 Hiroyuki Nagashima
Hiroyuki Nagashima Naoto Ishii
Naoto Ishii Takanori So
Takanori So
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 30 August 2018
Sec. T Cell Biology
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01986
This article is part of the Research Topic TRAF Proteins in Health and Disease View all 12 articles
There is growing evidence that tumor necrosis factor (TNF) receptor-associated factors (TRAFs) bind to unconventional membrane-bound receptors in many cell types and control their key signaling activity, in both positive and negative ways. TRAFs function in a variety of biological processes in health and disease, and dysregulation of TRAF expression or activity often leads to a patho-physiological outcome. We have identified a novel attribute of TRAF2 and TRAF5 in interleukin-6 (IL-6) receptor signaling in CD4+ T cells. TRAF2 and TRAF5 are highly expressed by naïve CD4+ T cells and constitutively bind to the signal-transducing receptor common chain gp130 via the C-terminal TRAF domain. The binding between TRAF and gp130 limits the early signaling activity of the IL-6 receptor complex by preventing proximal interaction of Janus kinases (JAKs) associated with gp130. In this reason, TRAF2 and TRAF5 in naïve CD4+ T cells negatively regulate IL-6-mediated activation of signal transducer and activator of transcription 3 (STAT3) that is required for the development of IL-17-secreting CD4+ TH17 cells. Indeed, Traf2-knockdown in differentiating Traf5−/− CD4+ T cells strongly promotes TH17 development. Traf5−/− donor CD4+ T cells exacerbate the development of neuroinflammation in experimental autoimmune encephalomyelitis (EAE) in wild-type recipient mice. In this review, we summarize the current understanding of the role for TRAF2 and TRAF5 in the regulation of IL-6-driven differentiation of pro-inflammatory CD4+ T cells, especially focusing on the molecular mechanism by which TRAF2 and TRAF5 inhibit the JAK-STAT pathway that is initiated in the IL-6 receptor signaling complex.
The tumor necrosis factor receptor-associated factor (TRAF) family molecules in mammals were initially discovered as cytoplasmic adaptor proteins interacting with one of the tumor necrosis factor receptor superfamily (TNFRSF) molecules, TNFR2 (1). TRAF molecules are also present in Caenorhabditis elegans and Drosophila melanogaster (2–4). There are six mammalian TRAF molecules, TRAF1 to TRAF6, which share a conserved C-terminal TRAF-C domain that accommodates a short stretch of amino acids found in the cytoplasmic tail of receptors. Mammalian TRAFs critically participate in the signal transduction by receptors, such as TNFRSF molecules, Toll-like receptors (TLRs), nucleotide binding-oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene (RIG)-I-like receptors (RLRs), interleukin receptors, interferon receptors, transforming growth factor-β (TGF-β) receptor, the T-cell receptor (TCR) and platelet receptors. TRAFs link these receptors to various signaling cascades that are important in health and disease (3, 5–12).
One of the TRAF family molecules, TRAF5, is highly expressed in lung and moderately expressed in thymus, spleen, and kidney (13). In contrast to mice deficient in Traf2, Traf3, or Traf6, which become runted and die prematurely, Traf5−/− mice are born at the expected Mendelian ratios and exhibit no obvious abnormalities (14). One important question to be resolved is how TRAF5 specifically regulates cellular responses that are different and separate from those regulated by other TRAF family molecules.
Upon antigen exposure, naïve CD4+ T cells differentiate into different effector CD4+ helper T cell (TH cell) subsets that control the functions of B cells, macrophages, and CD8+ cytotoxic T cells through cell-to-cell contact and/or by secreting specific effector cytokines. There is growing evidence that TRAFs recruited to the TCR, costimulatory TNFRSF molecules, and cytokine receptors control key signaling events in CD4+ T cells and are critical for the activation, differentiation, and survival of TH cells in both positive and negative manners (11).
Although it has been well recognized that TRAF molecules play essential roles in T cell biology, the detailed functions and their molecular mechanisms of action are still enigmatic. In this review, we will highlight a novel function of TRAF2 and TRAF5 in the regulation of CD4+ TH17 cell differentiation that is controlled by pro-inflammatory cytokine IL-6 and its receptor signaling complex.
The regulation of IL-6 receptor signaling by TRAF molecules was initially suggested by the observation that after culturing in IL-6-containing TH17 skewing condition in vitro, differentiating CD4+ T cells lacking Traf5 produced a higher amount of IL-17 than did wild-type counterparts. However, Traf5-deficiency had no significant role for the development of TH1, TH2, TH17, Treg cells in polarized in vitro cultures. Accordingly, Traf5−/− mice exhibited exacerbated TH17 cell-dependent neuroinflammation in a model of experimental autoimmune encephalomyelitis (EAE). The enhanced EAE phenotype was recapitulated in irradiated wild-type mice that had been transferred with Traf5−/− CD4+ T cells, demonstrating that TRAF5 expressed in CD4+ T cells negatively regulates the generation of pathogenic TH17 cells (15). These results strongly suggested that TRAF5 regulated IL-6 receptor signaling that is required for TH17 differentiation.
Indeed, Traf5−/− naïve CD4+ T cells stimulated with a complex of IL-6 and soluble IL-6R (IL-6–sIL-6R) without triggering of TCR and CD28 exhibited increased phosphorylation of JAK1 and STAT3. In addition, the retrovirally transduced Traf5 gene in CD4+ T cells suppressed the phosphorylation of STAT3 mediated by IL-6–sIL-6R (16, 17). The negative regulatory function of TRAF5 for STAT3 was also observed in primary CD8+ T cells, but not in macrophages. One of the possible reasons would be that the expression of Traf5 mRNA was almost five times lower in macrophages than in CD4+ and CD8+ T cells (15). These results strongly suggest that if a cell expresses substantial levels of endogenous TRAF5 and gp130, TRAF5 can repress IL-6 receptor signaling activity in this cell type. Importantly, TRAF5 exhibited no inhibitory role for the STAT3 phosphorylation mediated by signaling through IL-10 receptor or IL-21 receptor in CD4+ T cells, demonstrating the specific action of TRAF5 for IL-6 receptor signaling (15).
By using a BAF/B03 cell line that stably expresses gp130 (BAF-gp130), we examined the role for TRAF family molecules in IL-6 receptor signaling and found that not only TRAF5 but also TRAF2 inhibited STAT3 phosphorylation and cell proliferation mediated by IL-6–sIL-6R, while TRAF1, TRAF3, TRAF4, and TRAF6 did not. In accordance with this, TRAF2 displayed a similar activity as TRAF5 in terms of the regulation of IL-6 receptor signaling and TH17 development, which was confirmed by shRNA-mediated knockdown and overexpression of each Traf gene in differentiating wild-type CD4+ T cells. TRAF2 did not inhibit the STAT3 phosphorylation downstream of IL-21 receptor in CD4+ T cells (16), confirming the specificity of TRAF2 to the IL-6 receptor signaling. Thus, we concluded that both TRAF2 and TRAF5 work as negative regulators of the IL-6 receptor signaling pathway.
NF-κB-inducing kinase (NIK) is critical for TH17 development, and both TRAF2 and TRAF3 limit NIK activity through ubiquitin-dependent degradation (18–21). In this reason, it was possible that TRAF2 and TRAF3 might inhibit TH17 development via degradation of NIK. However, increasing or decreasing the expression of TRAF3 did not affect the sensitivity of the IL-6 receptor signaling and the development of TH17 cells (16). In addition, it is unclear how TRAF2 regulates the differentiation of naïve CD4+ T cells into TH17 cells (20). Thus, we concluded that TRAF2 regulation of NIK expression levels is not the mechanism to limit the development of TH17 cells.
Although naïve CD4+ T cells from Traf5−/− and wild-type mice produced equivalent amounts of IL-6 in response to antigen stimulation (15), Traf2−/− macrophages and Traf5−/− B cells produced more IL-6 in response to TLR stimulation (22, 23). Traf2−/− Tnfa−/− mice displayed an inflammatory disorder and had elevated levels of IL-6 in serum (20). Thus, TRAF2 and TRAF5 might contribute to the development of TH17 cells in vivo via negative regulation of IL-6 production.
While TRAF2 and TRAF5 seemed to exhibit a similar role for the IL-6 receptor signaling pathway, detailed analyses revealed that the inhibition kinetic of TRAF2 for the IL-6 receptor signaling was different from that of TRAF5 due to different expression kinetics of respective TRAF proteins in developing CD4+ T cells. TRAF5 was higly expressed by unactivated naive CD4+ T cells, and Traf5 mRNA and TRAF5 protein were rapidly disappeared within a few hours upon TCR triggering (16). This means that there is only a narrow window of time for the inhibition for IL-6 receptor signaling by TRAF5 in differentiating CD4+ T cells. In contrast to this, Traf2 mRNA and TRAF2 protein were stably detected during the course of CD4+ T cell development, implying that TRAF2 can continuously suppress IL-6 receptor signaling as long as both gp130 and IL-6R are expressed in differentiating CD4+ T cells. In comparison with the regulation of Traf2 and Traf5 mRNAs, the expression of Traf1, Traf3, Traf4, and Traf6 mRNAs in CD4+ T cells were oppositely regulated, and these mRNAs were rapidly upregulated after stimulation with TCR and CD28, demonstrating that the expression of respective TRAF molecules is differentially controlled in recently activated naïve CD4+ T cells. In addition to this regulatory mechanism of TRAF molecules, after triggering of TCR and CD28, gp130 and IL-6R expressed by naïve CD4+ T cells were downregulated in a time-dependent manner, and these molecules were hardly detected on the surface of activated CD4+ T cells at 48 h after activation. In agreement with these results, addition of IL-6–sIL-6R at later time points of TH differentiation could not effectively promote the development of TH17 cells, and retrovirus-mediated transduction of short hairpin RNA (shRNA) that targets Traf5 in differentiating wild-type CD4+ T cells could not enhance the production of IL-17. On the other hand, shRNA-mediated knockdown of Traf2 in differentiating CD4+ T cells further promoted the development of TH17 cells in both wild-type and Traf5−/− conditons (16). Thus, it is reasonable to conclude that the instructive signals from IL-6 receptor for TH17 development are restricted both by the negative action of TRAF2 and TRAF5 and by the expression levels of gp130, IL-6R and TRAF5 (Figure 1). The expression of TRAF5 protein can be regulated by the ubiquitin proteasome system (21, 24, 25), although it is not known whether this type of regulation of TRAF5 is ongoing in activated naïve CD4+ T cells.
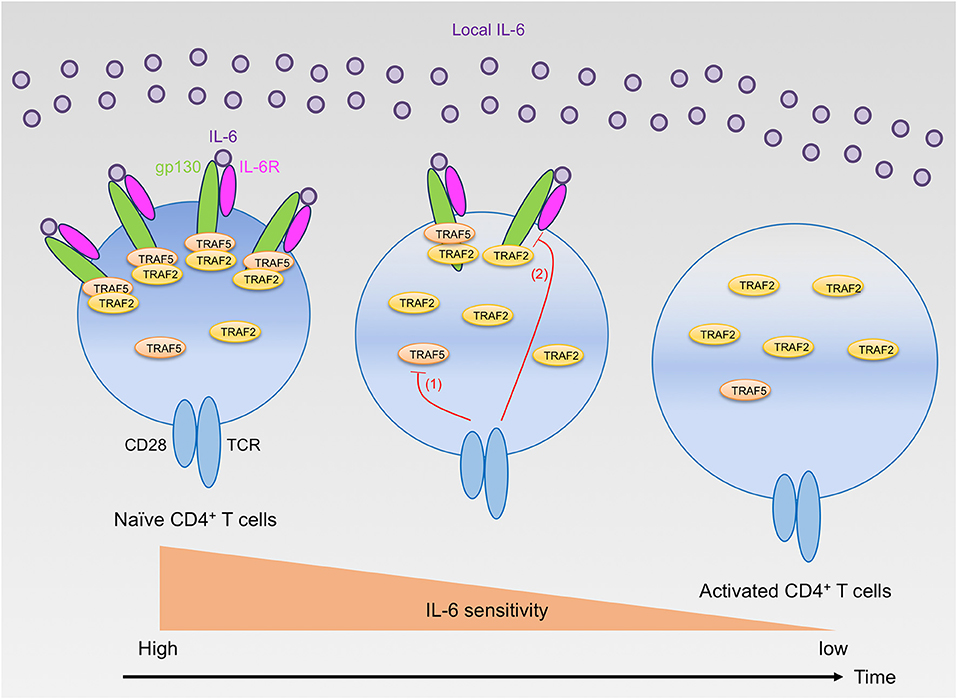
Figure 1. Regulation of IL-6 receptor signaling sensitivity by TRAF2 and TRAF5 in CD4+ T cells. Naïve CD4+ T cells highly express gp130, IL-6R, TRAF2, and TRAF5. Naïve CD4+ T cells can react to extracellular IL-6, but the signaling via the IL-6 receptor, IL-6R and gp130, is restrained by gp130-associated TRAF2 and TRAF5. After T cell activation by TCR and CD28, TRAF5 protein is rapidly downregulated (red line 1), while TRAF2 protein is maintained. Hence, TRAF5 limits the early IL-6 receptor signaling that is important for TH17 development. In contrast, TRAF2 can inhibit the signaling activity of the IL-6 receptor complex even in the later phase of TH17 differentiation. Moreover, the TCR and CD28 signaling also suppresses the expression of both gp130 and IL-6R (red line 2), and these receptor proteins are almost disappeared from the T cell surface within a few days after T cell activation. Therefore, activated CD4+ T cells lose their responsiveness to IL-6 and cannot receive the instructive IL-6 receptor signals required for TH17 development.
Naïve Traf5−/− CD4+ T cells expressed the same level of IL-6R and gp130 as wild-type naïve CD4+ T cells did, and the TRAF5 expression did not affect the STAT3 activation downstream of IL-10 receptor or IL-21 receptor in CD4+ T cells (15). Thus, we thought that TRAF5 directly regulated a key signaling process in the IL-6 receptor complex. Indeed, endogenously expressed TRAF5 constitutively bound to a cytoplasmic region of gp130 in primary CD4+ T cells. Co-immunoprecipitation assay using mutant proteins of TRAF5 and gp130 revealed that TRAF5 required its carboxy-terminal TRAF-C domain but not its amino-terminal RING/zinc-finger domains to interact with gp130 and that the TRAF5-C domain associated with a cytoplasmic region from residue 774 to residue 798 of gp130, gp130 (774-798), 774Val-Phe-Ser-Arg-Ser-Glu-Ser-Thr-Gln-Pro-Leu-Leu-Asp-Ser-Glu-Glu-Arg-Pro-Glu-Asp-Leu-Gln-Leu-Val-Asp798, which contains recognition elements for the TRAF-C domain (26–28). Similarly, TRAF2 bound to the same region in gp130 via the TRAF2-C domain. This cytoplasmic region of gp130 is highly conserved across various species including human (15, 16).
How do TRAF2 and TRAF5 negatively regulate IL-6 receptor signaling? Although the expression of TRAF5 did not inhibit the interaction between JAK1 and gp130, TRAF5 repressed the phosphorylation of JAK1, gp130, and STAT3 mediated by IL-6–sIL-6R. Therefore, it was hypothesized that TRAF5 limits the proximal interaction of JAK proteins and resulting their auto-phosphorylation by disturbing the optimal dimerization of gp130 upon interaction with IL-6 and IL-6R. By employing luciferase fragment complementation system using fusion proteins of JAK1 with either the N-terminal or the C terminal protein fragment of firefly luciferase, it was revealed that TRAF2 or TRAF5 suppressed JAK1-JAK1 interactions occurring after ligation of gp130 with IL-6–sIL-6R. Importantly, the JAK1-JAK1 interaction was intact in a mutant of gp130 (Δ774-798), which lacks the binding site for TRAF2 and TRAF5, even in the presence of TRAF2 or TRAF5. In addition, it was notable that RING and Zn finger domains of TRAF2 and TRAF5 are dispensable but TRAF-C domain is essential to suppress JAK1-JAK1 interactions to limit IL-6 receptor signaling (15, 17). This demonstrates that TRAF2 and TRAF5 require both TRAF-C domain and TRAF-binding region in gp130 to inhibit JAK1-JAK1 interactions and the following molecular events in the IL-6 receptor signaling pathway. Moreover, the expression of a peptide fragment of gp130 (769–800) fused with GFP in wild-type CD4+ T cells promoted the TH17 development (15), indicating that the peptide fragment of gp130 competitively inhibits the endogenous interaction between TRAFs and gp130. All of these data support the idea that TRAF2 and TRAF5 associated with gp130 via TRAF-C domains negatively regulate the JAK activation in the IL-6 receptor signaling complex that plays an essential role in initiating the JAK-STAT signaling pathway (Figure 2).
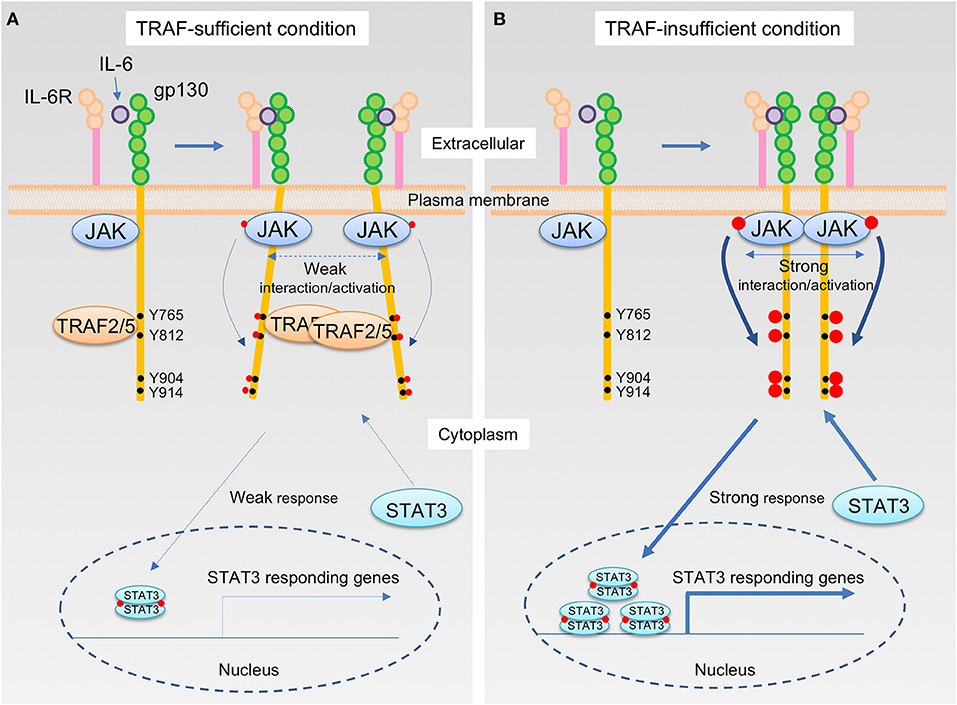
Figure 2. The IL-6 receptor signaling pathway that is regulated by TRAF2 and TRAF5. (A,B) Upon interaction of IL-6 with the IL-6R, the complex of IL-6 and IL-6R next binds to the IL-6 receptor common chain gp130, which leads to dimerization of gp130. Janus kinase (JAK) is constitutively bound to the intracellular domains of gp130, and thus this event brings JAKs into close proximity, inducing transphosphorylation of each JAK on a tyrosine residue, indicated in red circle, that stimulates kinase activity of JAKs. The activated JAKs then phosphorylate the cytoplasmic tail of gp130 on specific tyrosine residues, generating binding sites for signal transducer and activator of transcription (STAT) including STAT3. Recruitment of a STAT3 to the phosphorylated gp130 brings the STAT3 close to the activated JAK, which then the activated JAK phosphorylates a tyrosine residue of the STAT3. Phosphorylated STAT3 molecules form a dimer, and STAT3 dimers translocate to the nucleus, then induce the gene transcription involved in TH17 differentiation, including RAR-related orphan receptor-γt (RORγt) and IL-17. TRAF2 and TRAF5 constitutively bind to a cytoplasmic region of the gp130, which includes an amino acid sequence 774VFSRSESTQPLLDSEERPEDLQLVD798 and locates between first two out of four distal phosphorylated tyrosine motifs in gp130, Y765, Y812, Y904, and Y914, that are recognized by STAT3. For this reason, JAK interactions are interrupted by the presence of TRAF2 and/or TRAF5, and this event causes a weaker interaction/activation of JAKs and subsequent attenuated responses in the IL-6 receptor signaling pathway (A). On the other hand, in the absence of TRAF2 and/or TRAF5, a stronger association of JAKs facilitates an augmented JAK activation that leads to the enhanced STAT3 responses in the IL-6 receptor signaling pathway (B).
There has been some unsolved issues and controversy about the mechanisms regarding regulation of the IL-6 receptor signaling and the TH17 development mediated by TRAF2 and TRAF5. Firstly, when CD4+ T cells were stimulated with IL-6–sIL-6R instead of IL-6 alone, TRAF2 and TRAF5 could efficiently inhibit the IL-6 receptor signaling activity (15, 16). IL-6 trans signaling is activated via membrane-bound gp130 that interacts with a complex of sIL-6R and IL-6. IL-6 trans signaling or IL-6 cluster signaling plays a dominant role for priming pathogenic TH17 cells (29, 30). This suggests that TRAF2 and TRAF5 may preferentially restrain IL-6 trans signaling activity. Secondly, the identified TRAF-binding region in gp130 is located between first two phosphorylated-tyrosine (p-Tyr) motifs in gp130, and thus it is possible that TRAF2 and TRAF5 may inhibit the binding of STAT3 to these p-Tyr motifs via making steric hindrance in gp130 (15, 16). Thirdly, it is not clear how gp130-associated TRAF proteins inhibit the proximal JAK interaction in the receptor complex. The binding between gp130 and JAK1 occurred independently of the interaction between gp130 and TRAF5 (17). TRAFs may restrain the formation of gp130 dimer and inhibit the reposition process of associated JAKs (Figure 2). Fourthly, TRAF2 and TRAF5 might recruit a protein tyrosine phosphatase to the IL-6 receptor signaling complex to inhibit the JAK-STAT signaling, although this mechanism is utilized by TRAF3 expressed in B cells (31). Fifthly, TRAF5 might work as a positive regulator for RAR-related orphan receptor-γt (RORγt) activity and augment RORγt-mediated TH17 responses in a certain experimental setting (32), although this is inconsistent with the conclusion presented here.
It is now clear that TRAF family molecules control a wide range of signaling mediated by membrane-bound receptors in many cell types including CD4+ T cells. Findings highlighted here illustrate how TRAF2 and TRAF5 impact IL-6-mediated TH17 generation and TH17-driven immuno-pathology in vivo and in vitro and the molecular mechanisms by which TRAF2 and TRAF5 restrain IL-6 receptor signaling. The conclusion that TRAF2 and TRAF5 interacted with gp130 suppress proximal JAK-JAK interactions and resulting JAK phosphorylation in the receptor complex suggests that these TRAFs also regulate signals downstream of receptors for other IL-6 family cytokines that utilize gp130. Dysregulated TRAF5 expression might play an important role in autoimmune and inflammatory diseases in human (33). It will be absolutely important in the future to understand how TRAF2 and TRAF5 control signal transduction through unconventional cytokine receptors and to characterize its impact on immune responses and other relevant biological responses mediated by CD4+ T cells and other cell types.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work supported by JSPS KAKENHI grant numbers JP15H04640 (TS) and JP18H02572 (TS), the Daiichi-Sankyo Foundation of Life Science (TS), and the Tamura Science and Technology Foundation (TS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank members of Department of Microbiology and Immunology, Tohoku University Graduate School of Medicine for their assistance and help. We thank the Biomedical Research Core and the Institute for Animal Experimentation (Tohoku University Graduate School of Medicine) for technical support.
1. Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell (1994) 78:681–92. doi: 10.1016/0092-8674(94)90532-0
2. Grech A, Quinn R, Srinivasan D, Badoux X, Brink R. Complete structural characterisation of the mammalian and Drosophila TRAF genes: implications for TRAF evolution and the role of RING finger splice variants. Mol Immunol. (2000) 37:721–34. doi: 10.1016/S0161-5890(00)00098-5
3. Inoue J, Ishida T, Tsukamoto N, Kobayashi N, Naito A, Azuma S, et al. Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp Cell Res. (2000) 254:14–24. doi: 10.1006/excr.1999.4733
4. Wajant H, Henkler F, Scheurich P. The TNF-receptor-associated factor family: scaffold molecules for cytokine receptors, kinases and their regulators. Cell Signal. (2001) 13:389–400. doi: 10.1016/S0898-6568(01)00160-7
5. Chung JY, Park YC, Ye H, Wu H. (2002). All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci. 115, 679–88.
6. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. (2003) 21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126
7. Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. (2005) 23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839
8. Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. (2009) 9:271–85. doi: 10.1038/nri2526
9. Ha H, Han D, Choi Y. TRAF-mediated TNFR-family signaling. Curr Protoc Immunol. (2009) Chapter 11:Unit11.9D. doi: 10.1002/0471142735.im1109ds87
10. Xie P. TRAF molecules in cell signaling and in human diseases. J Mol signal. (2013) 8:7. doi: 10.1186/1750-2187-8-7
11. So T, Nagashima H, Ishii N. TNF Receptor-Associated Factor (TRAF) Signaling Network in CD4(+) T-Lymphocytes. Tohoku J Exp Med. (2015) 236:139–54. doi: 10.1620/tjem.236.139
12. Bishop GA. TRAF3 as a powerful and multitalented regulator of lymphocyte functions. J Leukoc Biol. (2016) 100:919–26. doi: 10.1189/jlb.2MR0216-063R
13. Ishida TK, Tojo T, Aoki T, Kobayashi N, Ohishi T, Watanabe T, et al. TRAF5, a novel tumor necrosis factor receptor-associated factor family protein, mediates CD40 signaling. Proc Nat Acad Sci USA. (1996) 93:9437–42. doi: 10.1073/pnas.93.18.9437
14. Nakano H, Sakon S, Koseki H, Takemori T, Tada K, Matsumoto M, et al. Targeted disruption of Traf5 gene causes defects in CD40- and CD27-mediated lymphocyte activation. Proc Nat Acad Sci USA. (1999) 96:9803–8. doi: 10.1073/pnas.96.17.9803
15. Nagashima H, Okuyama Y, Asao A, Kawabe T, Yamaki S, Nakano H, et al. The adaptor TRAF5 limits the differentiation of inflammatory CD4(+) T cells by antagonizing signaling via the receptor for IL-6. Nat Immunol. (2014) 15:449–56. doi: 10.1038/ni.2863
16. Nagashima H, Okuyama Y, Hayashi T, Ishii N, So T. TNFR-associated factors 2 and 5 differentially regulate the instructive IL-6 receptor signaling required for Th17 development. J Immunol. (2016) 196:4082–9. doi: 10.4049/jimmunol.1501610
17. Kimura M, Nagashima H, Okuyama Y, Ishii N, So T. TRAF2 and TRAF5 associated with the signal transducing receptor gp130 limit IL-6-driven transphosphorylation of JAK1 through the inhibition of proximal JAK-JAK interaction. Int Immunol. (2018) 30:291–99. doi: 10.1093/intimm/dxy029
18. Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity (2008) 28:391–401. doi: 10.1016/j.immuni.2008.01.009
19. Jin W, Zhou XF, Yu J, Cheng X, Sun SC. Regulation of Th17 cell differentiation and EAE induction by MAP3K NIK. Blood (2009) 113:6603–10. doi: 10.1182/blood-2008-12-192914
20. Lin WJ, Su YW, Lu YC, Hao Z, Chio II, Chen NJ, et al. Crucial role for TNF receptor-associated factor 2 (TRAF2) in regulating NFkappaB2 signaling that contributes to autoimmunity. Proc Nat Acad Sci USA. (2011) 108:18354–9. doi: 10.1073/pnas.1109427108
21. Yang XD, Sun SC. Targeting signaling factors for degradation, an emerging mechanism for TRAF functions. Immunol Rev. (2015) 266:56–71. doi: 10.1111/imr.12311
22. Buchta CM, Bishop GA. TRAF5 negatively regulates TLR signaling in B lymphocytes. J Immunol. (2014) 192:145–50. doi: 10.4049/jimmunol.1301901
23. Jin J, Xiao Y, Hu H, Zou Q, Li Y, Gao Y, et al. Proinflammatory TLR signalling is regulated by a TRAF2-dependent proteolysis mechanism in macrophages. Nat Commun. (2015) 6:5930. doi: 10.1038/ncomms6930
24. Bishop GA. The multifaceted roles of TRAFs in the regulation of B-cell function. Nat Rev Immunol. (2004) 4:775–86. doi: 10.1038/nri1462
25. Chen BB, Coon TA, Glasser JR, McVerry BJ, Zhao J, Zhao Y, et al. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat Immunol. (2013) 14:470–9. doi: 10.1038/ni.2565
26. Arch RH, Thompson CB. 4-1BB and Ox40 are members of a tumor necrosis factor (TNF)-nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclear factor kappaB. Mole Cell Biol. (1998) 18:558–65. doi: 10.1128/MCB.18.1.558
27. Park YC, Burkitt V, Villa AR, Tong L, Wu H. Structural basis for self-association and receptor recognition of human TRAF2. Nature (1999) 398:533–8. doi: 10.1038/19110
28. Ye H, Park YC, Kreishman M, Kieff E, Wu H. The structural basis for the recognition of diverse receptor sequences by TRAF2. Mole Cell. (1999) 4:321–30. doi: 10.1016/S1097-2765(00)80334-2
29. Jones GW, McLoughlin RM, Hammond VJ, Parker CR, Williams JD, Malhotra R, et al. Loss of CD4+ T cell IL-6R expression during inflammation underlines a role for IL-6 trans signaling in the local maintenance of Th17 cells. J Immunol. (2010) 184:2130–9. doi: 10.4049/jimmunol.0901528
30. Heink S, Yogev N, Garbers C, Herwerth M, Aly L, Gasperi C, et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat Immunol. (2017) 18:74–85. doi: 10.1038/ni.3632
31. Lin WW, Yi Z, Stunz LL, Maine CJ, Sherman LA, Bishop GA. The adaptor protein TRAF3 inhibits interleukin-6 receptor signaling in B cells to limit plasma cell development. Sci Signal. (2015) 8:ra88. doi: 10.1126/scisignal.aaa5157
32. Wang X, Yang J, Han L, Zhao K, Wu Q, Bao L, et al. TRAF5-mediated Lys-63-linked polyubiquitination plays an essential role in positive regulation of RORgammat in promoting IL-17A expression. J Biol Chem. (2015) 290:29086–94. doi: 10.1074/jbc.M115.664573
Keywords: TRAF5, TRAF2, IL-6, TH17, autoimmunity, inflammation
Citation: Nagashima H, Ishii N and So T (2018) Regulation of Interleukin-6 Receptor Signaling by TNF Receptor-Associated Factor 2 and 5 During Differentiation of Inflammatory CD4+ T Cells. Front. Immunol. 9:1986. doi: 10.3389/fimmu.2018.01986
Received: 29 June 2018; Accepted: 13 August 2018;
Published: 30 August 2018.
Edited by:
Tania H. Watts, University of Toronto, CanadaReviewed by:
Koji Yasutomo, Tokushima University, JapanCopyright © 2018 Nagashima, Ishii and So. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takanori So, dHNvQHBoYS51LXRveWFtYS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.