
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Immunol., 28 September 2018
Sec. Primary Immunodeficiencies
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02130
 Rohan Ameratunga1,2*
Rohan Ameratunga1,2*Common Variable Immunodeficiency Disorders (CVID) are the most frequent symptomatic primary immune deficiency (PID) in adults. Current estimates suggest a prevalence of ~1:25,000, although a recent study has suggested an even greater frequency than previous estimates (1). The majority of CVID patients suffer recurrent infections because of late onset antibody failure (LOAF) leading to immune system failure (ISF). Current criteria do not allow CVID to be diagnosed before 4 years although some patients have symptoms dating back to infancy. Most patients experience recurrent or severe bacterial infections and less commonly autoimmunity as a result of CVID. Some patients present with a sarcoidosis-like disorder or enteritis (2, 3). A proportion of patients with CVID have a prominent T cell defect leading to severe viral or opportunistic infections. These patients have been deemed to have late onset combined immunodeficiency (LOCID) (4). LOCID is currently separated from CVID although I have argued LOCID should be included as a subset of CVID (5).
In spite of major progress in the last decade, the genetic basis of CVID is unknown in most patients. A causative mutation has been identified in up to 30% (6). If a causative defect is identified, these patients are removed from the umbrella diagnosis of CVID and are reclassified as having a CVID-like disorder caused by a specific mutation. To fulfill a diagnosis, all current CVID criteria require exclusion of other immunodeficiencies including NFKB1, NFKB1, CTLA4 etc. It is however likely earlier series of patients with CVID included many with CVID-like disorders, whose mutations were undiscovered.
We have recently discovered new genetic defects in two NZ families with CVID-like disorders. In the first family, we have confirmed the existence of quantitative epistasis in humans (7). Epistasis is the non-linear, synergistic interaction of two or more genetic loci either leading to a much more severe disorder or a novel phenotype (8, 9). The existence of epistasis was first predicted by William Bateson in 1909 but has remained highly controversial because of the lack of well characterized examples in humans (10). In this family, the synergistic interaction of TNFRSF13B/TACI and TCF3 mutations resulted in a severe immunodeficiency and systemic lupus erythematosus (SLE) in the proband (Figure 1). Other members of the family who have various permutations of the two mutated genes had a milder phenotype, which was reflected in their in vitro B cell differentiation and antibody production studies (7).
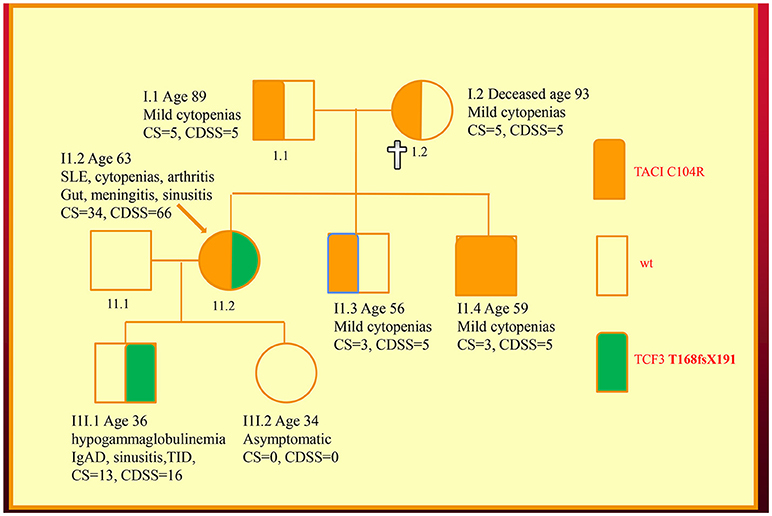
Figure 1. Family with a digenic CVID-like disorder caused by epistatic interactions of TNFRSF13B/TACI and TCF3 genes. The proband (arrow) suffers from both a severe immunodeficiency as well as SLE. Other members are as described in our previous publications including a mild symptomatic brother (II.3) with severe hypogammaglobulinemia caused by homozygous C104R mutations of the TNFRSF13B/TACI gene. CDSS, CVID disease severity score; CS, clinical score. The CS was suggested as means of determining eligibility for SCIG/IVIG but we have used it as a surrogate marker of disease severity (5, 7, 11).
We have also co-discovered NFKB1 mutations as a cause of a novel CVID-like disorder in the second family (Figure 2) (12). It was striking there was a very broad spectrum of phenotypes in this NZ family in spite of carrying the identical mutation. The recently deceased sister had a severe LOCID phenotype, while a 46-year-old asymptomatic brother carries the identical mutation (12). Other members of the family have widely varying phenotypes including recurrent infections or autoimmunity (5).
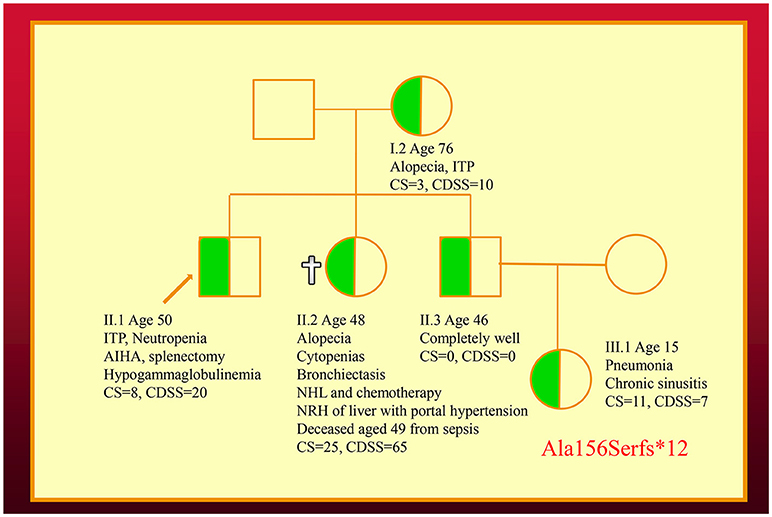
Figure 2. Family with an NFKB1 mutation. The proband (II.1) is shown with an arrow. Other family members are described in the text. Note that siblings of III.1 are not shown. CDSS, CVID disease severity score; CS, clinical score.
Here I present a disease severity score for CVID and CVID-like disorders (Table 1). There are potentially many clinical and theoretical advantages in developing a disease severity score in CVID and CVID-like disorders (Table 2). A very high score might suggest the presence of epistasis or LOCID. A diagnosis of LOCID will require confirmatory laboratory tests. A disease severity score may have clinical utility in allowing closer follow-up of severely affected patients in clinics and offering prognostic information. These potential advantages are discussed in more detail below.
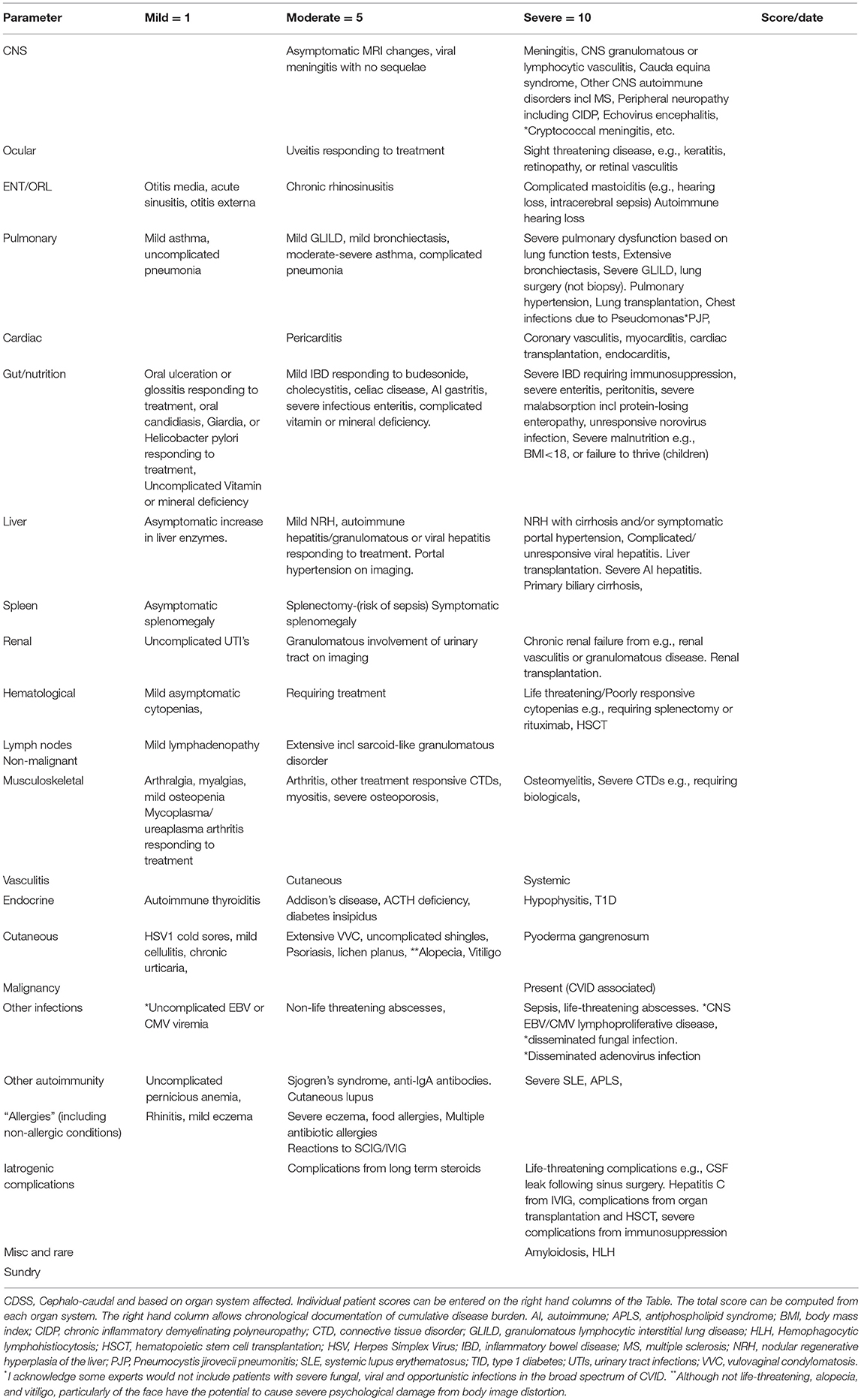
Table 1. An instrument for assessing CVID disease severity.
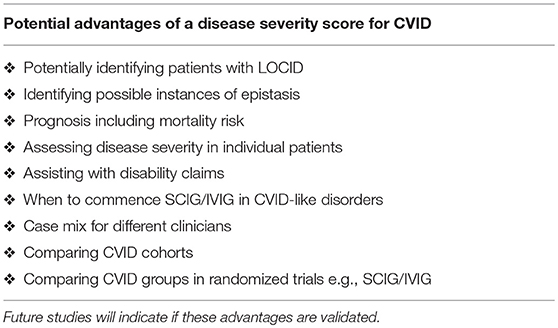
Table 2. Potential advantages of a disease severity score for CVID.
There has been a previous attempt at generating a disease severity score seven years ago (13). Here I develop these ideas further by focusing on target organ damage, which has been shown to determine prognosis. Because there is disagreement about the precise laboratory values which define CVID, (14) these have not been included in the CVID disease severity score (CDSS) in Table 1. Qualifying laboratory values or genetic mutations could be added if this disease severity instrument is to be used in a patient registry. To illustrate its utility, the CVID disease severity scores of the two families are presented here and compared with the clinical score.
The CDSS focuses primarily on cumulative organ damage as a result of infections, autoimmunity or inflammation. All of these are examples of ISF, as they indicate the presence of immunodeficiency or dysregulation of the immune system. Most of the sequelae (Table 1) quantified in the CDSS have been described in large cohorts of CVID patients, which as noted above, likely also included CVID-like patients (3, 15–19). It thus seems reasonable to apply this instrument to patients with CVID-like disorders also. To simplify the instrument, the severity of complications have been arbitrarily divided into three categories, mild, moderate, and severe. In general, mild manifestations can be easily treated and do not cause long-term morbidity. Moderate category conditions do cause short and long-term morbidity and may not be reversible. Conditions falling into the severe category are either life threatening or have the potential to cause severe disability such as visual loss or severe pulmonary dysfunction.
There is probably general agreement amongst experienced colleagues that some complications of CVID and CVID-like disorders are more severe than others (20). There is also support from previous studies indicating complications such as enteritis, malnutrition, cytopenias, and malignancy have a worse prognosis in patients with CVID (3, 18). Mortality is also increased in patients with functional or structural lung disease or hepatitis (16, 21). These have been placed in the severe column. In contrast, the worse prognosis with delayed diagnosis and early onset disease is likely to be reflected in higher organ damage scores in the CDSS. Early onset of disease may indicate a more aggressive disease trajectory while delay in diagnosis is likely to result in more severe organ damage from unmitigated infection and inflammation (3, 16).
As noted above, it is possible for patients to change categories if they deteriorate. A patient with mild bronchiectasis can progress to the severe category if the bronchiectasis becomes more extensive. Mild bronchiectasis is defined as causing few or no symptoms but is demonstrated on CT scans. I have not attempted to define the precise severity of bronchiectasis based on the number of lobes affected or extent of damage. If a patient has more than one complication in an organ system, each complication will receive a separate score e.g., a patient who has both severe bronchiectasis and severe interstitial lung disease will have a score of 20 (10+10). Similarly, if a patient suffers peritonitis, it is recorded only once in the GI complications list but not as an additional severe infection.
Recurrent uncomplicated pneumonias are not scored, as these are likely to be a reflection of underlying bronchiectasis, under treatment with SCIG/IVIG, chronic upper respiratory tract disease (22) or due another condition such as undiagnosed gastroesophageal reflux (20). In the absence of these predisposing factors, future studies will indicate if this group should be included in the moderate category. Similarly, recurrent acute otitis media and recurrent acute sinusitis are likely to reflect chronic rhinosinusitis.
Malignancy related to CVID was excluded from our diagnostic criteria for CVID, as it can be difficult to determine if malignancy is the cause or the result of CVID (23, 24). It has been included in this disease severity score, as its presence will clearly influence prognosis (25). Similarly, damage to some organ systems such as the CNS are unlikely to result in mild complications, most are likely to be life-threatening or have the potential to cause severe disability.
I have not included numbers of antibiotic courses, as this may vary with local practice. Some centers administer prophylactic antibiotics routinely. I have included Pseudomonas lung infections as this is often seen late in the course of CVID lung disease and is very difficult to treat. It is likely to be an important prognostic marker (22).
I have also included multiple antibiotic allergies as a moderate complication, as this will limit treatment options and therefore adversely affect prognosis (26). Patients are also at increased risk of morbidity if they have to be desensitized acutely to antibiotics. Total IgE is decreased in the majority of CVID patients (27). Asthma and rhinitis in the absence of bronchiectasis and chronic sinus disease however, appear to feature prominently in some series of CVID patients and may be an intrinsic part of the immune dysregulation of CVID.(28, 29) Most, (but not all) have negative skin tests or specific IgE to common aeroallergen (26).
Splenectomy appears to be surprisingly well tolerated in CVID patients and has been placed in the moderate category (30). The main prognostic issue is the risk of sepsis. Note that precise values for the sizes of an enlarged spleen and lymph nodes were suggested in a previous scoring system. I have avoided this, as current prognostic data does not appear to support such precise cut-offs at this time (13). Perhaps future cohorts may validate such precise approaches and the scoring system could be changed.
Treatment of CVID can lead to complications and this has been included in the disease severity score. In some cases treatment can lead to life-threatening sequelae such as CSF leaks following endoscopic sinus surgery or Hepatitis C from IVIG preparations. Iatrogenic complications have been kept separate, as it allows a clinician to discuss future therapy in the context of problems from previous treatment. Complications from solid organ transplantation and hematopoietic stem cell transplantation (HSCT) have been placed in this category. The CDSS may be useful in determining if there has been a decrease in disease severity following HSCT for CVID. Some complications may resolve while others may emerge after HSCT.
The CDSS has not included unrelated co-morbid conditions such as severe (atherosclerotic) coronary artery disease, which will clearly affect an individual's prognosis.
The recognized phenotypic spectrum of CVID (based on diagnostic criteria) and CVID-like disorders (based on the mutation) are expanding (8). The last row has been intentionally left blank for rare or new manifestations of CVID/CVID-like disorders. This will give some flexibility so new manifestations can be included in the future. It will also mean the maximum score for the CDSS may change over time. Any future studies using this instrument should define new criteria placed in the last row.
Disease severity instruments vary in their utility. Some instruments such as injury severity scores accurately predict mortality following trauma (31). These instruments are however less able to predict the severity of long-term disability in survivors of major trauma. Often, disease severity instruments evolve over time when new information becomes available from prospective studies. This is seen in SLE, where there have been multiple iterations of the original SLEDAI (SLE Disease Activity Index) and BILAG (British Isles Lupus Assessment Group) scoring systems (32).
The CDSS can be used either as an index of disease burden or an index of disease activity in CVID/ CVID-like disorders, where scores improve when complications such as ITP or AIHA are treated and are no longer active. I suggest however that the scores are added as an index of cumulative disease burden. This is more likely to reflect the prognostic trajectory of a patient with CVID. In rare CVID/ CVID-like patients with predominant autoimmune disease, it may be useful as a disease activity score, analogous to the SLEDAI.
There are thus competing goals when formulating disease severity scores. The table I have presented can be used clinically and can be updated on a regular basis, particularly if a patient's disorder is in evolution. Each complication can be dated in the right hand column to provide a comprehensive overview of an individual patient's progress over time.
The CDSS will be useful in busy clinics where the same patient is reviewed by different clinicians on separate visits. It may alert a new clinician to the likely severity of the CVID/ CVID-like disorder in an individual patient. It may help achieve consistency in how often patients are followed up in clinic. In general, patients with complex disorders or those rapidly deteriorating require close clinical supervision.
The CDSS will assist with determining the case mix of a clinician in a Clinical Immunology department. This will help with job sizing, as it will offer an objective assessment of the complexity of each clinician's workload. It will also serve as a check-list for junior colleagues in training. When a patient with CVID is seen for the first time, damage to each target organ should be carefully assessed. It may also be useful comparing disease burden in control vs. treatment arms in future randomized trials of new SCIG/IVIG products. It may help identify allocation bias when assessing outcomes of different treatments.
As I have shown here, a disease severity score is very useful in family studies. The CDSS correlates with the clinical phenotypes in both kindreds (Figures 1, 2). As can be seen in the first family, the digenic proband has the highest CDSS, consistent with the epistatic interactions of the TNFRSF13B/TACI and TCF3 mutations (7). Her son carrying only the TCF3 mutation has a higher score than other family members bearing either homozygous or heterozygous mutations of the TNFRSF13B/TACI gene. This indicates the TCF3 mutation has a much greater impact on disease severity compared to mutations of TNFRSF13B/TACI (9).
In this digenic family, the CDSS supports the separation of genes predisposing to CVID vs. those causing CVID-like disorders. In our diagnostic criteria, we have separated genes which cause CVID-like disorders (CTLA4, LRBA, NFKB1 etc.) from those which predispose to CVID (TNFRSF13B/TACI, BAFFR, TWEAK, MSH5) (23). As seen in the first family, there are substantial differences in the disease severity scores of family members bearing TNFRSF13B/TACI vs. TCF3 mutations, showing effective genotype-phenotype separation (9). If patients carrying genes predisposing to CVID have a high CDSS, this should prompt a search for a second causative mutation as seen in the first family.
In the second kindred, the proband's sister (II.2) with the LOCID sub-phenotype had the highest score compared with other family members (5). This reflects the severe damage to multiple organ systems caused by the T cell defect. A high CDSS should alert a clinician to the possibility of the LOCID sub-phenotype of CVID. A disease severity instrument can thus be useful in providing prognostic information (Table 2). Patients with multiple category 3 complications have a more severe disease burden and are likely to have an increased risk of mortality. Patients rapidly developing multiple complications may be candidates for alternative forms of treatment including HSCT (33, 34).
This instrument can thus be applied to patients with CVID-like disorders also, where a causative mutation is identified (8). Both CVID and CVID-like disorders appear to share analogous subphenotypes, such as the autoimmune variant or the infections only phenotype (5). In CVID-like disorders, the CDSS can be used to monitor asymptomatic family members carrying the same mutation. If they start developing symptoms, they may be candidates to begin SCIG/IVIG.
In the future, the CDSS could also be used to compare the severity of different mutations causing CVID-like disorders. Because of the effects of variable penetrance and expressivity, large cohorts of CVID-like patients will be needed to compare the CDSS of different mutations. Furthermore, age of onset and disease duration will need to be factored when comparing CDSS scores for different mutations. The precise location of the mutation might influence the phenotype. As seen in the brother (II.3) in the second family, it is becoming apparent that a proportion of patients carrying mutations of CVID-like disorders are asymptomatic (8). In contrast to patients bearing mutations of genes predisposing to CVID (TNFRSF13B/TACI, BAFFR, TWEAK, MSH5), the CDSS is likely to vary widely in patients bearing mutations of CVID-like genes.
Such a disease severity instrument can also be useful in supporting insurance and disability claims and also show the need for ongoing social support of these patients. Having a disease severity score may assist funders of SCIG/IVIG, as was originally proposed for the clinical score (11). Given the difficulties reliably assessing vaccine responses in CVID patients, (35, 36) clinical symptoms may be a more reliable marker of LOAF/ISF, as it reflects ISF (14).
This instrument may also be useful in dealing with patients with primary hypogammaglobulinemia who have not met indications for SCIG/IVIG replacement in a particular clinic. We have divided these patients into symptomatic (sHGUS) vs. asymptomatic (aHGUS) hypogammaglobulinemia of uncertain significance (23). In our practice symptoms are a major indication for SCIG/IVIG treatment, providing the patient also meets laboratory criteria for CVID (35).
For comparison, both the CDSS and the CS are shown in the family pedigrees (Figures 1, 2). In both of the families there appears to be close correlation between the CDSS and CS. The CDSS however covers many more affected organ systems than the CS, which is similar to a previously described list of 15 “unlucky complications” of CVID (13, 37). The CDSS may therefore be more sensitive than the CS or the list of 15 “unlucky complications.”
There are important caveats to any disease severity instrument (Table 3). There may be some intra-observer variability for example when scoring the severity of bronchiectasis. It is more likely there will inter-observer variability. Some clinicians may judge a complication as moderate, while others may assess it as severe. This may be less of an issue for an individual patient than when comparing patients. I also acknowledge there is heterogeneity within these complications: some patients with GLILD may respond to IVIG alone, while others may require more intense therapy. With better methods to quantify organ damage and response to treatment, a finer scale may provide reproducible data in the future.
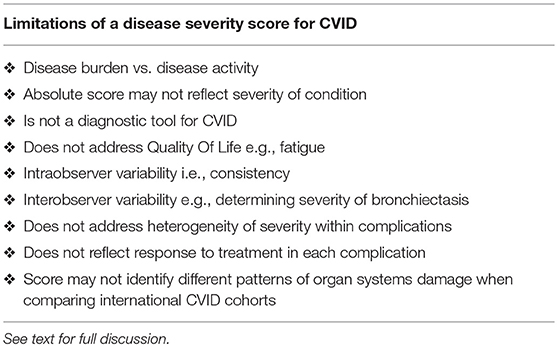
Table 3. Disadvantages of a disease severity score for CVID.
The absolute score may not necessarily reflect the severity of the condition in any given patient. Patients with chronic rhinosinusitis and treated ITP for example will have the identical score as a patient with end stage lung disease from bronchiectasis. Similarly, in the absence of sequelae, patients with otitis externa receive the same score as a patient with uncomplicated pneumonia. As noted above, patients with multiple severe complications (column 3) are however likely to have a higher disease burden with increased morbidity and mortality risk. This is seen in both the families presented here.
When comparing international cohorts, it may be more informative to compare scores for each complication. The absolute score may mask important differences including rates of bronchiectasis vs. autoimmunity etc. Such differences have been demonstrated in different CVID cohorts across Europe (3). It is important to note the CDSS cannot be used as diagnostic criteria for CVID. It is a disease severity score for patients with hypogammaglobulinemia, CVID and CVID-like disorders. Diagnostic criteria for CVID have been previously discussed and the reader is referred to these publications (14, 23, 38, 39).
This instrument does not address Quality of Life (QOL) in CVID. Fatigue is an important symptom affecting CVID patients but is not included in this instrument (40). Several studies have used established QOL surveys or have developed new instruments for patients with CVID and antibody deficiency (41–43). The disease severity score presented here could be complemented by the SF36 questionnaire or similar QOL instruments.
The strengths and weaknesses of this instrument will become apparent over time. It will be important to validate the CDSS with long-term prospective cohorts from around the globe. This will be helpful in assessing various aspects of validity and reliability of this instrument (32). I would be very pleased to receive feedback and suggestions for improving future iterations of this CVID disease severity instrument.
This manuscript complies with ethics standards. All studies have been undertaken with the consent of both families. These studies are approved by the NZ Ministry of Health Ethics committee and the ADHB ethics committee. There are no ethical impediments to publish this work.
The author confirms being the sole contributor of this work and has approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
I thank my patients for participating in our research studies for the benefit of others. I hope our discoveries and new concepts we have introduced will assist them and their families. I thank the A+ Trust, AMRF, ASCIA, and IDFNZ for grant support. Some opinions expressed in this article may not conform to current views of CVID or CVID-like disorders but I invite the reader to consider these arguments. I thank the reviewers for thorough review of this article and respectfully acknowledge differing opinions on some aspects of this article.
1. Selenius JS, Martelius T, Pikkarainen S, Siitonen S, Mattila E, Pietikainen R, et al. Unexpectedly high prevalence of common variable immunodeficiency in Finland. Front Immunol. (2017) 8:1190. doi: 10.3389/fimmu.2017.01190
2. Ameratunga R, Becroft DM, Hunter W. The simultaneous presentation of sarcoidosis and common variable immune deficiency. Pathology (2000) 32:280–2. doi: 10.1080/pat.32.4.280.282
3. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood (2008) 112:277–86. doi: 10.1182/blood-2007-11-124545
4. Malphettes M, Gerard L, Carmagnat M, Mouillot G, Vince N, Boutboul D, et al. Late-onset combined immune deficiency: a subset of common variable immunodeficiency with severe T cell defect. Clin Infect Dis. (2009) 49:1329–38. doi: 10.1086/606059
5. Ameratunga R, Ahn Y, Jordan A, Lehnert K, Brothers S, Woon ST. Keeping it in the family: the case for considering late onset combined immunodeficiency a subset of common variable immunodeficiency disorders. Exp Rev Clin Immunol. (2018) 14:549–56. doi: 10.1080/1744666X.2018.1481750
6. Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova JL, et al. Genetic Diagnosis Using Whole Exome Sequencing in Common Variable Immunodeficiency. Front Immunol. (2016) 7:220. doi: 10.3389/fimmu.2016.00220
7. Ameratunga R, Koopmans W, Woon ST, Leung E, Lehnert K, Slade CA, et al. Epistatic interactions between mutations of TACI (TNFRSF13B) and TCF3 result in a severe primary immunodeficiency disorder and systemic lupus erythematosus. Clin Trans Immunol. (2017) 6:e159. doi: 10.1038/cti.2017.41
8. Ameratunga R, Lehnert K, Woon ST, Gillis D, Bryant VL, Slade CA, et al. Review: diagnosing common variable immunodeficiency disorder in the era of genome sequencing. Clin Rev Allergy Immunol. (2018) 54:261–8. doi: 10.1007/s12016-017-8645-0
9. Ameratunga R, Woon ST, Bryant VL, Steele R, Slade C, Leung E, et al. Clinical implications of digenic inheritiance and epistasis in primary immunodeficiency disorders. Front Immunol. (2018) 8:1965. doi: 10.3389/fimmu.2017.01965
10. Bateson. Discussion on the influence of heredity on disease, with special reference to tuberculosis, cancer, and diseases of the nervous system: introductory address. Proc R Soc Med. (1909) 2:22–30.
11. Agarwal S, Cunningham-Rundles C. Treatment of hypogammaglobulinemia in adults: a scoring system to guide decisions on immunoglobulin replacement. J Allergy Clin Immunol. (2013) 131:1699–701. doi: 10.1016/j.jaci.2013.01.036
12. Fliegauf M, Bryant LV, Frede N, Slade C, Woon S-T, Lehnert K, et al. Haploinsufficiency of the NF-κB1 subunit p50 in common variable immunodeficiency. Am J Hum Genet. (2015) 97:389–403. doi: 10.1016/j.ajhg.2015.07.008
13. Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is Not CVID common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. (2011) 111:47–107. doi: 10.1016/B978-0-12-385991-4.00002-7
14. Ameratunga R, Brewerton M, Slade C, Jordan A, Gillis D, Steele R, et al. Comparison of diagnostic criteria for common variable immunodeficiency disorder. Front Immunol. (2014) 5:415. doi: 10.3389/fimmu.2014.00415
15. Gathmann B, Mahlaoui N, Gerard L, Oksenhendler E, Warnatz K, Schulze I, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. (2014) 134:116–26. doi: 10.1016/j.jaci.2013.12.1077
16. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood (2012) 119:1650–7. doi: 10.1182/blood-2011-09-377945
17. Oksenhendler E, Gerard L, Fieschi C, Malphettes M, Mouillot G, Jaussaud R, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. (2008) 46:1547–54. doi: 10.1086/587669
18. Cunningham-Rundles C. The many faces of common variable immunodeficiency. Hematology Am Soc Hematol Educ Program (2012) 2012:301–5. doi: 10.1182/asheducation-2012.1.301
19. Farmer JR, Ong MS, Barmettler S, Yonker LM, Fuleihan R, Sullivan KE, et al. Common variable immunodeficiency non-infectious disease endotypes redefined using unbiased network clustering in large electronic datasets. Front Immunol. (2017) 8:1740. doi: 10.3389/fimmu.2017.01740
20. Jolles S. The variable in common variable immunodeficiency: a disease of complex phenotypes. J Allergy Clin Immunol Pract. (2013) 1:545–56, quiz 57. doi: 10.1016/j.jaip.2013.09.015
21. Morimoto Y, Routes JM. Granulomatous disease in common variable immunodeficiency. Curr Allergy Asthma Rep. (2005) 5:370–5. doi: 10.1007/s11882-005-0008-x
22. Baumann U, Routes JM, Soler-Palacin P, Jolles S. The lung in primary immunodeficiencies: new concepts in infection and inflammation. Front Immunol. (2018) 9:1837. doi: 10.3389/fimmu.2018.01837
23. Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. (2013) 174:203–11. doi: 10.1111/cei.12178
24. Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for CVID. Exp Rev Clin Immunol. (2014) 10:183–6. doi: 10.1586/1744666X.2014.875274
25. Quinti I, Agostini C, Tabolli S, Brunetti G, Cinetto F, Pecoraro A, et al. Malignancies are the major cause of death in patients with adult onset common variable immunodeficiency. Blood (2012) 120:1953–4. doi: 10.1182/blood-2012-05-431064
26. Bjelac JA, Blanch MB, Fernandez J. Allergic disease in patients with common variable immunodeficiency at a tertiary care referral center. Ann Allergy Asthma Immunol. (2018) 120:90–2. doi: 10.1016/j.anai.2017.09.075
27. Lawrence MG, Palacios-Kibler TV, Workman LJ, Schuyler AJ, Steinke JW, Payne SC, et al. Low serum IgE is a sensitive and specific marker for common variable immunodeficiency (CVID). J Clin Immunol. (2018) 38:225–33. doi: 10.1007/s10875-018-0476-0
28. Urm SH, Yun HD, Fenta YA, Yoo KH, Abraham RS, Hagan J, et al. Asthma and risk of selective IgA deficiency or common variable immunodeficiency: a population-based case-control study. Mayo Clinic Proceed. (2013) 88:813–21. doi: 10.1016/j.mayocp.2013.05.021
29. Agondi RC, Barros MT, Rizzo LV, Kalil J, Giavina-Bianchi P. Allergic asthma in patients with common variable immunodeficiency. Allergy (2010) 65:510–5. doi: 10.1111/j.1398-9995.2009.02211.x
30. Wong GK, Goldacker S, Winterhalter C, Grimbacher B, Chapel H, Lucas M, et al. Outcomes of splenectomy in patients with common variable immunodeficiency (CVID): a survey of 45 patients. Clin Exp Immunol. (2013) 172:63–72. doi: 10.1111/cei.12039
31. Baker SP, O'Neill B, Haddon W Jr, Long WB. The injury severity score: a method for describing patients with multiple injuries and evaluating emergency care. J Trauma (1974) 14:187–96. doi: 10.1097/00005373-197403000-00001
32. Castrejon I, Tani C, Jolly M, Huang A, Mosca M. Indices to assess patients with systemic lupus erythematosus in clinical trials, long-term observational studies, and clinical care. Clin Exp Rheumatol. (2014) 32(5 Suppl. 85):S-85–95.
33. Tesi B, Priftakis P, Lindgren F, Chiang SC, Kartalis N, Lofstedt A, et al. Successful hematopoietic stem cell transplantation in a patient with LPS-responsive Beige-like anchor (LRBA) gene mutation. J Clin Immunol. (2016) 36:480–9. doi: 10.1007/s10875-016-0289-y
34. Slatter MA, Engelhardt KR, Burroughs LM, Arkwright PD, Nademi Z, Skoda-Smith S, et al. Hematopoietic stem cell transplantation for CTLA4 deficiency. J Allergy Clin Immunol. (2016) 138:615–9.e1. doi: 10.1016/j.jaci.2016.01.045
35. Ameratunga R, Storey P, Barker R, Jordan A, Koopmans W, Woon ST. Application of diagnostic and treatment criteria for common variable immunodeficiency disorder. Expert Rev Clin Immunol. (2015) 12:257–66. doi: 10.1586/1744666X.2016.1126509
36. Beck SC. Making sense of serotype-specific pneumococcal antibody measurements. Ann Clin Biochem. (2013) 50(Pt 6):517–9. doi: 10.1177/0004563213500241
37. Mokhtari M, Shakeri A, Mirminachi B, Abolhassani H, Yazdani R, Grimbacher B, et al. Important factors influencing severity of common variable immunodeficiency. Arch Iranian Med. (2016) 19:544–50.
38. Ameratunga R, Gillis D, Steele R. Diagnostic criteria for common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. (2016) 4:1017–8. doi: 10.1016/j.jaip.2016.02.023
39. Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International Consensus document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. (2016) 4:38–59. doi: 10.1016/j.jaip.2015.07.025
40. Hajjar J, Guffey D, Minard CG, Orange JS. Increased incidence of fatigue in patients with primary immunodeficiency disorders: prevalence and associations within the US immunodeficiency network registry. J Clin Immunol. (2017) 37:153–65. doi: 10.1007/s10875-016-0367-1
41. Rider NL, Kutac C, Hajjar J, Scalchunes C, Seeborg FO, Boyle M, et al. Health-related quality of life in adult patients with common variable immunodeficiency disorders and impact of treatment. J Clin Immunol. (2017) 37:461–75. doi: 10.1007/s10875-017-0404-8
42. Quinti I, Pulvirenti F, Giannantoni P, Hajjar J, Canter DL, Milito C, et al. Development and initial validation of a questionnaire to measure health-related quality of life of adults with common variable immune deficiency: the CVID_QoL questionnaire. J Allergy Clin Immunol Pract. (2016) 4:1169–79.e4. doi: 10.1016/j.jaip.2016.07.012
43. Ballow M, Conaway MR, Sriaroon P, Rachid RA, Seeborg FO, Duff CM, et al. Construction and validation of a novel disease-specific quality-of-life instrument for patients with primary antibody deficiency disease (PADQOL-16). J Allergy Clin Immunol. (2017) 139:2007–10.e8. doi: 10.1016/j.jaci.2016.11.029
Keywords: CVID, LOCID, CVID-like, CDSS, epistasis, digenic, PID
Citation: Ameratunga R (2018) Assessing Disease Severity in Common Variable Immunodeficiency Disorders (CVID) and CVID-Like Disorders. Front. Immunol. 9:2130. doi: 10.3389/fimmu.2018.02130
Received: 28 February 2018; Accepted: 29 August 2018;
Published: 28 September 2018.
Edited by:
Hans Dieter Ochs, University of Washington, United StatesReviewed by:
Klaus Warnatz, Albert-Ludwigs-Universität Freiburg, GermanyCopyright © 2018 Ameratunga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rohan Ameratunga, cm9oYW5hQGFkaGIuZ292dC5ueg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.