ongoing studies]. Therefore, peripheral blood samples should be collected at 1, 3, and 6 months after transplantation (Tables 2, 3). In our opinion, whenever possible, besides the classical CD19+CD24hiCD38hi Breg, other regulatory B cell subsets (71) should be evaluated. In addition, functional assays to evaluate the immunosuppressive capacity of reconstituted Breg populations are also desirable (72).
After transplant-induced immunological depletion, lymphocytes repopulate the immune space both through enhanced T cell neogenesis from the thymus and proliferation of residual non-depleted peripheral lymphocytes, in a biological process named homeostatic proliferation. At this condition, CD8+ T cells proliferate more than the CD4+ T cell population (73).
Abrahamsson et al. (39) showed that CD8+CD28−CD57+ T cell subsets were persistently increased in MS patients after AHSCT and were able to suppress CD4+ T cell proliferation with variable potency (17). CD8+CD28−CD57+ T cells have regulatory properties and their numbers are usually found decreased in AD (74) (Table 1). In this context, we suggest that CD8+CD28−CD57+ T regulatory T cells expand preferentially following peripheral immune homeostasis disruption, such as in the early post-AHSCT period. In addition, many other mechanistic studies have also demonstrated that this regulatory/suppressor T cell subset expands after transplantation, indicating its important role in AHSCT for AD (Table 1).
We have also recently found in SSc patients that homeostatic proliferation after AHSCT results in transient telomere attrition and increased numbers of senescent and exhausted CD8+CD28−CD57+ T cells. In addition, high expression of programmed cell death protein 1 (PD-1) in peripheral T cells is associated with better clinical outcomes after AHSCT (48).
In our opinion, this T cell population plays and important role in controlling autoimmunity early post-AHSCT. Future immune monitoring studies should better characterize their phenotype, their immunosuppressive capacity and possibly other mechanisms of action in AD patients undergoing AHSCT (Tables 2, 3). Our studies have suggested that these CD8+CD28−CD57+ T cells express PD-1 (73), and perhaps other immunoregulatory molecules, as supported by recent literature from other researcher groups (75).
During T cell activation, the PD1 (or CD279) molecule is expressed on the cell surface and may engage its ligand, the programmed cell death ligand 1 (PDL1; also known as CD274) and/or PDL2 (also known as CD273). Upon binding, positive signals are generated through TCR and CD28 (76). These co-inhibitory receptors act as immune checkpoints for effector T cells, which regulate the adaptive immune responses. In fact, mice deficient in PD-1 molecules are susceptible to development of autoimmunity and AD, as reviewed by Sharpe and Pauken (77). Moreover, PD-1 polymorphisms detected in human autoimmune disorders support a role for the PD-1 pathway in self-tolerance mechanisms (77). Of note, during viral infections, the PD-1 molecule is upregulated on the surface of T cells during acute homeostatic proliferation (78, 79).
Thangavelu et al. (80) showed that recent thymic emigrants deficient in PD-1, which were generated after transfer of PD-1 deficient hematopoietic stem cells into lymphopenic adult Rag-deficient mice, induced a systemic inflammatory disease (80). Therefore, under lymphopenic conditions, PD-1 signaling is essential for systemic self-tolerance. In addition, Ellestad et al. (81) demonstrated that the most important role of PD-1 pathway is not to promote Treg expansion, but to control T cell activation and proliferation in response to self-antigens (81). Therefore, in lymphopenic environments, the PD-1 pathway is essential to regulate T cell activation and proliferation and consequently to avoid development of autoimmunity and AD.
In this context, Arruda et al. (43) showed that MS patients treated with AHSCT had expansion of PD-1+CD19+ B-cells and PD-1+CD8+ T-cells early post-transplantation. Therefore, the PD-1 inhibitory pathway is considered an immune regulatory mechanism by which AHSCT restores auto-tolerance in patients with MS (45) and other AD (Table 1).
Our group has recently shown increased expression of PD-1 in T cells from SSc patients who responded to AHSCT. As a general mechanism to keep potentially autoreactive CD8+ T-cell clones under control after AHSCT, PD-1 expression may be a reliable immune marker of clinical response in SSc and MS patients after AHSCT (48) (Table 1). Based on these recent data, we recommend the evaluation of expression of PD1 on T and B cell subsets in future immune monitoring studies (Tables 2, 3). In addition, we suggest to evaluate the expression of Tim-3 (T cell immunoglobulin-3), Lag-3 (Lymphocyte activation gene-3 or CD223) and TIGIT (T cell immunoglobulin and ITIM domain), other co-inhibitory receptors with specialized functions in immune regulation, on T cells (82).
Elimination of autoreactive effector memory T cells in patients with AD is supposed to ameliorate the autoimmune aggression and diminish disease activity. Sun and colleagues showed reduced T cell responses to myelin basic protein in the reconstituted immune system of MS patients after AHSCT, when compared to pre-transplant levels (13) (Table 1).
Recently, we showed that pre-transplant frequencies of autoreactive CD8+ T cells in T1D patients predicted the duration of insulin independency after AHSCT (26). Additionally, T1D patients that remained insulin free for longer periods also had persistently lower frequencies of autoreactive CD8+ T cells compared with patients who resumed insulin after transplantation (26) (Table 1).
In our opinion, monitoring of antigen-specific autoreactivity should be performed routinely in immune monitoring studies, as reported previously (13, 17, 26). We acknowledge the enormous difficulties to quantify peripheral autoreactive CD4+ or CD8+ T cells based on the variety of autoantigens/peptides and class-I or class-II HLA molecules. However, collaborations should be settled with autoimmunity expert groups for monitoring autoreactive T cells on frozen PBMC patient samples or researchers could use commercially available HLA-tetramers.
We believe it would be very important to also investigate if late reactivation of the AD after AHSCT coincides with the increase of new autoreactive naive T cells produced by the reactivated thymus or/and with a decrease in immune regulatory networks (Treg and Breg cell numbers and/or function), triggered by environmental factors, leading to loss of the once achieved “fine immune balance.”
Current data indicate that more intense immunoablation is associated with more favorable clinical outcomes after AHSCT (26, 31, 32, 35, 45) due to greater depletion of autoreactive T and B cells. However, complete eradication of the existing autoreactive immunologic memory may not be possible, even under high-intensity myeloablative conditioning regimens, since memory T and B cells reside in the bone marrow, are present in other body tissues and may survive immunoablative regimens (51, 83).
Currently, it is not known if immunosuppressive agents can infiltrate the tissues beyond the bone marrow. In experimental models, it has been shown that T cell depletion by ATG is more efficient in the blood compared with peripheral lymphoid organs (84). Park and Kupper (83) have demonstrated that tissue resident memory T cells are not totally depleted in non-barrier tissues after high dose immunosuppressive therapy (83). Curiously, in patients with refractory Crohn's disease, it was recently shown that the TCR repertoire diversifies after AHSCT in their intestinal tissue. The authors showed significant resetting of the TCR repertoire, since only 20% of TCR sequences were detected pre-transplantation and also 6 and 12 months post-AHSCT periods (49), prior to the production of any new naive T cells.
So far, few disease-specific immune mechanisms of AHSCT have been reported. These mechanisms include quantification of disease-specific autoreactive T cells and their characterization (cytokine-profile, mRNA expression) (13, 16, 38); identification/quantification/functional characterization of different regulatory T cell subsets (33); and quantification of cytokine and other soluble markers related to disease pathogenesis (16, 17).
Darlington et al. (36) demonstrated that myelin-specific T cells reconstitute after AHSCT in all transplanted MS patients. These autoreactive T cells may expand from residual cells or may be newly generated by the thymus. Re-emerged myelin-specific T cells had the same Th1 and Th2 baseline profile, but Th17 cells decreased after transplantation, as demonstrated by lower RORγ expression and IL-17A serum concentration. In addition, levels of IL-1β and IL-6 were also decreased in MS patients after AHSCT (16). In MS patients, Muraro et al. (14) showed increased expression of Fas on CD4+ and CD8+ T cells after AHSCT (14). Moreover, the same group has shown that the pro-inflammatory CD8+CD161high mucosal-associated invariant T cell (MAIT) subset was depleted from the peripheral blood of MS patients after AHSCT (17) (Table 1).
Most reported studies have evaluated classical regulatory T cell subsets (Table 1) in the context of AHSCT for AD, but Zhang et al. (26) identified peripheral CD8+ T cells from SLE patients with sustained high FoxP3 expression and increased expression of CTLA-4, PD-1, PD-L1, latency-associated peptide (LAP), and CD103, when compared with pre-transplant CD8+ T cells. The CD8+ Treg subset reconstituted post-transplantation presented increased suppressive activity, both autoantigen-specific and non-specific, which was predominantly TGF-β dependent and contact-independent. Therefore, the generation of a new LAPhighCD103highCD8+ Treg subset was reported, improving the immune regulatory deficiency and correlating with clinical remission (33) (Table 1).
In summary, we cannot conclude that the immune mechanisms cited above are disease-specific because they have not been yet evaluated in all AD subtypes currently treated with AHSCT. For future immune monitoring studies, we suggest the evaluation of validated disease-specific biomarkers (closely related to disease pathogenesis) in patients with AD undergoing AHSCT therapy (85–93).
Recent mechanistic studies of AHSCT for AD are more complete and comprehensive. Higher numbers of transplanted patients, longer follow-up, different methodologies, and modern experimental approaches have contributed to the understanding of the reconstituting immune system. This mechanistic knowledge can be now reverted to the clinic, improving transplantation protocols and clinical outcomes. Moreover, the identification of new biomarkers may further enhance transplantation efficacy.
Biomarkers are urgently needed to improve disease diagnosis, to monitor disease activity and therapeutic responses, and to allow validation of new therapies (94–96). A biomarker is defined as a biological substance that can be quantified and dynamically evaluated in the context of normal biological or pathogenic processes and their progression, or in the context of clinical responses to therapeutic interventions. Biomarkers must be reproducible, robust and validated (94–96).
Mechanistic biomarkers may inform and validate how a particular treatment works. Over the past years, many mechanistic biomarkers of AHSCT for AD have been described and validated, based on different clinical outcomes (response and lack of response to treatment). In this context, increased PD-1 expression and expansion of Treg and Breg cells associate with a favorable clinical response to AHSCT in SSc, MS, and T1D patients (Table 1, Figure 1).
In addition, predictive biomarkers are disease-associated and indicate whether there is a chance of disease activation. They can also measure, at time of enrollment, the expected patient responsiveness to a specific treatment. Karnell et al. (15) proposed the increased numbers of memory CD4+ and CD8+ T cells at pre-transplantation as predictive biomarkers for response in MS patients. In addition, cumulative frequency of autoreactive islet-specific CD8+ T cells at pre-transplantation predicts the clinical outcome of AHSCT in T1D patients (26). After AHSCT, T1D patients with higher autoreactivity before transplantation reactivated disease earlier than patients with lower autoreactivity (26).
Biomarkers that predict the future outcome of a patient with a specific disease, and also the overall survival rates and clinical benefits from a therapeutic intervention, are called prognostic biomarkers. Several clinical studies demonstrated that remission of AD after AHSCT associates with increased TCR repertoire diversity due to thymic reactivation and to the decrease of effector and memory T cell numbers (14, 27, 32, 39, 40). In this context, thymic reactivation may be considered a prognostic biomarker, associated with positive clinical outcomes (Table 1, Figure 1).
Additional prognostic biomarkers have been defined for AHSCT in AD, such as increased Treg frequencies and reduced inflammatory cytokine levels (IFN-γ and IL-12) in responsive Crohn's disease patients (37), increased frequencies of T and B cells expressing PD-1 in responsive MS patients (62), reduced levels of inflammatory markers (TNFR2, CXCL10, and GAL-9) in responsive juvenile dermatomyositis patients, and increased Treg numbers in long-term responsive T1D patients (26). More recently, Arruda et al. (48) demonstrated that clinical improvement of SSc patients is related to increased numbers of newly generated Treg and Breg after AHSCT, as a result of coordinated thymic and bone marrow rebounds (31) (Table 1, Figure 1).
Since the publication of Sun et al. (13), we have increased our knowledge on mechanistic biomarkers of AHSCT (Table 1). It is now clear that most immune mechanisms are common to several AD and depend more on the conditioning regimen and quality of immune reconstitution, rather than only on disease pathogenesis (Table 1). Every parameter that affects the quality of immune reconstitution (e.g., infections, patient age, conditioning regimen, previous treatments) may affect clinical outcomes after AHSCT. Most patients respond to AHSCT and achieve long-term disease remission, however a subset of patients reactivate the AD after transplant (1, 4, 7–10) (Figure 1). Therefore, additional immune interventions are urgently warranted to improve AHSCT protocols.
In our opinion, we have reached another level of investigation in the field of AHSCT for AD. Future perspectives are protocol improvements (e.g., modifications in the conditioning regimens) and combined therapies (e.g., infusion of in vitro expanded immune regulatory cells, immune modulatory drugs). These may vary according to AD pathogenesis (Figure 1).
Strategies can be directed to improve specific immune regulatory mechanisms of AHSCT. For example, to increase the number and/or function of regulatory CD4+ or CD8+ T cell subsets in patients who did not respond sufficiently to the AHSCT as a single treatment, we could propose combined therapies with similar immune mechanisms of action, such as administration of intravenous immunoglobulin (IVIG) (97), vitamin D (98) or low-dose rapamycin (99), infusion of autologous expanded Treg (100), or infusion of autologous or allogeneic mesenchymal stromal cells (101).
The ultimate importance of immune monitoring evaluations in patients undergoing AHSCT for AD is their impact in the clinical setting and consequent contributions to improve safety and efficacy of clinical protocols. Here, we provide three examples of how immune monitoring evaluations have already impacted the AHSCT scenario.
In 2004, Sun et al. (13) showed that myelin-specific T cells were not detected in MS patients at early periods post-AHSCT but that they reconstituted 1 year post-transplantation. Similarly, our group has shown that type 1 diabetes patients with higher frequencies of autoreactive islet-specific T cells before transplantation reactivate the disease earlier than patients with lower frequencies of these cells (26). These data indicate that conditioning regimens might have different efficacy on different AD and/or patients depending on their immune status and/or level of autoreactivity before transplantation. Indeed, our study has shown that high dose immunosuppression was not strong enough to sufficiently deplete autoreactive islet-specific T cells (26). Based on these observations, our center has increased the immunoablative intensity of the conditioning regimen protocol from 200 mg/Kg of cyclophosphamide plus 4.5 mg/Kg of rabbit anti-thymocyte globulin (ATG), to 120 mg/Kg of cyclophosphamide plus 150 mg/Kg of fludarabine and 4.5 mg/Kg of ATG, to further increase T and B cell ablation.
In the second example, Dubinsky et al. (25) demonstrated that T-cell clones persisting in the peripheral blood after autologous hematopoietic SCT were undetectable in the CD34+ selected graft (16). We can therefore assume that the residual autoreactive cells, responsible for disease reactivation in some patients, did not emerge from the CD34+ autograft, but most probably survived the immunoablative regimen. In this context we recommend, for future mechanistic investigations, the evaluation of graft composition (all T and B naive and memory subsets, regulatory T and B cell subsets, autoreactive cells) after CD34+ cell mobilization for each transplanted AD disease, as well as its correlation with clinical outcomes.
Third, some studies have evaluated how CD34+ graft selection influences the immune reconstitution after AHSCT (1). Recently, Keever-Taylor et al. (102) showed that the manufacturing of autologous CD34+ cells in the “High-Dose Immunosuppression and Autologous Transplantation for Multiple Sclerosis” (HALT MS) and “Scleroderma: Cyclophosphamide or Transplantation” (SCOT) protocols was comparable across all transplantation centers and allowed successful granulocyte and platelet recoveries. On the other hand, Oliveira et al. (44) have demonstrated that CD34+ selection does not add benefit to the outcomes of transplanted SSc patients. These findings should be further confirmed by prospective randomized trials (22).
Immune monitoring and biobanking guidelines for AHSCT in patients with AD patients have been established by the European Blood and Marrow Transplantation (EBMT) group, in 2015 (12). They have provided very reliable recommendations for comparative research studies envisioning biomarker discovery (12). We believe EBMT guidelines for immune monitoring are still valid, especially for disease-specific evaluations. However, new data on the topic have been made available in the past 3 years, and we believe it is important to add them to this perspective, as additional/complementary recommendations (12) (Figure 1, Tables 2, 3).
Here, we encourage standardization of immune monitoring studies throughout transplantation centers worldwide associated with data registries of clinical and laboratory results, aiming to empower statistical analyses. Nowadays, with modern biobanking infrastructure and internationalization trends it should be easier to manage multicenter immune monitoring studies in order to identify potential and consistent biomarkers.
Discovery and development of biomarkers are definitely an unmet need in the field of AHSCT for AD. Some biomarkers can serve as early surrogates of eventual clinical outcomes or guide therapeutic decisions by enabling identification of individuals likely to not respond to this therapy (50, 85–87, 103).
In recent years, both traditional and next-generation applications, including large-scale transcriptomic, epigenomic, genomic, lipidomic, cytomic, and proteomic technologies have yielded a huge amount of new candidate biomarkers that correlate with different clinical phenotypes of AD. Researchers should now focus on discovering and developing such biomarkers that would allow the improvement of clinical protocols and therapeutic efficacy of AHSCT for AD (57–61).
We encourage the scientific community to discuss future therapeutic directions and to establish updated guidelines of immune monitoring studies in order to expand the field. The following points should be addressed in the future, to better understand and subsequently improve AHSCT-AD outcomes:
• Establish a standardized immune monitoring platform for AHSCT-AD clinical trials: (a) to harmonize immune reconstitution results allowing centers to gather data/results and perform meta-analysis; (b) to invest in biomarker discovery by modern technologies and appropriate biobanking logistics (see recommendations on Tables 2, 3, and Figure 1);
• Conduct additional multicenter clinical trials to harmonize clinical and also immune monitoring data, allowing significant, and conclusive results about AHSCT-AD efficacy and immune mechanisms;
• Establish standardized conditioning regimens for each AD, based on the recent experiences and clinical achievements from each group. Since conditioning regimens have great impact on immune reconstitution and potentially on clinical outcomes, there is an urgent need for standardization. Later, more personalized conditioning approaches may be allowed, based on the AD disease and patient immune status.
• Develop combined therapies to improve therapeutic efficacy of AHSCT in AD patients who do not respond sufficiently to transplantation as a single treatment (Figure 1).
This study has been approved by the Brazilian institutional review board, where the patients were enrolled, and complied with country-specific regulations. The study was conducted according to the Declaration of Helsinki and Good Practice Guidelines. All patients read and signed informed consents.
KM and MO wrote the manuscript. LA, JL-J, JdA, GdO performed literature review data analysis. All authors revised the manuscript and contributed to the final editing. All authors agreed with the content of the manuscript.
This work was supported by the São Paulo Research Foundation (Fundação de Amparo à Pesquisa do Estado de São Paulo—FAPESP, Centro de Terapia Celular/Center for Cell-Based Therapy, CTC-CEPID-FAPESP, No. 2013/08135-2).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to honor the memory of Professor Julio César Voltarelli. He was the founder and the coordinator of the Bone Marrow Transplantation Unit of the University Hospital at the Ribeirão Preto Medical School, University of São Paulo, Brazil. He passed away on March 21, 2012. His greatest legacy was the establishment of the clinical protocols of hematopoietic stem cell transplantation studies for autoimmune diseases in Brazil.
1. Swart JF, Delemarre EM, van Wijk F, Boelens J, Kuball J, van Laar JM, et al. Haematopoietic stem cell transplantation for autoimmune diseases. Nat Rev Rheumatol. (2017) 13:244–56. doi: 10.1038/nrrheum.2017.7
2. Muraro PA, Martin R, Mancardi GL, Nicholas R, Sormani MP, Saccardi R. Autologous haematopoietic stem cell transplantation for treatment of multiple sclerosis. Nat Rev Neurol. (2017) 13:391–405. doi: 10.1038/nrneurol.2017.81
3. Burt RK, Shah SJ, Dill K, Grant T, Gheorghiade M, Schroeder J, et al. Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomised phase 2 trial. Lancet (2011) 378:498–506. doi: 10.1016/S0140-6736(11)60982-3
4. Burt RK, Oliveira MC, Shah SJ, Moraes DA, Simoes B, Gheorghiade M, et al. Cardiac involvement and treatment-related mortality after non-myeloablative haemopoietic stem-cell transplantation with unselected autologous peripheral blood for patients with systemic sclerosis: a retrospective analysis. Lancet (2013) 381:1116–24. doi: 10.1016/S0140-6736(12)62114-X
5. van Laar JM, Farge D, Sont JK, Naraghi K, Marjanovic Z, Larghero J, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis. JAMA (2014) 311:2490–8. doi: 10.1001/jama.2014.6368
6. Mancardi GL, Sormani MP, Gualandi F, Saiz A, Carreras E, Merelli E, et al. Autologous hematopoietic stem cell transplantation in multiple sclerosis: a phase II trial. Neurology (2015) 84:981–8. doi: 10.1212/WNL.0000000000001329
7. Farge D, Burt RK, Oliveira MC, Mousseaux E, Rovira M, Marjanovic Z, et al. Cardiopulmonary assessment of patients with systemic sclerosis for hematopoietic stem cell transplantation: recommendations from the European Society for Blood and Marrow Transplantation Autoimmune Diseases Working Party and collaborating partners. Bone Marrow Transplant. (2017) 52:1495–03. doi: 10.1038/bmt.2017.56
8. Muraro PA, Pasquini M, Atkins HL, Bowen JD, Farge D, Fassas A, et al. Long-term outcomes after autologous hematopoietic stem cell transplantation for multiple sclerosis. JAMA Neurol. (2017) 74:459–69. doi: 10.1001/jamaneurol.2016.5867
9. Snowden JA, Badoglio M, Labopin M, Giebel S, McGrath E, Marjanovic Z, et al. Evolution, trends, outcomes, and economics of hematopoietic stem cell transplantation in severe autoimmune diseases. Blood Adv. (2017) 1:2742–55. doi: 10.1182/bloodadvances.2017010041
10. Snowden JA, Panés J, Alexander T, Allez M, Ardizzone S, Dierickx D, et al. Autologous haematopoietic stem cell transplantation (AHSCT) in severe Crohn's disease: a review on behalf of ECCO and EBMT. J Crohns Colitis (2018) 12:476–88. doi: 10.1093/ecco-jcc/jjx184
11. Sullivan KM, Goldmuntz EA, Keyes-Elstein L, McSweeney PA, Pinckney A, Welch B, et al. Myeloablative autologous stem-cell transplantation for severe scleroderma. N Engl J Med. (2018) 378:35–47. doi: 10.21430/M3SM4LTLH
12. Alexander T, Bondanza A, Muraro PA, Greco R, Saccardi R, Daikeler T, et al. SCT for severe autoimmune diseases: consensus guidelines of the European Society for Blood and Marrow Transplantation for immune monitoring and biobanking. Bone Marrow Transplant. (2015) 50:173–80. doi: 10.1038/bmt.2014.251
13. Sun W, Popat U, Hutton G, Zang YC, Krance R, Carrum G, et al. Characteristics of T-cell receptor repertoire and myelin-reactive T cells reconstituted from autologous haematopoietic stem-cell grafts in multiple sclerosis. Brain (2004) 127:996–1008. doi: 10.1093/brain/awh117
14. Muraro PA, Douek DC, Packer A, Chung K, Guenaga FJ, Cassiani-ingoni R, et al. Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J Exp Med. (2005) 201:805–16. doi: 10.1084/jem.20041679
15. Karnell FG, Lin D, Motley S, Duhen T, Lim N, Campbell DJ, et al. Reconstitution of immune cell populations in multiple sclerosis patients after autologous stem cell transplantation. Clin Exp Immunol. (2017) 189:268–78. doi: 10.1111/cei.12985
16. Li L, Shen S, Ouyang J, Hu Y, Hu L, Cui W, et al. Autologous hematopoietic stem cell transplantation modulates immunocompetenT cells and improves β-cell function in Chinese patients with new onset of type 1 diabetes. J Clin Endocrinol Metab. (2012) 97:1729–36. doi: 10.1210/jc.2011-2188
17. de Oliveira GL, Malmegrim KC, Ferreira AF, Tognon R, Kashima S, Couri CEB, et al. Up-regulation of fas and fasL pro-apoptotic genes expression in type 1 diabetes patients after autologous haematopoietic stem cell transplantation. Clin Exp Immunol. (2012) 168:291–302. doi: 10.1111/j.1365-2249.2012.04583.x
18. Malmegrim KC, de Azevedo JT, Arruda LC, Abreu JR, Couri CE, de Oliveira GL, et al. Immunological balance is associated with clinical outcome after autologous hematopoietic stem cell transplantation in type 1 diabetes. Front Immunol. (2017) 8:167. doi: 10.3389/fimmu.2017.00167
19. Farge D, Henegar C, Carmagnat M, Daneshpouy M, Marjanovic Z, Rabian C, et al. Analysis of immune reconstitution after autologous bone marrow transplantation in systemic sclerosis. Arthritis Rheum. (2005) 52:1555–63. doi: 10.1002/art.21036
20. Bohgaki T, Atsumi T, Bohgaki M, Furusaki A, Kondo M, Sato-Matsumura KC, et al. Immunological reconstitution after autologous hematopoietic stem cell transplantation in patients with systemic sclerosis: relationship between clinical benefits and intensity of immunosuppression. J Rheumatol. (2009) 36:1240–8. doi: 10.3899/jrheum.081025
21. Tsukamoto H, Nagafuji K, Horiuchi T, Mitoma H, Niiro H, Arinobu Y, et al. Analysis of immune reconstitution after autologous CD34+ stem/progenitor cell transplantation for systemic sclerosis: predominant reconstitution of Th1 CD4+ T cells. Rheumatology (2011) 50:944–52. doi: 10.1093/rheumatology/keq414
22. Baraut J, Grigore EI, Jean-Louis F, Khelifa SH, Durand C, Verrecchia F, et al. Peripheral blood regulatory T cells in patients with diffuse systemic sclerosis (SSc) before and after autologous hematopoietic SCT: a pilot study. Bone Marrow Transplant. (2014) 49:349–54. doi: 10.1038/bmt.2013.202
23. Arruda LC, Malmegrim KC, Lima-Júnior JR, Clave E, Dias JB, Moraes DA, et al. Immune rebound associates with a favorable clinical response to autologous HSCT in systemic sclerosis patients. Blood Adv. (2018b) 2:126–41. doi: 10.1182/bloodadvances.2017011072
24. Alexander T, Thiel A, Rosen O, Massenkeil G, Sattler A, Kohler S, et al. Depletion of autoreactive immunologic memory followed by autologous hematopoietic stem cell transplantation in patients with refractory SLE induces long-term remission through de novo generation of a juvenile and tolerant immune system. Blood (2017) 113:214–23. doi: 10.1182/blood-2008-07-168286
25. Dubinsky AN, Burt RK, Martin R, Muraro PA. T-cell clones persisting in the circulation after autologous hematopoietic SCT are undetectable in the peripheral CD34+ selected graft. Bone Marrow Transplant. (2010) 45:325–31. doi: 10.1038/bmt.2009.139
26. Zhang L, Bertucci AM, Ramsey-Goldman R, Burt RK, Datta SK. Regulatory T cell (Treg) subsets return in patients with refractory lupus following stem cell transplantation, and TGF–producing CD8+ treg cells are associated with immunological remission of lupus. J Immunol. (2009) 183:6346–58. doi: 10.4049/jimmunol.0901773
27. Wada H, Terasako K, Kamiya Y, Sato M, Kimura SI, Okuda S, et al. Immune recovery after autologous PBSC transplantation without in vitro graft manipulation for refractory systemic lupus erythematosus. Bone Marrow Transplant. (2011) 46:1450–4. doi: 10.1038/bmt.2010.332
28. Alexander T, Schneider S, Hoyer B, Cheng Q, Thiel A, Ziemer S, et al. Development and resolution of secondary autoimmunity after autologous haematopoietic stem cell transplantation for systemic lupus erythematosus: competition of plasma cells for survival niches? Ann Rheum Dis. (2013) 72:1102–5. doi: 10.1136/annrheumdis-2012-202729
29. Burt RK, Craig RM, Milanetti F, Quigley K, Gozdziak P, Bucha J, et al. Autologous nonmyeloablative hematopoietic stem cell transplantation in patients with severe anti-TNF refractory Crohn disease: long-term follow-up. Blood (2010) 116:6123–32. doi: 10.1182/blood-2010-06-292391
30. Clerici M, Cassinotti A, Onida F, Trabattoni D, Annaloro C, Della A, et al. Immunomodulatory effects of unselected haematopoietic stem cells autotransplantation in refractory Crohn's disease. Dig Liver Dis. (2011) 43:946–52. doi: 10.1016/j.dld.2011.07.021
31. de Kleer I. Autologous stem cell transplantation for autoimmunity induces immunologic self-tolerance by reprogramming autoreactive T cells and restoring the CD4+CD25+ immune regulatory network. Blood (2006) 107:1696–702. doi: 10.1182/blood-2005-07-2800
32. Brinkman DM, de Kleer IM, ten Cate R, van Rossum MA, Bekkering WP, Fasth A, et al. Autologous stem cell transplantation in children with severe progressive systemic or polyarticular juvenile idiopathic arthritis long-term followup of a prospective clinical trial. Arthritis Rheum. (2007) 56:2410–21. doi: 10.1002/art.22656
33. Wu Q, Pesenac AM, Stansfield A, Barge D, Helen E, Abinun M, et al. Immunological characteristics and T-cell receptor clonal diversity in children with systemic juvenile idiopathic arthritis undergoing T-cell-depleted autologous stem cell transplantation. Immunology (2014) 142:227–36. doi: 10.1111/imm.12245
34. Delemarre EM, van den Broek T, Mijnheer G, Meerding J, Wehrens EJ, Olek S, et al. Autologous stem cell transplantation aids autoimmune patients by functional renewal and TCR diversi fi cation of regulatory T cells. Blood (2016) 127:91–102. doi: 10.1182/blood-2015-06-649145
35. Szodoray P, Varoczy L, Papp G, Barath S, Nakken B, Szegedi G, et al. Immunological reconstitution after autologous stem cell transplantation in patients with refractory systemic autoimmune diseases. Scand J Rheumatol. (2012) 41:110–15. doi: 10.3109/03009742.2011.606788
36. Darlington PJ, Touil T, Doucet JS, Gaucher D, Zeidan J, Gauchat D, et al. Diminished Th17 (not Th1) responses underlie multiple sclerosis disease abrogation after hematopoietic stem cell transplantation. Ann Neurol. (2013) 73:341–54. doi: 10.1002/ana.23784
37. Enders FB, Delemarre EM, Kuemmerle-Deschner J, van der Torre P, Wulffraat NM, Prakken BP, et al. Autologous stem cell transplantation leads to a change in proinflammatory plasma cytokine profile of patients with juvenile dermatomyositis correlating with disease activity. Ann Rheum Dis. (2015) 74:315–17. doi: 10.1136/annrheumdis-2014-206287
38. Ye L, Li L, Wan B, Yang M, Hong J, Gu W, et al. Immune response after autologous hematopoietic stem cell transplantation in type 1 diabetes mellitus. Stem Cell Res Ther. (2017) 8:90. doi: 10.1186/s13287-017-0542-1
39. Abrahamsson SV, Angelini DF, Dubinsky AN, Morel E, Oh U, Jones JL, et al. Non-myeloablative autologous haematopoietic stem cell transplantation expands regulatory cells and depletes IL-17 producing mucosal-associated invariant T cells in multiple sclerosis. Brain (2013) 136(Pt 9):2888–903. doi: 10.1093/brain/awt182
40. Muraro PA, Robins H, Malhotra S, Howell M, Phippard D, Desmarais C, et al. Brief report T cell repertoire following autologous stem cell transplantation for multiple sclerosis. J Clin Invest. (2014) 124:1168–72. doi: 10.1172/JCI71691DS1
41. de Paula Sousa A, Malmegrim KC, Panepucci RA, Brum DS, Barreira AA, et al. Autologous hematopoietic stem cell transplantation reduces abnormalities in the expression of immune genes in multiple sclerosis. Clin Sci. (2014) 120:111–20. doi: 10.1042/CS20140095
42. Arruda LC, Lorenzi JC, Sousa AP, Zanette DL, Palma PV, Panepucci RA, et al. Autologous hematopoietic SCT normalizes miR-16,−155 and−142-3p expression in multiple sclerosis patients. Bone Marrow Transplant. (2015) 50:380–9. doi: 10.1038/bmt.2014.277
43. Arruda LC, Clave E, Moins-Teisserenc H, Douay C, Farge D, Toubert A. Resetting the immune response after autologous hematopoietic stem cell transplantation for autoimmune diseases. Curr Res Transl Med. (2016) 64:107–13. doi: 10.1016/j.retram.2016.03.004
44. Oliveira MC, Labopin M, Henes J, Moore J, Papa ND, Cras A, et al. Does ex vivo CD34+ positive selection influence outcome after autologous hematopoietic stem cell transplantation in systemic sclerosis patients? Bone Marrow Transplant. (2016) 51:501–5. doi: 10.1038/bmt.2015.299
45. Arruda LC, de Azevedo JT, de Oliveira GL, Scortegagna GT, Rodrigues ES, Palma et al. Immunological correlates of favorable long-term clinical outcome in multiple sclerosis patients after autologous hematopoietic stem cell transplantation. Clin Immunol. (2016) 169:47–57. doi: 10.1016/j.clim.2016.06.005
46. Cull G, Hall D, Fabis-Pedrini MJ, Carroll WM, Forster L, Robins F, et al. Lymphocyte reconstitution following autologous stem cell transplantation for progressive MS. Mult Scler J Exp Transl Clin. (2017) 3:2055217317700167. doi: 10.1177/2055217317700167
47. Zhang X, Ye L, Hu J, Tang W, Liu R, Yang M, et al. Acute response of peripheral blood cell to autologous hematopoietic stem cell transplantation in type 1 diabetic patient. PLoS One. (2012) 7:e31887. doi: 10.1371/journal.pone.0031887
48. Arruda LC, Lima-Júnior JR, Clave E, Moraes DA, Douay C, Fournier I, et al. Homeostatic proliferation leads to telomere attrition and increased PD-1 expression after autologous hematopoietic SCT for systemic sclerosis. Bone Marrow Transplant. (2018a) 53:1319–27. doi: 10.1038/s41409-018-0162-0
49. Le Bourhis L, Maria Corraliza A, Auzolle C, Ricart E, Hawkey CJ, Lindsay JO, et al. Resetting of the mucosal T cell repertoire after hematopoietic stem cell transplantation in refractory Crohn's disease. J Crohn's Colitis (2017) 11:S2–3. doi: 10.1093/ecco-jcc/jjx002.003
50. Váróczy L, Kovács I, Baráth S, Gyimesi E, Illés Á, Zeher M, et al. Changes in the number of CD8+ T lymphocytes in the peripheral blood of patients with various autoimmune diseases after autologous hematopoietic stem cell transplantations and their relations to the survival times. Arch Immunol Ther Exp. (2013) 61:421–6. doi: 10.1007/s00005-013-0241-2
51. Muraro PA, Douek DC. Renewing the T cell repertoire to arrest autoimmune aggression. Trends Immunol. (2006) 27:61–7. doi: 10.1016/j.it.2005.12.003
52. Anderson MS, Su MA. AIRE expands: new roles in immune tolerance and beyond. Nat Rev Immunol. (2016) 16:247–58. doi: 10.1038/nri.2016.9
53. Bluestone JA, Bour-Jordan H, Cheng M, Anderson M. T cells in the control of organ-specific autoimmunity. J Clin Invest. (2015) 125:2250–60. doi: 10.1172/JCI78089
54. Chan AY, Anderson MS. Central tolerance to self revealed by the autoimmune regulator. Ann NY Acad Sci. (2015) 1356:80–9. doi: 10.1111/nyas.12960
55. Douek DC, Vescio RA, Betts MR, Brenchley JM, Hill BJ, Zhang L, et al. Assessment of thymic output in adults after haematopoietic stem-cell transplantation and prediction of T-cell reconstitution. Lancet (2000) 355:1875–81. doi: 10.1016/S0140-6736(00)02293-5
56. Storek J, Geddes M, Khan F, Huard B, Helg C, Chalandon Y, et al. Reconstitution of the immune system after hematopoietic stem cell transplantation in humans. Semin Immunopathol. (2008) 30:425–37. doi: 10.1007/s00281-008-0132-5
57. Maecker HT, Lindstrom TM, Robinson WH, Utz PJ, Hale M, Boyd SD, et al. New tools for classification and monitoring of autoimmune diseases. Nat Rev Rheumatol. (2012) 8:317–28. doi: 10.1038/nrrheum.2012.66
58. Hoefer IE, Steffens S, Ala-Korpela M, Bäck M, Badimon L, Bochaton-Piallat ML, et al. Novel methodologies for biomarker discovery in atherosclerosis. Eur Heart J. (2015) 36:2635–42. doi: 10.1093/eurheartj/ehv236
59. Kanakry CG, Bakoyannis G, Perkins SM, McCurdy SR, Vulic A, Warren EH, et al. Plasma-derived proteomic biomarkers in human leukocyte antigen-haploidentical or human leukocyte antigen-matched bone marrow transplantation using post-transplantation cyclophosphamide. Haematologica (2017) 102:932–40. doi: 10.3324/haematol.2016.152322
60. Burel JG, Peters B. Discovering transcriptional signatures of disease for diagnosis versus mechanism. Nat Rev Immunol. (2018) 18:289–90. doi: 10.1038/nri.2018.26
61. Paczesny S. Biomarkers for posttransplantation outcomes. Blood (2018) 131:2193–204. doi: 10.1182/blood-2018-02-791509
62. Liston A, Gray DH. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol. (2014) 14:154–65. doi: 10.1038/nri3605
63. Rosenblum MD, Way SS, Abbas AK. Regulatory T cell memory. Nat Rev Immunol. (2016) 16:90–101. doi: 10.1038/nri.2015.1
64. Almeida ARM, Ciernik IF, Sallusto F, Lanzavecchia A. CD4+CD25+ Treg regulate the contribution of CD8+ T-cell subsets in repopulation of the lymphopenic environment. Eur J Immunol. (2010) 40:3478–88. doi: 10.1002/eji.201040600
65. Winstead CJ, Reilly CS, Moon JJ, Jenkins MK, Hamilton SE, Jameson SC, et al. CD4+CD25+Foxp3+ regulatory T cells optimize diversity of the conventional T cell repertoire during reconstitution from lymphopenia. J Immunol. (2010) 184:4749–60. doi: 10.4049/jimmunol.0904076
66. Finak G, Langweiler M, Jaimes M, Malek M, Taghiyar J, Korin Y, et al. Standardizing flow cytometry immunophenotyping analysis from the human immunophenotyping consortium. Sci Rep. (2016) 6:20686. doi: 10.1038/srep20686
67. Hasan M, Beitz B, Rouilly V, Libri V, Urrutia A, Duffy D, et al. Semi-automated and standardized cytometric procedures for multi-panel and multi-parametric whole blood immunophenotyping. Clin Immunol. (2015) 157:261–76. doi: 10.1016/j.clim.2014.12.008
68. Schlickeiser S, Streitz M, Sawitzki B. Standardized multi-color flow cytometry and computational biomarker discovery. Methods Mol Biol (2016) 1371:225–38. doi: 10.1007/978-1-4939-3139-2_15
69. Pockley AG, Lindsay JO, Foulds GA, Rutella S, Gribben JG, Alexander T, et al. Immune reconstitution after autologous hematopoietic stem cell transplantation in Crohn's disease: current status and future directions. a review on behalf of the EBMT autoimmune diseases working party and the autologous stem cell transplantation in refra. Front Immunol. (2018) 9:646. doi: 10.3389/fimmu.2018.00646
70. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. (2012) 30:221–41. doi: 10.1146/annurev-immunol-020711-074934
71. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity (2015) 42:607–12. doi: 10.1016/j.immuni.2015.04.005
72. Flores-Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, Isenberg DA, et al. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med. (2013) 5:173ra23. doi: 10.1126/scitranslmed.3005407
73. Zwang NA, Turka LA. Homeostatic expansion as a barrier to lymphocyte depletion strategies. Curr Opin Organ Transplant. (2014) 19:357–62. doi: 10.1097/MOT.0000000000000096
74. Vuddamalay Y, van Meerwijk JPM. CD28– and CD28lowCD8+ regulatory T cells: of mice and men. Front Immunol. (2017) 8:31. doi: 10.3389/fimmu.2017.00031
75. Cencioni MT, Magliozzi R, Nicholas R, Ali R, Malik O, Reynolds R, et al. Programmed death 1 is highly expressed on CD8+ CD57+ T cells in patients with stable multiple sclerosis and inhibits their cytotoxic response to Epstein-Barr virus. Immunology (2017) 152:660–76. doi: 10.1111/imm.12808
76. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. (2005) 23:515–48. doi: 10.1146/annurev.immunol.23.021704.115611
77. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. (2017) 18:153–67. doi: 10.1038/nri.2017.108
78. Radziewicz H, Dunham RM, Grakoui A. PD-1 tempers Tregs in chronic HCV infection. J Clin Invest. (2009) 119:450–3. doi: 10.1172/JCI38661
79. Lin S-J, Peacock CD, Bahl K, Welsh RM. Programmed death-1 (PD-1) defines a transient and dysfunctional oligoclonal T cell population in acute homeostatic proliferation. J Exp Med. (2007) 204:2321–33. doi: 10.1084/jem.20062150
80. Thangavelu G, Parkman JC, Ewen CL, Uwiera RR, Baldwin TA, Anderson CC. Programmed death-1 is required for systemic self-tolerance in newly generated T cells during the establishment of immune homeostasis. J Autoimmun. (2011) 36:301–12. doi: 10.1016/j.jaut.2011.02.009
81. Ellestad KK, Thangavelu G, Ewen CL, Boon L, Anderson CC. PD-1 is not required for natural or peripherally induced regulatory T cells: severe autoimmunity despite normal production of regulatory T cells. Eur J Immunol. (2014) 44:3560–72. doi: 10.1002/eji.201444688
82. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
83. Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med. (2015) 21:688–97. doi: 10.1038/nm.3883
84. Xia CQ, Chernatynskaya AV, Wasserfall CH, Wan S, Looney BM, Eisenbeis S, et al. Anti-thymocyte globulin (ATG) differentially depletes naïve and memory T cells and permits memory-type regulatory T cells in nonobese diabetic mice. BMC Immunol. (2012) 13:70. doi: 10.1186/1471-2172-13-70
85. Comabella M, Montalban X. Body fluid biomarkers in multiple sclerosis. Lancet Neurol. (2014) 13:113–26. doi: 10.1016/S1474-4422(13)70233-3
86. Robinson WH, Mao R. Biomarkers to guide clinical therapeutics in rheumatology? Curr Opin Rheumatol. (2016) 28:168–75. doi: 10.1097/BOR.0000000000000250
87. Wermuth PJ, Piera-Velazquez S, Rosenbloom J, Jimenez SA. Existing and novel biomarkers for precision medicine in systemic sclerosis. Nat Rev Rheumatol. (2018) 14:421–32. doi: 10.1038/s41584-018-0021-9
88. Katsarou A, Gudbjornsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Prim. (2017) 3:17016. doi: 10.1038/nrdp.2017.16
89. Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, et al. Systemic sclerosis. Nat Rev Dis Prim. (2015)1:15002. doi: 10.1038/nrdp.2015.2
90. Longo DL, Reich DS, Lucchinetti CF, Calabresi PA. Multiple Sclerosis. N Engl J Med. (2018) 378:169–80. doi: 10.1056/NEJMra1401483
91. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Prim. (2016) 2:16039. doi: 10.1038/nrdp.2016.39
92. Hinze C, Gohar F, Foell D. Management of juvenile idiopathic arthritis: hitting the target. Nat Rev Rheumatol (2015) 11:290–300. doi: 10.1038/nrrheum.2014.212
93. De Souza HS, Fiocchi C, Iliopoulos D. The IBD interactome: an integrated view of aetiology, pathogenesis and therapy. Nat Rev Gastroenterol Hepatol. (2017) 14:739–49. doi: 10.1038/nrgastro.2017.110
94. Alevizos I, Illei GG. MicroRNAs as biomarkers in rheumatic diseases. Nat Rev Rheumatol. (2010) 6:391–8. doi: 10.1038/nrrheum.2010.81
95. Colburn W. a. Biomarkers in drug discovery and development: from target identification through drug marketing. J Clin Pharmacol. (2003) 43:329–41. doi: 10.1177/0091270003252480
96. Frank R, Hargreaves R. Clinical biomarkers in drug discovery and development. Nat Rev Drug Discov. (2003) 2:566–80. doi: 10.1038/nrd1130
97. Trinath J, Hegde P, Sharma M, Maddur MS, Rabin M, Vallat JM, et al. Intravenous immunoglobulin expands regulatory T cells via induction of cyclooxygenase-2-dependent prostaglandin E2 in human dendritic cells. Blood (2013) 122:1419–27. doi: 10.1182/blood-2012-11-468264
98. Hayes CE, Hubler SL, Moore JR, Barta LE, Praska CE, Nashold FE. Vitamin D actions on CD4+ T cells in autoimmune disease. Front Immunol. (2015) 6:100. doi: 10.3389/fimmu.2015.00100
99. Furukawa A, Wisel SA, Tang Q. Impact of immune-modulatory drugs on regulatory T cell. Transplantation (2016) 100:2288–300. doi: 10.1097/TP.0000000000001379
100. Zhang D, Tu E, Kasagi S, Zanvit P, Chen Q, Chen W. Manipulating regulatory T cells: a promising strategy to treat autoimmunity. Immunotherapy (2015) 7:1201–11. doi: 10.2217/imt.15.79
101. Shi Y, Wang Y, Li Q, Liu K, Hou J, Shao C, et al. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nat Rev Nephrol. (2018) 14:493–507. doi: 10.1038/s41581-018-0023-5
102. Keever-Taylor CA, Heimfeld S, Steinmiller KC, Nash RA, Sullivan KM, Czarniecki CW, et al. Manufacture of autologous CD34+selected grafts in the NIAID-sponsored HALT-MS and SCOT multicenter clinical trials for autoimmune diseases. Biol Blood Marrow Transplant. (2017) 23:1463–72. doi: 10.1016/j.bbmt.2017.05.018
Keywords: hematopoietic stem cell transplantation, immune reconstitution, autoimmune diseases, biomarkers, immune tolerance, immunoregulation
Citation: Malmegrim KCR, Lima-Júnior JR, Arruda LCM, de Azevedo JTC, de Oliveira GLV and Oliveira MC (2018) Autologous Hematopoietic Stem Cell Transplantation for Autoimmune Diseases: From Mechanistic Insights to Biomarkers. Front. Immunol. 9:2602. doi: 10.3389/fimmu.2018.02602
Received: 08 March 2018; Accepted: 23 October 2018;
Published: 16 November 2018.
Edited by:
Anne Mary Dickinson, Newcastle University, United KingdomReviewed by:
Adriane Feijo Evangelista, Barretos Cancer Hospital, BrazilCopyright © 2018 Malmegrim, Lima-Júnior, Arruda, de Azevedo, de Oliveira and Oliveira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelen Cristina Ribeiro Malmegrim, a2VsZW5mYXJpYXNAZmNmcnAudXNwLmJy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.