 Fadil A. Bidmos
Fadil A. Bidmos Sara Siris
Sara Siris Camilla A. Gladstone
Camilla A. Gladstone Paul R. Langford
Paul R. Langford- Department of Medicine, Imperial College London, London, United Kingdom
The ongoing, and very serious, threat from antimicrobial resistance necessitates the development and use of preventative measures, predominantly vaccination. Polysaccharide-based vaccines have provided a degree of success in limiting morbidity from disseminated bacterial infections, including those caused by the major human obligate pathogens, Neisseria meningitidis, and Streptococcus pneumoniae. Limitations of these polysaccharide vaccines, such as partial coverage and induced escape leading to persistence of disease, provide a compelling argument for the development of protein vaccines. In this review, we briefly chronicle approaches that have yielded licensed vaccines before highlighting reverse vaccinology 2.0 and its potential application in the discovery of novel bacterial protein vaccine candidates. Technical challenges and research gaps are also discussed.
Background: Vaccine Discovery in the Pre-Whole Genome Sequencing (WGS) ERA
Precedent to advancements in genomics, vaccines were developed based on Pasteur's rules of vaccinology. A 230-fold serial passage of a bovine bacillus in bile medium produced the live attenuated Bacillus Calmette-Guerin (BCG) vaccine against Mycobacterium tuberculosis (MTB) (1). Then, a trivalent blend of poliovirus inactivated in < 0.5% formalin was used by Salk et al. as a safe and effective vaccine against poliovirus (2). The inability to culture some pathogens in vitro (owing to safety or lack of suitable culture conditions), extensive antigenic variability, and molecular mimicry limit the broad applicability of traditional culture-based techniques in the development of vaccines targeting other economically-important pathogens such as Mycobacterium leprae and Neisseria meningitidis.
The inadequacies of culture-based techniques caused a shift in focus to the use of subunit components as vaccine candidates. Identification of these subunit vaccine candidates was largely hypothesis-driven, targeted cellular components and were often well-known virulence factors: for example, the pertussis toxin and fimbriae in the acellular pertussis vaccine (3); and the meningococcal porin, PorA, in the epidemic-specific detergent-extracted outer membrane vesicle (OMV) vaccines of Chile, Norway, and New Zealand (4–6). With complex biosynthetic methods, bacterial capsular polysaccharides served as prime components of effective vaccines, used singly, for example, in the 23-valent pneumococcal polysaccharide vaccine, PPSV23 (7). In addition, these capsular antigens have been conjugated to carrier proteins in a dose-sensitive manner for enhanced immunogenicity, as found in the Haemophilus influenzae type b (Hib) (8), pneumococcal (9), and meningococcal (10) conjugate vaccines. The limited capacity of these hypothesis-driven studies to focus on only a handful of candidates at a time was costly in time, labor and financial terms. This is especially because of the large pool from which prospective candidates for individual bacterial pathogens are screened, coupled with the low likelihood of targets satisfying key vaccine candidacy criteria (abundantly-expressed, surface-exposed, functionally-immunogenic, and highly-conserved). Thus, alternative high throughput methods were sought to accelerate the pre-clinical vaccine development phase, especially in situations requiring rapid curtailment of disease transmission.
Whole Genomic and Proteomic Approaches
Reverse Vaccinology (RV)
The publication of the first complete bacterial genome sequence in 1995 [for H. influenzae (11)] heralded a revolution in approaches to vaccine development. By using genomic data and preset bioinformatic screens, putative surface-associated antigens of a pathogen were identified. The subsequent recombinant expression of these genes and immunization of animals with recombinant proteins, for the determination of active and passive levels of protection, provided data that substantiated or annulled the vaccine candidacy of selected antigens (12, 13). This “classical” RV approach led to the development of the multicomponent meningococcal serogroup B vaccine (4CMenB) (14). While 4CMenB has potential for cross-serogroup protection (15), it has been argued that pan-genomic in silico analysis is more appropriate because of the high degree of intraspecific diversity exhibited by many bacterial pathogens (16). Using this pan-genomic approach, Maione et al. (17) identified four protective antigens from the analysis of an octa-genomic panel derived from the most prevalent disease-causing Streptococcus agalactiae strains. The main attraction of RV lies in its applicability to any pathogen with WGS data and to which antibody-mediated immunity for protection against disease is crucial. Its use in the discovery of candidate antigens comprising vaccines targeting other bacterial pathogens, including the multidrug-resistant Acinetobacter baumanii, has been demonstrated (18–20). However, important non-classical surface-associated proteins may be missed due to the parameters of the bioinformatic screen(s).
Related to RV is the use of transcriptomics to identify novel vaccine antigens. For example, the comparative analysis of the meningococcal transcriptome in ex vivo human whole blood and in vitro nasopharyngeal colonization models revealed three antigens that were differentially regulated between invasive disease and asymptomatic colonization, and were thus subjects for further vaccine candidacy studies (21) However, this transcriptomics-based approach has not been widely employed.
Surfome and Secretome Analysis
Whole proteomic approaches, involving enzymatic processing of whole cells or extracellular exudates followed by liquid-chromatography mass spectrometry (LC-MS) or peptide fragment fingerprinting, also allow for high-throughput screening of the antigenic repertoire of a pathogen (22). The power of these proteomic methods in identifying rare protective antigens missed by the in silico screens of RV makes them appealing [as exemplified by the case of the cell wall-anchored antigen, SAN_1485, of S. agalactiae (23)]. Converse to RV, proteolytic digestion is more suited toward Gram-positive bacteria, since Gram-negative bacteria are more susceptible to proteolysis-induced cell lysis.
Reverse Vaccinology 2.0
The majority of currently-available bacterial vaccines provide protection by inducing pathogen-specific antibodies. Therefore, harnessing the antibody component of a potent human humoral response to disseminated infection is valuable for the identification of novel protective antigens. This approach, termed reverse vaccinology 2.0 (RV 2.0) (24, 25), relies on the isolation and recombinant expression of the variable regions of heavy (VH) and light (VL = κ or λ) chain genes of immunoglobulin (focus has centerd on IgG) using a variety of molecular tools. Enriched by the development of high-throughput technologies, the screening of large numbers of antibody-secreting cells (ASCs) is also advancing knowledge of host-pathogen interactive biology and auto-immunity (26, 27).
Monoclonal Antibody (mAb) Generation From ASCs
The first, and perhaps most crucial, phase of RV 2.0 is the cloning of human monoclonal antibodies (mAbs) from ASCs. Previously, immortalization of these ASCs via myeloma fusions or Epstein Barr virus (EBV) transformation were valuable to mAb production (28, 29). Because these were culture-based methods, the survival of all B-cells was not guaranteed and the omission of ASCs expressing antibodies cognate to crucial antigens was probable. Other techniques such as phage-display technology (30) and proteomic mining (31, 32) circumvent the unique issues affecting ASC immortalization techniques by focusing on recombinant antibody expression. However, the small proportion of antigen-specific antibodies (estimated at 10–15%) that are produced (33) because of the random pairing of VH and VL sequences make phage display and proteomic mining imprecise.
A more favored approach to mAb cloning is the single-cell sorting of ASCs into multi-well plates using flow cytometry, followed by the cloning of mAbs from each well (34, 35). To clone a high proportion of antigen-specific antibodies, this approach, termed expression cloning, requires blood sampling during the peak immune response and is thus more suited to short-lived plasmablasts (CD3−, CD14−, CD19+, CD20−, CD56−, CD27high, and CD38high), since higher circulating numbers of these are indicative of very recent history of infection (36). Notwithstanding, several studies have demonstrated its applicability to memory B-cells (37, 38). Further in vitro selection of antigen-specific plasmablasts or memory B-cells using eGFP-bound viral-like particles (39), labeled-antigen probes (40, 41) or in vivo antigen-specific plasmablast enrichment in irradiated SCID/beige chimera mice (42) enhance the pathogen-specific mAb output of the approach. Converse to phage display and proteomic mining technologies, expression cloning yields mAbs with natural, host-like VH+VL pairings. Further refinements to this elegant method include: substituting restriction endonuclease cloning with Gibson assembly to enhance cloning precision (43); assembly of both VH and VL fragments into a single expression vector (44); and succeeding cell sorting with paired-chain antibody repertoire sequencing, thereby encompassing all V gene families, including unique clones expressed at low frequencies (33, 45, 46).
Assessment of Recombinant mAb Function
Subsequent to cloning, the clinical relevance of mAbs is assessed in in vivo investigations of passive immunity (47) or in vitro functional assays: for example, the well-established viral neutralization (48) and serum bactericidal assays (49), some of which have provided data employed in vaccine licensure (50). The cognate antigens targeted by functional mAbs can subsequently be determined using protein array screens or classical immunoproteomic approaches.
Application of RV 2.0 to Viral Vaccine Development
The power of RV 2.0 (see Figure 1) in the identification of viral vaccine candidates has been demonstrated in several studies focussing on human cytomegalovirus (HCMV), respiratory syncytial virus (RSV), HIV, influenza and dengue viruses (25, 51). Some of these candidate antigens, discovered using RV 2.0, include a novel pentameric glycoprotein complex, the gHgLpUL128L pentamer, which induces high neutralizing titres against HCMV in mice (52) and the F protein of RSV stabilized to the prefusion conformation (53, 54). Accruing data from phase 1b/2a clinical trials show that a mAb (MEDI8897) reactive with prefusion F epitopes is effective when used prophylactically in preterm infants (55). Like MEDI8897, mAb MHAA4549A, cloned from a healthy vaccinee and which targets and neutralizes all known influenza A strains (56), demonstrated significant antiviral activity in a phase 2 human influenza A virus challenge (57). Thus, these studies have signified the use of RV 2.0 in producing broadly-neutralizing mAbs for post-infection prophylaxis in addition to identifying functionally-immunogenic vaccine candidates.
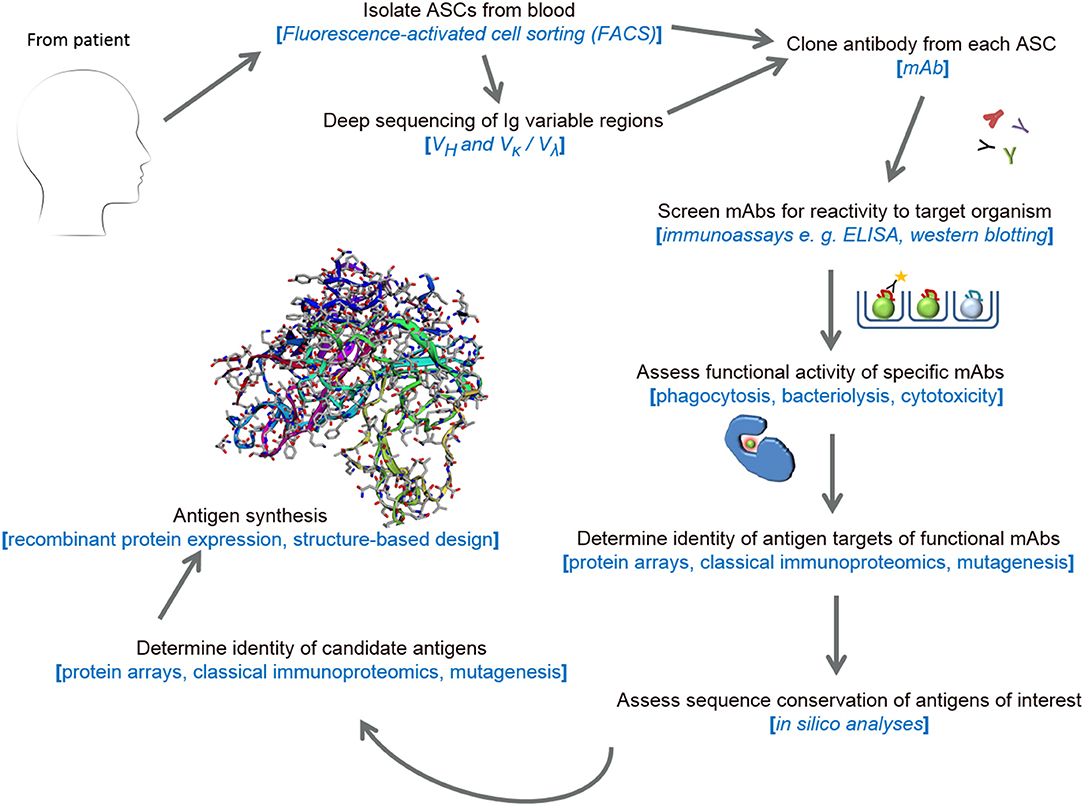
Figure 1. Schematic representation of RV 2.0.
Potential Application to Antibacterial Vaccine Discovery
Judging by the progress made with the development of novel and effective viral immunotherapies, RV 2.0 is showing promise and is equally applicable to bacterial vaccinology. RV 2.0 was employed by Lu et al. (58) to identify functional anti-Staphylococcus aureus mAbs induced during bacteraemia. A total of ten mAbs were produced, four of which enhanced opsonophagocytosis of Wood46, a S. aureus reference strain. While three of the four functional mAbs targeted S. aureus antigens with known identities, the fourth mAb reacted with a novel antigen. Recently, Zimmermann et al. (59) also demonstrated that functional anti-MTB surface antigen antibodies can be cloned from patient-derived plasmablasts of reactivated memory B-cell origins, providing further evidence for a role for antibodies in the modulation of potent immune responses toward MTB. Taken together with other studies investigating the importance of antibody-mediated neutralization of intracellular pathogens (60), a role for the vaccine-induced generation of antibodies against pathogens such as MTB and Chlamydia trachomatis, using antigens derived with RV 2.0 is, thus, evidenced. Similarly, Bidmos et al. (61) and Blum et al. (45) cloned functional antibodies from sufferers of meningococcal and Lyme disease, respectively; thus, underscoring the utility of the approach for identifying novel targets in different classes of bacteria. Continued use of RV 2.0 in bacterial vaccine discovery is, therefore, encouraged following the surmounting of technical challenges and filling of research gaps. In the following sections of this mini-review, emphasis will be placed on human mAb cloning and serological correlates of protection, since other related technical aspects of RV 2.0 such as recombinant protein expression, high-throughput sequencing of bacterial genomes and antibody repertoires, antigen identity determination and structure-based antigen design have been reviewed elsewhere (62–66).
Pathogen-Specific mAb Output
To identify novel antigens using the expression cloning method with precision, plasmablasts from patients convalescing from bacterial disease are required. Fundamental to the application of the expression cloning approach, therefore, is the determination of the magnitude and peak duration of the plasmablast response in these patients. The information on the duration of peak plasmablast circulation instructs optimum sampling time, which in turn impacts on the precision of pathogen-specific mAb generation. Studies assessing this magnitude of circulating plasmablast following bacterial infection have reported similar durations of peak response to those reported for primary or secondary viral infections [6–7 days for primary infections and ~10 days post-infection for secondary infections; reviewed in (36)]. Recently, Band et al. (67) reported a significant induction of differentiating (Ki-67+) plasmablasts in patients of nosocomial bacterial infections compared to healthy controls. This induction peaked between days 8 and 16 post-culture positivity in A. baumanii-infected patients reaching levels as high as 21% of the total lymphocyte population. Perhaps unsurprisingly, it was also observed that this induction was markedly different in individuals, reflecting differences in immunocompetence, as peak plasmablast levels ranged from: 1 to 21% among A. baumanii-infected patients; and 5–40% in Escherichia coli-infected patients. Consistent with the findings of Band et al. (67), a plasmablast response presented to: MTB infection in 38% of a patient cohort with levels ranging from 1 to 4% in those with strong serum IgG responses (59); S. aureus bacteraemia with mean levels of ~3.2% (1–7% range) (58); and up to 4% of circulating CD19+ cells in untreated sufferers of Lyme borreliosis (45).
The implications of these data for the precision of the pathogen-specific mAb output are considerable. Firstly, there is a paucity of information in published literature on how many plasmablasts are pathogen-specific (bacterial) following inductions in patients, owing to the unavailability of suitable molecular probes that will enhance the Fluorescent Activated Cell Sorting (FACS) gating strategy. In the absence of such data, strategies such as the Ig-capture based technique described by Pinder et al. (40) could be employed to enrich for specific plasmablasts. It is more likely that complex antigens (whole bacterial cells, OMV, or outer membrane preparations) would be more beneficial in these strategies, when adapted, compared to single-antigen probes in order to obtain a plasmablast population targeting a wider antigen pool. It is noteworthy that in cases where patients are subjected to immediate antibiotic therapy on hospital admission because of rapid progression of disease (e.g., septicaemia and meningitis), clinical isolates may be unobtainable, making the design of plasmablast enrichment probes difficult (also, the reason behind the unsuitability of memory B-cells in the absence of enrichment strategies). While clinical isolates from other disease sufferers could be utilized, they are non-ideal because rare mAb epitopes specific for the infecting strain will be missed. Secondly, considering differences in the magnitude of the plasmablast response and for logistic reasons (for example, restrictions on blood sample volume in pediatric cases), pooling of patient samples may be required for the generation of a highly-diverse plasmablast pool, targeting several antigens, (and their variants) of the same pathogen. This is especially necessary for pathogens in which certain antigens are immunodominant such as PorA of N. meningitidis, which may mask immunity to rare but equally protective antigens.
If a total plasmablast sort approach is warranted (i.e., inclusive of non-pathogen specific plasmablasts), an attractive option is the rational selection of over-represented VH+VL combinations for mAb cloning based on the assumption that overrepresentation of V families, specifically among plasmablasts, is an indicator of preferential usage in response to a pathogen. Adequate depth of sequencing is, however, required in order to avoid non-inclusion of clonal V families expressed at lower frequencies (58). In silico analysis should also include antibodies with similar complementarity-determining region H3 loops [key to antibody conformation and affinity (68)] in addition to the exploitation of de-noising algorithms, which would minimize the presence of errors introduced by sequencing (69).
In vitro Serological Correlates of Protection
Assessment of pathogen-specific mAb function is performed via standardized assays. Given the differences in biology of bacterial pathogens, these assays are specifically tailored to reflect mode of clearance of the pathogen from systemic circulation. Antibody-driven, complement-dependent bactericidal activity is measured in the standardized serum bactericidal assays designed for the meningococcus (49) while phagocytosis by neutrophils and macrophages enhanced by opsonic antibodies is assessed in the opsonophagocytic assays used in pneumococcal vaccine development (70). Similar assays have been employed in the assessment of functional immunity against Campylobacter jejuni, Group B Streptococcus, typhoidal and non-typhoidal Salmonella and Neisseria gonorrhoeae (71–75). While standardization of some of these pathogen-specific assays is pending, de novo design of in vitro correlates assessing functional activity of antibodies is not as straightforward for other pathogens, such as Bordetella pertussis (76). For facultative intracellular pathogens such as Francisella tularensis and MTB, for example, current correlate strategies in development are not suitable for assessments of mAb function as they involve peripheral blood lymphocytes only (77, 78). An added benefit of in vitro assessments of cloned mAb function, as a component of RV 2.0, is the needlessness of or significant reduction in usage of experimental animals. Efforts to develop and standardize in vitro correlates to assess mAb function are, hereby, merited.
Beyond bactericidal or opsonic functions, mAbs exhibit a variety of functions, including the modulation of cellular immune responses [extensively reviewed in Cooper (79), Amanna and Slifka (80)], which require assessment. These functions also include toxin neutralization (useful in pertussis and diphtheritic infections, for example) (81, 82) and increase in cellular cytotoxicity affecting intracellular pathogens such as C. trachomatis (83). Hence, non-bactericidal or non-opsonic mAbs, if exhibitive of these other functions, can still be utilized in other immunotherapeutic avenues.
Conclusion
With the increase in multidrug resistance among bacterial pathogens, the development of further effective preventive measures will be of significant benefit to public health. RV 2.0, a conceptually-advanced approach with the advantages of employing the natural host response (patient VH-VL combinations), relative speed, and reduction in animal use, has the potential to be a powerful tool in bacterial vaccine development. However, use of RV 2.0 is dependent on optimization of the technical aspects, and there are excellent prospects that this is achievable.
Author Contributions
FB and PL conceptualization. FB, SS, and CG writing—original draft. FB and PL writing—reviewing and editing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Mubarak Bidmos (Qatar University) and Dr. Victoria Wright (Imperial College London) for their critical but constructive review of the manuscript. PL and FB have received research grants from John and Michelle Bresnahan via MeningitisNow, and the Imperial College Confidence-in-Concept Award; and CG is funded by a BBSRC grant BB/R505742/1 from the National Productivity Investment Fund (NPIF), for work related to this manuscript.
References
1. Calmette A. Preventive vaccination against tuberculosis with BCG. Proc R Soc Med. (1931) 24:85–94.
2. Salk JE, Lewis LJ, Bennett BL, Ward EN, Krech U, Youngner JS. Antigenic activity of poliomyelitis vaccines undergoing field test. Am J Pub Health Nat Health (1955) 45:151–62. doi: 10.2105/AJPH.45.2.151
3. Olin P, Rasmussen F, Gustafsson L, Hallander HO, Heijbel H. Randomised controlled trial of two-component, three-component, and five-component acellular pertussis vaccines compared with whole-cell pertussis vaccine. Lancet (1997) 350:1569–77. doi: 10.1016/S0140-6736(97)06508-2
4. Boslego J, Garcia J, Cruz C, Zollinger W, Brandt B, Ruiz S, et al. Efficacy, safety, and immunogenicity of a meningococcal group B (15: P1.3) outer membrane protein vaccine in Iquique, Chile. Vaccine (1995) 13:821–9. doi: 10.1016/0264-410X(94)00037-N
5. Galloway Y, Stehr-Green P, McNicholas A, O'Hallahan J. Use of an observational cohort study to estimate the effectiveness of the New Zealand group B meningococcal vaccine in children aged under 5 years. Int J Epidemiol. (2008) 38, 413–418. doi: 10.1093/ije/dyn228
6. Rosenqvist E, Høiby EA, Wedege E, Bryn K, Kolberg J, Klem A, et al. Human antibody responses to meningococcal outer membrane antigens after three doses of the Norwegian group B meningococcal vaccine. Infect Immun. (1995) 63:4642–52.
7. Robbins JB, Austrian R, Lee CJ, Rastogi SC, Schiffman G, Henrichsen J, et al. Considerations for formulating the second-generation pneumococcal capsular polysaccharide vaccine with emphasis on the cross-reactive types within groups. J Infect Dis. (1983) 148:1136–59. doi: 10.1093/infdis/148.6.1136
8. Schneerson R, Barrera O, Sutton A, Robbins JB. Preparation, characterization, and immunogenicity of Haemophilus influenzae type b polysaccharide-protein conjugates. J Exp Med. (1980) 152:361–76. doi: 10.1084/jem.152.2.361
9. Poolman JT, Peeters CC, Van Den Dobbelsteen GP. The history of pneumococcal conjugate vaccine development: dose selection. Exp Rev Vaccines (2013) 12:1379–94. doi: 10.1586/14760584.2013.852475
10. Anderson EL, Bowers T, Mink CM, Kennedy DJ, Belshe RB, Harakeh H, et al. Safety and immunogenicity of meningococcal A and C polysaccharide conjugate vaccine in adults. Infect Immun. (1994) 62:3391–5.
11. Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science (1995) 269:496–512. doi: 10.1126/science.7542800
12. Pizza M, Scarlato V, Masignani V, Giuliani MM, Aricò B, Comanducci M, et al. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science (2000) 287:1816–20. doi: 10.1126/science.287.5459.1816
13. Mora M, Veggi D, Santini L, Pizza M, Rappuoli R. Reverse vaccinology. Drug Discov. Today (2003) 8:459–64. doi: 10.1016/S1359-6446(03)02689-8
14. Serruto D, Bottomley MJ, Ram S, Giuliani MM, Rappuoli R. The new multicomponent vaccine against meningococcal serogroup B, 4CMenB: immunological, functional and structural characterization of the antigens. Vaccine (2012) 30:B87–97. doi: 10.1016/j.vaccine.2012.01.033
15. Harris SL, Zhu D, Murphy E, McNeil LK, Wang X, Mayer LW, et al. Preclinical evidence for the potential of a bivalent fHBP vaccine to prevent Neisseria meningitidis serogroup C disease. Hum Vaccin. (2011) 7:68–74. doi: 10.4161/hv.7.0.14564
16. Mora M, Donati C, Medini D, Covacci A, Rappuoli R. Microbial genomes and vaccine design: refinements to the classical reverse vaccinology approach. Curr Opin Microbiol. (2006) 9:532–6. doi: 10.1016/j.mib.2006.07.003
17. Maione D, Margarit I, Rinaudo CD, Masignani V, Mora M, Scarselli M, et al. Identification of a universal Group B Streptococcus vaccine by multiple genome screen. Science (2005) 309:148–50. doi: 10.1126/science.1109869
18. Chiang MH, Sung WC, Lien SP, Chen YZ, Lo AFY, Huang JH, et al. Identification of novel vaccine candidates against Acinetobacter baumannii using reverse vaccinology. Hum. Vaccin Immunother. (2015) 11:1065–73. doi: 10.1080/21645515.2015.1010910
19. Meunier M, Guyard-Nicodème M, Hirchaud E, Parra A, Chemaly M, Dory D. Identification of novel vaccine candidates against Campylobacter through reverse vaccinology. J Immunol Res. (2016) 2016:5715790. doi: 10.1155/2016/5715790
20. Talukdar S, Zutshi S, Prashanth KS, Saikia KK, Kumar P. Identification of potential vaccine candidates against Streptococcus pneumoniae by reverse vaccinology approach. Appl Biochem Biotechnol. (2014) 172:3026–41. doi: 10.1007/s12010-014-0749-x
21. Hey A, Li MS, Hudson MJ, Langford PR, Kroll JS. Transcriptional profiling of Neisseria meningitidis interacting with human epithelial cells in a long-term in vitro colonization model. Infect Immun. (2013) 81:4149–59. doi: 10.1128/IAI.00397-13
22. Bittaye M, Cash P. Streptococcus pneumoniae proteomics: determinants of pathogenesis and vaccine development. Exp Rev Proteomics (2015) 12:607–21. doi: 10.1586/14789450.2015.1108844
23. Doro F, Liberatori S, Rodríguez-Ortega MJ, Rinaudo CD, Rosini R, Mora M, et al. Surfome analysis as a fast track to vaccine discovery identification of a novel protective antigen for Group B Streptococcus hypervirulent strain COH1. Mol Cell Proteomics (2009) 8:1728–37. doi: 10.1074/mcp.M800486-MCP200
25. Rappuoli R, Bottomley MJ, D'Oro U, Finco O, De Gregorio E. Reverse vaccinology 2.0: human immunology instructs vaccine antigen design. J Exp Med. (2016) 213:469–81. doi: 10.1084/jem.20151960
26. Di Niro R, Mesin L, Zheng NY, Stamnaes J, Morrissey M, Lee JH, et al. High abundance of plasma cells secreting transglutaminase 2–specific IgA autoantibodies with limited somatic hypermutation in celiac disease intestinal lesions. Nat Med. (2012) 18:441–5. doi: 10.1038/nm.2656
27. Amara K, Steen J, Murray F, Morbach H, Fernandez-Rodriguez BM, Joshua V, et al. Monoclonal IgG antibodies generated from joint-derived B cells of RA patients have a strong bias toward citrullinated autoantigen recognition. J Exp Med. (2013) 210:445–55. doi: 10.1084/jem.20121486
28. Steinitz M, Klein G, Koskimies S, Makel O. EB virus-induced B lymphocyte cell lines producing specific antibody. Nature (1977) 269:420–22. doi: 10.1038/269420a0
29. Kozbor D, Roder JC, Chang TH, Steplewski Z, Koprowski H. Human anti-tetanus toxoid monoclonal antibody secreted by EBV-transformed human B cells fused with murine myeloma. Hybridoma (1982) 1:323–8. doi: 10.1089/hyb.1.1982.1.323
30. Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Ann Rev Immunol. (1994) 12:433–55. doi: 10.1146/annurev.iy.12.040194.002245
31. Cheung WC, Beausoleil SA, Zhang X, Sato S, Schieferl SM, Wieler JS, et al. A proteomics approach for the identification and cloning of monoclonal antibodies from serum. Nat Biotechnol. (2012) 30:447–52. doi: 10.1038/nbt.2167
32. Sato S, Beausoleil SA, Popova L, Beaudet JG, Ramenani RK, Zhang X, et al. Proteomics-directed cloning of circulating antiviral human monoclonal antibodies. Nat Biotechnol. (2012) 30:1039–43. doi: 10.1038/nbt.2406
33. DeKosky BJ, Ippolito GC, Deschner RP, Lavinder JJ, Wine Y, Rawlings BM, et al. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat Biotechnol. (2013) 31:166–9. doi: 10.1038/nbt.2492
34. Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R, et al. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc. (2009) 4:372. doi: 10.1038/nprot.2009.3
35. Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods (2008) 329:112–24. doi: 10.1016/j.jim.2007.09.017
36. Fink K. Origin and function of circulating plasmablasts during acute viral infections. Front Immunol. (2012) 3:78. doi: 10.3389/fimmu.2012.00078
37. Scheid JF, Mouquet H, Feldhahn N, Seaman MS, Velinzon K, Pietzsch J, et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature (2009) 458:636–40. doi: 10.1038/nature07930
38. Muellenbeck MF, Ueberheide B, Amulic B, Epp A, Fenyo D, Busse CE, et al. Atypical and classical memory B cells produce Plasmodium falciparum neutralizing antibodies. J Exp Med. (2013) 210:389–99. doi: 10.1084/jem.20121970
39. Di Niro R, Mesin L, Raki M, Zheng NY, Lund-Johansen F, Lundin KE, et al. Rapid generation of rotavirus-specific human monoclonal antibodies from small-intestinal mucosa. J Immunol. (2010) 185:5377–83. doi: 10.4049/jimmunol.1001587
40. Pinder CL, Kratochvil S, Cizmeci D, Muir L, Guo Y, Shattock RJ, et al. Isolation and characterization of antigen-specific plasmablasts using a novel flow cytometry–based Ig capture assay. J Immunol. (2017) 199:4180–8. doi: 10.4049/jimmunol.1701253
41. Goodwin E, Gilman MS, Wrapp D, Chen M, Ngwuta JO, Moin SM, et al. Infants infected with respiratory syncytial virus generate potent neutralizing antibodies that lack somatic hypermutation. Immunity (2018) 48:339–49.e5. doi: 10.1016/j.immuni.2018.01.005
42. Lin Z, Chiang NY, Chai N, Seshasayee D, Lee WP, Balazs M, et al. In vivo antigen-driven plasmablast enrichment in combination with antigen-specific cell sorting to facilitate the isolation of rare monoclonal antibodies from human B cells. Nat Protoc. (2014) 9:1563–77. doi: 10.1038/nprot.2014.104
43. Ho IY, Bunker JJ, Erickson SA, Neu KE, Huang M, Cortese M, et al. Refined protocol for generating monoclonal antibodies from single human and murine B cells. J Immunol Methods (2016) 438:67–70. doi: 10.1016/j.jim.2016.09.001
44. Dodev TS, Karagiannis P, Gilbert AE, Josephs DH, Bowen H, James LK, et al. A tool kit for rapid cloning and expression of recombinant antibodies. Sci Rep. (2014) 4:5885. doi: 10.1038/srep05885
45. Blum LK, Adamska JZ, Martin DS, Rebman AW, Elliott SE, Cao RRL, et al. Robust B cell responses predict rapid resolution of Lyme disease. Front Immunol. (2018) 9:1634. doi: 10.3389/fimmu.2018.01634
46. Tan YC, Blum LK, Kongpachith S, Ju CH, Cai X, Lindstrom TM, et al. High-throughput sequencing of natively paired antibody chains provides evidence for original antigenic sin shaping the antibody response to influenza vaccination. Clin Immunol. (2014) 151:55–65. doi: 10.1016/j.clim.2013.12.008
47. Gupta P, Kamath AV, Park S, Chiu H, Lutman J, Maia M, et al. Preclinical pharmacokinetics of MHAA4549A, a human monoclonal antibody to influenza A virus, and the prediction of its efficacious clinical dose for the treatment of patients hospitalized with influenza A. mAbs 8:991–7. doi: 10.1080/19420862.2016.1167294
48. Mascola JR, D'Souza P, Gilbert P, Hahn BH, Haigwood NL, Morris L, et al. Recommendations for the design and use of standard virus panels to assess neutralizing antibody responses elicited by candidate Human Immunodeficiency Virus type 1 vaccines. J Virol. (2005) 79:10103–7. doi: 10.1128/JVI.79.16.10103-10107.2005
49. Borrow R, Aaberge IS, Santos GF, Eudey TL, Oster P, Glennie A, et al. Interlaboratory standardization of the measurement of serum bactericidal activity by using human complement against meningococcal serogroup B, strain 44/76-SL, before and after vaccination with the Norwegian MenBvac outer membrane vesicle vaccine. Clin Diagn Lab Immunol. (2005) 12:970–6. doi: 10.1128/CDLI.12.8.970-976.2005
50. Borrow R, Balmer P, Miller E. Meningococcal surrogates of protection — serum bactericidal antibody activity. Vaccine (2005) 23:2222–7. doi: 10.1016/j.vaccine.2005.01.051
51. Burton DR. What are the most powerful immunogen design vaccine strategies? Reverse vaccinology 2.0 shows great promise. Cold Spring Harb Perspect Biol. (2017) 9:a030262. doi: 10.1101/cshperspect.a030262
52. Kabanova A, Perez L, Lilleri D, Marcandalli J, Agatic G, Becattini S, et al. Antibody-driven design of a human cytomegalovirus gHgLpUL128L subunit vaccine that selectively elicits potent neutralizing antibodies. Proc Natl Acad Sci USA. (2014) 111:17965–17970. doi: 10.1073/pnas.1415310111
53. Krarup A, Truan D, Furmanova-Hollenstein P, Bogaert L, Bouchier P, Bisschop IJ, et al. A highly stable prefusion RSV F vaccine derived from structural analysis of the fusion mechanism. Nat Commun. (2015) 6:8143. doi: 10.1038/ncomms9143
54. McLellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science (2013) 340:1113–7. doi: 10.1126/science.1234914
55. Domachowske JB, Khan AA, Esser MT, Jensen K, Takas T, Villafana T, et al. Safety, tolerability, and pharmacokinetics of MEDI8897, an extended half-life single-dose respiratory syncytial virus prefusion F-targeting monoclonal antibody administered as a single dose to healthy preterm infants. Pediatr Infect Dis J. (2018) 37:886–92. doi: 10.1097/INF.0000000000001916
56. Nakamura G, Chai N, Park S, Chiang N, Lin Z, Chiu H, et al. An in vivo human-plasmablast enrichment technique allows rapid identification of therapeutic influenza A antibodies. Cell Host Microb. (2013) 14:93–103. doi: 10.1016/j.chom.2013.06.004
57. McBride JM, Lim JJ, Burgess T, Deng R, Derby MA, Maia M, et al. Phase 2 randomized trial of the safety and efficacy of MHAA4549A, a broadly neutralizing monoclonal antibody, in a human influenza A virus challenge model. Antimicrob Agents Chemother. (2017) 61:e01154–17. doi: 10.1128/AAC.01154-17
58. Lu DR, Tan YC, Kongpachith S, Cai X, Stein EA, Lindstrom TM, et al. Identifying functional anti-Staphylococcus aureus antibodies by sequencing antibody repertoires of patient plasmablasts. Clin Immunol. (2014) 152:77–89. doi: 10.1016/j.clim.2014.02.010
59. Zimmermann N, Thormann V, Hu B, Köhler AB, Imai-Matsushima A, Locht C, et al. Human isotype-dependent inhibitory antibody responses against Mycobacterium tuberculosis. EMBO Mol Med. (2016) 8:1325–39. doi: 10.15252/emmm.201606330
60. Olsen AW, Lorenzen EK, Rosenkrands I, Follmann F, Andersen P. Protective effect of vaccine promoted neutralizing antibodies against the intracellular pathogen Chlamydia trachomatis. Front Immunol. (2017) 8:1652. doi: 10.3389/fimmu.2017.01652
61. Bidmos FA, Nadel S, Screaton GR, Kroll JS, Langford PR. Cross-reactive bactericidal anti-meningococcal antibodies can be isolated from convalescing invasive meningococcal disease patients using reverse vaccinology 2.0. Front Immunol. (2018) 9:1621. doi: 10.3389/fimmu.2018.01621
62. Dormitzer PR, Ulmer JB, Rappuoli R. Structure-based antigen design: a strategy for next generation vaccines. Trends Biotechnol. (2008) 26:659–67. doi: 10.1016/j.tibtech.2008.08.002
63. Georgiou G, Ippolito GC, Beausang J, Busse CE, Wardemann H, Quake SR. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat Biotechnol. (2014) 32:158–68. doi: 10.1038/nbt.2782
64. Hess JL, Blazer L, Romer T, Faber L, Buller RM, Boyle MD. Immunoproteomics. J Chromatogr B (2005) 815:65–75. doi: 10.1016/j.jchromb.2004.07.047
65. Loman NJ, Constantinidou C, Chan JZ, Halachev M, Sergeant M, Penn CW, et al. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat Rev Microbiol. (2012) 10:599–606. doi: 10.1038/nrmicro2850
66. Young CL, Britton ZT, Robinson AS. Recombinant protein expression and purification: a comprehensive review of affinity tags and microbial applications. Biotechnol J. (2012) 7:620–34. doi: 10.1002/biot.201100155
67. Band VI, Ibegbu C, Kaur SP, Cagle SM, Trible R, Jones CL, et al. Induction of human plasmablasts during infection with antibiotic-resistant nosocomial bacteria. J Antimicrob Chemother. (2014) 69:1830–3. doi: 10.1093/jac/dku047
68. Weitzner BD, Dunbrack RL Jr, Gray JJ. The origin of CDR H3 structural diversity. Structure (2015) 23:302–11. doi: 10.1016/j.str.2014.11.010
69. Mysara M, Leys N, Raes J, Monsieurs P. IPED: a highly efficient denoising tool for Illumina MiSeq Paired-end 16S rRNA gene amplicon sequencing data. BMC Bioinformatics (2016) 17:192. doi: 10.1186/s12859-016-1061-2
70. Song JY, Moseley MA, Burton RL, Nahm MH. Pneumococcal vaccine and opsonic pneumococcal antibody. J Infect Chemother. (2013) 19:412–25. doi: 10.1007/s10156-013-0601-1
71. Boyd MA, Tennant SM, Saague VA, Simon R, Muhsen K, Ramachandran G, et al. Serum bactericidal assays to evaluate typhoidal and nontyphoidal Salmonella vaccines. Clin Vacc Immunol. (2014) 21:712–21. doi: 10.1128/CVI.00115-14
72. Le Doare K, Faal A, Jaiteh M, Sarfo F, Taylor S, Warburton F, et al. Association between functional antibody against Group B Streptococcus and maternal and infant colonization in a Gambian cohort. Vaccine (2017) 35:2970–8. doi: 10.1016/j.vaccine.2017.04.013
73. Necchi F, Saul A, Rondini S. Development of a high-throughput method to evaluate serum bactericidal activity using bacterial ATP measurement as survival readout. PLoS ONE (2017) 12:e0172163. doi: 10.1371/journal.pone.0172163
74. Piekarowicz A, Kłyz A, Majchrzak M. Oral immunization of rabbits with S. enterica Typhimurium expressing Neisseria gonorrhoeae filamentous phage Φ6 induces bactericidal antibodies against N. gonorrhoeae. Sci Rep. (2016) 6:22549. doi: 10.1038/srep22549
75. Sahin O, Zhang Q, Meitzler JC, Harr BS, Morishita TY, Mohan R. Prevalence, antigenic specificity, and bactericidal activity of poultry anti-Campylobacter maternal antibodies. Appl Environ Microbiol. (2001) 67:3951–7. doi: 10.1128/AEM.67.9.3951-3957.2001
76. Brookes C, Freire-Martin I, Cavell B, Alexander F, Taylor S, Persaud R, et al. Bordetella pertussis isolates vary in their interactions with human complement components. Emerg Microb Infect. (2018) 7:81. doi: 10.1038/s41426-018-0084-3
77. De Pascalis R, Hahn A, Brook HM, Ryden P, Donart N, Mittereder L, et al. A panel of correlates predicts vaccine-induced protection of rats against respiratory challenge with virulent Francisella tularensis. PLoS ONE (2018) 13:e0198140. doi: 10.1371/journal.pone.0198140
78. Kurtz SL, Elkins KL. Correlates of vaccine-induced protection against Mycobacterium tuberculosis revealed in comparative analyses of lymphocyte populations. Clin Vacc Immunol. (2015) 22:1096–108. doi: 10.1128/CVI.00301-15
79. Cooper AM. Cell-mediated immune responses in tuberculosis. Ann Rev Immunol. (2009) 27:393–422. doi: 10.1146/annurev.immunol.021908.132703
80. Amanna IJ, Slifka MK. Contributions of humoral and cellular immunity to vaccine-induced protection in humans. Virology (2011) 411:206–15. doi: 10.1016/j.virol.2010.12.016
81. Di Giovine P, Pinto, A, Ölander RM, Sesardic D, Stickings P, Berbers G, Bujko M. External quality assessment for the determination of diphtheria antitoxin in human serum. Clin Vacc Immunol. (2010) 17:1282–90. doi: 10.1128/CVI.00096-10
82. Millen SH, Bernstein DI, Connelly B, Ward JI, Chang SJ, Weiss AA. Antibody-mediated neutralization of pertussis toxin-induced mitogenicity of human peripheral blood mononuclear cells. Infect Immun. (2004) 72:615–20. doi: 10.1128/IAI.72.1.615-620.2004
Keywords: reverse vaccinology 2.0, human monoclonal antibodies, bacterial pathogens, vaccine candidate antigens, immunotherapy
Citation: Bidmos FA, Siris S, Gladstone CA and Langford PR (2018) Bacterial Vaccine Antigen Discovery in the Reverse Vaccinology 2.0 Era: Progress and Challenges. Front. Immunol. 9:2315. doi: 10.3389/fimmu.2018.02315
Received: 30 June 2018; Accepted: 17 September 2018;
Published: 08 October 2018.
Edited by:
Pedro A. Reche, Complutense University of Madrid, SpainReviewed by:
Sudheer Gupta, All India Institute of Medical Sciences Bhopal, IndiaPietro Speziale, Università degli Studi di Pavia, Italy
Paola Massari, Tufts University School of Medicine, United States
Copyright © 2018 Bidmos, Siris, Gladstone and Langford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fadil A. Bidmos, Zi5iaWRtb3NAaW1wZXJpYWwuYWMudWs=