
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 28 August 2018
Sec. Vaccines and Molecular Therapeutics
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01870
 Nadja Schubert1†
Nadja Schubert1† Katharina Lisenko1†
Katharina Lisenko1† Christian Auerbach2Anke Weitzmann1
Christian Auerbach2Anke Weitzmann1 Shanawaz Mohammed Ghouse1Lina Muhandes1Christa Haase1Tobias Häring1Livia Schulze1
Shanawaz Mohammed Ghouse1Lina Muhandes1Christa Haase1Tobias Häring1Livia Schulze1 David Voehringer3Florian Gunzer2
David Voehringer3Florian Gunzer2 Werner Müller4Thorsten B. Feyerabend5Hans-Reimer Rodewald5
Werner Müller4Thorsten B. Feyerabend5Hans-Reimer Rodewald5 Anne Dudeck1,6Axel Roers1*
Anne Dudeck1,6Axel Roers1*
Innate inflammatory responses are crucial for induction and regulation of T cell and antibody responses. Mast cell (MC)-deficient Kit mutant mice showed impaired adaptive immunity, suggesting that MCs provide essential adjuvant activities, and pharmacological MC activation was proposed as a new adjuvant principle. However, the Kit mutations result in complex alterations of the immune system in addition to MC deficiency. We revisited the role of MCs in vaccination responses using Mcpt5-Cre R26DTA/DTA and Cpa3Cre/+ mice that lack connective tissue MCs or all MCs, respectively, but feature an otherwise normal immune system. These animals showed no impairment of T and B cell responses to intradermal vaccination with protein antigen plus complete Freund’s adjuvant. Moreover, we demonstrate that the adjuvant effects of the MC secretagogue c48/80 in intradermal or mucosal immunization are independent of the presence of MCs. We hence find no evidence for a regulation by MCs of adaptive immune responses to protein antigens. The finding that immunological MC functions differ from those suggested by experiments in Kit mutants, emphasizes the importance of rigorous tests in Kit-independent MC-deficiency models.
Understanding the interplay of cellular and molecular factors involved in induction and regulation of adaptive immune responses is crucial to promote diverse areas of medicine including control of infectious disease, vaccine development, and immunotherapy of cancer. Dendritic cells (DCs), the most important antigen-presenting cells, represent the central switch controlling T cell, and thereby also B cell responses. DCs reside in peripheral tissues where they sample their environment. They sense microbial infection by means of pattern-recognition receptors (PRRs) which detect microbial structures [pathogen-associated molecular patterns (PAMPs)] as well as molecular changes associated with cell stress and cell death caused by the infection as a measure of how severe a threat an infection poses [danger-associated molecular patterns (DAMPs)] (1). DCs integrate PRR signals and, depending on the outcome of this process, migrate to draining LNs, upregulate their antigen-presentation machinery and increase expression of co-stimulatory molecules and cytokines, thereby providing essential signals for T cell activation and differentiation in the LN. The intensity and quality of PRR and cytokine signals the DC received in the tissue determines intensity and quality of the T cell response it drives in the LN (1).
Dendritic cells also respond to cytokines and other factors that tissue cells secrete upon detection of PAMPs or DAMPs. Mast cells (MCs) are tissue-resident hematopoietic cells that, like DCs, can express a wide array of PRRs and potentially release important immunostimulatory mediators. MCs are located in most tissues but their numbers are highest at inner and outer body surfaces suggesting a role in defense against environmental challenges. MCs were reported to be critical promoters of adaptive immunity that provide adjuvant effects through rapid provision of histamin and TNF, thereby boosting DC activation, DC migration to draining LNs, and lymphocyte homing to LNs (2–11). Moreover, MCs themselves were suggested to migrate to tissue-draining LNs and contribute to the initiation of adaptive immune responses by directly serving as antigen-presenting cells (12–18). Pharmacological MC activation was proposed as a novel adjuvant principle for vaccination (19). In keeping with these studies, important roles in diverse physiological and pathogenic immune responses have been attributed to MCs (20–25). However, these studies, which collectively built a view of the MC as an important regulator/amplifier of local innate responses that adjuvants adaptive immunity, were largely based on experiments in mutant mouse strains that are MC-deficient because of hypomorphic expression of the receptor tyrosin kinase Kit, the receptor for the growth factor stem cell factor. Since compromised Kit expression affects multiple hematopoietic and other lineages (26–29), phenotypes of Kit mutant strains are not necessarily the consequence of the absence of MCs. Reconstitution of Kit mutants with MCs differentiated from bone marrow cells in vitro (BMMCs) has been widely used as an argument that a phenotype was caused by MC deficiency (30). However, the many observed inconsistencies in results obtained in Kit mutants and BMMC reconstitutions therein, and Kit-independent MC-deficient mutants (see below) show that Kit mutants replete with BMMCs have been an unreliable experimental system. While Kit mutant strains were a common tool for the investigation of MC functions over the past decades, novel mouse models with intact Kit expression were recently developed, in which MC deficiency was achieved by different principles (31–34). Cpa3Cre/+ mice (32) express high levels of Cre recombinase that eliminate MCs and a fraction of basophils through Cre-mediated genotoxicity and a p53-dependent damage response (32). These animals are completely devoid of MCs and feature a reduction of basophil numbers to 40%. In Mcpt5-Cre R26DTA/DTA mice (31, 35), expression of Cre recombinase is controlled by the mast cell protease (Mcpt) 5 promoter and thereby restricted to connective tissue mast cells (CTMCs). Cre recombinase deletes a loxP-flanked stop cassette of the R26DTA knock in allele (36), which results in subsequent expression of diphtheria toxin A (DTA) selectively in CTMCs, which thereby kill themselves. As a consequence, Mcpt5-Cre R26DTA/DTA mice constitutively lack CTMCs, while mucosal MCs are unaffected. In both strains, IgE-mediated systemic anaphylaxis is abrogated, consistent with lack of MCs (32, 35). However, when other MC in vivo functions identified earlier in the Kit mutant models were revisited in the new, Kit-independent strains, phenotypes of the Kit mutants were not reproducible in many instances (31, 32, 34, 35, 37–46). These findings suggested that reduced Kit expression in the hematopoietic system rather than MC deficiency was responsible for some of the immunological phenotypes of the Kit mutant strains (40), and called for a systematic reproduction of key experiments in the new, Kit-independent MC-deficiency models. The aim of this study was to revisit the role of MCs in vaccination responses.
To study the role of MCs in adaptive immune responses, we used the Mcpt5-Cre R26DTA/DTA system (31, 35) to generate mice with selective deficiency for CTMCs. For maximum efficiency of MC depletion, we bred the ROSA26-DTA knock in allele (R26DTA) to homozygosity. Mcpt5-Cre R26DTA/DTA mice featured virtually complete absence of CTMCs in peritoneal cavity and skin without alteration of other hematopoietic cell subsets in peritoneum, skin, spleen, bone marrow, or blood as determined by flow cytometry and histology (Figure S1 in Supplementary Material). We had earlier shown highly efficient depletion of skin MCs on formalin-fixed, Giemsa-stained sections (47). As mucosal MCs of the intestinal tract can be metachromatically stained using special fixation procedures like Carnoy fixation but not after formalin fixation, we now performed additional Giemsa staining of Carnoy-fixed skin to exclude the presence of skin MC populations that fail to stain with conventional protocols (Figure S1 in Supplementary Material) and confirmed near complete absence of MCs in the skin of Mcpt5-Cre R26DTA/DTA mice.
We first investigated the impact of MCs on stimulation of the adaptive immune system by comparing LN hypertrophy in response to bacterial tissue infection in the presence or absence of MCs. McLachlan et al. described that injection of uropathogenic E. coli into the footpad of control mice resulted in a marked increase in total cellularity of the draining popliteal LN (3). LN hypertrophy was diminished in MC-deficient KitW/W-v mice; however, the response was restored by reconstitution of the animals with wild-type BMMCs. We injected the same uropathogenic E. coli strain J96 into the footpad of Mcpt5-Cre+R26DTA/DTA and Cre-negative littermates. Saline injected animals served as controls. 24 h after infection, the E. coli-injected Cre-negative controls displayed a substantial increase in popliteal LN size (data not shown) and total cellularity with increased numbers of T cells, B cells, and DCs compared with the saline injected group as expected (Figure 1A). To our surprise, LN hypertrophy was not reduced in E. coli-injected Mcpt5-Cre+R26DTA/DTA animals, but in fact, total cellularity and T cell, B cell, and DC numbers were even increased compared with the Cre-negative controls (Figure 1A). MCs were virtually absent in the infected footpad tissue of Mcpt5-Cre+R26DTA/DTA mice 24 h after infection as verified by histology (Figure 1B). Collectively, these data demonstrate that the recruitment of lymphocytes and DCs to LNs draining the footpad tissue inflamed by the innate response to bacterial infection did not require the presence of MCs in the infected tissue.
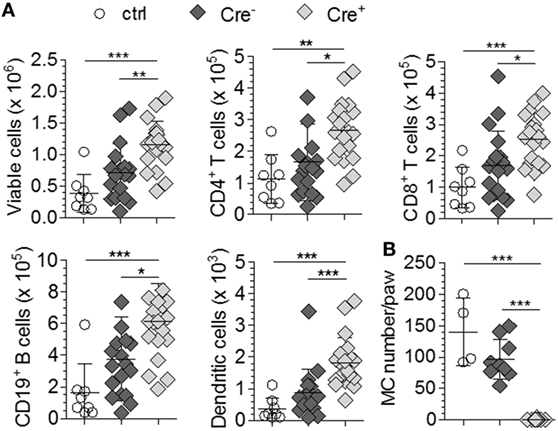
Figure 1. Hypertrophy of LNs draining E. coli-infected tissue does not depend on mast cells (MCs). (A) Total LN cellularity and absolute size of different cell populations of the ipsilateral popliteal LN analyzed 24 h after injection of 1 × 105 E. coli J96 into the hind footpad of Cre-negative R26DTA/DTA (n = 9) and Mcpt5-Cre+R26DTA/DTA mice (n = 10). Four Cre-negative control mice were injected with saline. Data from two independent experiments with similar results. Means ± SD are shown. (B) Numbers of MCs in E. coli J96-infected or saline-injected footpad tissue of the animals in (A) 24 h after infection as determined by histological analysis of Giemsa-stained tissue sections. Means ± SD are shown. In all cases, statistical analysis was performed using one-way ANOVA and Bonferroni’s multiple comparison test.
We first tested whether absence of MCs affects T cell responses. We used the model antigen 2W1S, a variant of peptide 52–68 from the MHC II I-Eα chain, which is not expressed and therefore immunogenic in C57BL/6 mice (48). Mcpt5-Cre+R26DTA/DTA and control mice were injected intradermally with this peptide along with CFA at the tail base. Inguinal LN cellularity and the antigen-specific T cell response were monitored thereafter.
The LNs of mice injected with CFA in addition to 2W1S peptide showed substantial increases in total cellularity compared with saline-injected controls 3, 7, and 21 days after immunization that reflected increased numbers of T cells, B cells, and DCs (Figure 2A; Figure S2A in Supplementary Material). Peptide alone had no effect on LN cellularity. As in the case of E. coli infection (see above, Figure 1), MC deficiency in Mcpt5-Cre+R26DTA/DTA mice did not impair LN hypertrophy responses to intradermal immunization with antigen plus adjuvant. On the contrary, the increase in LN cell numbers was even more pronounced in the absence of MCs.
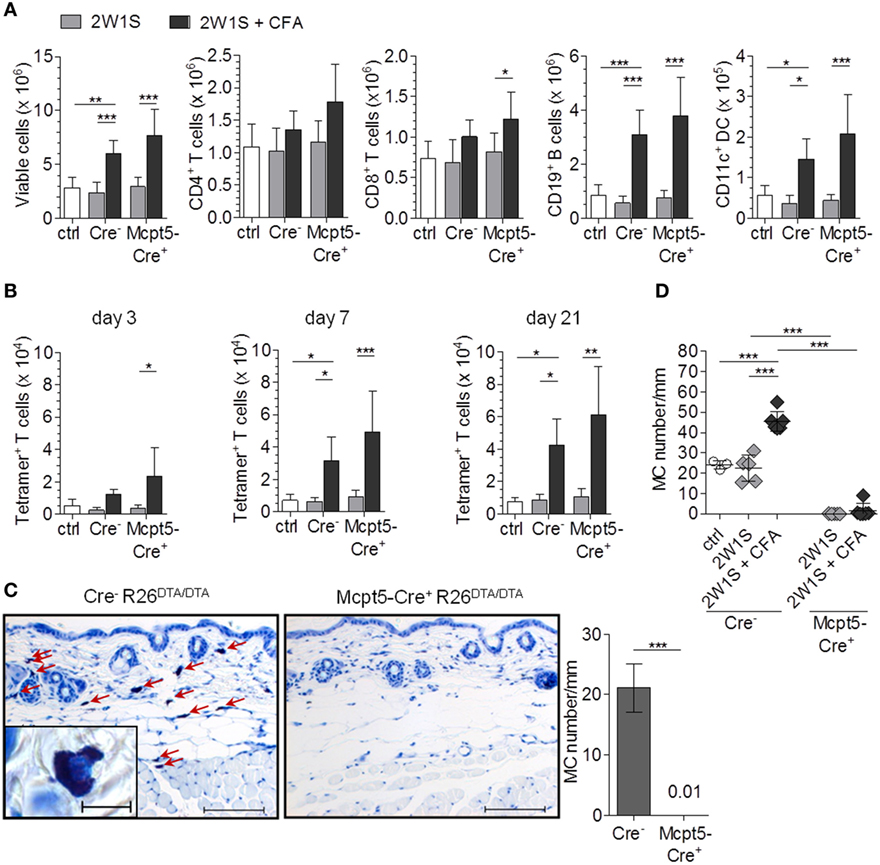
Figure 2. The antigen-specific T cell response is not diminished in mast cell (MC)-deficient mice. (A) Mcpt5-Cre+R26DTA/DTA mice and Cre-negative littermates were immunized with 2W1S peptide alone or peptide plus CFA by intradermal injection at the tail base (2W1S: n = 5 both groups; 2W1S/CFA: Cre− n = 11/Cre+ n = 12) and lymph node cellularity was assessed 3 days (see Figure S2 in Supplementary Material), 7 days, and 21 days (see Figure S2 in Supplementary Material) later. Saline-injected Cre-negative littermates served as additional controls. (B) Numbers of 2W1S-specific T cells in the LNs from A [time points as in (A)] were quantified by MHC II tetramer staining. (C,D) Efficient MC depletion of the dermal immunization site at the tail base was verified by histological analysis of Giemsa-stained skin sections in untreated mice (C) and at day 21 after immunization (D) (scale bar 100 µm, inset 10 µm). Dermal MCs were counted per 20-mm length of epidermis for each mouse. N = 7 per group at day 0; n = 4 Cre-negative saline controls, n = 5 2W1S both groups; n = 9 Cre−/n = 8 Cre+ 2W1S/CFA at day 21; means ± SD are shown. Data were collected from at least four independent experiments per time point. Statistical analysis was performed using one-way ANOVA and Bonferroni’s multiple comparison test (A,B,D) or unpaired two-sided Student’s t-test (C).
In parallel to LN hypertrophy, numbers of 2W1S peptide-specific CD4+ T cells were monitored by MHC class II tetramer labeling after immunization. For verification of the specificity of this staining, we immunized WT mice with 2W1S/CFA and stained total lymph node cells with 2W1S/MHC II tetramer-APC, flow cytometrically sorted tetramer-positive and tetramer-negative CD4+ T cells (Figure S2B in Supplementary Material) and restimulated them with 2W1S peptide in co-cultures with total LN cells of unimmunized WT mice. IFN-γ concentrations in the culture supernatant were quantified as a measure for T cell activation. High amounts of IFN-γ were released by tetramer-positive but not by tetramer-negative cells (Figure S2C in Supplementary Material), indicating that the tetramer indeed identified 2W1S-specific T cells. Robust increases of 2W1S-specific T cell numbers were induced by immunization with 2W1S peptide plus CFA. Importantly, this increase was not diminished but even was slightly more pronounced in MC-deficient compared with MC-competent Cre-negative control mice at all three time points analyzed (day 3, 7, and 21 after immunization, Figure 2B).
To verify MC deficiency at the dermal immunization site of Mcpt5-Cre+R26DTA/DTA mice, and exclude that the inflammatory adjuvant stimulus had led to reappearance of MCs, dermal tissue was sampled from untreated mice and at day 21 after immunization and MCs were counted in tissue sections. Figures 2C,D show that MCs were depleted with high efficiency in Mcpt5-Cre+R26DTA/DTA animals as expected. The local inflammatory response to CFA induced an increase in MC numbers in control but not Mcpt5-Cre+R26DTA/DTA mice.
Collectively, these results demonstrate that MCs are not required for the expansion of antigen-specific T cells in response to immunization with peptide plus adjuvant.
To study antibody responses in the presence or absence of MCs, Mcpt5-Cre+R26DTA/DTA and Cre-negative control mice were immunized intradermally with chicken ovalbumin (Ova) plus CFA at the tail base and serum was collected 10, 21, and 42 days later. Serum concentrations of Ova-specific IgG of major murine subclasses (IgG1, IgG2b, and IgG2c) were determined by ELISA (Figures 3A–C). Ova alone induced no or only moderate immune responses that were not diminished in the MC-deficient animals compared with control mice. Administration of Ova plus CFA resulted in robust increases in anti-Ova IgG1 and IgG2b already 10 days after immunization, while the IgG2c response began somewhat later. By day 21, anti-Ova IgG1 and IgG2b had increased further and ranged 3–4 orders of magnitude higher, compared with mice that received Ova alone, by day 42 after immunization. The responses of MC-deficient animals were in the same range or even slightly enhanced in comparison to MC-proficient Cre-negative controls at all time points analyzed.
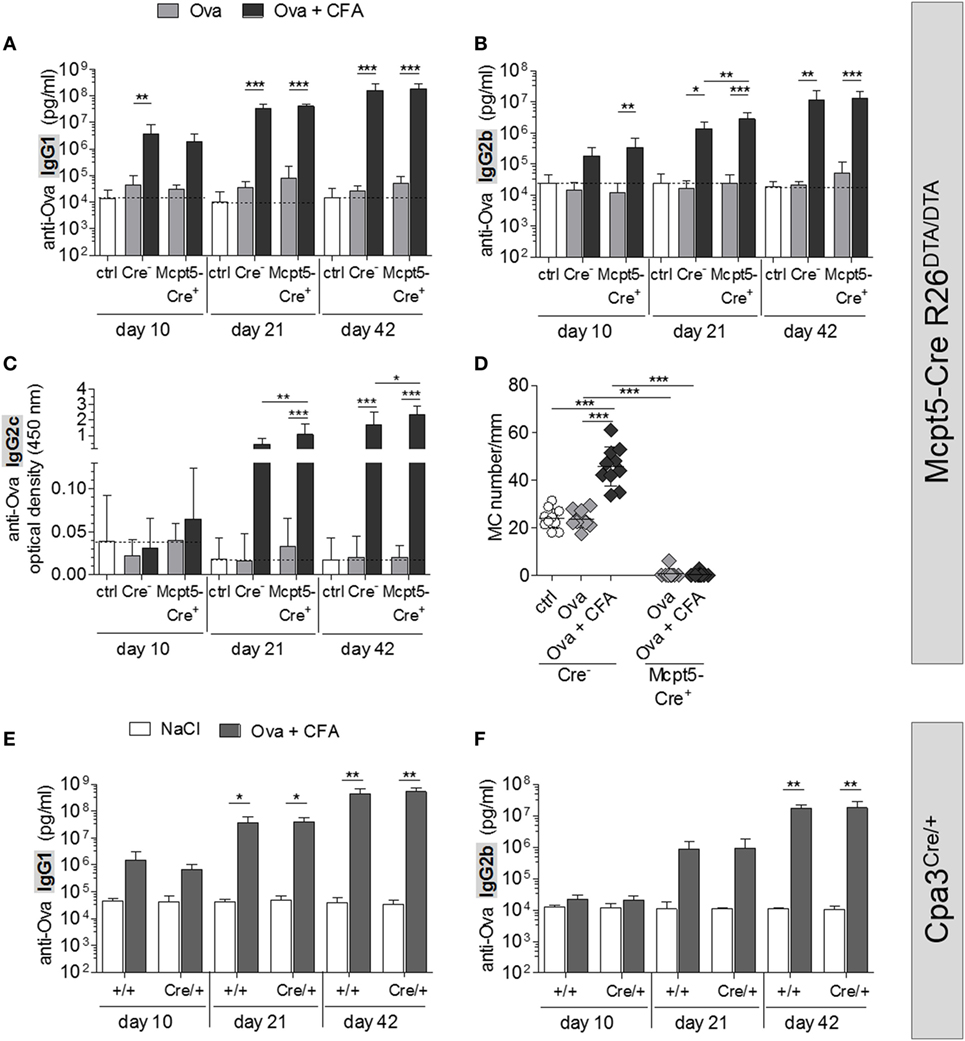
Figure 3. No impairment of humoral immune responses in mast cell (MC)-deficient mice. (A–C) Quantification of anti-Ova IgG1 (A), IgG2b (B), and IgG2c (C) by ELISA in the serum of saline treated Cre-controls (n = 12) and MC-deficient Mcpt5-Cre+R26DTA/DTA mice and Cre-negative littermates immunized intradermally with Ova alone or Ova plus CFA (Ova alone Cre+ n = 10, Cre− n = 10; Ova plus CFA Cre+ n = 11, Cre− n = 10). Two independent experiments were performed and yielded similar results. (D) MCs were counted in Giemsa-stained sections of tail base skin samples taken from each animal represented in (A–C) at day 42 after immunization per 20 mm length of epidermis. Means ± SD are shown. In all cases, statistical analysis was performed using one-way ANOVA and Bonferroni’s multiple comparison test. (E,F) Quantification of anti-Ova IgG1 (E) and IgG2b (F) by ELISA in the serum of saline treated controls (Cpa3+/+ n = 4, Cpa3Cre/+ n = 3) and MC-deficient Cpa3Cre/+ mice and MC-proficient Cpa3+/+ littermates immunized intradermally with Ova plus CFA (n = 7 both groups). A single experiment was performed.
To exclude that tissue inflammation induced by CFA resulted in recruitment of MCs at the immunization site even in Mcpt5-Cre+R26DTA/DTA mice, we counted MCs in Giemsa-stained sections of paraffin-embedded tail base skin, sampled from the injection site at day 42. As shown in Figure 3D, the CFA stimulus indeed resulted in a doubling of MC numbers at the immunization site in Cre–negative control mice, while MCs were absent or very low in number in Mcpt5-Cre+R26DTA/DTA mice without or with CFA administration. Using the Carnoy fixation protocol, however, we found a small population of residual MCs in LNs (but not skin, see Figure S1F in Supplementary Material) of Mcpt5-Cre+R26DTA/DTA mice that was not detectable with conventional formalin fixation.
To formally exclude that this small, residual population of LN MCs mediated potent immunostimulatory effects that might explain the absence of detectable immunological deficits in Mcpt5-Cre+R26DTA/DTA mice, we repeated immunizations in Cpa3Cre/+ mice, which are completely devoid of MCs (32). Absence of MCs in skin and LNs of this strain was verified by Giemsa staining of Carnoy-fixed sections of skin tissue obtained from untreated mice and of LN tissue sampled at the end of the experiment (Figures S3A,B in Supplementary Material). Cpa3Cre/+ mice and Cpa3+/+ littermate controls were immunized with Ova plus CFA at the tail base. Serum was collected 10, 21, and 42 days later and concentrations of Ova-specific IgG1 and IgG2b were determined by ELISA (Figures 3E,F). Administration of Ova plus CFA to Cpa3Cre/+ mice and littermate controls resulted in increases of anti-Ova IgG1 and IgG2b similar to the results obtained in Mcpt5-Cre R26DTA/DTA mice (Figures 3A,B). The intensity of this humoral immune response was not altered by the absence of MCs in Cpa3Cre/+ littermates.
We also studied the antigen-specific antibody response to another antigen, protective antigen (PA) of Bacillus anthracis in Mcpt5-Cre R26DTA/wt mice heterozygous for the R26DTA allele. In these mice, MCs in skin tissue of the tail base 42 days after immunization were less efficiently reduced to 40% of normal numbers or lower (Figure S3D in Supplementary Material), demonstrating the impact of the zygosity of the R26DTA allele. The reduction of MCs by 60% or more did not impair the PA-specific antibody response (Figure S3C in Supplementary Material).
Mast cell-derived TNF was reported to be a critical mediator of MC effects on adaptive immunity (7). In parallel to our experiments in mice lacking MCs, we directly addressed the role of TNF secretion by MCs using mice with normal MC numbers but highly efficient, selective inactivation of the TNF gene in CTMCs (49). Mcpt5-Cre TNFFL/FL and Cre-negative littermate control mice were injected with Ova plus CFA intradermally at the tail base and serum was collected at different time points. Figure S3E in Supplementary Material shows that Ova-specific IgG responses were not compromised in mice lacking TNF selectively in MCs compared with Cre-negative control mice at the 10 and 21 day time points. A slight (albeit statistically significant) reduction of specific IgG1 but not IgG2b was observed in the mutants compared to controls.
Collectively, we show that absence of MCs does not result in relevant impairment of antibody responses to immunization with protein antigen plus adjuvant.
Compound 48/80 (c48/80) triggers MC degranulation and was shown to induce hypertrophy of draining LNs upon injection into tissues and to potently enhance adaptive immune responses against protein antigens. c48/80 was also described to be an efficient, non-toxic adjuvant for administration along with immunizing antigen via mucosal surfaces (19, 50, 51). The adjuvant effects of c48/80 were reduced in MC-deficient KitW/W-v mice and reported to be dependent on MC-derived TNF (3, 7). Therefore, activation of TNF release from MCs by compounds that directly induce MC degranulation was proposed as a new principle of adjuvanting adaptive immune responses (19).
To investigate the adjuvant effects of c48/80 in our MC-deficiency model, we first injected c48/80 alone into the footpads of Mcpt5-Cre+R26DTA/DTA and Cre-negative littermate control mice and analyzed the draining popliteal LNs 24 h later. c48/80 induced significant LN hypertrophy in Cre-negative mice compared with saline-injected controls with increases in numbers of T cells, B cells, and DCs. Of note, LN hypertrophy was not reduced in MC-deficient Mcpt5–Cre+R26DTA/DTA animals but, on the contrary, was even enhanced compared with the responses of their MC-proficient littermates (Figure 4A). Highly efficient MC depletion in the injected tissue was verified histologically (Figure S4A in Supplementary Material). We next tested the effect of c48/80 in KitW/W-v and congenic littermate control Kit+/+ mice and reproduced the results of published studies (19) that described impaired c48/80-induced LN hypertrophy in KitW/W-v mice (Figure 4A).
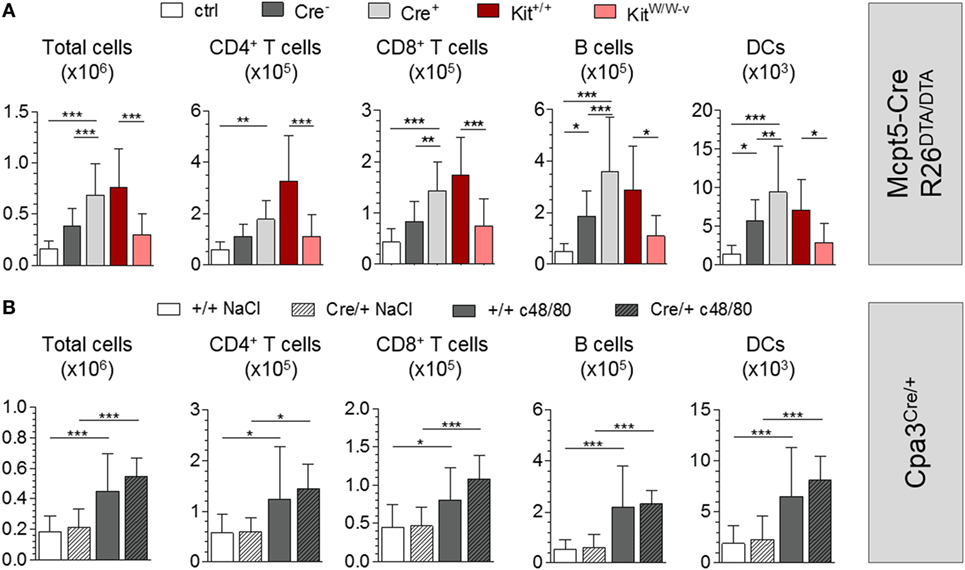
Figure 4. Mast cells (MCs) are not required for c48/80-mediated LN hypertrophy. (A) Cellularity of popliteal LNs 24 h after injection of c48/80 into footpads of Mcpt5-Cre+R26DTA/DTA and Cre-negative littermate control mice (n = 11 both groups) as well as KitW/W–v (n = 5) and congenic Kit+/+ control animals (n = 7) as determined by flow cytometry. Saline injected Cre-negative mice (n = 6) served as controls. Data from two independent experiments with similar results. (B) Cellularity of popliteal LNs 24 h after injection of c48/80 into footpads of Cpa3Cre/+ and Cpa3+/+ littermate control mice (Cpa3+/+ n = 16, Cpa3Cre/+ n = 11) as determined by flow cytometry. Saline was injected as control into one of both paws. A single experiment was performed. In all cases, means ± SD are shown and statistical analysis was performed using one-way ANOVA and Bonferroni’s multiple comparison test.
To exclude that residual LN MCs were responsible for the undiminished LN responses of Mcpt5-Cre+R26DTA/DTA mice, we repeated the c48/80 footpad injections in Cpa3Cre/+ mice and Cpa3+/+ littermate controls. Compared with the contralateral LNs draining the saline-injected footpad c48/80 induced significant LN hypertrophy including increased T cell, B cell, and DC numbers (Figure 4B). Confirming the results obtained in MC-deficient Mcpt5-Cre+R26DTA/DTA animals, LN hypertrophy was not reduced in Cpa3Cre/+ mice but was even slightly enhanced compared to MC-proficient Cpa3+/+ mice.
To investigate the impact of MC deficiency on c48/80-induced adaptive immune responses, we immunized Mcpt5-Cre R26DTA/DTA mice with antigen (2W1S peptide or Ova) alone or with antigen plus c48/80 intradermally at the tail base and analyzed the antigen-specific T cell and antibody responses. Numbers of 2W1S-specific CD4+ T cells identified by tetramer staining were increased in control animals that had received 2W1S peptide plus c48/80 compared to mice immunized with peptide alone (Figure 5A). This finding clearly demonstrates that c48/80 has potent adjuvant activity. However, this effect of c48/80 is independent of MCs since no reduction of this activity was observed in MC-deficient Mcpt5-Cre+R26DTA/DTA mice (Figure 5A). Immunization with Ova alone only modestly induced anti-Ova IgG1, while Ova-specific antibody concentrations increased by 2 orders of magnitude 42 days after injection of Ova plus c48/80 (Figure 5B), indicating robust adjuvant effects. MC deficiency in Mcpt5-Cre+R26DTA/DTA mice, however, did not compromise c48/80 adjuvant activity (Figure 5B). MC numbers were determined for each Mcpt5-Cre+R26DTA/DTA animal at the end of these experiments (Figure S4B in Supplementary Material).
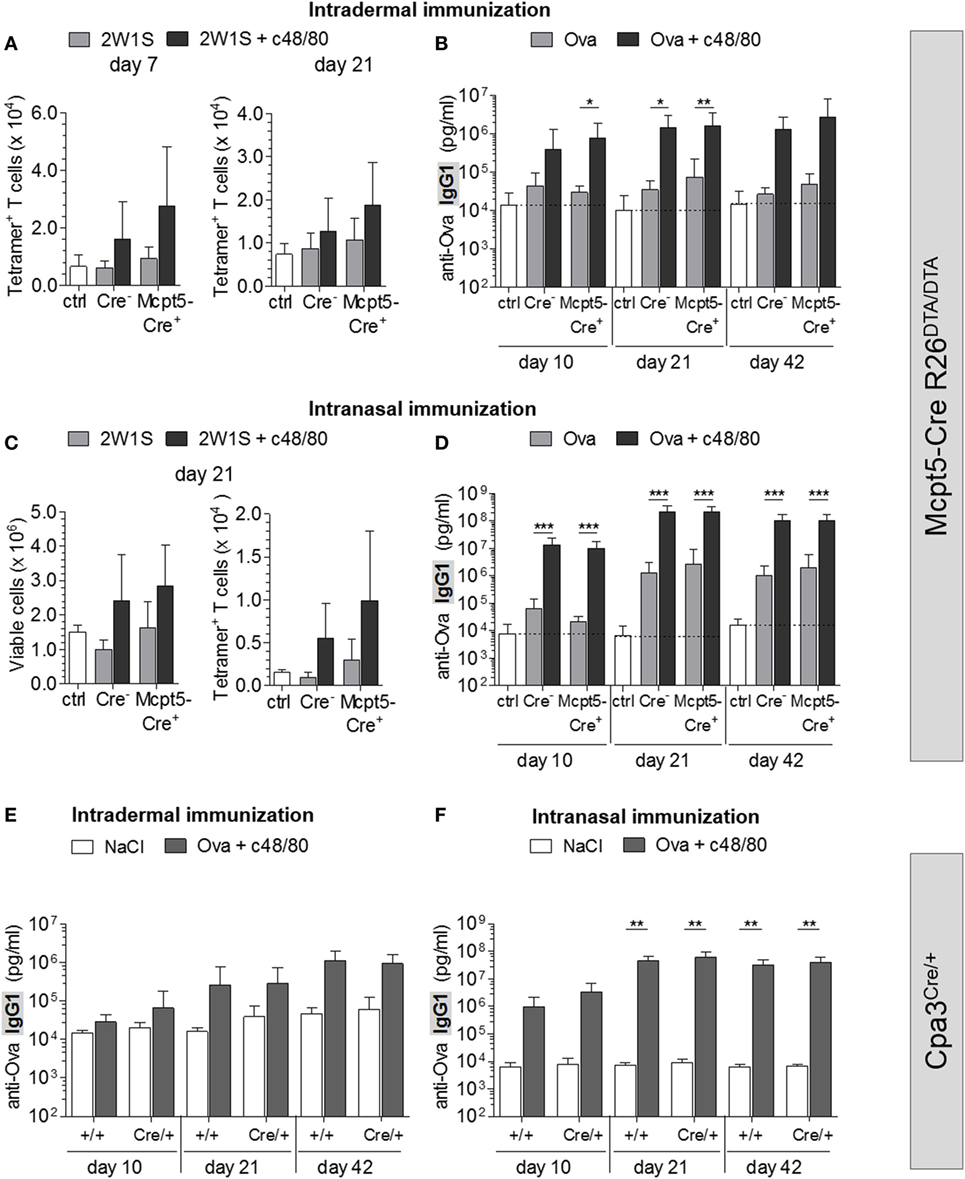
Figure 5. Mast cells (MCs) are dispensable for c48/80-induced antigen-specific T cell and antibody responses. (A,B) Cre-negative and Mcpt5-Cre+R26DTA/DTA mice were intradermally injected with antigen (2W1S or Ova) alone, with antigen plus c48/80 or with saline. (A) Total numbers of 2W1S-specific CD4+ T cells identified by tetramer staining in inguinal LNs 7 (saline n = 7; 2W1S alone n = 5 both groups; 2W1S plus c48/80 Cre+ n = 11, Cre− n = 10) or 21 days after immunization (saline n = 4; 2W1S alone n = 5 both groups; 2W1S plus c48/80 n = 7 both groups). (B) Quantification of anti-Ova IgG1 by ELISA in the serum of saline treated Cre-negative controls (n = 12) and mice immunized i.d. with Ova alone or with Ova plus c48/80 (Ova alone n = 10 both groups; Ova plus c48/80 Cre+ n = 9, Cre− n = 10). Data from two independent experiments with similar results. (C,D) Antigen alone (2W1S or Ova) or antigen plus c48/80 was administered into both nostrils of Cre-negative and Mcpt5-Cre+R26DTA/DTA mice. (C) Total cellularity and 2W1S-specific (tetramer+) CD4+ T cells were quantified flow cytometrically in draining mandibular LNs 21 days after immunization (saline n = 5; 2W1S alone Cre+ n = 6, Cre− n = 5; 2W1S plus c48/80 n = 6 both groups). (D) Quantification of anti-Ova IgG1 by ELISA in the serum of saline treated Cre-negative controls (n = 11) and mice immunized intranasally with Ova alone or with Ova plus c48/80 (Ova alone Cre+ n = 13, Cre− n = 12; Ova plus c48/80 n = 13 both groups). Data from three independent experiments that yielded similar results. (E) Quantification of anti-Ova IgG1 by ELISA in the serum of saline-treated control animals (Cpa3+/+ n = 4, Cpa3Cre/+ n = 3) and MC-deficient Cpa3Cre/+ mice and Cpa3+/+ littermates immunized intradermally with Ova plus c48/80 (Cpa3+/+ n = 7, Cpa3Cre/+ n = 6). A single experiment was performed. (F) Quantification of anti-Ova IgG1 by ELISA in the serum of saline treated controls (Cpa3+/+ n = 6, Cpa3Cre/+ n = 3) and mice immunized intranasally with Ova plus c48/80 (Cpa3+/+ n = 9, Cpa3Cre/+ n = 6). A single experiment was performed. In all cases, means ± SD are shown, and statistical analysis was performed using one-way ANOVA and Bonferroni’s multiple comparison test.
C48/80 has been described as a potent non-toxic adjuvant for immunization via the nasal mucosa, suggesting that the numerous connective tissue MCs surrounding the nasal-associated lymphatic tissue (NALT) can be activated by c48/80 to stimulate adaptive immunity (19). Intranasally administrated c48/80 was shown to induce degranulation of these CTMCs in the vicinity of the NALT (19). We verified by histology that this MC population was absent in the nasal mucosa of Mcpt5-Cre+R26DTA/DTA mice (Figure S4C in Supplementary Material). Efficient depletion of CTMCs in Mcpt5-Cre+R26DTA/DTA nasal tissue was further underpinned by quantification of mRNA of the CTMC-specific endogenous Mcpt5 gene, which we found undetectable or strongly reduced compared with control tissue (Figure S4E in Supplementary Material).
We applied antigen (2W1S or Ova) alone or along with c48/80 into the nostrils of MC-deficient Mcpt5-Cre+R26DTA/DTA mice and Cre-negative littermate control mice and analyzed the draining mandibular LNs by flow cytometry. While 2W1S peptide alone had no effects, mice that received 2W1S plus c48/80 featured increased numbers of total lymphocytes and DCs as well as 2W1S-specific (tetramer+) T cells in these LNs compared to saline-injected controls (Figure 5C). Likewise, administration of Ova along with c48/80 increased Ova-specific IgG1 about 100-fold compared with levels of mice that received Ova alone (Figure 5D). These findings further confirm the adjuvant qualities of c48/80. However, the stimulation of antigen-specific T and B cell responses by c48/80 was independent of the presence or absence of MCs (Figures 5C,D).
Administration of Ova without adjuvant via the nasal route resulted in modest IgG1 responses. Under such conditions of low intensity of danger signals, one might expect that MCs might be critical providers of adjuvant effects. However, also the antibody response to Ova alone was not reduced in MC-deficient versus control mice.
We made sure that MC deficiency of the nasal tissue persisted for the duration of the experiment (Figure S4D in Supplementary Material) and also excluded that the nasal mucosa contained mucosal type MCs (expressing Mcpt1) that in principle are not amenable to depletion in Mcpt5–Cre+R26DTA/DTA animals and might contribute to c48/80 adjuvant effects (Figure S4E in Supplementary Material).
To rule out effects of residual LN MCs in Mcpt5-Cre+R26DTA/DTA, we repeated intradermal and intranasal immunizations with Ova plus c48/80 in Cpa3Cre/+ and Cpa3+/+ littermate control mice. We showed that nasal tissue of Cpa3Cre/+ mice was devoid of MCs, by demonstrating the absence of Mcpt1 and Mcpt5 transcripts (Figure S4E in Supplementary Material). As in Mcpt5-Cre R26DTA/DTA mice, Ova plus c48/80 induced an increase in anti-Ova IgG1 in both groups. These responses were not diminished in MC-deficient Cpa3Cre/+ mice (Figures 5E,F).
Collectively, we demonstrate that presence or absence of MCs has no impact on the adjuvant effects of c48/80, which must therefore mediate these effects via other cell types.
The past two decades brought the exciting insight that adaptive immunity critically depends on innate immune responses of the tissues, which has tremendous impact on vaccine design and development of novel adjuvants (52, 53). Innate tissue responses determine, whether or not T and B cell responses are mounted and also regulate the effector class of the T and antibody response (1). The information on the tissue response is carried to the draining LN by DCs. Most tissue cells can contribute to the innate response in an infected tissue. MCs express several PRRs and are capable of releasing proinflammatory mediators (54, 55). Several of these, including the potently immunostimulatory cytokine TNF, are stored in the MCs’ secretory granules and can be rapidly released upon activation of the MC (55). TNF-containing MC particles were shown to reach draining LNs (2). The concept of the MC as an amplifier of innate immunity that provides adjuvant activity for adaptive responses was critically based on findings in Kit-mutant MC-deficient mice. LNs of KitW/W-v mice draining sites of E. coli infection showed reduced hypertrophy responses compared to LNs of wild-type mice (3). This phenotype was reversed by reconstitution of the infection site with in vitro differentiated wild-type, but not TNF-deficient MCs, which has been interpreted to suggest that MCs promote recruitment and retention of lymphocytes in LNs draining infected tissues by release of TNF (3). Impaired antigen-specific humoral responses were observed in KitW-sh/W-sh and KitW/W-v mice (7) and pharmacologic MC activation by MC secretagogues, including c48/80, was proposed as a new adjuvant principle in vaccination (19, 56).
However, using Mcpt5-Cre R26DTA/DTA mice that are profoundly deficient for connective tissue MCs but otherwise bear a normal immune system, we found no evidence for a MC function in adaptive immune responses. Compared with wild-type controls, the LN hypertrophy response to footpad infection with E. coli was unchanged in the MC-deficient animals, as were expansion of antigen-specific T cells and antibody responses upon vaccination with protein antigen plus CFA. The undiminished B cell response also indicated that T helper effector function was not compromised in the absence of MCs. We confirmed unimpaired humoral responses in the Cpa3Cre/+ mouse model of MC deficiency in which complete absence of all types of MCs was extensively documented (32). These mice also lack the small population of residual MCs that we detected in LNs of Mcpt5-Cre+R26DTA/DTA mice. Regarding compound c48/80, a well known stimulator of MC degranulation, we confirmed its adjuvant activity, but found that this adjuvant effect was not mediated by activation of MCs. C48/80 induced rapid LN hypertrophy that was unaffected by MC deficiency in Mcpt5-Cre+R26DTA/DTA or in Cpa3Cre/+ mice. Adaptive responses to intradermal or intranasal immunization with antigen were clearly enhanced by coadministration of c48/80 in MC-competent control mice. These responses were not reduced by the deficiency for MCs in Mcpt5-Cre+R26DTA/DTA or in Cpa3Cre/+ mice. Our results demonstrate that the adjuvant activity of c48/80 must be mediated by activating effects on cell types other than MCs. Compound 48/80 was shown to act on the Mrgprb2 receptor (57), which was reported to be expressed selectively in MCs (58). However, c48/80 impacts additional pathways also in non-MCs (59, 60) and the full spectrum of c48/80 effects outside the MC lineage is probably not fully understood. Our findings of undiminished adaptive responses despite MC deficiency are in line with the report by Feyerabend et al. of normal humoral responses, including immunoglobulin class switching and hypermutation, of Cpa3Cre/+ mice to i.p. immunization with antigen plus alum (32).
Our study adds to a long list of cases in which results in Kit mutant mouse strains did not reproduce in one of the novel mouse strains with Kit-independent selective MC deficiency but otherwise normal immune system, despite reported reversion of phenotypes of Kit mutants by reconstitution with in vitro differentiated MCs (31, 32, 34, 35, 37, 39, 40, 42, 44–46) [reviewed in Ref. (38, 41, 43)]. Collectively, our results strongly suggest that Kit mutants are unreliable models of MC deficiency, even when combined with MC reconstitutions. MC reconstitution with poorly defined in vitro differentiated cells, transferred into recipients that feature a broadly abnormal immune system and empty MC niches results in a complex situation that may be the cause of misleading data. While we formally cannot exclude a contribution of differences in genetic backgrounds to the discrepancies between our results in Mcpt5-Cre R26DTA/DTA and published studies using Kit-mutant strains, background differences were clearly ruled out as an explanation of conflicting results obtained in Kit mutants versus the Kit-independent MC-deficiency model Cpa3Cre/+ in another study (40). Moreover, adaptive immune responses are most commonly studied in C57BL/6 mice, which were in our case MC deficient.
The question of a MC contribution to adaptive immune responses has been addressed previously also in the context of autoimmunity. Experimental autoimmune encephalomyelitis (EAE) is a mouse model for multiple sclerosis in which CNS inflammation is induced by immunization of mice with brain-specific proteins plus CFA. While reduced EAE severity was observed in KitW/W-v mice (61, 62) and enhanced EAE was found in the KitW-sh/W-sh mice (63, 64), no alteration of EAE was detected in either of these strains in another study (65). Kit-independent MC deficiency in Cpa3Cre/+ mice had no effect on EAE (32). In a model of antibody-induced arthritis, KitW-sh/W-sh mice develop full disease, whereas KitW/W-v animals are resistant to experimental joint inflammation (66, 67), demonstrating that perturbations of the immune system due to hypomorphic Kit expression beyond their MC deficiency hamper interpretations of findings in Kit mutant mice. Kit-independent mouse models of MC cell deficiency did not reveal any role for MCs in this arthritis model (32, 68, 69). In collagen-induced arthritis (CIA), in which a breakdown of self-tolerance is induced by injection of collagen along with CFA into mice (70), a pathogenic contribution of MCs is conceivable on the level of induction of the adaptive anti-collagen response as well as on the level of the final organ damage. MCs functions in CIA were addressed in the KitW-sh/W-sh strain, which was found fully susceptible (71), while mice deficient for Mcpt 4 featured reduced arthritis scores (72). Based on experiments in the Kit-independent, inducible Mcpt5-Cre R26iDTR/iDTR MC-deficiency model on the moderately CIA susceptible background C57BL/6, Schubert et al. reported a role for MCs in CIA (68) and observed diminished anti-collagen T cell responses in the absence of MCs. A recent study reported a reduction of CIA in (C57BL/6 × DBA/1) F1 hybrid mice with inducible MC and basophil deficiency dependent on the time point of MC depletion (73). More recently, we backcrossed the Mcpt5-Cre-based MC-deficiency models to the CIA susceptible background DBA/1 for five generations and found no significant reduction of joint inflammation in these animals as yet (Figure S5 in Supplementary Material). Thus, more experiments are required to definitively settle the question of MC contributions to arthritis in this model.
Our results do not exclude that MCs play important roles in the induction of adaptive responses in contexts that remain to be identified. A special case of adaptive responses in which MC contributions play a role are pathogenic T cell responses to contact allergens. Such contact sensitizers are small chemicals that rapidly react with proteins and thereby render them immunogenic, a process called haptenization. The T cell response against the haptenized proteins depends on a rapid activation of an innate inflammatory response that these sensitizing chemicals induce (74). How contact allergens trigger this initial innate response remains unknown, but activation of MCs seems to play a role (31, 34, 49).
In line with a concept of MC- or basophil-mediated immune regulation, targeted inactivation of histamine receptors was shown to result in altered T cell polarization upon immunization with OVA and adjuvant (75). Moreover, effects of IgE-mediated MC activation on adaptive immunity are well documented (11). However, MC contributions to adaptive responses are much more restricted than previously concluded from experiments in Kit-mutant strains. MCs play no role in “standard” immune responses in situations with abundant danger signals for DC activation, but might serve to fill special functional gaps in the repertoire of mammalian innate responses. They might play a role for adaptive immunity under particular conditions of limiting danger signal intensities. The studies performed in Kit-independent MC-deficiency models indicate that potential functions of MCs in regulation of adaptive immunity, beyond pathophysiological functions in IgE-mediated allergic disease and delayed type hypersensitivity responses to exogenous chemicals, remain to be demonstrated.
All mice were housed at the Experimental Center, Medical Faculty Carl Gustav Carus, TU Dresden, under specific pathogen-free conditions. 8- to 21-week-old mice were used. Littermates were used as controls. All procedures were in accordance with institutional guidelines on animal welfare and were approved by the Landesdirektion Dresden (DD24-5131/207/2 and DD24-5131/207/33).
Ovalbumin (Catalog No. A2512), c48/80, and CFA (containing 1 mg/ml Mycobacterium tuberculosis) were obtained from Sigma-Aldrich. 2W1S peptide (sequence: EAWGALANWAVDSA) was kindly provided by Dr. J. B. McLachlan, New Orleans. Anthrax PA was obtained from List Biological Laboratories. All reagents were finally dissolved in sterile 0.9% saline. For intradermal immunization, each mouse was injected with 50 µl of a 10 ng/µl solution of the respective antigen (OVA, PA, or 2W1S peptide) with or without 0.6 µg/µl c48/80. Half of these 50 µl were injected intradermally at each side of the tail base. When CFA was used as an adjuvant, 0.5 µg antigen (OVA, PA, or 2W1S peptide) were dissolved in 25 µl saline and emulsified with 25 µl CFA/mouse (1:1 emulsion). Half of these 50 µl were injected at each side of the tail base. For mucosal immunization, mice were anesthetized with isoflurane and 10 µg of antigen or antigen plus 30 µg c48/80 were administered nasally at day 0, 7, and 14 in a total volume of 15 µl/mouse (7.5 µl into each nostril). For analysis of LN hypertrophy upon injection of c48/80, 2.4 µg/g body weight of c48/80 were dissolved in a volume of 40 µl. Half of this volume was injected subcutaneously into each hind footpad of one mouse. As negative controls, additional mice were injected with 20 µl of saline into each hind footpad. For analysis of LN hypertrophy in Cpa3Cre/+ mice, one hind footpad was injected with c48/80, the other with saline (20 µl each). Uropathogenic E. coli J96 (ATCC 700336) were purchased from LGC Standards (Wesel, Germany). From an overnight culture in Luria–Bertani (LB) broth, bacteria were grown again in LB broth to an A600 of 0.6 where they reached the exponential phase. Bacterial cultures yielded about 1 × 107 viable cells/ml. E. coli were washed twice and dissolved in sterile saline. 1 × 105 E. coli in a volume of 20 µl were injected into each hind footpad of one mouse. As negative controls, additional mice were injected with 20 µl of saline into each hind footpad.
Inguinal LNs were isolated at day 3, 7, or 21 after immunization with 2W1S plus adjuvant and single cell suspensions were generated by crushing between glass slides. APC-coupled 2W1S-MHC class II tetramer was kindly provided by Dr. J. B. McLachlan, Department of Microbiology and Immunology, Tulane University School of Medicine, New Orleans. Isolated cells were resuspended in 2% BSA/PBS and incubated with 2W1S-MHC II tetramer-APC in a final concentration of 10 nM for 1 h at room temperature in the dark. Cells were washed, stained with monoclonal antibodies in a volume of 50 µl for 30 min at 4°C and washed twice. Numbers of total cells, various cell types, and antigen-specific CD4+ T cells were determined using a Miltenyi MACSQuant® flow cytometer. After footpad injections, draining popliteal LNs were isolated 24 h later for flow cytometric analysis.
96-well plates (Nunc MaxiSorp™, VWR) were coated with Ova (2 ng/µl in 100 mM NaHCO3, 33.6 mM Na2CO3, pH 9.5) at 4°C over night. Plates were washed thrice with 0.05% Tween 20/PBS and blocked with 200 µl 2% skimmed milk powder/PBS for 1 h at 37°C. Plates were washed thrice. Standards of anti-Ova IgG1 and IgG2b (Thermo Scientific Fisher) ranging between 62,500 and 122 pg/ml were generated by serial twofold dilutions. 100 µl/well of standards and serum samples were pipetted into the Ova-coated plates in doublets and incubated 1 h at 37°C. Plates were washed thrice. Secondary antibodies (HRP-labeled goat anti-mouse IgG1, IgG2b, and IgG2c, Dianova) were diluted 1:10,000 (anti-IgG1 and -IgG2b) or 1:2,000 (anti-IgG2c) and 100 µl/well were incubated 1 h at 37°C. After washing thrice, 100 µl freshly mixed substrate (BD OptEIA™) per well were added and plates were incubated for 15–30 min at RT in the dark. Reactions were stopped by adding 50 µl 2N H2SO4/well. Optical density was measured at 450 nm (reference value 570 nm) using a Sunrise™ spectral photometer. Magellan™ software was used for non-linear regression analysis.
Tail base skin was fixed in 4% formalin for 1 week and embedded in paraffin. Heads of mice were freed from skin, muscles, eyes, and lower jaw, fixed in 4% formalin for 1 week and decalcified in Osteosoft (Merck) for 2 weeks. After removing incisors and neurocranium, tissue was embedded in paraffin. Hindpaws were isolated by cutting in the middle of the tibia and removing the skin. The paws were fixed in 4% formalin for 1 week, decalcified in Osteosoft for 2 weeks and embedded in paraffin. LNs were fixed in Carnoy’s solution (ethanol:chloroform:acetic acid 6:3:1) overnight followed by 8 h incubation in 100% ethanol and paraffin embedding. 5 µm paraffin sections were Giemsa-stained for MC quantification. Dermal MCs were counted per 20 mm horizontal length of epidermis. In frontal sections of the snout, MCs were counted in the connective tissue surrounding the NALT per 2 high power fields centered on the right and left NALT and the mean of MC numbers on 15 sections/mouse was calculated. In the hindpaws, MCs were quantified around the tarsal bones per high power field centered on the tarsal bone. MC numbers were quantified for each hind paw of one mouse and the mean was calculated. For cytospins, cells from the peritoneal cavity were isolated by flushing with 5 ml ice cold PBS. Pellets were resuspended in 2% BSA/PBS and 200 µl were loaded per cuvette and spun at 800 rpm for 2 min in a cytocentrifuge (Shandon). Air-dried slides were stained according to standard May-Grünwald and Giemsa protocols. Analysis was performed using Zeiss Axiovision Software.
Data are shown as mean ± SD unless stated otherwise. For two-group comparisons two-tailed Mann–Whitney test with 95% confidence interval was used. For multi-group comparisons, Kruskal–Wallis with Dunn’s post-test (significance level α = 0.05) was performed. Time-course data were analyzed by two-way ANOVA with Bonferroni post-tests. Chi square test was used to analyze CIA incidence. Significance levels: *p < 0.05, **p < 0.01, and ***p < 0.001.
All procedures were in accordance with institutional guidelines on animal welfare and were approved by the Landesdirektion Dresden (DD24-5131/207/2 and DD24-5131/207/33).
NS and KL designed and performed experiments. CA, AW, SG, LM, CH, TH, and LS performed experiments. DV generated R-DTA mice. TF and H-RR generated Cpa3Cre mice. FG and WM contributed to the design of the study. AD contributed to study design and discussed data. AR conceived and supervised the study. NS and AR wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We cordially thank J. B. McLachlan for providing APC-labeled 2W1S-MHC II-Tetramer and 2W1S peptide, and M. Maurer for providing WBB6F1-KitW/W-v mice. Excellent technical assistance by Christina Hiller and Anna Karutz is gratefully acknowledged. This work was supported by DFG grants Ro2133/7, Ro2133/5-1 and 5-2, and Ro2133/4 to AR, DFG grants Du1172/2, DU1172/3, and DU1172/4 to AD, and SFB643-B15 to DV, TF and H-RR were supported by SFB-TRR156-A07.
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01870/full#supplementary-material.
1. Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol (2015) 16(4):343–53. doi:10.1038/ni.3123
2. Kunder CA, St John AL, Li G, Leong KW, Berwin B, Staats HF, et al. Mast cell-derived particles deliver peripheral signals to remote lymph nodes. J Exp Med (2009) 206(11):2455–67. doi:10.1084/jem.20090805
3. McLachlan JB, Hart JP, Pizzo SV, Shelburne CP, Staats HF, Gunn MD, et al. Mast cell-derived tumor necrosis factor induces hypertrophy of draining lymph nodes during infection. Nat Immunol (2003) 4(12):1199–205. doi:10.1038/ni1005
4. Nakae S, Ho LH, Yu M, Monteforte R, Iikura M, Suto H, et al. Mast cell-derived TNF contributes to airway hyperreactivity, inflammation, and TH2 cytokine production in an asthma model in mice. J Allergy Clin Immunol (2007) 120(1):48–55. doi:10.1016/j.jaci.2007.02.046
5. Nakae S, Suto H, Berry GJ, Galli SJ. Mast cell-derived TNF can promote Th17 cell-dependent neutrophil recruitment in ovalbumin-challenged OTII mice. Blood (2007) 109(9):3640–8. doi:10.1182/blood-2006-09-046128
6. Nakae S, Suto H, Kakurai M, Sedgwick JD, Tsai M, Galli SJ. Mast cells enhance T cell activation: importance of mast cell-derived TNF. Proc Natl Acad Sci U S A (2005) 102(18):6467–72. doi:10.1073/pnas.0501912102
7. Shelburne CP, Nakano H, St John AL, Chan C, McLachlan JB, Gunn MD, et al. Mast cells augment adaptive immunity by orchestrating dendritic cell trafficking through infected tissues. Cell Host Microbe (2009) 6(4):331–42. doi:10.1016/j.chom.2009.09.004
8. St John AL, Chan CY, Staats HF, Leong KW, Abraham SN. Synthetic mast-cell granules as adjuvants to promote and polarize immunity in lymph nodes. Nat Mater (2012) 11(3):250–7. doi:10.1038/nmat3222
9. Caron G, Delneste Y, Roelandts E, Duez C, Herbault N, Magistrelli G, et al. Histamine induces CD86 expression and chemokine production by human immature dendritic cells. J Immunol (2001) 166(10):6000–6. doi:10.4049/jimmunol.166.10.6000
10. Amaral MM, Davio C, Ceballos A, Salamone G, Cañones C, Geffner J, et al. Histamine improves antigen uptake and cross-presentation by dendritic cells. J Immunol (2007) 179(6):3425–33. doi:10.4049/jimmunol.179.6.3425
11. Jawdat DM, Albert EJ, Rowden G, Haidl ID, Marshall JS. IgE-mediated mast cell activation induces Langerhans cell migration in vivo. J Immunol (2004) 173(8):5275–82. doi:10.4049/jimmunol.173.8.5275
12. Banovac K, Neylan D, Leone J, Ghandur-Mnaymneh L, Rabinovitch A. Are the mast cells antigen presenting cells? Immunol Invest (1989) 18(7):901–6. doi:10.3109/08820138909050768
13. Fox CC, Jewell SD, Whitacre CC. Rat peritoneal mast cells present antigen to a PPD-specific T cell line. Cell Immunol (1994) 158(1):253–64. doi:10.1006/cimm.1994.1272
14. Frandji P, Oskéritzian C, Cacaraci F, Lapeyre J, Peronet R, David B, et al. Antigen-dependent stimulation by bone marrow-derived mast cells of MHC class II-restricted T cell hybridoma. J Immunol (1993) 151(11):6318–28.
15. Frandji P, Tkaczyk C, Oskeritzian C, David B, Desaymard C, Mécheri S. Exogenous and endogenous antigens are differentially presented by mast cells to CD4+ T lymphocytes. Eur J Immunol (1996) 26(10):2517–28. doi:10.1002/eji.1830261036
16. Frandji P, Tkaczyk C, Oskéritzian C, Lapeyre J, Peronet R, David B, et al. Presentation of soluble antigens by mast cells: upregulation by interleukin-4 and granulocyte/macrophage colony-stimulating factor and downregulation by interferon-gamma. Cell Immunol (1995) 163(1):37–46. doi:10.1006/cimm.1995.1096
17. Gaudenzio N, Espagnolle N, Mars LT, Liblau R, Valitutti S, Espinosa E. Cell-cell cooperation at the T helper cell/mast cell immunological synapse. Blood (2009) 114(24):4979–88. doi:10.1182/blood-2009-02-202648
18. Kambayashi T, Allenspach EJ, Chang JT, Zou T, Shoag JE, Reiner SL, et al. Inducible MHC class II expression by mast cells supports effector and regulatory T cell activation. J Immunol (2009) 182(8):4686–95. doi:10.4049/jimmunol.0803180
19. McLachlan JB, Shelburne CP, Hart JP, Pizzo SV, Goyal R, Brooking-Dixon R, et al. Mast cell activators: a new class of highly effective vaccine adjuvants. Nat Med (2008) 14(5):536–41. doi:10.1038/nm1757
20. Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol (2010) 10(6):440–52. doi:10.1038/nri2782
21. Benoist C, Mathis D. Mast cells in autoimmune disease. Nature (2002) 420(6917):875–8. doi:10.1038/nature01324
22. Dawicki W, Marshall JS. New and emerging roles for mast cells in host defence. Curr Opin Immunol (2007) 19(1):31–8. doi:10.1016/j.coi.2006.11.006
23. Galli SJ, Nakae S, Tsai M. Mast cells in the development of adaptive immune responses. Nat Immunol (2005) 6(2):135–42. doi:10.1038/ni1158
24. Marshall JS. Mast-cell responses to pathogens. Nat Rev Immunol (2004) 4(10):787–99. doi:10.1038/nri1460
25. Marshall JS, Jawdat DM. Mast cells in innate immunity. J Allergy Clin Immunol (2004) 114(1):21–7. doi:10.1016/j.jaci.2004.04.045
26. Chervenick PA, Boggs DR. Decreased neutrophils and megakaryocytes in anemic mice of genotype W/W. J Cell Physiol (1969) 73(1):25–30. doi:10.1002/jcp.1040730104
27. Huizinga JD, Thuneberg L, Klüppel M, Malysz J, Mikkelsen HB, Bernstein A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature (1995) 373(6512):347–9. doi:10.1038/373347a0
28. Nigrovic PA, Gray DH, Jones T, Hallgren J, Kuo FC, Chaletzky B, et al. Genetic inversion in mast cell-deficient (Wsh) mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am J Pathol (2008) 173(6):1693–701. doi:10.2353/ajpath.2008.080407
29. Puddington L, Olson S, Lefrancois L. Interactions between stem cell factor and c-Kit are required for intestinal immune system homeostasis. Immunity (1994) 1(9):733–9. doi:10.1016/S1074-7613(94)80015-4
30. Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am J Pathol (2005) 167(3):835–48. doi:10.1016/S0002-9440(10)62055-X
31. Dudeck A, Dudeck J, Scholten J, Petzold A, Surianarayanan S, Köhler A, et al. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity (2011) 34(6):973–84. doi:10.1016/j.immuni.2011.03.028
32. Feyerabend TB, Weiser A, Tietz A, Stassen M, Harris N, Kopf M, et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity (2011) 35(5):832–44. doi:10.1016/j.immuni.2011.09.015
33. Lilla JN, Chen CC, Mukai K, BenBarak MJ, Franco CB, Kalesnikoff J, et al. Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood (2011) 118(26):6930–8. doi:10.1182/blood-2011-03-343962
34. Otsuka A, Kubo M, Honda T, Egawa G, Nakajima S, Tanizaki H, et al. Requirement of interaction between mast cells and skin dendritic cells to establish contact hypersensitivity. PLoS One (2011) 6(9):e25538. doi:10.1371/journal.pone.0025538
35. Peschke K, Weitzmann A, Heger K, Behrendt R, Schubert N, Scholten J, et al. IkappaB kinase 2 is essential for IgE-induced mast cell de novo cytokine production but not for degranulation. Cell Rep (2014) 8(5):1300–7. doi:10.1016/j.celrep.2014.07.046
36. Voehringer D, Liang HE, Locksley RM. Homeostasis and effector function of lymphopenia-induced “memory-like” T cells in constitutively T cell-depleted mice. J Immunol (2008) 180(7):4742–53. doi:10.4049/jimmunol.180.7.4742
37. Antsiferova M, Martin C, Huber M, Feyerabend TB, Förster A, Hartmann K, et al. Mast cells are dispensable for normal and activin-promoted wound healing and skin carcinogenesis. J Immunol (2013) 191(12):6147–55. doi:10.4049/jimmunol.1301350
38. Feyerabend TB, Gutierrez DA, Rodewald HR. Of mouse models of mast cell deficiency and metabolic syndrome. Cell Metab (2016) 24(1):1–2. doi:10.1016/j.cmet.2016.06.019
39. Gomez-Pinilla PJ, Farro G, Di Giovangiulio M, Stakenborg N, Némethova A, de Vries A, et al. Mast cells play no role in the pathogenesis of postoperative ileus induced by intestinal manipulation. PLoS One (2014) 9(1):e85304. doi:10.1371/journal.pone.0085304
40. Gutierrez DA, Muralidhar S, Feyerabend TB, Herzig S, Rodewald HR. Hematopoietic kit deficiency, rather than lack of mast cells, protects mice from obesity and insulin resistance. Cell Metab (2015) 21(5):678–91. doi:10.1016/j.cmet.2015.04.013
41. Katz HR, Austen KF. Mast cell deficiency, a game of kit and mouse. Immunity (2011) 35(5):668–70. doi:10.1016/j.immuni.2011.11.004
42. Paul C, Wolff S, Zapf T, Raifer H, Feyerabend TB, Bollig N, et al. Mast cells have no impact on cutaneous leishmaniasis severity and related Th2 differentiation in resistant and susceptible mice. Eur J Immunol (2016) 46(1):114–21. doi:10.1002/eji.201545613
43. Rodewald HR, Feyerabend TB. Widespread immunological functions of mast cells: fact or fiction? Immunity (2012) 37(1):13–24. doi:10.1016/j.immuni.2012.07.007
44. Schönhuber N, Seidler B, Schuck K, Veltkamp C, Schachtler C, Zukowska M, et al. A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat Med (2014) 20(11):1340–7. doi:10.1038/nm.3646
45. Willenborg S, Eckes B, Brinckmann J, Krieg T, Waisman A, Hartmann K, et al. Genetic ablation of mast cells redefines the role of mast cells in skin wound healing and bleomycin-induced fibrosis. J Invest Dermatol (2014) 134(7):2005–15. doi:10.1038/jid.2014.12
46. Chmelař J, Chatzigeorgiou A, Chung KJ, Prucnal M, Voehringer D, Roers A, et al. No role for mast cells in obesity-related metabolic dysregulation. Front Immunol (2016) 7:524. doi:10.3389/fimmu.2016.00524
47. Ghouse SM, Polikarpova A, Muhandes L, Dudeck J, Tantcheva-Poór I, Hartmann K, et al. Although abundant in tumor tissue, mast cells have no effect on immunological micro-milieu or growth of HPV-induced or transplanted tumors. Cell Rep (2018) 22(1):27–35. doi:10.1016/j.celrep.2017.12.010
48. Rees W, Bender J, Teague TK, Kedl RM, Crawford F, Marrack P, et al. An inverse relationship between T cell receptor affinity and antigen dose during CD4(+) T cell responses in vivo and in vitro. Proc Natl Acad Sci U S A (1999) 96(17):9781–6. doi:10.1073/pnas.96.17.9781
49. Dudeck J, Ghouse SM, Lehmann CH, Hoppe A, Schubert N, Nedospasov SA, et al. Mast-cell-derived TNF amplifies CD8(+) dendritic cell functionality and CD8(+) T cell priming. Cell Rep (2015) 13(2):399–411. doi:10.1016/j.celrep.2015.08.078
50. Gwinn WM, Johnson BT, Kirwan SM, Sobel AE, Abraham SN, Gunn MD, et al. A comparison of non-toxin vaccine adjuvants for their ability to enhance the immunogenicity of nasally-administered anthrax recombinant protective antigen. Vaccine (2013) 31(11):1480–9. doi:10.1016/j.vaccine.2013.01.012
51. Staats HF, Fielhauer JR, Thompson AL, Tripp AA, Sobel AE, Maddaloni M, et al. Mucosal targeting of a BoNT/A subunit vaccine adjuvanted with a mast cell activator enhances induction of BoNT/A neutralizing antibodies in rabbits. PLoS One (2011) 6(1):e16532. doi:10.1371/journal.pone.0016532
52. Hagan T, Nakaya HI, Subramaniam S, Pulendran B. Systems vaccinology: enabling rational vaccine design with systems biological approaches. Vaccine (2015) 33(40):5294–301. doi:10.1016/j.vaccine.2015.03.072
53. Pulendran B, Li S, Nakaya HI. Systems vaccinology. Immunity (2010) 33(4):516–29. doi:10.1016/j.immuni.2010.10.006
54. Yu Y, Blokhuis BR, Garssen J, Redegeld FA. Non-IgE mediated mast cell activation. Eur J Pharmacol (2016) 778:33–43. doi:10.1016/j.ejphar.2015.07.017
55. Wernersson S, Pejler G. Mast cell secretory granules: armed for battle. Nat Rev Immunol (2014) 14(7):478–94. doi:10.1038/nri3690
56. McGowen AL, Hale LP, Shelburne CP, Abraham SN, Staats HF. The mast cell activator compound 48/80 is safe and effective when used as an adjuvant for intradermal immunization with Bacillus anthracis protective antigen. Vaccine (2009) 27(27):3544–52. doi:10.1016/j.vaccine.2009.03.069
57. McNeil BD, Pundir P, Meeker S, Han L, Undem BJ, Kulka M, et al. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature (2015) 519(7542):237–41. doi:10.1038/nature14022
58. Dwyer DF, Barrett NA, Austen KF; Immunological Genome Project Consortium. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat Immunol (2016) 17(7):878–87. doi:10.1038/ni.3445
59. Bronner C, Wiggins C, Monté D, Märki F, Capron A, Landry Y, et al. Compound 48/80 is a potent inhibitor of phospholipase C and a dual modulator of phospholipase A2 from human platelet. Biochim Biophys Acta (1987) 920(3):301–5. doi:10.1016/0005-2760(87)90108-1
60. Schemann M, Kugler EM, Buhner S, Eastwood C, Donovan J, Jiang W, et al. The mast cell degranulator compound 48/80 directly activates neurons. PLoS One (2012) 7(12):e52104. doi:10.1371/journal.pone.0052104
61. Sayed BA, Walker ME, Brown MA. Cutting edge: mast cells regulate disease severity in a relapsing-remitting model of multiple sclerosis. J Immunol (2011) 186(6):3294–8. doi:10.4049/jimmunol.1003574
62. Secor VH, Secor WE, Gutekunst CA, Brown MA. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med (2000) 191(5):813–22. doi:10.1084/jem.191.5.813
63. Li H, Nourbakhsh B, Safavi F, Li K, Xu H, Cullimore M, et al. Kit (W-sh) mice develop earlier and more severe experimental autoimmune encephalomyelitis due to absence of immune suppression. J Immunol (2011) 187(1):274–82. doi:10.4049/jimmunol.1003603
64. Piconese S, Costanza M, Musio S, Tripodo C, Poliani PL, Gri G, et al. Exacerbated experimental autoimmune encephalomyelitis in mast-cell-deficient Kit W-sh/W-sh mice. Lab Invest (2011) 91(4):627–41. doi:10.1038/labinvest.2011.3
65. Bennett JL, Blanchet MR, Zhao L, Zbytnuik L, Antignano F, Gold M, et al. Bone marrow-derived mast cells accumulate in the central nervous system during inflammation but are dispensable for experimental autoimmune encephalomyelitis pathogenesis. J Immunol (2009) 182(9):5507–14. doi:10.4049/jimmunol.0801485
66. Cunin P, Penke LR, Thon JN, Monach PA, Jones T, Chang MH, et al. Megakaryocytes compensate for kit insufficiency in murine arthritis. J Clin Invest (2017) 127(5):1714–24. doi:10.1172/JCI84598
67. Zhou JS, Xing W, Friend DS, Austen KF, Katz HR. Mast cell deficiency in Kit(W-sh) mice does not impair antibody-mediated arthritis. J Exp Med (2007) 204(12):2797–802. doi:10.1084/jem.20071391
68. Schubert N, Dudeck J, Liu P, Karutz A, Speier S, Maurer M, et al. Mast cell promotion of T cell-driven antigen-induced arthritis despite being dispensable for antibody-induced arthritis in which T cells are bypassed. Arthritis Rheumatol (2015) 67(4):903–13. doi:10.1002/art.38996
69. Dellinger AL, Cunin P, Lee D, Kung AL, Brooks DB, Zhou Z, et al. Inhibition of inflammatory arthritis using fullerene nanomaterials. PLoS One (2015) 10(4):e0126290. doi:10.1371/journal.pone.0126290
70. Inglis JJ, Simelyte E, McCann FE, Criado G, Williams RO. Protocol for the induction of arthritis in C57BL/6 mice. Nat Protoc (2008) 3(4):612–8. doi:10.1038/nprot.2008.19
71. Pitman N, Asquith DL, Murphy G, Liew FY, McInnes IB. Collagen-induced arthritis is not impaired in mast cell-deficient mice. Ann Rheum Dis (2011) 70(6):1170–1. doi:10.1136/ard.2010.134528
72. Magnusson SE, Pejler G, Kleinau S, Abrink M. Mast cell chymase contributes to the antibody response and the severity of autoimmune arthritis. FASEB J (2009) 23(3):875–82. doi:10.1096/fj.08-120394
73. van der Velden D, Lagraauw HM, Wezel A, Launay P, Kuiper J, Huizinga TW, et al. Mast cell depletion in the preclinical phase of collagen-induced arthritis reduces clinical outcome by lowering the inflammatory cytokine profile. Arthritis Res Ther (2016) 18(1):138. doi:10.1186/s13075-016-1036-8
74. Martin SF. Allergic contact dermatitis: xenoinflammation of the skin. Curr Opin Immunol (2012) 24(6):720–9. doi:10.1016/j.coi.2012.08.003
Keywords: mast cells, adaptive immune responses, vaccination, adjuvant, compound 48/80
Citation: Schubert N, Lisenko K, Auerbach C, Weitzmann A, Ghouse SM, Muhandes L, Haase C, Häring T, Schulze L, Voehringer D, Gunzer F, Müller W, Feyerabend TB, Rodewald H-R, Dudeck A and Roers A (2018) Unimpaired Responses to Vaccination With Protein Antigen Plus Adjuvant in Mice With Kit-Independent Mast Cell Deficiency. Front. Immunol. 9:1870. doi: 10.3389/fimmu.2018.01870
Received: 13 April 2018; Accepted: 30 July 2018;
Published: 28 August 2018
Edited by:
Rashika El Ridi, Cairo University, EgyptReviewed by:
Beatrice Jahn-Schmid, Medizinische Universität Wien, AustriaCopyright: © 2018 Schubert, Lisenko, Auerbach, Weitzmann, Ghouse, Muhandes, Haase, Häring, Schulze, Voehringer, Gunzer, Müller, Feyerabend, Rodewald, Dudeck and Roers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Axel Roers, YXhlbC5yb2Vyc0B0dS1kcmVzZGVuLmRl
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.