 Elke M. Muntjewerff
Elke M. Muntjewerff Gina Dunkel
Gina Dunkel Mara J. T. Nicolasen
Mara J. T. Nicolasen Sushil K. Mahata
Sushil K. Mahata Geert van den Bogaart
Geert van den Bogaart- 1Department of Tumor Immunology, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Nijmegen, Netherlands
- 2VA San Diego Healthcare System, San Diego, CA, United States
- 3Department of Medicine, University of California at San Diego, La Jolla, CA, United States
- 4Department of Molecular Immunology, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Groningen, Netherlands
It is increasingly clear that inflammatory diseases and cancers are influenced by cleavage products of the pro-hormone chromogranin A (CgA), such as the 21-amino acids long catestatin (CST). The goal of this review is to provide an overview of the anti-inflammatory effects of CST and its mechanism of action. We discuss evidence proving that CST and its precursor CgA are crucial for maintaining metabolic and immune homeostasis. CST could reduce inflammation in various mouse models for diabetes, colitis and atherosclerosis. In these mouse models, CST treatment resulted in less infiltration of immune cells in affected tissues, although in vitro monocyte migration was increased by CST. Both in vivo and in vitro, CST can shift macrophage differentiation from a pro- to an anti-inflammatory phenotype. Thus, the concept is emerging that CST plays a role in tissue homeostasis by regulating immune cell infiltration and macrophage differentiation. These findings warrant studying the effects of CST in humans and make it an interesting therapeutic target for treatment and/or diagnosis of various metabolic and immune diseases.
Introduction
Inflammation-based diseases, such as chronic inflammation (Type 2 diabetes Mellitus (T2DM) and colitis), auto-immune diseases (rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE)), hypertension, tumor metastasis and development of severe cancers (myeloma, neuroendocrine tumors, lung, and breast cancer) (1–3) are major health problems. For instance, 415 million adults were globally affected by T2DM in 2015 and this caused 5.0 million deaths (4). For RA the current mortality rate is 2.2 million (5) and for SLE comorbidities including infection and cardiac malfunction account for 29% of all deaths (4, 6). The second leading cause of death worldwide is cancer and 1 in 6 people die to cancer, accounting for 8.8 million deaths in 2015 (4). The prevalence of all these diseases is increasing and, in many cases, sufficient therapies are not available. Recently, an interest in utilizing the body's own molecules to treat these diseases arose. An interesting candidate is the pro-hormone chromogranin A (CgA), which contributes to a balanced immune response. CgA is proteolytically cleaved, both intracellularly as well as extracellularly after its release, and this gives rise to several peptides (7, 8). These peptides exert a broad spectrum of regulatory functions among the metabolic, endocrine, cardiovascular and immune systems (9). It is becoming increasingly clear that one of these cleavage products, the bio-active peptide catestatin (CST: hCgA352−372) (10, 11), is particularly of interest, since it suppresses tissue inflammation and affects the immune system. Indeed the concept is emerging that CST plays an immunomodulatory role in macrophage differentiation and monocyte migration. This review will focus on the relatively new concept of modulating innate immunity by targeting CST, which may find applications in treatment of various inflammatory based diseases and cancer (1–3). We will review the effect of CST on infiltrating immune cells, tissue homeostasis and the role of CST in disease. Moreover, we will discuss remaining outstanding questions about the effects and molecular targets of CST, as well as further directions in research and therapeutic applications.
Cleavage Products of the Pro-Hormone Chromogranin A
Chromogranin A (CgA)
The human CgA gene is located on chromosome 14 (12, 13) and codes for a 439 amino acids long protein (14). As member of the granin family, CgA is characterized by an acidic pI, heat stability and 8-10 pairs of dibasic cleavage sites (15). Moreover, this 49 kDa protein has the capacity to form aggregates and the ability to bind calcium (Ca2+) with a high capacity, but low affinity (16). CgA was first identified as an acidic protein co-stored and co-released with ATP and catecholamines in chromaffin granules of neuroendocrine cells in the adrenal medulla (17, 18). CgA facilitates the storage in these granules of catecholamines and ATP at hyperosmotic concentrations in a non-diffusible form (17–21). Thereby CgA contributes to the biogenesis of secretory granules packed with condensed proteins, mostly (pro) hormones (22, 23) via recruitment of proteins involved in the formation and trafficking of vesicles, such as cytoskeleton-, GTP-, and Ca2+-binding proteins (24). The secretory granules route toward the cell periphery, where they mature and undergo calcium-controlled exocytosis (25–27). Upon an increase in Ca2+ concentration, CgA is co-released simultaneously with the stored hormones of the secretory granules via exocytosis (25–27). CgA is not only present in chromaffin cells, but has been detected in other secretory vesicles of endocrine, neuroendocrine and neuronal tissues (28–31) as well as in keratinocytes (32), myocardial cells (33–35), endothelial cells (36, 37), and macrophages (36). Interestingly, CgA is also present in cells of the pancreatic islet, secretory granules of glucagon containing α-cells and insulin producing β-cells, and may thereby modulate glucose metabolism (31, 38–42). This makes CgA particularly interesting in the context of metabolic diseases, such as diabetes. Patients suffering from carcinoids or other neuroendocrine tumors (25, 43–47), heart failure, renal failure, hypertension, RA, and IBD (48–54) display increased levels of circulating CgA, implicating an important role of CgA to influence human health and disease (3).
Cleavage Products of CgA
CgA can be proteolytically processed in various tissues and thereby serves as a precursor for several biological active peptides (Figure 1). The cleavage of CgA at its dibasic sites is performed by intra-granular and extra-cellular proteases, such as prohormone convertases 1 (PC1) (81), PC2 (81), furin (81), cysteine protease cathepsin L (CTSL) (82), the serine proteases plasmin (83, 84) and thrombin (85), as well as by kallikrein (86). Depending on the cleavage sites, post-translational modifications (glycosylation and phosphorylation) and proteolytic processing, CgA can result in the following six biological active peptides (9, 87). The first peptide identified was pancreastatin (PST) (hCgA250−301), which has an opposing effect to insulin (42, 58, 59). WE-14 (hCgA324−337) was identified in midgut carcinoid tumors and acts as an antigen for the highly diabetogenic CD4+ T cell clones (60–62). Chromofungin (hCgA47−66) has antimicrobial effects as well as effects on innate immune regulation (88, 89). Vasostatin (hCgA1−76) has a vasodilative and anti-angiogenic as well as antiadrenergic functions (55–57). Serpinin (hCgA402−439) regulates granule biogenesis (79) and acts as a myocardial ß agonist (80). Finally, the pleotropic peptide CST (hCgA352−372) has mainly anti-inflammatory effects (8, 90) and is the central focus of this review. The N-terminal 15 amino acid domain of bovine CST is called Cateslytin (bCgA344−358), which is the active domain of CST (76, 91). CgA is unique as several of its peptides exhibit opposing counter-regulatory effects for fine-tuning and maintaining metabolic homeostasis. As for cardiac function, this is regulated in rodents by the pro-adrenergic peptide serpinin (80) and both antiadrenergic peptides vasostatin and CST (66, 92). Likewise, angiogenesis is controlled by the antiangiogenic peptide vasostatin (85, 93) and the proangiogenic peptide CST (64, 85). Similarly, glucose homeostasis is maintained by pancreastatin (42, 58, 59, 94), which is an anti-insulin peptide and CST, which is a pro-insulin peptide (75). Although CgA processing has been reported to occur intracellularly inside the hormone-storage vesicles and extracellularly after its release in the blood, no systematic studies have been conducted to determine whether several proteolytic enzymes act at the same time to liberate all of the CgA peptides or act at different sites at different times in a tissue-specific manner. In addition, no attempts have been made so far to assess whether CgA peptides are generated in equal molar amounts or generated in response to physiological demands in different tissues. However, it has been reported that circulating concentrations of CgA peptides are different. For example, plasma vasostatin levels vary from 0.3 to 0.4 nM and CST circulates at 0.03 to 1.5 nM concentrations (9), which might represent different degrees of processing or rates of clearance from the circulation.
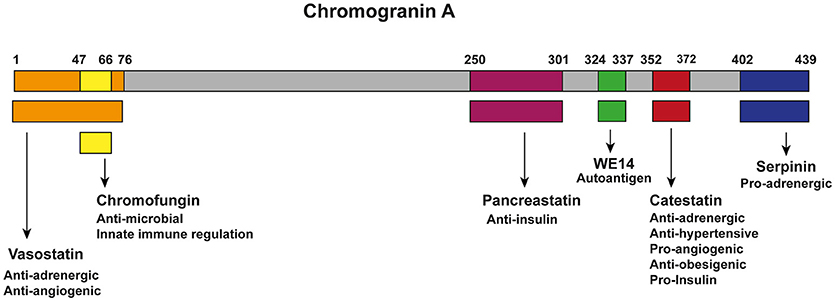
Figure 1. Chromogranin A and its bio-active peptides. Cleavage of CgA gives rise to six known biological active peptides: vasostatin (Orange; hCgA1−76), which has anti-adrenergic and anti-angiogenic functions (55–57); Chromofungin (Yellow; hCgA47−66) has antimicrobial effects as well as effects on innate immune regulation; pacreastatin (Purple; PST; hCgA250−301), which has anti-insulin functions (42, 58, 59); WE-14 (green; hCgA324−337), which acts as an autoantigen for the highly diabetogenic CD4+ T cell clones (60–62); CST (Red; hCgA352−372), which has pro-insulin, anti-obesigenic (63), pro-angiogenic (64, 65), anti-adrenergic, anti-hypersensitive (65–68), cardiomodulatory (8, 69–74) and anti-inflammatory functions (8, 32, 36, 75–78); serpinin (blue; hCgA402−439), which is pro-adrenergic and regulates granule biogenesis and acts as a myocardial ß agonist (79, 80).
The Pleotropic Peptide Catestatin (CST)
The CgA plasma levels range from 0.5 to 1 nM (9), whereas the physiological blood levels of CST range from 0.03 to 1.5 nM in healthy subjects (9, 95). At first CST was identified as a potent inhibitor of nicotine induced catecholamine release. As CST is secreted together with catecholamines, it can thereby function as an autocrine negative feedback-loop self-limiting further catecholamine secretion (10, 96, 97). Later, CST was found to play a role in the regulation of hypertension (65–68) and cardiac functions (8, 69–74), as well as in promoting angiogenesis (64, 85), decreasing obesity (63) and regulating innate immunity (8, 32, 36, 75–78). In line with this, alternated plasma levels of CST or its prohormone CgA have been observed in the context of various diseases. Plasma levels of CST are reduced in patients suffering from T2DM and hypertension (75, 95, 98), whereas elevated levels of the pro-hormone CgA have been detected in the plasma of patients with neuroendocrine tumors (25), hypertension (99, 100) and various inflammatory diseases, such as RA (6, 101, 102), SLE (6), inflammatory bowel disease (IBD) (53, 54, 103–105) as well as T1DM and T2DM (62, 106–109). This suggests that the lower levels of CST are caused by a dysregulation of proteolytic processing of CgA (98). The balance between processed peptides seems also important to counteract effects of the bio-active peptides. Since vasostatin has an anti-angiogenic effect (55–57), this might counteract the pro-angiogenic effect of CST (64, 85). Moreover, CgA-knockout (CgA-KO) mice develop an obese phenotype (42) as well as severe hypertension which could be rescued by intra-peritoneal injections of CST (67). Since hypertension is linked to diabetes, heart diseases and psoriasis (110), this indicates that CST might be important in various severe diseases. These findings support the hypothesis that the impaired processing of CgA might lead to lower CST levels which contributes to disease development. They also warrant further studies to elucidate the effects and mechanisms of CgA and its bio-active peptide products.
CST Contributes to Maintenance of Metabolic and Immune Homeostasis
CST Effects on Metabolism
In addition to its anti-inflammatory effects, CST also affects metabolism. Opposite to insulin, CST inhibits lipogenesis and increases lipolysis in adipose tissue by inhibition of the α2-adrenergic receptor and by enhancing leptin signaling (63). Simultaneously, it stimulates fatty acid uptake and breakdown in the liver, as reflected by increased expression of the genes involved in fatty acid oxidation upon intra-peritoneal CST injections in mice (63). In line with this, CST injections in CgA-KO mice resulted in decreased triglyceride levels in the plasma and reduced fat depot sizes by ~25% (63). These findings indicate that CST promotes lipid flux from adipose tissue to the liver for beta oxidation, which might explain the frequently observed weight gain in patients with inflammatory diseases, as these patients have lower plasma levels of CST (75, 95, 98).
Besides the effect on lipid metabolism, intra-peritoneal administration of CST improved glucose and insulin tolerance in Diet-induced obese (DIO) mice and insulin-resistant systemic CST-KO mice, that express a truncated version of CgA (75). This could be due to CST inhibiting gluconeogenesis in the liver, thereby lowering the production and release of glucose in the blood (75). This effect of CST could be mediated by the modulation of Kupffer-cells and monocyte-derived macrophages, since the effects of their cytokines are linked to glucose and insulin metabolism (75, 111) and for instance neutralization of TNF-α improves insulin sensitivity (112). Thus, CST can promote lipid and glucose metabolism, and thereby might help to prevent obesity and maintain homeostasis of metabolic functions (7, 63, 75). Although CST immunoreactivity has been detected in carcinoid tumors of the appendix, bronchus, stomach, small bowel and large bowel (113), its effects on cancer metabolism is yet to be investigated. However, insulin has been reported to promote cancer metabolism by upregulating pyruvate kinase M2 isoform (PKM2) expression and decreasing its activity, eventuating in amplification of cancer-metabolism-specific parameters like glucose uptake, lactate production, glycolytic pooling and macromolecular synthesis (114). In addition, several reports reveal increased cancer risk under hyperinsulinemic condition (115, 116). Since CST decreases insulin level in hyperinsulinemic as well as insulin-resistant DIO and CST-KO mice (75), we expect that CST would decrease tumor growth by decreasing expression of PKM2 and increasing its activity, which requires experimental validation. Interestingly, PST, another cleavage product of CgA, counteracts the metabolic and insulin sensitizing effects of CST (75). These anti-insulin actions of PST are likely important in maintaining homeostasis in glucose metabolism (7, 42, 94). The exact regulation of the proteolytic generation of CST and PST remains to be elucidated, but it could be coupled to metabolism via glycosylation because hyper-glycosylation of CgA is known to lead to reduced levels of CST (117). Thereby, the generation of CgA cleavage products might be regulated by sugar levels and this might play a role in progression of metabolic diseases, as for instance the increased blood glucose levels in T2DM might promote glycosylation of CgA.
CST Regulates Immune Homeostasis
CST contributes to the defense against infections in several ways (76, 77, 118). An initial study utilized 15 amino acids from the N-terminal end of bovine CST or cateslytin to demonstrate their antimicrobial activities (76). First, CST can directly act on invading microbes, as CST can penetrate the membrane of bacteria and fungi. At relatively high concentrations (>μM) it thereby directly can impair the growth of fungal pathogens (76). Moreover, CST can induce lysis of bacteria and helps to protect against infections following skin injuries in mice (32). Second, at least in vitro, CST can result in activation of neutrophils and mast cells which contribute to innate immune responses to infections (76–78, 118, 119). These effects may be restricted to local high CST concentrations, whereas systemic anti-inflammatory effects of CST have been best described in autoimmune diseases (Figure 2).
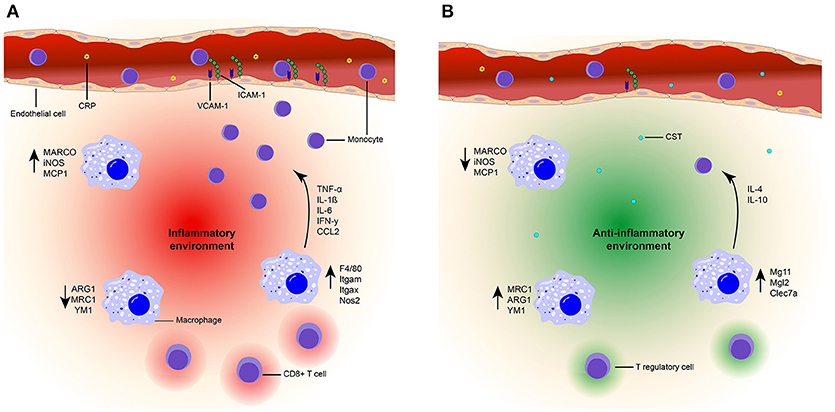
Figure 2. Model of the suppressive effects of CST on inflammation. (A) Inflammatory state. High plasma levels of CRP (yellow) are present in an inflammatory state. Due to the presence of chemokines (CCL2) and upregulation of integrin ligands [ICAM-1 (green) and VCAM-1 (blue)] on the endothelial cells (orange), increased monocyte (purple) infiltration is present at the inflammation site. An inflammatory environment (red) is created by the upregulation of pro-inflammatory markers (F4/80, Itgam, Itgax, NOS2, MARCO, iNOS, MCP1) in macrophages (blue). The production of inflammatory cytokines is also increased (TNF-a, IL-1β, IL-6, IFN-γ) and infiltration of CD8+ T cells occurs. (B) CST treated state. Treatment with CST (light blue) results in lower levels of circulating CRP (78, 120) and reduced expression of integrin ligands (36). Monocyte infiltration is reduced (36, 75, 78, 121) and macrophages are polarized toward an anti-inflammatory phenotype, characterized by upregulation of anti-inflammatory markers [MRC1, ARG1, YM1, Mg11, Mgl2, Clec7a (36, 75, 121)] and increased production of IL-10 and IL-4 (75, 121). Moreover, expression of pro-inflammatory genes and cytokines are reduced (36, 75, 78, 121). There will be no infiltration of CD8+ T cells and Tregs might be present. Together, this contributes to an anti-inflammatory environment (green) and improved tissue architecture (36, 78, 121).
In a colitis mouse model, intra-rectal injections with CST resulted in decreased serum levels of the acute phase reactant C-reactive protein (CRP) (78, 120) and suppressed activity of myeloperoxidase (MPO), which is a marker for granulocyte infiltration (78). As a result of these injections, the tissue architecture of the colon improved (78, 121). Moreover, in atherosclerotic mice (apolipoprotein E-deficient mice), intraperitoneal injection followed by continuous subcutaneous CST infusion significantly retarded atherosclerotic lesions by 40% in the entire surface area of the aorta (36). In both colitis and atherosclerosis models, the prevalence of macrophages and monocytes in inflamed tissues was reduced following administration of CST (36, 78, 121), thereby supporting the anti-inflammatory effects of CST. In DIO mice, the intra-peritoneal injection of CST inhibited the infiltration of monocytes in the liver and reduced CC-chemokine ligand 2 (CCL2)-induced chemotaxis of peritoneal macrophages (75). The molecular mechanisms by which CST affects monocyte and macrophage migration are still unclear. One possibility is that CST directly affects leukocyte migration. This is shown for monocytes, although in this case already low concentrations of CST (nM) promoted migration in an in vitro chemotaxis assay (122). The reasons underlying this discrepancy between in vitro and in vivo experiments is unclear, but could be due to CST affecting other chemokines (such as CCL2) present in the in vivo situation. Moreover, CST is also pro-angiogenic (64, 85) which might reduce its anti-inflammatory effect when present on its own. Another possibility is that CST affects the integrins that affect leukocyte extravasation. This possibility is supported by the finding that CST can reduce expression levels of the integrin ligands intracellular adhesion molecule 1 (ICAM-1) and vascular CAM-1 (VCAM-1) in endothelial cells, which correlate with lymphocyte extravasation (36). Finally, CST might reduce monocyte infiltration in inflammatory tissues by lowering the production of pro-inflammatory cytokines and chemokines by macrophages due to altered macrophage differentiation (75, 78).
Considering the effects of CST treatment on THP-1 cells (a human monocyte cell line), it seems that CST does not affect the overall differentiation of monocytes to macrophages. This was shown by consistent expression of the macrophage marker CD68 by THP-1 cells under CST treatment. However, CST steers the polarization of differentiation into less pro- and more anti-inflammatory phenotypes (36). CST treatment of THP-1 derived macrophages resulted in elevated levels of anti-inflammatory macrophage markers (mannose receptor C-type 1, (MRC1)) and reduced levels of pro-inflammatory macrophage markers (macrophage receptor with collagenous domain, (MARCO)) (36). Additionally, the gene expression levels of the pro-inflammatory macrophage markers inducible nitric oxygen synthase (iNOS) and monocyte chemoattractant protein 1 (Mcp1) were reduced upon intra-rectal injection of CST in a reactivated colitis mouse model, as well as in vitro in LPS stimulated peritoneal and colon macrophages (78, 121). In both the mouse model and in vitro, CST treatment resulted in decreased levels of pro-inflammatory cytokines (IL-6, IL-1β, TNF-α) (78, 121). In the reactivated colitis mice model, CST promoted expression of several anti-inflammatory genes (IL-10, Arg1, and Ym1) of the macrophages in the colon (121). Moreover, a reduction of pro-inflammatory gene expression (TNF-α, F4/80, Itgam, Itgax, Ifng, Nos2, and Ccl2) was detected in isolated Kupffer cells and monocyte derived macrophages of DIO mice after intra-peritoneal injections with CST, whereas levels of anti-inflammatory genes were increased (IL-10, Mgl1, IL-4, Arg1, and Mrc1) (75). These results could be confirmed in isolated macrophages treated in vitro with CST (75). In the DIO mouse model, intra-peritoneal injections with CST also reduced plasma levels of pro-inflammatory cytokines and chemokines (TNF-α, INF-γ, and CCL2) (75). Taken together, these findings indicate that CST shifts macrophage differentiation from a pro- to a more anti-inflammatory phenotype. Since adipose tissue macrophages (ATMs) have antigen-presenting capacities this shift could influence the adaptive immune response (123). Interestingly, CD8 deficient mice show a decrease in macrophage infiltration and adipose tissue (124), suggesting that CD8+ T cells infiltration precedes macrophage accumulation in inflammation. So, CST reduces inflammation, macrophage infiltration and might even influence the adaptive immune response by affecting Treg infiltration and decreasing CD8+ T cell infiltration, but that would need to be validated in future experiments.
Although it is increasingly clear that CST exerts anti-inflammatory effects on macrophages, the underlying mechanisms are still largely unknown. A key open question is to which receptor CST binds to exert its effect. This could either be a plasma membrane receptor as well as an intracellular target, since CST can penetrate the cell membrane of neutrophils (76, 77). Another question is which signaling pathways are influenced by CST. Based on experiments with inhibitors in mast cells, CST treatment leads to cellular activation by mobilizing intracellular Ca2+ and inducing the production of pro-inflammatory cytokines/chemokines (GM-CSF, CCL2, CCL3, and CCL4) via a mechanism possibly involving G-proteins, phospholipase C and the mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) (119). However, it is unknown whether these pathways are also responsible for the anti-inflammatory signaling in macrophages by CST.
Clinical Implications of CST
Given its roles in metabolic regulation and immune homeostasis, CST has potential clinical applications as a diagnostic marker and even as a therapeutic target. For example, lower levels of CST have been reported in the blood of patients suffering from T2DM, suggesting that it might be a diagnostic marker for this disease (75, 95, 98). However, it might be more useful to study CST levels relative to other cleavage products of CgA, considering that some of these cleavage products counteract the activities of CST. For instance, PST exerts opposing effects on insulin sensitivity and glucose metabolism compared to CST (58), and increased levels of PST can contribute to T2DM (41). The observed lower levels of CST might well be caused by dysregulation of proteolytic processing of CgA (98), since this could result in a higher ratio of PST to CST. Indeed, an altered processing of CgA has been observed in the microenvironment of tumors. Here CgA cleavage products lead to proangiogenic activity, as cleavage of the N- and C-terminal regions of CgA can activate antiangiogenic (vasostatin) and proangiogenic sites (CST), respectively (1). Further supporting the notion that CgA and its cleavage products can be diagnostic markers for various diseases, is that elevated levels of CgA have been detected in the plasma of patients with neuroendocrine tumors (25), hypertension (99, 100) and various inflammatory diseases, such as RA (6, 101, 102), SLE (6), IBD (53, 54, 103–105) as well as T1DM and T2DM (62, 106–109). However, not all assays used in the aforementioned studies allow to discern full-length from proteolytically processed CgA (125) and it would be very interesting to compare this to levels of unprocessed CgA and its cleavage products.
As described above, studies in mouse disease models have indicated that CST can be used as a therapeutic agent for treatment of various diseases, such as colitis, atherosclerosis and diabetes (42, 75, 78, 98, 121, 125). In particular in T2DM, CST is a promising drug candidate, since it basically targets all characteristics of T2DM and modulates both inflammation and metabolism by lowering blood glucose levels, improving insulin sensitivity and secretion as well as by reducing systemic inflammation (3, 126). Especially the ability of CST to shift macrophage polarization toward an anti-inflammatory phenotype makes it a strong therapeutic candidate for a range of inflammatory diseases, such as chronic inflammation (gastritis and colitis), auto-immune diseases (RA and SLE), hypertension, cancers and even inflammation-induced tumor metastasis (9, 25, 127).
Concluding Remarks
As discussed in this review, CST can decrease inflammation by reducing immune infiltration in inflamed tissues and altering macrophages differentiation into an anti-inflammatory phenotype (42, 75, 78, 98, 121, 125). These effects are already observed at concentrations in the nM range, which corresponds to physiological levels of circulating CST (9). By lowering the production of pro-inflammatory cytokines, CST may suppress inflammatory immune responses and/or might promote the dissolvement of inflammation. As a result, CST could prevent chronic states of inflammation and inhibit exaggerated inflammatory responses normally leading to tissue damage. Although CST exerts primarily anti-inflammatory effects, other cleavage products of CgA have opposing pro-inflammatory effects. Disbalances in the levels of circulating CgA-derived peptides might therefore contribute to various diseases (3). Detection and distinguishing of CgA cleavage products with current ELISA-based assays are imperfect, requiring more sensitive mass spectrometry-based assays instead. The mechanism by which CST (and other CgA cleavage products) is removed from the circulation remains unknown; amongst others, its receptor-binding partners need to be identified for instance by immune precipitation followed by proteomics. These data are required to fully understand the effects of CST and other CgA cleavage products.
Due to their effects on immune homeostasis, CST and other CgA-derived peptides are promising targets for diagnosis and therapy of diseases with an inflammatory component, such as diabetes, cancer and RA. A caveat is that almost all current studies on CgA have been conducted in mice and rats. Translating the findings from rodent to man will be essential and will help understanding and designing future diagnostic and therapeutic strategies.
Author Contributions
EM and GD wrote the manuscript. MN assisted in the literature search. SM and GvdB participated in discussion and reviewed/edited the manuscript.
Funding
GvdB is funded by a Career Development Award from the Human Frontier Science Program, the NWO Gravitation Programme 2013 (ICI-024.002.009), and a Vidi grant from the Netherlands Organization for Scientific Research (NWO-ALW VIDI 864.14.001). SM was supported by a grant from the US Department of Veterans Affairs (I01BX000323).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank IEL editor Dr. Lucy Robinson for comments on an earlier version that greatly improved the manuscript and we thank A.W. van der Burgh for his graphic design skills.
References
1. Bianco M, Gasparri AM, Colombo B, Curnis F, Girlanda S, Ponzoni M, et al. Chromogranin A is preferentially cleaved into proangiogenic peptides in the bone marrow of multiple myeloma patients. Cancer Res. (2016) 76:1781–91. doi: 10.1158/0008-5472.can-15-1637
2. Cotesta D, Caliumi C, Alò P, Petramala L, Reale MG, Masciangelo R, et al. High plasma levels of human chromogranin A and adrenomedullin in patients with pheochromocytoma. Tumori (2005) 91:53–8.
3. Loh YP, Cheng Y, Mahata SK, Corti A, Tota B. Chromogranin A and derived peptides in health and disease. J Mol Neurosci. (2012) 48:347–56. doi: 10.1007/s12031-012-9728-2
4. NoFerlay JB, Bray F, Pisani PPD. GLOBOCAN 2012: Cancer Incidence, Mortality and Prevalence Worldwide. IARC Cancer Section of Cancersurveillance. Lyon: IARC Press (2018) Available online at: http://www-dep.iarc.fr/.
5. Wolfe F, Mitchell DM, Sibley JT, Fries JF, Bloch DA, Williams CA, et al. The mortality of rheumatoid arthritis. Arthritis Rheum. (1994) 37:481–94. doi: 10.1002/art.1780370408
6. Capellino S, Lowin T, Angele P, Falk W, Grifka J, Straub RH. Increased chromogranin A levels indicate sympathetic hyperactivity in patients with rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol. (2008) 35:91–9. Available online at: www.jrheum.org/content/35/1/91.long
7. Bandyopadhyay GK, Mahata SK. Chromogranin A regulation of obesity and peripheral insulin sensitivity. Front Endocrinol (Lausanne) (2017) 8:20. doi: 10.3389/fendo.2017.00020
8. Mahata SK, Kiranmayi M, Mahapatra NR. Catestatin: a master regulator of cardiovascular functions. Curr Med Chem. (2018) 25:1352–74. doi: 10.2174/0929867324666170425100416
9. Corti A, Marcucci F, Bachetti T. Circulating chromogranin A and its fragments as diagnostic and prognostic disease markers. Pflügers Arch Eur J Physiol. (2017) 470:199–210. doi: 10.1007/s00424-017-2030-y
10. Mahata SK, O'Connor DT, Mahata M, Yoo SH, Taupenot L, Wu H, et al. Novel autocrine feedback control of catecholamine release. A discrete chromogranin a fragment is a noncompetitive nicotinic cholinergic antagonist. J Clin Invest. (1997) 100:1623–33. doi: 10.1172/jci119686
11. Wen G, Mahata SK, Cadman P, Mahata M, Ghosh S, Mahapatra NR, et al. Both rare and common polymorphisms contribute functional variation at CHGA, a regulator of catecholamine physiology. Am J Hum Genet. (2004) 74:197–207. doi: 10.1086/381399
12. Murray SS, Deaven LL, Burton DW, O'Connor DT, Mellon PL, Deftos LJ. The gene for human chromogranin A (CgA) is located on chromosome 14. Biochem Biophys Res Commun. (1987) 142:141–6. doi: 10.1016/0006-291x(87)90462-1
13. Modi WS, Levine MA, Seuanez HN, Dean M, O'Brien SJ. The human chromogranin A gene: chromosome assignment and RFLP analysis. Am J Hum Genet. (1989) 45:814–8.
14. Konecki DS, Benedum UM, Gerdes HH, Huttner WB. The primary structure of human chromogranin A and pancreastatin. J Biol Chem. (1987) 262:17026–30.
15. Winkler H, Fischer-Colbrie R. The chromogranins A and B: the first 25 years and future perspectives. Neuroscience (1992) 49:497–528. doi: 10.1016/0306-4522(92)90222-n
16. Videen JS, Mezger MS, Chang YM, O'Connor DT. Calcium and catecholamine interactions with adrenal chromogranins. Comparison of driving forces in binding and aggregation. J Biol Chem. (1992) 267:3066–73.
17. Winkler H, Westhead E. The molecular organization of adrenal chromaffin granules. Neuroscience (1980) 5:1803–23. doi: 10.1016/0306-4522(80)90031-7
18. Haigh JR, Parris R, Phillips JH. Free concentrations of sodium, potassium and calcium in chromaffin granules. Biochem J. (1989) 259:485–91. doi: 10.1042/bj2590485
19. Berneis KH, Goetz U, Da Prada M, Pletscher A. Interaction of aggregated catecholamines and nucleotides with intragranular proteins. Naunyn Schmiedebergs Arch Pharmacol. (1973) 277:291–6. doi: 10.1007/bf00505667
20. Kopell WN, Westhead EW. Osmotic pressures of solutions of ATP and catecholamines relating to storage in chromaffin granules. J Biol Chem. (1982) 257:5707–10.
21. Borges R, Díaz-Vera J, Domínguez N, Arnau MR, Machado JD. Chromogranins as regulators of exocytosis. J Neurochem. (2010) 114:335–43. doi: 10.1111/j.1471-4159.2010.06786.x
22. Kim T, Tao-Cheng J-H, Eiden LE, Loh YP. Chromogranin A, an “On/Off” switch controlling dense-core secretory granule biogenesis. Cell (2001) 106:499–509. doi: 10.1016/s0092-8674(01)00459-7
23. Taupenot L, Harper K, Mahapatra NR, Parmer RJ, Mahata SK, O'Connor DT. Identification of a novel sorting determinant for the regulated pathway in the secretory protein chromogranin A. J Cell Sci. (2002) 115:4827–41. doi: 10.1242/jcs.00140
24. Elias S, Delestre C, Ory S, Marais S, Courel M, Vazquez-Martinez R, et al. Chromogranin A induces the biogenesis of granules with calcium- and actin-dependent dynamics and exocytosis in constitutively secreting cells. Endocrinology (2012) 153:4444–56. doi: 10.1210/en.2012-1436
25. O'Connor DT, Bernstein KN. Radioimmunoassay of chromogranin a in plasma as a measure of exocytotic sympathoadrenal activity in normal subjects and patients with pheochromocytoma. N Engl J Med. (1984) 311:764–70. doi: 10.1056/nejm198409203111204
26. Takiyyuddin MA, Cervenka JH, Hsiao RJ, Barbosa JA, Parmer RJ, O'Connor DT. Chromogranin A. Storage and release in hypertension. Hypertension (1990) 15:237–46. doi: 10.1161/01.hyp.15.3.237
27. Takiyyuddin MA, Cervenka JH, Sullivan PA, Pandian MR, Parmer RJ, Barbosa JA, et al. Is physiologic sympathoadrenal catecholamine release exocytotic in humans? Circulation (1990) 81:185–95. doi: 10.1161/01.cir.81.1.185
28. Mahata SK, Mahata M, Marksteiner J, Sperk G, Fischer-Colbrie R, Winkler H. Distribution of mRNAs for chromogranins A and B and secretogranin II in rat brain. Eur J Neurosci. (1991) 3:895–904. doi: 10.1111/j.1460-9568.1991.tb00101.x
29. Montero-Hadjadje M, Vaingankar S, Elias S, Tostivint H, Mahata SK, Anouar Y. Chromogranins A and B and secretogranin II: evolutionary and functional aspects. Acta Physiol. (2007) 192:309–24. doi: 10.1111/j.1748-1716.2007.01806.x
30. Bartolomucci A, Possenti R, Mahata SK, Fischer-Colbrie R, Loh YP, Salton SRJ. The extended granin family: structure, function, and biomedical implications. Endocr Rev. (2011) 32:755–97. doi: 10.1210/er.2010-0027
31. Cohn DV, Elting JJ, Frick M, Elde R. Selective localization of the parathyroid secretory protein-I/adrenal medulla chromogranin A protein family in a wide variety of endocrine cells of the rat. Endocrinology (1984) 114:1963–74. doi: 10.1210/endo-114-6-1963
32. Radek KA, Lopez-Garcia B, Hupe M, Niesman IR, Elias PM, Taupenot L, et al. The neuroendocrine peptide catestatin is a cutaneous antimicrobial and induced in the skin after injury. J Invest Dermatol. (2008) 128:1525–34. doi: 10.1038/sj.jid.5701225
33. Biswas N, Curello E, O'Connor DT, Mahata SK. Chromogranin/secretogranin proteins in murine heart: myocardial production of chromogranin A fragment catestatin (Chga364–384). Cell Tissue Res. (2010) 342:353–61. doi: 10.1007/s00441-010-1059-4
34. Pieroni M, Corti A, Tota B, Curnis F, Angelone T, Colombo B, et al. Myocardial production of chromogranin A in human heart: a new regulatory peptide of cardiac function. Eur Heart J. (2007) 28:1117–27. doi: 10.1093/eurheartj/ehm022
35. Hansen LH, Darkner S, Svendsen JH, Henningsen K, Pehrson S, Chen X, et al. Chromogranin A in the mammalian heart: expression without secretion. Biomark Med. (2017) 11:541–5. doi: 10.2217/bmm-2017-0052
36. Kojima M, Ozawa N, Mori Y, Takahashi Y, Watanabe-Kominato K, Shirai R, et al. Catestatin prevents macrophage-driven atherosclerosis but not arterial injury–induced neointimal hyperplasia. Thromb Haemost. (2018) 118:182–94. doi: 10.1160/th17-05-0349
37. Helle KB, Metz-Boutigue MH, Cerra MC, Angelone T. Chromogranins: from discovery to current times. Pflügers Arch Eur J Physiol. (2017) 470:143–54. doi: 10.1007/s00424-017-2027-6
38. Wilson BS, Lloyd RV. Detection of chromogranin in neuroendocrine cells with a monoclonal antibody. Am J Pathol. (1984) 115:458–68.
39. Ehrhart M, Grube D, Bader MF, Aunis D, Gratzl M. Chromogranin A in the pancreatic islet: cellular and subcellular distribution. J Histochem Cytochem. (1986) 34:1673–82. doi: 10.1177/34.12.2878021
40. Lukinius A, Stridsberg M, Wilander E. Cellular expression and specific intragranular localization of chromogranin A, chromogranin B, and synaptophysin during ontogeny of pancreatic islet cells: an ultrastructural study. Pancreas (2003) 27:38–46. doi: 10.1097/00006676-200307000-00006
41. O'Connor DT, Cadman PE, Smiley C, Salem RM, Rao F, Smith J, et al. Pancreastatin: multiple actions on human intermediary metabolism in vivo, variation in disease, and naturally occurring functional genetic polymorphism. J Clin Endocrinol Metab. (2005) 90:5414–25. doi: 10.1210/jc.2005-0408
42. Gayen JR, Saberi M, Schenk S, Biswas N, Vaingankar SM, Cheung WW, et al. A novel pathway of insulin sensitivity in chromogranin A null mice. J Biol Chem. (2009) 284:28498–509. doi: 10.1074/jbc.m109.020636
43. O'Connor DT, Deftos LJ. Secretion of chromogranin A by peptide-producing endocrine neoplasms. N Engl J Med. (1986) 314:1145–51. doi: 10.1056/NEJM198605013141803
44. Gregorc V, Spreafico A, Floriani I, Colombo B, Ludovini V, Pistola L, et al. Prognostic value of circulating chromogranin A and soluble tumor necrosis factor receptors in advanced nonsmall cell lung cancer. Cancer (2007) 110:845–53. doi: 10.1002/cncr.22856
45. Corti A. Chromogranin A and the tumor microenvironment. Cell Mol Neurobiol. (2010) 30:1163–70. doi: 10.1007/s10571-010-9587-8
46. Corti A, Gasparri A, Chen FX, Pelagi M, Brandazza A, Sidoli A, et al. Characterisation of circulating chromogranin A in human cancer patients. Br J Cancer (1996) 73:924–32.
47. Bernini GP, Moretti A, Ferdeghini M, Ricci S, Letizia C, D'Erasmo E, et al. A new human chromogranin “A” immunoradiometric assay for the diagnosis of neuroendocrine tumours. Br J Cancer (2001) 84:636–42. doi: 10.1054/bjoc.2000.1659
48. Ceconi C, Ferrari R, Bachetti T, Opasich C, Volterrani M, Colombo B, et al. Chromogranin A in heart failure. A novel neurohumoral factor and a predictor for mortality. Eur Heart J. (2002) 23:967–74. doi: 10.1053/euhj.2001.2977
49. Røsjø H, Masson S, Latini R, Flyvbjerg A, Milani V, La Rovere MT, et al. Prognostic value of chromogranin A in chronic heart failure: data from the GISSI-Heart Failure trial. Eur J Heart Fail. (2010) 12:549–56. doi: 10.1093/eurjhf/hfq055
50. O'Connor DT, Pandlan MR, Carlton E, Cervenka JH, Hslao RJ. Rapid radioimmunoassay of circulating chromogranin A: in vitro stability, exploration of the neuroendocrine character of neoplasia, and assessment of the effects of organ failure. Clin Chem (1989) 35:1631–7.
51. Giampaolo B, Angelica M, Antonio S. Chromogranin “A” in normal subjects, essential hypertensives and adrenalectomized patients. Clin Endocrinol (Oxf). (2002) 57:41–50. doi: 10.1046/j.1365-2265.2002.01557.x
52. Tombetti E, Colombo B, Di Chio MC, Sartorelli S, Papa M, Salerno A, et al. Chromogranin-A production and fragmentation in patients with Takayasu arteritis. Arthritis Res Ther. (2016) 18:187. doi: 10.1186/s13075-016-1082-2
53. Sciola V, Massironi S, Conte D, Caprioli F, Ferrero S, Ciafardini C, et al. Plasma chromogranin a in patients with inflammatory bowel disease. Inflamm Bowel Dis. (2009) 15:867–71. doi: 10.1002/ibd.20851
54. Zissimopoulos A, Vradelis S, Konialis M, Chadolias D, Bampali A, Constantinidis T, et al. Chromogranin A as a biomarker of disease activity and biologic therapy in inflammatory bowel disease: a prospective observational study. Scand J Gastroenterol. (2014) 49:942–49. doi: 10.3109/00365521.2014.920910
55. Aardal S, Helle KB, Elsayed S, Reed RK, Serck-Hanssen G. Vasostatins, comprising the N-terminal domain of chromogranin A, suppress tension in isolated human blood vessel segments. J Neuroendocrinol. (1993) 5:405–12. doi: 10.1111/j.1365-2826.1993.tb00501.x
56. Tota B, Mazza R, Angelone T, Nullans G, Metz-Boutigue MH, Aunis D, et al. Peptides from the N-terminal domain of chromogranin A (vasostatins) exert negative inotropic effects in the isolated frog heart. Regul Pept. (2003) 114:123–30. doi: 10.1016/s0167-0115(03)00112-5
57. Corti A, Mannarino C, Mazza R, Angelone T, Longhi R, Tota B. Chromogranin A N-terminal fragments vasostatin-1 and the synthetic CGA 7–57 peptide act as cardiostatins on the isolated working frog heart. Gen Comp Endocrinol. (2004) 136:217–24. doi: 10.1016/j.ygcen.2003.12.012
58. Tatemoto K, Efendić S, Mutt V, Makk G, Feistner GJ, Barchas JD. Pancreastatin, a novel pancreatic peptide that inhibits insulin secretion. Nature (1986) 324:476–8. doi: 10.1038/324476a0
59. Bandyopadhyay GK, Lu M, Avolio E, Siddiqui JA, Gayen JR, Wollam J, et al. Pancreastatin-dependent inflammatory signaling mediates obesity-induced insulin resistance. Diabetes (2014) 64:104–16. doi: 10.2337/db13-1747
60. Curry WJ, Shaw C, Johnston CF, Thim L, Buchanan KD. Isolation and primary structure of a novel chromogranin A-derived peptide, WE-14, from a human midgut carcinoid tumour. FEBS Lett. (1992) 301:319–21. doi: 10.1016/0014-5793(92)80266-j
61. Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. (2010) 11:225–31. doi: 10.1038/ni.1844
62. Jin N, Wang Y, Crawford F, White J, Marrack P, Dai S, et al. N-terminal additions to the WE14 peptide of chromogranin A create strong autoantigen agonists in type 1 diabetes. Proc Natl Acad Sci USA. (2015) 112:13318–23. doi: 10.1073/pnas.1517862112
63. Bandyopadhyay GK, Vu CU, Gentile S, Lee H, Biswas N, Chi N-W, et al. Catestatin (chromogranin A352–372) and novel effects on mobilization of fat from adipose tissue through regulation of adrenergic and leptin signaling. J Biol Chem. (2012) 287:23141–51. doi: 10.1074/jbc.m111.335877
64. Theurl M, Schgoer W, Albrecht K, Jeschke J, Egger M, Beer AGE, et al. The neuropeptide catestatin acts as a novel angiogenic cytokine via a basic fibroblast growth factor-dependent mechanism. Circ Res. (2010) 107:1326–35. doi: 10.1161/circresaha.110.219493
65. Gayen JR, Zhang K, RamachandraRao SP, Mahata M, Chen Y, Kim H-S, et al. Role of reactive oxygen species in hyperadrenergic hypertension: biochemical, physiological, and pharmacological evidence from targeted ablation of the chromogranin A (Chga) gene. Circ Cardiovasc Genet. (2010) 3:414–25. doi: 10.1161/circgenetics.109.924050
66. Avolio E, Mahata SK, Mantuano E, Mele M, Alò R, Facciolo RM, et al. Antihypertensive and neuroprotective effects of catestatin in spontaneously hypertensive rats: interaction with GABAergic transmission in amygdala and brainstem. Neuroscience (2014) 270:48–57. doi: 10.1016/j.neuroscience.2014.04.001
67. Mahapatra NR, O'Connor DT, Vaingankar SM, Hikim APS, Mahata M, Ray S, et al. Hypertension from targeted ablation of chromogranin A can be rescued by the human ortholog. J Clin Invest. (2005) 115:1942–52. doi: 10.1172/jci24354
68. Biswas N, Gayen J, Mahata M, Su Y, Mahata SK, O'Connor DT. Novel peptide isomer strategy for stable inhibition of catecholamine release: application to hypertension. Hypertension (2012) 60:1552–9. doi: 10.1161/hypertensionaha.112.202127
69. Angelone T, Quintieri AM, Brar BK, Limchaiyawat PT, Tota B, Mahata SK, et al. The antihypertensive chromogranin A peptide catestatin acts as a novel endocrine/paracrine modulator of cardiac inotropism and lusitropism. Endocrinology (2008) 149:4780–93. doi: 10.1210/en.2008-0318
70. Gayen JR, Gu Y, O'Connor DT, Mahata SK. Global disturbances in autonomic function yield cardiovascular instability and hypertension in the chromogranin A null mouse. Endocrinology (2009) 150:5027–35. doi: 10.1210/en.2009-0429
71. Penna C, Alloatti G, Gallo MP, Cerra MC, Levi R, Tullio F, et al. Catestatin improves post-ischemic left ventricular function and decreases ischemia/reperfusion injury in heart. Cell Mol Neurobiol. (2010) 30:1171–9. doi: 10.1007/s10571-010-9598-5
72. Angelone T, Quintieri AM, Pasqua T, Gentile S, Tota B, Mahata SK, et al. Phosphodiesterase type-2 and NO-dependent S-nitrosylation mediate the cardioinhibition of the antihypertensive catestatin. Am J Physiol Circ Physiol. (2012) 302:H431–42. doi: 10.1152/ajpheart.00491.2011
73. Angelone T, Quintieri AM, Pasqua T, Filice E, Cantafio P, Scavello F, et al. The NO stimulator, Catestatin, improves the Frank–Starling response in normotensive and hypertensive rat hearts. Nitric Oxide (2015) 50:10–9. doi: 10.1016/j.niox.2015.07.004
74. Penna C, Pasqua T, Amelio D, Perrelli M-G, Angotti C, Tullio F, et al. Catestatin increases the expression of anti-apoptotic and pro-angiogenetic factors in the post-ischemic hypertrophied heart of SHR. PLoS ONE (2014) 9:e102536. doi: 10.1371/journal.pone.0102536
75. Ying W, Mahata S, Bandyopadhyay GK, Zhou Z, Wollam J, Vu J, et al. Catestatin inhibits obesity-induced macrophage infiltration and inflammation in the liver and suppresses hepatic glucose production, leading to improved insulin sensitivity. Diabetes (2018) 67:841–8. doi: 10.2337/db17-0788
76. Briolat J, Wu SD, Mahata SK, Gonthier B, Bagnard D, Chasserot-Golaz S, et al. New antimicrobial activity for the catecholamine release-inhibitory peptide from chromogranin A. Cell Mol Life Sci. (2005) 62:377–85. doi: 10.1007/s00018-004-4461-9
77. Zhang D, Shooshtarizadeh P, Laventie BJ, Colin DA, Chich JF, Vidic J, et al. Two chromogranin a-derived peptides induce calcium entry in human neutrophils by calmodulin-regulated calcium independent phospholipase A2. PLoS ONE (2009) 4:e4501. doi: 10.1371/journal.pone.0004501
78. Rabbi MF, Labis B, Metz-Boutigue MH, Bernstein CN, Ghia J-E. Catestatin decreases macrophage function in two mouse models of experimental colitis. Biochem Pharmacol. (2014) 89:386–98. doi: 10.1016/j.bcp.2014.03.003
79. Koshimizu H, Cawley NX, Kim T, Yergey AL, Loh YP. Serpinin: a novel chromogranin A-derived, secreted peptide up-regulates protease nexin-1 expression and granule biogenesis in endocrine cells. Mol Endocrinol. (2011) 25:732–44. doi: 10.1210/me.2010-0124
80. Tota B, Gentile S, Pasqua T, Bassino E, Koshimizu H, Cawley NX, et al. The novel chromogranin A-derived serpinin and pyroglutaminated serpinin peptides are positive cardiac β-adrenergic-like inotropes. FASEB J. (2012) 26:2888–98. doi: 10.1096/fj.11-201111
81. Eskeland NL, Zhou A, Dinh TQ, Wu H, Parmer RJ, Mains RE, et al. Chromogranin A processing and secretion: specific role of endogenous and exogenous prohormone convertases in the regulated secretory pathway. J Clin Invest. (1996) 98:148–56. doi: 10.1172/jci118760
82. Biswas N, Rodriguez-Flores JL, Courel M, Gayen JR, Vaingankar SM, Mahata M, et al. Cathepsin L colocalizes with chromogranin A in chromaffin vesicles to generate active peptides. Endocrinology (2009) 150:3547–57. doi: 10.1210/en.2008-1613
83. Jiang Q, Taupenot L, Mahata SK, Mahata M, O'Connor DT, Miles LA, et al. Proteolytic cleavage of chromogranin A (CgA) by plasmin. Selective liberation of a specific bioactive CgA fragment that regulates catecholamine release. J Biol Chem. (2001) 276:25022–9. doi: 10.1074/jbc.M101545200
84. Biswas N, Vaingankar SM, Mahata M, Das M, Gayen JR, Taupenot L, et al. Proteolytic cleavage of human chromogranin A containing naturally occurring catestatin variants: differential processing at catestatin region by plasmin. Endocrinology (2008) 149:749–57. doi: 10.1210/en.2007-0838
85. Crippa L, Bianco M, Colombo B, Gasparri AM, Ferrero E, Loh YP, et al. A new chromogranin A-dependent angiogenic switch activated by thrombin. Blood (2012) 121:392–402. doi: 10.1182/blood-2012-05-430314
86. Benyamin B, Maihofer AX, Schork AJ, Hamilton BA, Rao F, Schmid-Schönbein GW, et al. Identification of novel loci affecting circulating chromogranins and related peptides. Hum Mol Genet. (2016) 26:233–42. doi: 10.1093/hmg/ddw380
87. Gadroy P, Stridsberg M, Capon C, Michalski JC, Strub JM, Van Dorsselaer A, et al. Phosphorylation and O-glycosylation sites of human chromogranin A (CGA79-439) from urine of patients with carcinoid tumors. J Biol Chem. (1998) 273:34087–97. doi: 10.1074/JBC.273.51.34087
88. Eissa N, Hussein H, Kermarrec L, Elgazzar O, Metz-Boutigue MH, Bernstein CN, et al. Chromofungin (CHR: CHGA47-66) is downregulated in persons with active ulcerative colitis and suppresses pro-inflammatory macrophage function through the inhibition of NF-κB signaling. Biochem Pharmacol. (2017) 145:102–13. doi: 10.1016/j.bcp.2017.08.013
89. Eissa N, Hussein H, Kermarrec L, Grover J, Metz-Boutigue MH, Bernstein CN, et al. Chromofungin ameliorates the progression of colitis by regulating alternatively activated macrophages. Front Immunol. (2017) 8:1131. doi: 10.3389/fimmu.2017.01131
90. Mahata SK, Mahata M, Fung MM, O'Connor DT. Catestatin: a multifunctional peptide from chromogranin A. Regul Pept. (2010) 162:33–43. doi: 10.1016/j.regpep.2010.01.006
91. Crivellato E, Nico B, Ribatti D. The chromaffin vesicle: advances in understanding the composition of a versatile, multifunctional secretory organelle. Anat Rec Adv Integr Anat Evol Biol. (2008) 291:1587–602. doi: 10.1002/ar.20763
92. Cerra MC, Iuri L, Angelone T, Corti A, Tota B. Recombinant N–terminal fragments of chromogranin–a modulate cardiac function of the Langendorff–perfused rat heart. Basic Res Cardiol. (2006) 101:43–52. doi: 10.1007/s00395-005-0547-2
93. Pike SE, Yao L, Jones KD, Cherney B, Appella E, Sakaguchi K, et al. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J Exp Med. (1998) 188:2349–56.
94. Sánchez-Margalet V, González-Yanes C, Najib S, Santos-Álvarez J. Metabolic effects and mechanism of action of the chromogranin A-derived peptide pancreastatin. Regul Pept. (2010) 161:8–14. doi: 10.1016/j.regpep.2010.02.005
95. O'Connor DT, Kailasam MT, Kennedy BP, Ziegler MG, Yanaihara N, Parmer RJ. Early decline in the catecholamine release-inhibitory peptide catestatin in humans at genetic risk of hypertension. J Hypertens. (2002) 20:1335–45. doi: 10.1097/00004872-200207000-00020
96. Mahata SK, Mahata M, Wakade A, O'Connor DT. Primary structure and function of the catecholamine release inhibitory peptide catestatin (chromogranin A344–364): identification of amino acid residues crucial for activity. Mol Endocrinol. (2000) 14:1525–35. doi: 10.1210/me.14.10.1525
97. Mahata SK, Mahapatra NR, Mahata M, Wang TC, Kennedy BP, Ziegler MG, et al. Catecholamine secretory vesicle stimulus-transcription coupling in vivo. J Biol Chem. (2003) 278:32058–67. doi: 10.1074/jbc.m305545200
98. O'Connor DT, Zhu G, Rao F, Taupenot L, Fung MM, Das M, et al. Heritability and genome-wide linkage in US and Australian twins identify novel genomic regions controlling chromogranin A: implications for secretion and blood pressure. Circulation (2008) 118:247–57. doi: 10.1161/circulationaha.107.709105
99. Takiyyuddin MA, De Nicola L, Gabbai FB, Dinh TQ, Kennedy B, Ziegler MG, et al. Catecholamine secretory vesicles. Augmented chromogranins and amines in secondary hypertension. Hypertension (1993) 21:674–9. doi: 10.1161/01.hyp.21.5.674
100. O'Connor DT, Takiyyuddin M, Printz M, Dinh T, Barbosa J, Rozansky D, et al. Catecholamine storage vesicle protein expression in genetic hypertension. Blood Press (1999) 8:285–95. doi: 10.1080/080370599439508
101. Di Comite G, Previtali P, Rossi CM, Dell'Antonio G, Rovere-Querini P, Praderio L, et al. High blood levels of chromogranin A in giant cell arteritis identify patients refractory to corticosteroid treatment. Ann Rheum Dis. (2009) 68:293–5. doi: 10.1136/ard.2007.086587
102. Comite DG, Rossi CM, Marinosci A, Lolmede K, Baldissera E, Aiello P, et al. Circulating chromogranin A reveals extra-articular involvement in patients with rheumatoid arthritis and curbs TNF-alpha-elicited endothelial activation. J Leukoc Biol. (2009) 85:81–7. doi: 10.1189/jlb.0608358
103. Sidhu R, Drew K, McAlindon ME, Lobo AJ, Sanders DS. Elevated serum chromogranin a in irritable bowel syndrome (IBS) and inflammatory bowel disease (IBD): a shared model for pathogenesis? Inflamm Bowel Dis. (2010) 16:361–540. doi: 10.1002/ibd.20982
104. Wagner M, Stridsberg M, Peterson CGB, Sangfelt P, Lampinen M, Carlson M. Increased fecal levels of chromogranin A, chromogranin B, and secretoneurin in collagenous colitis. Inflammation (2013) 36:855–61. doi: 10.1007/s10753-013-9612-4
105. Eissa N, Hussein H, Hendy GN, Bernstein CN, Ghia J-E. Chromogranin-A and its derived peptides and their pharmacological effects during intestinal inflammation. Biochem Pharmacol. (2018) 152:315–26. doi: 10.1016/j.bcp.2018.04.009
106. Li Y, Zhou L, Li Y, Zhang J, Guo B, Meng G, et al. Identification of autoreactive CD8+ T cell responses targeting chromogranin A in humanized NOD mice and type 1 diabetes patients. Clin Immunol. (2015) 159:63–71. doi: 10.1016/j.clim.2015.04.017
107. Kogawa EM, Grisi DC, Falcão DP, Amorim IA, Rezende TMB, da Silva ICR, et al. Salivary function impairment in type 2 diabetes patients associated with concentration and genetic polymorphisms of chromogranin A. Clin Oral Investig. (2016) 20:2083–95. doi: 10.1007/s00784-015-1705-z
108. Kogawa EM, Grisi DC, Falcão DP, Amorim IA, Rezende TMB, da Silva ICR, et al. Impact of glycemic control on oral health status in type 2 diabetes individuals and its association with salivary and plasma levels of chromogranin A. Arch Oral Biol. (2016) 62:10–9. doi: 10.1016/j.archoralbio.2015.11.005
109. Moin ASM, Cory M, Choi J, Ong A, Dhawan S, Dry SM, et al. Increased chromogranin A–positive hormone-negative cells in chronic pancreatitis. J Clin Endocrinol Metab. (2018) 103:2126–35. doi: 10.1210/jc.2017-01562
110. Salihbegovic EM, Hadzigrahic N, Suljagic E, Kurtalic N, Sadic S, Zejcirovic A, et al. Psoriasis and high blood pressure. Med Arch. (2015) 69:13–5. doi: 10.5455/medarh.2015.69.13-15
111. Jager J, Aparicio-Vergara M, Aouadi M. Liver innate immune cells and insulin resistance: the multiple facets of Kupffer cells. J Intern Med. (2016) 280:209–20. doi: 10.1111/joim.12483
112. Hotamisligil G, Shargill N, Spiegelman B. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science (1993) 259:87–91. doi: 10.1126/science.7678183
113. Prommegger R, Ensinger C, Adlassnig C, Vaingankar S, Mahata SK, Marksteiner J, et al. Catestatin-A novel neuropeptide in carcinoid tumors of the appendix. Anticancer Res. (2004) 24:311–6. Available online at: http://ar.iiarjournals.org/content/24/1/311.long
114. Iqbal MA, Siddiqui FA, Gupta V, Chattopadhyay S, Gopinath P, Kumar B, et al. Insulin enhances metabolic capacities of cancer cells by dual regulation of glycolytic enzyme pyruvate kinase M2. Mol Cancer (2013) 12:1–12. doi: 10.1186/1476-4598-12-72
115. Gallagher EJ, LeRoith D. Minireview: IGF, insulin, and cancer. Endocrinology (2011) 152:2546–51. doi: 10.1210/en.2011-0231
116. Balkau B, Kahn HS, Courbon D, Eschwège E, Ducimetière P. Hyperinsulinemia predicts fatal liver cancer but is inversely associated with fatal cancer at some other sites: the Paris prospective study. Diabetes Care (2001) 24:843–9. doi: 10.2337/diacare.24.5.843
117. Ottesen AH, Carlson CR, Louch WE, Dahl MB, Sandbu RA, Johansen RF, et al. Glycosylated chromogranin A in heart failure: implications for processing and cardiomyocyte calcium homeostasis. Circ Hear Fail. (2017) 10:e003675. doi: 10.1161/CIRCHEARTFAILURE.116.003675
118. Deng Z, Xu C. Role of the neuroendocrine antimicrobial peptide catestatin in innate immunity and pain. Acta Biochim Biophys Sin (Shanghai) (2017) 49:967–72. doi: 10.1093/abbs/gmx083
119. Aung G, Niyonsaba F, Ushio H, Kajiwara N, Saito H, Ikeda S, et al. Catestatin, a neuroendocrine antimicrobial peptide, induces human mast cell migration, degranulation and production of cytokines and chemokines. Immunology (2011) 132:527–39. doi: 10.1111/j.1365-2567.2010.03395.x
120. Pei Z, Ma D, Ji L, Zhang J, Su J, Xue W, et al. Usefulness of catestatin to predict malignant arrhythmia in patients with acute myocardial infarction. Peptides (2014) 55:131–5. doi: 10.1016/j.peptides.2014.02.016
121. Rabbi MF, Eissa N, Munyaka PM, Kermarrec L, Elgazzar O, Khafipour E, et al. Reactivation of intestinal inflammation is suppressed by catestatin in a murine model of colitis via M1 macrophages and not the gut microbiota. Front Immunol. (2017) 8:985. doi: 10.3389/fimmu.2017.00985
122. Egger M, Beer AGE, Theurl M, Schgoer W, Hotter B, Tatarczyk T, et al. Monocyte migration: A novel effect and signaling pathways of catestatin. Eur J Pharmacol. (2008) 598:104–11. doi: 10.1016/j.ejphar.2008.09.016
123. Morris DL, Cho KW, Delproposto JL, Oatmen KE, Geletka LM, Martinez-Santibanez G, et al. Adipose tissue macrophages function as antigen-presenting cells and regulate adipose tissue CD4+ T cells in mice. Diabetes (2013) 62:2762–72. doi: 10.2337/db12-1404
124. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. (2009) 15:914–20. doi: 10.1038/nm.1964
125. Broedbaek K, Hilsted L. Chromogranin A as biomarker in diabetes. Biomark Med. (2016) 10:1181–9. doi: 10.2217/bmm-2016-0091
126. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. (2011) 11:98–107. doi: 10.1038/nri2925
Keywords: catestatin, immune modulation, macrophages, anti-inflammatory, inflammatory disease, chromogranin A
Citation: Muntjewerff EM, Dunkel G, Nicolasen MJT, Mahata SK and van den Bogaart G (2018) Catestatin as a Target for Treatment of Inflammatory Diseases. Front. Immunol. 9:2199. doi: 10.3389/fimmu.2018.02199
Received: 17 July 2018; Accepted: 05 September 2018;
Published: 04 October 2018.
Edited by:
Heiko Mühl, Goethe-Universität Frankfurt am Main, GermanyReviewed by:
Teresa Pasqua, Università della Calabria, ItalyBhalchandra Mirlekar, University of North Carolina at Chapel Hill, United States
Michal Amit Rahat, Technion–Israel Institute of Technology, Israel
Copyright © 2018 Muntjewerff, Dunkel, Nicolasen, Mahata and van den Bogaart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geert van den Bogaart, Zy52YW4uZGVuLmJvZ2FhcnRAcnVnLm5s
Sushil K. Mahata, c21haGF0YUB1Y3NkLmVkdQ==