 Shinjini Chakraborty
Shinjini Chakraborty Markus Huber-Lang
Markus Huber-Lang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 24 September 2018
Sec. Molecular Innate Immunity
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02050
This article is part of the Research Topic The Role of Complement in Health and Disease View all 50 articles
The overpowering effect of trauma on the immune system is undisputed. Severe trauma is characterized by systemic cytokine generation, activation and dysregulation of systemic inflammatory response complementopathy and coagulopathy, has been immensely instrumental in understanding the underlying mechanisms of the innate immune system during systemic inflammation. The compartmentalized functions of the innate and adaptive immune systems are being gradually recognized as an overlapping, interactive and dynamic system of responsive elements. Nonetheless the current knowledge of the complement cascade and its interaction with adaptive immune response mediators and cells, including T- and B-cells, is limited. In this review, we discuss what is known about the bridging effects of the complement system on the adaptive immune system and which unexplored areas could be crucial in understanding how the complement and adaptive immune systems interact following trauma.
Well-known ancient physician and philosopher Hippocrates from Kos (ca. 460–370 B.C.) had defined some interventions in trauma care, which had been radical at that time, eventually, some of them proving to be clinically relevant (1). Hippocratic Medicine followed the humoral pathology and defined the “four humors” or fluids of the body (among them blood), which had to be rebalanced to achieve healing. Almost as a foresight of the complement response after trauma, Hippocrates clearly pointed out the importance of humoral components in the pathophysiological course of diseases, thus being the first to indicate the involvement of the fluid phase and its potential relevance. Drawing on this age-old concept, complement activation was formerly defined as “disseminated intravascular multiple systems activation” following trauma as in a major burn injury (2). The cellular immune system with “pre-programmed responses” had been proposed to be of less importance after severe tissue injury compared to the mediator systems of complement components and other opsonins (3). Supporting this observation, activation of the humoral systems, including the coagulation and complement systems, was described to occur early after multiple injury because complement activation products, the anaphylatoxins C4a and C3a, were elevated and associated with injury severity and infectious complications (4, 5). These preliminary observations stimulated a growing interest in trauma-associated complement activation, which extended from temporal to spatial activation tendencies. Two studies are of relevance in this regard. In one clinical study, early after trauma (< 48 h) including multiple fractures, blunt abdominal trauma and blunt chest trauma with rib fractures, high C3a/C3 ratio was useful to segregate adult respiratory distress syndrome (ARDS) from non-ARDS patients (6). In a later study, plasma samples from patients were obtained as early as 30 min post-trauma comprising of penetrating injury or severe head injury and with a mean injury severity score (ISS) of 17, without any resuscitative fluid replacement therapy. Here it was found that the soluble terminal complement complex (TCC) sC5b-9 and Bb (activated factor B) were associated with high ISS (7). On comparing the two studies, several similarities and discrepancies can be summarized. Both studies identified an early activation of the alternative pathway, however, opposing in whether classical complement activation preceded or followed alternative pathway activation. Interesting to note in here is the difference of time-points in the two studies (30 min vs. <48 h post-trauma) and the fluid resuscitation that was received by patients in the former study as opposed to none received when blood samples were collected for the latter. Apart from this temporal association, the site and pattern of injury could also modulate activated complement factors and complexes differentially, as was described in a small cohort study proposing that the appearance of the TCC and C3dg as early as 24 h after trauma was mainly seen in patients who had suffered a thorax trauma (8).
When complement activation products were individually implicated in trauma-induced local and systemic trauma complications, several groups found that early C3a generation and depletion of C3 with a resultant increase in the C3a/C3 ratio were associated with the development of acute lung injury, ARDS, sepsis, multiple organ dysfunction and consequently a poor prognosis (6, 9–12). Likewise, anaphylatoxin C5a appears in the circulation of humans within 20 min post polytrauma and its levels have been related to the mortality rate (10). Exposure to C5a may result in significantly delayed neutrophil apoptosis early after trauma, thereby causing enhanced host damage through the recruitment and accumulation of neutrophils at the injury site (13). The underlying causes of rapid local and systemic complement activation via all pathways may be manifold and some remain speculative, for example, tissue hypoperfusion with acidic (micro)environments, natural antibodies, reactive oxygen species, coagulation-complement crosstalk, release of non-specific proteases with subsequent cleavage of complement components, exposure to pathogen-associated molecular patterns (PAMPs) in a synchronic shock situation, artificial surfaces like catheter materials and transfusion of blood products, including complement components in fresh frozen plasma among others (14–18). Uncontrolled complement activity can be further augmented by dysregulated levels of complement regulators, including C4b-binding protein and factor I (10). Additionally, an early depletion of central complement components causing trauma-induced complementopathy with declining complement hemolytic activity and manifestation of signs of immunodeficiency have been reported (10, 19).
While the complement system itself maybe dysregulated in the post-traumatic pathophysiology, activation of various other serine proteases, including proteases of apoptotic systems, like granzyme B and cathepsin D, may intensify the extent of complement activity. Damaged cells can also activate factor VII-activating proteases which, together with other proteases, rapidly cleave the complement factors C3 and C5 and generate the respective anaphylatoxins C3a and C5a (Figure 1) (16, 20). In addition to the autonomous action of the complement system after trauma, this ancient fluid-phase innate immune system is intimately associated with the cellular immune responses (13, 21–23). C5a functions not only as an effective chemoattractant for neutrophils, but also upregulates various adhesion molecules on the endothelium, evoking the classical signs of inflammation. C5a exposure leads to various acute defensive functions in neutrophils, including enhancement of phagocytic activity, mounting of an oxidative burst, further inflammatory mediator release and generation of neutrophil extracellular traps. On the contrary, after severe trauma or during prolonged systemic inflammatory conditions that promote complement activation, C5a may become “too much of a good thing” (24, 25). C5a-activated neutrophils have been shown to induce endothelial and mesothelial autologous tissue destruction, for example, of the human omentum, which performs several basic innate and adaptive immune functions (26). Recent studies have indicated that C5a also results in a profound morphological polarization of neutrophils and immunometabolic changes by enhancement of the glycolytic flux (27, 28). C5a was able to generate an alkalotic intracellular milieu in neutrophils and a local lactic acidosis microenvironment despite the absence of an ischemic condition or oxygen debt (27). Yet, it remains unclear to what extent these effects may contribute to known metabolic changes and acidosis after severe trauma. By contrast, deficiency in complement-related factors have also been reported in trauma. Blood neutrophils from trauma patients, which appeared unresponsive to C5a, displayed an impaired oxidative burst activity, less C5aR expression, increased C3b binding activity, and clinically more (infectious) complications (Figure 1) (29). Chemotactic desensitization of neutrophils to C5a was observed in patients with multiple trauma, burn injury or sepsis, indicating dysfunctional danger associated molecular pattern (DAMP) sensing by neutrophils upon excessive complement exposure (29, 30). In accordance with this, loss of C5aR1, C5aR2, and C3aR on neutrophils after multiple injury was found clinically and was correlated to infectious complications and multiple organ dysfunction (31–33). An integrated clinico-transcriptomic approach investigating RNA from leukocytes revealed that lower C5-expression on day 1 after polytrauma was associated with the development of nosocomial infections (34). Monocytes and macrophages are also major players in the orchestrated post-traumatic immune response and are highly responsive to complement factors. C5a is capable of priming peripheral blood mononuclear cells via p38 mitogen-activated protein kinase (MAPK) pathway activation (Figure 1), which in turn results in a significantly enhanced inflammatory mediator release upon secondary exposure to PAMPs (35). In previous studies, the migratory responsiveness of monocytes to C5a but not to fMLP had been found to be suppressed, with a maximum depression from 5 to 7 days after major injury (36, 37). The authors proposed these findings as an explanation for the predisposition of trauma patients to infection and poor wound healing.
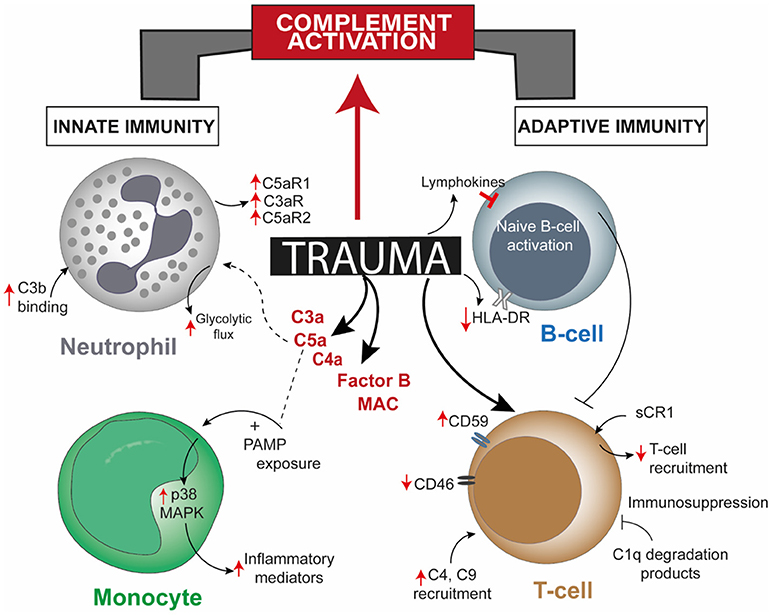
Figure 1. Overview of post-trauma complement activation bridging altered cellular responses of the innate and the adaptive immune system. Trauma alters one or more cellular responses of neutrophils, monocytes, and B-cells and by directly modulating complement regulatory proteins on T-cells, apart from robust activation of complement factors. Though there is a direct effect of trauma on T-cells and B-cells, involvement of mediating complement factors are unreported. CR1, complement receptor 1; HLA-DR, human leukocyte antigen—antigen D Related; MAPK, mitogen-activated protein kinase; PAMP, pathogen-associated molecular pattern.
When the “first line of defense” represented by physiological barriers, including the skin, is injured, the “second line of defense,” represented by innate immune cells and the complement system is challenged. Proteomic analyses of wound fluids after (surgical) trauma revealed the presence of key complement components including C3 and factor B, in addition to a discrete proteomic leukocyte signature, indicating that as part of the second line of defense, both leukocytes and complement implement the clearance of wound debris and regenerative processes (38). Currently, the role of complement in regard to the “third line of defense,” as formed by the adaptive immune system, is less well known, more so in the context of trauma.
The complement system and the adaptive immune responses after trauma have been described separately in various literature. Nonetheless, post-traumatic complement activation and its effects on lymphocyte phenotype and function remain elusive. At present, few studies mainly involving trauma injury like traumatic brain injury (TBI), burn injury, and splenectomy after blunt abdominal trauma, have brought the complement system and the adaptive immunity on to a common platform. Injury to the central and peripheral nervous system like TBI, peripheral nerve injury and spinal cord injury (SCI) have offered suitable opportunities to partially understand the basis of complement—adaptive immunity cross-talk. In a murine SCI model, IgM was proven to drive not only the recognition of neoepitopes on damaged cells, but also facilitated on-site complement activation and C3d deposition correlating with injury severity (39). This relation of complement activation to the injury was further bolstered through multiple studies, like, controlled brain contusion performed on strain-specific rats was found to have significant local C3 expression and high T-cell infiltration 3 days thereafter (40, 41). Implicating that mature T- and B-cells could be a pre-requisite for the generation of activated complement factors, Rag1(-/-) mice used to perform a closed head injury study had significantly decreased C3a levels compared to wild-type (WT) mice with no intergroup difference in injury severity (42). Thus, due to this experiential dependence of injury severity on complement activation, healing could also be achieved with complement inhibition. In post reperfused stroke it was seen that treatment with C3aR antagonist diminished infiltration of C3aR-expressing T-cells at the ischemic site, helping in neurogenesis (43). Pain responses due to peripheral nerve injury induced by partial ligation of the rat sciatic nerve could also be attenuated by intraperitoneal injection of soluble sCR1, with diminished macrophage and T-cell recruitment at the injury site (Figure 1) (44). However, the scope of these studies was limited as they did not investigate the cause/s of the observed effects.
Blunt abdominal injury is another example of trauma injury where patients frequently undergo splenectomy. The function of the complement system in attuning adaptive immune responses in the spleen has been underscored by two studies where the role of dendritic cells (DCs) and B-cell responses due to complement factors or receptors in the splenic marginal zone (MZ) has been described. The uptake and transfer of self-antigens by DCs to secondary lymphoid organs serves the crucial purpose of tolerance induction. However, proven through in vitro studies, CD8α+ DCs resident in splenic MZ can also actively uptake and internalize circulating iC3b opsonized apoptotic leukocytes and these cells require complement receptor (CR) 3 and not CR4 for this purpose (45). Coming to B-cell mediated responses, C4 deficient mice had higher immune complex localization in the splenic MZ and impaired antibody response and class-switching, which was restored when antigen was directed to the splenic MZ (46). Hence, complement seemed to play a role in modulating self-antigen localization such that peripheral B-cell tolerance is maintained. Splenectomy post-trauma affected immune function in terms of reduced T-cell response to phytohemagglutinin, decreased number of lymphocytes, decreased IgM levels, and no changes in C3, C4, and C5 levels (47). Contradicting this former study, other studies concluded that either serum IgM levels did not vary (48) or there was an increase in B-cell population (49). The commonality among them being the unchanged levels of complement factors, a stark demerit of these conclusions was that activated complement fragments were not measured, rendering one inconclusive as to what exact role the complement system might have had in insinuating the adaptive immune responses. In a later study including polytrauma patients, investigation of complement regulatory surface proteins on lymphocytes from the patients showed significantly high CD59 expression 120 and 240 h post trauma and significantly decreased CD46 expression up to 48 h after trauma, with or without splenectomy (Figure 1) (31). Whether an impairment or augmentation of lymphocyte activity is a cause of deregulated complement activation post-trauma was confirmed from burn injury studies, like, generation of C1q degradation peptides in burn patients having an immunosuppressive effect on lymphocytes (50). Serum obtained from major burn injury patients, when subjected to complement inactivating temperatures in vitro could affect mitogen-associated lymphocyte blastogenesis, establishing the fact that complement is putatively necessary for lymphocyte development in a trauma event (51). Extending on this concept, in a pig burn wound model (described as 8 burn wounds inflicted for 20 s with a 170°C heated copper rod over a 4 × 4 cm area on two flanks), systemic C3 increased significantly from day 9 and up to 60 days post injury, C4 increment was delayed after burn and a concomitant increase in T-cell infiltration at the wound site was seen on day 3 which declined 21 days post burn injury (52). Additionally, a local increase in C3 and C4 was observed 9 and 4 days post burn respectively, though both decreased after 21 and 9 days, respectively.
As evident from the paucity of relevant studies is that the causality of the complement—adaptive immunity interaction after trauma is still missing. In the following discussion, we focus on aspects of adaptive immunity, from antigen presentation to T- and B-cell functions, which have been proven to be under complement-mediated regulation and vice versa, and how such mechanisms are of importance in the “traumatic” context.
Antigen presentation is the first and foremost step in priming lymphocytes for their effector functions. This includes processing of exogenous foreign particles, which are in turn presented by major histocompatibility complex (MHC) class II to CD4+ T-cells and endogenous foreign particles are presented by MHC class I to CD8+ T-cells. MHC class II molecules are principally expressed on professional antigen presenting cells (APCs) e.g., macrophages, DCs and B-cells, while all nucleated cells express MHC class I on their surface. However, in addition to conventional antigen-presentation modes, MHC class I can also cross-present, i.e., exogenous antigens can be presented on MHC class I of professional APCs (53). Reduced antigen-presentation functions were initially reported in post-injury macrophages, further having been frequently described in trauma studies (54, 55). For example, reduced antigen presentation and interleukin (IL)-12 and interferon (IFN)-ɤ production after surgical trauma, a diminished population of HLA-DR+ monocytes early in trauma patients and attenuated IL-15 production by DCs following trauma hemorrhage have been reported (56–58). TBI and its impact could be lessened by targeted inhibition of class II-associated invariant peptide, an essential component in MHC antigen presentation (59). Therefore, antigen processing, presentation and co-stimulation of T-cells could well be under the regulation of activated complement factors, particularly in the event of excessive complement activation which has been observed after trauma. Few studies from the last decade could help one get an idea of the role played by complement factors in the uptake of antigens, self or non-self, through DCs and their effect in generating T-cell responses. The extensively convoluted pathophysiology of trauma can contribute to cellular apoptosis as seen in the spleen, thymus, and the gut and also in neurons after TBI and SCI (60–63) and requires resolution of unwarranted proinflammatory responses. Conversely, the said physiological mechanism can also yield in an imbalance of the inflammatory responses so generated. Suppression of proinflammatory response was identified to be mediated by DCs when a study found C1q and mannose binding lectin as receptors aiding apoptotic cell uptake by immature DCs (iDCs) (64). In connection to this, apoptotic cell removal by microglial cells, which are central nervous system tissue resident DCs, brings about inhibition of IL-6, IL-1α, TNF-α, and IL-1β as opsonization with C1q targets apoptotic cells to DC (65). Another study described that C1q opsonized apoptotic cell uptake by iDCs reduced their surface expression of CD86 costimulatory molecule along with diminished subsequent Th1 and Th17 cell proliferation, generating a contracted inflammatory response (66). This may explain the observed delayed development of immune paralysis in trauma patients, giving rise to poor prognosis as well as the susceptibility to sepsis (67). In a human study, severely injured patients have demonstrated monocyte deactivation, decreased HLA-DR expression on B-cells and dramatically influenced T-cell activation (68). Furthermore, a study showed increase in splenic T-cell and decrease in splenic B-cell 26S proteasome activity concomitant with changes in NF-κB expression after trauma-hemorrhage, suggesting the essential involvement of proteasomes in the regulation of signal transduction in splenic T- and B- cells (69). However, this study did not further describe if the antigen presentation machinery in B-cells is affected anyhow, as proteasome degradation is a key step in cytosolic antigen processing pathway of antigen presenting cells (53).
A complement-enriched environment is necessary for antigen presentation and T-cell differentiation, which is strongly dependent on the type of complement activation products generated (70, 71). Self-antigen presentation after degradation of apoptotic cells is guided by activated C3, contributing to MHC class II antigen processing and presentation (72). In several studies, the role of CR1 and 2 in prolonged antigen presentation have been described for antigen-specific B-cells and macrophage-mediated antigen presentation to T-cells (73–77). Studies have also attempted to describe the mechanistic pathway of antigen-loading efficiency of tetanus toxin-opsonized by C3b and C4b particles (78–80), though the type of antigen spoken hereof is of pathogenic source and may not necessarily explain mechanisms applicable in the context of trauma. Yet, the dynamics of C3 fragment mediated opsonization in purportedly regulating antigen processing is an intriguing premise. C3 activation fragments bound to antigen can delay its degradation in the cytosol by evading its fusion with lysosomes and maintain the antigen-loaded internalized cargo within DCs, a process which modulates T-cell responses to self-antigens (72). Additionally, antigens complexed with C3b were shown to form more stable MHC class II complex, modulating the antigen processing pathway in the late endocytic stage, the loading of the antigen onto MHC and the presentation of the antigen to T-cells resulting in T-cell proliferation (72, 78, 79, 81, 82). Furthermore, high intracellular cAMP in C3aR- or C3-deficient APCs impaired antigen presentation (83). While antigen presentation is facilitated by complement activation products, DC maturation could also be inhibited by iC3b, which exerts its effect by increased extracellular signal-regulated kinase 1/2 (ERK 1/2) phosphorylation and reduced p38 MAPK phosphorylation, resulting in decreased IL-12p70 and increased IL-10 (84). Even anaphylatoxin C5a is a potent effector in priming lymphocytes, because modified C5a peptide EP54 (response-selective agonist peptide) was identified as an adjuvant in DC-mediated T-cell priming (85). In addition to activated C3 and C5 products, elucidated effects of another complement factor C1q have been shown to drive DC-mediated functional activation of T-cells and cross-presentation of exogenous antigen to CD8+ T-cells resulting in greater CD8+ T-cell proliferation (86, 87). The gradual momentum gained by these evidences puts forward a need to confirm the same in trauma. While different experimental confirmations have brought forth the importance of complement activation and degradation products on antigen processing and presentation pathways, in vivo and in vitro analyses could help in further elaborating how high levels of C3 and C5 activation and/or degradation products generated during trauma could affect these downstream mechanisms.
Accumulating evidences supporting a possible interaction between the complement system and T-cells have been discussed in some reviews extensively (88–91). In this section we will discuss the relevant studies and recent developments with respect to complement—adaptive immunity crosstalk from the perspective of trauma-immunology.
Trauma severity and its outcome appear to be associated with elevated levels of the anaphylatoxins C3a, C4a, and C5a as well as other activated factors, including factor B and TCC (7, 92). Therefore, these intermediates could affect the resolution of the trauma response when adaptive immunity is involved. Invariably, there are obvious differences between sterile inflammation (like trauma) and infectious inflammation (like sepsis), which although can show similar effects, may or may not employ identical pathways to the resultant observed effect. Therefore, the origin of the antigens involved to enunciate the immune response is important. These antigens can be foreign (e.g., PAMPs), self yet foreign or DAMPs, or self-antigens. In PAMP-driven sepsis mice, treatment with anti-C5a antibodies displayed significantly reduced IL-17 levels compared to control-treated sepsis mice (93). C5a can also increase IL12+ DC migration from the peritoneal cavity to the periphery and could prime both Th1 and Th17 T-cells (94). Delving deeper into the exact effect of Th17 T-cells it was seen that, elevated level of IL-17 is generally observed in sepsis and this can be abrogated by a Rho-kinase inhibitor designating it as a participating downstream signaling mediator (95). Th17 cells mediating proinflammatory responses in trauma have been already reviewed extensively, and a few recent studies have investigated into their modulation by the complement system like elevated recruitment of the proinflammatory Th17-cell cohort in a trauma setting, whose inhibition proved to be protective and supported healing (66, 96–98). Interestingly, the DAMP-driven trauma and its associated Th17 responses is caught up in various contradictory results, albeit without the clarification of an intervening involvement of the complement system. When Inatsu et al., obtained peripheral blood mononuclear cells (PBMCs) from third degree thermal injury patients to culture and stimulate these PBMCs with Candida albicans hyphae in vitro, they found that PBMCs from burn patients had impaired Th17 differentiation (99). Notwithstanding this reported impairment in Th17 phenotype acquisition, subsequent murine studies showed in third degree burn model that Th17 cells were highly recruited at the site of wound as early as 3 h (100) and even 7 days post-injury (101). Few clinical and murine studies could support this conclusion as well for example increased circulating population of CD4+ Th17 T-cells 5 days after trauma in patients with an ISS > 20 (102) and greater IL-17 production by splenocytes in mice which were given an acute scald burn injury of 5 s at 95°C (103). Apart from systemic effects, the local response to trauma hemorrhage was found to effectively increase the Treg:Th17 ratio in the mesenteric lymph nodes, highly likely due to the concomitant decrement observed in circulatory CD103+ DCs (104). A recent clinical study where 114 trauma hemorrhagic shock (THS) and 50 control patients were recruited also showed that, THS patients who developed sepsis later on had lower Th17:Treg cell ratio (105). As sepsis is a common development in later time-points post-trauma, the relevance of IL-17 as a prognostic marker (67) and the contextual relevance of complementopathy in regulating this response could be further validated. IL-17 has also been identified to predict organ damage through computational network analysis of systemic inflammatory markers in blunt injury patients and neutralizing IL-17A attenuated organ damage in a trauma/hemorrhage mouse model (106). Therefore, it is a prerequisite to disentangle the gradually building yet convoluted concept of how exactly Th17 responses are under intricate modulation/s following trauma. This could be dependent on the time and type of injury, local to systemic differences and the inherent inconsistencies found in human and murine systems, apart from the proven role of complement in sepsis as opposed to the unexplored one in trauma. Complement factors and its regulatory components could also be one of those modulators in trauma injury and its yet untapped functions is an opportunity for further investigations.
ɤδ T-cells have been recently associated in a few trauma-related burn injury and fracture studies for example in regulation of fracture healing, high recruitment of ɤδ T-cells at the site of the wound, wound healing through modulation of myeloid cells and higher neutrophil recruitment in the lungs after trauma-hemorrhage (107–109). A recent study demonstrated how mitochondrial DAMPs can directly interact with ɤδ T-cells and induce synthesis of proinflammatory cytokines, including IL-1β and IL-6 (110). Though unavailable in the context of trauma, complement system's role has been elaborated in PAMP-driven systemic inflammatory response like sepsis. ɤδ T-cells obtained from cecal-ligation puncture (CLP) mice and transferred to recipient CLP mice followed by in vivo treatment with anti-C5a improved survival rates of the recipients (93). When these T-cells were obtained from anti-C5a pre-treated CLP mice and transferred to recipient CLP littermates, the recipients displayed reduced survival, indicating that the pathogenic role of ɤδ T-cells was modulated by C5a (93). Extending upon the effect of C5a, increased C5a generation was shown to stimulate neutrophil release of histones, which in turn can induce lymphocyte apoptosis (111). This study used WT and C5aR1 knockout (C5aR1-/-) mice to induce CLP-sepsis and stained apoptotic cells in the spleen by TUNEL, whereby WT CLP-sepsis mice had up to 5 times higher apoptotic cells per view field. The complement—lymphocyte connection could be confirmed for other complement factors as well, like, functionally defective Tregs, along with defective DCs and impaired B-cell switching were found in a 2 year old male patient with C3 deficiency (112). Hence, the concomitant over-activation of complement factors and following consumptive effect that trauma has on them could very well impede the functioning of the adaptive immune system. A seldom explored complement regulatory protein in traumatic conditions is the decay acceleration factor (DAF), which exerted a protective effect in a pig hemorrhage model and was seen to be upregulated in human neutrophils early after polytrauma (31, 113). Imitating central nervous system inflammation by injecting myelin oligodendrocyte glycoprotein (35–55) peptide showed that Daf1 transgenic mice with a higher cell-surface DAF expression displayed an attenuated disease phenotype and reduced antigen-specific Th1 and Th17 responses, further emphasizing on the role of DAF in T-cell-mediated proinflammatory functions (114). Hence, the robust involvement of complement factors in generation of T-cell responses could affect the mechanisms that are modulated downstream and the evidences explaining these potential mechanisms have been a recent addition to our understanding.
Severe tissue trauma and hemorrhagic shock (HS) is often associated with a significant oxygen deficit as reflected by lactate acidosis and a drop in the base excess (13). The decrease in pH can be manifested systemically and be even more pronounced locally. Low pH environments are known to activate key complement factors and thereby generate C3 and C5 activation products (115) which in principle can contribute to the complement activation and modulation of lymphocyte intracellular mechanisms. Metabolic effects of activated complement at lower concentrations functionally modulated CD4+ T-cells in terms of glutamine utilization and enhanced oxidative capacity of T-cells; whereas a higher concentration of complement resulted in cell death by ATP depletion (116). A combination of TCC and immune complexes trigger CD4+ T-cell activation by inducing phosphorylation of ζ-chain, ZAP-70, Syk, Src, and Lck along with the arrangement of the actin cytoskeleton and effector T-cell functions (117, 118). Upon stimulation, T-cells in C3 knock-out mice have also been shown to display attenuated T-bet transcription factor expression, a factor which determines lineage specificity in CD4+ T-cells (71). Nevertheless, the complement—T-cell association is not just restricted to this. In the last few years, the intimate association of complement mediated signals in T-cell phenotype acquisition has been elaborated through a series of important studies. It was seen that, absence of C5aR and C3aR signaling caused CD4+ T-cells to acquire a Treg cell phenotype in the absence of DCs, through induction of TGF-β and downregulation of complement factor expression by T-cells (119). Thus, autocrine complement activity of T-helper cells gained relevance with a following study showing the generation of intracellular C3a to be driven primarily by cathepsin-L protease synthesized by T-cells (120). A subsequent more recent study in this context showed how T-cell effector function results from intracellular metabolic alterations, modulated by intracellular synthesis of complement products and surface-expressed CD46 (121). Intracellular generation of C3 cleavage products and binding of C3b to CD46 drives the upregulation of glucose and amino-acid transporters and ragulator complex protein LAMTOR which affects cell growth (121). Even intracellular activation of C5 and its effect on NLRP3 inflammasome signaling are necessary for Th1 cell IFN-ɤ synthesis (122). Considering these evidences, it would be interesting to see how and where these mechanisms are affected, when the aforementioned downregulation of CD46 occurs on T-cells following trauma (31) and the role of intracellular C5aR1 in CD4+ T-cells thereafter.
Raad et al. has extensively reviewed how autoantibody generation is a common observation after central nervous system trauma (103). The review discusses in depth the origin of such autoantigen synthesis, which may result due to generation of autoreactive T-cells or even B-cell hyperactivation. Here, the role of the complement system in expediting lymphocyte sensitization and their responses to self-antigens generated following profuse tissue damage after trauma is left to be explored in detail. Ischemia-reperfusion (IR) injury is a common consequence of trauma where the role of complement has been confirmed together with humoral immune responses (51, 52). Clonally specific B-cells and the activated classical complement pathway were shown to be involved in injury pathogenesis, with IgM implicated as the key mediator (53). Additionally, the relevance of CR2 has been demonstrated in inducing IgG and IgM antibody production and their concomitant effects in IR injury pathogenesis (54). Inhibition of spleen tyrosine kinase (Syk), a membrane signaling protein found in B- and T-cells, elicited protective effects by reducing C3 and IgM deposition in tissues in a model of mesenteric IR injury with remote lung injury (55). Opposing existing concepts, C3 was subsequently shown not to be involved in IR injury, whereby a study revealed that surface-expressed CR2 on marginal-zone B-cells did not require C3 activation products to generate IR injury-induced antibodies (56). While nonspecific and specific humoral immune responses have been evaluated in burn injury and SCI, additional observations have been made in major trauma models (123–125). A study by Faist et al. evaluated B-cell function in 30 patients following major trauma over a 21-day period (126). Although the number of circulating B-cells in the trauma patients was not decreased following injury, the number of terminally maturing CIg+ B-cells were significantly decreased compared to controls up to 21 days post-trauma. B-cells obtained from the recruited patients in this study were analyzed in vitro and synthesis of IgA, IgM, and IgG by these B-cells were significantly reduced on day 1 after trauma. Later, IgG synthesis decreased on day 3 though total IgG levels were supra-normal. IgM synthesis was diminished throughout the study as compared to the controls (126). This post-traumatic IgM deficiency has also been described in other studies, a fact which is supported by the lack of the lymphokine IL-2 found in patients with multiple trauma (127, 128). Additionally, B-cell derived autoantibodies after SCI are co-localized with C1q on injured neurons, though it remains unclear whether complement activation modulates the production of these autoantibodies (50).
The complement system is furthermore involved in the maintenance of homeostasis by clearing debris, apoptotic bodies, and immune complexes (129). It regulates effector functions of natural antibodies, and C3 cleavage products participate in the opsonization and transport of antigen to the B-cell compartments of the secondary lymphoid tissue (130–132). A detailed role of complement regulators has been realized of late, like CR2/CD21 and CR1/CD35 expression by follicular DCs (FDCs), which function to retain the antigen in the lymphoid follicles required for the activation, proliferation, and antibody generation of B-cells (133, 134). CR1/2 and complement C3 support the localization of IgM-immune complexes in the splenic MZ (135), where B-cells concentrate the IgM-immune complexes onto FDCs. Supporting this, decreased MZ B-cells dramatically diminish the amount of IgM-immune complex on FDCs. The localization of antigen on FDCs has been implicated in optimizing the formation of germinal centre, memory B-cell formation, somatic hypermutation, and IgG class switching (135). Furthermore, in the FDCs of the peripheral lymphoid tissues, including the spleen and lymph nodes, CR1 and CR2 assist in autoreactive B-cell clone anergy by binding and presenting the self-antigen to these B-cells (136). This clearly points toward the differential expression of these two antagonistic complement regulators during the development of human B-cells, a phenomenon which may influence the maintenance of peripheral B-cell tolerance. Deficiencies in upstream complement molecules, including C1q, C2, and C4, may result in inefficient clearance, in the absence of which these products become a source of autoantigens for B-cells and a probable source of an autoimmune response (137). In addition to complement receptors, attachment of C4b to self-antigen and localization of these complexes to CD35 on stromal cells within the bone marrow also regulate the selection of potentially autoreactive B-cells (138). B-cell maturation and self-tolerance being the major foreground of the few players from the complement arsenal, B-cell class switching was shown to be regulated by C3, as C3-deficient patients had abnormal variation in IgG isotypes, including high IgG3, low IgG2, and no IgG4 response (139). These isotypes can in turn modulate the type of adaptive immune responses generated, having an overall effect in subsequent inflammatory responses generated in the said individuals. This indicates but does not yet prove how complement imbalance post-trauma could contribute to altered B-cell responses though further studies could help explain the same.
The antigen receptors on B-cells, similar to T-cells, are present as multiprotein complexes. Binding to their cognate ligand initiates an intracellular signaling cascade involving translocation to the nucleus and changes in gene expression that dictate the response of the lymphocyte. C3d has been extensively described in mediating signaling pathways, thus linking the innate to the adaptive immunity (140). In this regard, CR1 and CR2 are the best characterized complement regulators that mediate complement-dependent B-cell signaling. C3d interacts with both CR2 and the B-cell-receptor (BCR), causing activation and proliferation of B-cells and the production of specific antibodies. Studies have demonstrated that C3d can bind to CR2, which mobilizes the B-cell signaling complex consisting of CR2, CD19, CD81, and leu-13. Studies in mice lacking either CD19 or CR2 revealed that the CD19-CR2 complex is essential for proper immune function. This B-cell signaling complex confers important co-receptor activities to the BCR. The BCR consists of a non-signaling membrane immunoglobulin and a transmembrane immunoglobulin heterodimer Igα and Igβ bearing immunoreceptor tyrosine activation motifs (ITAMs). BCR stimulation activates two classes of tyrosine kinases: Src-family kinase (Src-protein-tyrosine kinase [PTK], including Lyn, Fyn, Blk, or Lck) and Syk kinase (141). Active Src-PTKs facilitate the phosphorylation and consequent activation of Syk. Activated Syk phosphorylates numerous intracellular substrates mediating B-cell activation, including the ITAMs of Igα and Igβ, and various adaptor proteins, including B-cell linker protein [BLNK or SH2-domain containing leukocyte protein of 65 kDa (SLP-65)]; which in turn leads to the recruitment of Bruton's tyrosine kinase (Btk) and phospholipase C-ɤ(PLC-ɤ) (142–146). Subsequent phosphorylation of PLC-ɤ generates IP3. This molecule orchestrates a cascade of events resulting in Ca2+ release from intracellular stores and activation and nuclear localization of the nuclear factor of activated T-cells (NFAT) and nuclear factor “kappa-light-chain-enhancer” of activated B-cells (NF-κB) pathways (147–152).
CD19 is the only molecule within the B-cell signaling complex capable of intracellular signaling. It acts as a membrane adaptor molecule by recruiting signaling intermediaries, including Vav and phosphotidylinositol-3 kinase (PI3K), which enhance Ca2+ flux, activation of ERK 1/2, and ultimately B-cell proliferation (153). C3d-opsonized Ag can co-ligate BCR with CR2, consequently improving antigen uptake (74, 154). This complex recruits several copies of CR2 and the B-cell signaling complex, upregulating intracellular Ca2+ mobilization and in turn decreasing the threshold of B-cell activation (4). In conclusion, C3d-CR2 binding resulting in CR2-BCR co-ligation reduces the threshold for cell activation by 10- to 100-fold (155) BCR-CR2 co-ligation is also able to inhibit first apoptosis signal (Fas)-mediated apoptosis. In this context, co-ligation results in increased expression levels of anti-apoptotic proteins, including Bcl-2 and the cellular FADD-like interleukin-1-ß-converting enzyme inhibitory protein short form. Invariably, the increase in Bcl-2 appears to inhibit Apaf-1-mediated apoptosis (156).
Whereas the role of human CR2 in B-cell activation is relatively well-established, much less is known about the exact function of CR1 (CD35). In contrast to CR2, CR1 has been described to mediate inhibitory signals (157–159). CR1 ligands include aggregated C3 and aggregated C3(H2O), which in contrast do not bind to CR2. Furthermore, CR1 binds to the complement components C3b and C4b and has co-factor activities in their cleavage by factor I to iC3b, a substrate for CD21, and iC4b, respectively (160). It was demonstrated that CR1 strongly reduced the intracellular Ca2+ level and phosphorylation of cytoplasmic proteins. Importantly, this inhibitory activity also occurs in the presence of the costimulatory cytokines IL-2 and IL-15 (157).
In trauma, impaired cellular function, including B-cell activation and antibody production, has been described. A recent study showed that the absence of CR1 and CR2 was protective toward secondary damages following closed head injury, including reduced IgM deposition at the injury site (161), though it was not sufficient to gather how are the downstream signaling mechanisms regulated to confer that protection. Further studies could attempt at deconstructing this yet unexplored premise.
Considerable knowledge regarding interaction of the complement system with the adaptive immune system has accumulated over time. Nevertheless, there is a scope for experimental and clinical studies to provide an insight into the biology of complement and B- and T-cell interactions particularly after tissue trauma. It is well established that complement receptors are involved in B- and T-cell signaling and function. In the context of trauma, the broadly described role of CR2 in B-cell-signaling cascades could be investigated upon. While CR2 mediates activation of B-cell signaling, CR1 has B-cell inhibitory functions. In addition to CR1 and CR2, studies have also revealed a minor expression of CR3 and CR4 on B- and T-lymphocytes (162). Their expression on leukocytes, especially macrophages has important functions in inflammatory conditions, including tissue trauma and infection, where CR3 and CR4 enhance antimicrobial functions by potentiating leukocyte cell adhesion (163), though their function on B- and T-lymphocytes remains undetermined. Because CR3 is expressed by cytotoxic T-cells, it can be speculated that this receptor may play an important role in lymphocyte functions (162). Like complement regulatory proteins, the receptors for the anaphylatoxins (C3aR, C5aR) appear to be crucial for proper cellular functions of monocytes and neutrophils, though their respective roles in adaptive immune cells have not been fully understood. An early study found C3aR on neutrophils, monocytes, and eosinophils, but not on B- lymphocytes, whereas another study found expression of both anaphylatoxins on tonsillar B-cells (164, 165). While a recent study found C3aR expression both at the mRNA and protein levels within the B-cell (166), to date it remains unclear whether they express C5aR (167).
Several studies have gone on to describe the direct and indirect involvement of the complement system in modulating B-cell responses, albeit not as a consequence of trauma. For example, C3 is required for memory B-cell differentiation and locally generated C5a by peritoneal macrophages is required for chemokine ligand 13 synthesis which is further needed for B1-cell homeostasis (168, 169). Activation of the BCR increased the amount of intracellular C3aR though it could not be detected on the cell surface even after activation. Additionally, B-cell-derived C3 and C3aR appear to have a positive impact on allogeneic stimulation of T-cells (166). Studies of T-lymphocytes have confirmed surface expression of C3aR and C5aR, and revealed that C3aR and C5aR signaling have an important impact on functions of T-cell subpopulations, including Th1 and Th17 responses (170). The effects of trauma on lymphocytes has been barely investigated. Only a limited number of publications indicate that B- and T-cell functions are impaired after trauma, but currently no study exists to clarify the role of the complement system in it. This could indicate that different forms of trauma may affect different pathways of complement and lymphocyte activation, but we do not have any evidence to explain which mechanisms are involved in this putative functional cross-talk. In this context, DAMPs appear to be undermines T-cell survival. Severe injury with consequent tissue damage leads to excessive release of self-DAMPs, including histones (21). Histones released by the effect of C5a on neutrophils have also been described to induce T-cell apoptosis (Figure 2). Therefore, it may be rational to propose that post-traumatic enhanced histone levels is an indirect effect of C5a anaphylatoxin and contribute to T-cell death by apoptosis. Additional investigation is desirable to understand whether post-traumatic C3aR and C5aR signaling is affected. Regarding cells of the innate immune system, C5a can induce C5aR1-mediated morphological changes of neutrophils together with altered chemotactic activity and neutrophils from septic shock patients even displayed decreased C5aR1 expression (28, 171). Activation of C5aR has been described to also enhance proinflammatory Th1 responses and to reduce anti-inflammatory Th2 responses, which should be also addressed in the context of trauma. Because trauma induces a shift toward the Th2 phenotype, it may be speculated that C3aR and C5aR signaling is impaired or their expression is decreased (Figure 2).
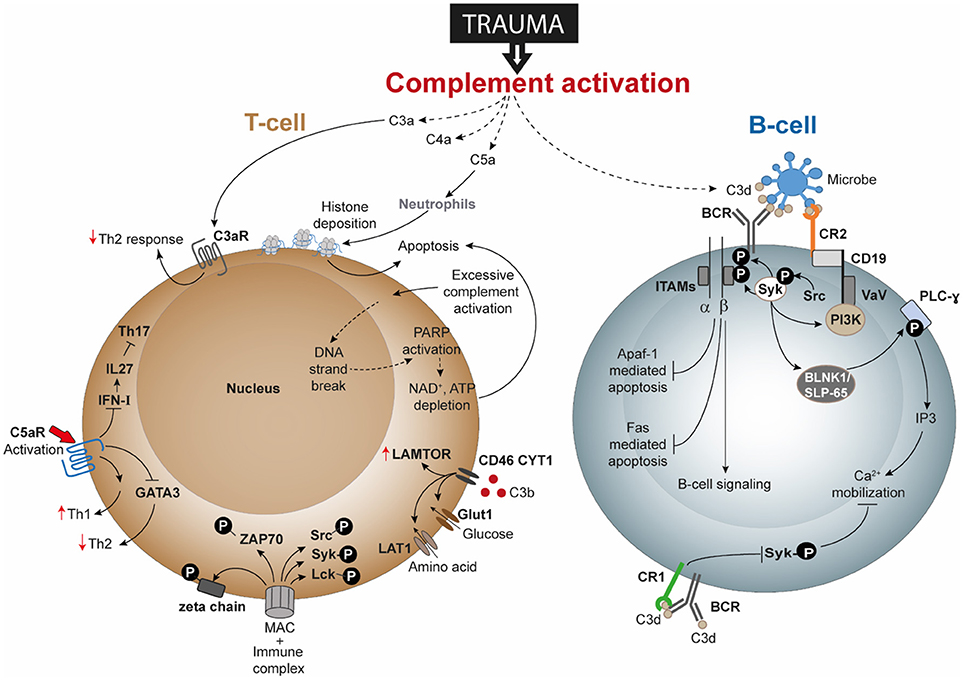
Figure 2. Speculated T-cell and B-cell responses following complement activation after trauma. Activation and dysregulation of complement factors and receptors following trauma may modulate key pathways in T-cells and B-cells. Trauma associated generation of activated complement products have been depicted with dotted lines. All these pathways are discussed in text and are potential areas where trauma research can focus upon. Apaf-1, Apoptotic protease activating factor 1; ATP, adenosine triphosphate; BCR, B-cell receptor; BLNK1/SLP-65, B cell linker protein 1; CD, cluster of differentiation; DNA, deoxyribonucleic acid; FAS: Fas cell surface death receptor; GATA3, GATA3 binding protein; Glut1, Glucose transporter 1; IFN-I, type-I interferon; IL, interleukin; IP3, inositol trisphosphate; ITAMs, immunoreceptor tyrosine-based activation motifs; LAMTOR, lysosomal adaptor and mitogen-activated protein kinase and mammalian target of rapamycin [mTOR] activator/regulator; LAT1, L-type amino acid transporter 1; Lck, Tyrosine-protein kinase Lck; MAC, membrane attack complex; NAD, nicotinamide adenine dinucleotide; PARP, poly (ADP-ribose) polymerase; PI3K, phosphoinositide 3-kinase; PLC-ɤ, phospholipase C- ɤ; Src, Proto-oncogene tyrosine-protein kinase Src; Syk, spleen tyrosine kinase; Th, T helper; VaV, Proto-oncogene vav; ZAP-70, Zeta-chain-associated protein kinase 70.
With regard to lymphocyte signaling, T-cells have been described to generate intracellular C3 and C5, which in turn can be cleaved and activated and are able to bind to receptors, including C3aR, C5aR, and CD46, thus altering signaling, cell growth, and T-cell responses. What remains unclear is whether autocrine- and/or systemically-generated C3 and C5 cleavage products (e.g., by trauma) may play predominant roles in signaling. It is well established that trauma generates excessive complement activation products. Because complement interferes with receptors on different T-cell subpopulations, including Th1, Th2 and Tregs, it is important to investigate how trauma induces changes to T-cell signaling, function and activity. In this regard, CD46 appears to represent an important receptor in the regulation of T-cell activation and function. For Th1 responses, C3b binding to CD46 and subsequent signaling has also been described to play a critical role in cytokine generation including IFN-ɤ, IL-10, and granzyme-B production in Th1 cells and in their metabolic processes (121, 172). Kolev et al. further established that changes in metabolism influence the resulting CD4+ T-cell functions (i.e., IFN-ɤ and IL-10 production), which may also be influenced in trauma (121). Excessive C3b generation after trauma may alter CD46 function or even expression on T-cell and consequently the functioning of T-cells. Following trauma, the presence of CD46 was found to be decreased on T-lymphocytes (31). Trauma may additionally change the concentrations of intracellular C3 and C5 and their activation, so that an autocrine loop alters CD46 signaling and thus T-cell activation. Further investigations could analyze whether excessive complement activation products after trauma influence T-cell survival, because complement products have been described to induce ATP depletion with subsequent T-cell apoptosis (Figure 2).
Nonetheless, no study to date has provided a mechanistic explanation of a trauma-induced impact on B-cell signaling. One interesting research field may be inhibitory receptors bearing immunoreceptor tyrosine-based inhibitory motifs (ITIMs), which are required for the regulation of B-cell immune responses. To date, numerous inhibitory receptors including CD22 and FcRII have been characterized as possessing one or more ITIMs within their cytoplasmic domain, which generate and transduce inhibitory signals (173). It may be speculated that after trauma, B-cell activation is impaired through a compensatory mechanism to protect the host from further immune reactions mediated by the adaptive system. It could also be possible that in addition to complement interaction with CR1 and CR2, the complement components may interact with these inhibitory receptors on B-cells, reinforcing inhibitory B-cell signaling and thus impairing B-cell activation. Further investigations in relation to the interaction of complement components with other receptors, particularly those with ITIMs could be considered.
Regarding complement therapeutic strategies, several specific and effective approaches have been recently evaluated for their clinical use in inflammatory disorders, including trauma, HS, and sepsis (21). The majority of these investigations focused on the influence of trauma on innate immune responses, including on neutrophil and macrophage function, but less, if at all, on the modulation of adaptive immunity. Therefore, it remains to be investigated to what extent neutrophils, macrophages and also adaptive immune cells contribute to metabolic changes after trauma and whether C5a as a “metabolic switch” may be influenced by specific inhibition (27). C5 cleavage could be inhibited ex vivo by small peptide inhibitors, which require future in vivo testing for their clinical potential (174). Whereas systemic inflammatory processes after trauma or during infection appear to significantly benefit from central complement blockades, there is no overall benefit for all injured tissues (22, 175, 176). For example, C5aR1 blockade failed to improve uneventful fracture healing, where femoral osteotomy was performed in mice following which immediate and post-12 h, a C5aR antagonist treatment was given (175). Early blockade of C5aR ameliorated both, the IL-6 levels and neutrophil recruitment at the site of injury, but late blockade did not effect in the same. Therefore, the compartmentalized immune response after trauma, involving most likely both innate and adaptive immune reactions, appears to require a specific complement intervention in the future with both adequate spatial and temporal resolution (177). Elaborating on the temporal aspect, the treatment with anti-C5a early post-blunt chest trauma in rats ameliorated subsequent leukocytosis, reduced white blood cell recruitment in the lung, and also decreased trauma induced TNF-α levels (178). As for local effects, 12 h after blunt chest trauma coupled with sepsis induction, C5 deficient mice exhibited decrease in cytokines IL-6 and monocyte chemotactic protein-1 levels in the lung tissue (179). Local healing processes are actively under the regulation of C5a, as its cognate receptor C5aR1 deficiency (C5aR1-/-) affected fracture healing in a mouse model of femoral fracture (180). A remarkable finding in this same study was that, 14 days after fracture in C5aR2 deficient mice, the recruitment of CD8+ T-cells was significantly higher compared to C5aR1-/- mice with fracture, indicating the differences in the elicited function of C5aR1 and C5aR2. Hence, targeted inhibition of complement at the damaged-tissue site is already partly realized. For example, the chimeric CR2-fH construct (mTT30), which can inhibit neuroinflammatory response after traumatic brain injury (181). Similarly, other complement blockers like TCC inhibitors C6 antisense oligonucleotide (OMCI) (182) and CD59-2a-CRIg (183) could turn out to be promising inhibitors and as a result, modulators of the adaptive immune responses. However, in the future, such damage-targeted principles need clinical translation and must not only consider the innate immune response but also the adaptive immune response after trauma, thereby aiming to improve any observed B- and T-cell dysfunction. Another emerging field represents vaccines that influence B- and T-cells in the context of prevention or treatment of infectious morbidity and mortality after traumatic injury, including soft-tissue wounds, human or animal bites, or after splenectomy (184). New effective therapeutics on the C3 level, for example, by compstatin as a small peptide inhibitor and on the C5 level, for example, by NDT9513727 as a non-peptide inverse agonist of C5aR1, need to be tested in the trauma context not only on the innate immune responses but also on the adaptive immune changes (185–187). In the clinic, dysfunctional adaptive immunity after severe tissue trauma requires improved and timely diagnosis. A first approach is represented by clear definition of the persistent inflammation–immunosuppressive catabolism syndrome with its innate and adaptive immune-suppressive characteristics (188). Up until now, the underlying mechanisms remain unclear and need to be elucidated to improve the long-term quality of life of a patient after severe trauma. Overall, complement may represent an innovative therapeutic target in trauma and critically ill patients, because it principally bridges innate and adaptive immunity.
SC is the first author, EK is the second and MH-L is the Senior author of the paper.
This work is supported by grants from the German Research Foundation (DFG) to MH-L. (INST 40/479-1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Dr. Stephanie Denk for preparing the figures for this review.
Apaf-1, Apoptotic protease activating factor 1; APC, antigen presenting cell; ARDS, Adult respiratory distress syndrome; ATP, adenosine triphosphate; BCR, B-cell receptor; BLK, B-cell lymphocyte kinase; BLNK, B cell linker protein; Btk, Bruton's tyrosine kinase; cAMP, cyclic adenosine monophosphate; CD, cluster of differentiation; Cig, cytoplasmatic Ig; CLP, cecal ligation and puncture; CR, complement receptor; CR2-fH, Complement Receptor 2- factor H fusion protein; DAF, decay-accelerating factor; DAMP, damage-associated molecular pattern; DC, dendritic cell; DNA, deoxyribonucleic acidEP-54, response-selective molecular agonist peptide; ERK(1/2), extracellular signaling-regulated kinase; Fas, first apoptosis signal; FcRII, Fc gamma receptor; FDC, follicular dendritic cell; fMLP, N-Formyl-L-methionyl-L-leucyl-L-phenylalanine; GLUT1, Glucose transporter 1; HLA-DR, human leukocyte antigen – antigen D Related; Ig, immunoglobulin; IL, interleukin; IP3, inositol trisphosphate; IR, ischemia-reperfusion; ITAM, immunoreceptor tyrosine-based activation motif; ITIM, immunoreceptor tyrosine-based inhibitory motif; LAMTOR, lysosomal adaptor and mitogen-activated protein kinase and mammalian target of rapamycin [mTOR] activator/regulator; LAT1, L-type amino acid transporter 1; Lck, Tyrosine-protein kinase Lck; MAC, membrane attack complex; MAPK, mitogen-activated protein kinase; MHC, major histocompatibility complex; MZ, marginal zone; NAD, nicotinamide adenine dinucleotide; NF-κB, nuclear factor κ-light-chain-enhancer of activated B cells; NLRP3, NACHT, LRR and PYD domains-containing protein 3; PAMP, pathogen-associated molecular pattern; PARP, poly (ADP-ribose) polymerase; PI3K, phosphoinositide 3-kinase; PLC, phospholipase C; PTK, protein-tyrosin-kinase; Rag1, recombination activating gene 1; RNA, ribonucleic acid; sCR1, soluble complement receptor 1; Src, Proto-oncogene tyrosine-protein kinase Src; Syk, spleen tyrosine kinase; TBI, traumatic brain injury; TCC, terminal complement complex; TGF-β, transforming growth factor beta; Th, T helper lymphocytes; Treg, T regulatory lymphocytes; TUNEL, Terminal deoxynucleotidyl transferase dUTP nick end labeling; WT, wild-type; ZAP-70, Zeta-chain-associated protein kinase 70.
1. Kleisiaris CF, Sfakianakis C, Papathanasiou IV. Health care practices in ancient Greece: the hippocratic ideal. J Med Ethics Hist Med. (2014) 7:6.
2. Zuckerman L, Caprini JA, Lipp V, Vagher JP. Disseminated intravascular multiple systems activation (DIMSA) following thermal injury. J Trauma. (1978) 18:432–9. doi: 10.1097/00005373-197806000-00008
3. Herman CM. The patient with multiple organ system failure: immunologic monitoring. Adv Shock Res. (1982) 8:195–207.
4. Risberg B, Medegard A, Heideman M, Gyzander E, Bundsen P, Oden M, et al. Early activation of humoral proteolytic systems in patients with multiple trauma. Crit Care Med. (1986) 14:917–25. doi: 10.1097/00003246-198611000-00001
5. Heideman M, Hugli TE. Anaphylatoxin generation in multisystem organ failure. J Trauma. (1984) 24:1038–43. doi: 10.1097/00005373-198412000-00006
6. Zilow G, Sturm JA, Rother U, Kirschfink M. Complement activation and the prognostic value of C3a in patients at risk of adult respiratory distress syndrome. Clin Exp Immunol. (1990) 79:151–7. doi: 10.1111/j.1365-2249.1990.tb05171.x
7. Ganter MT, Brohi K, Cohen MJ, Shaffer LA, Walsh MC, Stahl GL, et al. Role of the alternative pathway in the early complement activation following major trauma. Shock (2007) 28:29–34. doi: 10.1097/shk.0b013e3180342439
8. Fosse E, Mollnes TE, Aasen AO, Trumpy JH, Stokke T. Complement activation following multiple injuries. Acta Chir Scand. (1987) 153:325–30.
9. Zilow G, Joka T, Obertacke U, Rother U, Kirschfink M. Generation of anaphylatoxin C3a in plasma and bronchoalveolar lavage fluid in trauma patients at risk for the adult respiratory distress syndrome. Crit Care Med. (1992) 20:468–73. doi: 10.1097/00003246-199204000-00006
10. Burk AM, Martin M, Flierl MA, Rittirsch D, Helm M, Lampl L, et al. Early complementopathy after multiple injuries in humans. Shock. (2012) 37:348–54. doi: 10.1097/SHK.0b013e3182471795
11. Roumen RM, Redl H, Schlag G, Zilow G, Sandtner W, Koller W, et al. Inflammatory mediators in relation to the development of multiple organ failure in patients after severe blunt trauma. Crit Care Med. (1995) 23:474–80. doi: 10.1097/00003246-199503000-00010
12. Hecke F, Schmidt U, Kola A, Bautsch W, Klos A, Kohl J. Circulating complement proteins in multiple trauma patients–correlation with injury severity, development of sepsis, and outcome. Crit Care Med. (1997) 25:2015–24. doi: 10.1097/00003246-199712000-00019
13. Keel M, Trentz O. Pathophysiology of polytrauma. Injury (2005) 36:691–709. doi: 10.1016/j.injury.2004.12.037
14. Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. (2006) 12:682–7. doi: 10.1038/nm1419
15. Hoth JJ, Wells JD, Jones SE, Yoza BK, McCall CE. Complement mediates a primed inflammatory response after traumatic lung injury. J Trauma Acute Care Surg. (2014) 76:601–8. doi: 10.1097/TA.0000000000000129
16. Kanse SM, Gallenmueller A, Zeerleder S, Stephan F, Rannou O, Denk S, et al. Factor VII-activating protease is activated in multiple trauma patients and generates anaphylatoxin C5a. J Immunol. (2012) 188:2858–65. doi: 10.4049/jimmunol.1103029
17. Huber-Lang M, Ekdahl KN, Wiegner R, Fromell K, Nilsson B. Auxiliary activation of the complement system and its importance for the pathophysiology of clinical conditions. Semin Immunopathol. (2018) 40:87–102. doi: 10.1007/s00281-017-0646-9
18. Zimmermann T, Laszik Z, Nagy S, Kaszaki J, Joo F. The role of the complement system in the pathogenesis of multiple organ failure in shock. Prog Clin Biol Res. (1989) 308:291–7.
19. Grob P, Holch M, Fierz W, Glinz W, Geroulanos S. Immunodeficiency after major trauma and selective surgery. Pediatr Infect Dis J. (1988) 7:S37–S42. doi: 10.1097/00006454-198805001-00009
20. Huber-Lang M, Denk S, Fulda S, Erler E, Kalbitz M, Weckbach S, et al. Cathepsin D is released after severe tissue trauma in vivo and is capable of generating C5a in vitro. Mol Immunol. (2012) 50:60–5. doi: 10.1016/j.molimm.2011.12.005
21. Huber-Lang M, Lambris JD, Ward PA. Innate immune responses to trauma. Nat Immunol. (2018) 19:327–41. doi: 10.1038/s41590-018-0064-8
22. Huber-Lang M, Kovtun A, Ignatius A. The role of complement in trauma and fracture healing. Semin Immunol. (2013) 25:73–8. doi: 10.1016/j.smim.2013.05.006
23. Hietbrink F, Koenderman L, Rijkers G, Leenen L. Trauma: the role of the innate immune system. World J Emerg Surg. (2006) 1:15. doi: 10.1186/1749-7922-1-15
24. Gallinaro R, Cheadle WG, Applegate K, Polk HC Jr. The role of the complement system in trauma and infection. Surg Gynecol Obstet. (1992) 174:435–40.
25. Gerard C. Complement C5a in the sepsis syndrome–too much of a good thing? N Engl J Med. (2003) 348:167–9. doi: 10.1056/NEJMcibr022995
26. Lohde E, Raude H, Luck M, Kraas E, Lierse W. Complement activated granulocytes can cause autologous tissue destruction in man. Mediators Inflamm. (1992) 1:177–81. doi: 10.1155/S0962935192000279
27. Denk S, Neher MD, Messerer DAC, Wiegner R, Nilsson B, Rittirsch D, et al. Complement C5a functions as a master switch for the ph balance in neutrophils exerting fundamental immunometabolic effects. J Immunol. (2017) 198:4846–54. doi: 10.4049/jimmunol.1700393
28. Denk S, Taylor RP, Wiegner R, Cook EM, Lindorfer MA, Pfeiffer K, et al. Complement C5a-induced changes in neutrophil morphology during inflammation. Scand J Immunol. (2017) 86:143–55. doi: 10.1111/sji.12580
29. Solomkin JS, Cotta LA, Ogle JD, Brodt JK, Ogle CK, Satoh PS, et al. Complement-induced expression of cryptic receptors on the neutrophil surface: a mechanism for regulation of acute inflammation in trauma. Surgery (1984) 96:336–44.
30. Tennenberg SD, Jacobs MP, Solomkin JS. Complement-mediated neutrophil activation in sepsis- and trauma-related adult respiratory distress syndrome. clarification with radioaerosol lung scans. Arch Surg. (1987) 122:26–32. doi: 10.1001/archsurg.1987.01400130032004
31. Amara U, Kalbitz M, Perl M, Flierl MA, Rittirsch D, Weiss M, et al. Early expression changes of complement regulatory proteins and C5A receptor (CD88) on leukocytes after multiple injury in humans. Shock (2010) 33:568–75. doi: 10.1097/SHK.0b013e3181c799d4
32. Huber-Lang M, Sarma JV, Rittirsch D, Schreiber H, Weiss M, Flierl M, et al. Changes in the novel orphan, C5a receptor (C5L2), during experimental sepsis and sepsis in humans. J Immunol. (2005) 174:1104–10. doi: 10.4049/jimmunol.174.2.1104
33. Rittirsch D, Redl H, Huber-Lang M. Role of complement in multiorgan failure. Clin Dev Immunol. (2012) 2012:962927. doi: 10.1155/2012/962927
34. Rittirsch D, Schoenborn V, Lindig S, Wanner E, Sprengel K, Gunkel S, et al. Improvement of prognostic performance in severely injured patients by integrated clinico-transcriptomics: a translational approach. Crit Care. (2015) 19:414. doi: 10.1186/s13054-015-1127-y
35. Schaeffer V, Cuschieri J, Garcia I, Knoll M, Billgren J, Jelacic S, et al. The priming effect of C5a on monocytes is predominantly mediated by the p38 MAPK pathway. Shock (2007) 27:623–30. doi: 10.1097/SHK.0b013e31802fa0bd
36. Antrum RM, Solomkin JS. Complement activation products and monocyte migratory function in trauma. Curr Surg. (1985) 42:301–3.
37. Antrum RM, Solomkin JS. Monocyte dysfunction in severe trauma: evidence for the role of C5a in deactivation. Surgery (1986) 100:29–37.
38. Bekeschus S, Lackmann JW, Gumbel D, Napp M, Schmidt A, Wende K. A Neutrophil proteomic signature in surgical trauma wounds. Int J Mol Sci. (2018) 19:E761:doi: 10.3390/ijms19030761
39. Narang A, Qiao F, Atkinson C, Zhu H, Yang X, Kulik L, et al. Natural IgM antibodies that bind neoepitopes exposed as a result of spinal cord injury, drive secondary injury by activating complement. J Neuroinflammation (2017) 14:120. doi: 10.1186/s12974-017-0894-6
40. Al NF, Lindblom R, Strom M, Guerreiro-Cacais AO, Parsa R, Aeinehband S, et al. Strain influences on inflammatory pathway activation, cell infiltration and complement cascade after traumatic brain injury in the rat. Brain Behav Immun. (2013) 27:109–22. doi: 10.1016/j.bbi.2012.10.002
41. Bellander BM, Lidman O, Ohlsson M, Meijer B, Piehl F, Svensson M. Genetic regulation of microglia activation, complement expression, and neurodegeneration in a rat model of traumatic brain injury. Exp Brain Res. (2010) 205:103–14. doi: 10.1007/s00221-010-2342-z
42. Weckbach S, Neher M, Losacco JT, Bolden AL, Kulik L, Flierl MA, et al. Challenging the role of adaptive immunity in neurotrauma: Rag1(-/-) mice lacking mature B and T cells do not show neuroprotection after closed head injury. J Neurotrauma. (2012) 29:1233–42. doi: 10.1089/neu.2011.2169
43. Ducruet AF, Zacharia BE, Sosunov SA, Gigante PR, Yeh ML, Gorski JW, et al. Complement inhibition promotes endogenous neurogenesis and sustained anti-inflammatory neuroprotection following reperfused stroke. PLoS ONE (2012) 7:e38664. doi: 10.1371/journal.pone.0038664
44. Li M, Peake PW, Charlesworth JA, Tracey DJ, Moalem-Taylor G. Complement activation contributes to leukocyte recruitment and neuropathic pain following peripheral nerve injury in rats. Eur J Neurosci. (2007) 26:3486–500. doi: 10.1111/j.1460-9568.2007.05971.x
45. Morelli AE, Larregina AT, Shufesky WJ, Zahorchak AF, Logar AJ, Papworth GD, et al. Internalization of circulating apoptotic cells by splenic marginal zone dendritic cells: dependence on complement receptors and effect on cytokine production. Blood (2003) 101:611–20. doi: 10.1182/blood-2002-06-1769
46. Zachrau B, Finke D, Kropf K, Gosink HJ, Kirchner H, Goerg S. Antigen localization within the splenic marginal zone restores humoral immune response and IgG class switch in complement C4-deficient mice. Int Immunol. (2004) 16:1685–90. doi: 10.1093/intimm/dxh159
47. Downey EC, Shackford SR, Fridlund PH, Ninnemann JL. Long-term depressed immune function in patients splenectomized for trauma. J Trauma. (1987) 27:661–3. doi: 10.1097/00005373-198706000-00010
48. Tsai MC, Lin SL, Chuang CY. Changes in T-lymphocyte subpopulations in patients splenectomized for trauma. J Formos Med Assoc. (1991) 90:240–3.
49. Walusimbi MS, Dominguez KM, Sands JM, Markert RJ, McCarthy MC. Circulating cellular and humoral elements of immune function following splenic arterial embolisation or splenectomy in trauma patients. Injury (2012) 43:180–3. doi: 10.1016/j.injury.2011.05.028
50. Ninnemann JL, Ozkan AN. The immunosuppressive activity of C1q degradation peptides. J Trauma. (1987) 27:119–22.
51. Ferrara JJ, Dyess DL, Luterman A, Peterson RD, Curreri PW. In vitro effects of complement inactivation upon burn-associated cell-mediated immunosuppression. Am Surg. (1990) 56:571–4.
52. Korkmaz HI, Ulrich MMW, van Wieringen WN, Vlig M, Emmens RW, Meyer KW, et al. The Local and systemic inflammatory response in a pig burn wound model with a pivotal Role for complement. J Burn Care Res. (2017) 38:e796–806. doi: 10.1097/BCR.0000000000000486
53. Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. (2012) 12:557–69. doi: 10.1038/nri3254
54. Stephan RN, Saizawa M, Conrad PJ, Dean RE, Geha AS, Chaudry IH. Depressed antigen presentation function and membrane interleukin-1 activity of peritoneal macrophages after laparotomy. Surgery (1987) 102:147–54.
55. Ayala A, Ertel W, Chaudry IH. Trauma-induced suppression of antigen presentation and expression of major histocompatibility class II antigen complex in leukocytes. Shock (1996) 5:79–90. doi: 10.1097/00024382-199602000-00001
56. Kawasaki T, Fujimi S, Lederer JA, Hubbard WJ, Choudhry MA, Schwacha MG, et al. Trauma-hemorrhage induces depressed splenic dendritic cell functions in mice. J Immunol. (2006) 177:4514–20. doi: 10.4049/jimmunol.177.12.8877-a
57. Kawasaki T, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. Effect of interleukin-15 on depressed splenic dendritic cell functions following trauma-hemorrhage. Am J Physiol Cell Physiol. (2009) 296:C124–30. doi: 10.1152/ajpcell.00447.2008
58. Hietbrink F, Koenderman L, Althuizen M, Pillay J, Kamp V, Leenen LP. Kinetics of the innate immune response after trauma: implications for the development of late onset sepsis. Shock (2013) 40:21–7. doi: 10.1097/SHK.0b013e318295a40a
59. Tobin RP, Mukherjee S, Kain JM, Rogers SK, Henderson SK, Motal HL, et al. Traumatic brain injury causes selective, CD74-dependent peripheral lymphocyte activation that exacerbates neurodegeneration. Acta Neuropathol Commun. (2014) 2:143. doi: 10.1186/s40478-014-0143-5
60. Grootjans J, Hodin CM, de Haan JJ, Derikx JP, Rouschop KM, Verheyen FK, et al. Level of activation of the unfolded protein response correlates with Paneth cell apoptosis in human small intestine exposed to ischemia/reperfusion. Gastroenterology (2011) 140:529–39. doi: 10.1053/j.gastro.2010.10.040
61. Tiesi G, Reino D, Mason L, Palange D, Tomaio JN, Deitch EA. Early trauma-hemorrhage-induced splenic and thymic apoptosis is gut-mediated and toll-like receptor 4-dependent. Shock (2013) 39:507–13. doi: 10.1097/SHK.0b013e318293d020
62. Wan C, Chen J, Hu B, Zou H, Li A, Guo A, et al. Downregulation of UBE2Q1 is associated with neuronal apoptosis in rat brain cortex following traumatic brain injury. J Neurosci Res. (2014) 92:1–12. doi: 10.1002/jnr.23305
63. Wu J, Kharebava G, Piao C, Stoica BA, Dinizo M, Sabirzhanov B, et al. Inhibition of E2F1/CDK1 pathway attenuates neuronal apoptosis in vitro and confers neuroprotection after spinal cord injury in vivo. PLoS ONE. (2012) 7:e42129. doi: 10.1371/journal.pone.0042129
64. Nauta AJ, Castellano G, Xu W, Woltman AM, Borrias MC, Daha MR, et al. Opsonization with C1q and mannose-binding lectin targets apoptotic cells to dendritic cells. J Immunol. (2004) 173:3044–50. doi: 10.4049/jimmunol.173.5.3044
65. Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem. (2010) 112:733–43. doi: 10.1111/j.1471-4159.2009.06494.x
66. Clarke EV, Weist BM, Walsh CM, Tenner AJ. Complement protein C1q bound to apoptotic cells suppresses human macrophage and dendritic cell-mediated Th17 and Th1 T cell subset proliferation. J Leukoc Biol. (2015) 97:147–60. doi: 10.1189/jlb.3A0614-278R
67. Bosmann M, Ward PA. Therapeutic potential of targeting IL-17 and IL-23 in sepsis. Clin Transl Med. (2012) 1:4. doi: 10.1186/2001-1326-1-4
68. Ditschkowski M, Kreuzfelder E, Majetschak M, Obertacke U, Schade UF, Grosse-Wilde H. Reduced B cell HLA-DR expression and natural killer cell counts in patients prone to sepsis after injury. Eur J Surg. (1999) 165:1129–33. doi: 10.1080/110241599750007630
69. Samy TS, Schwacha MG, Chung CS, Cioffi WG, Bland KI, Chaudry IH. Proteasome participates in the alteration of signal transduction in T and B lymphocytes following trauma-hemorrhage. Biochim Biophys Acta (1999) 1453:92–104. doi: 10.1016/S0925-4439(98)00089-1
70. Sandor N, Pap D, Prechl J, Erdei A, Bajtay Z. A novel, complement-mediated way to enhance the interplay between macrophages, dendritic cells and T lymphocytes. Mol Immunol. (2009) 47:438–48. doi: 10.1016/j.molimm.2009.08.025
71. Pekkarinen PT, Vaali K, Junnikkala S, Rossi LH, Tuovinen H, Meri S, et al. A functional complement system is required for normal T helper cell differentiation. Immunobiology (2011) 216:737–43. doi: 10.1016/j.imbio.2010.10.004
72. Baudino L, Sardini A, Ruseva MM, Fossati-Jimack L, Cook HT, Scott D, et al. C3 opsonization regulates endocytic handling of apoptotic cells resulting in enhanced T-cell responses to cargo-derived antigens. Proc Natl Acad Sci USA. (2014) 111:1503–8. doi: 10.1073/pnas.1316877111
73. Heesters BA, Chatterjee P, Kim YA, Gonzalez SF, Kuligowski MP, Kirchhausen T, et al. Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity (2013) 38:1164–75. doi: 10.1016/j.immuni.2013.02.023
74. Prechl J, Baiu DC, Horvath A, Erdei A. Modeling the presentation of C3d-coated antigen by B lymphocytes: enhancement by CR1/2-BCR co-ligation is selective for the co-ligating antigen. Int Immunol. (2002) 14:241–7. doi: 10.1093/intimm/14.3.241
75. Hess MW, Schwendinger MG, Eskelinen EL, Pfaller K, Pavelka M, Dierich MP, et al. Tracing uptake of C3dg-conjugated antigen into B cells via complement receptor type 2:(CR2, CD21). Blood (2000) 95 2617–23.
76. Kerekes K, Prechl J, Bajtay Z, Jozsi M, Erdei A. A further link between innate and adaptive immunity: C3 deposition on antigen-presenting cells enhances the proliferation of antigen-specific T cells. Int Immunol. (1998) 10:1923–30. doi: 10.1093/intimm/10.12.1923
77. Kerekes K, Cooper PD, Prechl J, Jozsi M, Bajtay Z, Erdei A. Adjuvant effect of gamma-inulin is mediated by C3 fragments deposited on antigen-presenting cells. J Leukoc Biol. (2001) 69:69–74. doi: 10.1189/jlb.69.1.69
78. Serra VA, Cretin F, Pepin E, Gabert FM, Marche PN. Complement C3b fragment covalently linked to tetanus toxin increases lysosomal sodium dodecyl sulfate-stable HLA-DR dimer production. Eur J Immunol. (1997) 27:2673–9. doi: 10.1002/eji.1830271029
79. Cretin FC, Serra VA, Villiers MB, Laharie AM, Marche PN, Gabert FM. C3b complexation diversifies naturally processed T cell epitopes. Mol Immunol. (2007) 44:2893–9. doi: 10.1016/j.molimm.2007.01.013
80. Arvieux J, Yssel H, Colomb MG. Antigen-bound C3b and C4b enhance antigen-presenting cell function in activation of human T-cell clones. Immunology (1988) 65:229–35.
81. Rey-Millet CA, Villiers CL, Gabert FM, Chesne S, Colomb MG. C3b covalently associated to tetanus toxin modulates TT processing and presentation by U937 cells. Mol Immunol. (1994) 31:1321–7. doi: 10.1016/0161-5890(94)90050-7
82. Jacquier-Sarlin MR, Gabert FM, Villiers MB, Colomb MG. Modulation of antigen processing and presentation by covalently linked complement C3b fragment. Immunology (1995) 84:164–70.
83. Li K, Anderson KJ, Peng Q, Noble A, Lu B, Kelly AP, et al. Cyclic AMP plays a critical role in C3a-receptor-mediated regulation of dendritic cells in antigen uptake and T-cell stimulation. Blood (2008) 112:5084–94. doi: 10.1182/blood-2008-05-156646
84. Leng H, Ma L, Luo X, Kang K. Reversal of iC3b-inhibited dendritic cell differentiation via inhibition of the extracellular signal-regulated mitogen-activated protein kinase promotes CD4(+) T cell proliferation. J Photochem Photobiol B (2012) 111:50–8. doi: 10.1016/j.jphotobiol.2012.03.010
85. Hegde GV, Meyers-Clark E, Joshi SS, Sanderson SD. A conformationally-biased, response-selective agonist of C5a acts as a molecular adjuvant by modulating antigen processing and presentation activities of human dendritic cells. Int Immunopharmacol. (2008) 8:819–27. doi: 10.1016/j.intimp.2008.01.031
86. Baruah P, Dumitriu IE, Malik TH, Cook HT, Dyson J, Scott D, et al. C1q enhances IFN-gamma production by antigen-specific T cells via the CD40 costimulatory pathway on dendritic cells. Blood (2009) 113:3485–93. doi: 10.1182/blood-2008-06-164392
87. van MN, de Jong JM, Schuurhuis DH, van der Voort EI, Camps MG, Huizinga TW, et al. A novel role of complement factor C1q in augmenting the presentation of antigen captured in immune complexes to CD8+ T lymphocytes. J Immunol. (2007) 178:7581–6. doi: 10.4049/jimmunol.178.12.7581
88. Morgan BP, Marchbank KJ, Longhi MP, Harris CL, Gallimore AM. Complement: central to innate immunity and bridging to adaptive responses. Immunol Lett. (2005) 97:171–9. doi: 10.1016/j.imlet.2004.11.010
89. Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. (2004) 5:981–6. doi: 10.1038/ni1113
90. Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. (2010) 20:34–50. doi: 10.1038/cr.2009.139
91. Clarke EV, Tenner AJ. Complement modulation of T cell immune responses during homeostasis and disease. J Leukoc Biol. (2014) 96:745–56. doi: 10.1189/jlb.3MR0214-109R
92. Kossmann T, Stahel PF, Morganti-Kossmann MC, Jones JL, Barnum SR. Elevated levels of the complement components C3 and factor B in ventricular cerebrospinal fluid of patients with traumatic brain injury. J Neuroimmunol. (1997) 73:63–9. doi: 10.1016/S0165-5728(96)00164-6
93. Xu R, Wang R, Han G, Wang J, Chen G, Wang L, et al. Complement C5a regulates IL-17 by affecting the crosstalk between DC and gammadelta T cells in CLP-induced sepsis. Eur J Immunol. (2010) 40:1079–88. doi: 10.1002/eji.200940015
94. Ma N, Xing C, Xiao H, Wang Y, Wang K, Hou C, et al. C5a regulates IL-12+ DC migration to induce pathogenic Th1 and Th17 cells in sepsis. PLoS ONE (2013) 8:e69779. doi: 10.1371/journal.pone.0069779
95. Hasan Z, Palani K, Zhang S, Lepsenyi M, Hwaiz R, Rahman M, et al. Rho kinase regulates induction of T-cell immune dysfunction in abdominal sepsis. Infect Immun. (2013) 81:2499–506. doi: 10.1128/IAI.00126-13
96. Rendon JL, Choudhry MA. Th17 cells: critical mediators of host responses to burn injury and sepsis. J Leukoc Biol. (2012) 92:529–38. doi: 10.1189/jlb.0212083
97. Yuan Y, Ren J, Cao S, Zhang W, Li J. Exogenous C3 protein enhances the adaptive immune response to polymicrobial sepsis through down-regulation of regulatory T cells. Int Immunopharmacol. (2012) 12:271–7. doi: 10.1016/j.intimp.2011.11.022
98. Holloway TL, Rani M, Cap AP, Stewart RM, Schwacha MG. The association between the Th-17 immune response and pulmonary complications in a trauma ICU population. Cytokine (2015) 76:328–33. doi: 10.1016/j.cyto.2015.09.003
99. Inatsu A, Kogiso M, Jeschke MG, Asai A, Kobayashi M, Herndon DN, et al. Lack of Th17 cell generation in patients with severe burn injuries. J Immunol. (2011) 187:2155–61. doi: 10.4049/jimmunol.1003235
100. Sasaki JR, Zhang Q, Schwacha MG. Burn induces a Th-17 inflammatory response at the injury site. Burns (2011) 37:646–51. doi: 10.1016/j.burns.2011.01.028
101. Rani M, Zhang Q, Schwacha MG. Burn wound gammadelta T-cells support a Th2 and Th17 immune response. J Burn Care Res. (2014) 35:46–53. doi: 10.1097/01.bcr.0000440705.91099.cc
102. Wenderfer SE, Ke B, Hollmann TJ, Wetsel RA, Lan HY, Braun MC. C5a receptor deficiency attenuates T cell function and renal disease in MRLlpr mice. J Am Soc Nephrol. (2005) 16:3572–82. doi: 10.1681/ASN.2005040373
103. Pawaria S, Ramani K, Maers K, Liu Y, Kane LP, Levesque MC, et al. Complement component C5a permits the coexistence of pathogenic Th17 cells and type I IFN in lupus. J Immunol. (2014) 193:3288–95. doi: 10.4049/jimmunol.1401322
104. Morishita K, Coimbra R, Langness S, Eliceiri BP, Costantini TW. Neuroenteric axis modulates the balance of regulatory T cells and T-helper 17 cells in the mesenteric lymph node following trauma/hemorrhagic shock. Am J Physiol Gastrointest Liver Physiol. (2015) 309 G202–8. doi: 10.1152/ajpgi.00097.2015
105. Gupta DL, Bhoi S, Mohan T, Galwnkar S, Rao DN. Coexistence of Th1/Th2 and Th17/Treg imbalances in patients with post traumatic sepsis. Cytokine (2016) 88:214–21. doi: 10.1016/j.cyto.2016.09.010
106. Abboud A, Namas RA, Ramadan M, Mi Q, Almahmoud K, Abdul-Malak O, et al. Computational analysis supports an early, type 17 cell-associated divergence of blunt trauma survival and mortality. Crit Care Med. (2016) 44 e1074–81. doi: 10.1097/CCM.0000000000001951
107. Colburn NT, Zaal KJ, Wang F, Tuan RS. A role for gamma/delta T cells in a mouse model of fracture healing. Arthritis Rheum. (2009) 60:1694–703. doi: 10.1002/art.24520
108. Rani M, Zhang Q, Schwacha MG. Gamma delta T cells regulate wound myeloid cell activity after burn. Shock (2014) 42:133–41. doi: 10.1097/SHK.0000000000000176
109. Rani M, Zhang Q, Oppeltz RF, Schwacha MG. Gamma delta T cells regulate inflammatory cell infiltration of the lung after trauma-hemorrhage. Shock (2015) 43:589–97. doi: 10.1097/SHK.0000000000000358
110. Schwacha MG, Rani M, Nicholson SE, Lewis AM, Holloway TL, Sordo S, et al. Dermal gammadelta T-Cells can be activated by mitochondrial damage-associated molecular patterns. PLoS ONE (2016) 11:e0158993. doi: 10.1371/journal.pone.0158993
111. Grailer JJ, Fattahi F, Dick RS, Zetoune FS, Ward PA. Cutting edge: critical role for C5aRs in the development of septic lymphopenia in mice. J Immunol. (2015) 194:868–72. doi: 10.4049/jimmunol.1401193
112. Ghannam A, Pernollet M, Fauquert JL, Monnier N, Ponard D, Villiers MB, et al. Human C3 deficiency associated with impairments in dendritic cell differentiation, memory B cells, and regulatory T cells. J Immunol. (2008) 181:5158–66. doi: 10.4049/jimmunol.181.7.5158
113. Dalle Lucca JJ, Simovic M, Li Y, Moratz C, Falabella M, Tsokos GC. Decay-accelerating factor mitigates controlled hemorrhage-instigated intestinal and lung tissue damage and hyperkalemia in swine. J Trauma. (2011) 71:S151–60. doi: 10.1097/TA.0b013e318221aa4c
114. Li Q, Huang D, Nacion K, Bu H, Lin F. Augmenting DAF levels in vivo ameliorates experimental autoimmune encephalomyelitis. Mol Immunol. (2009) 46:2885–91. doi: 10.1016/j.molimm.2009.07.003
115. Fishelson Z, Horstmann RD, Muller-Eberhard HJ. Regulation of the alternative pathway of complement by pH. J Immunol. (1987) 138:3392–5.
116. Bacurau RF, O'Toole CE, Newsholme P, Costa Rosa LF. Sub-lethal concentrations of activated complement increase rat lymphocyte glutamine utilization and oxidation while lethal concentrations cause death by a mechanism involving ATP depletion. Cell Biochem Funct. (2002) 20:183–90. doi: 10.1002/cbf.943
117. Chauhan AK, Moore TL. T cell activation by terminal complex of complement and immune complexes. J Biol Chem. (2011) 286:38627–37. doi: 10.1074/jbc.M111.266809
118. Chauhan AK, Moore TL. Immune complexes and late complement proteins trigger activation of Syk tyrosine kinase in human CD4(+) T cells. Clin Exp Immunol. (2012) 167:235-45. doi: 10.1111/j.1365-2249.2011.04505.x
119. Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity (2008) 28:425–35. doi: 10.1016/j.immuni.2008.02.001
120. Liszewski MK, Kolev M, Le FG, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity (2013) 39:1143–57. doi: 10.1016/j.immuni.2013.10.018
121. Kolev M, Dimeloe S, Le FG, Navarini A, Arbore G, Povoleri GA, et al. Complement regulates nutrient influx and metabolic reprogramming during th1 cell responses. Immunity (2015) 42:1033–47. doi: 10.1016/j.immuni.2015.05.024
122. Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T helper. 1:immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science (2016) 352:aad1210. doi: 10.1126/science.aad1210
123. Valvis SM, Waithman J, Wood FM, Fear MW, Fear VS. The immune response to skin trauma is dependent on the etiology of injury in a mouse model of burn and excision. J Invest Dermatol. (2015) 135:2119–28. doi: 10.1038/jid.2015.123
124. Held KS, Lane TE. Spinal cord injury, immunodepression, and antigenic challenge. Semin Immunol. (2014) 26:415–20. doi: 10.1016/j.smim.2014.03.003
125. Oropallo MA, Goenka R, Cancro MP. Spinal cord injury impacts B cell production, homeostasis, and activation. Semin Immunol. (2014) 26:421–7. doi: 10.1016/j.smim.2014.09.014
126. Faist E, Ertel W, Baker CC, Heberer G. Terminal B-cell maturation and immunoglobulin (Ig) synthesis in vitro in patients with major injury. J Trauma (1989) 29:2–9. doi: 10.1097/00005373-198901000-00002
127. Puyana JC, Pellegrini JD, De AK, Kodys K, Silva WE, Miller CL. Both T-helper-1- and T-helper-2-type lymphokines are depressed in posttrauma anergy. J Trauma (1998) 44:1037–45. doi: 10.1097/00005373-199806000-00017
128. Gallagher G, Willdridge J. Evidence that interleukin 2 actively promotes both cell division and immunoglobulin secretion in human B cells. Int Arch Allergy Appl Immunol. (1988) 86 337–42. doi: 10.1159/000234594
129. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
130. Gonzalez SF, Kuligowski MP, Pitcher LA, Roozendaal R, Carroll MC. The role of innate immunity in B cell acquisition of antigen within LNs. Adv Immunol. (2010) 106:1–19. doi: 10.1016/S0065-2776(10)06001-3
131. Gonzalez SF, Lukacs-Kornek V, Kuligowski MP, Pitcher LA, Degn SE, Turley SJ, et al. Complement-dependent transport of antigen into B cell follicles. J Immunol. (2010) 185:2659–64. doi: 10.4049/jimmunol.1000522
132. Gonzalez SF, Degn SE, Pitcher LA, Woodruff M, Heesters BA, Carroll MC. Trafficking of B cell antigen in lymph nodes. Annu Rev Immunol. (2011) 29:215–33. doi: 10.1146/annurev-immunol-031210-101255
133. Fang Y, Xu C, Fu YX, Holers VM, Molina H. Expression of complement receptors. (1998) 1:and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response. J Immunol. 160 5273–9.
134. Hlavacek WS, Percus JK, Percus OE, Perelson AS, Wofsy C. Retention of antigen on follicular dendritic cells and B lymphocytes through complement-mediated multivalent ligand-receptor interactions: theory and application to HIV treatment. Math Biosci. (2002) 176:185–202. doi: 10.1016/S0025-5564(02)00091-3
135. Ferguson AR, Youd ME, Corley RB. Marginal zone B cells transport and deposit IgM-containing immune complexes onto follicular dendritic cells. Int Immunol. (2004) 16:1411–22. doi: 10.1093/intimm/dxh142
136. Truedsson L, Bengtsson AA, Sturfelt G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity (2007) 40:560–6. doi: 10.1080/08916930701510673
137. Vignesh P, Rawat A, Sharma M, Singh S. Complement in autoimmune diseases. Clin Chim Acta (2017) 465:123–30. doi: 10.1016/j.cca.2016.12.017
138. Prodeus AP, Goerg S, Shen LM, Pozdnyakova OO, Chu L, Alicot EM, et al. A critical role for complement in maintenance of self-tolerance. Immunity (1998) 9:721–31. doi: 10.1016/S1074-7613(00)80669-X
139. Pekkarinen PT, Heikkila N, Kisand K, Peterson P, Botto M, Daha MR, et al. Dysregulation of adaptive immune responses in complement C3-deficient patients. Eur J Immunol. (2015) 45:915–21. doi: 10.1002/eji.201444948
140. Toapanta FR, Ross TM. Complement-mediated activation of the adaptive immune responses: role of C3d in linking the innate and adaptive immunity. Immunol Res. (2006) 36:197–210. doi: 10.1385/IR:36:1:197
141. Kurosaki T, Takata M, Yamanashi Y, Inazu T, Taniguchi T, Yamamoto T, et al. Syk activation by the Src-family tyrosine kinase in the B cell receptor signaling. J Exp Med. (1994) 179:1725–9. doi: 10.1084/jem.179.5.1725
142. Fu C, Turck CW, Kurosaki T, Chan AC. BLNK: a central linker protein in B cell activation. Immunity (1998) 9:93–103. doi: 10.1016/S1074-7613(00)80591-9
143. Goitsuka R, Fujimura Y, Mamada H, Umeda A, Morimura T, Uetsuka K, et al. BASH, a novel signaling molecule preferentially expressed in B cells of the bursa of Fabricius. J Immunol. (1998) 161:5804–8.
144. Wienands J, Schweikert J, Wollscheid B, Jumaa H, Nielsen PJ, Reth M. SLP-65: a new signaling component in B lymphocytes which requires expression of the antigen receptor for phosphorylation. J Exp Med. (1998) 188:791–5. doi: 10.1084/jem.188.4.791
145. Rolli V, Gallwitz M, Wossning T, Flemming A, Schamel WW, Zurn C, et al. Amplification of B cell antigen receptor signaling by a Syk/ITAM positive feedback loop. Mol Cell. (2002) 10:1057–69. doi: 10.1016/S1097-2765(02)00739-6
146. Marshall AJ, Niiro H, Yun TJ, Clark EA. Regulation of B-cell activation and differentiation by the phosphatidylinositol 3-kinase and phospholipase Cgamma pathway. Immunol Rev. (2000) 176:30–46.
147. Hashimoto S, Iwamatsu A, Ishiai M, Okawa K, Yamadori T, Matsushita M, et al. Identification of the SH2 domain binding protein of Bruton's tyrosine kinase as BLNK–functional significance of Btk-SH2 domain in B-cell antigen receptor-coupled calcium signaling. Blood. (1999) 94:2357–64.
148. Kurosaki T, Tsukada S. BLNK: connecting Syk and Btk to calcium signals. Immunity (2000) 12:1–5. doi: 10.1016/S1074-7613(00)80153-3
149. Su YW, Zhang Y, Schweikert J, Koretzky GA, Reth M, Wienands J. Interaction of SLP adaptors with the SH2 domain of Tec family kinases. Eur J Immunol. (1999) 29:3702–11.
150. Gerondakis S, Siebenlist U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb Perspect Biol. (2010) 2:a000182. doi: 10.1101/cshperspect.a000182
151. Rothstein TL, Zhong X, Schram BR, Negm RS, Donohoe TJ, Cabral DS, et al. Receptor-specific regulation of B-cell susceptibility to Fas-mediated apoptosis and a novel Fas apoptosis inhibitory molecule. Immunol Rev. (2000) 176:116–33.
152. de Gorter DJ, Vos JC, Pals ST, Spaargaren M. The B cell antigen receptor controls AP-1 and NFAT activity through Ras-mediated activation of Ral. J Immunol. (2007) 178:1405–14. doi: 10.4049/jimmunol.178.3.1405
153. Carter RH, Spycher MO, Ng YC, Hoffman R, Fearon DT. Synergistic interaction between complement receptor type. 2:and membrane IgM on B lymphocytes. J Immunol. (1988) 141 457–63.
154. Cherukuri A, Cheng PC, Pierce SK. The role of the CD19/CD21 complex in B cell processing and presentation of complement-tagged antigens. J Immunol. (2001) 167:163–72. doi: 10.4049/jimmunol.167.1.163
155. Carter RH, Fearon DT. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science (1992) 256:105–7.
156. Mongini PK, Jackson AE, Tolani S, Fattah RJ, Inman JK. Role of complement-binding CD21/CD19/CD81 in enhancing human B cell protection from Fas-mediated apoptosis. J Immunol. (2003) 171:5244–54. doi: 10.4049/jimmunol.171.10.5244
157. Jozsi M, Prechl J, Bajtay Z, Erdei A. Complement receptor type 1 (CD35) mediates inhibitory signals in human B lymphocytes. J Immunol. (2002) 168:2782–8. doi: 10.4049/jimmunol.168.6.2782
158. Kremlitzka M, Polgar A, Fulop L, Kiss E, Poor G, Erdei A. Complement receptor type 1 (CR1, CD35) is a potent inhibitor of B-cell functions in rheumatoid arthritis patients. Int Immunol. (2013) 25:25–33. doi: 10.1093/intimm/dxs090
159. Kremlitzka M, Macsik-Valent B, Polgar A, Kiss E, Poor G, Erdei A. Complement receptor type 1 suppresses human B cell functions in SLE patients. J Immunol Res. (2016) 2016:5758192. doi: 10.1155/2016/5758192
160. Killick J, Morisse G, Sieger D, Astier AL. Complement as a regulator of adaptive immunity. Semin Immunopathol. (2018) 40:37–48. doi: 10.1007/s00281-017-0644-y
161. Neher MD, Rich MC, Keene CN, Weckbach S, Bolden AL, Losacco JT, et al. Deficiency of complement receptors CR2/CR1 in Cr2(-)/(-) mice reduces the extent of secondary brain damage after closed head injury. J Neuroinflammation (2014) 11:95. doi: 10.1186/1742-2094-11-95
162. Wagner C, Hansch GM, Stegmaier S, Denefleh B, Hug F, Schoels M. The complement receptor 3, CR3 (CD11b/CD18), on T lymphocytes: activation-dependent up-regulation and regulatory function. Eur J Immunol. (2001) 31:1173–80. doi: 10.1002/1521-4141(200104)31:4<1173::AID-IMMU1173>3.0.CO;2-9
163. Lukacsi S, Nagy-Balo Z, Erdei A, Sandor N, Bajtay Z. The role of CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in complement-mediated phagocytosis and podosome formation by human phagocytes. Immunol Lett. (2017) 189:64–72. doi: 10.1016/j.imlet.2017.05.014
164. Zwirner J, Gotze O, Begemann G, Kapp A, Kirchhoff K, Werfel T. Evaluation of C3a receptor expression on human leucocytes by the use of novel monoclonal antibodies. Immunology (1999) 97:166–72. doi: 10.1046/j.1365-2567.1999.00764.x
165. Martin U, Bock D, Arseniev L, Tornetta MA, Ames RS, Bautsch W, et al. The human C3a receptor is expressed on neutrophils and monocytes, but not on B or T lymphocytes. J Exp Med. (1997) 186:199–207. doi: 10.1084/jem.186.2.199
166. Kremlitzka M, Csati Z Erdei. Expression and function of C3 and C3AR in human B cells - a novel cross-talk between complement, TLRs and adaptive immunity. Mol Immunol. (2014) 61:278.
167. Soruri A, Kim S, Kiafard Z, Zwirner J. Characterization of C5aR expression on murine myeloid and lymphoid cells by the use of a novel monoclonal antibody. Immunol Lett. (2003) 88:47–52. doi: 10.1016/S0165-2478(03)00052-X
168. Jimenez-Reinoso A, Marin AV, Subias M, Lopez-Lera A, Roman-Ortiz E, Payne K, et al. Human plasma C3 is essential for the development of memory B, but not T, lymphocytes. J Allergy Clin Immunol. (2018) 141:1151–4. doi: 10.1016/j.jaci.2017.09.037
169. Broker K, Figge J, Magnusen AF, Manz RA, Kohl J, Karsten CM. A novel role for C5a in B-1 cell homeostasis. Front Immunol. (2018) 9:258. doi: 10.3389/fimmu.2018.00258
170. Kwan WH, van der Touw W, Paz-Artal E, Li MO, Heeger PS. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med. (2013) 210:257–68. doi: 10.1084/jem.20121525
171. Unnewehr H, Rittirsch D, Sarma JV, Zetoune F, Flierl MA, Perl M, et al. Changes and regulation of the C5a receptor on neutrophils during septic shock in humans. J Immunol. (2013) 190:4215–25. doi: 10.4049/jimmunol.1200534
172. Boothby MR, Raybuck AL, Cho SH. Complementing T Cells' functions: bringing in metabolism matters. Immunity (2015) 42:977–9. doi: 10.1016/j.immuni.2015.06.008
173. Pritchard NR, Smith KG. B cell inhibitory receptors and autoimmunity. Immunology (2003) 108:263–73. doi: 10.1046/j.1365-2567.2003.01592.x
174. Paredes RM, Reyna S, Vernon P, Tadaki DK, Dallelucca JJ, Sheppard F. Generation of complement molecular complex C5b-9 (C5b-9) in response to poly-traumatic hemorrhagic shock and evaluation of C5 cleavage inhibitors in non-human primates. Int Immunopharmacol. (2018) 54:221–5. doi: 10.1016/j.intimp.2017.10.033
175. Ehrnthaller C, Huber-Lang M, Kovtun A, Rapp AE, Kemmler J, Gebhard F, et al. C5aR inhibition in the early inflammatory phase does not affect bone regeneration in a model of uneventful fracture healing. Eur J Med Res. (2016) 21:42. doi: 10.1186/s40001-016-0236-7
176. Huber-Lang M, Ignatius A, Brenner RE. Role of complement on broken surfaces after Trauma. Adv Exp Med Biol. (2015) 865:43–55. doi: 10.1007/978-3-319-18603-0_3
177. Groeneveld KM, Leenen LP, Koenderman L, Kesecioglu J. Immunotherapy after trauma: timing is essential. Curr Opin Anaesthesiol. (2011) 24:219–23. doi: 10.1097/ACO.0b013e32834401e0
178. Flierl MA, Perl M, Rittirsch D, Bartl C, Schreiber H, Fleig V, et al. The role of C5a in the innate immune response after experimental blunt chest trauma. Shock (2008) 29:25–31. doi: 10.1097/shk.0b013e3180556a0b
179. Kalbitz M, Karbach M, Braumueller S, Kellermann P, Gebhard F, Huber-Lang M, et al. Role of complement C5 in experimental blunt chest trauma-induced septic acute lung injury (ALI). PLoS ONE (2016) 11:e0159417. doi: 10.1371/journal.pone.0159417
180. Kovtun A, Bergdolt S, Hagele Y, Matthes R, Lambris JD, Huber-Lang M, et al. Complement receptors C5aR1 and C5aR2 act differentially during the early immune response after bone fracture but are similarly involved in bone repair. Sci Rep. (2017) 7:14061. doi: 10.1038/s41598-017-14444-3
181. Rich MC, Keene CN, Neher MD, Johnson K, Yu ZX, Ganivet A, et al. Site-targeted complement inhibition by a complement receptor 2-conjugated inhibitor (mTT30) ameliorates post-injury neuropathology in mouse brains. Neurosci Lett. (2016) 617:188–94. doi: 10.1016/j.neulet.2016.02.025
182. Fluiter K, Opperhuizen AL, Morgan BP, Baas F, Ramaglia V. Inhibition of the membrane attack complex of the complement system reduces secondary neuroaxonal loss and promotes neurologic recovery after traumatic brain injury in mice. J Immunol. (2014) 192:2339–48. doi: 10.4049/jimmunol.1302793
183. Ruseva MM, Ramaglia V, Morgan BP, Harris CL. An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice. Proc Natl Acad Sci USA. (2015) 112:14319–24. doi: 10.1073/pnas.1513698112
184. Howdieshell TR, Heffernan D, Dipiro JT. Surgical infection society guidelines for vaccination after traumatic injury. Surg Infect. (2006) 7:275–303. doi: 10.1089/sur.2006.7.275
185. Mastellos DC, Yancopoulou D, Kokkinos P, Huber-Lang M, Hajishengallis G, Biglarnia AR, et al. Compstatin: a C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur J Clin Invest. (2015) 45:423–40. doi: 10.1111/eci.12419
186. Robertson N, Rappas M, Dore AS, Brown J, Bottegoni G, Koglin M, et al. Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nature (2018) 553:111–4. doi: 10.1038/nature25025
187. van GM, Ricklin D, Denk S, Halbgebauer R, Braun CK, Schultze A, et al. Protective effects of the complement inhibitor compstatin cp40 in hemorrhagic shock. Shock (2018). doi: 10.1097/SHK.0000000000001127. [Epub ahead of print].
Keywords: complement, trauma, innate immunity, adaptive immunity, T-cells, B-cells
Citation: Chakraborty S, Karasu E and Huber-Lang M (2018) Complement After Trauma: Suturing Innate and Adaptive Immunity. Front. Immunol. 9:2050. doi: 10.3389/fimmu.2018.02050
Received: 30 May 2018; Accepted: 20 August 2018;
Published: 24 September 2018.
Edited by:
Nicole Thielens, UMR5075 Institut de Biologie Structurale (IBS), FranceReviewed by:
Cordula M. Stover, University of Leicester, United KingdomCopyright © 2018 Chakraborty, Karasu and Huber-Lang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Huber-Lang, bWFya3VzLmh1YmVyLWxhbmdAdW5pa2xpbmlrLXVsbS5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.