
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 27 September 2018
Sec. Autoimmune and Autoinflammatory Disorders
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02091
 Qiong Wang1,2,3†Xin Zhou2†Yongjian Zhao1,3,4Jun Xiao2Yao Lu2Qi Shi1,3,4Yongjun Wang1,3,4,5*
Qiong Wang1,2,3†Xin Zhou2†Yongjian Zhao1,3,4Jun Xiao2Yao Lu2Qi Shi1,3,4Yongjun Wang1,3,4,5* Hongyan Wang2*
Hongyan Wang2* Qianqian Liang1,3,4*
Qianqian Liang1,3,4*Background: Rheumatoid arthritis (RA) is a chronic autoimmune disorder, characterized by an increased number of M1-like macrophages in the joints. Polyphyllin I (PPI), one of the main components in the Rhizoma of Paris polyphyllin, displays a selective inhibitory effect on various tumor cells. Here we sought to investigate the anti-rheumatoid arthritis effects and mechanisms of PPI on macrophages in vivo and in vitro.
Materials and Methods: In vitro, primary bone marrow-derived macrophages (BMMs) and peritoneal elucidated macrophages (PEMs) were stimulated by lipopolysaccharide (LPS) and Interferon (IFN)-γ and then treated with PPI. We determined the degree of activation of IKKα/β and p65, two key mediators of the NF-κB-mediated inflammatory pathway, by measuring their phosphorylated forms by Western blot. The p65 nuclear localization was detected by immunofluorescent staining. Further, a NF-κB-linked luciferase reporter plasmid, as well as those expressing key mediators of the Toll-like receptor 4 pathway, such as myeloid differentiation primary response 88 (MYD88), interleukin-1 receptor (IL-1R) associated kinase (IRAK)-1, TNF receptor associated factors (TRAF)-6, Transforming growth factor-b–activated kinase 1 (TAK1) and p65, were used to identify the mechanism by which PPI achieves its inhibitory effects on macrophage-mediated inflammation. Moreover, a NF-κB inhibitor, p65-targeted siRNAs, and a p65 plasmid were further used to validate the anti-inflammatory mechanism of PPI. In vivo, PPI (1 mg/kg) was administered intragastrically one time a day for 7 weeks starting on the 42nd day after the first immunization with collagen in a collagen-induced arthritis (CIA) mouse model. Micro-computed Tomography scanning, histological examination, F4/80 and iNOS double immunofluorescent staining and CD4 immunohistochemical staining were performed to determine the effect of PPI treatment on joint structure and inflammation in this model.
Results: PPI reduced the inflammatory cytokines production of PEMs stimulated by LPS/IFN-γ, inhibited the phosphorylation of IKKα/β and p65, and prevented p65 nuclear localization. The NF-κB luciferase assay showed that the target of PPI was closely related to the NF-κB pathway. Moreover, NF-κB inhibition, siRNA-mediated knockdown of p65, and p65 overexpression eliminated PPI's inhibitory effect. In addition, PPI attenuated the bone erosion and synovitis, as well as M1-like macrophage and T cell infiltration, in the ankle joint of the CIA model.
Conclusion: PPI demonstrated effective amelioration of synovial inflammation in the ankle joint of CIA mice while suppressing NF-κB-mediated production of pro-inflammatory effectors in activated macrophages.
Rheumatoid arthritis (RA) is a chronic autoimmune disease, which is characterized by elevated inflammatory cells infiltration into the synovial joints, eventually resulting in cartilage and bone damage (1, 2). Synovial macrophages are strongly associated with RA severity and are one of the most plentiful cells in the synovial membrane of humans and animals with RA (3, 4). Indeed, disease severity worsens as the number of macrophage increases in the joints, while effective therapy is associated with a decreasing number of macrophages (5, 6). Indeed, it has been reported that macrophages depletion in the joints could reduce the severity of synovial inflammation in a collagen-induced arthritis (CIA) mouse model (7).
Evidence suggests that activated macrophages can critically drive the progression of RA by producing pro-inflammatory cytokines and chemokines, such as interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α), which results in the destruction of articular cartilage and sub-chondral bone (8–11). It was reported that the level of IFN-γ is positively correlated with the severity of the disease (12). And TNF-α over-expression in mice leads to RA-like symptoms, while treatment with anti-TNF-α antibodies, such as etanercept and infliximab, can effectively prevent disease progression and protect the joints in the CIA model (13). Collectively, activated macrophages are identified as a treatment target in RA (14).
NF-κB signaling is one of the key transcriptional pathways in RA. In macrophages, the activation of NF-κB in response to Toll-like receptor (TLR) ligands, such as LPS, results in the transactivation of a number of cytokines, chemokines, and other pro-inflammatory mediators (15, 16), and it was reported that TLR4 was activated in RA (16). In the canonical pathway, strong stimuli such as LPS, IFN-γ, and TNF-α can induce the phosphorylation of inhibitor of NF-κB (IκB) kinase (IKK), which mediates the serine phosphorylation and degradation of IκBα, and leads to the release of NF-κB. Liberated NF-κB translocates into the nucleus to regulate the transcription of target genes, including TNF-α, interleukin (IL)-1β, IL-6, and inducible nitric oxide synthase (iNOS) (15–17). Therefore, the blockade of the NF-κB signaling pathway is considered a vital strategy for controlling inflammatory responses in RA (17, 18).
Polyphyllin I (PPI, Figure 1A), a small molecular monomer extracted from Rhizoma of Paris polyphyllin, displays a selective inhibitory effect on various tumor cells, including osteosarcoma cells (19, 20), lung cancer cells (21), ovarian cancer cells (22, 23), breast cancer cells (24), and myeloma cells(25). The chemical name of PPI is diosgenyl a-L-rhamnopyranosyl-(1–2)-[b-L-ara-binofuranosyl-(1–4)-b-D-glucopyranoside] with a molecular weight of 855.02 Da, and the chemical formula is C44H70O16 (26). Previously, PPI was reported to down-regulate the constitutive phosphorylation of p65, an NF-κB subunit, in the hepatocellular carcinoma cell line HepG2 (27) and in osteosarcoma cells (20). Those studies demonstrate that PPI has the possibility to affect the NF-κB signaling pathway in several tumor cells. However, whether PPI has therapeutic effects in the CIA mouse model and the relevance to the innate immune system through NF-κB signaling has not been reported previously.
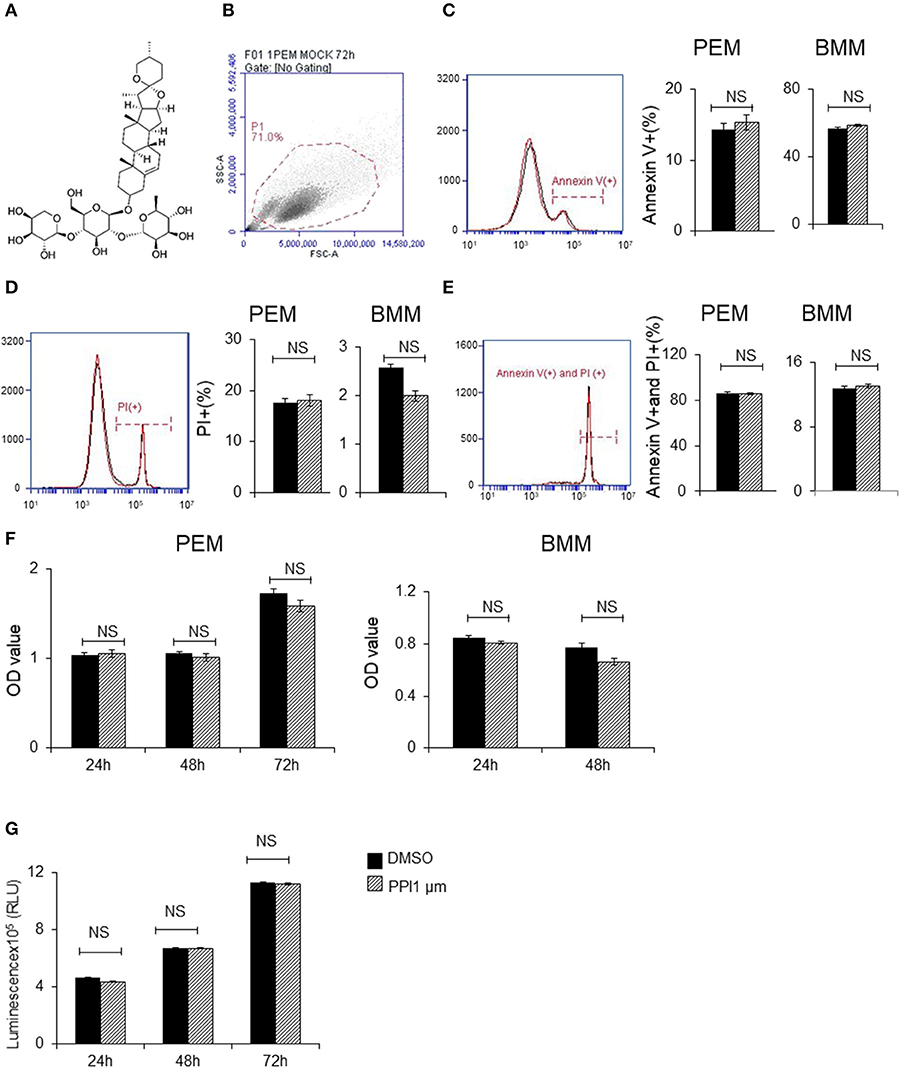
Figure 1. Polyphyllin I does not affect apoptosis or proliferation of macrophages. (A) Chemical structure of Polyphyllin I (PPI) (CAS: 50773-41-6). (B–E) Cell apoptosis of PEMs and BMMs treated with PPI (1 μM) or the same volume DMSO determined by FITC-conjugated anti-Annexin V antibody and PE-propidium (PI) followed by flow cytometry. Data represent mean ± SEM of three pooled experiments. NS, P > 0.05, Student's t-test. (F) Cell growth of PEMs and BMMs treated with PPI (1 μM) or the same volume DMSO determined by CKK8 assay. Data represent mean ± SEM of four pooled experiments. NS, P > 0.05, Student's t-test. (G) Cell viability of Raw 264.7 cells treated with PPI (1 μM) or the same volume DMSO determined by CellTiter-Glo. Data represent mean ± SEM of four pooled experiments. NS, P > 0.05, Student's t-test.
In this study, we sought to investigate the effect of PPI in the CIA mouse model in vivo and its mechanism of action in macrophages in vitro.
We purchased C57BL/6 and DBA/1J mice from Shanghai Slac Laboratory Animal Co. Ltd. We used 6–10 weeks old mice in all experiments. The study protocol was officially approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (approval numbers 1608-023). All animal procedures were strictly carried out in accordance with the approved guidelines.
We used a random number table to randomly divide female DBA/1J mice into three groups, including the Mock group, Model + sodium carboxyl methyl cellulose (CMC-Na, which acted as a carrier for PPI) group and Model + PPI group.
Among them, the Mock group consisted of normal mice without collagen injection, while the Model + PPI group and the Model + CMC-Na group represented collagen-induced arthritis (CIA) mice and a carrier control, respectively. Bovine type II collagen (2 mg/ml, Chondrex, cat. # 20021) was fully emulsified with an equal volume of complete Freund Adjuvant (4 mg/ml M. tuberculosis, Chondrex, cat. # 7001). CIA mice were made by subcutaneously injected the emulsion 100 μl at the base of the tail. Generally, the incidence of arthritis was > 90% at 42–56 days.
In the Model + PPI group, PPI (1 mg/kg body weight) was suspended in 0.5% CMC-Na (1 mg/ml) and intragastrically given to CIA mice from day(D) 42 to D91 post first immunization, once a day, while the Mock and Model + CMC-Na group were intragastrically administrated the same volume of CMC-Na as the PPI group. PPI was purchased from the ChemFaces Biochemical Co., Ltd. (cat. # CFN90255).
Clinical arthritis was evaluated by the following scale: grade 0, normal (no swelling); grade 1, mild, but definite redness and swelling of the ankle or wrist, or apparent redness and swelling limited to individual digits, regardless of the number of affected digits; grade 2, moderate redness and swelling of ankle or wrist; grade 3, severe redness and swelling of the entire paw including digits; grade 4, maximally inflamed limb with involvement of multiple joints (28, 29). Each limb was graded with a score of 0 to 4, with a maximum possible score of 16 for each mouse. Two independent examiners blinded to the treatment group assessed the clinical score once a week.
Primary macrophages, such as bone marrow-derived macrophages (BMMs) and peritoneal elucidated macrophages (PEMs), and two cell lines (Raw 264.7 and 293T) were used in our study. BMMs were obtained from the tibias and femur bones of 8–12 weeks old C57/BL6 mice. After red blood cell lysis, all the cells were seeded into 12-well plates (2 million cells per well) in macrophage inducing medium for 5 days with half medium change two times at the third and fourth day. The BMMs were prepared for use after suspending the cells into the complete 1640 medium overnight. The complete 1640 medium was 1640 medium (Invitrogen, Grand Island, CA, USA) plus 10% (vol/vol) fetal bovine serum (FBS) and penicillin and streptomycin (P/S; 100 U/ml), while the macrophage inducing medium is the complete 1640 mediums plus 20 ng/ml murine M-CSF (Peprotech, cat. # 315-02). PEMs were isolated by repeated washing of the abdominal cavity after injecting 3 ml of 3% BBLTM Thioglycollate Medium Brewer Modified medium (BD, REF, USA; cat. # 211716) into 8–12 weeks old C57/BL6 mice in 4 days. 293T and Raw 264.7 cells were obtained from ATCC (ATCC® Number is CRL-11268™ and TIB-71™ respectively). PEMs, 293T, and RAW264.7 were cultured by completed DMEM medium consisted of DMEM medium (Invitrogen, Grand Island, CA, USA), 10% (vol/vol) FBS and 100 U/ml P/S.
Two p65 siRNAs (AGAAGACAUUGAGGUGUAUTT (5′-3′), p65#1 and GAAGAAGAGUCCUUUCAAUTT (5′-3′), p65#2) and the negative control siRNA were transfected into PEMs by Lipofectamine® RNAiMAX Transfection Reagen (cat. # 13778075) strictly under the manufacturer's instructions. Sixty hours after transfection, PPI was added into the medium, 3 h later, LPS/IFN-γ was added into the medium.
Total RNA was prepared by using Trizol (Invitrogen) and the cDNAs were generated by PrimeScriptTM RT reagent Kit (cat. # RR047A) according to the manufacturer's instructions. The relative mRNA expression of IL-1β (mouse), IL-6 (mouse), TNF-α (mouse), and NOS2 (mouse), hCCL5 (human), hCXCL10 (human), CD40 (mouse) and CD86 (mouse) were measured by qPCR CFX96 machine (Bio-rad). HieffTM qPCR SYBR Green Master Mix was purchased from Shanghai Yeasen Biological Technology Co.Ltd. The β-actin acted as a normalization control for all of the mRNAs listed above. The primers for qRT-PCR were shown in Table 1.
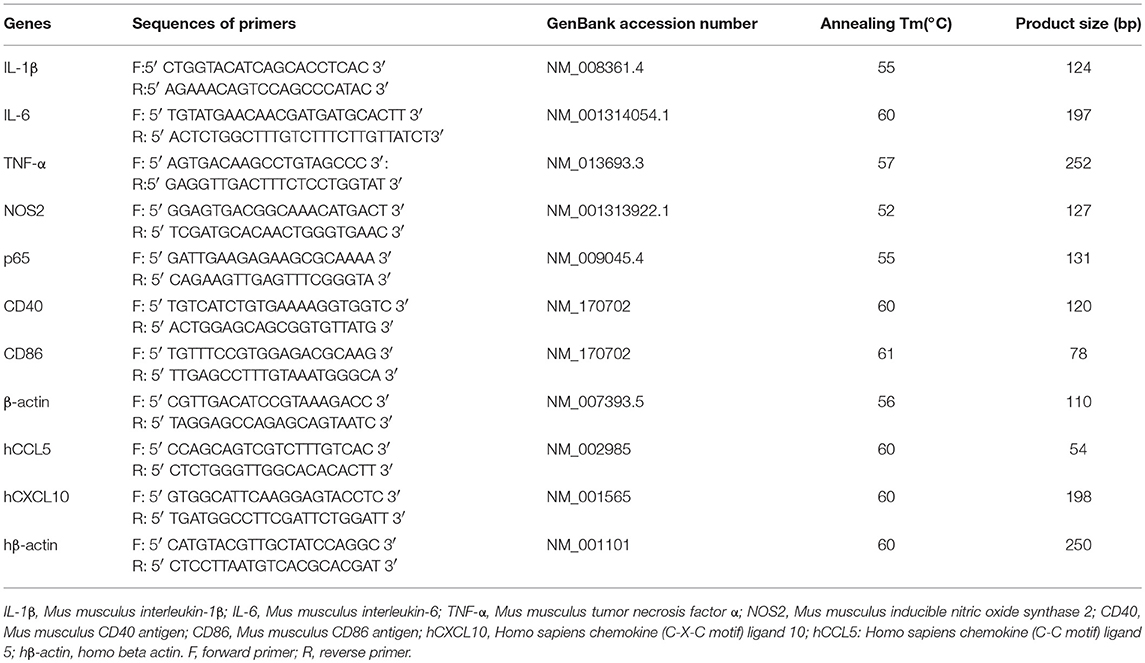
Table 1. Sequences of Primers Used in the Real-Time Polymerase Chain Reaction.
The ankle joints, kidney and liver were obtained when mice were sacrificed. We soaked the joints in 10% formalin for 48 h after removing the skin and muscle. Then all the left joints were decalcified by a 10% EDTA solution over 21 days. After a series of dehydration, waxing and embedding, the joints were cut into 5 micrometer (μm)-thick slices. The slices were stained by alcian blue/orange G (ABOG) or tartrate-resistant acid phosphatase (TRAP) for histologic analysis.
The samples of kidney and liver tissue were fixed in 10% formalin for 24 h, followed by a series of dehydration, waxing and embedding. And 5 μm sections were cut for hematoxylin and eosin (H&E) staining.
The slices from ankle joint samples were incubated with anti-F4/80 (Abcam, cat. # ab6640) and anti-iNOS (Abcam, cat. # ab15323) primary antibodies overnight at 4°C. The secondary antibody included Goat-anti-Rat IgG H&L (Alexa Fluor® 647; Abcam. cat. # ab150167) and Goat-Anti-Rabbit IgG H&L (Alexa Fluor® 488; Abcam. cat. # ab150077). And the nuclei were stained by DAPI (4',6-diamidino-2-phenylindole, sigma, cat. # D9564). The images were recorded by an Olympus BX-51 microscope.
For immunofluorescence, PEMs were seeded on round coverslip in 24-well plates. After stimulation at certain time points, cells were fixed by 4% paraformaldehyde for 10 min, permeabilized by 0.5% Triton-X, and blocked by 3% Bovine Serum Albumin (BSA) for 30 min at room temperature. The cells on the coverslip were incubated with anti-NF-κB p65 antibody (Abcam, cat. # ab16502) overnight at 4°C and goat anti-rabbit IgG Alexa Fluor® 488 (Abcam. cat. # ab150077) for 1 h at room temperature. The nuclei were stained by Bisbenzimide H 33342 Trihydrochloride (Hoechst; Sigma Aldrich, CAS: # 23491-52-3). Images were recorded by an Olympus BX-51 microscope.
Immunohistochemistry was performed in accordance with the kit's instructions (Zhongshan Jinqiao PV-9000 Universal Two-Step Test Kit) and the slices were incubated with anti-CD3 antibody (Abcam, cat. # ab56313) overnight at 4°C.
Serum aspartate aminotransferase (AST) was tested by AST reagent kit (Rate method), Serum alanine aminotransferase (ALT) was examined by ALT reagent kit (Rate method), Serum creatinine (CRE) was tested by CRE reagent kit (Enzymatic method) and blood urea nitrogen (UREA) was detected by UREA reagent kit (Enzymatic method). All the measurements were strictly performed according to the manufacturer's instructions (Shanghai Shensuo UNF medical diagnostic articles Co., LTD).
Cell lysate was prepared by sodium dodecyl sulfate (SDS)-loading buffer lysing and boiled for 10 min, then separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a Polyvinylidene fluoride (PVDF) membrane. After being blocked in tris-buffered saline and Tween 20 (TBST) with 5% BSA for 1 h at room temperature, the membrane was incubated with primary antibodies, such as Phospho-IKKα (Ser176)/IKKβ (Ser177) Antibody (cat. # 2688), Phospho-NF-κB p65 (Ser536) (93H1) Rabbit mAb (cat. # 3033), Phospho-SAPK/JNK (Thr183/Tyr185) (81E11) Rabbit mAb, (cat. # 4668), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP® Rabbit mAb (cat. # 4370), Phospho-p38 MAPK (Thr180/Tyr182) (D3F9) XP® Rabbit mAb (cat. # 4511), NF-κB p65 (D14E12) XP® Rabbit mAb (cat. # 8242) or HA-Tag (C29F4) Rabbit mAb (cat. # 3724) overnight at 4°C, and corresponding horseradish peroxidase-labeled secondary antibodies (cat. # 7074), while GAPDH (D16H11) XP® Rabbit mAb (cat. #5174) and β-Actin (8H10D10) Mouse mAb (cat. #3700) coupling horseradish peroxidase itself. All antibodies were purchased from Cell Signaling Technology. Detection was performed with ECL substrate (Thermo Scientific, West Palm Beach, FL, USA). The digital images were recorded by miniature chemiluminescence imaging machine (SAGECREATION, Beijing, China).
pNFκB-luc was purchased from Beyotime Biotechnology (cat. # D2206-1μg). The plasmid was constructed by inserting NF-κB nucleus DNA binding sites into pGL6 plasmid at the multiple cloning sites, which could be used to detect the activation level of NF-κB with high sensitivity. pRL-SV40-C (Beyotime Biotechnology, cat. # D2768-1μg) is a reporter plasmid which can be used to detect the Renilla luciferase reporter gene in mammalian cells.
After transfecting the NF-κB and Renilla luciferase reporters into 293T cells for 8 h by Lipofectamine 2000 reagent (Invitrogen, cat. # 11668-027) according to the manufacturer's instructions (30), we changed the medium with the complete DMEM medium containing different doses of PPI (0, 0.25, 0.5, and 1 μM), 3 h later, added human TNF-α (PeproTech, cat. # 300-01A) 20 ng/ml for another 33 h. The double luciferase was detected by the Dual-Luciferase® Reporter Assay System (Promega, cat. # E1910).
3 × HA-tagged human Myd88 (myeloid differentiation primary response 88, NM_001172567), TRAF6 (TNF receptor associated factor 6, NM_145803), IRAK1 (Interleukin 1 receptor associated kinase 1, NM_001569), TAK1 (TGF beta-activated kinase 1, AF218074.1) and p65 (RELA proto-oncogene, NF-kB subunit, NM_021975) was cloned into pcDNA™3.1(+) (Invitrogen, cat. # V79020) at the Multiple Cloning Site. Plasmids expressing Myd88, TRAF6, IRAK1, TAK1, p65, or vector were transfected into 293T cells together with pNFκB-luc and Renilla to measure the relative luciferase reading by PEI (1 μg/μl) (Polysciences, cat. # 23966-2). The double luciferase was also detected by the Dual-Luciferase® Reporter Assay System (Promega, cat. # E1910).
Pre-coating 48-well plate with anti-CD3 (Bio X cell, cat.# BE0001-1, 1 μg/ml) and anti-CD28 (Bio X cell, cat.# BE0015-5, 1 μg/ml) antibody dissolved into coating buffer (PBS + 10 mM NaHCO3, pH = 9.0) at 4°C overnight. Spleen cells from C57/BL6 mice were harvested and seeded as 2 million cells/well in a 48-well plate. Th1 cells (IFN-γ positive) were induced by co-cultured with IL-12 (10 ng/ml) and IL-2 (10 U/ml) for 48 h, while Treg cells (Foxp3-positive) were induced by co-culture with TGF-β (5 ng/ml) and IL-2 (10 U/ml) for 48 h. Then the population was tested by fluorescence activated cell sorting (FACS).
PEMs or BMMs were used for cell apoptosis analysis, while 0.8 × 106 cells were seeded into 12-well plate with 1 ml completed DMEM medium. After 12 h of incubation with 1μM PPI, the cells were harvested for staining. Anti-Annexin V (eBioscience, cat: # BMS306APC-20) for flow cytometry were purchased from eBioscience. Propidium iodide (PI) was acquired from Sigma (cat. # P4170). Staining processes were carried out according to the manufacturer's instructions. Briefly, fresh cells were stained with anti-Annexin V-APC antibody for 40 min at 4°C, after washing the cells were suspended with 200 μL FACS buffer (PBS+2%FBS+2‰ sodium azide), then incubated with 0.5 μL PI (100 μg/ml). After 70 μm filter, samples were processed by Accuri C6 (BD Biosciences, San Jose, CA, USA).
For the CD4 and Foxp3 FACS, the cells were stained with FITC Rat Anti-Mouse CD4 (BD Pharmingen™, cat. # 553047) for 40 min at 4°C and then fixed and permeabilized by eBioscience™ Foxp3/Transcription Factor Staining Buffer Set (Invitrogen™, cat. # 00-5523-00). After 40 min of CD4 antibody staining, the cells were fixed by 100 μl 1 × fix buffer for 40 min at 4°C, then washed by 1 × washing buffer, then incubated by Alexa Fluor® 647 Mouse anti-Human FoxP3 (BD Pharmingen™, cat. # 561184) for 40 min at 4°C. For IFN-γ FACS, the fix buffer was 2% paraformaldehyde and the washing buffer was FACS buffer.
The ELISA kit of IL-1β, IL-6 and TNF-α were from NeoBioScience and the NO test kit (Griess method) was from Beyotime Biotechnology. To measure IL-1β concentration, SL1344 was added in the supernatant for 15 min to produce mature IL-1β. All test were carried out strictly under the manufactures' instructions.
Right ankle joints were fixed in 10% formalin for 48 h, washed in phosphate-buffered saline (PBS) for 2 h and then soaked in 75% ethanol, scanned by micro-CT system (Scanco VIVA CT80, SCANCO Medical AG, Switzerland). The scanning parameters were as follows: pixel size 15.6 μm, tube voltage 55 kV, tube current 72 μA, integration time 200 ms. The cross-section images were then reconstructed and realigned in 3D, the bone volume (BV) of astragalus were measured and a density threshold was set from 370 to 1000 as Bone by μCT Evaluation program V6.6 (Scanco Medical AG, Switzerland). A stack of 340–441 cross-sections was reconstructed, with an inter slice distance of 1 pixel (15.6 μm), corresponding to a reconstructed height of 5.3–6.9 mm, recreating the ankle joints.
Statistical analysis was performed by Graphpad Prism (Version 6.0). Data represent as mean ± standard error of mean (SEM). Statistical significance is determined by unpaired two-tailed Student's t-test (or nonparametric test) with 95% confidence intervals for two groups comparison or one-way Analysis of variance (ANOVA) or nonparametric for 3 groups. As to 3 groups with different times points, we applied two-way ANOVA (or nonparametric) comparison. A P-value < 0.05 was considered statistically significant.
To avoid the toxic effects of PPI, we firstly examined the effect of PPI on PEMs and BMMs in vitro. We found PPI did not affect the early apoptosis or late apoptosis or the death of PEMs or BMMs after incubation for 72 h (Figures 1B–E). In addition, we examined the cell growth activity by CCK-8 kit, and found that PPI did not show any effect on PEMs or BMMs after incubation for 24, 48, or 72 h compared with the equivalent volume of DMSO (Figure 1F). We also investigated the effect of PPI on the cell growth of RAW264.7 cells for 24, 48, and 72 h by Cell Titer-Glo® Luminescent Cell Viability Assay, and obtained similar results that 1 μM PPI did not significantly affect the cell viability of RAW264.7 cells at any time point (Figure 1G).
It has been clearly demonstrated that IL-1β and TNF-α, IL-6 are crucial pro-inflammatory cytokines in response to LPS/IFN-γ, while iNOS is a classical pro-inflammatory M1 marker (31–35). BMMs were pretreated with PPI (0.5 and 1 μM) for 3 h, then treated with LPS (1 μg/mL)/IFN-γ (100 ng/ml) for another 6 h. We found that PPI decreased the mRNA expression of IL-1β, TNF-α, IL-6, and iNOS induced by LPS/IFN-γ activated macrophages in a concentration-dependent manner (Figures 2A–D). Furthermore, we also tested the concentration of IL-1β, TNF-α, and IL-6 in the supernatant of BMMs by ELISA after the pretreatment of PPI (0.25, 0.5, and 1 μM) for 3 h and LPS/IFN-γ treatment for another 6 h. We also investigated nitric oxide (NO) production in the conditioned medium of BMMs after 3 h of PPI pretreatment (0.25, 0.5, and 1μM) following LPS/IFN-γ treatment for 24 h. We found that PPI significantly decreased the production of IL-1β, TNF-α, IL-6, and NO by BMMs in a concentration-dependent manner (Figures 2E–H).
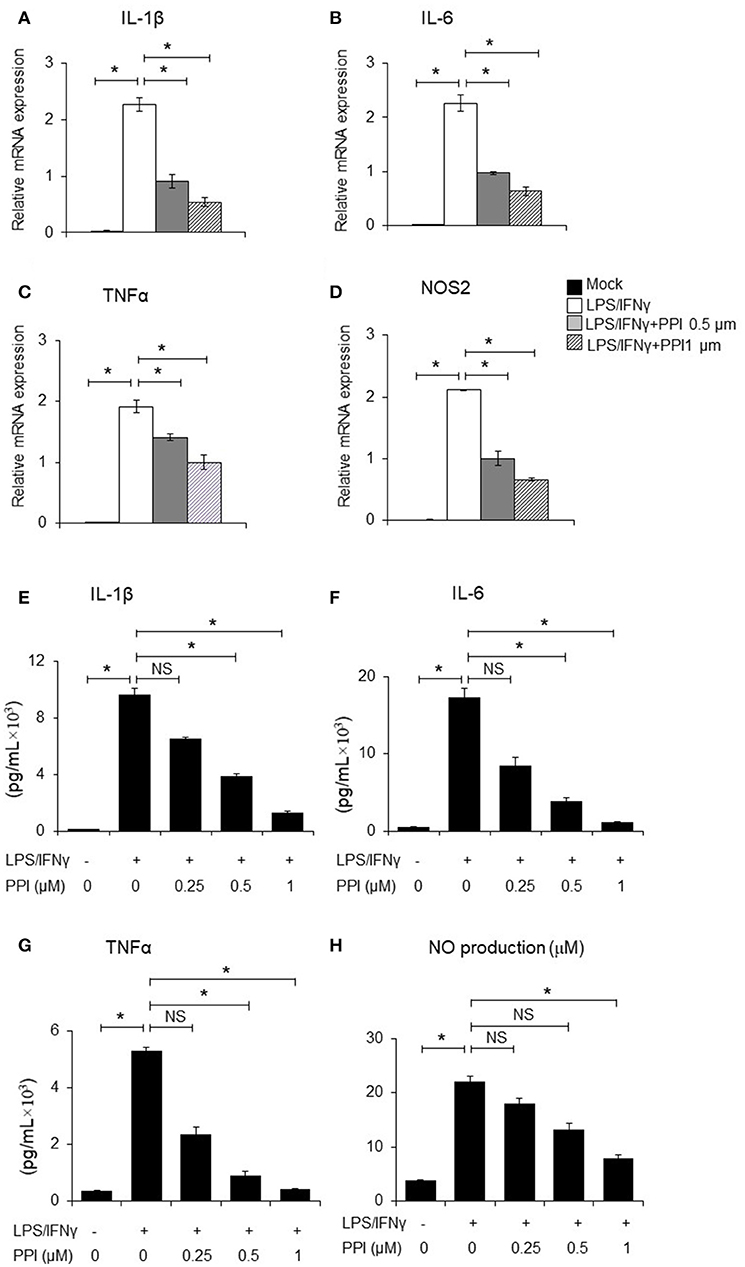
Figure 2. PPI ameliorated the inflammatory cytokines production of BMMs stimulated by LPS/IFN-γ. (A–D) IL-1β, IL-6, TNF-α, and NOS2 mRNA expression of BMMs stimulated with LPS (1 μg/ml) /IFN-γ (100 ng/ml) for 6 h in the presence of PPI (0, 0.5, or 1 μM) pretreated for 3 h. Representative data of at least 3 mice. Data represent mean ± SEM. NS, P > 0.05, *P < 0.05, One-way analysis of variance (ANOVA). (E–H) ELISA in the supernatants of BMMs stimulated with LPS/ IFN-γ for 6 h (IL-1β, IL-6, and TNF-α) and 24 h (NO) in the presence of PPI (0.25, 0.5, or 1 μM) pretreated for 3 h. Data represents mean ± SEM of four pooled experiments. NS, P > 0.05, *P < 0.05, One-way ANOVA.
Both NF-κB and MAPK (JNK 1/2, p38 MAPK, and ERK 1/2) are critical downstream mediators of TLR signaling and participate in regulating pro-inflammatory mediators and cytokines production (36–38). Thus, we examined whether PPI regulates the activation of MAPKs or NF-κB signaling by LPS/IFN-γ-stimulated macrophages. We found that the phosphorylation of IKKα/β, p65, JNK 1/2, p38 MAPK, and ERK 1/2 of BMMs were markedly increased after LPS/IFN-γ stimulation at 15 and 30 min. Treatment of PPI (1 μM) significantly inhibited LPS/IFN-γ-induced phosphorylation of IKKα/β and p65, without any effect on the phosphorylation of JNK 1/2, p38 MAPK and ERK 1/2 in BMMs (Figure 3A). In addition, an immunofluorescence assay revealed that PPI significantly reduced the level of p65 in the nucleus of PEMs induced by LPS/IFN-γ (Figure 3B).
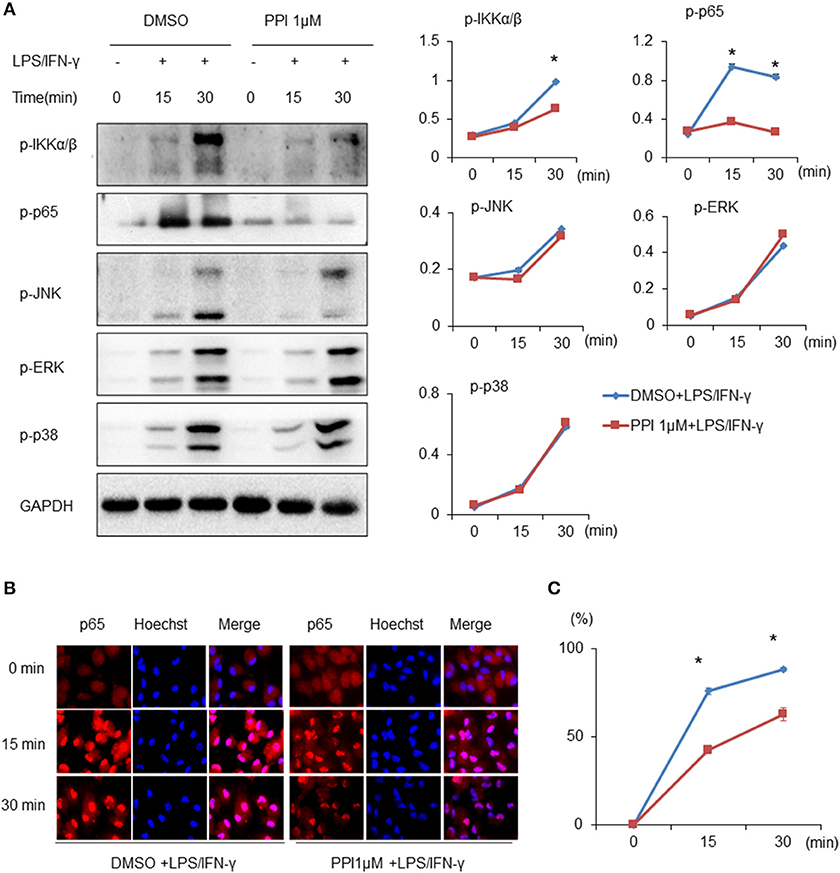
Figure 3. PPI ameliorated the phosphorylation of IKKα/β and p65, and p65 nuclear accumulation, without any effect on MAPK signaling under the stimulation of LPS/IFN-γ. (A) Immunoblots for phosphorylation of IKKα/β, p65, JNK, ERK, p38, and the protein level of GAPDH in cell lysates of BMMs stimulated with LPS (1 μg/ml)/IFN-γ (100 ng/ml) for 15, 30, and 60 min in the presence of PPI (1μM) or the same volume DMSO pretreated for 3 h. Blot is representative of three independent experiments. The normalized density of was shown on the right. Data represent mean ± SEM of three pooled experiments. *P < 0.05, two-way ANOVA. (B, C) PEMs were unstimulated or stimulated with LPS (1 μg/ml) /IFN-γ (100 ng/ml) for 15, 30, and 60 min in the presence or absence of PPI pretreatment for 3 h. The cells were stained with anti-p65 (in red) and Hoechst (in blue). Representative 100 × images. More than 300 cells from each sample were measured. The values are the mean ± SEM. The experiment was repeated at least three times. *P < 0.05, two-way ANOVA.
To further confirm the effect of PPI on NF-κB signaling, we used an NF-κB luciferase gene reporter assay. Briefly, after transfection with NF-κB luciferase and Renilla plasmids into 293T cells for 6 h, PPI (0, 0.25, 0.5, 1 μM) was added into the culture medium for 3 h, and then the human TNF-α (20ng/ml) was added for another 15 h. The NF-κB luciferase activity was detected by the double luciferase system. We found that PPI diminished TNF-α-stimulated NF-κB luciferase activation in a dose-dependent manner (Figure 4A). Since there are no TLRs on 293T cells, we used human TNF-α to mimic the activation of NF-κB in 293T cells.
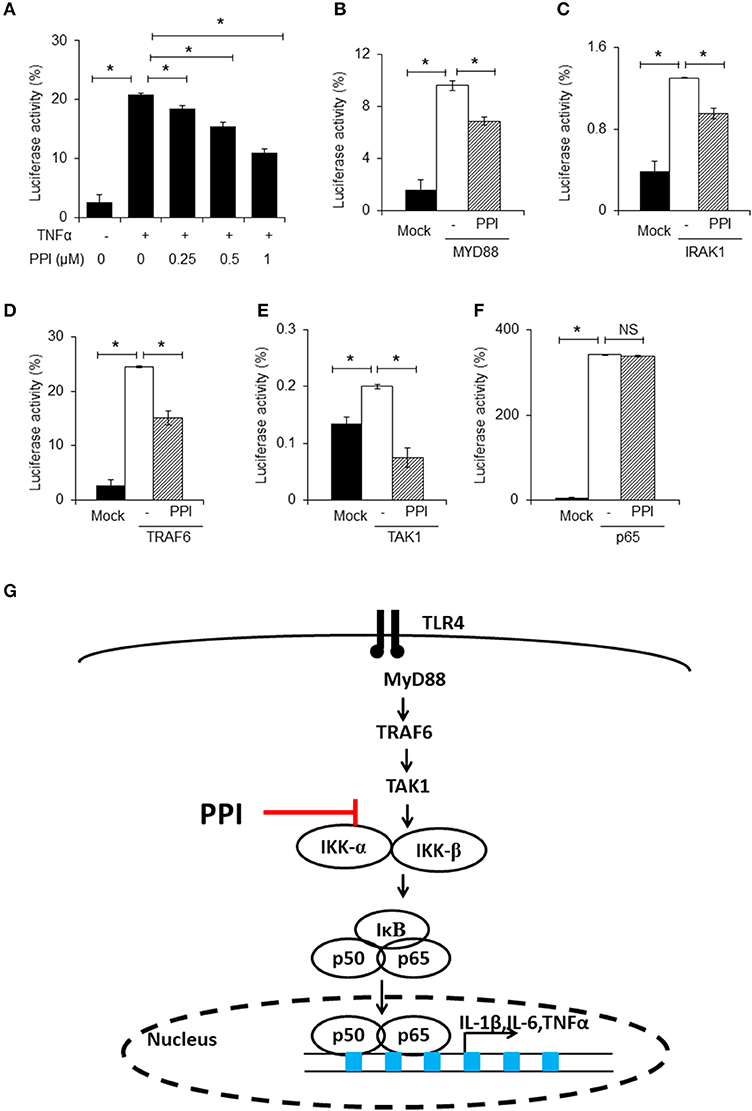
Figure 4. PPI inhibited NF-κB luciferase activity. (A) NF-κB and Renilla luciferase plasmid were co-transfected into 293T cells. 8 h later, the cells were incubated with different doses of PPI (0, 0.25, 0.5, 1 μM) for 3 h, then treated with human TNF-α (20 ng/ml) for another 15 h, and the NF-κB and Renilla luciferase was detected by using Dual Luciferase® Reporter Gene Aassy Kit. Data represent mean ± SEM of three pooled experiments. *P < 0.05, one-way ANOVA. (B–F) The 293T cells, after transfection with MYD88, IRAK1, TRAF6, TAK1 or vector plasmids together with the NF-κB luciferase and Renilla plasmid for 8 h, was cultured with PPI (1 μM) for another 24 h, and the double luciferase was detected. Data represent mean ± SEM of three pooled experiments. NS, P > 0.05, *P < 0.05, one-way ANOVA. (G) Diagram illustrating the inhibitory effect of PPI on the TLR4 pathway.
In the next step, we separately transfected MYD88, IRAK1, TRAF6, TAK1, or p65 expression plasmids into 293T cells, which were transfected with NF-κB luciferase and Renilla plasmids at the same time, to mimic the activated state of the NF-κB pathway. We found that PPI could inhibit NF-κB transcription when MYD88, IRAK1, TRAF6, and TAK1 were overexpressed, but could not block NF-κB activation when p65 was overexpressed (Figures 4B–F). The findings are summarized in Figure 4G.
To further investigate whether NF-κB signaling contributes to PPI-inhibited inflammatory cytokines expression, we firstly blocked NF-κB signaling by treating BMMs with the NF-κB inhibitor BAY117082 (Sigma, CAS:19542-67-7, 5 μM), followed by treatment with PPI. We then treated the cells with LPS/IFN-γ and measured the mRNA expression of IL-1β, IL-6 and iNOS. We found that NF-κB inhibition decreased LPS/IFN-γ-induced inflammatory cytokines expression, and PPI did not significantly inhibit LPS/IFN-γ-induced inflammatory cytokines expression in the presence of the NF-κB inhibitor (Figures 5A–C).
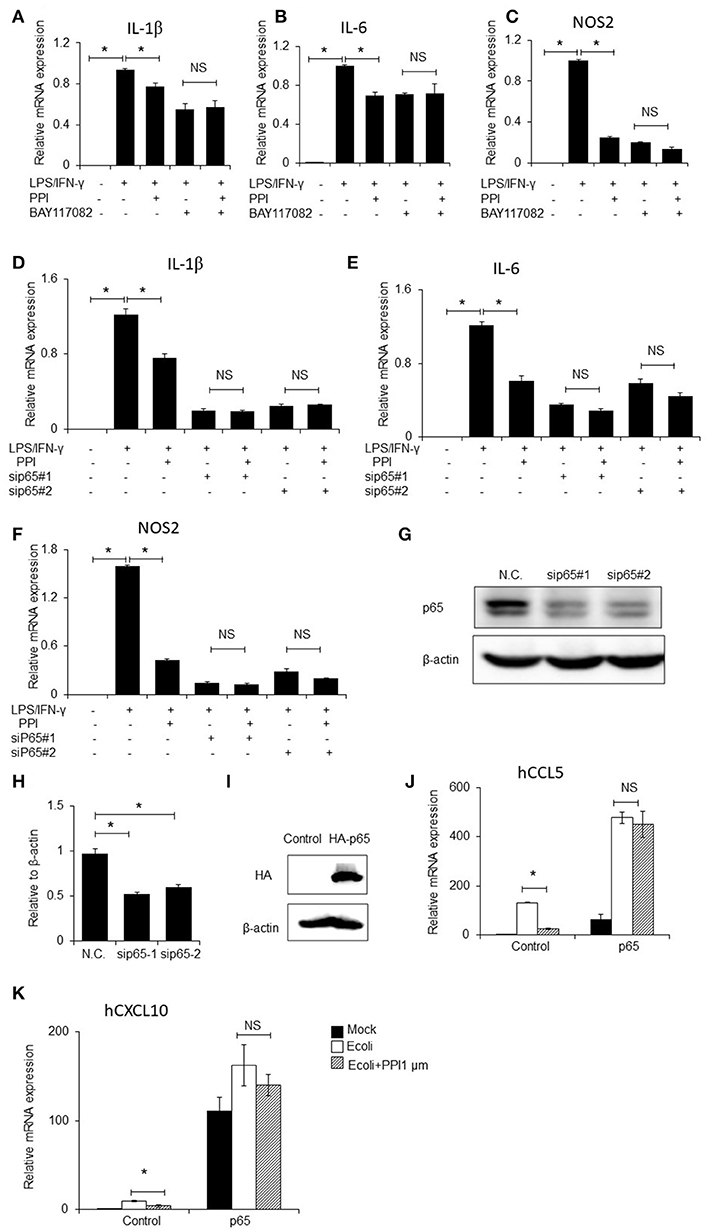
Figure 5. NF-κB inhibitor, p65 siRNAs and overexpression of p65 abolishes the inhibitory effect of PPI. (A–C) IL-1β, IL-6, and NOS2 mRNA expression of BMMs were treated with the NF-κB inhibitor BAY117082 (5μM) and PPI (1 μM) for 3 h and LPS/IFN-γ for another 6 h. Data represent mean ± SEM of three pooled experiments. NS, P > 0.05, *P < 0.05, one-way ANOVA. (D–F) IL-1β, IL-6, and NOS2 mRNA expression of PEMs transfected with siP65#1 and siP65#2 for 60 h, then with PPI (1 μM) or the same volume DMSO for 3h, LPS/IFN-γ for another 6 h. Data represent mean ± SEM of three pooled experiments. NS, P > 0.05, *P < 0.05, one-way ANOVA. (G) Immunoblots for p65 of PEMs transfected with siP65 for 60 h. Blot is representative of three independent experiments. (H) The density of p65 was normalized against β-actin. Data represent mean ± SEM of three pooled experiments, *P < 0.05, one-way ANOVA. (I–K) 293T cells were transfected with p65 plasmid for 36h, following PPI (1μM) for 3h and E.coli (5 × 107 CFU/ml) for another 6 h, immunoblots for p65, and CCL5, and CXCL10 mRNA expression were determined by qPCR. Blot is representative of three independent experiments. Data represent mean ± SEM of three pooled experiments. *P < 0.05, NS, P > 0.05, one-way ANOVA.
We next developed two p65 siRNAs, and transfected them into PEMs, followed by treatment with PPI and LPS/IFN-γ. qPCR results indicated that PPI significantly reduced LPS/IFN-γ-induced IL-1β, IL-6, and iNOS mRNA expression in the treated PEMs, but PPI could not significantly inhibit LPS/IFN-γ induced inflammatory cytokines expression when the cells were transfected with the two p65 siRNAs (Figures 5D–F). The ability of the two siRNAs to knock down p65 expression was confirmed by Western blot (Figures 5G, H). In addition, we transfected p65-3 × HA-pcDNA3.1 or the vector plasmid into 293T cells for 36 h, added PPI for another 3 h then stimulated 293T by E.coli (5 × 107 CFU/ml) for another 6 h, and found that PPI could not inhibit the expression of CCL5 or CXCL10 mRNA expression induced by E.coli when p65 was overexpressed (Figures 5I–K).
In order to determine whether PPI can affect other signaling pathways stimulated by LPS/IFN-γ, we examined the mRNA and protein expression of CD40 and CD86, which are triggered by LPS/IFN-γ but does not require NF-κB activation. We pretreated PEMs or BMMs with PPI (0.5 or 1 μM) for 3 h, then LPS/IFN-γ for another 6 h, and found the mRNA expression of CD40 and CD86 increased under the stimulation of LPS/IFN-γ, while the up-regulation was not significantly reduced at any concentration of PPI (Figures 6A, B). We also investigated the protein expression of CD86 by FACS after PPI pretreatment for 3 h and LPS/IFN-γ treatment for another 24 h, and found that PPI could not decrease CD86 protein level under the stimulation of LPS/IFN-γ (Figures 6C–G).
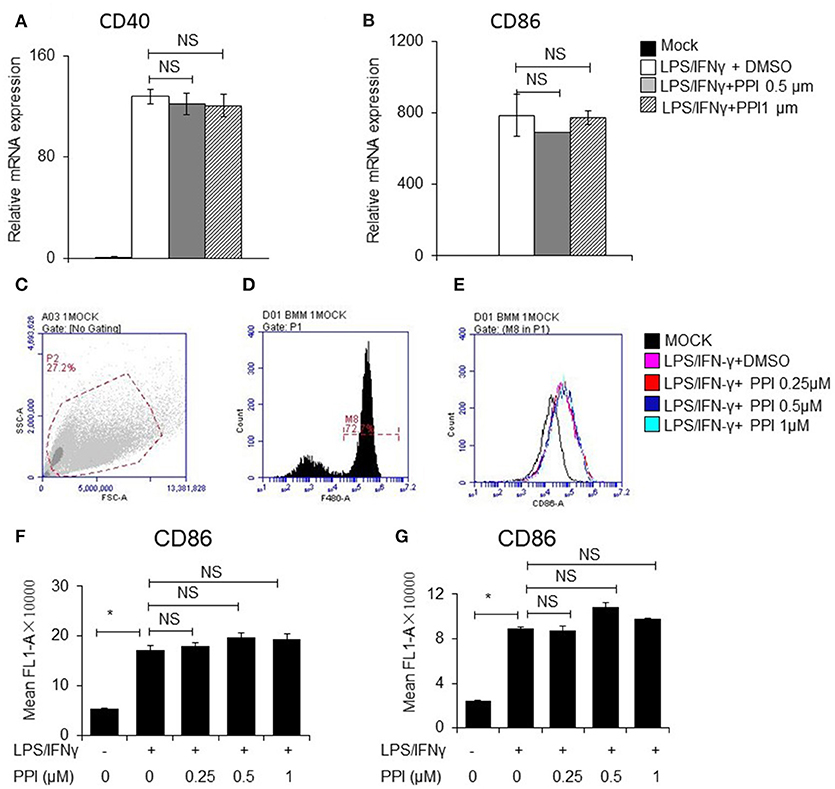
Figure 6. PPI did not affect CD40 and CD86 expression triggered by LPS/IFN-γ, which does not require NFκB activation. (A, B) CD40 and CD86 mRNA expression of BMMs was pretreated with PPI (0.25, 0.5, or 1 μM) or the same volume DMSO for 3 h, then stimulated with LPS (1 μg/ml) /IFN-γ (100 ng/ml) for another 6 h. Data represent mean ± SEM. NS, P > 0.05, one-way ANOVA. (C–G) BMMs or PEMs were pretreated with PPI (0.25, 0.5, or 1 μM) or the same volume DMSO for 3 h, then treated with LPS (1μg/ml)/IFN-γ (100ng/ml) for another 24 h, CD86 level were examined by flow cytometry. Data represent mean ± SEM. NS, P > 0.05, *P < 0.05, one-way ANOVA.
To determine the in vivo effect of PPI on RA, we treated CIA mice with PPI. Because PPI is insoluble in water, we utilized CMC-Na to suspend PPI, and used this vehicle alone as a negative control. We found that PPI treatment did not significantly cause weight loss in CIA mice (Figure 7A). Evaluation of clinically relevant endpoints showed that the severity score in the PPI-treated group was significantly less than the control group at day 77, when the score reached a peak in the control group (Model + CMC-Na; Figure 7B). Histological examination and histomorphometric analysis showed that ankle joints from the Model + CMC-Na group had severe inflammation and extensive cartilage and bone damage, and the inflammatory synovium area was significantly reduced in ankles of PPI-treated mice (Figures 7C–F). Three-dimensional micro-CT revealed that the bone volume of astragalus was significantly reduced in the Model + CMC-Na group, and the bone volume of talus in the Model +PPI group was significantly greater than the Model + CMC-Na group (Figures 7C, G). TRAP staining showed that the Model +PPI group had fewer osteoclasts around the astragalus in the ankle joints than the Model + CMC-Na group (Figures 7C, H).
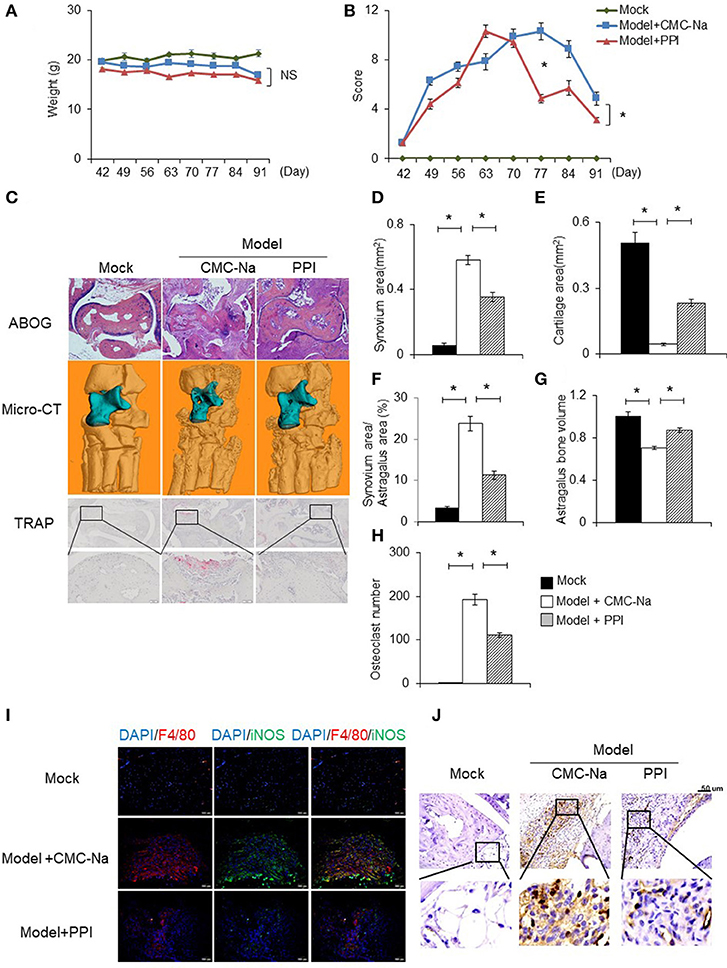
Figure 7. PPI attenuated collagen induced rheumatoid arthritis and protected bone erosion. CIA mice were treated intragastrically with CMC-Na with or without PPI (10 μg/kg) daily from day 42 after immunization to day 91. All mice were sacrificed on day 91. (A, B) The weight and arthritis swelling severity scores in CIA mice were recorded weekly from day 42 to day 91 after immunization. Data represent mean ± SEM of two pooled experiments, n = 4 for Mock, n = 7 for Model+CMC-Na and n = 7 for Model+PPI group. NS, P > 0.05, *P < 0.05, two-way ANOVA. (C) Representive images of ABOG-staining (4×), micro computed tomography three–dimensional and TRAP staining images (4×) of ankle joints. In micro computed tomography three–dimensional ankle joints, the blue part indicates astragalus, n = 4 for Mock, n = 7 for Model+CMC-Na and n = 6 for Model+PPI group. (D–H) Synovial area in the ankle joint, the cartilage area of the astragalus, the synovial area/astragalus area and astragalus bone volume, and osteoclasts number of the joint were calculated. Data represent mean ± SEM of two pooled experiments, n = 4 for Mock, n = 7 for Model+CMC-Na and 6 for Model+PPI group. *P < 0.05, one-way ANOVA. (I) Double immunofluorescence staining with anti-F4/80 (red) and anti-iNOS (green) antibodies at ankle sections show that decreased activated macrophages (double positive of F4/80 and iNOS) in the synovium of PPI-treated ankle joints, representative images (20×) from the Mock group (n = 4), the Model+CMC-Na (n = 7) and the Model+PPI group (n = 6). (J) Representative images of immunohistochemical staining with anti-CD3 antibody in the synovium around astragalus, bar indicates 50 μm, n = 4 for Mock, n = 7 for Model+CMC-Na and n = 6 for Model+PPI group.
To confirm the in vivo anti-inflammatory effect of PPI in CIA mice was through inhibition of the inflammatory response of macrophages, double immunofluorescence (IF) staining on ankle tissue sections was performed with labeled anti-bodies against the M1-specific antigen F4/80 and iNOS (39–44). Both positive staining of F4/80 and iNOS indicate an inflammatory response was activated at macrophages. The results demonstrated that abundant double-positive macrophages existed in the ankle joint of CIA mice, while PPI significantly reduced their presence (Figure 7I).
These data suggested that PPI ameliorated infiltration of inflammatory macrophages into the ankle joint of CIA mice. In order to determine whether the beneficial effect of PPI in the CIA model also comes from its action on T cells, we did immunohistochemical staining with an anti-CD3 antibody and found that CD3-positive cells were potently decreased in the PPI group compared with the Model + CMC-Na group (Figure 7J). To further confirm the effect of PPI on T cell differentiation in vitro, we treated T cells with PPI (0.5 and 1 μM) and did not find any effect on Th1 or Tregs differentiation (Figures 8A–F).
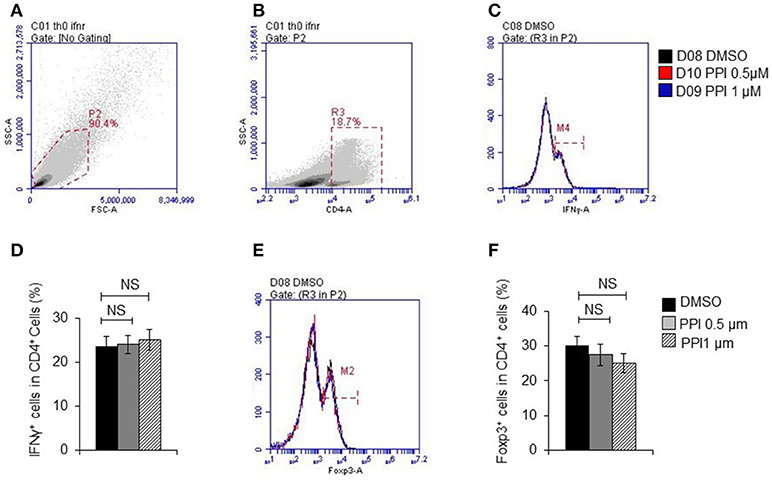
Figure 8. PPI did not show effect on T cell differentiation in vitro. (A) Representative images of initial gate of whole spleen cells population. (B) Representative images of CD4 positive cells. (C) IFNγ positive cells in CD4 positive cells. (D) Percentage of IFNγ positive cells in CD4 positive cells from 4 mice, (E) Foxp3 positive cells in CD4 positive cells. (F) Percentage of Foxp3 positive cells in CD4 positive cells from 4 mice. Data represent mean ± SEM. NS, P > 0.05, one-way ANOVA.
The liver and kidney tissue, as well as peripheral blood, of Mock, Model + CMC-Na and Model +PPI group mice were obtained to examine hepatocyte/glomerulus morphological characteristics and injury in CIA mice in vivo. The hematoxylin and eosin (H&E) staining of the liver samples showed that the PPI group had normal hepatic lobules and hepatocytes compared to the Mock group, while the serum ALT and AST test indicated that hepatocytes in the PPI group did not show evidence of injury to that organ (Figures 9 A–D). The H&E staining of the kidney showed that the glomerulus were normal in the PPI group, and the UREA and CRE test showed that the PPI group had a normal filtration function compared with the Mock group (Figures 9A,E,F).
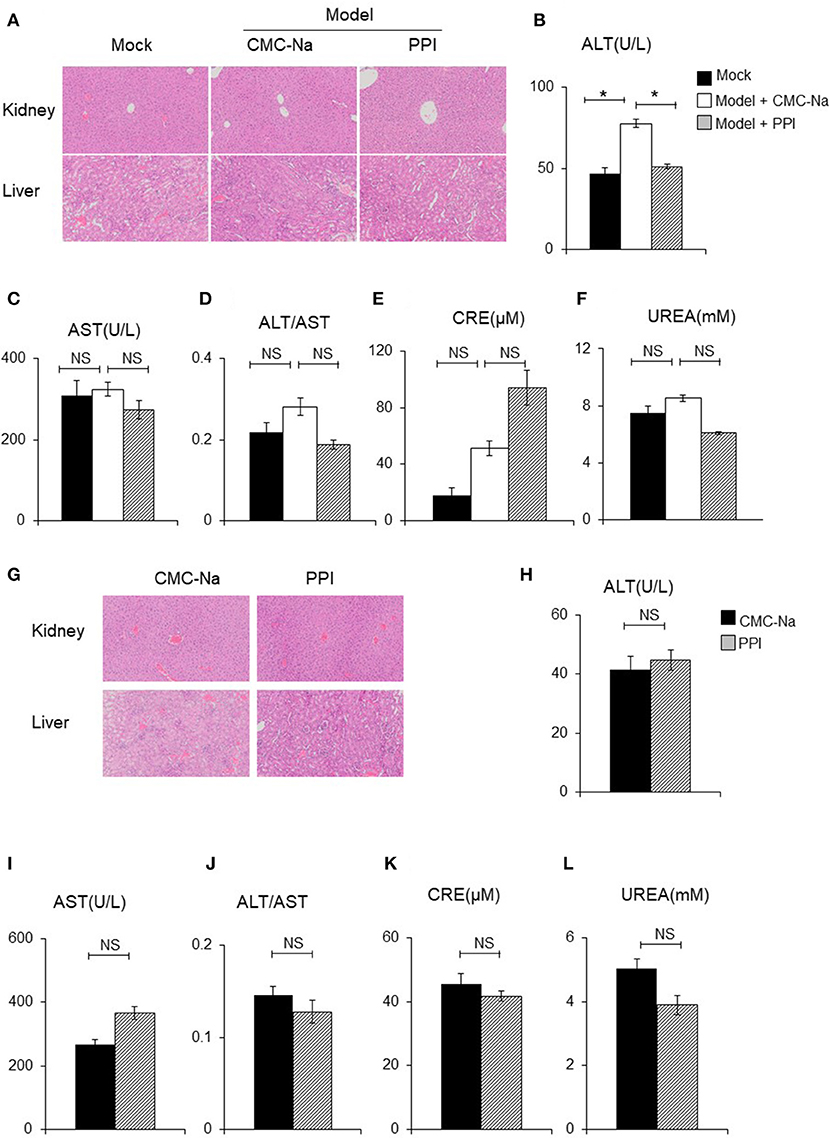
Figure 9. PPI did not show any damage effect on liver and kidney of CIA and normal mice. (A) The hematoxylin-eosin (H&E) staining of kidney and liver of CIA mice treated by CMC-Na with or without PPI (4×). (B, C) Serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT). (D) The ratio of ALT/AST. (E, F) Serum creatinine (CRE) and blood urea nitrogen (UREA) were detected by CRE reagent kit or UREA reagent kit. (B–F) Data represent mean ± SEM of two pooled experiments, n = 4 for Mock, n = 7 for Model+CMC-Na and n = 6 for Model+PPI group. NS, P > 0.05, one-way ANOVA. (G) H&E staining of the kidney and liver of normal mice treated with CMC-Na with or without PPI for 7 weeks (4×). (H–L) Serum level of AST, ALT, ALT/AST, UREA, and CRE of normal mice treated by CMC-Na with or without PPI. Data represent mean ± SEM of one pooled experiments. n = 5 for the CMC-Na and n = 5 for the PPI group, NS, P > 0.05, *P < 0.05, Student's t-test.
Next we tested whether PPI could cause hepatocyte injury or reduce glomerular filtration in the healthy mice. Ten C57/BL6 female mice were randomly divided into a normal group and PPI-treatment group by a random number table. For the PPI treatment group, PPI was intragastrically infused at a dose of 1 mg/kg once a day for 7 weeks, while the control group was administered with the carrier (CMC-Na) in the same volume. All the mice were sacrificed to harvest serum, liver, and kidney samples. Similar to the observation of CIA mice, PPI did not show any evidence of hepatocyte or glomerular injury as determined by H&E staining, as well as ALT, AST, UREA, and CRE tests (Figures 9G–L).
In the current study, PPI was found to attenuate NF-κB signaling in activated macrophages in vitro and joint inflammation of CIA mice in vivo.
RA is a chronic autoimmune inflammatory disease characterized by massive infiltration of diverse immune cells resulting in cartilage and bone destruction (45). In particular, numerous activated macrophages accumulate in the synovial inflammatory infiltrate and pannus cartilage interface, possess extensive pro-inflammatory, destructive, and remodeling capacities and are actively involved in the chronic inflammation and joint destruction (14, 46, 47). There are two types of macrophages, the classical activated (M1-like) and the alternatively activated (M2-like). The M1-like macrophage stimulated by LPS/IFN-γ or TNF-α/IFN-γ are featured by iNOS production, whereas the M2-like macrophage stimulated by IL-4/13 is marked by arginase expression (48). Generally, M1-like polarized macrophages are involved in resistance to pathogens while M2-like polarized macrophages help with tissue repair as well as parasite resistance, tumor growth and invasion (49). Previous studies found that in active RA, high concentrations of M1-like polarized macrophages localize in the synovium (50).
LPS is an outer membrane polysaccharide of Gram-negative bacteria, which can trigger the immune response via binding TLR4 and initiates a signaling cascade to activate the NF-κB and MAPK signaling pathways in macrophages and stimulate the production of inflammatory cytokines, chemokines and mediators (51, 52). LPS has been applied widely to accentuate or reactivate arthritis in different animal models (53–56). The effects of drug therapy on LPS-induced production of inflammatory cytokines and activated signaling pathways in macrophages are considered important for the exploration and evaluation of RA therapy (52). As it has been reported that IFN-γ is positively correlated with the severity of RA, we used LPS and IFN-γ in our study to stimulate primary bone marrow-derived macrophages and peritoneal elucidated macrophages to mimic macrophage activation in RA, and we found that PPI significantly suppressed inflammatory cytokines and mediator expression in this system, suggesting an anti-inflammatory potential of PPI.
Previously, PPI was reported to decrease the phosphorylation of the NF-κB subunit p65 and its downstream target gene expression in the hepatocellular carcinoma cell line HepG2 (27) and human osteosarcoma cells (20), and reduce protein expression of p65 in human lung cancer cells (57). And as many previous studies suggest that the blockage of NF-κB signaling pathways is a crucial strategy to control inflammatory responses of RA (12, 15–17, 58), this led us to hypothesize that PPI might have anti-inflammatory effects in RA and, if so, by inhibiting NF-κB signaling in the macrophage. Here, we found that pretreatment with PPI suppressed LPS/IFN-γ-induced phosphorylation of p-IKKα/β and p-p65, and p65 nuclear accumulation, while also inhibiting TNF-α-induced NF-κB activation. These results suggest that PPI has an inhibitory effect on NF-κB signaling in macrophages. In order to validate that the anti-inflammatory effect of PPI on LPS/IFN-γ-activated macrophages is indeed through NF-κB signaling, we used a pharmacological NF-κB signaling inhibitor, two p65 siRNAs and one p65 overexpression plasmid. To exclude the effect of PPI on other signaling pathways under LPS/IFN-γ regulation, we examined CD40 and CD86 mRNA and protein expression after LPS/IFN-γ stimulation and PPI co-treatment, and found that PPI did not decrease the production of these two markers.
On the other hand, PPI was reported to activate c-Jun expression and the JNK signaling pathway in the ovarian cancer cell line HO-8910PM (22), glioma cells (59), and human lung cancer cells (57), but we found that PPI had no effect on JNK phosphorylation. It is well known that besides NF-κB, ERK, and p38-MAPK are also important downstream mediators of LPS-TLR4 signaling and pro-inflammatory cytokines production in activated macrophage (31–35). Here, we find that PPI has no notable effect on the phosphorylation of ERK and p38-MAPK.
TLRs play a critical role in the generation of both innate and adaptive immunity and can drive inflammation. In response to LPS, the innate immune system is activated through TLRs on macrophages, resulting in the initiation and persistence of inflammation in RA (53, 60–62). After stimulation and dimerization, the TLR signaling pathways except TLR3, recruit the adaptor molecule MyD88, which recruits downstream signaling mediators, such as IRAK-1, IRAK4, and TRAF6 to form receptor complexes (63, 64). Such complexes further activate the phosphorylation of TAK1 and NF-κB, and finally the transcription of inflammatory cytokines and mediators (65). In the present study, in order to further define the mechanism of PPI action, MYD88, IRAK1, TRAF6, TAK1, and p65 on NF-κB signaling pathway was overexpressed to mimic the activation of TLR4 since the 293T cells lack of TLR4. We found that PPI inhibited NF-κB transcriptional activation when those factors above were transfected except for p65, suggesting PPI plays a role in an anti-NF-κB effect at the downstream of TAK1.
It was reported that daily intravenous injection of PPI (2.73 mg/kg body weight) for 10 days in nude mice bearing MCF-7 cells effectively decreased tumor growth without significant toxicity of the heart and liver (66). In our study, intragastric administration of PPI (1 mg/kg body weight) once a day for 7 weeks in a CIA model in DBA/1 mice attenuated joint score, synovial inflammation, bone erosion and the degree of iNOS-high macrophages in the ankle joints, without any signs of hepatocyte or glomerular injury. These results indicate that PPI effectively reduced the symptoms of synovial inflammation in the CIA mouse model without any signs of liver or kidney injury, while also suggesting PPI as a potential new drug therapy for RA.
In summary, we demonstrate here for the first time, to the best of our knowledge, that PPI effectively ameliorates synovial inflammation in the ankle joint of CIA mice and suppresses pro-inflammatory mediator production of activated macrophage without any signs of liver or kidney injury in vivo. We also found that this compound can inhibit NF-κB signaling-mediated production of pro-inflammatory cytokines in BMMs and PEMs stimulated by LPS/IFN-γ. Together, these results suggest PPI as a new avenue for therapy of RA.
The data supporting the conclusions of this article are included within the article.
QW and XZ performed most of the experiments, analyzed the data and participated in the manuscript draft. YZ and JX helped feed the mice, finished mice treatment and the sample processing work. YL performed the inhibitor and siRNA experiment. QS, YW, and HW provided scientific input and helped with manuscript editing. QL designed the study, and drafted and finalized the manuscript. All authors read and approved the final manuscript.
This work was sponsored by research grants from National Natural Science Foundation (81330085 to QS, 81822050, 81571617 to HW and 81673990 to QL), Shanghai TCM Medical Center of Chronic Disease (2017ZZ01010 to YW), The program for innovative research team of ministry of science and technology of China (2015RA4002 to YW), Research project of Shanghai science and Technology Commission (17401971100 to QL), Changning District Guanghua Excellent PI Project (No. 2016-01 to YW), Longyi innovation team program (LYCX-01 to YW), the Program for Innovative Research Team of Ministry of Education of China (to YW) and Ministry of Science and Technology of China (No. 2016YFD00200 to HW).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer PT and handling Editor declared their shared affiliation at the time of review.
The authors would like to acknowledge professor Lian-ping Xing form Department of Pathology and Laboratory Medicine, University of Rochester Medical Center, Rochester, New York for her experience and advice of this study.
RA, Rheumatoid arthritis; PPI, Polyphyllin I; CIA, collagen induced arthritis; IFN-γ, interferon-γ; TNF-α, Tumor necrosis factor-α; TLRs, Toll-like receptor;IκB, inhibitor of NF-κB; IKK, inhibitor of NF-κB kinase; IL, interleukin; iNOS, inducible nitric oxide synthase; IACUC, Institutional Animal Care and Use Committee; CMC-Na, sodium carboxyl methyl cellulose; BMMs, bone marrow-derived macrophages; PEMs, peritoneal elucidated macrophages; ABOG, Alcian blue-Hematoxylin-Orange G; HE, hematein eosin; AST, serum aspartate aminotransferase; ALT, serum alanine aminotransferase; UREA, blood urea nitrogen; CRE, Serum creatinine; micro-CT, Micro computed Tomography; BV, bone volume; IF, immunofluorescence; M1, macrophage classical activation; M2, alternative activation; IRAK, IL-1R associated kinase; TRAF, TNF receptor associated factor; LPS, lipopolysaccharides; nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB); Myeloid differentiation primary response 88 (MYD88), IRAK1, interleukin-1 receptor (IL-1R) associated kinase 1; TRAF6, TNF receptor associated factors 6; TAK1, Transforming growth factor-b–activated kinase 1; FBS, fetal bovine serum; P/S, penicillin and streptomycin; M-CSF, Macrophage colony-stimulating factor; Quantitative Real-time-PCR (qPCR); DAPI, 4',6-diamidino-2-phenylindole; BSA, Bovine Serum Albumin; SDS, sodium dodecyl sulfate; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; PVDF, Polyvinylidene fluoride; TBST, tris-buffered saline and Tween 20; SEM, standard error of mean; ANOVA, Analysis of variance; ELISA, enzyme-linked immunosorbent assay; NO, nitric oxide.
1. Hinks A, Eyre S, Ke X, Barton A, Martin P, Flynn E, et al. Overlap of disease susceptibility loci for rheumatoid arthritis and juvenile idiopathic arthritis. Ann Rheum Dis. (2010) 69:1049–53. doi: 10.1136/ard.2009.110650
2. Reynolds G, Gibbon JR, Pratt AG, Wood MJ, Coady D, Raftery G, et al. Synovial CD4+ T-cell-derived GM-CSF supports the differentiation of an inflammatory dendritic cell population in rheumatoid arthritis. Ann Rheum Dis. (2016) 75:899–907. doi: 10.1136/annrheumdis-2014-206578
3. Choi S, You S, Kim D, Choi SY, Kwon HM, Kim HS, et al. Transcription factor NFAT5 promotes macrophage survival in rheumatoid arthritis. J Clin Invest. (2017) 127:954–69. doi: 10.1172/JCI87880
4. Yeo L, Adlard N, Biehl M, Juarez M, Smallie T, Snow M, et al. Expression of chemokines CXCL4 and CXCL7 by synovial macrophages defines an early stage of rheumatoid arthritis. Ann Rheum Dis. (2016) 75:763–71. doi: 10.1136/annrheumdis-2014-206921
5. Tak PP, Smeets TJ, Daha MR, Kluin PM, Meijers KA, Brand R, et al. Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity. Arthritis Rheum. (1997) 40:217–25. doi: 10.1002/art.1780400206
6. Mulherin D, Fitzgerald O, Bresnihan B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum. (1996) 39:115–24. doi: 10.1002/art.1780390116
7. Li J, Hsu HC, Yang P, Wu Q, Li H, Edgington LE, et al. Treatment of arthritis by macrophage depletion and immunomodulation: testing an apoptosis-mediated therapy in a humanized death receptor mouse model. Arthritis Rheum (2012) 64:1098–109. doi: 10.1002/art.33423
8. Alves CH, Farrell E, Vis M, Colin EM, Lubberts E. Animal models of bone loss in inflammatory arthritis: from cytokines in the bench to novel treatments for bone loss in the bedside-a comprehensive review. Clin Rev Allergy Immunol. (2016) 51:27–47. doi: 10.1007/s12016-015-8522-7
9. Schuerwegh AJ, Van Offel JF, Stevens WJ, Bridts CH, De Clerck LS. Influence of therapy with chimeric monoclonal tumour necrosis factor-alpha antibodies on intracellular cytokine profiles of T lymphocytes and monocytes in rheumatoid arthritis patients. Rheumatology (2003) 42:541–8. doi: 10.1093/rheumatology/keg171
10. Kim WJ, Kang YJ, Koh EM, Ahn KS, Cha HS, Lee WH. LIGHT is involved in the pathogenesis of rheumatoid arthritis by inducing the expression of pro-inflammatory cytokines and MMP-9 in macrophages. Immunology (2005) 114:272–9. doi: 10.1111/j.1365-2567.2004.02004.x
11. Pope RM. Apoptosis as a therapeutic tool in rheumatoid arthritis. Nat Rev Immunol. (2002) 2:527–35. doi: 10.1038/nri846
12. Drexler SK, Kong PL, Wales J, Foxwell BM. Cell signalling in macrophages, the principal innate immune effector cells of rheumatoid arthritis. Arthritis Res Ther. (2008) 10:216. doi: 10.1186/ar2481
13. Deng GM, Zheng L, Chan FK, Lenardo M. Amelioration of inflammatory arthritis by targeting the pre-ligand assembly domain of tumor necrosis factor receptors. Nature Medicine (2005) 11:1066–72. doi: 10.1038/nm1304
14. Wang Y, Han CC, Cui D, Li Y, Ma Y, Wei W. Is macrophage polarization important in rheumatoid arthritis? Int Immunopharmacol. (2017) 50:345–52. doi: 10.1016/j.intimp.2017.07.019
15. Fearon U, Canavan M, Biniecka M, Veale DJ. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat Rev Rheumatol. (2016) 12:385–97. doi: 10.1038/nrrheum.2016.69
16. Huang Q, Ma Y, Adebayo A, Pope RM. Increased macrophage activation mediated through toll-like receptors in rheumatoid arthritis. Arthritis Rheum (2007) 56:2192–201. doi: 10.1002/art.22707
17. Min SY, Yan M, Du Y, Wu T, Khobahy E, Kwon SR, et al. Intra-articular nuclear factor-kappaB blockade ameliorates collagen-induced arthritis in mice by eliciting regulatory T cells and macrophages. Clin Exp Immunol. (2013) 172:217–27. doi: 10.1111/cei.12054
18. Gao LN, Feng QS, Zhang XF, Wang QS, Cui YL. Tetrandrine suppresses articular inflammatory response by inhibiting pro-inflammatory factors via NF-kappaB inactivation. J Orthop Res. (2016) 34:1557–68. doi: 10.1002/jor.23155
19. Chang J, Li Y, Wang X, Hu S, Wang H, Shi Q, et al. Polyphyllin I suppresses human osteosarcoma growth by inactivation of Wnt/beta-catenin pathway in vitro and in vivo. Sci Rep. (2017) 7:7605. doi: 10.1038/s41598-017-07194-9
20. Chang J, Wang H, Wang X, Zhao Y, Zhao D, Wang C, et al. Molecular mechanisms of Polyphyllin I-induced apoptosis and reversal of the epithelial-mesenchymal transition in human osteosarcoma cells. J Ethnopharmacol. (2015) 170:117–27. doi: 10.1016/j.jep.2015.05.006
21. Lou W, Chen Y, Zhu KY, Deng H, Wu T, Wang J. Polyphyllin I overcomes EMT-associated resistance to erlotinib in lung cancer cells via IL-6/STAT3 pathway inhibition. Biol Pharm Bull. (2017) 40:1306–13. doi: 10.1248/bpb.b17-00271
22. Gu L, Feng J, Xu H, Luo M, Su D. Polyphyllin I inhibits proliferation and metastasis of ovarian cancer cell line HO-8910PM in vitro. J Tradit Chin Med. (2013) 33:325–33. doi: 10.1016/S0254-6272(13)60174-0
23. Gu L, Feng J, Zheng Z, Xu H, Yu W. Polyphyllin I inhibits the growth of ovarian cancer cells in nude mice. Oncol Lett. (2016) 12:4969–74. doi: 10.3892/ol.2016.5348
24. Li GB, Fu RQ, Shen HM, Zhou J, Hu XY, Liu YX, et al. Polyphyllin I induces mitophagic and apoptotic cell death in human breast cancer cells by increasing mitochondrial PINK1 levels. Oncotarget (2017) 8:10359–74. doi: 10.18632/oncotarget.14413
25. Liang Y, Li X, He X, Qiu X, Jin XL, Zhao XY, et al. Polyphyllin I induces cell cycle arrest and apoptosis in human myeloma cells via modulating beta-catenin signaling pathway. Eur J Haematol. (2016) 97:371–8. doi: 10.1111/ejh.12741
26. Yue G, Wei J, Qian X, Yu L, Zou Z, Guan W, et al. Synergistic anticancer effects of polyphyllin I and evodiamine on freshly-removed human gastric tumors. PLoS ONE (2013) 8:e65164. doi: 10.1371/journal.pone.0065164
27. Han W, Hou G, Liu L. Polyphyllin I (PPI) increased the sensitivity of hepatocellular carcinoma HepG2 cells to chemotherapy. Int J Clin Exp Med. (2015) 8:20664–9.
28. Sumariwalla PF, Cao Y, Wu HL, Feldmann M, Paleolog EM. The angiogenesis inhibitor protease-activated kringles 1-5 reduces the severity of murine collagen-induced arthritis. Arthritis Res Ther. (2003) 5:R32–39. doi: 10.1186/ar608
29. Gao B, Calhoun K, Fang D. The proinflammatory cytokines IL-1beta and TNF-α induce the expression of Synoviolin, an E3 ubiquitin ligase, in mouse synovial fibroblasts via the Erk1/2-ETS1 pathway. Arthritis Res Ther. (2006) 8:R172. doi: 10.1186/ar2081
30. Zhang Y, Lu Y, Ma L, Cao X, Xiao J, Chen J, et al. Activation of vascular endothelial growth factor receptor-3 in macrophages restrains TLR4-NF-κB signaling and protects against endotoxin shock. Immunity (2014) 40:501–14. doi: 10.1016/j.immuni.2014.01.013
31. Lin CY, Lee CH, Chang YW, Wang HM, Chen CY, Chen YH. Pheophytin a inhibits inflammation via suppression of LPS-induced nitric oxide synthase-2, prostaglandin E2, and interleukin-1beta of macrophages. Int J Mol Sci. (2014) 15:22819–34. doi: 10.3390/ijms151222819
32. Zenker S, Panteleev-Ivlev J, Wirtz S, Kishimoto T, Waldner MJ, Ksionda O, et al. A key regulatory role for Vav1 in controlling lipopolysaccharide endotoxemia via macrophage-derived IL-6. J Immunol. (2014) 192:2830–6. doi: 10.4049/jimmunol.1300157
33. Liu L, Yang C, Shen J, Huang L, Lin W, Tang H. GABRA3 promotes lymphatic metastasis in lung adenocarcinoma by mediating upregulation of matrix metalloproteinases. Oncotarget (2016) 7:32341–50. doi: 10.18632/oncotarget.8700
34. Awad AS, You H, Gao T, Cooper TK, Nedospasov SA, Vacher J, et al. Macrophage-derived tumor necrosis factor-alpha mediates diabetic renal injury. Kidney Int. (2015) 88:722–33. doi: 10.1038/ki.2015.162
35. Liu L, Zhao Y, Xie K, Sun X, Gao Y, Wang Z. Estrogen-induced nongenomic calcium signaling inhibits lipopolysaccharide-stimulated tumor necrosis factor alpha production in macrophages. PLoS ONE (2013) 8:e83072. doi: 10.1371/journal.pone.0083072
36. Nakayama M, Niki Y, Kawasaki T, Takeda Y, Horiuchi K, Sasaki A, et al. Enhanced susceptibility to lipopolysaccharide-induced arthritis and endotoxin shock in interleukin-32 alpha transgenic mice through induction of tumor necrosis factor alpha. Arthritis Res Ther. (2012) 14:R120. doi: 10.1186/ar3850
37. Ehlting C, Ronkina N, Bohmer O, Albrecht U, Bode KA, Lang KS, et al. Distinct functions of the mitogen-activated protein kinase-activated protein (MAPKAP) kinases MK2 and MK3. MK2 mediates lipopolysaccharide-induced signal transducers and activators of transcription 3 (STAT3) activation by preventing negative regulatory effects of MK3. J Biol Chem. (2011) 286:24113–24. doi: 10.1074/jbc.M111.235275
38. Li P, Fan JB, Gao Y, Zhang M, Zhang L, Yang N, et al. miR-135b-5p inhibits LPS-induced TNFalpha production via silencing AMPK phosphatase Ppm1e. Oncotarget (2016) 7:77978–86. doi: 10.18632/oncotarget.12866
39. Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. (1981) 11:805–15. doi: 10.1002/eji.1830111013
40. Hume DA, Gordon S. Mononuclear phagocyte system of the mouse defined by immunohistochemical localization of antigen F4/80. Identification of resident macrophages in renal medullary and cortical interstitium and the juxtaglomerular complex. J Exp Med. (1983) 157:1704–9.
41. Hume DA, Halpin D, Charlton H, Gordon S. The mononuclear phagocyte system of the mouse defined by immunohistochemical localization of antigen F4/80: macrophages of endocrine organs. Proc Natl Acad Sci USA. (1984) 81:4174–7. doi: 10.1073/pnas.81.13.4174
42. Lu M, Dai Y, Xu M, Zhang C, Ma Y, Gao P, et al. The attenuation of 14-3-3zeta is involved in the caffeic acid-blocked lipopolysaccharide-stimulated inflammatory response in RAW264.7 macrophages. Inflammation (2017) 40:1753–60. doi: 10.1007/s10753-017-0618-1
43. Udompong S, Mankhong S, Jaratjaroonphong J, Srisook K. Involvement of p38 MAPK and ATF-2 signaling pathway in anti-inflammatory effect of a novel compound bis[(5-methyl)2-furyl](4-nitrophenyl)methane on lipopolysaccharide-stimulated macrophages. Int Immunopharmacol (2017) 50:6–13. doi: 10.1016/j.intimp.2017.05.015
44. Weng P, Zhang XT, Sheng Q, Tian WF, Chen JL, Yuan JJ, et al. Caveolin-1 scaffolding domain peptides enhance anti-inflammatory effect of heme oxygenase-1 through interrupting its interact with caveolin-1. Oncotarget (2017) 8:40104–14. doi: 10.18632/oncotarget.16676
45. McInnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet (2017) 389:2328–37. doi: 10.1016/S0140-6736(17)31472-1
46. Lee JH, Kim B, Jin WJ, Kim HH, Ha H, Lee ZH. Pathogenic roles of CXCL10 signaling through CXCR3 and TLR4 in macrophages and T cells: relevance for arthritis. Arthritis Res Ther. (2017) 19:163. doi: 10.1186/s13075-017-1353-6
47. Weyand CM, Zeisbrich M, Goronzy JJ. Metabolic signatures of T-cells and macrophages in rheumatoid arthritis. Curr Opin Immunol. (2017) 46:112–20. doi: 10.1016/j.coi.2017.04.010
48. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
49. Udalova I, Mantovani A, Feldmann M. Macrophage heterogeneity in the context of rheumatic diseases. Nat Rev Rheumatol. (2016) 12:472–85. doi: 10.1038/nrrheum.2016.91
50. Vandooren B, Noordenbos T, Ambarus C, Krausz S, Cantaert T, Yeremenko N, et al. Absence of a classically activated macrophage cytokine signature in peripheral spondylarthritis, including psoriatic arthritis. Arthritis Rheum (2009) 60:966–75. doi: 10.1002/art.24406
51. Park SY, Lee SW, Baek SH, Lee CW, Lee WS, Rhim BY, et al. Suppression of PU.1-linked TLR4 expression by cilostazol with decrease of cytokine production in macrophages from patients with rheumatoid arthritis. Br J Pharmacol. (2013) 168:1401–11. doi: 10.1111/bph.12021
52. Abdollahi-Roodsaz S, Joosten LA, Roelofs MF, Radstake TR, Matera G, Popa C, et al. Inhibition of Toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum (2007) 56:2957–67. doi: 10.1002/art.22848
53. Hou Y, Lin H, Zhu L, Liu Z, Hu F, Shi J, et al. Lipopolysaccharide increases the incidence of collagen-induced arthritis in mice through induction of protease HTRA-1 expression. Arthritis Rheum (2013) 65:2835–46. doi: 10.1002/art.38124
54. Terato K, Ye XJ, Miyahara H, Cremer MA, Griffiths MM. Induction by chronic autoimmune arthritis in DBA/1 mice by oral administration of type II collagen and Escherichia coli lipopolysaccharide. Br J Rheumatol. (1996) 35:828–38. doi: 10.1093/rheumatology/35.9.828
55. Yoshino S, Yamaki K, Taneda S, Yanagisawa R, Takano H. Reactivation of antigen-induced arthritis in mice by oral administration of lipopolysaccharide. Scand J Immunol. (2005) 62:117–22. doi: 10.1111/j.1365-3083.2005.01647.x
56. Yoshino S, Ohsawa M. The role of lipopolysaccharide injected systemically in the reactivation of collagen-induced arthritis in mice. Br J Pharmacol. (2000) 129:1309–14. doi: 10.1038/sj.bjp.0703166
57. Li L, Wu J, Zheng F, Tang Q, Wu W, Hann SS. Inhibition of EZH2 via activation of SAPK/JNK and reduction of p65 and DNMT1 as a novel mechanism in inhibition of human lung cancer cells by polyphyllin I. J Exp Clin Cancer Res. (2016) 35:112. doi: 10.1186/s13046-016-0388-x
58. Han EJ, Kim HY, Lee N, Kim NH, Yoo SA, Kwon HM, et al. Suppression of NFAT5-mediated inflammation and chronic arthritis by novel kappaB-binding Inhibitors. EBioMedicine (2017) 18:261–73. doi: 10.1016/j.ebiom.2017.03.039
59. Liu J, Zhang Y, Chen L, Yu F, Li X, Dan T, et al. Polyphyllin I induces G2/M phase arrest and apoptosis in U251 human glioma cells via mitochondrial dysfunction and the JNK signaling pathway. Acta Biochim Biophys Sin. (2017) 49:479–86. doi: 10.1093/abbs/gmx033
60. Abdollahi-Roodsaz S, Joosten LA, Helsen MM, Walgreen B, van Lent PL, van den Bersselaar LA, et al. Shift from toll-like receptor 2 (TLR-2) toward TLR-4 dependency in the erosive stage of chronic streptococcal cell wall arthritis coincident with TLR-4-mediated interleukin-17 production. Arthritis Rheum (2008) 58:3753–64. doi: 10.1002/art.24127
61. Roelofs MF, Boelens WC, Joosten LA, Abdollahi-Roodsaz S, Geurts J, Wunderink LU, et al. Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J Immunol. (2006) 176:7021–7. doi: 10.4049/jimmunol.176.11.7021
62. Sacre SM, Andreakos E, Kiriakidis S, Amjadi P, Lundberg A, Giddins G, et al. The Toll-like receptor adaptor proteins MyD88 and Mal/TIRAP contribute to the inflammatory and destructive processes in a human model of rheumatoid arthritis. Am J Pathol. (2007) 170:518–25. doi: 10.2353/ajpath.2007.060657
63. Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity (1997) 7:837–47. doi: 10.1016/S1074-7613(00)80402-1
64. Burns K, Martinon F, Esslinger C, Pahl H, Schneider P, Bodmer JL, et al. MyD88, an adapter protein involved in interleukin-1 signaling. J Biol Chem. (1998) 273:12203–9. doi: 10.1074/jbc.273.20.12203
65. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. (2004) 4:499–511. doi: 10.1038/nri1391
Keywords: polyphyllin I, collagen-induced arthritis, primary macrophages, NF-κB, inflammation
Citation: Wang Q, Zhou X, Zhao Y, Xiao J, Lu Y, Shi Q, Wang Y, Wang H and Liang Q (2018) Polyphyllin I Ameliorates Collagen-Induced Arthritis by Suppressing the Inflammation Response in Macrophages Through the NF-κB Pathway. Front. Immunol. 9:2091. doi: 10.3389/fimmu.2018.02091
Received: 22 February 2018; Accepted: 24 August 2018;
Published: 27 September 2018.
Edited by:
J. Michelle Kahlenberg, University of Michigan, United StatesReviewed by:
Stefania Gallucci, Temple University, United StatesCopyright © 2018 Wang, Zhou, Zhao, Xiao, Lu, Shi, Wang, Wang and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongjun Wang, eWp3YW5nODg4OEAxMjYuY29t
Hongyan Wang, aG9uZ3lhbndhbmdAc2liY2IuYWMuY24=
Qianqian Liang, bGlhbmdxaWFucWlhbnRjbUAxMjYuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.