 Veron Ramsuran
Veron Ramsuran Rodger Ewy
Rodger Ewy Hoang Nguyen
Hoang Nguyen Smita Kulkarni
Smita Kulkarni- 1Centre for the AIDS Programme of Research in South Africa, KwaZulu-Natal Research Innovation and Sequencing Platform, School of Laboratory Medicine and Medical Sciences, Nelson R. Mandela School of Medicine, College of Health Sciences, University of KwaZulu-Natal, Durban, South Africa
- 2Genetics Department, Texas Biomedical Research Institute, San Antonio, TX, United States
The clinical outcomes of infections are highly variable among individuals and are determined by complex host-pathogen interactions. Genome-wide association studies (GWAS) are powerful tools to unravel common genetic variations that are associated with disease risk and clinical outcomes. However, GWAS has only rarely revealed information on the exact genetic elements and their effects underlying an association because the majority of the hits are within non-coding regions. Some of the variants or the linked polymorphisms are now being discovered to have functional significance, such as regulatory elements in the promoter and enhancer regions or the microRNA binding sites in the 3′untranslated region of the protein-coding genes, which influence transcription, RNA stability, and translation of the protein-coding genes. However, only 3% of the entire transcriptome is protein-coding, signifying that non-coding RNAs represent most of the transcripts. Thus, a large portion of previously identified intergenic GWAS single nucleotide polymorphisms (SNPs) is in the non-coding RNAs. The non-coding RNAs form a large-scale regulatory network across the transcriptome, greatly expanding the complexity of gene regulation. Accumulating evidence also suggests that the “non-coding” genome regions actively regulate the highly dynamic three dimensional (3D) chromatin structures, which are critical for genome function. Epigenetic modulation like DNA methylation and histone modifications further affect chromatin accessibility and gene expression adding another layer of complexity to the functional interpretation of genetic variation associated with disease outcomes. We provide an overview of the current information on the influence of variation in these “untranslated” regions of the human genome on infectious diseases. The focus of this review is infectious disease-associated polymorphisms and gene regulatory mechanisms of pathophysiological relevance.
Introduction
The influence of host genetic polymorphisms on inter-individual variation in disease outcomes has been investigated through candidate gene sequencing and the large scale Genome-wide association studies (GWAS). However, these studies have rarely revealed information on the exact genetic elements underlying an association because majority of the hits are in intergenic regions, some of which are now being discovered to have functional significance. This lack of functional information limits their use as biomarkers or potential targets for therapy.
Despite the high resolution and specialized genotypic platforms and the wealth of data, which allows genotype imputations (1), causal genetic variations have remained elusive. Surveys of several published GWAS indicate a multitude of disease-associated polymorphisms in enriched in non-coding regions (2), regulatory elements (3) and expression quantitative trait loci (eQTL) (3, 4), suggesting that their associations with diseases are due to their involvement in regulation of gene expression. Only about 10% of the disease associated single nucleotide polymorphisms (SNPs) in GWAS affect protein-coding sequences (2). The majority of non-coding SNPs are in the deoxyribonuclease I (DNase I) hypersensitive sites (2). These SNPs may affect binding of transcription factors to the promoter and enhancer regions and transcription of the downstream genes. It is complicated to delineate functions of enhancer region polymorphisms as they can be defined mostly by the epigenomic profile like methylation, chromatin accessibility, histone modifications and expression of enhancer RNAs (eRNAs)(2, 5–7). 3D chromatin structure has only begun to be studied since the turn of the millennium beginning with the development of the chromosome conformation capture (8). Growing literature indicates that disease associated SNPs in the non-coding genome contribute to gene regulation through 3D interactions (9, 10). A small percent of SNPs in the 3′untranslated region (3′UTR) influence post-transcriptional mRNA decay or translation by altering the microRNA (miRNA) binding sites.
Recently, another class of non-coding RNAs, termed long non-coding RNAs (lncRNAs) have been discovered. The lncRNAs regulate a number of cellular and developmental processes (11–13). The majority of the transcriptome is made up of these non-coding RNAs. Thus, it is predicted that a sizable portion of previously identified intergenic GWAS SNPs could be marking variation in function or expression of non-coding RNAs (14, 15).
Here we will review the influence of variation in the non-coding genome on transcriptional or post-transcriptional regulation of gene expression and infectious disease outcomes.
Promoter Regulatory Elements
Polymorphisms in the DNA regulatory regions modulate the epigenetic alterations and transcription factor binding resulting in variations in gene expression and immune responses. Cytokines, chemokines and their receptors are the key regulators of immune response and inflammation. Promoter polymorphisms in these genes are associated with several infectious diseases. Interleukin 10 (IL-10) promoter polymorphism is associated with increased mortality in severe sepsis (16), susceptibility to chronic hepatitis C virus (HCV) infection, resistance to antiviral therapy (17), and predisposition to Epstein Barr virus (EBV) infection (18). IL-8 promoter polymorphisms is associated with IL-8 release and incidence of virus bronchiolitis (19). Genomic variations in the promoters of cytokines and other innate immune genes have been linked to susceptibility to Mycobacterium tuberculosis (M.tb) infection (20). Promoter variations in the chemokine (21) and chemokine receptor genes are known to influence the course of HIV infection (22–24). Polymorphism in a TF binding site of HLA-C promoter associated with HIV control (25).
Some of the gene regulatory polymorphisms alter the DNA methylation pattern. A methyl group is added to the nucleotide cytosine, which is followed by a guanine to form a CpG dinucleotide (26). Short stretches of DNA with frequent CpG dinucleotides termed CpG islands are mainly located near the promoters of genes. Variation in the promoter methylation of CCR5 (27) and human leukocyte antigen (HLA-A) genes have been shown to significantly impact outcomes of human immunodeficiency virus (HIV) infection (28).
Splicing
Precise splicing of mRNAs is critical for its translation and functioning of the resulting protein. Alternative splicing is often employed by the cells to generate transcript diversity (29, 30). Splicing is orchestrated by the complex interaction between spliceosomes and intronic splicing signals. Spliceosomes are complexes of small ribonucleoproteins (snRNPs), which interact with intronic splicing signals like donor and acceptor sites, polypyrimidine tract, branch points like enhancers and silencers of splicing. Sequence variation in these splicing signals affect mRNA processing. A wide range (15–60%) of the human disease related polymorphisms are predicted to alter splicing (31). A SNP in the acceptor site of an antiviral enzyme OAS1 associates with the level of OAS1 activity and susceptibility to viral infections (32). Intron region polymorphism in ULK1 associate with decreased expression of the gene, compromised immune responses and associate with increase M.tb replication in the latently infected patients leading to the development of pulmonary TB (33). The SNPs in the splice sites of PLCXD3 showed significant association with prion mediated sporadic Creutzfeldt-Jakob's Disease (34).
MicroRNA
MicroRNAs are small, 22 nucleotide RNAs associated with RNA-induced silencing complexes (RISC) and target specific mRNAs for degradation or inhibition of translation. The genomic variation in miRNA or the miRNA binding site in the target genes have been implicated in the differential susceptibility and clinical manifestations of infectious disease. The SNPs in host miRNA loci have been associated with susceptibility to leprosy, clearance of hepatitis B virus (HBV), human cytomegalovirus (hCMV) infection (35–39), the prion mediated spontaneous Creutzfeldt-Jakob's Disease and fatal familial insomnia (40).
A mutation in the miRNA binding site can disrupt binding to miRNA to its target thus allowing the target to be expressed at higher levels. MicroRNA binding site polymorphisms have been implicated in susceptibility or prognosis of infection. The functional effect of some of these associations have been validated. High levels of HLA-C mRNA and cell surface expression associate with control HIV viremia and slower progression to acquired immunodeficiency syndrome (AIDS) (41, 42). The allele specific expression variation of HLA-C is partly explained by a polymorphic miR-148a binding site encoded in the 3′ UTR of HLA-C. The alleles with disrupted miRNA binding site escape regulation by miR-148a, are expressed at higher levels and associate with lower HIV viral loads (43). A functional SNP, within the 3′ UTR of IFNL3 is in a binding site of HCV-induced cellular miRNAs. The allele, which allows escape of miRNA mediated downregulation associates with an increase in IFNL3 miRNA expression and showed significant association with natural and therapy-induced HCV clearance (44).
Host miRNAs target the HIV transcripts and inhibit translation resulting in silencing of HIV gene expression in resting CD4+ T cells (45), whereas HCV requires host miR-122 for replication (46). Pathogens deregulate the host miRNA expression to their advantage, such as in Zika virus in astrocytes (47), Mycobacterium leprae as well as M. tb infection (48, 49); or directly target the host transcriptome with a miRNA encoded in their own genome, such as in Rotavirus (50).
Long Non-Coding RNAs
The long non-coding RNAs are >200 bp transcripts without protein-coding potential. LncRNAs have been shown to enhance or repress the transcription of protein-coding genes, including immune-related genes (51–53), implicating a role for lncRNAs in disease outcomes through gene regulation. The majority of lncRNAs are found in low abundance and localized in the nucleus (54), where they participate in transcriptional regulation through diverse mechanisms (55, 56). However, some localize to the cytoplasm, where they function as competitive endogenous RNAs that can act as “sponges” for miRNAs (57). Thus, the lncRNAs form a large-scale regulatory network across the transcriptome (58). LncRNA gene variations are associated with disease outcomes, and molecular mechanisms have been delineated for a few in cancer (59) and autoimmune disease (60). A SNP in a lncRNA expressed in neutrophils was associated with doubled risk of Pneumococcal bacteremia in Kenyan children (46). Polymorphisms in lncRNAs are associated with tuberculosis susceptibility (61). A variant in the lncRNA gene has been shown to confer susceptibility to HBV-related carcinoma in Chinese population (62). Several viruses encode lncRNAs in their genome (63) and modulate host responses. A few known examples are arthropod-borne flaviviruses (64), Kaposi's sarcoma-associated herpesvirus (65) and HIV (66–68). Pathogens like influenza A (69), HCV (70), adenovirus (71), herpes simplex virus [HSV-1] (72), HIV (13, 73–75) and M.tb (76–80) alter expression of the host lncRNAs upon infection. An interferon-independent host lncRNA promotes viral replication by modulating cellular metabolism (81). The expression and functional variations within the lncRNA genes and their role in regulating immune response and inflammation is an area of intense research and likely to discover novel pathways of host-pathogen interactions.
Multidimensional Regulation and Complex Gene Interactions
The variation in gene expression levels are determined by several transcriptional, post-transcriptional and post-translational processes (Figure 1). A single variant or haplotype may not be the sole contributor to disease associations. This is exemplified by a few genetic associations for which molecular mechanisms have been delineated.
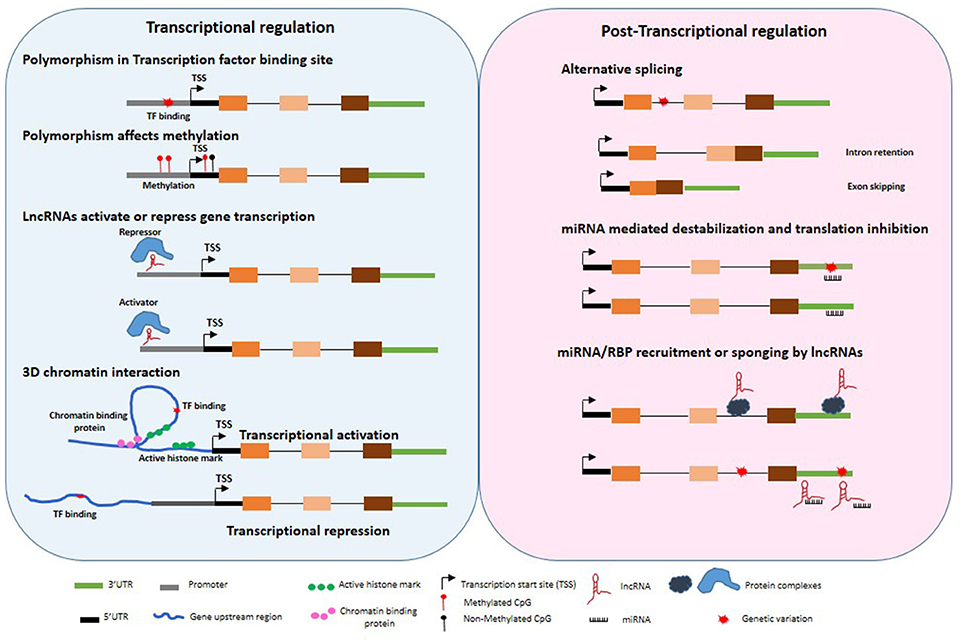
Figure 1. Untranslated gene variations influence regulation of gene expression. Disease associated polymorphisms may alter methylation, transcription factor binding in the gene promoter regions, recruitment of repressor or activators, 3 dimensional chromatin structure, splicing, miRNA binding to 3′UTR, transcriptional and post-transcriptional regulation of target genes through variation in lncRNA expression and function.
GWAS implicated multiple linked variations at the interferon lambda (IFNL) in HCV clearance (82, 83). A 3′UTR variant in IFNL3 disrupting miRNA binding site and increasing stability of IFNL3 mRNA was found to be protective (44). A strongly linked dinucleotide variant, which introduces a premature stop codon in another interferon gene, IFNL4 associated with HCV clearance (84). Thus linked genotypes conferring high expression of IFNλ3 and truncated, non-functional IFNλ4 associated with HCV clearance. This paradoxical association of expression of the full length antiviral protein IFNλ4 with HCV persistence remained unexplained. Recently, it was found that the full length IFNλ4 isoform is poorly expressed due to aberrant splicing resulting in intron retention and a weak polyadenylation signal (85). The mRNA instability genotype in the IFNL3 3′UTR is in strong linkage disequilibrium (LD) with the full length expression genotype of IFNL4 suggesting that the persistence of HCV is likely attributed to the impaired expression and anti-viral function of IFNλ3.
The HLA genes located on human chromosome 6 are extremely polymorphic, exhibit strong LD and allele-specific expression patterns. The nucleotide diversity observed within the MHC region, particularly within the regulatory region upstream of the initiation codon of certain class I genes, has been observed about 20-fold higher than the genome average (86). Variations within the HLA-C 3′UTR (43) and promoter (25) showed a strong influence on HLA-C mRNA and protein expression, which associates with HIV control. Both the causal variations in the 3′UTR and promoter only partially account for the allele specific gene expression variation, indicating that yet unknown variation either within the HLA-C gene region or elsewhere in the genome is responsible for a large part of HLA-C expression.
Interplay Between Genomic Variations and Epigenomic Modulations
Specific epigenetic changes could result in switching on or off certain genes, and determines which proteins are transcribed. In some cases, several factors have been shown to contribute to causal phenotype in addition to the genomic variation. A recent study investigated role of a SNP in 5′UTR of the gene encoding an antiviral protein, Interferon-induced transmembrane protein 3 (IFITM3), which inhibits entry of influenza virus into the host cell. The risk allele was found to disrupt a CpG site, decrease binding of a transcription factor, interferon regulatory factor (IRF3) and increase binding of CCCTC-binding factor (CTCF) leading to lower expression of IFITM3 expression, altered methylation and also affected the expression of IFITM3 neighboring genes important in anti-viral response. CTCF is a versatile protein important in maintaining topologically associating domains of chromatin and contributing to 3D chromatin structure. CTCF has also been shown to function as an “insulator” blocking chromatin positioning and inhibiting interaction of enhancer with promoters. Increased CTCF binding associated with the IFITM3 risk allele and disruption of neighboring gene expression patterns indicative of CTCF boundary activity.
DNA methylation and histone modification are just two of the epigenetic mechanisms widely described (87). Examining certain genes, we observe the balancing act between genomics and epigenomics within infectious diseases. One such example is the HLA-A gene, where specific HLA-A alleles are observed to possess individual HIV allelic effects, based on the peptide presented (88). However, more recently it has been shown HLA-A alleles possess distinct allele-specific promoter methylation and mRNA expression levels (89). Furthermore, examining nearly 10,000 HIV patients across diverse ethnic backgrounds, Ramsuran et al showed that HLA-A mRNA expression levels play an important role in HIV viral control (28). The patients with high expression of HLA-A showed impaired HIV control. Unlike the protective effect of high HLA-C expression on HIV viral control and contrary to conventional understanding, elevated levels of HLA-A do not protect against HIV but rather lead to accelerated disease progression by increasing cell surface expression of a natural killer (NK) ligand, HLA-E and stronger inhibition of NK cells (Figure 2). HLA-E serves as a ligand for the strong inhibitory NK receptor CD94/NKG2A. A signal peptide derived from the leader sequence of HLA-A, -B and -C molecules stabilizes HLA-E expression on the cell surface (90). The signal peptide has a variation at position 2 (residue−21) resulting in either a methionine (-21M) or threonine (-21T). A methionine (-21M) stabilizes and increases HLA-E expression on the cell surface, while the presence of−21T results in lower HLA-E expression. High expression of HLA-A provides elevated levels of HLA-A derived signal peptide increasing HLA-E expression resulting in strong NK inhibition and impaired killing of HIV infected cells. Presence of HLA-B alleles with−21M results in strong “licensing” of NK cells and further exacerbates the inhibitory effects of high HLA-A expression on NK cell activity and results in poor HIV control. Thus, variable HLA-A expression levels in combination with either−21M or −21T HLA-B alleles lead to diverse HIV outcomes (28).
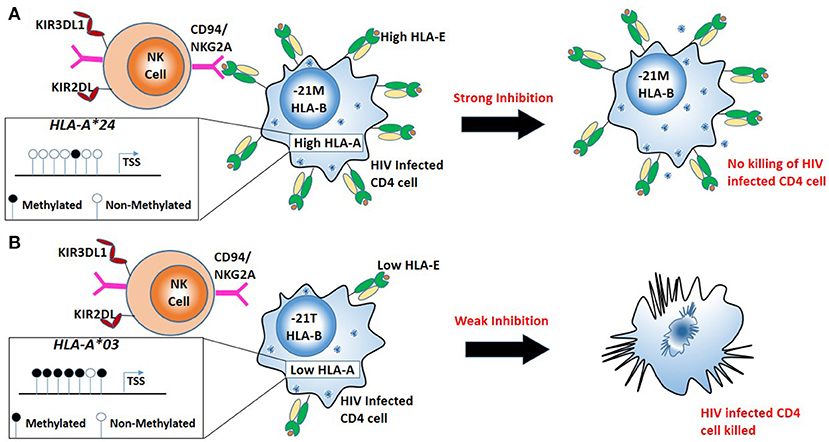
Figure 2. Combination of high HLA-A allele expression and HLA-B alleles mediating in robust “licensing” of NK cells result in strong NK cell inhibition and result in poor HIV control. (A) Less promoter methylation results in a high expression HLA-A (e.g., HLA-A*24). High HLA-A expression with the−21M genotype at HLA-B leads to increased cell surface expression of HLA-E, robust NK cell licensing through CD94/NKG2A increased inhibition of NK cells. Strong inhibition of NK cells leads to escape of HIV infected cells from NK mediated killing. (B) High promoter methylation results in low expression of HLA-A (e.g., HLA-A*03). Low expression HLA-A allele with−21T genotype at HLA-B results in lower HLA-E on the surface resulting in little CD94/NKG2A licensing, NK cells mediated killing HIV infected cells.
Variability within the HLA-A peptide binding region (exons 2 and 3) of the genomic sequence allows specific alleles to slow or accelerate HIV disease progression. However, epigenetic mechanisms alter the expression levels of these alleles, which also causes delayed or rapid HIV progression. This balance between genetic and epigenetic events makes this region one of the most complex multifactorial sites associating with a range of diseases.
3D Chromatin Structure
Over half of the significant associations in the published GWAS were reported in genome expanses lacking annotated genes in the large regions (>500 kb) flanking associated SNP. Such regions, termed “gene deserts” have been implicated in the regulation of genome function through their influence on intricate 3D genome structure. The 3D architecture of the genome facilitates spatial juxtaposition of and interaction between distant loci, which contributes to gene regulation. A SNP on chromosome 8q24 in a gene desert of 1.2Mb has been shown to affect long range (>300Kb) interactions of a cancer gene, Myc (9, 10). SNPs associated with type 2 diabetes have been implicated in 3D genome interactions leading to impaired interferon signal responses (10). Although 3D chromatin structure changes after infection have been reported in a few cases (91–95), studies on 3D genome structure are underrepresented in the field of infectious diseases.
Perspective and Future Directions
Gene association studies and GWAS have identified several genetic variants, which influence outcomes of complex human disease including infections. However, the majority of the disease-associated variants map to the non-coding regions of the genome. Discovery and functional validation of causal variants and the specific regulatory mechanisms are needed to advance understanding of the molecular pathways and their possible manipulation by therapy. Current literature reveals a number of efforts in determining the causal SNPs from the variants identified in the GWAS and linked SNPs. Disease-associated SNPs are further investigated to determine if they are marking coding-region variants through strong LD or are likely to alter promoter and enhancer region, splicing, miRNA binding, or lncRNA expression and function. The possible candidates are examined in a variety of in silico and molecular and biochemical analyses, including prediction of transcription factor or miRNA binding sites, chromatin or RNA immunoprecipitations, nucleic acid sequencing and mass-spectrometry to identify DNA, RNA or protein factors interacting with the genomic variation or the resulting RNA and protein products. Loss or gain of function studies are carried out to determine functional consequences of the RNA or protein target. Distinct regions in the genome (promoter, 3′UTR, intron, miRNA, lncRNA) require diverse molecular and biochemical techniques. Although these focused, gene-specific studies discover novel molecular mechanisms of gene regulation, a fresh approach is needed to detangle multilayered gene regulatory pathways and complex gene interactions, which are often cell-type and context-specific (infected vs. uninfected).
Interpretation of the functional consequences of a disease associated gene variation can be complex, especially in case of the intergenic SNPs as it may affect none or multiple genes and pathways through the expression of non-coding RNAs and chromatin organization. CRISPR/Cas9 mediated editing of the SNP or the putative target region is being increasingly used to address the ambiguity. Cell line based models are more permissive for such manipulations. However, these models may not represent the complex environment of cells and tissues. Organoid models are being developed to recapitulate the tissue environment and interaction. Multiple genetic variants co-operatively regulate a phenotype (96) Editing of a single gene will not discern this combinatorial effect. Further evolution of gene editing techniques to simultaneously target multiple regulatory regions is needed of the gene editing techniques to simultaneously target multiple regulatory regions. Gene editing techniques are even harder to implement in case of long non-coding RNAs. Manipulation of gene expression through short interfering RNAs (siRNA, shRNAs) and nucleases are limited by off-target effects. Alternatively approaches of nuclease deficient CRISPR/Cas9 (dCas9) mediated inhibition or activation of gene expression are being increasingly applied specially in the field of non-coding RNAs. Epigenetic modulations are hard to recapitulate as the causal and the target regions could be spread over a large genomic distance. The engineered systems utilizing dCas9 collectively called “epigenetic toggle switches” are being used to mimic deletion of enhances or recruitment of epigenetic repressors (97). Full implications of variations that influence 3D chromatin structure are yet to be realized in part due to difficulties in precisely capturing and mapping interactions between distinct regions of chromatin. However, the field has exploded with multiple complementary techniques like Hi-C, ChIA-PET, 3D-FISH, which help capture both the short and the long-range interactions (98). Integrated investigations of disease-associated genetic variations, context-specific gene expression, spatial organization of chromatin and epigenetic alterations in primary human cells, genomic and transcriptomic analysis at tissue and single cell level are needed to unravel the role of untranslated regions in susceptibility to infections.
Author Contributions
VR and SK conceptualized the framework and all authors contributed to writing, reviewing and editing of the review.
Funding
VR is funded by the South African Medical Research Council Flagship Award (grant number MRC-RFA-UFSP-01-2013/UKZN-HIVEPI) and by Sub-Saharan African Network for TB/HIV Research Excellence (SANTHE) – a DELTAS Africa Initiative programme (grant number DEL-15-006). RE, HN, and SK are supported by the Texas Biomedical Research Institute. This work is supported in part by NIH AI140956-01 (SK).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. (2010) 11:499–511. doi: 10.1038/nrg2796
2. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science (2012) 337:1190–5. doi: 10.1126/science.1222794
3. Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet. (2009) 10:184–94. doi: 10.1038/nrg2537
4. Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature (2012) 482:390–4. doi: 10.1038/nature10808
5. Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH roadmap epigenomics mapping consortium. Nat Biotechnol. (2010) 28:1045–8. doi: 10.1038/nbt1010-1045
6. Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature (2012) 489:57–74. doi: 10.1038/nature11247
7. Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature (2015) 518:317–30. doi: 10.1038/nature14248
8. Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science (2002) 295:1306–11. doi: 10.1126/science.1067799
9. Ahmadiyeh N, Pomerantz MM, Grisanzio C, Herman P, Jia L, Almendro V, et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci USA (2010) 107:9742–6. doi: 10.1073/pnas.0910668107
10. Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature (2011) 470:264–8. doi: 10.1038/nature09753
11. Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. (2009) 23:1494–504. doi: 10.1101/gad.1800909
12. Nagano T, Fraser P. No-nonsense functions for long noncoding RNAs. Cell (2011) 145:178–81. doi: 10.1016/j.cell.2011.03.014
13. Zhang Q, Chen CY, Yedavalli VS, Jeang KT. NEAT1 long noncoding RNA and paraspeckle bodies modulate HIV-1 posttranscriptional expression. MBio (2013) 4:e00596–12. doi: 10.1128/mBio.00596-12
14. Kumar V, Westra HJ, Karjalainen J, Zhernakova DV, Esko T, Hrdlickova B, et al. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet. (2013) 9:e1003201. doi: 10.1371/journal.pgen.1003201
15. Ricano-Ponce I, Wijmenga C. Mapping of immune-mediated disease genes. Annu Rev Genomics Hum Genet. (2013) 14:325–53. doi: 10.1146/annurev-genom-091212-153450
16. Stanilova SA, Miteva LD, Karakolev ZT, Stefanov CS. Interleukin-10-1082 promoter polymorphism in association with cytokine production and sepsis susceptibility. Intensive Care Med. (2006) 32:260–6. doi: 10.1007/s00134-005-0022-4
17. Vidigal PG, Germer JJ, Zein NN. Polymorphisms in the interleukin-10, tumor necrosis factor-alpha, and transforming growth factor-beta1 genes in chronic hepatitis C patients treated with interferon and ribavirin. J Hepatol. (2002) 36:271–7. doi: 10.1016/S0168-8278(01)00243-4
18. Helminen ME, Kilpinen S, Virta M, Hurme M. Susceptibility to primary Epstein-Barr virus infection is associated with interleukin-10 gene promoter polymorphism. J Infect Dis. (2001) 184:777–80. doi: 10.1086/322987
19. Hull J, Thomson A, Kwiatkowski D. Association of respiratory syncytial virus bronchiolitis with the interleukin 8 gene region in UK families. Thorax (2000) 55:1023–7. doi: 10.1136/thorax.55.12.1023
20. Azad AK, Sadee W, Schlesinger LS. Innate immune gene polymorphisms in tuberculosis. Infect Immun. (2012) 80:3343–59. doi: 10.1128/IAI.00443-12
21. McDermott DH, Beecroft MJ, Kleeberger CA, Al-Sharif FM, Ollier WE, Zimmerman PA, et al. Chemokine RANTES promoter polymorphism affects risk of both HIV infection and disease progression in the Multicenter AIDS Cohort Study. AIDS (2000) 14:2671–8. doi: 10.1097/00002030-200012010-00006
22. Martin MP, Dean M, Smith MW, Winkler C, Gerrard B, Michael NL, et al. Genetic acceleration of AIDS progression by a promoter variant of CCR5. Science (1998) 282:1907–11.
23. McDermott DH, Zimmerman PA, Guignard F, Kleeberger CA, Leitman SF, and Murphy PM. CCR5 promoter polymorphism and HIV-1 disease progression. Multicenter AIDS Cohort Study (MACS). Lancet (1998) 352:866–70.
24. Mummidi S, Ahuja SS, Gonzalez E, Anderson SA, Santiago EN, Stephan KT, et al. Genealogy of the CCR5 locus and chemokine system gene variants associated with altered rates of HIV-1 disease progression. Nat Med. (1998) 4:786–93. doi: 10.1038/nm0798-786
25. Vince N, Li H, Ramsuran V, Naranbhai V, Duh FM, Fairfax BP, et al. HLA-C level is regulated by a polymorphic Oct1 binding site in the HLA-C promoter region. Am J Hum Genet. (2016) 99:1353–8. doi: 10.1016/j.ajhg.2016.09.023
26. Bird AP. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. (1980) 8:1499–504. doi: 10.1093/nar/8.7.1499
27. Gornalusse GG, Mummidi S, Gaitan AA, Jimenez F, Ramsuran V, Picton A, et al. Epigenetic mechanisms, T-cell activation, and CCR5 genetics interact to regulate T-cell expression of CCR5, the major HIV-1 coreceptor. Proc Natl Acad Sci USA (2015) 112:E4762–71. doi: 10.1073/pnas.1423228112
28. Ramsuran V, Naranbhai V, Horowitz A, Qi Y, Martin MP, Yuki Y, et al. Elevated HLA-a expression impairs HIV control through inhibition of NKG2A-expressing cells. Science (2018) 359:86–90. doi: 10.1126/science.aam8825
29. Wang Z, and Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA (2008) 14:802–13. doi: 10.1261/rna.876308
30. Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature (2010) 463:457–63. doi: 10.1038/nature08909
31. Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. (2007) 8:749–61. doi: 10.1038/nrg2164
32. Bonnevie-Nielsen V, Field LL, Lu S, Zheng DJ, Li M, Martensen PM, et al. Variation in antiviral 2',5'-oligoadenylate synthetase (2'5'AS) enzyme activity is controlled by a single-nucleotide polymorphism at a splice-acceptor site in the OAS1 gene. Am J Hum Genet. (2005) 76:623–33. doi: 10.1086/429391
33. Horne DJ, Graustein AD, Shah JA, Peterson G, Savlov M, Steele S, et al. Human ULK1 variation and susceptibility to mycobacterium tuberculosis infection. J Infect Dis. (2016) 214:1260–7. doi: 10.1093/infdis/jiw347
34. Bishop MT, Sanchez-Juan P, Knight RS. Splice site SNPs of phospholipase PLCXD3 are significantly associated with variant and sporadic Creutzfeldt-Jakob disease. BMC Med Genet. (2013) 14:91. doi: 10.1186/1471-2350-14-91
35. Cheong JY, Shin HD, Kim YJ, and Cho SW. Association of polymorphism in MicroRNA 219-1 with clearance of hepatitis B virus infection. J Med Virol. (2013) 85:808–14. doi: 10.1002/jmv.23551
36. Cezar-de-Mello PF, Toledo-Pinto TG, Marques CS, Arnez LE, Cardoso CC, Guerreiro LT, et al. Pre-miR-146a (rs2910164 G>C) single nucleotide polymorphism is genetically and functionally associated with leprosy. PLoS Negl Trop Dis. (2014) 8:e3099. doi: 10.1371/journal.pntd.0003099
37. Yu SJ, Kim JW, Lee JH, Yoon JH, Lee HS, Cheong JY, et al. Association of a microRNA-323b polymorphism with the persistence of hepatitis B virus infection by the enhancement of viral replication. J Viral Hepat. (2014) 21:853–9. doi: 10.1111/jvh.12215
38. Misra MK, Mishra A, Pandey SK, Kapoor R, Sharma RK, and Agrawal S. Genetic variation in Micro-RNA genes of host genome affects clinical manifestation of symptomatic Human Cytomegalovirus infection. Hum Immunol. (2015) 76:765–9. doi: 10.1016/j.humimm.2015.09.035
39. Al-Qahtani AA, Al-Anazi MR, Nazir N, Wani K, Abdo AA, Sanai FM, et al. Association of single nucleotide polymorphisms in microRNAs with susceptibility to hepatitis B virus infection and HBV-related liver complications: a study in a Saudi Arabian population. J Viral Hepat. (2017) 24:1132–42. doi: 10.1111/jvh.12749
40. Gao C, Shi Q, Wei J, Zhou W, Xiao K, Wang J, et al. The associations of two SNPs in miRNA-146a and one SNP in ZBTB38-RASA2 with the disease susceptibility and the clinical features of the Chinese patients of sCJD and FFI. Prion (2018) 12:34–41. doi: 10.1080/19336896.2017.1405885
41. Thomas R, Apps R, Qi Y, Gao X, Male V, O'HUigin C, et al. HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat Genet. (2009b) 41:1290–4. doi: 10.1038/ng.486
42. Apps R, Qi Y, Carlson JM, Chen H, Gao X, Thomas R, et al. Influence of HLA-C expression level on HIV control. Science (2013) 340:87–91. doi: 10.1126/science.1232685
43. Kulkarni S, Savan R, Qi Y, Gao X, Yuki Y, Bass SE, et al. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature (2011) 472:495–8. doi: 10.1038/nature09914
44. McFarland AP, Horner SM, Jarret A, Joslyn RC, Bindewald E, Shapiro BA, et al. The favorable IFNL3 genotype escapes mRNA decay mediated by AU-rich elements and hepatitis C virus-induced microRNAs. Nat Immunol. (2014) 15:72–9. doi: 10.1038/ni.2758
45. Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, et al. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med. (2007) 13:1241–7. doi: 10.1038/nm1639
46. Yu Y, Scheel TKH, Luna JM, Chung H, Nishiuchi E, Scull MA, et al. miRNA independent hepacivirus variants suggest a strong evolutionary pressure to maintain miR-122 dependence. PLoS Pathog. (2017) 13:e1006694. doi: 10.1371/journal.ppat.1006694
47. Kozak RA, Majer A, Biondi MJ, Medina SJ, Goneau LW, Sajesh BV, et al. MicroRNA and mRNA dysregulation in astrocytes infected with zika virus. Viruses (2017) 9:E297. doi: 10.3390/v9100297
48. Soares CT, Trombone APF, Fachin LRV, Rosa PS, Ghidella CC, Ramalho RF, et al. Differential expression of MicroRNAs in leprosy skin lesions. Front Immunol. (2017) 8:1035. doi: 10.3389/fimmu.2017.01035
49. von Both U, Berk M, Agapow PM, Wright JD, Git A, Hamilton MS et al. Mycobacterium tuberculosis exploits a molecular Off switch of the immune system for intracellular survival. Sci Rep. (2018) 8:661. doi: 10.1038/s41598-017-18528-y
50. Zhou Y, Geng P, Liu Y, Wu J, Qiao H, Xie Y, et al. Rotavirus-encoded virus-like small RNA triggers autophagy by targeting IGF1R via the PI3K/Akt/mTOR pathway. Biochim Biophys Acta (2018) 1864:60–8. doi: 10.1016/j.bbadis.2017.09.028
51. Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science (2013) 341:789–92. doi: 10.1126/science.1240925
52. Gomez JA, Wapinski OL, Yang YW, Bureau JF, Gopinath S, Monack DM, et al. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-gamma locus. Cell (2013) 152:743–54. doi: 10.1016/j.cell.2013.01.015
53. Archin NM, Bateson R, Tripathy MK, Crooks AM, Yang KH, Dahl NP, et al. HIV-1 expression within resting CD4+ T cells after multiple doses of vorinostat. J Infect Dis (2014) 210:728–35. doi: 10.1093/infdis/jiu155
54. Guttman M, Rinn JL, and Modular regulatory principles of large non-coding RNAs Nature (2012) 482:339–46. doi: 10.1038/nature10887
55. Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. (2013) 14:699–712. doi: 10.1038/nrm3679
56. Sallam T, Jones MC, Gilliland T, Zhang L, Wu X, Eskin A, et al. Feedback modulation of cholesterol metabolism by the lipid-responsive non-coding RNA LeXis. Nature (2016) 534:124–8. doi: 10.1038/nature17674
57. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell (2011) 146:353–8. doi: 10.1016/j.cell.2011.07.014
58. Kumar MS, Armenteros-Monterroso E, East P, Chakravorty P, Matthews N, Winslow MM, et al. HMGA2 functions as a competing endogenous RNA to promote lung cancer progression. Nature (2014) 505:212–7. doi: 10.1038/nature12785
59. Ling H, Spizzo R, Atlasi Y, Nicoloso M, Shimizu M, Redis RS, et al. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. (2013) 23:1446–61. doi: 10.1101/gr.152942.112
60. Castellanos-Rubio A, Fernandez-Jimenez N, Kratchmarov R, Luo X, Bhagat G, Green PH, et al. A long noncoding RNA associated with susceptibility to celiac disease. Science (2016) 352:91–5. doi: 10.1126/science.aad0467
61. Zhao Z, Zhang M, Ying J, Hu X, Zhang J, Zhou Y, et al. Significance of genetic polymorphisms in long non-coding RNA AC079767.4 in tuberculosis susceptibility and clinical phenotype in Western Chinese Han population. Sci Rep. (2017) 7:965. doi: 10.1038/s41598-017-01163-y
62. Liu Y, Pan S, Liu L, Zhai X, Liu J, Wen J, et al. A genetic variant in long non-coding RNA HULC contributes to risk of HBV-related hepatocellular carcinoma in a Chinese population. PLoS ONE (2012) 7:e35145. doi: 10.1371/journal.pone.0035145
63. Tycowski KT, Guo YE, Lee N, Moss WN, Vallery TK, Xie M, et al. Viral noncoding RNAs: more surprises. Genes Dev. (2015) 29:567–84. doi: 10.1101/gad.259077.115
64. Moon SL, Anderson JR, Kumagai Y, Wilusz CJ, Akira S, Khromykh AA, et al. A noncoding RNA produced by arthropod-borne flaviviruses inhibits the cellular exoribonuclease XRN1 and alters host mRNA stability. RNA (2012) 18:2029–40. doi: 10.1261/rna.034330.112
65. Sztuba-Solinska J, Rausch JW, Smith R, Miller JT, Whitby D, Le Grice SFJ. Kaposi's sarcoma-associated herpesvirus polyadenylated nuclear RNA: a structural scaffold for nuclear, cytoplasmic and viral proteins. Nucleic Acids Res. (2017) 45:6805–21. doi: 10.1093/nar/gkx241
66. Kobayashi-Ishihara M, Yamagishi M, Hara T, Matsuda Y, Takahashi R, Miyake A, et al. HIV-1-encoded antisense RNA suppresses viral replication for a prolonged period. Retrovirology (2012) 9:38. doi: 10.1186/1742-4690-9-38
67. Saayman S, Ackley A, Turner AW, Famiglietti M, Bosque A, Clemson M, et al. An HIV-encoded antisense long noncoding RNA epigenetically regulates viral transcription. Mol Ther. (2014) 22:1164–75. doi: 10.1038/mt.2014.29
68. Zapata JC, Campilongo F, Barclay RA, DeMarino C, Iglesias-Ussel MD, Kashanchi F, et al. The human immunodeficiency virus 1 ASP RNA promotes viral latency by recruiting the polycomb repressor complex 2 and promoting nucleosome assembly. Virology (2017) 506:34–44. doi: 10.1016/j.virol.2017.03.002
69. Winterling C, Koch M, Koeppel M, Garcia-Alcalde F, Karlas A, Meyer TF. Evidence for a crucial role of a host non-coding RNA in influenza A virus replication. RNA Biol. (2014) 11:66–75. doi: 10.4161/rna.27504
70. Carnero E, Barriocanal M, Prior C, Pablo Unfried J Segura, V Guruceaga, E et al. Long noncoding RNA EGOT negatively affects the antiviral response and favors HCV replication. EMBO Rep. (2016) 17:1013–28. doi: 10.15252/embr.201541763
71. Zhao H, Chen M, Lind SB, Pettersson U. Distinct temporal changes in host cell lncRNA expression during the course of an adenovirus infection. Virology (2016) 492:242–50. doi: 10.1016/j.virol.2016.02.017
72. Hu B, Huo Y, Chen G, Yang L, Wu D, Zhou J. Functional prediction of differentially expressed lncRNAs in HSV-1 infected human foreskin fibroblasts. Virol J. (2016) 13:137. doi: 10.1186/s12985-016-0592-5
73. Chang ST, Sova P, Peng X, Weiss J, Law GL, Palermo RE, et al. (2011). Next-generation sequencing reveals HIV-1-mediated suppression of T cell activation and RNA processing and regulation of noncoding RNA expression in a CD4+ T cell line. MBio 2:e00134−11. doi: 10.1128/mBio.00134-11
74. Peng X, Sova P, Green RR, Thomas MJ, Korth MJ, Proll S, et al. Deep sequencing of HIV-infected cells: insights into nascent transcription and host-directed therapy. J Virol. (2014) 88:8768–82. doi: 10.1128/JVI.00768-14
75. Trypsteen W, Mohammadi P, Van Hecke C, Mestdagh P, Lefever S, Saeys Y, et al. Differential expression of lncRNAs during the HIV replication cycle: an underestimated layer in the HIV-host interplay. Sci Rep. (2016) 6:36111. doi: 10.1038/srep36111
76. Fu Y, Yi Z, Li J, Li R. Deregulated microRNAs in CD4+ T cells from individuals with latent tuberculosis versus active tuberculosis. J Cell Mol Med. (2014) 18:503–13. doi: 10.1111/jcmm.12205
77. Yi Z, Li J, Gao K, Fu Y. Identifcation of differentially expressed long non-coding RNAs in CD4+ T cells response to latent tuberculosis infection. J Infect. (2014) 69:558–68. doi: 10.1016/j.jinf.2014.06.016
78. Wang Y, Zhong H, Xie X, Chen CY, Huang D, Shen L, et al. Long noncoding RNA derived from CD244 signaling epigenetically controls CD8+ T-cell immune responses in tuberculosis infection. Proc Natl Acad Sci USA (2015) 112:E3883–92. doi: 10.1073/pnas.1501662112
79. Yang X, Yang J, Wang J, Wen Q, Wang H, He J, et al. Microarray analysis of long noncoding RNA and mRNA expression profiles in human macrophages infected with Mycobacterium tuberculosis. Sci Rep. (2016) 6:38963. doi: 10.1038/srep38963
80. Fu Y, Xu X, Xue J, Duan W, Yi Z. Deregulated lncRNAs in B Cells from Patients with Active Tuberculosis. PLoS ONE (2017) 12:e0170712. doi: 10.1371/journal.pone.0170712
81. Wang P, Xu J, Wang Y, Cao X. An interferon-independent lncRNA promotes viral replication by modulating cellular metabolism. Science (2017) 358:1051–5. doi: 10.1126/science.aao0409
82. Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature (2009) 461:399–401. doi: 10.1038/nature08309
83. Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O'Huigin C, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature (2009a) 461:798–801. doi: 10.1038/nature08463
84. Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. (2013) 45:164–71. doi: 10.1038/ng.2521
85. Hong M, Schwerk J, Lim C, Kell A, Jarret A, Pangallo J, et al. Interferon lambda 4 expression is suppressed by the host during viral infection. J Exp Med. (2016) 213:2539–52. doi: 10.1084/jem.20160437
86. Ramsuran V, Hernandez-Sanchez PG, O'HUigin C, Sharma G, Spence N, Augusto DG, et al. Sequence and phylogenetic analysis of the untranslated promoter regions for HLA Class I Genes. J Immunol. (2017) 198:2320–9. doi: 10.4049/jimmunol.1601679
87. Macdonald WA. Epigenetic mechanisms of genomic imprinting: common themes in the regulation of imprinted regions in mammals plants, and insects. Genet Res Int. (2012) 2012:585024. doi: 10.1155/2012/585024
88. Carrington M, Bashirova AA, McLaren PJ. On stand by: host genetics of HIV control. AIDS (2013) 27:2831–9. doi: 10.1097/01.aids.0000432536.85335.c8
89. Ramsuran V, Kulkarni S, O'Huigin C, Yuki Y, Augusto DG, Gao X, et al. Epigenetic regulation of differential HLA-A allelic expression levels. Hum Mol Genet. (2015) 24:4268–75. doi: 10.1093/hmg/ddv158
90. Braud VM, Allan DS, O'Callaghan CA, Soderstrom K, D'Andrea A, Ogg GS, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature (1998) 391:795–9.
91. Li C, Shi Z, Zhang L, Huang Y, Liu A, Jin Y, et al. Dynamic changes of territories 17 and 18 during EBV-infection of human lymphocytes. Mol Biol Rep. (2010) 37:2347–54. doi: 10.1007/s11033-009-9740-y
92. Hellert J, Weidner-Glunde M, Krausze J, Lunsdorf H, Ritter C, Schulz TF, et al. The 3D structure of Kaposi sarcoma herpesvirus LANA C-terminal domain bound to DNA. Proc Natl Acad Sci USA (2015) 112:6694–9. doi: 10.1073/pnas.1421804112
93. Grant MJ, Loftus MS, Stoja AP, Kedes DH, Smith MM. Superresolution microscopy reveals structural mechanisms driving the nanoarchitecture of a viral chromatin tether. Proc Natl Acad Sci USA (2018) 115:4992–7. doi: 10.1073/pnas.1721638115
94. Lupey-Green LN, Caruso LB, Madzo J, Martin KA, Tan Y, Hulse M, et al. (2018). PARP1 stabilizes CTCF binding and chromatin structure to maintain epstein barr virus latency type. J Virol. 92:e00755-18. doi: 10.1128/JVI.00755-18
95. Reichel A, Stilp AC, Scherer M, Reuter N, Lukassen S, Kasmapour B, et al. Chromatin-Remodeling factor SPOC1 acts as a cellular restriction factor against human cytomegalovirus by repressing the major immediate early promoter. J Virol. (2018) doi: 10.1128/JVI.00342-18
96. Corradin O, Saiakhova A, Akhtar-Zaidi B, Myeroff L, Willis J, Cowper-Sal lari R, et al. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res. (2014) 24:1–13. doi: 10.1101/gr.164079.113
97. Gao X, Tsang JC, Gaba F, Wu D, Lu L, Liu P. Comparison of TALE designer transcription factors and the CRISPR/dCas9 in regulation of gene expression by targeting enhancers. Nucleic Acids Res. (2014) 42:e155. doi: 10.1093/nar/gku836
Keywords: methylation, promoter, microRNA, lncRNA, polymorphism
Citation: Ramsuran V, Ewy R, Nguyen H and Kulkarni S (2018) Variation in the Untranslated Genome and Susceptibility to Infections. Front. Immunol. 9:2046. doi: 10.3389/fimmu.2018.02046
Received: 03 April 2018; Accepted: 20 August 2018;
Published: 07 September 2018.
Edited by:
Eduardo Jose Melo Dos Santos, Universidade Federal do Pará, BrazilReviewed by:
Pei Xu, Sun Yat-sen University, ChinaSophie Limou, National Cancer Institute at Frederick, United States
Arman Bashirova, Leidos Biomedical Research, Inc., United States
Copyright © 2018 Ramsuran, Ewy, Nguyen and Kulkarni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Smita Kulkarni, c2t1bGthcm5pQHR4YmlvbWVkLm9yZw==