 Elena Pipi
Elena Pipi Saba Nayar1†
Saba Nayar1† David H. Gardner
David H. Gardner Serena Colafrancesco
Serena Colafrancesco Francesca Barone
Francesca Barone
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 September 2018
Sec. Inflammation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01952
This article is part of the Research Topic Autoantibodies View all 89 articles
Tertiary lymphoid structures (TLS) are frequently observed in target organs of autoimmune diseases. TLS present features of secondary lymphoid organs such as segregated T and B cell zones, presence of follicular dendritic cell networks, high endothelial venules and specialized lymphoid fibroblasts and display the mechanisms to support local adaptive immune responses toward locally displayed antigens. TLS detection in the tissue is often associated with poor prognosis of disease, auto-antibody production and malignancy development. This review focuses on the contribution of TLS toward the persistence of the inflammatory drive, the survival of autoreactive lymphocyte clones and post-translational modifications, responsible for the pathogenicity of locally formed autoantibodies, during autoimmune disease development.
The polyclonal expansion of autoreactive B cells is a cardinal feature of autoimmune conditions. Whether directed against a single antigen or playing part in a poly-specific response, autoreactive B cells support the persistence of the autoimmune process and, in several cases are directly pathogenic.
The development of an autoreactive B cell repertoire during the natural history of the autoimmune condition is regulated by the process of affinity maturation against single or multiple autoantigens that occurs within the inner part of the B cell follicles, classically within secondary lymphoid organs (SLOs) (1). Formation of B cell follicles and germinal centers (GC) has been also described in ectopic or tertiary lymphoid structures (TLS) in a process defined “ectopic lymphoneogenesis.” TLS form at target organs of chronic inflammatory/autoimmune process, localized infections and in the areas surrounding solid tumors (2–11). The prognostic value of these structures is debated. TLS formation in target organs autoimmune disease is classically associated with disease persistence and worst clinical manifestations. In solid tumors TLS have instead been associated with the generation of an anti-tumor response, however in some cases the ability of tumor cells to induce T regulatory cells (Treg) and suppress the host immune response has been described Table 1.
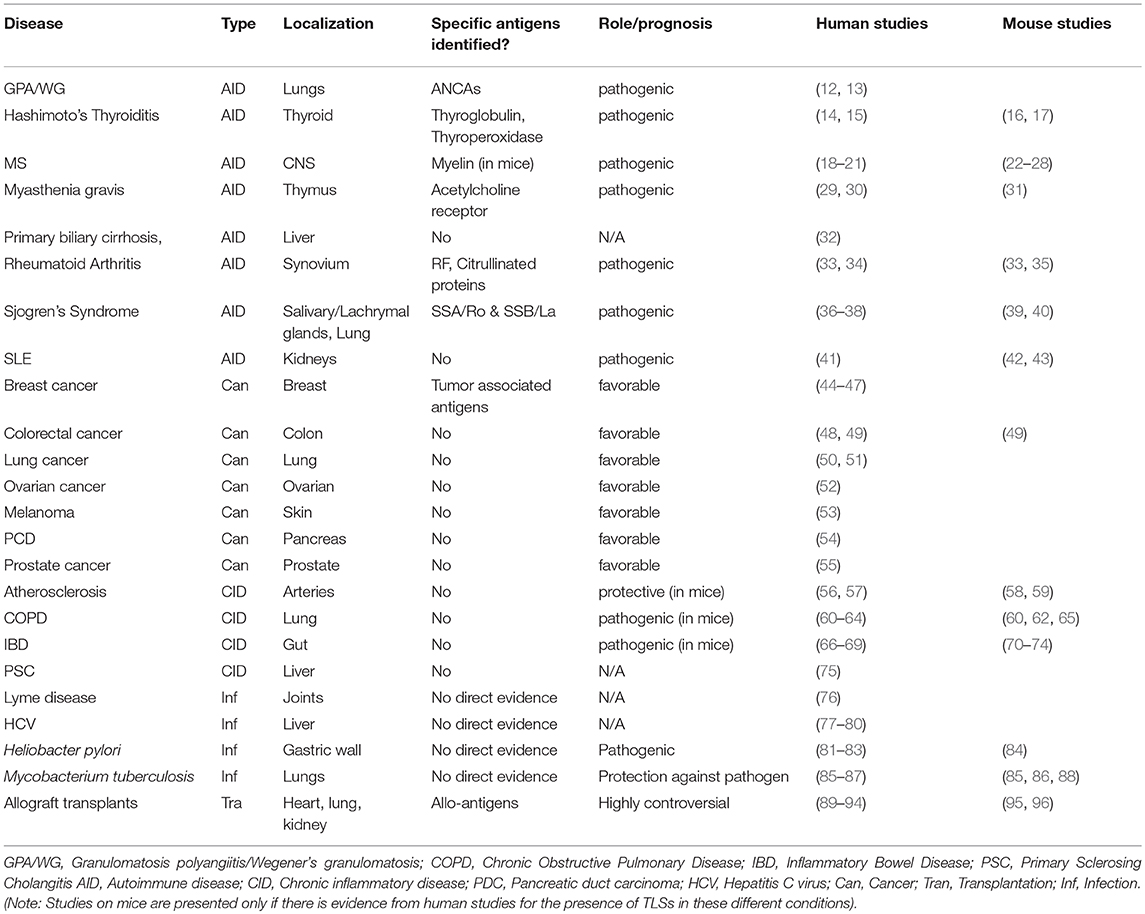
Table 1. TLS in different conditions.
Often indicated as “tertiary lymphoid organs,” TLS fail to adhere to the proper definition of organs as they lack a stable structural organization, including a capsule, and are better classified as “tertiary lymphoid structures” or TLS (97). TLS are not present in embryonic life and form in adult life to support local aggregation of lymphocytes at the target organ of disease. Other terms including “ectopic lymphoid structures” (ELS) or “ectopic germinal centers”. The latter, however, should only be used when GC formation is determined histologically within the ectopic lymphoid tissue (97–101). The cross-over between TLS and SLO is the subject of debate and has been reviewed by ourselves and others in recent publications (9, 98).
The term “tertiary lymphoid” tissue in the literature dates back to 1992 and was introduced by Louis Picker and Eugene Butcher (102) to describe the formation of extra-lymphoid sites, where memory lymphocytes and/or precursors can be re-stimulated by antigen to induce further clonal expansion or terminal effector responses. By definition, TLS arise in tissues whose main function is other than the generation of immune cells or the initiation of an adaptive immune response. This excludes the bone marrow and thymus, (as primary lymphoid organs) and spleen, lymph nodes and Peyer's patches (which are defined as SLOs). The kidneys, heart, pancreas, synovium and salivary glands are not embryologically predisposed to host the presence of lymphoid tissue therefore lymphocyte assembly at these sites should be considered TLS. The liver provides a hematopoietic function during embryonic development (103) however, this function is lost postnatally, thus including this organ among those that host TLS in adult life (97).
TLS form in response to a series of pro-inflammatory cytokines and TNF receptor family members following the local cross-talk between inflammatory immune cells and resident stromal cells. Fibroblasts, perivascular myo-fibroblasts and resident mesenchymal cells have been differently implicated in TLS development (39, 75, 104–112). Their role has been recently reviewed elsewhere (97, 113, 114). Probably evolved before SLO, TLS might have developed in ectopic tissues to fulfill the survival need of aggregated leucocytes, prior to placentation and development of SLOs. As such, the ability of TLS to be initiated independently from Lymphotoxin (LT) upon the expression of inflammatory cytokines and in absence of lymphoid tissue inducer cells (LTi) might have remained as heritage of their developmental ancestry (97).
Physiologically, the generation of a humoral response requires the physical interaction of naïve B cells with antigen experienced T cells within the confined space of a microenvironment rich in survival and chemotactic factors (115). Lymphocytes are recruited from the bloodstream to the SLO in response to a chemotactic gradient that regulates cell positioning and interactions (116, 117). CXCL13 and CCL19/CCL21, ligands for the chemokine receptors CXCR5 and CCR7, respectively, regulate the recirculation of naïve B cells between the inner part of the B cell follicle to the outer area of the T/B cell boundary (118), thus enabling the contact of B cells with antigen-experienced, activated T cells (119). Within the follicles, antigen-experienced B cells migrate toward the dark zone of the GC, a highly hypoxic CXCL12-rich area. Within this area they become highly proliferating centroblasts and upregulate the enzyme activation-induced cytidine deaminase (AID) (120, 121), that regulates the introduction of single base-pair substitutions of antibody gene segments in the immunoglobulin (Ig) variable-region genes, in a process defined as somatic hypermutation (SHM) (122).
Following SHM, B cells stop proliferating and undergo the process of affinity maturation (123). Differentiated, non-dividing B cells (centrocytes) upregulate CXCR5 and migrate along the CXCL13 gradient toward the GC light zone (120), herby establishing connections with the network of follicular dendritic cells (FDC) that provide survival factors (124, 125) and antigen presentation via the CR2 receptor (125, 126). Within the light zone, centrocytes also encounter mature T follicular helper cells (Tfh), known to provide signals for selection and terminal differentiation into long-lived plasma cells or memory B cells (127–130). Once exited from the GC, affinity matured B cells undergo the process of class switch recombination (CSR), that regulates isotype switching and ultimate effector function of the immunoglobulins (Igs). This latter process is also regulated by AID (130–142) (Figure 1A).
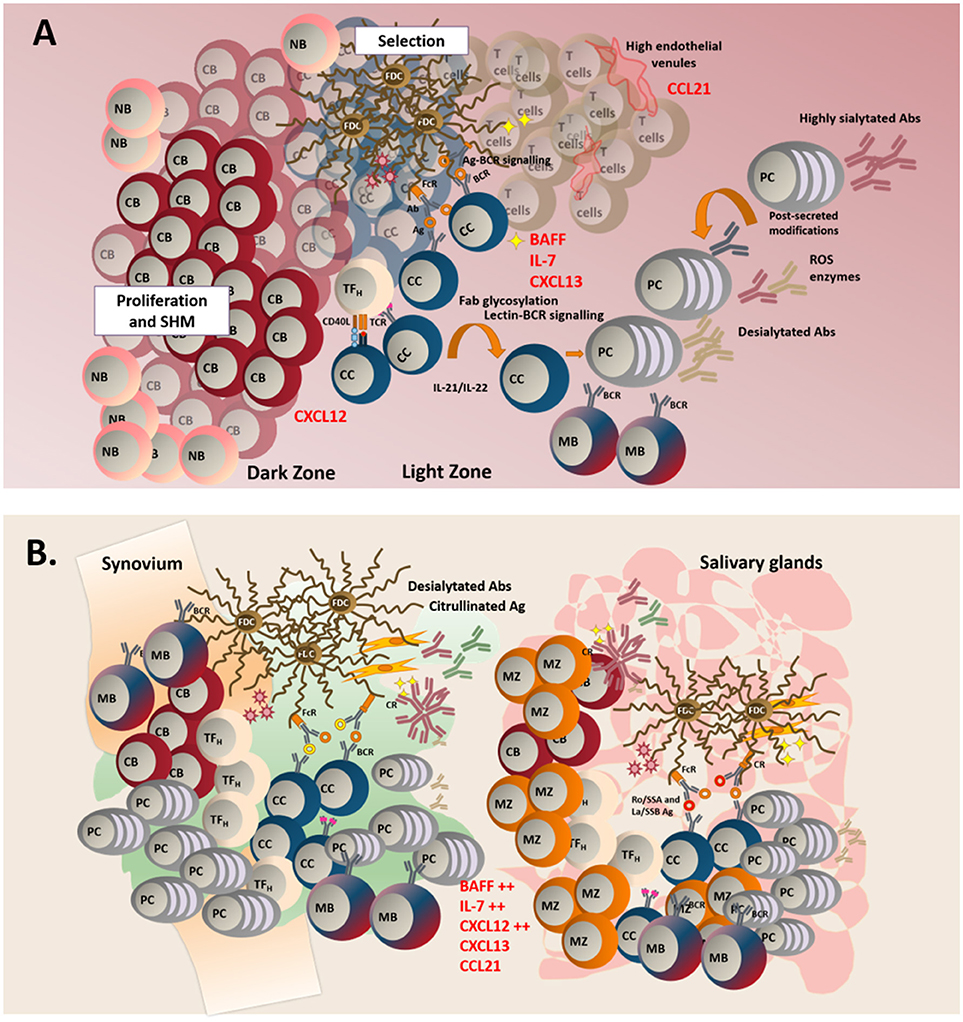
Figure 1. (A) In TLS, naïve B cells (NB), enter the follicle to initiate a classical germinal center reaction. In the dark zone, centroblasts (CB) proliferate and acquire somatic hypermutations in their variable region. In the light zone, centrocytes (CC) are selected after their interaction with specific antigen found on the surface of follicular dendritic cells (FDCs). Lectin-BCR signaling can potentially result in enhanced selection of B cells. Failure to receive survival signals from either TFH (T follicular helper cells) or FDCs leads to B cell apoptosis. Successful affinity maturation results in either mature B cell (MB) of plasma cells generation. GC microenvironment can control the outcome of the immune response by regulating the glycosylation profile of the antibodies. (B) TLS display a less organized anatomical structure and a predominant infiltration of MB and marginal zone B cells (MB). Aberrant production of survival and chemoattractant signals is observed at these sites.
This organizational program in SLOs is maintained by the anatomical differentiation of specialized, resident stromal cells that regulate migration and functional activation of the immune cells in the different part of the follicle (138, 143–149). The development of this stromal network and the signals required for his homeostasis have been reviewed elsewhere (150). TLS display a similar anatomical structure to support naïve B and T cell recirculation, including the expression of homeostatic lymphoid chemokines CXCL13, CCL21 and CCL19 and the molecular complex peripheral node addressin (PNAd) (97, 98, 151, 152). However, the complex anatomical compartmentalization displayed in SLO is rarely acquired in TLS. While the majority of reports on TLS describe a certain degree of T/B cell segregation, vascular/stromal cell specialization and expression of lymphoid chemokines, the presence of a more complex organization of the TLS and the formation of functional GC is highly variable within and amongst diseases (4, 153–155). In TLS that form during chronic autoimmune processes, the establishment of such disorganized microenvironment, rich in survival factors and pro-inflammatory cytokines, but likely missing key checkpoints for autoreactive cells screening, is likely responsible for the local generation of pathogenic autoantibody specificities and oncogenic mutations, ultimately favoring disease progression (1, 9, 97, 98).
In 1996, Nancy Ruddle described the presence of a “structural chronic inflammatory process” caused by ectopic production of lymphotoxin, in the context of chronic inflammation of the pancreas (156). Since then, TLS formation has been associated with a localized process of inflammation at sites of infection, autoimmunity, cancer, and allograft rejection. The ultimate pathogenic role of TLS is still debated (98, 151) and most likely depends on the context, organ and type of disease. For the scope of this review we will focus on the role of TLS in supporting the autoimmune process in chronic autoimmune conditions and we will discuss the role of TLS in Rheumatoid Arthritis (RA) and primary Sjögren's Syndrome (pSS) (33, 36, 135, 151, 157–162).
RA is the most common rheumatic autoimmune condition, affecting 0.5–1% of the global adult population. The pathological features of the disease include severe inflammation of the synovial membrane that, in some cases, leads to tissue destruction and subchondral bone erosions (163–166). Histologically, the disease can be classified in 3 main histopathological subtypes: a lymphoid type, mainly characterized by T and B cell aggregates that form TLS; a myeloid type, characterized by diffuse infiltration of prevalent monocyte and macrophages; and the fibroid type, defined by scarce or no immune cell infiltration and prevalent synovial fibroblast hyperplasia (151).
The presence of a “…marked infiltration of chronic inflammatory cells (lymphocytes or plasma cells predominating) with tendency to form “lymphoid nodules” was recognized already in the 1957 RA classification criteria (167). In 1972, Munthe and Natvig suggested that the RA synovial membrane is similar to an active lymphoid organ, containing many lymphoid follicles with GC that undergo local division and differentiation into plasma cells with restricted Ig production (168). Later, Steere and colleagues described “elements found in normal organized lymphoid tissue” in synovial lesions from both RA and Lyme disease patients (169); suggesting that the formation of GC-like structures in the synovium is not specific for RA and can be driven by the local antigenic stimulation. It took, however, more than 40 years after these first descriptions to introduce the concept that B-cell affinity maturation could arise within the inflamed synovium (170). It is now accepted that TLS are present in less than half of RA patients who display so called “lymphoid” synovitis (151) and that, in those patients, the presence of TLS is associated with differential prognosis and disease manifestations (151). TLS formation in the synovia have been also identified in patients with psoriatic arthritis (171) and ankylosing spondylitis (172, 173).
A similar phenomenon of leucocyte aggregation in lymphoid like structures occurs in the salivary glands of patients affected by pSS, a disease characterized by chronic inflammation of the exocrine glands, with progressive loss of function (sicca syndrome) and systemic activation of the humoral response (174). Excessive B cell hyperactivity and extra-glandular manifestation are observed in ~30% of pSS patients and an increased risk for lymphoma development has been described in this condition. In 1974, Chused et al. first described the presence of lymphoid-like structures in the salivary glands of patients with pSS (175). This was followed by the report of local antigenic stimulation within GC-like structures in the salivary glands (176) and, 10 years later, by the description of FDC network formation within the aggregates (177). In 1998, Stott and colleagues provided the first experimental evidence of an antigen-driven GC response, defined by clonal B cell proliferation and clone hypermutation within the salivary gland inflammatory foci (37), and, since then, several features associated with lymphoneogenesis have been reported within pSS aggregates (157, 178).
It is now recognized that during pSS, TLS form in the minor salivary and/or parotid gland in around 30–40% of patients (151) and those structures host a phenomenon of oligoclonal B cell expansion and SHM of the Ig variable genes (37). The formation of TLS in pSS salivary glands correlates with increased B cell hyperactivity, the presence of anti-SSA and anti-SSB autoantibodies, hypergammaglobulinemia and cryoglobulinemia, supporting the hypothesis that TLS persistence contributes to disease progression in pSS (179). Our group has contributed to these reports, describing both the expression of lymphoid chemokines and of AID within highly organized aggregates that harbor in the salivary gland of patients with pSS and MALT lymphoma (135, 180). The relationship between TLS formation and disease progression in pSS is still debated. TLS detection has been associated with high antibody titer, systemic manifestations and lymphoma development. However, the direct correlation between GC formation in the salivary glands and lymphoma formation has not been demonstrated, suggesting that the development of GC+ TLS within the salivary glands represent one of the stages in the process of lymphomagenesis but is not per se sufficient to induce lymphoma (135, 154, 161, 180–182).
In order to better understand the pathogenic effect that TLS play in disease it is important to dissect the elements, present within these structures that contribute to their function and persistence in the tissue.
There is enough evidence to support the hypothesis that TLS form to provide an immune response against locally displayed antigens. There are suggestions that TLS formation is an antigen (Ag)-driven process. In the mucosal associated lymphoid tissue that forms during Helicobacter Pilori gastritis antigen clearance following antibiotic treatment impacts on TLS maintenance and progression to lymphoma (183), similarly inducible bronchial associated lymphoid tissue can dissolve upon antigen clearance (184). Maffia and colleagues explored the properties of Ag presentation within TLS (58, 185) demonstrating that Ag presentation is regulated by a random process of diffusion, rather than selective Ag uptake by DCs. Those data are reinforced by the anatomical structure of TLS where conduits, able to support Ag movement and APC migration have been described (186). In this context, the absence of a capsule could favor not only the initial Ag delivery in the tissue, but the progressive accumulation of new antigen specificities during the course of the immune response, favoring the persistence of these structures in the tissue.
During a classical immune response, the antigens are collected by antigen presenting cells in the periphery and moved, via a complex network of lymphatic vessels, to draining lymph nodes (LNs) (187–189). LN space is pre-formed during the embryonic development and anatomically set before the generation of the immune response to accommodate optimal interaction between APC, Ag and immune cells. Differently by SLOs, TLS organization is not anatomically predisposed to organize such a response and Ag presentation is often provided by non-immune cells, such as stromal cells and epithelial cells (190–193).
Lack of Ag drainage could mechanistically explain TLS formation. TLS form spontaneously in the lungs of mice deficient for CCR7, a chemokine receptor required for the migration of antigen-charged dendritic cells (DCs) to draining lymph nodes (194). However, the reconstitution of these animals with CCR7-sufficient cells is enough to re-establish the physiological delivery of the antigen to the lymph node and to induce TLS resolution in the tissue. This evidence appears to suggest that an intrinsic defect in DCs is sufficient to trigger TLS establishment. However, it is not clear whether this phenomenon could be also supported by a defect of lymphatic drainage from the inflamed tissue.
The expansion of a functional network of lymphatic vessels is required for appropriate antigen delivery to the SLOs. There are several reports describing the dramatic remodeling of the lymphatic vessels during inflammation, whereby the activation of NF-κB pathway supported by the expression of LT, IL-1 and TNFα, stimulates the expression of Prox1 and increases the transcripts for the VEGF-R3, both of which are factors involved in lymphoangiogenesis (195–201). TLS lack the presence of an organized lymphatic system such as the one described in SLOs (152). However, the expansion of the lymphatic vascular system has been observed in these structures, in response to the same cytokine milieu that regulates the maturation of the non-vascular stroma at these sites (97, 105). It is not clear whether these newly formed vascular structures are, however, able to establish viable connections with pre-existing lymphatics. The failure to do so would prevent efficient drainage of the antigen to the SLOs and support the excessive antigenic stimulation in the peripheral tissue (89, 202–206).
Lymphangiogenesis associated with tertiary lymphoid structure (TLS) has been reported in numerous studies. Defects in lymphangiogenesis in RA present with a reduction in lymphatic flow, absence of lymphatic pulse and collapse of draining LNs is observed during disease and is associated with flare onsets as has been shown in vivo and ex vivo studies performed by Schwarz and colleagues (207). Accordingly, effective therapeutic approaches in RA, including anti-TNF and B cell have been associated with the expansion of the lymphatic bed (208) and increase in cell drainage from the synovium (209).
In a model of pSS our group demonstrated that during TLS assembly an expansion of the lymphatic vascular network takes place and this is regulated by the sequential engagement of IL-7 and LTβR signaling; suggesting the presence of a natural pro-resolving mechanism for lymphocyte exit from the tissues during TLS establishment (105).
The open questions related to the mobilization of Ag loaded APC to the draining SLOs could be addressed in the future by inducing TLS formation and tracking the movement of labeled antigen-loaded DCs across vessels. The possibility to interfere pharmacologically or genetically with the process of lymphoangiogenesis and with the molecules responsible for cell migration across these structures, is likely to elucidate this complex phenomenon and to provide evidences on the role of aberrant antigen presentation and vascular disturbances in TLS establishment and persistence.
Both RA and pSS are characterized by antibody production against a discrete set of autoantigens and a large body of research in this area has been focused around the identification of antigen specificities within the TLS in the context of these diseases. The presence of citrullinated proteins has been reported within the synovia of RA patients by Baeten (210) and others, and associated with the local expression of the enzyme peptidyl arginine deiminase (PAD) in patients characterized by high systemic and local levels of anti-citrullinated antibodies (APCA) (211). This report fails to demonstrate the presence of the citrullinated proteins within the synovial TLS and is in disagreement with other studies reporting the detection of citrullinated proteins in non-RA synovium lacking classical TLS (212); casting doubts on the exclusive association between citrullinated protein expression and TLS development in RA. Additional evidence that associate the presence of TLS with the generation of auto-antibody specificity against citrullinated peptides (but not necessarily local display of the defined antigen) will be discussed in a different section of this review.
Stronger evidences supporting the link between TLS and local auto-antigen presentation have been provided in pSS. Ro/SSA 52 kDa, Ro/SSA 60 kDa and La/SSB belong to a intracellular complex of RNA binding proteins that is physiologically involved in the intrinsic response to viruses (213). The aberrant expression of Ro and La has been reported in pSS patients upon cellular apoptosis or extracellular transport in vesicles (214–216). Moreover, the presence of anti-Ro52/TRIM21 specific plasma cells has been demonstrated, at the boundaries of well-organized TLS in pSS salivary glands, establishing a clear connection between local antigen presentation and TLS formation in this disease (158). The presence of extractable nuclear antigen (ENA) antibodies against these two ribonucleoproteins is pathognomonic for Sjogren's and associated with more severe systemic manifestation and worst prognosis (214, 216, 217).
We have recently reviewed the role of non-haematopoietic cells in TLS establishment and organization (97, 98) and for the scope of this issue focused on autoantibodies, we will limit the discussion in this manuscript to the lymphocytic compartment.
Whilst mainly constituted of B cells and associated with aberrant humoral responses and GC formation, TLS establishment and maintenance strongly relies on T cells. In humans, the presence of a shared TCR specificity among different follicles in the RA synovium, has been described, suggesting the presence of a common antigen for different TLS that form within the synovial tissue (218). In line with this finding, depletion of CD8+ T cells in human synovium-SCID mouse chimeras hinders the formation on TLS (218).
Recently, efforts have been made to identify the cells and signals required for TLS establishment and a series of reports have highlighted the important role of IL-17+ T cells. Th17 cells are required for iBALT formation (219) and for TLS establishment in a model of experimental autoimmune encephalomyelitis (EAE); the latter, dependent on the production of LT-αβ, IL-17 and IL-22 (22, 23, 220, 221). In human renal allograft rejection, Th17 cells have been shown to promote ectopic GC formation in an IL-21 dependent manner (222). Aberrant differentiation of Th17 cells in the absence of IL-27 has been also associated with aberrant TLS formation in an experimental model of arthritis and in a model of pSS (223, 224).
Our group has recently demonstrated the requirement of IL-22 producing T cells in the early phases of TLS establishment in murine salivary glands (39). In this model, IL-22 production, similarly to the IL-17 production in the lungs and brain, appears to regulate, independently but also in synergy with lymphotoxin and TNF, the ectopic production of lymphoid chemokines that defines TLS formation (97). These studies demonstrate that T cells, and in particular Th17/Th22 cells, play an important role in shaping the constituents of TLS in a manner that can support subsequent B cell recruitment and germinal center formation.
Whether TLS provide a site of aggregation for naïve T cell is not clear and whilst naïve T cell recruitment and priming has been reported within TLS that form in pancreatic tissue in NOD mice (225), it is more likely that effector T cells and central memory recirculate in these structures, in particular in the earliest phases of TLS assembly. On the contrary it is now well accepted that TLS function as a site for functional T cell polarization. TLS maintenance appears to hinge on the functional relationship between T-follicular helper cells and regulatory T cell populations. T cells displaying a Tfh phenotype have been described in TLS, where they are expected to regulate the GC reaction and the activation of resident proliferating B cells (1). In the TLS that form outside the arterial wall and control the atherosclerotic plaque development, the presence of Tfh correlates with the organization and maintenance of the ectopic B cell clusters (226). Functional interference of the Tfh by ICOS-L blockade results in decreased TLS formation and aberrant atherosclerotic plaque formation. The opposite effect is obtained by depletion of T regulatory cells, previously demonstrated to play a critical role in the homeostatic control of the TLS and in the atherosclerotic process (58).
The developmental program of Tfh in TLS is debated. There are suggestions that this population in TLS derives from a population of peripheral CXCR5+ T cells that migrate to the peripheral tissue following the newly established CXCL13 gradient. These circulating CXCR5+ cells do not bear classical signs of activation and would, by definition, preferentially migrate to SLOs; however, the local differentiation of HEVs and the expression of PNAd (the ligand for L-selectin) supports their homing to the TLS (227). Others suggest that, within TLS, Tfh locally differentiate from other T cell subpopulations, including Th17. In support of this hypothesis, in EAE, Th17 cells appear to acquire some characteristics of Tfh including the expression of CXCR5, ICOS and Bcl6 (23). Similarly, within the inflamed joints of RA patients, a population of PD1hiCXCR5−CD4+ T cells termed “peripheral helpers” has been described that appear to fulfill the function of Tfh within the periphery (228). In pSS the expansion of Tfh cells has been reported and correlates with the increasing frequency of memory B and plasma cells in the tissue and blood (229, 230).
Genetic manipulation in conditional knockouts is currently in use to induce TLS formation in mice deficient for specific T cell populations and will allow better definition of cellular requirements for TLS formation.
Classically, fully established TLS are mainly characterized by B cell infiltration and the inversion of the B/T cell ratio within TLS has been used as an index of disease severity (231, 232). In SLO, naïve B cells are known to receive antigen education and co-stimulation; however, whether a similar phenomenon would regularly occur in TLS is debated. Patients with pSS display altered peripheral blood B cell frequencies with a predominance of CD27− naïve B cells, diminished frequencies/absolute numbers of CD27+ memory B cells in the periphery, and an enrichment of mature B cells in the salivary glands (233, 234). The presence of CD20+CD27+ B cells and plasmablasts is a consistent finding in pSS salivary glands biopsies (235). Whilst we have reported the presence of IgD+ naïve B cells, in particular in large TLS (180), memory B cells remain the predominant component of the infiltrates (180, 236). This casts doubts over the possibility that naïve B cells are primed within the TLS (235). In support of this hypothesis, bona fide GC B cells (CD10+CD21+/−CD24+/−CD27−CD38+IgD− that express AID) are rarely found within the B cell aggregates of TLS, that are mainly inhabited by CD10−CD21+CD24+CD27±CD38−IgD+ marginal zone-like type II transitional B cells (159) (Figure 1B).
The connection between the marginal zone (MZ) and TLS establishment is also not clear. There are several evidences supporting the involvement of MZ B cells in autoimmunity, including reports of preferential SHM and B cell proliferation in MRL Fas/lpr mice spleen (237) and the presence of RF+ cells in the splenic marginal sinus bridging channels (238). The low threshold of BCR activation, the numerous effector functions of MZ B cells and the link between autoimmunity, TLS and MZ lymphoma development in pSS suggests a direct involvement of this population in TLS pathology (239, 240). However, the origin of the MZ-like B cells and the relationship between those and the ectopic GC has not been proven. In humans, MZ B cells are allegedly able to recirculate and carry a highly mutated B cell receptor (241–243), thus suggesting a post-GC origin of this population. This is not the case in mice, where MZ B cells are stable and permanently located in the spleen (242–244). Interestingly, however, MZ-B cells in humans share some phenotypic features of transitional B cells, a highly autoreactive B cell population that emerge from the BM and mature inside the spleen before entering the follicle (245–248), suggesting the possibility that transitional immature autoreactive cells are inhabiting the ectopic follicles. The recirculation pattern and screening of transitional B cells has been described from Spencer and co-authors in an elegant work that describes the migration and BCR editing of this population in the gut-associated lymphoid tissue (GALT) (245). This process, aimed at modifying the specificity of autoreactive clones, is altered in systemic lupus erythematosus (SLE), resulting in the expansion of the autoreactive B cell repertoire (245). In diseases characterized by TLS formation, such as pSS and RA, this recirculation pathway could be also altered, favoring the migration of autoreactive clones from the lymphoid organs to the TLS. Hereby, the aberrant expression of survival factors and chemokines would support clonal expansion in the absence of BCR editing and support persistence of autoimmunity.
The use of mass cytometry on digested tissue and sections are needed to better characterize in humans the phenotype and functional features of the B cells inhabiting the TLS. The use of transgenic mice engineered to track cells in vivo (249) will be useful in inducible models of TLS to perform migration studies in vivo.
More than simply acting as a hub for lymphocyte migration, TLS have also been shown to provide critical survival signals for incoming lymphocytes and differentiated long-lived plasma cells such as BAFF, IL-7, and CXCL12 (98, 250). The persistence of TLS in the tissue, despite peripheral B cell depletion of post Rituximab, has been reported in RA (251), SS and lymphoma (252) and, more recently in peri-bronchial TLS described in two patients with cystic fibrosis and chronic Pseudomonas aeruginosa infection treated with B cell depletion therapy before transplantation. The reason for this persistence most probably resides on the excess survival factors, such as B cell activating factor (BAFF) or IL-7 present within the TLS that protects tissue infiltrating cells.
BAFF is a potent B-cell survival factor produced within SLO GCs and in the periphery by fibroblasts and epithelial cells (159, 248–253) Excess BAFF is known to rescue self-reactive B-cells from peripheral deletion and allows their entry into forbidden follicular and marginal zone niches (253). The connection between BAFF, MZ B cells, loss of tolerance and TLS emerged from studies in mice transgenic for BAFF (BAFF-Tg), that develop a lupus-like syndrome followed by infiltration of MZ-like B cells within salivary glands TLS (254). Interestingly, BAFF-Tg asplenic mice that lack MZ-B and B1a cells, but retain normal B1b cell numbers, develop lupus nephritis but lack TLS in the salivary glands, suggesting that both BAFF and MZ-B cells are required for TLS establishment in this model (255).
Other lymphoid survival cytokines including IL-7 have been described in association with TLS establishment in chronic diseases (162, 256–258). Gene expression levels of IL-7, IL-7 receptor (both IL-7Rα and IL-2Rγ subunits) and its downstream signaling gene JAK3 are significantly elevated in RA patient biopsies displaying TLS (259). Similarly, engagement of the IL-7/IL-7R axis has been linked to formation of TLS in salivary glands and associated with pSS pathology (22, 23, 33, 36, 37, 39, 58, 75, 89, 102–112, 112–262). Among other critical homeostatic functions, IL-7 can abrogate the suppressive ability of Treg, altering the balance between pro-inflammatory effector cells vs. suppressive T cells (162, 256, 258). Consistent with these observations, in vivo studies demonstrated the ability of IL-7 to induce TLS formation (263–265). The reciprocal expression of T and B cell survival factors in TLS is somehow strictly regulated by the critical balance between infiltrating T and B cells, probably in response to gradients of lymphotoxin and TNF family members. The mechanism regulating this production and the resulting segregation of lymphocytes in T or B cell rich areas is still under investigation (266).
We and others have provided evidence that a functional GC response takes place within these structures. This supports the concept that even if TLS do not initiate disease they are involved in its progression. In particular, we have demonstrated that AID is expressed in pSS salivary gland TLS in association with networks of follicular dendritic cells (135) and that its expression is retained in the large GCs found in parotid pSS-MALT lymphomas. On the contrary, neoplastic B cells are found to be consistently negative for AID expression (135). AID expression in GC B cells controls susceptibility to apoptosis, ultimately regulating the magnitude of the GC response (267). In SLO, low levels of AID expression have been associated with defective somatic hypermutation and decreased peripheral B cell tolerance (268). AID expression in TLS is consistently low (as compared to SLOs), thus potentially explaining the aberrant survival and lack of selection of autoreactive B-cell clones in ectopic GCs.
Other data have been generated supporting the functional role of TLS in sustaining the generation of novel antibody specificities. Transplantation of TLS from pSS salivary glands infected with Epstein-Barr virus (EBV) into SCID mice have been shown to support the production of anti-Ro 52/anti-La 48 and anti-EBV antibodies and the survival of autoreactive B cell clones (158). Similar data have been produced for RA. The presence of CD138+ plasma cells, characterized by immune reactivity against citrullinated fibrinogen, has been described within AID+/CD21+ follicular structures (33). Moreover, the survival of these clones in a transfer model of human biopsies in SCID mice, alongside the detection of gamma-Cmu circular transcripts in synovial grafts, has been reported. These observations provide evidence that synovial TLS represents an independent compartment for B cell maturation (33).
The contribution of the immune response that arises within TLS toward disease severity, including the production of autoantibodies, remains controversial (151). Nonetheless there are substantial evidences in support of local antibody production within the inflamed synovium and convincing documentation that the synovial microenvironment could independently favor the production of RA specific antibodies (33)
Mellors et al. firstly described the presence of “plasma B cells” that are able to react with FITC-labeled human IgG, interpreting this result as evidence of synovial production of rheumatoid factors (RF) by tissue-resident plasma cells (269). The first solid indication of local IgG production in RA is dated to 1968 with the report of Ig synthesis in rheumatoid synovium in vitro (270). Further studies supported this observation suggesting that gene selection, usage of kappa/lambda chains and class switching follows a non-stochastic process in the RA synovium (168). Similarly, the enrichment in RF+ B cells producing mono-reactive, affinity matured, class switched antibodies in the RA synovium is highly suggestive of a local process of affinity maturation (271–273). On the contrary, clones producing mono-reactive RF have not been obtained from the synovial tissue of patients with osteoarthritis, where TLS do not form, supporting the link between chronic autoimmune diseases and TLS (271–275).
The production of anti-citrullinated protein antibodies (ACPA) has been firmly associated with RA development (276) and there are convincing evidences that these specificities can be locally produced in the RA synovium within the TLS (277, 278). Both anti-cyclic citrullinated peptide (CCP) antibodies (279) and anti-CCP producing B cells (280) have been detected in the synovial fluid of RA patients and antibodies against different citrullinated RA candidate antigens (vimentin, type II collagen, fibrinogen and α-enolase) appear to be enriched in the joint compared to paired serum (281). Notably, the presence of anti-CCP antibodies in the synovium has been also reported in RA seronegative patients (279, 282), thus highlighting the dissociation between the systemic and local autoimmune response. In support of this notion, the presence of FcRL4+ ACPA producing IgA-B has been reported in the synovium, but not in the blood of RA patients (283). This observation provides an indication that inflammatory joints provide a specific microenvironment able to shape and influence B cell immune phenotype and output.
The ability of TLS to sustain the whole autoimmune process in the absence of SLO is debated. However, cloning of the local B cell repertoire isolated from inflamed organs bearing TLS is highly suggestive of the presence of a functional and SLO-independent process of affinity maturation. Terminally differentiated CD20−CD38+ cells, rheumatoid factor (RF) producing B cells have been detected in the inflamed joints of RA patients (284). Moreover, clonal analysis has provided evidence of an antigen-dependent process of SHM, selection and isotype switching in TLS positive RA synovium, indicating that a dominant antigen-specific local immune response shapes the synovial plasma cell repertoire (170, 285–290). Similarly, in pSS, the multiclonal expansion of B cells within the salivary glands has been described. Expansion of B cell clones bearing Humkv325, a conserved V kappa gene usually associated with lymphomas, was described previously in 1989 (291). Additional studies further supported the notion that an antigen-driven germinal center-type B cell response and somatic hypermutation occurs within the salivary glands (37, 292, 293). The presence of clones that expand and mature in the TLS does not prove that the autoimmune process is initiated within the TLS, or that the presence of TLS is causative of disease. However, a certain degree of antigen-experience and affinity maturation of the B cell repertoire undoubtedly occurs within TLS (33, 135, 153, 160, 294–296) and, whilst the causal role of these structures in disease initiation cannot be proved, TLS certainly display the ability to host and perpetuate the autoimmune process. Production of Ig and RF has been shown in other tissues, in addition to the synovium, including rheumatoid pericardium (297), pleura (298), muscles (299), and in the inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of RA (219). The presence of sputum autoantibodies in the absence of systemic seropositivity, and the increased autoantibody:total Ig ratio in the sputum (300) suggest that lymphoid tissue present in the bronchi of RA lungs can also act as sites of antibody development.
Independent IgG and IgM synthesis has been also described in pSS salivary glands (301) with later studies confirming the presence of RF+ clones in ~43% of patients with pSS (302) and with the ability of salivary gland infiltrating B cells to secrete antibodies specific for the Ro52/TRIM21, Ro60 and La autoantigens (36, 179, 217, 303). In vitro expression of recombinant antibodies derived from either newly emigrant/transitional mature naïve B cells from pSS patients and healthy individuals confirmed the presence of high frequencies of autoreactive antibodies in both populations. This suggests a general defective peripheral B cell tolerance in this condition (304).
Analysis of Ig levels in different compartments (blood, saliva) has further contributed to our understanding of the ability of TLS to independently produce antibodies. Increased levels of IgA, but not IgG- and IgM-RF, has been detected in the saliva of patients with pSS (305). A study on isotype distribution of anti-Ro/SS-A and anti-La/SS-B antibodies in the plasma and saliva of patients with pSS demonstrated a correlation between the focus score (the measured degree of salivary gland inflammation) and autoantibody titers in saliva or blood. This report establishes a pathogenic link between locally displayed autoantigens, presence of antigen specific B cells in the inflamed tissue and autoreactive Ig levels (306).
It is becoming increasingly clear that antibody post-translational modifications, in particular glycosylation, can influence their function and pathogenicity. However, a relationship between the pathogenic microenvironment established in the TLS and the progressive acquisition of pathogenic post-translational modifications has not been demonstrated.
Glycosylation is the process by which glycans are attached to proteins, lipids and other molecules, thereby altering their structure and influencing their biological activity. Whilst IgG presents a single conserved N glycosylation site within the Fc region, other subclasses are more heavily glycosylated (307). IgG Fc glycosylation determines the binding of the globulins to their receptors, FcRs type I (FcgammaRs) and II (SIGN-R1, DC-SIGN, DCIR, CD22, and CD23), thereby influencing Ig downstream pro-inflammatory, anti-inflammatory or immunomodulatory effects (308, 309). In addition to conserved IgG Fc glycans, ~15–25% of serum IgG contain glycans within the Fab domain. Intriguingly, the attachment sites for N-glycans to the Fab portion is determined by the process of somatic hypermutation and, accordingly, Fab glycosylation could influence antibody binding, activity, half-life, formation of immunocomplexes and strength of BCR signaling [extensively reviewed in (310)].
The presence of altered glycosylation in RA was suggested in the 1970s, but it wasn't until 1985 when two studies from Oxford and Japan demonstrated different galactosylation profiles between normal individuals and patients with RA or OA (311). Later, Axford and colleagues reported the presence of reduced circulating B cell galactosyltransferase activity in RA (312), which was later confirmed in other studies (313–315). Other post-translational modifications have been described in RA. Several studies have demonstrated the presence of an altered overall glycosylation status within specific Ig subclasses (316) that can be detected before disease onset (317). This correlates with measures of disease activity (318, 319) and decreased sialylation of RF-IgG, but not in non-RF-IgG (318, 320, 321).
More recently, the degree of IgG glycosylation has been used to monitor treatment effectiveness (321) and, whilst no differences have been observed in the Fc glycosylation pattern between ACPA-IgG1 and total IgG1 in arthralgia patients, a decrease in galactose residues have been observed in patients with preclinical synovitis before the onset of RA; a change probably supported by the increasingly inflammatory microenvironment (322). The increased presence of agalactosylated IgG in the synovial fluid as compared to serum samples of RA has also been reported (323). Finally, Scherer et al. recently demonstrated the presence of autoreactive IgG in synovial fluid with decreased number of galactosylation and sialylation sites as compared to serum. This latter difference appeared to be specific for autoreactive specificities as no difference was observed in total IgG glycosylation (324).
Elevated levels of asialylated IgG have been detected in 60% of pSS patients and those appear to correlate with clinical manifestations, such as Raynaud's phenomenon and arthritis. A strong correlation with rheumatoid factor or IgA-containing immune complexes was reported (325). Based on IgG galactosylation, the pSS patients can be classified into two groups: one with comparable galactosylation status as in RA patients with the presence of RF, and the other similar to healthy individuals, and RF seronegative (326).
More recently, studies on Fab glycosylation and disease have been performed. Corsiero and colleagues reported the relationship between increased molecular weight of anti-NET antibodies and the presence of N-glycans onto the Fab domain of autoreactive clones in RA, suggesting that the process of SHM occurring in the synovium is responsible for the acquisition of-N glycosylation sites (286). Acquisition of N-glycosylation sites and subsequent enrichment in Fab-glycans in the variable domain of ACPA-IgG has been further confirmed (327, 328). On a similar note, it has been reported that there is a selective increase in Fab-N glycosylation sites in ACPA specific clones. However, the presence of those glycans didn't appear to significantly alter the antigen binding of the APCA. Accordingly, in silico analysis suggested that the added glycans were not located on the antibody binding sites (329). Moreover, an increased frequency of N-glycans in the Fab ACPA domain, but in association with altered antibody affinity, has been also reported (330). Interestingly, this enrichment was more prominent on ACPA isolated from synovial fluid compared with peripheral blood (264), providing evidence that the local microenvironment influences the immunoglobulin glycosylation pattern. Hamza et al. recently reported the high prevalence of acquired IgG N-glycosylation sites in pSS suggesting a hypothesis that in pSS, the selection pressures that shape the antibody repertoire in the parotid glands results from an antigen-independent mechanism and is driven by interactions between glycosylated B cell receptors and lectins within the microenvironment (328). In summary, the glycan composition can have different associations with the disease, depending on the site of glycosylation. Decreased and increased glycosylation for the Fc and Fab portion, respectively, have been associated with RA and SS.
A relationship between post-translational modifications and antibody pathogenicity has been proposed. Leader et al. reported the presence of agalactosylated IgG in synovial immunocomplexes, suggesting a pathogenic role for agalactosylated IgG (331). However, the relationship between glycosylation and RF activity is debated (318, 332). The presence of N-linked glycosylation sites within the Fc portion of target IgG has been also shown to markedly reduce RF binding in vitro (333) whilst the ability of rheumatoid factors to selectively bind hypogalactosylated IgG has been suggested (334). In mice, desialylated but not sialylated immune complexes appear to enhance osteoclastogenesis in vitro (335). Accordingly, artificial sialylation of anti-type II collagen antibodies, including ACPAs, but not other IgG can supress the development of collagen-induced arthritis (CIA) (320, 336).
A potential pathogenic role of IgG glycosylation in pSS pathogenesis, to our knowledge, has not been addressed yet. A recent study pointed out that the Fc-mucin binding is enhanced when antibodies are agalactosylated, offering a mechanistic concept for increased binding on mucosal surfaces of the inflammatory agalocysylated antibodies and potential antibody pathogenicity (337).
Agalactosylated IgG levels were not found to be correlated in twin pairs indicating a low influence from genetic factors for IgG glycosylation (338). However, four loci contained genes for glycosyltransferases (ST6GAL1, B4GALT1, FUT8, and MGAT3) have been highlighted in genome-wide association studies for loci associated with IgG N-glycosylation (339). There is evidence to support the notion that the microenvironment can influence Fc IgG glycosylation. A recent study illustrates the ability of CpG, IFN-gamma and IL-21 to increase Fc-linked galactosylation and reduce bisecting N-acetylglucosamine levels (340). Stimulation of a mouse B cell lymphoma line with IL-4 and IL-5, but not LPS, has been shown to significantly decrease the terminal glycosylation of secreted IgA (341) and IgM (342), but substantially increase the terminal glycosylation of MHC Class-I (342), suggesting that the glycosylation machinery works in a protein-specific manner.
A mechanistic link between inflammation and post-translational modification has been recently established by G. Schett's group in a manuscript illustrating the ability of IL-21 and IL-22 to regulate the expression of α2,6-sialyltransferase-1 in newly differentiating plasma cells, thus controlling the glycosylation profile of secreted IgG (320). Interestingly, both T cell-independent B cell activation (343) and tolerance induction with T cell-dependent protein antigens (344) results in the production of sialylated IgG. However, T cell independent vaccination seems to result in a stronger induction of sialylated antigen specific antibodies (345).
IgG glycosylation can also be controlled at an extracellular level. IgG sialylation has been reported in the bloodstream, through secreted ST6Gal1 (326). S-glycosyltransferases have also been shown to alter the IgG molecule at sites of inflammation with local platelets serving as nucleotide-sugar donors (346). Other reports link the process of altered glycosylation to a post-secretory degradative process involving oxygen free radicals (347). All together these reports suggest the possibility that the site of antibody synthesis can profoundly affect the post-translational profile of the immunoglobulins.
Due to technological limitations, the extent of the disease-related glycan alterations and the role of these modifications in disease pathophysiology has not been thoroughly addressed. A novel microfluidic-based method to identify trace sulphated IgG N-glycans as biomarkers for rheumatoid arthritis has been recently described (348) and high-throughput methods for analysing IgG glycosylation have also been introduced (349). These tools have been only used in selected populations and their application on a larger scale could, in the future, unveil differences and patterns not yet captured.
To our knowledge, there has been no attempt to use these stratification tools in the context of TLS associated pathologies. The differential profile of glycosylation observed in Ig isolated, respectively from serum and synovial compartments suggest the fascinating hypothesis that SLO and TLS differentially regulate these post-translational modifications (323, 324, 350). However, the possibility that Ig derived from SLO and TLS present substantially different “sugary fingerprints” and that those patterns correlate with a certain degree of tissue involvement and disease progression has still to be proven.
If the concept of an association between progressive post-translational modifications of the Ig repertoire and continuous antigen exposure within a highly inflammatory environment is true, we should be able to detect progressive accumulation of Ig in patients undergoing malignant transformation through the course of autoimmunity. The occurrence of non-Hodgkin's lymphoma (NHL) is pathogenically linked to TLS and represents the most serious complication of pSS, but not RA (351).
B cell VH and VL gene analysis for pSS patients with lymphoma revealed several point mutations in the germline genes and intra-clonal sequence heterogeneity, in line with an ongoing somatic hypermutation process sustaining lymphoma growth (352, 353). It is believed that the emergence of monoclonal RF B cells within the polyclonal infiltrate of the salivary gland TLS represents a key developmental step in lymphomagenesis. Chromosomal abnormalities and mutation eventually converge in these B clones that present a proliferative advantage, ultimately converting them into malignant clones. Indeed, there is a strong bias for RF specific B cells in the salivary gland MALT-type lymphoma (354–356), whilst alternative analysis of the B cell repertoire in micro dissected labial salivary glands could not convincingly demonstrate predominance of RF reactivity in the infiltrating clones (357). The relationship between GC formation and lymphomagenesis has been recently challenged and further studies will be required to clarify the pathogenic link between TLS persistence and emergency of malignant clones (161, 182, 358, 359).
The high incidence of acquired N-glycosylation sites found in follicular lymphoma (360) would be suggestive of a similar phenomenon in pSS associated MALT lymphoma; however, contrary to these expectations, patients with MALT lymphoma present low frequency of N-glycosylation sites (161, 182, 358, 359, 361). Longitudinal analysis of the glycosylation and sialylation profile in patients with TLS undergoing lymphoma transformations are needed to address these questions.
In conclusion, TLS formation can be easily considered as a hallmark of tissue autoimmunity. In the past few years a large body of work has been generated aimed at dissecting key aspects of TLS biology, however, many areas have to be further addressed. The inter-dependency between SLOs and TLS has to be better dissected in order to understand whether these immune hubs are functional, both in the early phases as tolerance is broken and, later, during disease progression. The signals regulating migration pathways and differentiation of immune cells within the TLS should also be investigated in vivo with the prospective to target these pathways therapeutically. A better knowledge around the signals involved in TLS establishment and maturation, but in particular, the mechanisms regulating GC formation and regulation should be acquired. Moreover, a specific effort should be made to dissect the functional role of TLS GCs in the development of lymphoma. Finally, key questions should be answered around the cross-talk between the TLS and their surrounding environment, dissecting the permissive factors for TLS formation and persistence in the tissue.
The acquired knowledge on the role of non-haematopoietic stromal cells in TLS biology has been critically important in explaining why these structures are resistant to classical immune cell therapy. In the future, potential combined therapy could be utilized to interfere with the microenvironment alongside targeting immune cells in TLS associated disease that is non-responsive to classical immunosuppression.
EP and FB defined the content of the manuscript, contributed to literature search and manuscript writing. SN, DG and SC contributed to literature search and manuscript writing. CS contributed to manuscript writing. EP and FB created graphical illustrations. All authors approved the final version of the manuscript.
FB is supported by ARUK.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol. (2014) 14:447–62. doi: 10.1038/nri3700
2. Engelhard VH, Rodriguez AB, Mauldin IS, Woods AN, Peske JD, Slingluff, et al. Immune cell infiltration and tertiary lymphoid structures as determinants of antitumor immunity. J Immunol. (2018) 200:432–42. doi: 10.4049/jimmunol.1701269
3. Teillaud JL, Dieu-Nosjean MC. Tertiary lymphoid structures: an anti-tumor school for adaptive immune cells and an antibody factory to fight cancer? Front Immunol. (2017) 8:830. doi: 10.3389/fimmu.2017.00830
4. Jing F, Choi EY. Potential of Cells and Cytokines/Chemokines to Regulate Tertiary Lymphoid Structures in Human Diseases. Immune Netw. (2016) 16:271–80. doi: 10.4110/in.2016.16.5.271
5. Alsughayyir J, Pettigrew GJ, Motallebzadeh R. Spoiling for a fight: b lymphocytes as initiator and effector populations within tertiary lymphoid organs in autoimmunity and transplantation. Front Immunol. (2017) 8:1639. doi: 10.3389/fimmu.2017.01639
6. Sautes-Fridman C, Lawand M, Giraldo NA, Kaplon H, Germain C, Fridman WH, et al. Tertiary lymphoid structures in cancers: prognostic value, regulation, and manipulation for therapeutic intervention. Front Immunol. (2016) 7:407. doi: 10.3389/fimmu.2016.00407
7. Germain C, Gnjatic S, Dieu-Nosjean MC. Tertiary lymphoid structure-associated b cells are key players in anti-tumor immunity. Front Immunol. (2015) 6:67. doi: 10.3389/fimmu.2015.00067
8. Mitsdoerffer M, Peters A. Tertiary lymphoid organs in central nervous system autoimmunity. Front Immunol. (2016) 7:451. doi: 10.3389/fimmu.2016.00451
9. Jones GW, Hill DG, Jones SA. Understanding immune cells in tertiary lymphoid organ development: it is all starting to come together. Front Immunol. (2016) 7:401. doi: 10.3389/fimmu.2016.00401
10. Buettner M, Lochner M. Development and function of secondary and tertiary lymphoid organs in the small intestine and the colon. Front Immunol. (2016) 7:342. doi: 10.3389/fimmu.2016.00342
11. Neyt K, Perros F, GeurtsvanKessel CH, Hammad H, Lambrecht BN. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol. (2012) 33:297–305. doi: 10.1016/j.it.2012.04.006
12. Voswinkel J, Assmann G, Held G, Pitann S, Gross WL, Holl-Ulrich K, et al. Single cell analysis of B lymphocytes from Wegener's granulomatosis: B cell receptors display affinity maturation within the granulomatous lesions. Clin Exp Immunol. (2008) 154:339–45. doi: 10.1111/j.1365-2249.2008.03775.x
13. Voswinkel J, Mueller A, Kraemer JA, Lamprecht P, Herlyn K, Holl-Ulrich K, et al. B lymphocyte maturation in Wegener's granulomatosis: a comparative analysis of VH genes from endonasal lesions. Ann Rheum Dis. (2006) 65:859–64. doi: 10.1136/ard.2005.044909
14. Armengol MP, Cardoso-Schmidt CB, Fernandez M, Ferrer X, Pujol-Borrell R, Juan M. Chemokines determine local lymphoneogenesis and a reduction of circulating CXCR4+ T and CCR7 B and T lymphocytes in thyroid autoimmune diseases. J Immunol. (2003) 170:6320–8. doi: 10.4049/jimmunol.170.12.6320
15. Armengol MP, Juan M, Lucas-Martin A, Fernandez-Figueras MT, Jaraquemada D, Gallart T, et al. Thyroid autoimmune disease: demonstration of thyroid antigen-specific B cells and recombination-activating gene expression in chemokine-containing active intrathyroidal germinal centers. Am J Pathol. (2001) 159:861–73. doi: 10.1016/S0002-9440(10)61762-2
16. Furtado GC, Marinkovic T, Martin AP, Garin A, Hoch B, Hubner W, et al. Lymphotoxin beta receptor signaling is required for inflammatory lymphangiogenesis in the thyroid. Proc Natl Acad Sci USA. (2007) 104:5026–31. doi: 10.1073/pnas.0606697104
17. Martin AP, Coronel EC, Sano G, Chen SC, Vassileva G, Canasto-Chibuque C, et al. A novel model for lymphocytic infiltration of the thyroid gland generated by transgenic expression of the CC chemokine CCL21. J Immunol. (2004) 173:4791–8. doi: 10.4049/jimmunol.173.8.4791
18. Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain (2011) 134:2755–71. doi: 10.1093/brain/awr182
19. Lovato L, Willis SN, Rodig SJ, Caron T, Almendinger SE, Howell OW, et al. Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain (2011) 134:534–41. doi: 10.1093/brain/awq350
20. Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain (2007) 130:1089–104. doi: 10.1093/brain/awm038
21. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. (2004) 14:164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x
22. Pikor NB, Astarita JL, Summers-Deluca L, Galicia G, Qu J, Ward LA, et al. Integration of Th17- and lymphotoxin-derived signals initiates meningeal-resident stromal cell remodeling to propagate neuroinflammation. Immunity (2015) 43:1160–73. doi: 10.1016/j.immuni.2015.11.010
23. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity (2011) 35:986–96. doi: 10.1016/j.immuni.2011.10.015
24. Lehmann-Horn K, Wang SZ, Sagan SA, Zamvil SS, von Budingen HC. B cell repertoire expansion occurs in meningeal ectopic lymphoid tissue. JCI Insight. (2016) 1:e87234. doi: 10.1172/jci.insight.87234
25. Dang AK, Tesfagiorgis Y, Jain RW, Craig HC, Kerfoot SM. Meningeal infiltration of the spinal cord by non-classically activated B cells is associated with Chronic disease course in a spontaneous B cell-dependent model of CNS Autoimmune disease. Front Immunol. (2015) 6:470. doi: 10.3389/fimmu.2015.00470
26. Kuerten S, Schickel A, Kerkloh C, Recks MS, Addicks K, Ruddle NH, et al. Tertiary lymphoid organ development coincides with determinant spreading of the myelin-specific T cell response. Acta Neuropathol. (2012) 124:861–73. doi: 10.1007/s00401-012-1023-3
27. Bettelli E, Baeten D, Jager A, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest. (2006) 116:2393–402. doi: 10.1172/JCI28334
28. Magliozzi R, Columba-Cabezas S, Serafini B, Aloisi F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle-like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. (2004) 148:11–23. doi: 10.1016/j.jneuroim.2003.10.056
29. Hill ME, Shiono H, Newsom-Davis J, Willcox N. The myasthenia gravis thymus: a rare source of human autoantibody-secreting plasma cells for testing potential therapeutics. J Neuroimmunol. (2008) 201-202:50–56. doi: 10.1016/j.jneuroim.2008.06.027
30. Sims GP, Shiono H, Willcox N, Stott DI. Somatic hypermutation and selection of B cells in thymic germinal centers responding to acetylcholine receptor in myasthenia gravis. J Immunol. (2001) 167:1935–44. doi: 10.4049/jimmunol.167.4.1935
31. Robinet M, Villeret B, Maillard S, Cron MA, Berrih-Aknin S, Le Panse R. Use of toll-like receptor agonists to induce ectopic lymphoid structures in myasthenia gravis mouse models. Front Immunol. (2017) 8:1029. doi: 10.3389/fimmu.2017.01029
32. Sharifi S, Murphy M, Loda M, Pinkus GS, Khettry U. Nodular lymphoid lesion of the liver: an immune-mediated disorder mimicking low-grade malignant lymphoma. Am J Surg Pathol. (1999) 23:302–8. doi: 10.1097/00000478-199903000-00009
33. Humby F, Bombardieri M, Manzo A, Kelly S, Blades MC, Kirkham B, et al. Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med. (2009) 6:e1. doi: 10.1371/journal.pmed.0060001
34. Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O'Fallon WM, et al. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. (2001) 167:1072–80. doi: 10.4049/jimmunol.167.2.1072
35. Croia C, Serafini B, Bombardieri M, Kelly S, Humby F, Severa M, et al. Epstein-Barr virus persistence and infection of autoreactive plasma cells in synovial lymphoid structures in rheumatoid arthritis. Ann Rheum Dis. (2013) 72:1559–68. doi: 10.1136/annrheumdis-2012-202352
36. Salomonsson S, Jonsson MV, Skarstein K, Brokstad KA, Hjelmstrom P, Wahren-Herlenius M, et al. Cellular basis of ectopic germinal center formation and autoantibody production in the target organ of patients with Sjogren's syndrome. Arthritis Rheum. (2003) 48:3187–201. doi: 10.1002/art.11311
37. Stott DI, Hiepe F, Hummel M, Steinhauser G, Berek C. Antigen-driven clonal proliferation of B cells within the target tissue of an autoimmune disease. The salivary glands of patients with Sjogren's syndrome J Clin Invest. (1998) 102:938–46. doi: 10.1172/JCI3234
38. Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD. Inducible bronchus-associated lymphoid tissue. (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest. (2006) 116:3183–94. doi: 10.1172/JCI28756
39. Barone F, Nayar S, Campos J, Cloake T, Withers DR, Toellner KM, et al. IL-22 regulates lymphoid chemokine production and assembly of tertiary lymphoid organs. Proc Natl Acad Sci USA. (2015) 112:11024–9. doi: 10.1073/pnas.1503315112
40. Bombardieri M, Barone F, Lucchesi D, Nayar S, van den Berg WB, Proctor G, et al. Inducible tertiary lymphoid structures, autoimmunity, and exocrine dysfunction in a novel model of salivary gland inflammation in C57BL/6 mice. J Immunol. (2012) 189:3767–76. doi: 10.4049/jimmunol.1201216
41. Chang A, Henderson SG, Brandt D, Liu N, Guttikonda R, Hsieh C, et al. In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J Immunol. (2011) 186:1849–60. doi: 10.4049/jimmunol.1001983
42. Dorraji SE, Hovd AK, Kanapathippillai P, Bakland G, Eilertsen GO, Figenschau SL, et al. Mesenchymal stem cells and T cells in the formation of Tertiary Lymphoid Structures in Lupus Nephritis. Sci Rep. (2018) 8:7861. doi: 10.1038/s41598-018-26265-z
43. Kang S, Fedoriw Y, Brenneman EK, Truong YK, Kikly K, Vilen BJ. BAFF induces tertiary lymphoid structures and positions t cells within the glomeruli during lupus nephritis. J Immunol. (2017) 198:2602–11. doi: 10.4049/jimmunol.1600281
44. Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4+ follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest. (2013) 123:2873–92. doi: 10.1172/JCI67428
45. Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie JJ, et al. Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res. (2011) 71:5678–87. doi: 10.1158/0008-5472.CAN-11-0431
46. Kotlan B, Simsa P, Teillaud JL, Fridman WH, Toth J, McKnight M, et al. Novel ganglioside antigen identified by B cells in human medullary breast carcinomas: the proof of principle concerning the tumor-infiltrating B lymphocytes. J Immunol. (2005) 175:2278–85. doi: 10.4049/jimmunol.175.4.2278
47. Nzula S, Going JJ, Stott DI. Antigen-driven clonal proliferation, somatic hypermutation, and selection of B lymphocytes infiltrating human ductal breast carcinomas. Cancer Res. (2003) 63:3275–80.
48. Coppola D, Nebozhyn M, Khalil F, Dai H, Yeatman T, Loboda A, et al. Unique ectopic lymph node-like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am J Pathol. (2011) 179:37–45. doi: 10.1016/j.ajpath.2011.03.007
49. Bergomas F, Grizzi F, Doni A, Pesce S, Laghi L, Allavena P, et al. Tertiary intratumor lymphoid tissue in colo-rectal cancer. Cancers. (Basel). (2011) 4:1–10. doi: 10.3390/cancers4010001
50. de Chaisemartin L, Goc J, Damotte D, Validire P, Magdeleinat P, Alifano M, et al. Characterization of chemokines and adhesion molecules associated with T cell presence in tertiary lymphoid structures in human lung cancer. Cancer Res. (2011) 71:6391–9. doi: 10.1158/0008-5472.CAN-11-0952
51. Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol. (2008) 26:4410–7. doi: 10.1200/JCO.2007.15.0284
52. Kroeger DR, Milne K, Nelson BH. Tumor-infiltrating plasma cells are associated with tertiary lymphoid Structures, cytolytic T-cell responses, and superior prognosis in ovarian cancer. Clin Cancer Res. (2016) 22:3005–15. doi: 10.1158/1078-0432.CCR-15-2762
53. Martinet L, Le Guellec S, Filleron T, Lamant L, Meyer N, Rochaix P, et al. High endothelial venules. (HEVs) in human melanoma lesions: major gateways for tumor-infiltrating lymphocytes. Oncoimmunology (2012) 1:829–39. doi: 10.4161/onci.20492
54. Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br J Cancer (2015) 112:1782–90. doi: 10.1038/bjc.2015.145
55. Garcia-Hernandez ML, Uribe-Uribe NO, Espinosa-Gonzalez R, Kast WM, Khader SA, Rangel-Moreno J. A unique cellular and molecular microenvironment is present in tertiary lymphoid organs of patients with spontaneous prostate cancer regression. Front Immunol. (2017) 8:563. doi: 10.3389/fimmu.2017.00563
56. Houtkamp MA, de Boer OJ, van der Loos CM, van der Wal AC, Becker AE. Adventitial infiltrates associated with advanced atherosclerotic plaques: structural organization suggests generation of local humoral immune responses. J Pathol. (2001) 193:263–9. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH774>3.0.CO;2-N
57. Ramshaw AL, Parums DV. Immunohistochemical characterization of inflammatory cells associated with advanced atherosclerosis. Histopathology (1990) 17:543–52. doi: 10.1111/j.1365-2559.1990.tb00794.x
58. Hu D, Mohanta SK, Yin C, Peng L, Ma Z, Srikakulapu P, et al. Artery tertiary lymphoid organs control aorta immunity and protect against atherosclerosis via vascular smooth muscle cell lymphotoxin beta receptors. Immunity (2015) 42:1100–15. doi: 10.1016/j.immuni.2015.05.015
59. Grabner R, Lotzer K, Dopping S, Hildner M, Radke D, Beer M, et al. Lymphotoxin beta receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE-/- mice. J Exp Med. (2009) 206:233–48. doi: 10.1084/jem.20080752
60. Seys LJ, Verhamme FM, Schinwald A, Hammad H, Cunoosamy DM, Bantsimba-Malanda C, et al. Role of B Cell-activating factor in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2015) 192:706–18. doi: 10.1164/rccm.201501-0103OC
61. Polverino F, Cosio BG, Pons J, Laucho-Contreras M, Tejera P, Iglesias A, et al. B cell-activating factor. An orchestrator of lymphoid follicles in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2015) 192:695–705. doi: 10.1164/rccm.201501-0107OC
62. Roos AB, Sanden C, Mori M, Bjermer L, Stampfli MR, Erjefalt JS. IL-17A is elevated in end-stage chronic obstructive pulmonary disease and contributes to cigarette smoke-induced lymphoid neogenesis. Am J Respir Crit Care Med. (2015) 191:1232–41. doi: 10.1164/rccm.201410-1861OC
63. Van Pottelberge G R, Bracke KR, Van den Broeck S, Reinartz SM, van Drunen CM, Wouters EF, et al. Plasmacytoid dendritic cells in pulmonary lymphoid follicles of patients with COPD. Eur Respir J. (2010) 36:781–91. doi: 10.1183/09031936.00140409
64. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. (2004) 350:2645–53. doi: 10.1056/NEJMoa032158
65. Bracke KR, Verhamme FM, Seys LJ, Bantsimba-Malanda C, Cunoosamy DM, Herbst R, et al. Role of CXCL13 in cigarette smoke-induced lymphoid follicle formation and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2013) 188:343–55. doi: 10.1164/rccm.201211-2055OC
66. Carlsen HS, Baekkevold ES, Johansen FE, Haraldsen G, Brandtzaeg P. B cell attracting chemokine 1 (CXCL13) and its receptor CXCR5 are expressed in normal and aberrant gut associated lymphoid tissue. Gut (2002) 51:364–71. doi: 10.1136/gut.51.3.364
67. Kaiserling E. Newly-formed lymph nodes in the submucosa in chronic inflammatory bowel disease. Lymphology (2001) 34:22–9.
68. Fujimura Y, Kamoi R, Iida M. Pathogenesis of aphthoid ulcers in Crohn's disease: correlative findings by magnifying colonoscopy, electron microscopy, and immunohistochemistry. Gut (1996) 38:724–32. doi: 10.1136/gut.38.5.724
69. Surawicz CM, Belic L. Rectal biopsy helps to distinguish acute self-limited colitis from idiopathic inflammatory bowel disease. Gastroenterology (1984) 86:104–13.
70. Olivier BJ, Cailotto C, van der Vliet J, Knippenberg M, Greuter MJ, Hilbers FW, et al. Vagal innervation is required for the formation of tertiary lymphoid tissue in colitis. Eur J Immunol. (2016) 46:2467–80. doi: 10.1002/eji.201646370
71. McNamee EN, Masterson JC, Jedlicka P, Collins CB, Williams IR, Rivera-Nieves J. Ectopic lymphoid tissue alters the chemokine gradient, increases lymphocyte retention and exacerbates murine ileitis. Gut. (2013) 62:53–62. doi: 10.1136/gutjnl-2011-301272
72. Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. J Exp Med. (2011) 208:125–34. doi: 10.1084/jem.20100052
73. Kawamura T, Kanai T, Dohi T, Uraushihara K, Totsuka T, Iiyama R, et al. Ectopic CD40 ligand expression on B cells triggers intestinal inflammation. J Immunol. (2004) 172:6388–97. doi: 10.4049/jimmunol.172.10.6388
74. Spahn TW, Herbst H, Rennert PD, Lugering N, Maaser C, Kraft M, et al. Induction of colitis in mice deficient of Peyer's patches and mesenteric lymph nodes is associated with increased disease severity and formation of colonic lymphoid patches. Am J Pathol. (2002) 161:2273–82. doi: 10.1016/S0002-9440(10)64503-8
75. Grant AJ, Goddard S, Ahmed-Choudhury J, Reynolds G, Jackson DG, Briskin M, et al. Hepatic expression of secondary lymphoid chemokine (CCL21) promotes the development of portal-associated lymphoid tissue in chronic inflammatory liver disease. Am J Pathol. (2002) 160:1445–55. doi: 10.1016/S0002-9440(10)62570-9
76. Ghosh S, Steere AC, Stollar BD, Huber BT. In situ diversification of the antibody repertoire in chronic Lyme arthritis synovium. J Immunol. (2005) 174:2860–9. doi: 10.4049/jimmunol.174.5.2860
77. Racanelli V, Sansonno D, Piccoli C, D'Amore FP, Tucci FA, Dammacco F. Molecular characterization of B cell clonal expansions in the liver of chronically hepatitis C virus-infected patients. J Immunol. (2001) 167:21–9. doi: 10.4049/jimmunol.167.1.21
78. Murakami J, Shimizu Y, Kashii Y, Kato T, Minemura M, Okada K, et al. Functional B-cell response in intrahepatic lymphoid follicles in chronic hepatitis C. Hepatology (1999) 30:143–50. doi: 10.1002/hep.510300107
79. Magalini AR, Facchetti F, Salvi L, Fontana L, Puoti M, Scarpa A. Clonality of B-cells in portal lymphoid infiltrates of HCV-infected livers. J Pathol. (1998) 185:86–90. doi: 10.1002/(SICI)1096-9896(199805)185:1<86::AID-PATH59>3.0.CO;2-R
80. Sansonno D, De Vita S, Iacobelli AR, Cornacchiulo V, Boiocchi M, Dammacco F. Clonal analysis of intrahepatic B cells from HCV-infected patients with and without mixed cryoglobulinemia. J Immunol. (1998) 160:3594–601.
81. Mazzucchelli L, Blaser A, Kappeler A, Scharli P, Laissue JA, Baggiolini M, et al. BCA-1 is highly expressed in Helicobacter pylori-induced mucosa-associated lymphoid tissue and gastric lymphoma. J Clin Invest. (1999) 104:R49–54. doi: 10.1172/JCI7830
82. Zaitoun AM. The prevalence of lymphoid follicles in Helicobacter pylori associated gastritis in patients with ulcers and non-ulcer dyspepsia. J Clin Pathol. (1995) 48:325–9. doi: 10.1136/jcp.48.4.325
83. Genta RM, Hamner HW. The significance of lymphoid follicles in the interpretation of gastric biopsy specimens. Arch Pathol Lab Med. (1994) 118:740–3.
84. Winter S, Loddenkemper C, Aebischer A, Rabel K, Hoffmann K, Meyer TF, et al. The chemokine receptor CXCR5 is pivotal for ectopic mucosa-associated lymphoid tissue neogenesis in chronic Helicobacter pylori-induced inflammation. J Mol Med. (Berl). (2010) 88:1169–80. doi: 10.1007/s00109-010-0658-6
85. Slight SR, Rangel-Moreno J, Gopal R, Lin Y, Fallert Junecko BA, Mehra S, et al. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J Clin Invest. (2013) 123:712–26. doi: 10.1172/JCI65728
86. Tsai MC, Chakravarty S, Zhu G, Xu J, Tanaka K, Koch C, et al. Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell Microbiol. (2006) 8:218–32. doi: 10.1111/j.1462-5822.2005.00612.x
87. Ulrichs T, Kosmiadi GA, Trusov V, Jorg S, Pradl L, Titukhina M, et al. Human tuberculous granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defence in the lung. J Pathol. (2004) 204:217–28. doi: 10.1002/path.1628
88. Maglione PJ, Xu J, Chan J. B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. J Immunol. (2007) 178:7222–34. doi: 10.4049/jimmunol.178.11.7222
89. Kerjaschki D, Regele HM, Moosberger I, Nagy-Bojarski K, Watschinger B, Soleiman A, et al. Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J Am Soc Nephrol. (2004) 15:603–12. doi: 10.1097/01.ASN.0000113316.52371.2E
90. Sato M, Hirayama S, Matsuda Y, Wagnetz D, Hwang DM, Guan Z, et al. Stromal activation and formation of lymphoid-like stroma in chronic lung allograft dysfunction. Transplantation (2011) 91:1398–405. doi: 10.1097/TP.0b013e31821b2f7a
91. Cheng J, Torkamani A, Grover RK, Jones TM, Ruiz DI, Schork NJ, et al. Ectopic B-cell clusters that infiltrate transplanted human kidneys are clonal. Proc Natl Acad Sci USA. (2011) 108:5560–5. doi: 10.1073/pnas.1101148108
92. Thaunat O, Patey N, Caligiuri G, Gautreau C, Mamani-Matsuda M, Mekki Y, et al. Chronic rejection triggers the development of an aggressive intragraft immune response through recapitulation of lymphoid organogenesis. J Immunol. (2010) 185:717–28. doi: 10.4049/jimmunol.0903589
93. Thaunat O, Patey N, Gautreau C, Lechaton S, Fremeaux-Bacchi V, Dieu-Nosjean MC, et al. B cell survival in intragraft tertiary lymphoid organs after rituximab therapy. Transplantation (2008) 85:1648–53. doi: 10.1097/TP.0b013e3181735723
94. Zarkhin V, Kambham N, Li L, Kwok S, Hsieh SC, Salvatierra O, et al. Characterization of intra-graft B cells during renal allograft rejection. Kidney Int. (2008) 74:664–73. doi: 10.1038/ki.2008.249
95. Li W, Bribriesco AC, Nava RG, Brescia AA, Ibricevic A, Spahn JH, et al. Lung transplant acceptance is facilitated by early events in the graft and is associated with lymphoid neogenesis. Mucosal Immunol. (2012) 5:544–54. doi: 10.1038/mi.2012.30
96. Brown K, Sacks SH, Wong W. Tertiary lymphoid organs in renal allografts can be associated with donor-specific tolerance rather than rejection. Eur J Immunol. (2011) 41:89–96. doi: 10.1002/eji.201040759
97. Barone F, Gardner DH, Nayar S, Steinthal N, Buckley CD, Luther SA. Stromal fibroblasts in tertiary lymphoid structures: a novel target in chronic inflammation. Front Immunol. (2016) 7:477. doi: 10.3389/fimmu.2016.00477
98. Buckley CD, Barone F, Nayar S, Benezech C, Caamano J. Stromal cells in chronic inflammation and tertiary lymphoid organ formation. Annu Rev Immunol. (2015) 33:715–45. doi: 10.1146/annurev-immunol-032713-120252
99. Barone F, Nayar S, Buckley CD. The role of non-hematopoietic stromal cells in the persistence of inflammation. Front Immunol. (2012) 3:416. doi: 10.3389/fimmu.2012.00416
100. Carragher DM, Rangel-Moreno J, Randall TD. Ectopic lymphoid tissues and local immunity. Semin Immunol. (2008) 20:26–42. doi: 10.1016/j.smim.2007.12.004
101. Jones GW, Jones SA. Ectopic lymphoid follicles: inducible centres for generating antigen-specific immune responses within tissues. Immunology (2016) 147:141–51. doi: 10.1111/imm.12554
102. Picker LJ, Butcher EC. Physiological and molecular mechanisms of lymphocyte homing. Ann Rev Immunol. (1992) 10:561–91. doi: 10.1146/annurev.iy.10.040192.003021
103. Golub R, Cumano A. Embryonic hematopoiesis. Blood Cells Mol Dis. (2013) 51:226–31. doi: 10.1016/j.bcmd.2013.08.004
104. Manzo A, Bugatti S, Caporali R, Prevo R, Jackson DG, Uguccioni M, et al. CCL21 expression pattern of human secondary lymphoid organ stroma is conserved in inflammatory lesions with lymphoid neogenesis. Am J Pathol. (2007) 171:1549–62. doi: 10.2353/ajpath.2007.061275
105. Nayar S, Campos J, Chung MM, Navarro-Nunez L, Chachlani M, Steinthal N, et al. bimodal expansion of the lymphatic vessels is regulated by the sequential expression of il-7 and lymphotoxin alpha1beta2 in newly formed tertiary lymphoid structures. J Immunol. (2016) 197:1957–67. doi: 10.4049/jimmunol.1500686
106. Dutertre CA, Clement M, Morvan M, Schakel K, Castier Y, Alsac JM, et al. Deciphering the stromal and hematopoietic cell network of the adventitia from non-aneurysmal and aneurysmal human aorta. PLoS ONE (2014) 9:e89983. doi: 10.1371/journal.pone.0089983
107. Cupovic J, Onder L, Gil-Cruz C, Weiler E, Caviezel-Firner S, Perez-Shibayama C, et al. Central Nervous System Stromal Cells Control Local CD8(+) T Cell Responses during Virus-Induced Neuroinflammation. Immunity (2016) 44:622–33. doi: 10.1016/j.immuni.2015.12.022
108. Barone F, Bombardieri M, Manzo A, Blades MC, Morgan PR, Challacombe SJ, et al. Association of CXCL13 and CCL21 expression with the progressive organization of lymphoid-like structures in Sjogren's syndrome. Arthritis Rheum. (2005) 52:1773–84. doi: 10.1002/art.21062
109. Manzo A, Paoletti S, Carulli M, Blades MC, Barone F, Yanni G, et al. Systematic microanatomical analysis of CXCL13 and CCL21 in situ production and progressive lymphoid organization in rheumatoid synovitis. Eur J Immunol. (2005) 35:1347–59. doi: 10.1002/eji.200425830
110. Park YE, Woo YJ, Park SH, Moon YM, Oh HJ, Kim JI, et al. IL-17 increases cadherin-11 expression in a model of autoimmune experimental arthritis and in rheumatoid arthritis. Immunol Lett. (2011) 140:97–103. doi: 10.1016/j.imlet.2011.07.003
111. Lotzer K, Dopping S, Connert S, Grabner R, Spanbroek R, Lemser B, et al. Mouse aorta smooth muscle cells differentiate into lymphoid tissue organizer-like cells on combined tumor necrosis factor receptor-1/lymphotoxin beta-receptor NF-kappaB signaling. Arterioscler Thromb Vasc Biol. (2010) 30:395–402. doi: 10.1161/ATVBAHA.109.191395
112. Luther SA, Bidgol A, Hargreaves DC, Schmidt A, Xu Y, Paniyadi J, et al. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J Immunol. (2002) 169:424–33. doi: 10.4049/jimmunol.169.1.424
113. Stranford S, Ruddle NH. Follicular dendritic cells, conduits, lymphatic vessels, and high endothelial venules in tertiary lymphoid organs: parallels with lymph node stroma. Front Immunol. (2012) 3:350. doi: 10.3389/fimmu.2012.00350
114. Ruddle NH. Lymphatic vessels and tertiary lymphoid organs. J Clin Invest. (2014) 124:953–9. doi: 10.1172/JCI71611
115. Janeway Jr CA, Bottomly K. Signals and signs for lymphocyte responses. Cell (1994) 76:275–85. doi: 10.1016/0092-8674(94)90335-2
116. Ansel KM, Ngo VN, Hyman PL, Luther SA, Forster R, Sedgwick JD, et al. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature (2000) 406:309–14. doi: 10.1038/35018581
117. Cyster JG, Ngo VN, Ekland EH, Gunn MD, Sedgwick JD, Ansel KM. Chemokines and B-cell homing to follicles. Curr Top Microbiol Immunol. (1999) 246, 87–92; discussion 93. doi: 10.1007/978-3-642-60162-0_11
118. Pereira JP, Kelly LM, Cyster JG. Finding the right niche: B-cell migration in the early phases of T-dependent antibody responses. Int Immunol. (2010) 22:413–9. doi: 10.1093/intimm/dxq047
119. Wolniak KL, Shinall SM, Waldschmidt TJ. The germinal center response. Crit Rev Immunol. (2004) 24:39–65. doi: 10.1615/CritRevImmunol.v24.i1.20
120. Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. (2004) 5:943. doi: 10.1038/ni1100
121. Bannard O, Horton RM, Allen CD, An J, Nagasawa T, Cyster JG. Germinal center centroblasts transition to a centrocyte phenotype according to a timed program and depend on the dark zone for effective selection. Immunity (2013) 39:912–24. doi: 10.1016/j.immuni.2013.08.038
122. Nagaoka H, Muramatsu M, Yamamura N, Kinoshita K, Honjo T. Activation-induced deaminase. (AID)-directed hypermutation in the immunoglobulin Sμ region: implication of AID involvement in a common step of class switch recombination and somatic hypermutation. J Exp Med. (2002) 195:529–34. doi: 10.1084/jem.20012144
123. MacLennan IC. Germinal centers. Ann Rev Immunol. (1994) 12:117–39. doi: 10.1146/annurev.iy.12.040194.001001
124. Haberman AM, Shlomchik MJ. Reassessing the function of immune-complex retention by follicular dendritic cells. Nat Rev Immunol. (2003) 3:757. doi: 10.1038/nri1178
125. Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, et al. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. J Exp Med. (2011) 208:2497–510. doi: 10.1084/jem.20111449
126. Allen CD, Cyster JG. Follicular dendritic cell networks of primary follicles and germinal centers: Phenotype and function. Semin Immunol. (2008) 20:14-25. doi: 10.1016/j.smim.2007.12.001
127. Zhang Y, Garcia-Ibanez L, Toellner KM. Regulation of germinal center B-cell differentiation. Immunol Rev. (2016) 270, 8–19. doi: 10.1111/imr.12396
128. Vinuesa CG, Cyster JG. How T cells earn the follicular rite of passage. Immunity (2011) 35:671–80. doi: 10.1016/j.immuni.2011.11.001
129. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular Helper T Cells. Annu Rev Immunol. (2016) 34:335–68. doi: 10.1146/annurev-immunol-041015-055605
130. Ise W, Fujii K, Shiroguchi K, Ito A, Kometani K, Takeda K, et al. T follicular helper cell-germinal center B cell interaction strength regulates entry into plasma cell or recycling germinal center cell fate. Immunity (2018) 48:702–15 e4. doi: 10.1016/j.immuni.2018.03.027
131. Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell. (1991) 67:1121–9. doi: 10.1016/0092-8674(91)90289-B
132. Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature (1991) 354:389–92. doi: 10.1038/354389a0
133. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase. (AID), a potential RNA editing enzyme. Cell (2000) 102:553–63. doi: 10.1016/S0092-8674(00)00078-7
134. Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. (1999) 274:18470–6. doi: 10.1074/jbc.274.26.18470
135. Bombardieri M, Barone F, Humby F, Kelly S, McGurk M, Morgan P, et al. Activation-induced cytidine deaminase expression in follicular dendritic cell networks and interfollicular large B cells supports functionality of ectopic lymphoid neogenesis in autoimmune sialoadenitis and MALT lymphoma in Sjogren's syndrome. J Immunol. (2007) 179:4929–38. doi: 10.4049/jimmunol.179.7.4929
136. Eisen HN, Siskind GW. Variations in Affinities of Antibodies during the Immune Response. Biochemistry. (1964) 3:996–1008. doi: 10.1021/bi00895a027
137. Allen CD, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity. (2007) 27:190–202. doi: 10.1016/j.immuni.2007.07.009
138. Moser K, Tokoyoda K, Radbruch A, MacLennan I, Manz RA. Stromal niches, plasma cell differentiation and survival. Curr Opin Immunol. (2006) 18:265–70. doi: 10.1016/j.coi.2006.03.004
139. Oropallo MA, Cerutti A. Germinal center reaction: antigen affinity and presentation explain it all. Trends Immunol. (2014) 35:287–9. doi: 10.1016/j.it.2014.06.001
140. Mesin L, Ersching J, Victora GD. Germinal center B cell dynamics. Immunity (2016) 45:471–82. doi: 10.1016/j.immuni.2016.09.001
141. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell (2010) 143:592–605. doi: 10.1016/j.cell.2010.10.032
142. Bannard O, Cyster JG. Germinal centers: programmed for affinity maturation and antibody diversification. Curr Opin Immunol. (2017) 45:21–30. doi: 10.1016/j.coi.2016.12.004
143. Cupedo T, Lund FE, Ngo VN, Randall TD, Jansen W, Greuter MJ, et al. Initiation of cellular organization in lymph nodes is regulated by non-B cell-derived signals and is not dependent on CXC chemokine ligand 13. J Immunol. (2004) 173:4889–96. doi: 10.4049/jimmunol.173.8.4889
144. Rodda LB, Bannard O, Ludewig B, Nagasawa T, Cyster JG. Phenotypic and morphological properties of germinal center dark zone Cxcl12-expressing reticular cells. J Immunol. (2015) 195:4781–91. doi: 10.4049/jimmunol.1501191
145. Roozendaal R, Mebius RE. Stromal cell-immune cell interactions. Annu Rev Immunol. (2011) 29:23–43. doi: 10.1146/annurev-immunol-031210-101357
146. Cyster JG, Ansel KM, Reif K, Ekland EH, Hyman PL, Tang HL, et al. Follicular stromal cells and lymphocyte homing to follicles. Immunol Rev. (2000) 176:181–93. doi: 10.1034/j.1600-065X.2000.00618.x
147. Kranich J, Krautler NJ. How Follicular Dendritic Cells Shape the B-Cell Antigenome. Front Immunol. (2016) 7:225. doi: 10.3389/fimmu.2016.00225
148. Denton AE, Linterman MA. Stromal networking: cellular connections in the germinal centre. Curr Opin Immunol. (2017) 45:103–11. doi: 10.1016/j.coi.2017.03.001
149. Yi T, Wang X, Kelly LM, An J, Xu Y, Sailer AW, et al. Oxysterol gradient generation by lymphoid stromal cells guides activated B cell movement during humoral responses. Immunity (2012) 37:535–48. doi: 10.1016/j.immuni.2012.06.015
150. Hughes CE, Benson RA, Bedaj M, Maffia P. Antigen-presenting cells and antigen presentation in tertiary lymphoid organs. Front Immunol. (2016) 7:481. doi: 10.3389/fimmu.2016.00481
151. Bombardieri M, Lewis M, Pitzalis C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat Rev Rheumatol. (2017) 13:141–54. doi: 10.1038/nrrheum.2016.217
152. Ruddle NH. High endothelial venules and lymphatic vessels in tertiary lymphoid organs: characteristics, functions, and regulation. Front Immunol. (2016) 7:491. doi: 10.3389/fimmu.2016.00491
153. Corsiero E, Nerviani A, Bombardieri M, Pitzalis C. Ectopic lymphoid structures: powerhouse of autoimmunity. Front Immunol. (2016) 7:430. doi: 10.3389/fimmu.2016.00430
154. Bombardieri M, Pitzalis C. Ectopic lymphoid neogenesis and lymphoid chemokines in Sjogren's syndrome: at the interplay between chronic inflammation, autoimmunity and lymphomagenesis. Curr Pharm Biotechnol. (2012) 13:1989–96. doi: 10.2174/138920112802273209
155. Weyand CM, Goronzy JJ. Ectopic germinal center formation in rheumatoid synovitis. Ann N Y Acad Sci. (2003) 987:140–9. doi: 10.1111/j.1749-6632.2003.tb06042.x
156. Kratz A, Campos-Neto A, Hanson MS, Ruddle NH. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. J Exp Med. (1996) 183:1461–72. doi: 10.1084/jem.183.4.1461
157. Amft N, Curnow SJ, Scheel-Toellner D, Devadas A, Oates J, Crocker J, et al. Ectopic expression of the B cell-attracting chemokine BCA-1. (CXCL13) on endothelial cells and within lymphoid follicles contributes to the establishment of germinal center-like structures in Sjogren's syndrome. Arthritis Rheum. (2001) 44:2633–41. doi: 10.1002/1529-0131(200111)44:11<2633::AID-ART443>3.0.CO;2-9
158. Croia C, Astorri E, Murray-Brown W, Willis A, Brokstad KA, Sutcliffe N, et al. Implication of Epstein-Barr virus infection in disease-specific autoreactive B cell activation in ectopic lymphoid structures of Sjogren's syndrome. Arthritis Rheumatol. (2014) 66:2545–57. doi: 10.1002/art.38726
159. Daridon C, Pers JO, Devauchelle V, Martins-Carvalho C, Hutin P, Pennec YL, et al. Identification of transitional type II B cells in the salivary glands of patients with Sjogren's syndrome. Arthritis Rheum. (2006) 54:2280–8. doi: 10.1002/art.21936
160. Hansen A, Lipsky PE, Dorner T. B cells in Sjogren's syndrome: indications for disturbed selection and differentiation in ectopic lymphoid tissue. Arthritis Res Ther. (2007) 9:218. doi: 10.1186/ar2210
161. Theander E, Vasaitis L, Baecklund E, Nordmark G, Warfvinge G, Liedholm R, et al. Lymphoid organisation in labial salivary gland biopsies is a possible predictor for the development of malignant lymphoma in primary Sjogren's syndrome. Ann Rheum Dis. (2011) 70:1363–8. doi: 10.1136/ard.2010.144782
162. Finke D, Schmutz S. Interleukin 7-induced lymphoid neogenesis in arthritis: recapitulation of a fetal developmental programme? Swiss Med Wkly. (2008) 138:500–5.
163. Martin L. Rheumatoid arthritis: symptoms, diagnosis, and management. Nurs Times. (2004) 100:40–4.
164. Tak P. P., Bresnihan B. (2000). The pathogenesis and prevention of joint damage in rheumatoid arthritis: advances from synovial biopsy and tissue analysis. Arthritis Rheum 43:2619–33. doi: 10.1002/1529-0131(200012)43:12<2619::AID-ANR1>3.0.CO;2-V
165. Targonska-Stepniak B. [Rheumatoid arthritis as a connective tissue disease]. Wiad Lek. (2018) 71:47–51.
166. Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid arthritis. Nat Rev Dis Primers. (2018) 4:18001. doi: 10.1038/nrdp.2018.1
167. Ropes MW, Bennett GA, Cobb S, Jacox R, Jessar RA. Proposed diagnostic criteria for rheumatoid arthritis. Ann Rheum Dis. (1957) 16:118–25. doi: 10.1136/ard.16.1.118
168. Munthe E, Natvig JB. Immunglobulin classes, subclasses and complexes of IgG rheumatoid factor in rheumatoid plasma cells. Clin Exp Immunol. (1972) 12:55–70.
169. Steere AC, Duray PH, Butcher EC. Spirochetal antigens and lymphoid cell surface markers in Lyme synovitis. Comparison with rheumatoid synovium and tonsillar lymphoid tissue. Arthritis Rheum. (1988) 31:487–95. doi: 10.1002/art.1780310405
170. Schroder AE, Greiner A, Seyfert C, Berek C. Differentiation of B cells in the nonlymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc Natl Acad Sci USA. (1996) 93:221–5. doi: 10.1073/pnas.93.1.221
171. Canete JD, Santiago B, Cantaert T, Sanmarti R, Palacin A, Celis R, et al. Ectopic lymphoid neogenesis in psoriatic arthritis. Ann Rheum Dis. (2007) 66:720–6. doi: 10.1136/ard.2006.062042
172. Voswinkel J, Weisgerber K, Pfreundschuh M, Gause A. B lymphocyte involvement in ankylosing spondylitis: the heavy chain variable segment gene repertoire of B lymphocytes from germinal center-like foci in the synovial membrane indicates antigen selection. Arthritis Res. (2001) 3:189–95. doi: 10.1186/ar297
173. Krenn V, Hensel F, Kim HJ, Souto Carneiro MM, Starostik P, Ristow G, et al. Molecular IgV(H) analysis demonstrates highly somatic mutated B cells in synovialitis of osteoarthritis: a degenerative disease is associated with a specific, not locally generated immune response. Lab Invest. (1999) 79:1377–84.
174. Campos J, Hillen MR, Barone F. Salivary Gland Pathology in Sjogren's Syndrome. Rheum Dis Clin North Am. (2016) 42:473–83. doi: 10.1016/j.rdc.2016.03.006
175. Chused TM, Hardin JA, Frank MM, Green I. Identification of cells infiltrating the minor salivary glands in patients with Sjogren's syndrome. J Immunol. (1974) 112:641–8.
176. Nair PN, Schroeder HE. Duct-associated lymphoid tissue. (DALT) of minor salivary glands and mucosal immunity. Immunology (1986) 57:171–80.
177. Aziz KE, McCluskey PJ, Wakefield D. Characterisation of follicular dendritic cells in labial salivary glands of patients with primary Sjogren syndrome: comparison with tonsillar lymphoid follicles. Ann Rheum Dis. (1997) 56:140–3. doi: 10.1136/ard.56.2.140
178. Xanthou G, Polihronis M, Tzioufas AG, Paikos S, Sideras P, Moutsopoulos HM. “Lymphoid” chemokine messenger RNA expression by epithelial cells in the chronic inflammatory lesion of the salivary glands of Sjogren's syndrome patients: possible participation in lymphoid structure formation. Arthritis Rheum (2001) 44:408–18. doi: 10.1002/1529-0131(200102)44:2<408::AID-ANR60>3.0.CO;2-0
179. Salomonsson S, Wahren-Herlenius M. Local production of Ro/SSA and La/SSB autoantibodies in the target organ coincides with high levels of circulating antibodies in sera of patients with Sjogren's syndrome. Scand J Rheumatol. (2003) 32:79–82. doi: 10.1080/03009740310000076
180. Barone F, Bombardieri M, Rosado MM, Morgan PR, Challacombe SJ, De Vita S, et al. CXCL13, CCL21, and CXCL12 expression in salivary glands of patients with Sjogren's syndrome and MALT lymphoma: association with reactive and malignant areas of lymphoid organization. J Immunol. (2008) 180:5130–40. doi: 10.4049/jimmunol.180.7.5130
181. Alunno A, Leone MC, Giacomelli R, Gerli R, Carubbi F. Lymphoma and lymphomagenesis in primary sjogren's syndrome. Front Med. (Lausanne). (2018) 5:102. doi: 10.3389/fmed.2018.00102
182. Haacke EA, van der Vegt B, Vissink AF, Spijkervet KL, Bootsma HF, Kroese GM. Germinal centres in diagnostic labial gland biopsies of patients with primary Sjogren's syndrome are not predictive for parotid MALT lymphoma development. Ann Rheum Dis. (2017) 76:1781–4. doi: 10.1136/annrheumdis-2017-211290
183. Pereira MI, Medeiros JA. Role of Helicobacter pylori in gastric mucosa-associated lymphoid tissue lymphomas. World J Gastroenterol. (2014) 20:684–98. doi: 10.3748/wjg.v20.i3.684
184. Halle S, Dujardin HC, Bakocevic N, Fleige H, Danzer H, Willenzon S, et al. Induced bronchus-associated lymphoid tissue serves as a general priming site for T cells and is maintained by dendritic cells. J Exp Med. (2009) 206:2593–601. doi: 10.1084/jem.20091472
185. Macritchie N, Grassia G, Sabir SR, Maddaluno M, Welsh P, Sattar N, et al. Plasmacytoid dendritic cells play a key role in promoting atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. (2012) 32:2569–79. doi: 10.1161/ATVBAHA.112.251314
186. Link A, Hardie DL, Favre S, Britschgi MR, Adams DH, Sixt M, et al. Association of T-zone reticular networks and conduits with ectopic lymphoid tissues in mice and humans. Am J Pathol. (2011) 178:1662–75. doi: 10.1016/j.ajpath.2010.12.039
187. Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol. (2006) 7:344–53. doi: 10.1038/ni1330
188. Junt T, Scandella E, Ludewig B. Form follows function: lymphoid tissue microarchitecture in antimicrobial immune defence. Nat Rev Immunol. (2008) 8:764–75. doi: 10.1038/nri2414
189. Neely HR, Flajnik MF. Emergence and evolution of secondary lymphoid organs. Annu Rev Cell Dev Biol. (2016) 32:693–711. doi: 10.1146/annurev-cellbio-111315-125306
190. Lukacs-Kornek V, Turley SJ. Self-antigen presentation by dendritic cells and lymphoid stroma and its implications for autoimmunity. Curr Opin Immunol. (2011) 23:138–45. doi: 10.1016/j.coi.2010.11.012
191. Malhotra D, Fletcher AL, Turley SJ. Stromal and hematopoietic cells in secondary lymphoid organs: partners in immunity. Immunol Rev. (2013) 251:160–76. doi: 10.1111/imr.12023
192. Turley SJ, Fletcher AL, Elpek KG. The stromal and haematopoietic antigen-presenting cells that reside in secondary lymphoid organs. Nat Rev Immunol. (2010) 10:813–25. doi: 10.1038/nri2886
193. van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. (2010) 10:664–74. doi: 10.1038/nri2832
194. Kocks JR, Davalos-Misslitz AC, Hintzen G, Ohl L, Forster R. Regulatory T cells interfere with the development of bronchus-associated lymphoid tissue. J Exp Med. (2007) 204:723–34. doi: 10.1084/jem.20061424
195. Alitalo K. The lymphatic vasculature in disease. Nat Med. (2011) 17:1371–80. doi: 10.1038/nm.2545
196. Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature (2005) 438:946–53. doi: 10.1038/nature04480
197. Flister MJ, Wilber A, Hall KL, Iwata C, Miyazono K, Nisato RE, et al. Inflammation induces lymphangiogenesis through up-regulation of VEGFR-3 mediated by NF-kappaB and Prox1. Blood (2010) 115:418–29. doi: 10.1182/blood-2008-12-196840
198. Mounzer RH, Svendsen OS, Baluk P, Bergman CM, Padera TP, Wiig H, et al. Lymphotoxin-alpha contributes to lymphangiogenesis. Blood (2010) 116:2173–82. doi: 10.1182/blood-2009-12-256065
199. Kunder CA, St John AL, Abraham SN. Mast cell modulation of the vascular and lymphatic endothelium. Blood (2011) 118:5383–93. doi: 10.1182/blood-2011-07-358432
200. Cursiefen C, Chen L, Borges LP, Jackson D, Cao J, Radziejewski C, et al. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J Clin Invest. (2004) 113:1040–50. doi: 10.1172/JCI20465
201. Hamrah P, Chen L, Zhang Q, Dana MR. Novel expression of vascular endothelial growth factor receptor. (VEGFR)-3 and VEGF-C on corneal dendritic cells. Am J Pathol. (2003) 163:57–68. doi: 10.1016/S0002-9440(10)63630-9
202. Thaunat O, Kerjaschki D, Nicoletti A. Is defective lymphatic drainage a trigger for lymphoid neogenesis? Trends Immunol. (2006) 27:441–5. doi: 10.1016/j.it.2006.08.003
203. Burman A, Haworth O, Hardie DL, Amft EN, Siewert C, Jackson DG, et al. A chemokine-dependent stromal induction mechanism for aberrant lymphocyte accumulation and compromised lymphatic return in rheumatoid arthritis. J Immunol. (2005) 174:1693–700. doi: 10.4049/jimmunol.174.3.1693
204. von der Weid PY, Rehal S, Ferraz JG. Role of the lymphatic system in the pathogenesis of Crohn's disease. Curr Opin Gastroenterol. (2011) 27:335–41. doi: 10.1097/MOG.0b013e3283476e8f
205. Kajiya K, Detmar M. An important role of lymphatic vessels in the control of UVB-induced edema formation and inflammation. J Invest Dermatol. (2006) 126:919–21. doi: 10.1038/sj.jid.5700126
206. Wilkinson LS, Edwards JC. Demonstration of lymphatics in human synovial tissue. Rheumatol Int. (1991) 11:151–5. doi: 10.1007/BF00332553
207. Bouta EM, Wood RW, Brown EB, Rahimi H, Ritchlin CT, Schwarz EM. In vivo quantification of lymph viscosity and pressure in lymphatic vessels and draining lymph nodes of arthritic joints in mice. J Physiol. (2014) 592:1213–23. doi: 10.1113/jphysiol.2013.266700
208. Polzer K, Baeten D, Soleiman A, Distler J, Gerlag DM, Tak PP, et al. Tumour necrosis factor blockade increases lymphangiogenesis in murine and human arthritic joints. Ann Rheum Dis. (2008) 67:1610–6. doi: 10.1136/ard.2007.083394
209. Li J, Ju Y, Bouta EM, Xing L, Wood RW, Kuzin I, et al. Efficacy of B cell depletion therapy for murine joint arthritis flare is associated with increased lymphatic flow. Arthritis Rheum. (2013) 65:130–8. doi: 10.1002/art.37709
210. Baeten D, Peene I, Union A, Meheus L, Sebbag M, Serre G, et al. Specific presence of intracellular citrullinated proteins in rheumatoid arthritis synovium: relevance to antifilaggrin autoantibodies. Arthritis Rheum. (2001) 44:2255–62.
211. De Rycke L, Nicholas AP, Cantaert T, Kruithof E, Echols JD, Vandekerckhove B, et al. Synovial intracellular citrullinated proteins colocalizing with peptidyl arginine deiminase as pathophysiologically relevant antigenic determinants of rheumatoid arthritis-specific humoral autoimmunity. Arthritis Rheum. (2005) 52:2323–30. doi: 10.1002/art.21220
212. Vossenaar ER, Smeets TJ, Kraan MC, Raats JM, van Venrooij WJ, Tak PP. The presence of citrullinated proteins is not specific for rheumatoid synovial tissue. Arthritis Rheum. (2004) 50:3485–94. doi: 10.1002/art.20584
213. Keene JD. Molecular structure of the La and Ro autoantigens and their use in autoimmune diagnostics. J Autoimmun. (1989) 2:329–34. doi: 10.1016/0896-8411(89)90160-1
214. Tzioufas AG, Hantoumi I, Polihronis M, Xanthou G, Moutsopoulos HM. Autoantibodies to La/SSB in patients with primary Sjogren's syndrome (pSS) are associated with upregulation of La/SSB mRNA in minor salivary gland biopsies (MSGs). J Autoimmun. (1999) 13:429–34. doi: 10.1006/jaut.1999.0333
215. de Wilde PC, Kater L, Bodeutsch C, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Aberrant expression pattern of the SS-B/La antigen in the labial salivary glands of patients with Sjogren's syndrome. Arthritis Rheum. (1996) 39:783–91. doi: 10.1002/art.1780390510
216. Barcellos KS, Nonogaki S, Enokihara MM, Teixeira MS, Andrade LE. Differential expression of Ro/SSA 60 kDa and La/SSB, but not Ro/SSA 52 kDa, mRNA and protein in minor salivary glands from patients with primary Sjogren's syndrome. J Rheumatol. (2007) 34:1283–92.
217. Tengner P, Halse AK, Haga HJ, Jonsson R, Wahren-Herlenius M. Detection of anti-Ro/SSA and anti-La/SSB autoantibody-producing cells in salivary glands from patients with Sjogren's syndrome. Arthritis Rheum. (1998) 41:2238–48. doi: 10.1002/1529-0131(199812)41:12<2238::AID-ART20>3.0.CO;2-V
218. Kang YM, Zhang X, Wagner UG, Yang H, Beckenbaugh RD, Kurtin PJ, et al. CD8 T cells are required for the formation of ectopic germinal centers in rheumatoid synovitis. J Exp Med. (2002) 195:1325–36. doi: 10.1084/jem.20011565
219. Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol. (2011) 12:639–46. doi: 10.1038/ni.2053
220. Chiang EY, Kolumam GA, Yu X, Francesco M, Ivelja S, Peng I, et al. Targeted depletion of lymphotoxin-alpha-expressing TH1 and TH17 cells inhibits autoimmune disease. Nat Med. (2009) 15:766–73. doi: 10.1038/nm.1984
221. Chiang EY, Kolumam G, McCutcheon KM, Young J, Lin Z, Balazs M, et al. In vivo depletion of lymphotoxin-alpha expressing lymphocytes inhibits xenogeneic graft-versus-host-disease. PLoS ONE (2012) 7:e33106. doi: 10.1371/journal.pone.0033106
222. Deteix C, Attuil-Audenis V, Duthey A, Patey N, McGregor B, Dubois V, et al. Intragraft Th17 infiltrate promotes lymphoid neogenesis and hastens clinical chronic rejection. J Immunol. (2010) 184:5344–51. doi: 10.4049/jimmunol.0902999
223. Lucchesi D, Pontarini E, Coleby R, Jones GW, Hill DG, Pitzalis C, et al. Interleukin-27 Regulates the magitude of the ectopic germinal centre response in a viral inducible model of sialadenitis. Ann Rheumatic Dis. (2018) 77. doi: 10.1136/annrheumdis-2018-EWRR2018.96
224. Jones GW, Bombardieri M, Greenhill CJ, McLeod L, Nerviani A, Rocher-Ros V, et al. Interleukin-27 inhibits ectopic lymphoid-like structure development in early inflammatory arthritis. J Exp Med. (2015) 212:1793–802. doi: 10.1084/jem.20132307
225. Lee Y, Chin RK, Christiansen P, Sun Y, Tumanov AV, Wang J, et al. Recruitment and activation of naive T cells in the islets by lymphotoxin beta receptor-dependent tertiary lymphoid structure. Immunity. (2006) 25:499–509. doi: 10.1016/j.immuni.2006.06.016
226. Clement M, Guedj K, Andreata F, Morvan M, Bey L, Khallou-Laschet J, et al. Control of the T follicular helper-germinal center B-cell axis by CD8+ regulatory T cells limits atherosclerosis and tertiary lymphoid organ development. Circulation (2015) 131:560–70. doi: 10.1161/CIRCULATIONAHA.114.010988
227. Schmitt N, Bentebibel SE, Ueno H. Phenotype and functions of memory Tfh cells in human blood. Trends Immunol. (2014) 35:436–42. doi: 10.1016/j.it.2014.06.002
228. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature (2017) 542:110–4. doi: 10.1038/nature20810
229. Jin L, Yu D, Li X, Yu N, Li X, Wang Y, et al. CD4+CXCR5+ follicular helper T cells in salivary gland promote B cells maturation in patients with primary Sjogren's syndrome. Int J Clin Exp Pathol. (2014) 7:1988–96.
230. Kang KY, Kim HO, Kwok SK, Ju JH, Park KS, Sun DI, et al. Impact of interleukin-21 in the pathogenesis of primary Sjogren's syndrome: increased serum levels of interleukin-21 and its expression in the labial salivary glands. Arthritis Res Ther. (2011) 13:R179. doi: 10.1186/ar3504
231. Voulgarelis M, Tzioufas AG. Pathogenetic mechanisms in the initiation and perpetuation of Sjogren's syndrome. Nat Rev Rheumatol. (2010) 6:529–37. doi: 10.1038/nrrheum.2010.118
232. Christodoulou MI, Kapsogeorgou EK, Moutsopoulos HM. Characteristics of the minor salivary gland infiltrates in Sjogren's syndrome. J Autoimmun. (2010) 34:400–7. doi: 10.1016/j.jaut.2009.10.004
233. Bohnhorst JO, Thoen JE, Natvig JB, Thompson KM. Significantly depressed percentage of CD27+ (memory) B cells among peripheral blood B cells in patients with primary Sjogren's syndrome. Scand J Immunol. (2001) 54:421–7. doi: 10.1046/j.1365-3083.2001.00989.x
234. Bohnhorst JO, Bjorgan MB, Thoen JE, Jonsson R, Natvig JB, Thompson KM. Abnormal B cell differentiation in primary Sjogren's syndrome results in a depressed percentage of circulating memory B cells and elevated levels of soluble CD27 that correlate with Serum IgG concentration. Clin Immunol. (2002) 103:79–88. doi: 10.1006/clim.2002.5199
235. Larsson A, Bredberg A, Henriksson G, Manthorpe R, Sallmyr A. Immunohistochemistry of the B-cell component in lower lip salivary glands of Sjogren's syndrome and healthy subjects. Scand J Immunol. (2005) 61:98–107. doi: 10.1111/j.0300-9475.2005.01540.x
236. Hansen A, Odendahl M, Reiter K, Jacobi AM, Feist E, Scholze J, et al. Diminished peripheral blood memory B cells and accumulation of memory B cells in the salivary glands of patients with Sjogren's syndrome. Arthritis Rheum. (2002) 46:2160–71. doi: 10.1002/art.10445
237. William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science (2002) 297:2066–70. doi: 10.1126/science.1073924
238. William J, Euler C, Shlomchik MJ. Short-lived plasmablasts dominate the early spontaneous rheumatoid factor response: differentiation pathways, hypermutating cell types, and affinity maturation outside the germinal center. J Immunol. (2005) 174:6879–87. doi: 10.4049/jimmunol.174.11.6879
239. Nocturne G, Mariette X. B cells in the pathogenesis of primary Sjogren syndrome. Nat Rev Rheumatol. (2018) 14:133–45. doi: 10.1038/nrrheum.2018.1
240. Nocturne G, Mariette X. Sjogren Syndrome-associated lymphomas: an update on pathogenesis and management. Br J Haematol. (2015) 168:317–27. doi: 10.1111/bjh.13192
241. Reynaud CA, Descatoire M, Dogan I, Huetz F, Weller S, Weill JC. IgM memory B cells: a mouse/human paradox. Cell Mol Life Sci. (2012) 69:1625–34. doi: 10.1007/s00018-012-0971-z
242. Steiniger B, Timphus EM, Barth PJ. The splenic marginal zone in humans and rodents: an enigmatic compartment and its inhabitants. Histochem Cell Biol. (2006) 126:641–8. doi: 10.1007/s00418-006-0210-5
243. Vossenkamper A, Spencer J. Transitional B cells: how well are the checkpoints for specificity understood? Arch Immunol Ther Exp. (Warsz). (2011) 59:379–84. doi: 10.1007/s00005-011-0135-0
244. Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol. (2005) 5:606–16. doi: 10.1038/nri1669
245. Vossenkamper A, Blair PA, Safinia N, Fraser LD, Das L, Sanders TJ, et al. A role for gut-associated lymphoid tissue in shaping the human B cell repertoire. J Exp Med. (2013) 210:1665–74. doi: 10.1084/jem.20122465
246. Mietzner B, Tsuiji M, Scheid J, Velinzon K, Tiller T, Abraham K, et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc Natl Acad Sci USA. (2008) 105:9727–32. doi: 10.1073/pnas.0803644105
247. Meffre E, Wardemann H. B-cell tolerance checkpoints in health and autoimmunity. Curr Opin Immunol. (2008) 20:632–8. doi: 10.1016/j.coi.2008.09.001
248. Bemark M, Holmqvist J, Abrahamsson J, Mellgren K. Translational Mini-Review Series on B cell subsets in disease. Reconstitution after haematopoietic stem cell transplantation - revelation of B cell developmental pathways and lineage phenotypes. Clin Exp Immunol. (2012) 167:15–25. doi: 10.1111/j.1365-2249.2011.04469.x
249. Tomura M, Yoshida N, Tanaka J, Karasawa S, Miwa Y, Miyawaki A, et al. Monitoring cellular movement in vivo with photoconvertible fluorescence protein “Kaede” transgenic mice. Proc Natl Acad Sci USA. (2008) 105:10871–6. doi: 10.1073/pnas.0802278105
250. Szyszko EA, Brokstad KA, Oijordsbakken G, Jonsson MV, Jonsson R, Skarstein K. Salivary glands of primary Sjogren's syndrome patients express factors vital for plasma cell survival. Arthritis Res Ther. (2011) 13:R2. doi: 10.1186/ar3220
251. Vos K, Thurlings RM, Wijbrandts CA, van Schaardenburg D, Gerlag DM, Tak PP. Early effects of rituximab on the synovial cell infiltrate in patients with rheumatoid arthritis. Arthritis Rheum. (2007) 56:772–8. doi: 10.1002/art.22400
252. Quartuccio L, Fabris M, Moretti M, Barone F, Bombardieri M, Rupolo M, et al. Resistance to rituximab therapy and local BAFF overexpression in Sjogren's syndrome-related myoepithelial sialadenitis and low-grade parotid B-cell lymphoma. Open Rheumatol J. (2008) 2:38–43. doi: 10.2174/1874312900802010038
253. Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. (2004) 20:785–98. doi: 10.1016/j.immuni.2004.05.010
254. Groom J, Kalled SL, Cutler AH, Olson C, Woodcock SA, Schneider P, et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjogren's syndrome. J Clin Invest. (2002) 109:59–68. doi: 10.1172/JCI0214121
255. Fletcher CA, Groom JR, Woehl B, Leung H, Mackay C, Mackay F. Development of autoimmune nephritis in genetically asplenic and splenectomized BAFF transgenic mice. J Autoimmun. (2011) 36:125–34. doi: 10.1016/j.jaut.2010.12.002
256. Hartgring SA, Bijlsma JW, Lafeber FP, van Roon JA. Interleukin-7 induced immunopathology in arthritis. Ann Rheum Dis. (2006) 65(Suppl. 3):iii69–74. doi: 10.1136/ard.2006.058479
257. Hartgring SA, Willis CR, Alcorn D, Nelson LJ, Bijlsma JW, Lafeber FP, et al. Blockade of the interleukin-7 receptor inhibits collagen-induced arthritis and is associated with reduction of T cell activity and proinflammatory mediators. Arthritis Rheum. (2010) 62:2716–25. doi: 10.1002/art.27578
258. Huang HY, Luther SA. Expression and function of interleukin-7 in secondary and tertiary lymphoid organs. Semin Immunol. (2012) 24:175–89. doi: 10.1016/j.smim.2012.02.008
259. Timmer TC, Baltus B, Vondenhoff M, Huizinga TW, Tak PP, Verweij CL, et al. Inflammation and ectopic lymphoid structures in rheumatoid arthritis synovial tissues dissected by genomics technology: identification of the interleukin-7 signaling pathway in tissues with lymphoid neogenesis. Arthritis Rheum. (2007) 56:2492–502. doi: 10.1002/art.22748
260. Harada S, Yamamura M, Okamoto H, Morita Y, Kawashima M, Aita T, et al. Production of interleukin-7 and interleukin-15 by fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheum. (1999) 42:1508–16. doi: 10.1002/1529-0131(199907)42:7<1508::AID-ANR26>3.0.CO;2-L
261. Van Roon JA, Verweij MC, Wijk MW, Jacobs KM, Bijlsma JW, Lafeber FP. Increased intraarticular interleukin-7 in rheumatoid arthritis patients stimulates cell contact–dependent activation of CD4+ T cells and macrophages. Arthritis Rheumatol. (2005) 52:1700–10. doi: 10.1002/art.21045
262. Hikida M, Nakayama Y, Yamashita Y, Kumazawa Y, Nishikawa SI, Ohmori H. Expression of recombination activating genes in germinal center B cells: involvement of interleukin 7 (IL-7) and the IL-7 receptor. J Exp Med. (1998) 188:365–72. doi: 10.1084/jem.188.2.365
263. Watanabe M, Ueno Y, Yajima T, Okamoto S, Hayashi T, Yamazaki M, et al. Interleukin 7 transgenic mice develop chronic colitis with decreased interleukin 7 protein accumulation in the colonic mucosa. J Exp Med. (1998) 187:389–402. doi: 10.1084/jem.187.3.389
264. Uehira M, Matsuda H, Hikita I, Sakata T, Fujiwara H, Nishimoto H. The development of dermatitis infiltrated by gamma delta T cells in IL-7 transgenic mice. Int Immunol. (1993) 5:1619–27. doi: 10.1093/intimm/5.12.1619
265. Meier D, Bornmann C, Chappaz S, Schmutz S, Otten LA, Ceredig R, et al. Ectopic lymphoid-organ development occurs through interleukin 7-mediated enhanced survival of lymphoid-tissue-inducer cells. Immunity. (2007) 26:643–54. doi: 10.1016/j.immuni.2007.04.009
266. Hillen MR, Radstake TR, Hack CE, van Roon JA. Thymic stromal lymphopoietin as a novel mediator amplifying immunopathology in rheumatic disease. Rheumatology (Oxford). (2015) 54:1771–9. doi: 10.1093/rheumatology/kev241
267. Zaheen A, Boulianne B, Parsa JY, Ramachandran S, Gommerman JL, Martin A. AID constrains germinal center size by rendering B cells susceptible to apoptosis. Blood (2009) 114:547–54. doi: 10.1182/blood-2009-03-211763
268. Cantaert T, Schickel JN, Bannock JM, Ng YS, Massad C, Delmotte FR, et al. Decreased somatic hypermutation induces an impaired peripheral B cell tolerance checkpoint. J Clin Invest. (2016) 126:4289–302. doi: 10.1172/JCI84645
269. Mellors RC, Heimer R, Corcos J, Korngold L. Cellular Origin of Rheumatoid Factor. J Exp Med. (1959) 110:875–86. doi: 10.1084/jem.110.6.875
270. Smiley JD, Sachs C, Ziff M. In vitro synthesis of immunoglobulin by rheumatoid synovial membrane. J Clin Invest. (1968) 47:624–32. doi: 10.1172/JCI105758
271. Randen I, Brown D, Thompson KM, Hughes-Jones N, Pascual V, Victor K, et al. Clonally related IgM rheumatoid factors undergo affinity maturation in the rheumatoid synovial tissue. J Immunol. (1992) 148:3296–301.
272. Thompson KM, Borretzen M, Randen I, Forre O, Natvig JB. V-gene repertoire and hypermutation of rheumatoid factors produced in rheumatoid synovial inflammation and immunized healthy donors. Ann N Y Acad Sci. (1995) 764:440–9. doi: 10.1111/j.1749-6632.1995.tb55861.x
273. Koopman WJ, Schrohenloher RE, Crago SS, Spalding DM, Mestecky J. IgA rheumatoid factor synthesis by dissociated synovial cells. Characterization and relationship to IgM rheumatoid factor synthesis. Arthritis Rheum. (1985) 28:1219–27. doi: 10.1002/art.1780281105
274. Hakoda M, Ishimoto T, Hayashimoto S, Inoue K, Taniguchi A, Kamatani N, et al. Selective infiltration of B cells committed to the production of monoreactive rheumatoid factor in synovial tissue of patients with rheumatoid arthritis. Clin Immunol Immunopathol. (1993) 69:16–22. doi: 10.1006/clin.1993.1144
275. Thompson KM, Randen I, Borretzen M, Forre O, Natvig JB. Variable region gene usage of human monoclonal rheumatoid factors derived from healthy donors following immunization. Eur J Immunol. (1994) 24:1771–8. doi: 10.1002/eji.1830240808
276. van Venrooij WJ, van Beers JJ, Pruijn GJ. Anti-CCP antibodies: the past, the present and the future. Nat Rev Rheumatol. (2011) 7:391–8. doi: 10.1038/nrrheum.2011.76
277. Amara K, Steen J, Murray F, Morbach H, Fernandez-Rodriguez BM, Joshua V, et al. Monoclonal IgG antibodies generated from joint-derived B cells of RA patients have a strong bias toward citrullinated autoantigen recognition. J Exp Med. (2013) 210:445–55. doi: 10.1084/jem.20121486
278. Masson-Bessiere C, Sebbag M, Durieux JJ, Nogueira L, Vincent C, Girbal-Neuhauser E, et al. In the rheumatoid pannus, anti-filaggrin autoantibodies are produced by local plasma cells and constitute a higher proportion of IgG than in synovial fluid and serum. Clin Exp Immunol. (2000) 119:544–52. doi: 10.1046/j.1365-2249.2000.01171.x
279. Caspi D, Anouk M, Golan I, Paran D, Kaufman I, Wigler I, et al. Synovial fluid levels of anti-cyclic citrullinated peptide antibodies and IgA rheumatoid factor in rheumatoid arthritis, psoriatic arthritis, and osteoarthritis. Arthritis Rheum. (2006) 55:53–6. doi: 10.1002/art.21691
280. Reparon-Schuijt C C, van Esch WJ, van Kooten C, Schellekens GA, de Jong BA, van Venrooij WJ, et al. Secretion of anti-citrulline-containing peptide antibody by B lymphocytes in rheumatoid arthritis. Arthritis Rheum. (2001) 44:41–7. doi: 10.1002/1529-0131(200101)44:1<41::AID-ANR6>3.0.CO;2-0
281. Snir O, Widhe M, Hermansson M, von Spee C, Lindberg J, Hensen S, et al. Antibodies to several citrullinated antigens are enriched in the joints of rheumatoid arthritis patients. Arthritis Rheum. (2010) 62:44–52. doi: 10.1002/art.25036
282. Spadaro A, Riccieri V, Scrivo R, Alessandri C, Valesini G. Anti-cyclic citrullinated peptide antibody determination in the synovial fluid of patients with rheumatoid arthritis: comment on the article by Caspi et al. Arthritis Rheum. (2006) 55:681–2; author reply 682. doi: 10.1002/art.22113
283. Amara K, Clay E, Yeo L, Ramskold D, Spengler J, Sippl N, et al. Immunoglobulin characteristics and RNAseq data of FcRL4+ B cells sorted from synovial fluid and tissue of patients with rheumatoid arthritis. Data Brief. (2017) 13:356–70. doi: 10.1016/j.dib.2017.06.009
284. Reparon-Schuijt C C, van Esch WJ, van Kooten C, Levarht EW, Breedveld FC, Verweij CL. Functional analysis of rheumatoid factor-producing B cells from the synovial fluid of rheumatoid arthritis patients. Arthritis Rheum (1998) 41:2211–20. doi: 10.1002/1529-0131(199812)41:12<2211::AID-ART17>3.0.CO;2-O
285. Van Esch WJ, Reparon-Schuijt CC, Hamstra HJ, Van Kooten C, Logtenberg T, Breedveld FC, et al. Human IgG Fc-binding phage antibodies constructed from synovial fluid CD38+ B cells of patients with rheumatoid arthritis show the imprints of an antigen-dependent process of somatic hypermutation and clonal selection. Clin Exp Immunol. (2003) 131:364–76. doi: 10.1046/j.1365-2249.2003.02068.x
286. Corsiero E, Bombardieri M, Carlotti E, Pratesi F, Robinson W, Migliorini P, et al. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann Rheum Dis. (2016) 75:1866–75. doi: 10.1136/annrheumdis-2015-208356
287. Gause A, Gundlach K, Carbon G, Daus H, Trumper L, Pfreundschuh M. Analysis of VH gene rearrangements from synovial B cells of patients with rheumatoid arthritis reveals infiltration of the synovial membrane by memory B cells. Rheumatol Int. (1997) 17:145–50. doi: 10.1007/s002960050026
288. Gause A, Gundlach K, Zdichavsky M, Jacobs G, Koch B, Hopf T, et al. The B lymphocyte in rheumatoid arthritis: analysis of rearranged V kappa genes from B cells infiltrating the synovial membrane. Eur J Immunol. (1995) 25:2775–82. doi: 10.1002/eji.1830251010
289. Voswinkel J, Trumper L, Carbon G, Hopf T, Pfreundschuh M, Gause A. Evidence for a selected humoral immune response encoded by VH4 family genes in the synovial membrane of a patient with rheumatoid arthritis. (RA). Clin Exp Immunol. (1996) 106:5–12. doi: 10.1046/j.1365-2249.1996.d01-806.x
290. Voswinkel J, Weisgerber K, Pfreundschuh M, Gause A. The B lymphocyte in rheumatoid arthritis: recirculation of B lymphocytes between different joints and blood. Autoimmunity (1999) 31:25–34. doi: 10.3109/08916939908993856
291. Kipps TJ, Tomhave E, Chen PP, Fox RI. Molecular characterization of a major autoantibody-associated cross-reactive idiotype in Sjogren's syndrome. J Immunol. (1989) 142:4261–8.
292. Gellrich S, Rutz S, Borkowski A, Golembowski S, Gromnica-Ihle E, Sterry W, et al. Analysis of V(H)-D-J(H) gene transcripts in B cells infiltrating the salivary glands and lymph node tissues of patients with Sjogren's syndrome. Arthritis Rheum. (1999) 42:240–7. doi: 10.1002/1529-0131(199902)42:2<240::AID-ANR5>3.0.CO;2-I
293. Maier-Moore JS, Koelsch KA, Smith K, Lessard CJ, Radfar L, Lewis D, et al. Antibody-secreting cell specificity in labial salivary glands reflects the clinical presentation and serology in patients with Sjogren's syndrome. Arthritis Rheumatol. (2014) 66:3445–56. doi: 10.1002/art.38872
294. Manzo A, Bombardieri M, Humby F, Pitzalis C. Secondary and ectopic lymphoid tissue responses in rheumatoid arthritis: from inflammation to autoimmunity and tissue damage/remodeling. Immunol Rev. (2010) 233:267–85. doi: 10.1111/j.0105-2896.2009.00861.x
295. Astorri E, Bombardieri M, Gabba S, Peakman M, Pozzilli P, Pitzalis C. Evolution of ectopic lymphoid neogenesis and in situ autoantibody production in autoimmune nonobese diabetic mice: cellular and molecular characterization of tertiary lymphoid structures in pancreatic islets. J Immunol. (2010) 185:3359–68. doi: 10.4049/jimmunol.1001836
296. Le Pottier L, Devauchelle V, Fautrel A, Daridon C, Saraux A, Youinou P, et al. Ectopic germinal centers are rare in Sjogren's syndrome salivary glands and do not exclude autoreactive B cells. J Immunol. (2009) 182:3540–7. doi: 10.4049/jimmunol.0803588
297. Spalding DM, Haber P, Schrohenloher RE, Koopman WJ. Production of immunoglobulin and rheumatoid factor by lymphoid cells in rheumatoid pericardium. Arthritis Rheum. (1985) 28:1071–4. doi: 10.1002/art.1780280917
298. Halla JT, Koopman WJ, Schrohenloher RE, Darby WL, Heck LW. Local synthesis of IgM and IgM rheumatoid factor in rheumatoid pleuritis. J Rheumatol. (1983) 10:204–9.
299. Halla JT, Koopman WJ, Fallahi S, Oh SJ, Gay RE, Schrohenloher RE. Rheumatoid myositis. Clinical and histologic features and possible pathogenesis. Arthritis Rheum. (1984) 27:737–43. doi: 10.1002/art.1780270703
300. Willis VC, Demoruelle MK, Derber LA, Chartier-Logan CJ, Parish MC, Pedraza IF, et al. Sputum autoantibodies in patients with established rheumatoid arthritis and subjects at risk of future clinically apparent disease. Arthritis Rheum. (2013) 65:2545–54. doi: 10.1002/art.38066
301. Talal N, Asofsky R, Lightbody P. Immunoglobulin synthesis by salivary gland lymphoid cells in Sjogren's syndrome. J Clin Invest. (1970) 49:49–54. doi: 10.1172/JCI106221
302. Anderson LG, Cummings NA, Asofsky R, Hylton MB, Tarpley TMJr, Tomasi TBJr, et al. Salivary gland immunoglobulin and rheumatoid factor synthesis in Sjogren's syndrome. Natural history and response to treatment Am J Med. (1972) 53:456–63. doi: 10.1016/0002-9343(72)90141-6
303. Halse A, Harley JB, Kroneld U, Jonsson R. Ro/SS-A-reactive B lymphocytes in salivary glands and peripheral blood of patients with Sjogren's syndrome. Clin Exp Immunol. (1999) 115:203–7. doi: 10.1046/j.1365-2249.1999.00778.x
304. Glauzy S, Sng J, Bannock JM, Gottenberg JE, Korganow AS, Cacoub P, et al. Defective Early B Cell Tolerance Checkpoints in Sjogren's Syndrome Patients. Arthritis Rheumatol. (2017) 69:2203–8. doi: 10.1002/art.40215
305. Markusse HM, Otten HG, Vroom TM, Smeets TJ, Fokkens N, Breedveld FC. Rheumatoid factor isotypes in serum and salivary fluid of patients with primary Sjogren's syndrome. Clin Immunol Immunopathol. (1993) 66:26–32. doi: 10.1006/clin.1993.1004
306. Halse AK, Marthinussen MC, Wahren-Herlenius M, Jonsson R. Isotype distribution of anti-Ro/SS-A and anti-La/SS-B antibodies in plasma and saliva of patients with Sjogren's syndrome. Scand J Rheumatol. (2000) 29:13–9. doi: 10.1080/030097400750001752
307. Maverakis E, Kim K, Shimoda M, Gershwin ME, Patel F, Wilken R, et al. Glycans in the immune system and The Altered Glycan Theory of Autoimmunity: a critical review. J Autoimmun. (2015) 57:1–13. doi: 10.1016/j.jaut.2014.12.002
308. Bruckner C, Lehmann C, Dudziak D, Nimmerjahn F. Sweet SIGNs: IgG glycosylation leads the way in IVIG-mediated resolution of inflammation. Int Immunol. (2017) 29:499–509. doi: 10.1093/intimm/dxx053
309. Pincetic A, Bournazos S, DiLillo DJ, Maamary J, Wang TT, Dahan R, et al. Type, I., and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol. (2014) 15:707–16. doi: 10.1038/ni.2939
310. van de Bovenkamp, F S, Hafkenscheid L, Rispens T, Rombouts Y. The Emerging Importance of IgG Fab Glycosylation in immunity. J Immunol. (2016) 196:1435–41. doi: 10.4049/jimmunol.1502136
311. Parekh RB, Dwek RA, Sutton BJ, Fernandes DL, Leung A, Stanworth D, et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature (1985) 316:452–7. doi: 10.1038/316452a0
312. Axford JS, Mackenzie L, Lydyard PM, Hay FC, Isenberg DA, Roitt IM. Reduced B-cell galactosyltransferase activity in rheumatoid arthritis. Lancet (1987) 2:1486–8. doi: 10.1016/S0140-6736(87)92621-3
313. Keusch J, Lydyard PM, Berger EG, Delves PJ. B lymphocyte galactosyltransferase protein levels in normal individuals and in patients with rheumatoid arthritis. Glycoconj J. (1998) 15:1093–7. doi: 10.1023/A:1006957711557
314. Furukawa K, Matsuta K, Takeuchi F, Kosuge E, Miyamoto T, Kobata A. Kinetic study of a galactosyltransferase in the B cells of patients with rheumatoid arthritis. Int Immunol. (1990) 2:105–12. doi: 10.1093/intimm/2.1.105
315. Axford JS. Decreased B-cell galactosyltransferase activity in rheumatoid arthritis. Br J Rheumatol. (1988) 27(Suppl. 2):170. doi: 10.1093/rheumatology/XXVII.suppl_2.170
316. Kratz EM, Borysewicz K, Katnik-Prastowska I. Terminal monosaccharide screening of synovial immunoglobulins G and A for the early detection of rheumatoid arthritis. Rheumatol Int. (2010) 30:1285–92. doi: 10.1007/s00296-009-1139-5
317. Tomana M, Schrohenloher R, Bennett P, Del Puente A, Koopman W. Occurrence of deficient galactosylation of serum IgG prior to the onset of rheumatoid arthritis. Rheumatol Int. (1994) 13:217–20. doi: 10.1007/BF00290198
318. Matsumoto A, Shikata K, Takeuchi F, Kojima N, Mizuochi T. Autoantibody activity of IgG rheumatoid factor increases with decreasing levels of galactosylation and sialylation. J Biochem. (2000) 128:621–8. doi: 10.1093/oxfordjournals.jbchem.a022794
319. Parekh RB, Roitt IM, Isenberg DA, Dwek RA, Ansell BM, Rademacher TW. Galactosylation of IgG associated oligosaccharides: reduction in patients with adult and juvenile onset rheumatoid arthritis and relation to disease activity. Lancet (1988) 1:966–9. doi: 10.1016/S0140-6736(88)91781-3
320. Pfeifle R, Rothe T, Ipseiz N, Scherer HU, Culemann S, Harre U, et al. Regulation of autoantibody activity by the IL-23-TH17 axis determines the onset of autoimmune disease. Nat Immunol. (2017) 18:104–13. doi: 10.1038/ni.3579
321. Gindzienska-Sieskiewicz E, Radziejewska I, Domyslawska I, Klimiuk PA, Sulik A, Rojewska J, et al. Changes of glycosylation of IgG in rheumatoid arthritis patients treated with methotrexate. Adv Med Sci. (2016) 61:193–7. doi: 10.1016/j.advms.2015.12.009
322. Rombouts Y, Ewing E, van de Stadt LA, Selman MH, Trouw LA, Deelder AM, et al. Anti-citrullinated protein antibodies acquire a pro-inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann Rheum Dis. (2015) 74:234–41. doi: 10.1136/annrheumdis-2013-203565
323. Tsuchiya N, Endo T, Matsuta K, Yoshinoya S, Takeuchi F, Nagano Y, et al. Detection of glycosylation abnormality in rheumatoid IgG using N-acetylglucosamine-specific Psathyrella velutina lectin. J Immunol. (1993) 151:1137–46.
324. Scherer HU, van der Woude D, Ioan-Facsinay A, el Bannoudi H, Trouw LA, Wang J, et al. Glycan profiling of anti-citrullinated protein antibodies isolated from human serum and synovial fluid. Arthritis Rheum. (2010) 62:1620–9. doi: 10.1002/art.27414
325. Youinou P, Pennec YL, Casburn-Budd R, Dueymes M, Letoux G, Lamour A. Galactose terminating oligosaccharides of IgG in patients with primary Sjogren's syndrome. J Autoimmun. (1992) 5:393–400. doi: 10.1016/0896-8411(92)90151-F
326. Kuroda Y, Nakata M, Makino A, Matsumoto A, Ohashi K, Itahashi K, et al. Structural studies on IgG oligosaccharides of patients with primary Sjogren's syndrome. Glycoconj J. (2002) 19:23–31. doi: 10.1023/A:1022528829799
327. Kempers AC, Hafkenscheid L, Dorjee AL, Moutousidou E, van de Bovenkamp FS, Rispens T, et al. The extensive glycosylation of the ACPA variable domain observed for ACPA-IgG is absent from ACPA-IgM. Ann Rheum Dis. (2017) 77:1087–8. doi: 10.1136/annrheumdis-2017-211533
328. Hamza N, Hershberg U, Kallenberg CG, Vissink A, Spijkervet FK, Bootsma H, et al. Ig gene analysis reveals altered selective pressures on Ig-producing cells in parotid glands of primary Sjogren's syndrome patients. J Immunol. (2015) 194:514–21. doi: 10.4049/jimmunol.1302644
329. Lloyd KA, Steen J, Titcombe PJ, Mueller DL, Klareskog L, Malmström V, et al. 08.19 Variable domain n-linked glycosylation is a key feature of monoclonal acpa-igg. Ann Rheum Dis. (2017) 76:A82–3. doi: 10.1136/annrheumdis-2016-211055.19
330. Rombouts Y, Willemze A, van Beers JJ, Shi J, Kerkman PF, van Toorn L, et al. Extensive glycosylation of ACPA-IgG variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann Rheum Dis. (2016) 75:578–85. doi: 10.1136/annrheumdis-2014-206598
331. Leader KA, Lastra GC, Kirwan JR, Elson CJ. Agalactosyl IgG in aggregates from the rheumatoid joint. Br J Rheumatol. (1996) 35:335–41. doi: 10.1093/rheumatology/35.4.335
332. Falkenburg WJJ, Kempers AC, Dekkers G, Ooijevaar-de Heer P, Bentlage AE, Vidarsson G, et al. Rheumatoid factors do not preferentially bind to ACPA-IgG or IgG with altered galactosylation. Rheumatology. (Oxford) (2017) 56:2025–30. doi: 10.1093/rheumatology/kex284
333. al-Balaghi S, Abedi-Valugerdi M, Moller E. Binding specificities of a polyreactive and a monoreactive human monoclonal IgG rheumatoid factor: role of oligosaccharides. Scand J Immunol. (1996) 44:470–7. doi: 10.1046/j.1365-3083.1996.d01-338.x
334. Soltys AJ, Hay FC, Bond A, Axford JS, Jones MG, Randen I, et al. The binding of synovial tissue-derived human monoclonal immunoglobulin M rheumatoid factor to immunoglobulin G preparations of differing galactose content. Scand J Immunol. (1994) 40:135–43. doi: 10.1111/j.1365-3083.1994.tb03442.x
335. Harre U, Lang SC, Pfeifle R, Rombouts Y, Fruhbeisser S, Amara K, et al. Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nat Commun. (2015) 6:6651. doi: 10.1038/ncomms7651
336. Ohmi Y, Ise W, Harazono A, Takakura D, Fukuyama H, Baba Y, et al. Sialylation converts arthritogenic IgG into inhibitors of collagen-induced arthritis. Nat Commun. (2016) 7:11205. doi: 10.1038/ncomms11205
337. Gunn B, Schneider J, Shansab M, Bastian AR, Fahrbach K, Smith A, Mahan A, et al. Enhanced binding of antibodies generated during chronic HIV infection to mucus component MUC16. Mucosal Immunol. (2016) 9:1549–58. doi: 10.1038/mi.2016.8
338. Azuma K, Shinzaki S, Asazawa H, Kuroki E, Kawamoto S, Kamada Y, et al. Twin studies on the effect of genetic factors on serum agalactosyl immunoglobulin G levels. Biomed Rep. (2014) 2:213–6. doi: 10.3892/br.2014.216
339. Lauc G, Huffman JE, Pucic M, Zgaga L, Adamczyk B, Muzinic A, et al. Loci associated with N-glycosylation of human immunoglobulin G show pleiotropy with autoimmune diseases and haematological cancers. PLoS Genet. (2013) 9:e1003225. doi: 10.1371/journal.pgen.1003225
340. Wang J, Balog CI, Stavenhagen K, Koeleman CA, Scherer HU, Selman MH, et al. Fc-glycosylation of IgG1 is modulated by B-cell stimuli. Mol Cell Proteomics. (2011) 10:M110.004655. doi: 10.1074/mcp.M110.004655
341. Chintalacharuvu SR, Emancipator SN. The glycosylation of IgA produced by murine B cells is altered by Th2 cytokines. J Immunol. (1997) 159:2327–33.
342. Chintalacharuvu SR, Emancipator SN. Differential glycosylation of two glycoproteins synthesized by murine B cells in response to IL-4 plus IL-5. Cytokine (2000) 12:1182–8. doi: 10.1006/cyto.2000.0699
343. Hess C, Winkler A, Lorenz AK, Holecska V, Blanchard V, Eiglmeier S, et al. T cell-independent B cell activation induces immunosuppressive sialylated IgG antibodies. J Clin Invest. (2013) 123:3788–96. doi: 10.1172/JCI65938
344. Oefner CM, Winkler A, Hess C, Lorenz AK, Holecska V, Huxdorf M, et al. Tolerance induction with T cell-dependent protein antigens induces regulatory sialylated IgGs. J Allergy Clin Immunol. (2012) 129:1647–55 e13. doi: 10.1016/j.jaci.2012.02.037
345. Kao D, Lux A, Schaffert A, Lang R, Altmann F, Nimmerjahn F. IgG subclass and vaccination stimulus determine changes in antigen specific antibody glycosylation in mice. Eur J Immunol. (2017) 47:2070–9. doi: 10.1002/eji.201747208
346. Pagan JD, Kitaoka M, Anthony RM. Engineered sialylation of pathogenic antibodies in vivo attenuates Autoimmune disease. Cell (2018) 172, 564–577 e13. doi: 10.1016/j.cell.2017.11.041
347. Griffiths HR, Lunec J. The effects of oxygen free radicals on the carbohydrate moiety of IgG. FEBS Lett. (1989) 245:95–9. doi: 10.1016/0014-5793(89)80199-1
348. Wang JR, Gao WN, Grimm R, Jiang S, Liang Y, Ye H, et al. A method to identify trace sulfated IgG N-glycans as biomarkers for rheumatoid arthritis. Nat Commun. (2017) 8:631. doi: 10.1038/s41467-017-00662-w
349. Trbojevic-Akmacic I, Vilaj M, Lauc G. High-throughput analysis of immunoglobulin G glycosylation. Expert Rev Proteomics. (2016) 13:523–34. doi: 10.1080/14789450.2016.1174584
350. Hafkenscheid L, Bondt A, Scherer HU, Huizinga TW, Wuhrer M, Toes RE, et al. Structural analysis of variable domain glycosylation of anti-citrullinated protein antibodies in rheumatoid arthritis reveals the presence of highly sialylated glycans. Mol Cell Proteomics. (2017) 16:278–87. doi: 10.1074/mcp.M116.062919
351. Royer B, Cazals-Hatem D, Sibilia J, Agbalika F, Cayuela JM, Soussi T, et al. Lymphomas in patients with Sjogren's syndrome are marginal zone B-cell neoplasms, arise in diverse extranodal and nodal sites, and are not associated with viruses. Blood (1997) 90:766–75.
352. Lasota J, Miettinen MM. Coexistence of different B-cell clones in consecutive lesions of low-grade MALT lymphoma of the salivary gland in Sjogren's disease. Mod Pathol. (1997) 10:872–8.
353. Bahler DW, Miklos JA, Swerdlow SH. Ongoing Ig gene hypermutation in salivary gland mucosa-associated lymphoid tissue-type lymphomas. Blood (1997) 89:3335–44.
354. Bende RJ, Aarts WM, Riedl RG, de Jong D, Pals ST, van Noesel CJ. Among B cell non-Hodgkin's lymphomas, MALT lymphomas express a unique antibody repertoire with frequent rheumatoid factor reactivity. J Exp Med. (2005) 201:1229–41. doi: 10.1084/jem.20050068
355. De Re V, De Vita S, Gasparotto D, Marzotto A, Carbone A, Ferraccioli G, et al. Salivary gland B cell lymphoproliferative disorders in Sjogren's syndrome present a restricted use of antigen receptor gene segments similar to those used by hepatitis C virus-associated non-Hodgkins's lymphomas. Eur J Immunol. (2002) 32:903–10. doi: 10.1002/1521-4141(200203)32:3<903::AID-IMMU903>3.0.CO;2-D
356. Martin T, Weber JC, Levallois H, Labouret N, Soley A, Koenig S, et al. Salivary gland lymphomas in patients with Sjogren's syndrome may frequently develop from rheumatoid factor B cells. Arthritis Rheum. (2000) 43:908–16. doi: 10.1002/1529-0131(200004)43:4<908::AID-ANR24>3.0.CO;2-K
357. Bende RJ, Slot LM, Hoogeboom R, Wormhoudt TA, Adeoye AO, Guikema JE, et al. Stereotypic rheumatoid factors that are frequently expressed in mucosa-associated lymphoid tissue-type lymphomas are rare in the labial salivary glands of patients with Sjogren's syndrome. Arthritis Rheumatol. (2015) 67:1074–83. doi: 10.1002/art.39002
358. Risselada AP, Kruize AA, Bijlsma JW. Clinical features distinguishing lymphoma development in primary Sjogren's Syndrome–a retrospective cohort study. Semin Arthritis Rheum. (2013) 43:171–7. doi: 10.1016/j.semarthrit.2013.03.001
359. Voulgarelis M, Dafni UG, Isenberg DA, Moutsopoulos HM. Malignant lymphoma in primary Sjogren's syndrome: a multicenter, retrospective, clinical study by the European Concerted Action on Sjogren's Syndrome. Arthritis Rheum. (1999) 42:1765–72. doi: 10.1002/1529-0131(19990842:8<1765::AID-ANR28>3.0.CO;2-V
360. Zhu D, McCarthy H, Ottensmeier CH, Johnson P, Hamblin TJ, Stevenson FK. Acquisition of potential N-glycosylation sites in the immunoglobulin variable region by somatic mutation is a distinctive feature of follicular lymphoma. Blood (2002) 99:2562–8. doi: 10.1182/blood.V99.7.2562
361. Zhu D, Ottensmeier CH, Du MQ, McCarthy H, Stevenson FK. Incidence of potential glycosylation sites in immunoglobulin variable regions distinguishes between subsets of Burkitt's lymphoma and mucosa-associated lymphoid tissue lymphoma. Br J Haematol. (2003) 120:217–22. doi: 10.1046/j.1365-2141.2003.04064.x
Keywords: tertiary lymphoid structures (TLS), autoantibodies, germinal center response, glycosylation, B-cells
Citation: Pipi E, Nayar S, Gardner DH, Colafrancesco S, Smith C and Barone F (2018) Tertiary Lymphoid Structures: Autoimmunity Goes Local. Front. Immunol. 9:1952. doi: 10.3389/fimmu.2018.01952
Received: 11 February 2018; Accepted: 07 August 2018;
Published: 12 September 2018.
Edited by:
Ralf J. Ludwig, Universität zu Lübeck, GermanyReviewed by:
Andreas Habenicht, Ludwig-Maximilians-Universität München, GermanyCopyright © 2018 Pipi, Nayar, Gardner, Colafrancesco, Smith and Barone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Barone, Zi5iYXJvbmVAYmhhbS5hYy51aw==
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.