 Rebecca A. Drummond
Rebecca A. Drummond Luis M. Franco2
Luis M. Franco2 Michail S. Lionakis
Michail S. Lionakis- 1Fungal Pathogenesis Section, Laboratory of Clinical Immunology and Microbiology (LCIM), National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Bethesda, MD, United States
- 2Laboratory of Immune System Biology (LISB), National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Bethesda, MD, United States
CARD9 is a signaling adaptor protein that is involved in the transduction of signals from a variety of innate pattern recognition receptors, including the C-type lectin receptors and intracellular NOD receptors and nucleic acid sensors. As a result, CARD9 has been shown in animal models to be an important regulator of immunity to bacteria, fungi, and viruses. Studies in humans with autosomal recessive CARD9 deficiency have indicated a highly specific role for this molecule in the activation of antifungal immune responses in the central nervous system, the oral mucosa, and the skin. Moreover, CARD9-dependent functions have recently been indicated to modulate the development of autoimmunity, inflammatory bowel diseases, and cancer. In this mini-review, we highlight the recent studies that have identified several novel functions of CARD9 in various disease contexts, and we summarize the contemporary understanding of the genetics and immunology of human CARD9 deficiency.
Introduction
Innate recognition of microbes by pattern recognition receptors (PRRs) is a critical first step in the defense against infection. To activate antimicrobial immunity, PRRs must initiate intracellular signaling cascades which control cellular responses, such as cytokine production, phagocytosis, and assembly of microbial killing complexes. Many PRRs use signaling molecules and adaptor proteins that are shared with other members of the same PRR family. As a result, deficiency of a shared signaling or adaptor protein often results in immune dysfunctions that are more severe than a deficiency in a single receptor and can have profound consequences for the control of infections (1).
Pathogenic fungi are predominantly recognized by the C-type lectin receptor (CLR) and toll-like receptor (TLR) families, of which many members couple to the signaling adaptor proteins CARD9 and MyD88, respectively, in order to activate their functions and initiate defense against fungal invasion (2). Although both CLRs and TLRs bind to and activate antifungal immune responses, the phenotypic consequences of human deficiencies in either of the two shared signaling adaptors have demonstrated that CARD9 plays a specific, superior role in the control of fungal diseases in humans compared to that of MyD88. Indeed, patients with genetic deficiency in CARD9 exhibit a primary immunodeficiency disorder (PID) which manifests as an extreme susceptibility to fungal infections, but not bacterial, viral, or parasitic infections, and is the only PID described to date that specifically predisposes to fungal diseases without other infectious or non-infectious sequelae (3). In contrast, MyD88 deficiency results in the development of life-threatening pyogenic bacterial infections without the spontaneous development of fungal disease (4). The highly specific susceptibility to fungi in CARD9-deficient patients further extends to the fungal species and organs affected, indicating that CARD9 is required for organ-specific antifungal immune responses of which we are only now beginning to define. Furthermore, animal models and genome-wide analyses of human single-nucleotide polymorphisms (SNPs) have indicated that CARD9 may also function to promote immunity to other pathogens and contribute toward autoimmune and hyperinflammatory disorders. Thus, CARD9 is a multi-functional signaling protein involved in many aspects of the immune system.
In this review, we discuss recent research that has unveiled the many possible functional roles of CARD9, predominantly defined using animal models. Finally, we discuss the genetics of human CARD9 deficiency and highlight the critical role for this molecule in antifungal immune surveillance in humans.
The Multi-Functional Roles of CARD9
CARD9 Is a Critical Activator of Antifungal Immune Responses
Many of the prototypical PRRs that recognize common components of fungal cell walls are members of the CLR family, and include Dectin-1, Dectin-2, Dectin-3, Mincle, and the Mannose Receptor (CD206). Many of these receptors initiate intracellular signaling cascades that are CARD9-dependent, the best studied of which is the Syk-dependent pathway downstream of Dectin-1 (Figure 1A). In brief, ligation of Dectin-1 by β-glucan (the fungal ligand for this receptor) results in recruitment of Syk kinase and the formation of the CBM signalosome, composed of CARD9, BCL10, and MALT1 (5). Activation of CARD9 function requires Vav proteins, of which there are three isoforms (Vav1, Vav2, and Vav3). Deletion of Vav1-3 in mice results in a phenocopy of CARD9-deficiency, indicating that Vav-mediated activation of CARD9 is a critical step in the induction of protective antifungal immunity. In line with this, human VAV3 SNPs are enriched in patient cohorts with candidemia (6). Activated CARD9 then leads to the production of inflammatory mediators, such as IL-6, IL-12, GM-CSF, TNF, and IL-1β via the activation of NFκB and ERK, the latter of which occurs via a RASGRF1–H-Ras pathway (7). These CARD9-dependent signaling pathways are regulated by Rubicon, a protein best known for its functions in autophagy (8). Rubicon competitively binds to CARD9 which results in the disassembly of the CBM complex, thus switching off signaling and preventing excessive inflammatory responses. However, modulation of Rubicon expression levels using lentiviral vectors in mice demonstrated that a reduction in Rubicon expression could help promote fungal clearance and survival by enhancing CARD9-dependent antifungal immune responses, at least in acute infection models (8). Compared to other PRR families, the molecular signaling events that occur upon CLR ligation are poorly defined. Yet, understanding these pathways will be critical to determine how iatrogenic interventions that manipulate these pathways affect vulnerable patients. For example, the recently introduced Syk inhibitors for hematological malignancies and graft-versus-host disease (9, 10), may predispose to the development of dangerous invasive fungal infections in patient cohorts already at risk for these diseases.
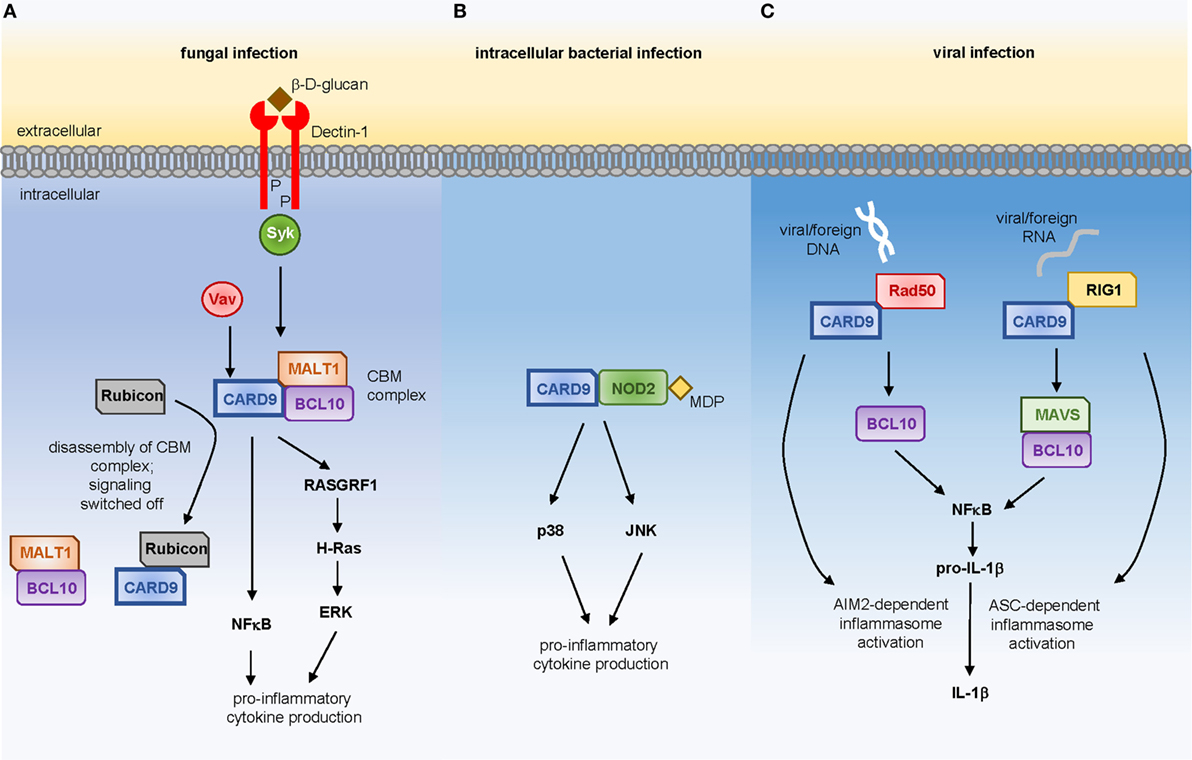
Figure 1. CARD9-dependent signaling in response to (A) fungal ligands via Dectin-1, (B) muramyl-dipeptide derived from intracellular bacterial pathogens, or (C) viral DNA or RNA.
Animals with a genetic deletion of Card9 are susceptible to challenge with a variety of fungal species, including Candida albicans, Aspergillus fumigatus, Cryptococcus neoformans, and some rarer dematiaceous fungi (11–14). In each of these models, Card9−/− mice generally exhibit reduced inflammatory cytokine production, which contributes to the inability to control fungal growth within infected organs. Protective antifungal cytokines that appear critically dependent on CARD9 function include IL-6, TNF, and IL-1β. In humans, CARD9 deficiency predisposes to a smaller range of pathogenic fungi (discussed below) indicating that CARD9-dependent functions in humans may be more context-dependent than in the mouse.
Despite the central position of CARD9 in the CLR signaling pathway, ablation of CARD9 function does not result in complete abrogation of antifungal immune responses (in either mice or human), indicating that CARD9-dependent mechanisms are required only for certain protective processes that may be specific to the cell type in question. Indeed, the dependency on CARD9 for NFκB activation has been shown to vary among different types of myeloid cells (7), including neutrophils, macrophages, and dendritic cells (DCs) (13). Neutrophils are the most important effector cell in the defense against systemic C. albicans infections, since neutropenia is a significant independent risk factor for the development of and suffering worse outcome from these infections in humans, and neutrophil depletion significantly reduces survival to C. albicans challenge in mice (2, 15). Many intrinsic neutrophil functions, including phagocytosis, production of reactive oxygen species (ROS), and chemotaxis appear largely independent of CARD9 for a variety of fungal species, shown with ex vivo studies using human neutrophils (16–19). In contrast, for ROS-independent fungal killing, CARD9 is required for the killing of unopsonized yeast cells as part of a PI3Kγ-dependent pathway, whereas killing of opsonized C. albicans yeast largely requires FcRγ and PKC and is independent of CARD9 (20). These two distinct fungal killing pathways utilized by neutrophils helps to explain the observation that neutrophils isolated from CARD9-deficient patients exhibit a selective killing defect toward unopsonized yeast (18, 19). This defect may partly contribute toward the fungal central nervous system (CNS)-tropism observed in CARD9-deficient patients (see below), since opsonization is naturally low in the CNS and thus CARD9-dependent killing of unopsonized cells would be particularly critical at this site. However, human neutrophil killing against the invasive filamentous Candida forms observed in tissue appears intact, whether opsonized or unopsonized (19). In addition, we and others have shown that a critical function of CARD9 in antifungal defense is promoting neutrophil recruitment specifically into fungal-infected organs via CXC chemokine production in both mice and humans (19, 21), which appears to largely contribute toward the organ-specific manifestation of fungal diseases observed in CARD9-deficient humans.
In experimental C. albicans CNS infections in mice, CARD9 promotes neutrophil recruitment in a fungal- and brain-specific manner (19). In this situation, CARD9 is required for the appropriate induction of CXC chemokines in the CNS by resident macrophages (i.e., microglia) and glia cells, as well as recruited neutrophils. Therefore, CARD9 controls local inflammatory chemokine production in addition to a neutrophil-intrinsic positive feedback chemotaxis loop in the brain (19). CARD9-dependent production of neutrophil-attracting chemokines is also evident during pulmonary infection with A. fumigatus in mice (14), and we recently reported CARD9-deficient patients who developed extrapulmonary A. fumigatus infection that was associated with a lack of neutrophil accumulation at the infected site (21). Moreover, reduced production of CXC chemokines resulting in a lack of neutrophil recruitment has been described in chronically fungal-infected subcutaneous tissue in experimental phaeohyphomycosis models (22). Therefore, CARD9 appears to be a central regulator of neutrophil recruitment to specific organs during invasive fungal infection.
CARD9 and Anti-Bacterial Immunity
Stimulation of Card9−/− myeloid cells with purified bacterial products has revealed that CARD9 is required for inflammatory cytokine production in response to specific bacterial stimuli. IL-6 production following stimulation with peptidoglycan or muramyl-dipeptide (MDP), predominant components of Gram-positive bacterial cell walls, is highly dependent on CARD9 whereas responses to LPS are CARD9-independent (23). Recognition of MDP is largely controlled by the intracellular NOD2 receptor, to which CARD9 couples in order to drive activation of p38 and JNK kinases (Figure 1B), thus promoting immunity to intracellular bacterial pathogens including Listeria monocytogenes (23) and Mycobacterium tuberculosis (24).
In addition to activating innate immune responses to intracellular bacteria, other studies have indicated that CARD9 may be involved in the induction of adaptive immunity to these pathogens. Mouse T-cells specific for flagellin were shown to require Syk-CARD9 signaling for their activation, which was mechanistically linked to TLR5-dependent antigen presentation by CARD9-expressing DCs (25). Polarization to the Th17 lineage also depends on CARD9 in the context of gastrointestinal (GI) bacterial infection (26), similar to what has been described for the generation of Th17 responses in the fungal-infected oral mucosa (27). Moreover, humoral immunity mediated by B-cells has also been shown to be affected by deletion of Card9, since bacterial-specific IgG production during GI infection is significantly reduced in Card9−/− animals (28). Thus, animal models have demonstrated that Card9 can play distinct protective roles in immunity to intracellular bacterial pathogens, however, the relevance of these functions in humans remains to be determined given that CARD9-deficient patients do not appear susceptible to bacterial infections.
CARD9 and Antiviral Immunity
Studies delineating the molecular pathways controlling IL-1β activation and secretion by myeloid cells in response to viral infection have identified CARD9 as a key regulator of these responses (Figure 1C). Following viral infection or transfection with foreign DNA, CARD9 directly interacts with Rad50, a cytosolic DNA sensor, and these CARD9-Rad50 complexes subsequently recruit BCL10 to promote IL-1β secretion. Using a vaccinia virus infection model, Card9−/− animals were shown to produce reduced levels of IL-1β following infection which negatively impacted the ability of these animals to generate an appropriate antiviral CD8+ T-cell response (29). CARD9 is also required for immunity to retroviruses by transducing RIG-1-dependent signals via MAVS and BCL10 to activate NFκB and transcription of pro-IL-1β, which in turn is processed into active IL-1β by ASC-dependent activation of the inflammasome (30). As shown for antifungal immunity, Rubicon can also regulate these CARD9-dependent antiviral functions, since enhancing Rubicon expression levels was found to significantly skew host immunity in favor of pathogen resistance (8).
CARD9 and Autoimmunity
Recent studies have demonstrated potential novel roles of CARD9 in autoimmune diseases using animal models, providing interesting new insights into how this protein functions in organ-specific inflammation. During experimental uveitis, Mincle-mediated activation of CARD9 is required for the recruitment of pathogenic Th1 and Th17 cells (31), and it was later shown that fungal antigens could exacerbate the development of this disease and this occurred via Dectin-2-CARD9 signaling (32). In line with the critical role for CARD9 in neutrophil recruitment, Card9−/− animals showed reduced production of neutrophil-targeted chemokines, such as CXCL1, CXCL2, and CCL3, resulting in decreased accumulation of neutrophils to the arthritic joint (33). As a result, Card9−/− animals are protected from autoantibody-induced arthritis, and this protection was mapped to neutrophils using neutrophil-specific Card9-deficient mice (33). Population-based human studies have indicated that CARD9 genetic variation may modulate the risk of development of inflammatory bowel diseases (IBD; discussed below), ankylosing spondylitis (34), IgA nephropathy (35), and primary sclerosing cholangitis (36), and more studies are required to determine the role of the CLR/CARD9 axis in organ-specific autoimmune disease development.
CARD9 and Cancer
CARD9-dependent signaling in tumor development and metastasis has been revealed in several recent studies using animal models and cell lines. Patients with IBD are at risk for the development of colonic cancers. Human SNPs in CARD9 have been identified as risk factors for IBD by several GWAS-based studies (discussed below), and animal models have shown that CARD9 promotes the production of pro-inflammatory cytokines IL-1β and IL-22 in the gut during active colitis, which in turn drives colonic tumor growth (37). Other work has additionally shown that metastasis of colonic tumor cells to the liver depends on CARD9 signaling. CARD9 is highly expressed by tumor-infiltrating macrophages and has been shown to be a critical modulator of the polarization of these cells toward a highly inflammatory metastatic-inducing phenotype (38).
In addition to colonic cancer, CARD9 signaling has also been implicated in the inappropriate activation of renal cell carcinoma (RCC) cells. Mutation affecting the tumor suppressor gene VHL (most often somatic, but occasionally involving germline cells) is a major hallmark of RCC, which in turn causes inappropriate activation of NFκB and c-Jun. CARD9 is a mechanistic link between VHL inactivation and the activation of these inflammatory transcription factors (39, 40). Direct interaction between CARD9 and VHL is required for C-terminal phosphorylation of CARD9 by the kinase CK2, which limits CARD9 activity and NFκB activation. Reducing CARD9 expression with a silencing RNA approach in VHL−/− cells lowered NFκB activity to wild-type levels, and significantly reduced the tumorigenic potential of these cells (40). Similarly, the loss of VHL promotes an additional pro-inflammatory CARD9-dependent pathway that results in the activation of JNK signaling and c-Jun activity (39). Collectively, these studies provide the intriguing possibility that inappropriate CARD9 activation contributes to the development of certain cancers and may be a novel target of future therapies for these diseases.
The Genetics and Clinical Spectrum of Human CARD9 Mutations
Despite the wide range of CARD9-dependent functions identified in animal models and the potential influence of this protein in several disease scenarios, loss-of-function mutations in human CARD9 have unequivocally demonstrated the importance of CARD9 in antifungal immunity. Human CARD9 deficiency is characterized by the spontaneous development of fungal infections that predominantly localize to the oral mucosa, CNS, bone, and subcutaneous tissues, and often involves specific families of pathogenic fungi including Candida species (CNS, bone, and mucosal disease) and dark-walled molds and yeast-like fungi (e.g., Aspergillus, Exophiala, and Phialophora) that localize to the CNS, skin, bone, and abdominal organs (3, 21, 41–43). The underlying mechanisms that cause susceptibility to fungal diseases in CARD9-deficient patients is not well understood, and could be related to the poor production of inflammatory cytokines and chemokines in response to fungal agonists (13, 18, 19). Indeed, a cohort of French-Canadian CARD9-deficient patients were successfully treated with recombinant GM-CSF therapy, which the authors show corrected defective GM-CSF responses and ERK signaling in CARD9-deficient myeloid cells (43), while another study used G-CSF therapy to correct defective IL-17 responses in a CARD9-deficient patient (44). These studies indicate that replacing cytokines that are classically associated with antifungal defense may be an appropriate therapy option for human CARD9 deficiency. However, we recently showed that these approaches may not be effective for all patients which are potentially related to the variety of CARD9 mutations observed (see below). Our data indicated that although different CARD9 mutations disrupt GM-CSF production by myeloid cells, the impact of these mutations on ERK activation varied with mutation and appeared to correlate with the efficacy of GM-CSF therapy in these patients (45). Thus, treatment options for human CARD9-deficiency are still limited, and further investigation into the mechanisms that cause fungal susceptibility in these patients is warranted. In addition, the organ- and fungal species-specific nature of CARD9-deficiency disease is also poorly understood. As discussed above, we and others have shown that defects in neutrophil recruitment are likely the major contributing factor toward the organ-specific nature of the disease, however, other contributing roles of tissue-resident macrophages, stromal cells, and/or additional functional deficits in recruited inflammatory cells remain to be fully explored.
Although many CARD9-deficient patients have similar clinical presentations, there is diversity in the genetic mutations underlying the condition [see Ref. (3) for a table of all reported human CARD9 mutations]. More than 15 missense and nonsense mutations in CARD9 have now been described in the coiled-coil and CARD domains, as well as the promoter region (3). Inactivation of both alleles appears to be necessary for the occurrence of disease, so the condition follows an autosomal recessive mode of inheritance. De novo variants in CARD9 have also been reported in patients with debilitating fungal infections of the eyes, bone, and skin (46, 47). Interestingly, although some of the reported CARD9 mutations have been identified in unrelated patients, there are instances where the same mutation does not give rise to a similar clinical phenotype (3). For example, homozygosity for the Q298X mutation has been reported to give rise to Candida meningoencephalitis and deep dermatophytosis in different patients (41, 48), while patients with very similar clinical presentations can have mutations in different parts of the CARD9 gene (47). Therefore, it is currently unclear which mutations predispose to which clinical phenotypes and whether there is any overlap, and this will require more clinical descriptions of CARD9-deficient patients and further investigations into CARD9-dependent immune signaling and functions, especially in humans.
Like many other genes that are associated with Mendelian traits, human CARD9 is intolerant to sequence variation. To quantify gene-level intolerance to functional genetic variation, Petrovski et al. proposed a Residual Variation Intolerance Score (49). Of the nine CARD genes for which a score was calculated in that study, only three (CARD9, CARD10, and CARD11) had negative scores, suggesting low tolerance to functional genetic variation. In fact, CARD9 had lower tolerance to variation than 82% of the 16,956 genes for which a score was calculated in the study.
In addition to deleterious mutations which predispose to fungal infection, there have been reports of genetic variants of CARD9 in humans that associate with autoimmune disease. The CARD9 single-nucleotide polymorphism S12N (rs4077515) was recently shown to be highly enriched in patients with allergic bronchopulmonary aspergillosis, which the authors linked to RelB activation by this CARD9 variant that subsequently activated IL-5 production by alveolar macrophages and drove pathogenic eosinophil recruitment and Th2 responses within the lung (50). Another example is the CARD9 variant in which the C-terminal region is truncated (51, 52). One of these truncated variants of CARD9 (S12NΔ11 or CARD9Δ11) is strongly associated with protection against IBD, which is mechanistically linked to the inability of these variants to be activated by TRIM62, a ubiquitin ligase that is required for CARD9 activation and subsequent pro-inflammatory cytokine production. While the lack of TRIM62-mediated activation is protective in the context of inappropriate intestinal inflammation and may represent a target for therapeutic intervention in IBD (53), these mutations would be predicted to negatively affect antifungal immunity, since Trim62−/− animals are unable to control fungal growth and succumb to infection significantly faster compared to their wild-type counterparts (52). Intriguingly, in human cell lines, CARD9Δ11 variants act in a dominant-negative fashion (52), raising the possibility of the existence of humans who may be heterozygous for dominant-negative CARD9 mutations in the C-terminal region and yet may present with functional CARD9-deficiency. This possibility should be considered by clinicians looking for the underlying cause of an extreme fungal infection in an otherwise immunocompetent patient.
While the CARD9Δ11 variant is protective in IBD, other lines of evidence suggest a role for CARD9 in the pathogenesis of the disease. Specifically, there are other CARD9 variants that are enriched in patient cohorts with IBD (51) indicating that dysregulated CARD9 function can have profound consequences for immune homeostasis in the gut. The CARD9 human SNP, S12N, is associated with increased CARD9 mRNA expression and IBD development (51, 54), but not fungemia (55), thus supporting the notion that CARD9-dependent functions in the immune system are context-dependent. However, many studies analyzing the functions of CARD9 in the context of GI inflammation have discovered links between the development of GI disorders and fungal commensals, indicating that CARD9-dependent functions in innate fungal defense may also be important in gut-related diseases. For example, CARD9 deficiency has been associated with an over-representation of fungi within the microbiota (56), and there have been reports of CARD9-deficient patients developing colitis caused by invasive intestinal infection with Candida glabrata (48), and most recently by the β-glucan-containing microalgae Prototheca zopfii (57). These studies suggest that, at least partially, CARD9 may function to control fungal microbes in the gut and prevent infection-related disease in these tissues, which might explain the strong association between human CARD9 genetic variants and the development of IBD-related disorders.
Concluding Remarks
The significance of CARD9 and its signaling partners in antifungal defense has been an important realization for the field in the past two decades. A comprehensive understanding of the CARD9-dependent signaling pathways and their relevance for disease processes will be required to (1) utilize CARD9 as a future therapeutic target and (2) prevent and treat possible side-effects of immune-based therapies for cancer and autoimmunity. In particular, the non-fungal-related functions of CARD9 warrant further study, especially to understand the relevance of these functions in humans who carry genetic variants of CARD9, which are currently only partially defined yet have the potential to significantly influence CARD9-dependent functions. The study of human CARD9 deficiency has yielded novel insights into how this adaptor signaling molecule functions to protect against invasive fungal diseases and, importantly, these findings point to potential new avenues for the development of immune-based treatments for invasive fungal infections, which represent a global clinical challenge.
Author Contributions
All authors contributed in writing the mini-review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Disease, National Institutes of Health.
References
1. Lionakis MS, Netea MG, Holland SM. Mendelian genetics of human susceptibility to fungal infection. Cold Spring Harb Perspect Med (2014) 4:1–21. doi:10.1101/cshperspect.a019638
2. Lionakis MS, Iliev ID, Hohl TM. Immunity against fungi. JCI Insight (2017) 2(11):e93156. doi:10.1172/jci.insight.93156
3. Drummond RA, Lionakis MS. Mechanistic insights into the role of C-type lectin receptor/CARD9 signaling in human antifungal immunity. Front Cell Infect Microbiol (2016) 6:39. doi:10.3389/fcimb.2016.00039
4. von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science (2008) 321:691–6. doi:10.1126/science.1158298
5. Drummond RA, Saijo S, Iwakura Y, Brown GD. The role of Syk/CARD9 coupled C-type lectins in antifungal immunity. Eur J Immunol (2011) 41:276–81. doi:10.1002/eji.201041252
6. Roth S, Bergmann H, Jaeger M, Yeroslaviz A, Neumann K, Koenig P-A, et al. Vav proteins are key regulators of Card9 signaling for innate antifungal immunity. Cell Rep (2016) 17:2572–83. doi:10.1016/j.celrep.2016.11.018
7. Jia X-M, Tang B, Zhu L-L, Liu Y-H, Zhao X-Q, Gorjestani S, et al. CARD9 mediates Dectin-1-induced ERK activation by linking Ras-GRF1 to H-Ras for antifungal immunity. J Exp Med (2014) 211:2307–21. doi:10.1084/jem.20132349
8. Yang C-S, Rodgers M, Min C-K, Lee J-S, Kingeter L, Lee J-Y, et al. The autophagy regulator rubicon is a feedback inhibitor of CARD9-mediated host innate immunity. Cell Host Microbe (2012) 11:277–89. doi:10.1016/j.chom.2012.01.019
9. Liu D, Mamorska-Dyga A. Syk inhibitors in clinical development for hematological malignancies. J Hematol Oncol (2017) 10:145. doi:10.1186/s13045-017-0512-1
10. Flynn R, Allen JL, Luznik L, MacDonald KP, Paz K, Alexander KA, et al. Targeting Syk-activated B cells in murine and human chronic graft-versus-host disease. Blood (2015) 125:4085–94. doi:10.1182/blood-2014-08-595470
11. Yamamoto H, Nakamura Y, Sato K, Takahashi Y, Nomura T, Miyasaka T, et al. Defect of CARD9 leads to impaired accumulation of gamma interferon-producing memory phenotype T cells in lungs and increased susceptibility to pulmonary infection with Cryptococcus neoformans. Infect Immun (2014) 82:1606–15. doi:10.1128/IAI.01089-13
12. Wu W, Zhang R, Wang X, Song Y, Liu Z, Han W, et al. Impairment of immune response against dematiaceous fungi in Card9 knockout mice. Mycopathologia (2016) 181(9–10):631–42. doi:10.1007/s11046-016-0029-0
13. Gross O, Gewies A, Finger K, Schafer M, Sparwasser T, Peschel C, et al. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature (2006) 442:651–6. doi:10.1038/nature04926
14. Jhingran A, Kasahara S, Shepardson KM, Junecko BAF, Heung LJ, Kumasaka DK, et al. Compartment-specific and sequential role of MyD88 and CARD9 in chemokine induction and innate defense during respiratory fungal infection. PLoS Pathog (2015) 11:e1004589. doi:10.1371/journal.ppat.1004589
15. Lionakis MS, Levitz SM. Host control of fungal infections: lessons from basic studies and human cohorts. Annu Rev Immunol (2018) 36:157–91. doi:10.1146/annurev-immunol-042617-053318
16. Li X, Cullere X, Nishi H, Saggu G, Durand E, Mansour MK, et al. PKC-δ activation in neutrophils promotes fungal clearance. J Leukoc Biol (2016) 100(3):581–8. doi:10.1189/jlb.4A0915-405R
17. Liang P, Wang X, Wang R, Wan Z, Han W, Li R. CARD9 deficiencies linked to impaired neutrophil functions against Phialophora verrucosa. Mycopathologia (2015) 179(5–6):347–57. doi:10.1007/s11046-015-9877-2
18. Drewniak A, Gazendam RP, Tool ATJ, van Houdt M, Jansen MH, van Hamme JL, et al. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood (2013) 121:2385–92. doi:10.1182/blood-2012-08-450551
19. Drummond RA, Collar AL, Swamydas M, Rodriguez CA, Lim JK, Mendez LM, et al. CARD9-dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PLoS Pathog (2015) 11:e1005293. doi:10.1371/journal.ppat.1005293
20. Gazendam RP, van Hamme JL, Tool ATJ, van Houdt M, Verkuijlen PJJH, Herbst M, et al. Two independent killing mechanisms of Candida albicans by human neutrophils: evidence from innate immunity defects. Blood (2014) 124:590–7. doi:10.1182/blood-2014-01-551473
21. Rieber N, Gazendam RP, Freeman AF, Hsu AP, Collar AL, Sugui JA, et al. Extrapulmonary Aspergillus infection in patients with CARD9 deficiency. JCI Insight (2016) 1(17):e89890. doi:10.1172/jci.insight.89890
22. Wang X, Zhang R, Wu W, Song Y, Wan Z, Han W, et al. Impaired specific antifungal immunity in CARD9-deficient patients with Phaeohyphomycosis. J Invest Dermatol (2018) 138:607–17. doi:10.1016/j.jid.2017.10.009
23. Hsu Y-MS, Zhang Y, You Y, Wang D, Li H, Duramad O, et al. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol (2007) 8:198–205. doi:10.1038/ni1426
24. Dorhoi A, Desel C, Yeremeev V, Pradl L, Brinkmann V, Mollenkopf HJ, et al. The adaptor molecule CARD9 is essential for tuberculosis control. J Exp Med (2010) 207:777–92. doi:10.1084/jem.20090067
25. Atif SM, Lee S-J, Li L-X, Uematsu S, Akira S, Gorjestani S, et al. Rapid CD4(+) T-cell responses to bacterial flagellin require dendritic cell expression of Syk and CARD9. Eur J Immunol (2015) 45:513–24. doi:10.1002/eji.201444744
26. Sokol H, Conway KL, Zhang M, Choi M, Morin B, Cao Z, et al. Card9 mediates intestinal epithelial cell restitution, T-helper 17 responses, and control of bacterial infection in mice. Gastroenterology (2013) 145:591–601. doi:10.1053/j.gastro.2013.05.047
27. Bishu S, Hernández-Santos N, Simpson-Abelson MR, Huppler AR, Conti HR, Ghilardi N, et al. The adaptor CARD9 is required for adaptive but not innate immunity to oral mucosal Candida albicans infections. Infect Immun (2014) 82:1173–80. doi:10.1128/IAI.01335-13
28. Lamas B, Michel M-L, Waldschmitt N, Pham H-P, Zacharioudaki V, Dupraz L, et al. Card9 mediates susceptibility to intestinal pathogens through microbiota modulation and control of bacterial virulence. Gut (2017) 1–9. doi:10.1136/gutjnl-2017-314195
29. Roth S, Rottach A, Lotz-Havla AS, Laux V, Muschaweckh A, Gersting SW, et al. Rad50-CARD9 interactions link cytosolic DNA sensing to IL-1[beta] production. Nat Immunol (2014) 15:538–45. doi:10.1038/ni.2888
30. Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1β production. Nat Immunol (2009) 11:63. doi:10.1038/ni.1824
31. Lee EJ, Brown BR, Vance EE, Snow PE, Silver PB, Heinrichs D, et al. Mincle activation and the Syk/Card9 signaling axis are central to the development of autoimmune disease of the eye. J Immunol (2016) 196(7):3148–58. doi:10.4049/jimmunol.1502355
32. Brown BR, Lee EJ, Snow PE, Vance EE, Iwakura Y, Ohno N, et al. Fungal-derived cues promote ocular autoimmunity through a Dectin-2/Card9-mediated mechanism. Clin Exp Immunol (2017) 190:293–303. doi:10.1111/cei.13021
33. Nemeth T, Futosi K, Sitaru C, Ruland J, Mocsai A. Neutrophil-specific deletion of the CARD9 gene expression regulator suppresses autoantibody-induced inflammation in vivo. Nat Commun (2016) 7:11004. doi:10.1038/ncomms11004
34. Pointon JJ, Harvey D, Karaderi T, Appleton LH, Farrar C, Stone MA, et al. Elucidating the chromosome 9 association with AS; CARD9 is a candidate gene. Genes Immun (2010) 11:490–6. doi:10.1038/gene.2010.17
35. Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet (2014) 46:1187–96. doi:10.1038/ng.3118
36. Janse M, Lamberts LE, Franke L, Raychaudhuri S, Ellinghaus E, MuriBoberg K, et al. Three ulcerative colitis susceptibility loci are associated with primary sclerosing cholangitis and indicate a role for IL2, REL and CARD9. Hepatology (2011) 53:1977–85. doi:10.1002/hep.24307
37. Bergmann H, Roth S, Pechloff K, Kiss EA, Kuhn S, Heikenwalder M, et al. Card9-dependent IL-beta regulates IL-22 production from group 3 innate lymphoid cells and promotes colitis-associated cancer. Eur J Immunol (2017) 47:1342–53. doi:10.1002/eji.201646765
38. Yang M, Shao JH, Miao YJ, Cui W, Qi YF, Han JH, et al. Tumor cell-activated CARD9 signaling contributes to metastasis-associated macrophage polarization. Cell Death Differ (2014) 21:1290–302. doi:10.1038/cdd.2014.45
39. An JB, Liu HR, Magyar CE, Guo YC, Veena MS, Srivatsan ES, et al. Hyperactivated JNK is a therapeutic target in pVHL-deficient renal cell carcinoma. Cancer Res (2013) 73:1374–85. doi:10.1158/0008-5472.CAN-12-2362
40. Yang H, Minamishima YA, Yan Q, Schlisio S, Ebert BL, Zhang X, et al. pVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF-kappa B agonist Card9 by CK2. Mol Cell (2007) 28:15–27. doi:10.1016/j.molcel.2007.09.010
41. Lanternier F, Pathan S, Vincent QB, Liu L, Cypowyj S, Prando C, et al. Deep dermatophytosis and inherited CARD9 deficiency. N Engl J Med (2013) 369:1704–14. doi:10.1056/NEJMoa1208487
42. Glocker EO, Hennigs A, Nabavi M, Schaffer AA, Woellner C, Salzer U. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med (2009) 361:1727–35. doi:10.1056/NEJMoa0810719
43. Gavino C, Hamel N, Zeng JB, Legault C, Guiot M-C, Chankowsky J, et al. Impaired RASGRF1/ERK-mediated GM-CSF response characterizes CARD9 deficiency in French-Canadians. J Allergy Clin Immunol (2015) 137(4):1178.e–88.e. doi:10.1016/j.jaci.2015.09.016
44. Celmeli F, Oztoprak N, Turkkahraman D, Seyman D, Mutlu E, Frede N, et al. Successful granulocyte colony stimulating factor treatment of relapsing Candida albicans meningoencephalitis caused by CARD9 deficiency. Pediatr Infect Dis J (2015) 35(4):428–31. doi:10.1097/inf.0000000000001028
45. Drummond RA, Zahra FT, Natarajan M, Swamydas M, Hsu AP, Wheat LJ, et al. GM-CSF therapy in human CARD9 deficiency. J Allergy ClinImmunol (2018). doi:10.1016/j.jaci.2018.05.025
46. Jones N, Garcez T, Newman W, Denning D. Endogenous Candida endophthalmitis and osteomyelitis associated with CARD9 deficiency. BMJ Case Rep (2016) 2016:1–3. doi:10.1136/bcr-2015-214117
47. Wang X, Wang W, Lin Z, Wang X, Li T, Yu J, et al. CARD9 mutations linked to subcutaneous phaeohyphomycosis and TH17 cell deficiencies. J Allergy Clin Immunol (2014) 133:905.e–8.e. doi:10.1016/j.jaci.2013.09.033
48. Lanternier F, Mahdaviani SA, Barbati E, Chaussade H, Koumar Y, Levy R, et al. Inherited CARD9 deficiency in otherwise healthy children and adults with Candida species-induced meningoencephalitis, colitis, or both. J Allergy Clin Immunol (2015) 135:1558–68. doi:10.1016/j.jaci.2014.12.1930
49. Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet (2013) 9:e1003709. doi:10.1371/journal.pgen.1003709
50. Xu X, Xu J-F, Zheng G, Lu H-W, Duan J-L, Rui W, et al. CARD9S12N facilitates the production of IL-5 by alveolar macrophages for the induction of type 2 immune responses. Nat Immunol (2018) 19:547–60. doi:10.1038/s41590-018-0112-4
51. Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet (2011) 43:1066–U50. doi:10.1038/ng.952
52. Cao Z, Conway KL, Heath RJ, Rush JS, Leshchiner ES, Ramirez-Ortiz ZG, et al. Ubiquitin ligase TRIM62 regulates CARD9-mediated anti-fungal immunity and intestinal inflammation. Immunity (2015) 43:715–26. doi:10.1016/j.immuni.2015.10.005
53. Leshchiner ES, Rush JS, Durney MA, Cao Z, Dančík V, Chittick B, et al. Small-molecule inhibitors directly target CARD9 and mimic its protective variant in inflammatory bowel disease. Proc Natl Acad Sci U S A (2017) 114:11392–7. doi:10.1073/pnas.1705748114
54. Beaudoin M, Goyette P, Boucher G, Lo KS, Rivas MA, Stevens C, et al. Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet (2013) 9(9):e1003723. doi:10.1371/journal.pgen.1003723
55. Rosentul DC, Plantinga TS, Oosting M, Scott WK, Edwards DRV, Smith PB, et al. Genetic variation in the Dectin-1/CARD9 recognition pathway and susceptibility to candidemia. J Infect Dis (2011) 204:1138–45. doi:10.1093/infdis/jir458
56. Lamas B, Richard ML, Leducq V, Pham H-P, Michel M-L, Da Costa G, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med (2016) 22(6):598–605. doi:10.1038/nm.4102
Keywords: CARD9, fungi, primary immunodeficiency, C-type lectin receptors, candidiasis, neutrophils
Citation: Drummond RA, Franco LM and Lionakis MS (2018) Human CARD9: A Critical Molecule of Fungal Immune Surveillance. Front. Immunol. 9:1836. doi: 10.3389/fimmu.2018.01836
Received: 01 May 2018; Accepted: 25 July 2018;
Published: 06 August 2018
Edited by:
Frederic Bornancin, Novartis (Switzerland), SwitzerlandReviewed by:
Yuval Itan, Icahn School of Medicine at Mount Sinai, United StatesRuben Martinez-Barricarte, Rockefeller University, United States
Copyright: © 2018 Drummond, Franco and Lionakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michail S. Lionakis, bGlvbmFraXNtQG5pYWlkLm5paC5nb3Y=
†Present address: Rebecca A. Drummond, Institute of Immunology and Immunotherapy, Institute of Microbiology and Infection, University of Birmingham, Birmingham, United Kingdom