 Janine E. Melsen1*
Janine E. Melsen1* Gertjan Lugthart1
Gertjan Lugthart1 Carly Vervat1Szymon M. Kielbasa2
Carly Vervat1Szymon M. Kielbasa2 Sander A. J. van der Zeeuw2Henk P. J. Buermans3Monique M. van Ostaijen-ten Dam1
Sander A. J. van der Zeeuw2Henk P. J. Buermans3Monique M. van Ostaijen-ten Dam1 Arjan C. Lankester1
Arjan C. Lankester1 Marco W. Schilham1
Marco W. Schilham1
- 1Department of Pediatrics, Leiden University Medical Center, Leiden, Netherlands
- 2Department of Biomedical Data Sciences, Leiden University Medical Center, Leiden, Netherlands
- 3Department of Human Genetics, Leiden Genome Technology Center, Leiden University Medical Center, Leiden, Netherlands
Human lymphoid tissues harbor, in addition to CD56bright and CD56dim natural killer (NK) cells, a third NK cell population: CD69+CXCR6+ lymphoid tissue (lt)NK cells. The function and development of ltNK cells remain poorly understood. In this study, we performed RNA sequencing on the three NK cell populations derived from bone marrow (BM) and blood. In ltNK cells, 1,353 genes were differentially expressed compared to circulating NK cells. Several molecules involved in migration were downregulated in ltNK cells: S1PR1, SELPLG and CD62L. By flow cytometry we confirmed that the expression profile of adhesion molecules (CD49e−, CD29low, CD81high, CD62L−, CD11c−) and transcription factors (Eomeshigh, Tbetlow) of ltNK cells differed from their circulating counterparts. LtNK cells were characterized by enhanced expression of inhibitory receptors TIGIT and CD96 and low expression of DNAM1 and cytolytic molecules (GZMB, GZMH, GNLY). Their proliferative capacity was reduced compared to the circulating NK cells. By performing gene set enrichment analysis, we identified DUSP6 and EGR2 as potential regulators of the ltNK cell transcriptome. Remarkably, comparison of the ltNK cell transcriptome to the published human spleen-resident memory CD8+ T (Trm) cell transcriptome revealed an overlapping gene signature. Moreover, the phenotypic profile of ltNK cells resembled that of CD8+ Trm cells in BM. Together, we provide transcriptional and phenotypic data that clearly distinguish ltNK cells from both the CD56bright and CD56dim NK cells and substantiate the view that ltNK cells are tissue-resident cells, which are functionally restrained in killing and have low proliferative activity.
Introduction
The recent identification of tissue-resident lymphocytes in both human and mice contributes to our extending knowledge on the heterogeneity of innate and adaptive lymphocyte populations. One of the key markers expressed by the majority of these non-circulating lymphocytes is CD69, previously known as an early activation marker. Nowadays, it has become clear that CD69 is associated with tissue-residency because it promotes internalization of sphingosine-1-phosphate receptor 1 (S1PR1) (1–3). Expression of S1PR1 enables migration of lymphocytes towards the S1P gradient, which is higher in blood compared to tissues (4, 5).
Subsets of the human innate natural killer (NK) cells exhibit a tissue-resident phenotype as demonstrated in various organs, including the uterus, liver, tonsils, bone marrow (BM), spleen, and lymph nodes (6–12). CD69+ tissue-resident NK cells are characterized by a distinct chemokine receptor and adhesion molecule repertoire (13, 14). For instance, CXCR6 is highly expressed by tissue-resident NK cells in lymphoid tissues and liver, while CD49a is characteristic for uterus-resident NK cells (6, 7, 9, 12). The remaining non-resident (circulating) NK cells are subdivided into two populations based on CD56 and CD16 expression: CD56brightCD16−/+ and CD56dimCD16+ (15). The CD56brightCD16−/+ NK cells are potent cytokine producers (IFN-γ, TNF-α), while the CD56dimCD16+ NK cells have a high capacity to kill infected and transformed cells (16–18). The function of tissue-resident NK cells is still unknown, since they are neither good producers of IFN-γ nor potent killers (7, 12).
Among the human tissue-resident lymphocytes of the adaptive immune system, the tissue-resident memory (Trm) cells are extensively investigated and have been identified in skin, spleen, tonsil, lung, liver, lymph node, salivary glands, and intestines (19–25). Trm cells exhibit a unique molecular program, which distinguishes them from their circulating counterparts (22, 23, 25–29). Local environmental cues appear to play a role in the development of Trm; numerous murine studies provided evidence for a developmental pathway in which KLRG1− CD8+ effector T cells give rise to Trm cells under influence of local cues, such as TGF-β and IL-15 (27, 30–32). In contrast to the Trm cells, transcriptional regulation and developmental requirements for tissue-resident NK cells are poorly understood.
In this study, we demonstrated that lymphoid tissue-resident (lt)NK cells in BM possess a transcriptional profile, which distinguished them from the two circulating NK cell subsets. In addition, by the use of published data, we found that lymphoid tissue-resident CD69+CD8+ Trm cells share a transcriptional and phenotypic profile with ltNK cells. Together, we provide a comprehensive molecular framework of the conventional CD56bright and CD56dim NK cells as well as the tissue-resident ltNK cells and provide a core gene signature, which might be involved in promoting tissue-residency.
Materials and Methods
Tissue and Ethics Statement
With approval of the institutional review board (P08.001), residual paired BM and blood samples from four healthy BM donors were used for RNA sequencing after informed consent was provided. Validation of the RNA sequence data by flow cytometry was performed by analyzing residual BM samples of healthy donors (n = 18) and was evaluated anonymously in accordance with Dutch national ethical and professional guidelines (http://www.federa.org).
Purification of NK Cell Populations
Mononuclear cells (MNCs) were isolated by Ficoll-Isopaque (Leiden University Medical Center Pharmacy, Leiden, Netherlands) density gradient centrifugation, and NK cells were enriched using the MACS untouched NK cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to manufacturers’ instructions. Lymphocytes were stained for surface markers with the antibodies listed in Table S1 in Supplementary Material. The detailed staining procedure is provided in the supplemental methods. The following NK cell populations were isolated from the BM: NKG2A+ ltNK cells, NKG2A− ltNK cells, CD56bright and CD56dim NK cells. From blood, the CD56bright, NKG2A+CD56dim, and NKG2A−CD56dim NK cells were isolated. The populations were FACS-purified on an ARIAIII cell sorter [Becton Dickinson (BD), Franklin Lakes, NJ, USA]. The gating strategy is depicted in Figure 1A. The NK cell populations were collected in NucleoSpin RA1 lysis buffer (Machery Nagel, Düren, Germany) and stored at −80°C prior to analyses. The total numbers of cells analyzed for each purified NK cell population are provided in Table S2 in Supplementary Material.
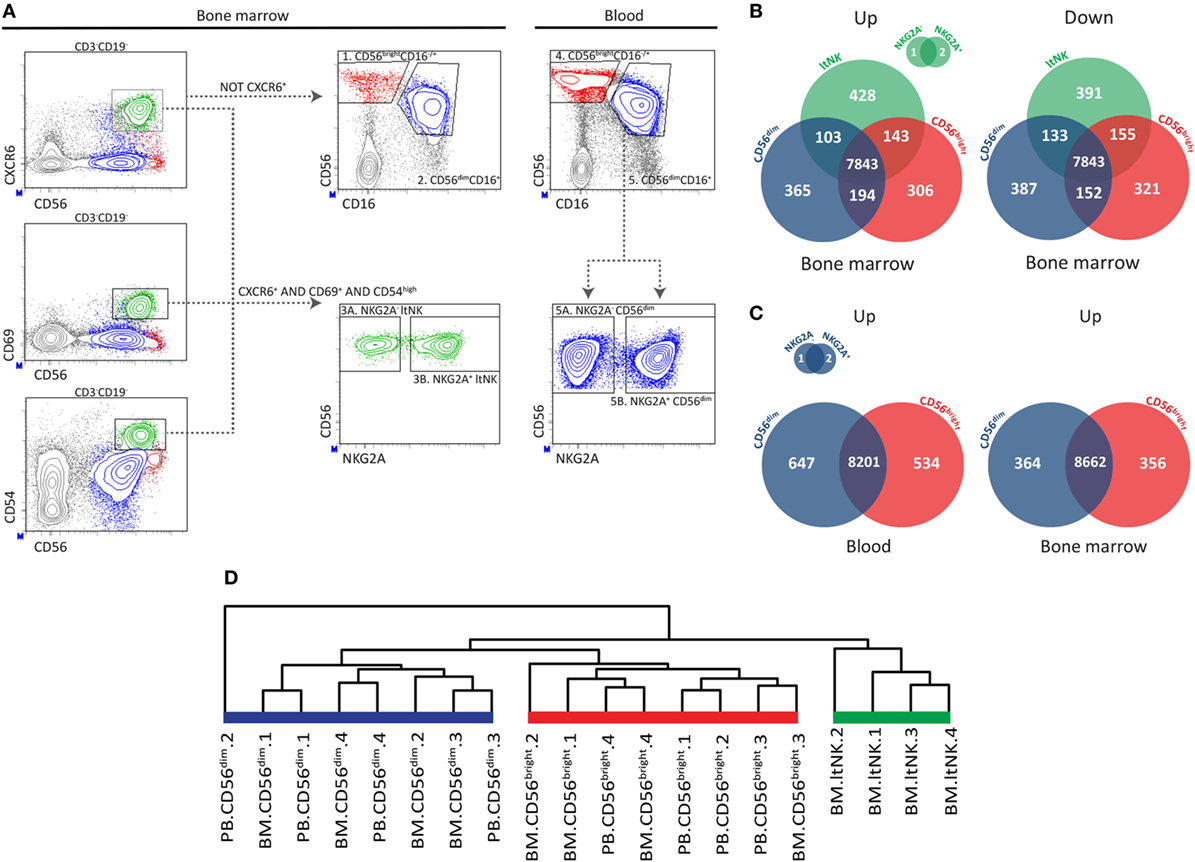
Figure 1. RNA sequence analysis confirms distinctiveness of CD56bright, CD56dim, and ltNK cells. (A) The applied gating strategy to isolate the different natural killer (NK) cell subsets from bone marrow (BM) and blood is depicted. A representative sample of BM NK cell enriched mononuclear cells is shown. Lymphocytes were gated based on forward and sideward scatter and the doublets were excluded. Residual B and T cells were excluded by selecting CD3−CD19− lymphocytes. LtNK cells were defined as CD56+CXCR6+CD69+CD54+. CD56+CXCR6− NK cells were divided into CD56brightCD16+/− and CD56dimCD16+ NK cells. The blood-derived CD56dim and BM derived ltNK cells were further divided into a NKG2A+ and NKG2A− fraction. (B,C) Venn-diagrams illustrating the number of differentially expressed genes (up- and downregulated) between the NK cell subsets in blood and BM. (D) Unsupervised hierarchical clustering of the individual donor samples of BM and blood (PB) derived CD56bright, CD56dim, and ltNK cells. The clustering is based on the expression pattern of all the 9,382 genes, which were included in the differential expression analysis.
RNA Sequence
Total RNA was extracted using the NucleoSpin RNA XS kit (Macherey Nagel) and converted to cDNA and pre-amplified using the SMARTer Ultra Low RNA kit (Clontech Laboratories, Mountain View, CA, USA). A sequencing library was generated from 1 ng amplified cDNA using the Illumina Nextera XT kit (Illumina, San Diego, CA, USA). All 28 samples were pooled and sequenced on two lanes of the HiSeq2500 system (Illumina). A detailed description of the methods used to perform RNA sequencing and map the reads are provided in the supplemental methods. RNA paired-end reads were aligned to the Homo Sapiens reference genome version hg19 using GSNAP. The read counts were normalized for library size with the “voom” function of the limma package (33). Genes with averaged normalized counts below 4 counts per million of uniquely mapped reads (CPM) were excluded from further analysis. To determine the differentially expressed genes, a linear model was fitted to each gene and empirical Bayes moderated t-statistics were applied. Genes were considered as significantly differentially expressed if the false discovery rate (FDR) was below the 0.05 threshold. Unsupervised hierarchical clustering was performed using the hclust function in R (Version 0.99.903, R Foundation for Statistical Computing, Vienna, Austria). Heatmaps were generated using the heatmap3 package (34, 35).
RNA Sequence Validation by Flow Cytometry
To validate the differentially expressed genes at the protein level, MNCs isolated from blood and BM were stained with fluorochrome-labeled antibodies in PBS (Braun, Melsungen, Germany) containing 0.1% NaN3 (LUMC Pharmacy) and 0.5% bovine serum albumin (BSA, Sigma-Aldrich, St. Louis, MO, USA). When unconjugated anti-CXCR6 was used, a three-step staining procedure was used as described under the Section “Purification of NK Cell Populations” in supplemental methods. To determine intracellular expression of Eomes and Tbet, MNCs were fixated and permeabilized using the FOXP3 transcription factor staining kit, as described in the protocol (Invitrogen, Carlsbad, CA, USA). The antibodies used are listed in Table S3 in Supplementary Material. Data were acquired on a LSRII flow cytometer (BD) using FACS Diva software (v8.0, BD). In the extracellular and intracellular staining protocols, respectively DAPI (25 ng/ml, Sigma-Aldrich) and Fixable Viability Dye eFluor 455UV (1:1,000, eBioscience, San Diego, CA, USA), were used to exclude dead cells. Kaluza analysis software (v1.5, Beckman Coulter, Brea, CA, USA) was used for post-acquisition analysis. Density plots were generated using FACS Diva software (v8.0). NK cells were defined as living CD45+CD19−CD3−CD7+CD56+ lymphocytes. Within this cell population, ltNK cells were identified as CD69+CXCR6+ or CD69+CD54high. The remaining NK cells were divided into CD56brightCD16−/+ and CD56dimCD16+ NK cells, based on the levels of CD56 and CD16 expression. CD8+ memory T cells were defined as CD3+CD8+CCR7−CD45RA+/− lymphocytes within the live gate (Figure S1 in Supplementary Material). CD69 was used to identify CD8+ tissue-resident memory T cells. Fluorescence minus one controls were used as a negative control.
In Vitro Assays
To determine, LIGHT, CD30L, and IFN-γ expression, MNCs from BM were cultured in AIM-V (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% human serum and stimulated with recombinant human IL12 (10 ng/ml, PeproTech, Rocky Hill, NJ, USA), recombinant human IL15 (10 ng/ml, CellGenix, Freiburg, Germany), and recombinant human IL18 (20 ng/ml, MBL International, Woburn, MA, USA), or a combination of phorbol myristate acetate (PMA, 12.5 ng/ml, Sigma-Aldrich), and ionomycin (1 μg/ml, Sigma-Aldrich). BD Golgistop (1:1,500, BD) was added after 1 h of culture. After 4 h of stimulation, cells were harvested and stained for surface markers (Table S3 in Supplementary Material). To stain intracellular IFN-γ, cells were subsequently fixated with 4% paraformaldehyde and permeabilized with saponin, as previously described (Table S3 in Supplementary Material) (36). To study the proliferative capacity ltNK (CD49e−CD56+CD69+CXCR6+), CD56bright (CD49e+CD56bright) and CD56dim (CD49e+CD56dimCD16+) NK cells were purified and cultured for 6 days in the presence of IL2 (1,000 IU/ml, Chiron, Emryville, CA, USA), IL15 (10 ng/ml), or IL21 (10 ng/ml, PeproTech). After 6 days, intracellular Ki67 expression was determined. For this purpose, NK cells were fixated and permeabilized using the FOXP3 transcription factor staining kit (Table S3 in Supplementary Material). The counts of CD56+ NK cells after culture were assessed by flow cytometry.
Gene Set Enrichment Analysis
To determine whether certain gene sets were enriched in the ltNK cell population, CAMERA (limma package) analysis was applied using the normalized expression values of 9,382 genes (37). Gene set collections C2 (curated gene sets), C3 (motif gene sets), C5 (GO gene sets), and C7 (immunologic signatures), derived from the Molecular Signatures Database (MSigDB v6.0) were included. Two analyses were performed: ltNK versus CD56bright and ltNK versus CD56dim. Gene sets that were significantly enriched (FDR < 0.05) in both analyses are described in Table S4A in Supplementary Material. The combined Z-scores of target gene expression were calculated as previously described (38). To compare ltNK cells and CD8+ tissue-resident memory T cells isolated from spleen and lung, the normalized counts dataset of GSE94964 was downloaded (25). A log2 normalization was applied and genes with a mean log2 value < 2 were removed. Subsequently, the log2 FC was calculated of genes expressed in both T cells and NK cells. Plots were generated by use of the ggplot2 package (35).
Statistical Tests
To compare pairwise protein expression data or combined Z scores between ltNK, CD56bright, and CD56dim NK cells, one-way ANOVA test was applied. Tukey’s correction was applied to correct for multiple testing. CD69+ and CD69− memory T cells were compared using a paired t-test. P-values below 0.05 were considered as statistically significant. Statistics and scatter dot plots were generated using GraphPad Prism software (v7.00, Graphpad, La Jolla, CA, USA). Statistics of the RNA sequencing were calculated as described under “RNA sequence.”
Results
CD56bright, CD56dim, and ltNK Cells Constitute Three Distinct NK Cell Populations
Previously, we reported that CD69+CXCR6+ lymphoid tissue (lt)NK cells from BM, spleen, and lymph node are phenotypically and functionally distinct from circulating CD56bright and CD56dim NK cells (12). To find clues on the function and development of ltNK cells, we compared the transcriptional signatures of ltNK cells and the two conventional circulating NK cell subsets. From this point onward, the term ltNK cells refers to BM ltNK cells. NK cells from BM and blood of four healthy donors were first enriched using magnetic beads. Subsequently, NK cell populations were FACS-purified (from BM: 1. CD56bright, 2. CD56dim, 3. ltNK and from blood: 4. CD56bright, 5. CD56dim). LtNK cells were selected based on combined expression of CD69, CXCR6, and CD54 (Figure 1A). The blood-derived CD56dim and BM-derived ltNK cells were initially separated into a NKG2A+ and NKG2A− fraction (Figure 1A). When comparing these fractions, only three genes (of 9,382 genes included) had a significantly differential expression (Figures 1B,C). Therefore, we pooled the expression data proportionally from the NKG2A+ and NKG2A− fractions in subsequent analyses.
We first evaluated differences in gene expression levels between the ltNK cells and the conventional NK (non-ltNK) cells within the BM. In ltNK cells, 674 genes were significantly upregulated while 679 genes were downregulated compared with CD56bright and/or CD56dim NK cells (Figure 1B). LtNK cells expressed 152 genes at the highest level and 194 at the lowest level of all NK cell subsets (Figure 1B). Moreover, unsupervised hierarchical clustering of the individual samples, based on expression of all 9,382 genes, demonstrated that the ltNK cells clustered (Figure 1D). Considering the high number of differentially expressed genes and the cluster formed by ltNK cells, the ltNK cells truly represent a distinct subset, rather than just resembling one of the circulating populations transiently trafficking through lymphoid tissues.
Conventional NK cell subsets in both lymphoid tissues and blood are phenotypically similar (12). This prompted us to hypothesize that the CD56bright and CD56dim NK cells in BM represent the circulating NK cells. Indeed, the transcriptome analysis revealed that only 16 and 76 genes of the total 9,382 genes were significantly differentially expressed in marrow derived versus circulating CD56dim and CD56bright NK cells, respectively. Correspondingly, circulating CD56dim and CD56bright NK cells cluster together with the BM derived CD56dim and CD56bright NK cells, respectively (Figure 1D). Comparison of the CD56dim with CD56bright NK cells from blood revealed that 1,181 (647 + 534) of the 9,382 genes were differentially expressed (Figure 1C). Comparing the same populations in BM yielded 720 (364 + 356) genes of which 74% overlapped with the differentially expressed genes in blood (Figure 1C). Despite the inevitable blood contamination of BM aspirates, our findings suggest that CD56bright and CD56dim NK cells are two distinct subsets, which do not differ between blood and marrow.
LtNK Cells Are EomeshighTbetlow
To evaluate whether the transcriptional network differs between the NK cell subsets, transcription factor profiles of the NK cell populations in BM (Figure 2A) and blood were generated (Figure S2 in Supplementary Material). EOMES and TBX21 (Tbet) were the highest and lowest expressed by ltNK cells, respectively (Figure 2A). In line with this, ltNK cells had an EomeshighTbetlow phenotype. Eomes is often used to discriminate NK cells (Eomes+) from the helper innate lymphoid cells (Eomes−), confirming that ltNK cells belong to the NK cell lineage (Figure 2B) (39). Human liver-resident CXCR6+ NK cells were previously found to be EomeshighTbetlow as well (8, 40). In both murine and human NK cells, ZEB2 transcript levels increase during the process of NK cell maturation (41). mRNA levels of ZEB2 in ltNK cells were equal to CD56bright NK cells and lower than in CD56dim NK cells (Figure 2C).
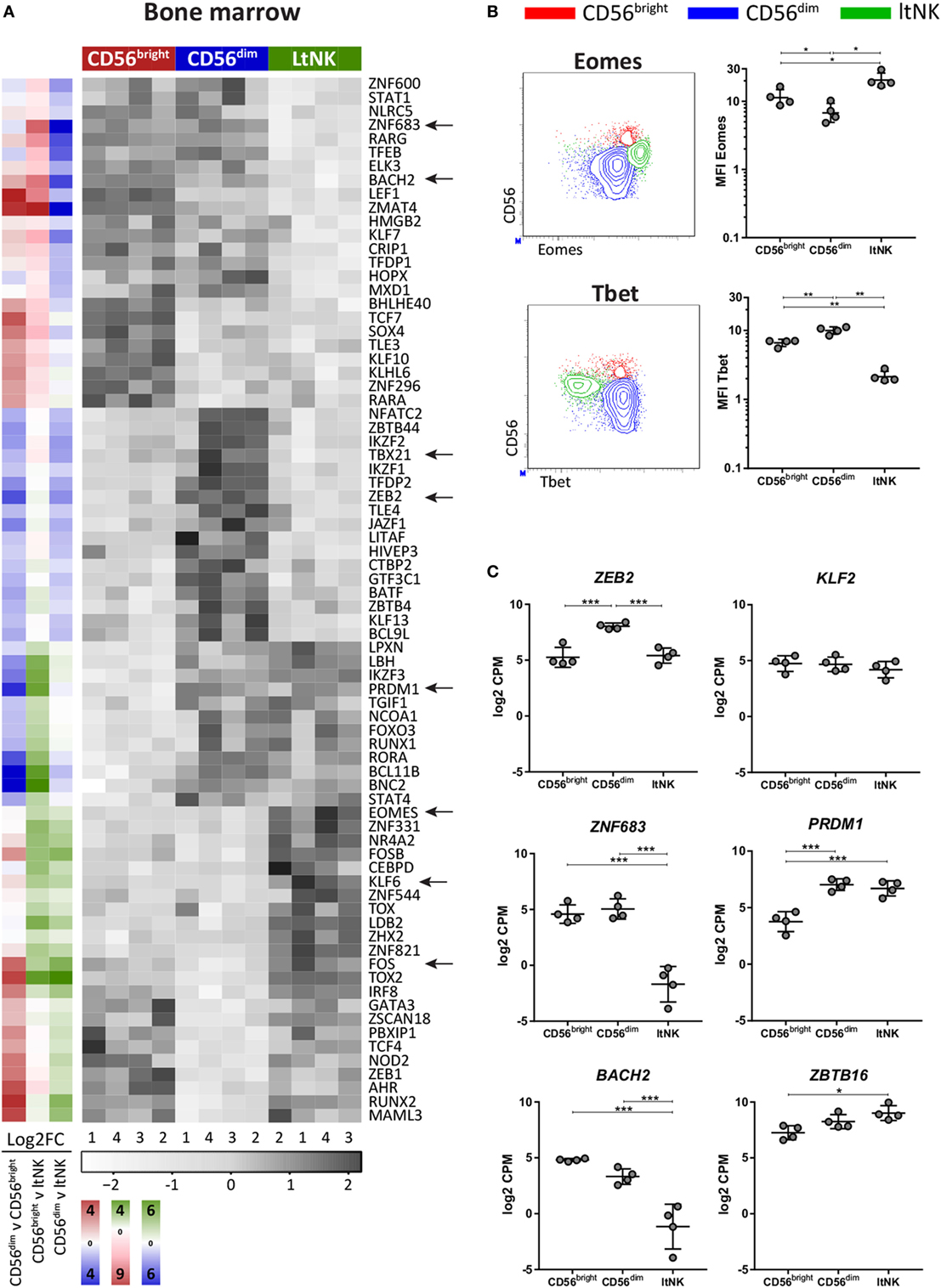
Figure 2. LtNK cells are EomeshighTbetlow. (A) Heatmap illustrates normalized mRNA expression values of transcription factors, which have the highest or lowest mRNA expression [false discovery rate (FDR) <0.05] in 1 of the 3 bone marrow (BM)-derived natural killer (NK) cell subsets. The column side bars represent the log2-fold change (FC) of gene expression levels in one NK cell subset versus another. The color indicates in which NK cell population the gene is expressed at the highest level (green = ltNK, red = CD56bright, blue = CD56dim). The color intensity represents the magnitude of the FC. (B) Eomes and Tbet expression of CD56bright (red), CD56dim (blue), and ltNK cells (green), as determined by flow cytometry. Shown are representative dot plots of BM-derived NK cells. MFI, mean fluorescence intensity. *P < 0.05, **P < 0.01, by one-way ANOVA. (C) Normalized mRNA expression levels of selected transcription factors. *FDR < 0.05, ***FDR < 0.001. CPM, counts per million of uniquely mapped reads. FDR, false discovery rate. Error bars represent mean ± SD in (B,C).
Several transcription factors are associated with development of tissue-resident lymphocytes. Because Kruppel-like factor 2 (KLF2) upregulates S1PR1 and CD62L expression, downregulation of KLF2 supports tissue-residency (42, 43). We found only a minor non-significant difference in KLF2 expression between ltNK cells and circulating NK cells (Figure 2C). Maintenance of murine liver-resident NK cells is dependent on Zfp683 (Hobit) while maintenance of conventional NK cells is not (26). This contradicts human NK cells: ZNF683 (HOBIT) was expressed at lower levels in ltNK cells, while higher levels were observed in CD56bright and CD56dim NK cells (Figure 2C), as was previously shown by flow cytometry on the latter two populations from blood (44). PRDM1 (Blimp1), which regulates maintenance of murine tissue-resident T cells did not differ in expression between ltNK and CD56dim NK cells (Figure 2C) (26). However, the transcriptional repressor of PRDM1, BACH2, was lower expressed in ltNK cells compared to the circulating NK cells, while ZBTB16, which encodes PLZF, a repressor of BACH2, was elevated (Figure 2C) (45, 46).
Adhesion Molecule Profile of ltNK Cells Reveals Tissue-Resident Features
To obtain clues on environmental interactions mediated by ltNK cells, we analyzed the differentially expressed genes encoding surface molecules and visualized the result in heatmaps (Figure 3A; Figure S2 in Supplementary Material). Among the surface molecules up- and downregulated in ltNK cells compared to circulating NK cells, we identified several molecules associated with tissue-residency. S1PR1, the receptor for S1P, which promotes tissue egress was expressed at the lowest mRNA level of all NK cell subsets (Figures 3A,B). SELPLG (CD162, Selectin P ligand) and SELL (CD62L, l-selectin), both involved in lymphocyte recruitment from blood to tissues via interaction with vessel endothelium, were downregulated as well (Figures 3A,B). By flow cytometry, we confirmed that CD62L was not expressed by ltNK cells (Figure 3C).
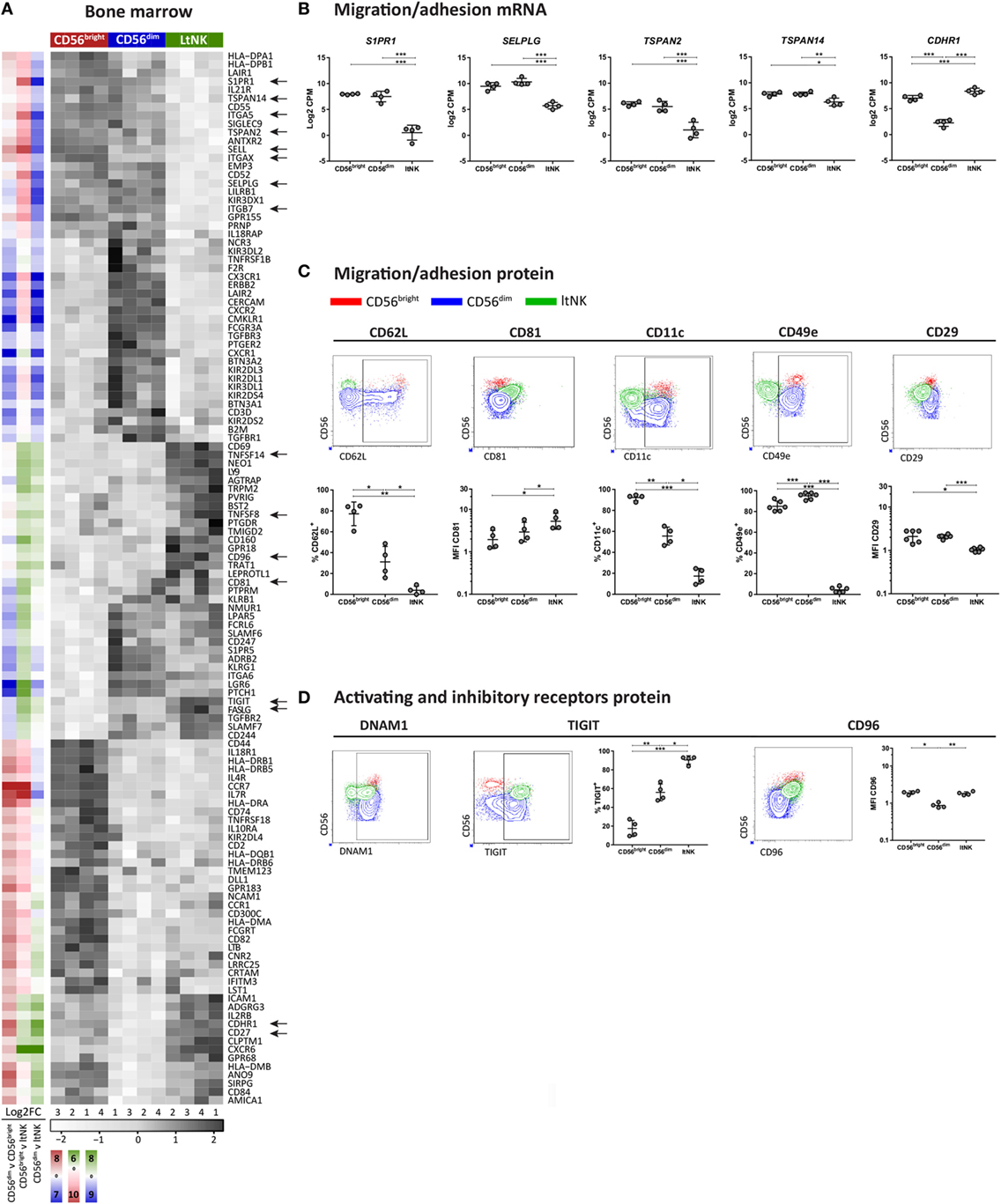
Figure 3. Surface molecule profile of ltNK cells reveals altered adhesion molecule repertoire. (A) Heatmap shows scaled mRNA expression values of genes, which encode surface molecules and have the highest or lowest mRNA expression [false discovery rate (FDR) < 0.05] in 1 of the 3 bone marrow (BM)-derived natural killer (NK) cell subsets. The column side bars represent the log2-fold change (FC) of gene expression levels in one NK cell subset versus another. The color indicates in which NK cell population the gene is expressed at the highest level (green = ltNK, red = CD56bright, blue = CD56dim). The color intensity represents the magnitude of the FC. (B) Normalized mRNA expression levels of differentially expressed surface molecules involved in migration/adhesion. *FDR < 0.05, **FDR < 0.01, ***FDR < 0.001. CPM, counts per million of uniquely mapped reads; FDR, false discovery rate. (C,D) Validation of mRNA expression of surface molecules by flow cytometry. Shown are representative dot plots of BM-derived NK cells. MFI, mean fluorescence intensity. *P < 0.05, **P < 0.01, ***P < 0.001, by one-way ANOVA. Mean ± SD are shown in (B–D).
Other adhesion molecules in the list of differentially expressed surface molecules were the tetraspanins TSPAN2↓, TSPAN14↓, and CD81↑ (TSPAN28), the integrins ITGAX↓ (CD11c), ITGB7↓, and ITGA5↓ (CD49e), and the cadherin CDHR1↑ (Figures 3A,B). Consistent with the RNA sequence data, CD81 protein was expressed at a higher intensity on the cell surface of ltNK cells compared with the conventional NK cells (Figure 3C). CD11c expression was indeed significantly lower on ltNK cells compared with CD56bright and CD56dim NK cells. Expression of ITGB7 did not differ between ltNK and circulating NK cells (Figure S3A in Supplementary Material). Remarkably, while the vast majority of conventional NK cells expressed CD49e, ltNK cells lacked this integrin on their surface (Figure 3C). The dimerizing partner of CD49e, ITGB1 (CD29) was also expressed at a significantly lower intensity on ltNK cells compared with the conventional NK cells (Figure 3C). Overall, the adhesion molecule profile of ltNK cells differs significantly from both the CD56bright and CD56dim NK cells.
LtNK Cells Express the Inhibitory Receptors TIGIT and CD96 and Lack Cytolytic Proteins
Natural killer cell effector function depends on signals derived from activating and inhibitory receptors and on the potential of NK cells to modulate other cells by expressing ligands or secrete effector molecules. Genes encoding TNF superfamily members were highly expressed by ltNK cells: CD27, TNFSF14 (LIGHT), TNFSF8 (CD30L), and FASLG (Figure 3A). Earlier, we demonstrated the high protein expression of CD27 on ltNK cells (12). However, LIGHT and CD30L were not detected on the surface of resting NK cells (Figure S3B in Supplementary Material). Although PMA/ionomycin stimulation induced LIGHT expression, mainly on the CD56bright and ltNK cell subset, CD30L expression could not be induced (Figure S3B in Supplementary Material). In addition, the surface molecule transcriptome of ltNK cells contained various receptors, which are known to inhibit NK cell activity. TIGIT and CD96 are inhibitory receptors, which engage the same ligands as the stimulatory receptor DNAM1 (CD226) and were expressed at high levels in ltNK cells (Figure 3A). DNAM1 is expressed by 40% of the ltNK cells, while all conventional NK cells express DNAM1 (Figure 3D) (12). Flow cytometry confirmed that nearly 100% of ltNK cells express TIGIT, in contrast to CD56dim (mean 56%) and CD56bright (mean 18%) NK cells (Figure 3D). The CD96 expression of ltNK cells was comparable to the CD56bright NK cell population (Figure 3D). In line with the inhibitory receptor repertoire, we previously reported that ltNK cells do not efficiently lyse K562 target cells (12). Furthermore, the genes encoding the cytolytic molecules granzyme B (GZMB), granzyme H (GZMH), and granulysin (GNLY) were expressed at the lowest level by ltNK cells (Figures 4A,B). At the protein level, resting ltNK cells did not express granzyme B, but did express perforin albeit at a lower level than CD56dim NK cells (12). Collectively, ltNK cells express the inhibitory receptors TIGIT and CD96 and are not endowed with a full cytotoxic machinery.
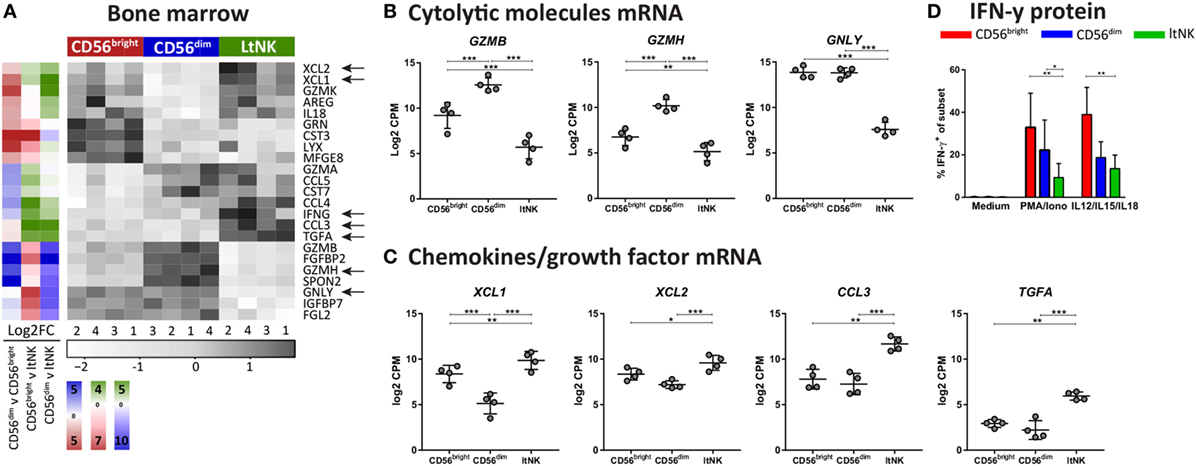
Figure 4. Effector molecule profile of ltNK cells highlights low killing capacity. (A) Heatmap depicts normalized mRNA expression levels of genes, which encode effector molecules and have the highest or lowest expression in 1 of the 3 bone marrow (BM) derived natural killer (NK) cell subsets [false discovery rate (FDR) < 0.05]. The column side bars represent the log2-fold change (FC) of gene expression levels in one NK cell subset versus another. The color indicates in which NK cell population the gene is expressed at the highest level (green = ltNK, red = CD56bright, blue = CD56dim). (B,C) Normalized mRNA expression values of differentially expressed (B) cytolytic molecules (granzyme B, granzyme H, and granulysin) and (C) chemokines (XCL1, XCL2, CCL3) and the growth factor TGFα. CPM, counts per million of uniquely mapped reads. *FDR < 0.05, **FDR < 0.01, ***FDR < 0.001. FDR, false discovery rate. (D) Intracellular IFN-γ expression of CD56bright, CD56dim, and ltNK cells. BM mononuclear cells (n = 7) were stimulated for 4 h with PMA/ionomycin or the combination of IL12, IL15, and IL18. After 1 h, Golgistop was added to the culture. *P < 0.05, **P < 0.01, by one-way ANOVA. Mean ± SD are shown in (B–D).
High Transcript Levels of IFNG in ltNK Cells Cannot Be Recapitulated at the Protein Level In Vitro
Potentially, other secreted effector molecules are involved in ltNK cell functioning. Elevated transcript levels of XCL1, XCL2, CCL3, and TGFα were detected in ltNK cells (Figures 4A,C). Strikingly, IFNG mRNA was also enriched in ltNK cells. This suggests that ltNK cells can rapidly modulate the immune response by producing IFN-γ upon triggering, as was earlier demonstrated for CD8+ tissue-resident memory T cells in spleen and lung (23, 25). However, we were not able to activate IFN-γ production in ltNK cells. Strong short-term stimulation with PMA/ionomycin or IL12/IL15/IL18 did not result in a high percentage of IFN-γ producing ltNK cells (Figure 4D). Therefore, triggering of IFN-γ production in ltNK cells may require different stimuli than classically used for CD56bright and CD56dim NK cells.
Gene Set Enrichment Analysis (GSEA) Reveals Potential Inducers of ltNK Cell Transcriptome
To obtain clues on over- or underrepresented pathways in ltNK cells, we performed a GSEA by use of CAMERA and the Broad institute gene set collections. In addition, GSEA can uncover molecules, which might induce the genetic program of ltNK cells by analyzing expression of the downstream target genes. Only gene sets, which were significantly up- or downregulated in ltNK cells compared to both CD56bright and CD56dim NK cells (FDR < 0.05) were further analyzed. We first selected the gene sets, which were upregulated in ltNK cells versus the circulating NK cells (Table S4A in Supplementary Material). Among those gene sets, we identified epidermal growth factor (EGF) signaling and BM stromal cell stimulation (Figure 5A). EGF has been shown to induce a network of transcription factors, including NR4A2, FOS, JUN, KLF6, and ZFP63L1, which were all expressed the highest by ltNK cells (Figure 2A) (47). Moreover, SPRY1 and SPRY2, two inhibitors of EGF and other growth factor receptor tyrosine kinase signaling were also enriched in ltNK cells, which is suggestive of an inhibitory feedback loop (48). These results favor the hypothesis of a growth factor induced transcriptional program in ltNK cells, potentially mediated by cells from the BM microenvironment.
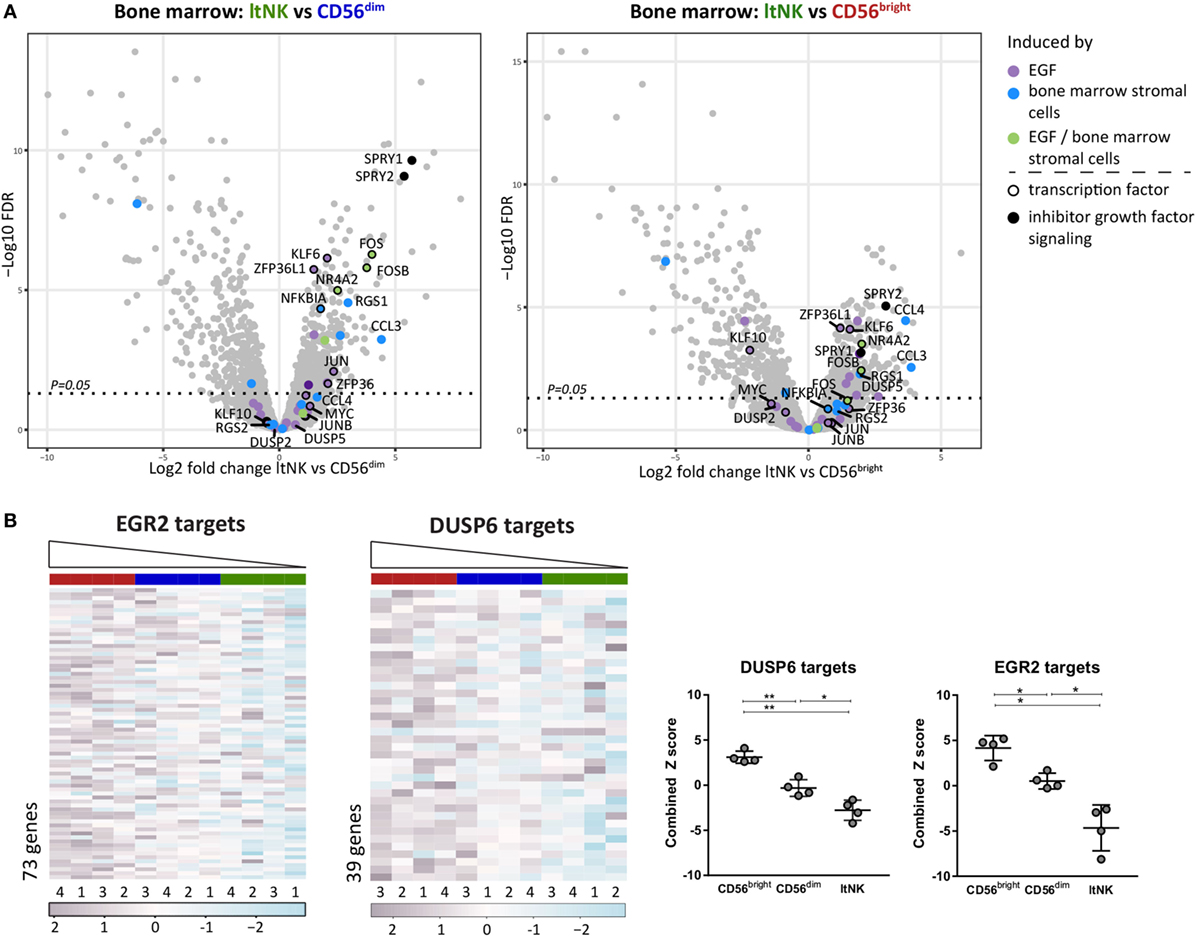
Figure 5. Gene set enrichment analysis (GSEA) reveals potential inducers of ltNK cell transcriptome. (A) The gene sets epidermal growth factor (EGF) signaling (M16311) and bone marrow (BM) stimulation (M5929) were enriched in ltNK cells, as identified by GSEA using CAMERA and the Broad institute gene set collections. The log2 fold change of ltNK versus CD56bright or ltNK versus CD56dim is plotted against the false discovery rate (FDR) in volcanoplots. Genes that were induced by either EGF, BM stromal cells, or both are depicted. SPRY1 and SPRY2 are two inhibitors of growth factor signaling. FDR, false discovery rate. (B) GSEA revealed that targets that are repressed by EGR2 (M12804) or DUSP6 (M7339) are downregulated in ltNK cells. Heatmaps show the normalized expression values of the corresponding genes, which are repressed by EGR2 (n = 73) and DUSP6 (n = 39). The column order is based on the combined Z score of each donor natural killer cell population (high → low). The combined Z score is a quantification of the overall expression level of the target genes. Error bars represent mean ± SD. *P < 0.05, **P < 0.01, by one-way ANOVA.
Second, we selected the downregulated gene sets, which included many transcription factors (Table S4B in Supplementary Material). Among the transcription factor gene sets, three downregulated gene sets included genes, which expression was negatively affected by the responsible transcription factor, indicating that the corresponding transcription factors are more active in ltNK cells: EGR2, BCL3, and YBX1. The expression of the target genes of these three transcription factors was indeed the lowest in the ltNK subset, as quantified by the combined Z score (Figure 5B; Figure S4 in Supplementary Material). EGR2 was earlier reported to be higher expressed by lung-resident CD8+ memory T cells than by circulating effector memory CD8+ T cells (23). In addition, another set of genes, which expression is inhibited by the dual-specificity phosphatase DUSP6 (inhibitor of MAP kinase signaling), were downregulated in ltNK cells (M7339, Figure 5B). Recently, DUSP6 has been identified as a core tissue-resident memory T cell molecule (25). These results imply that EGR2 and DUSP6 are candidate molecules involved in induction or maintenance of ltNK cells.
LtNK Cells Have Less Proliferative Capacity Than CD56bright and CD56dim NK Cells
The downregulated gene sets of the GSEA also included many cell-cycle related genes (Table S4C in Supplementary Material). The hallmark data set “G2M checkpoint” represents genes involved in progression through the cell division cycle. Expression of those genes was the lowest in ltNK cells and the highest in CD56bright NK cells (Figure 6A). The same was true for expression of the targets of E2F (“Hallmark E2F targets”), a family of transcription factors, which regulates the cell cycle (Figure 6A). To assess their proliferative capacity, we sorted ltNK, CD56bright, and CD56dim NK cells from BM and stimulated the cells with IL2, IL15, or IL21 (Figure 6B). In agreement, ltNK cells had the lowest expansion and lowest intracellular Ki67 expression upon stimulation with IL2 and IL15. As previously described, the CD56bright NK cells were highly proliferative (49).
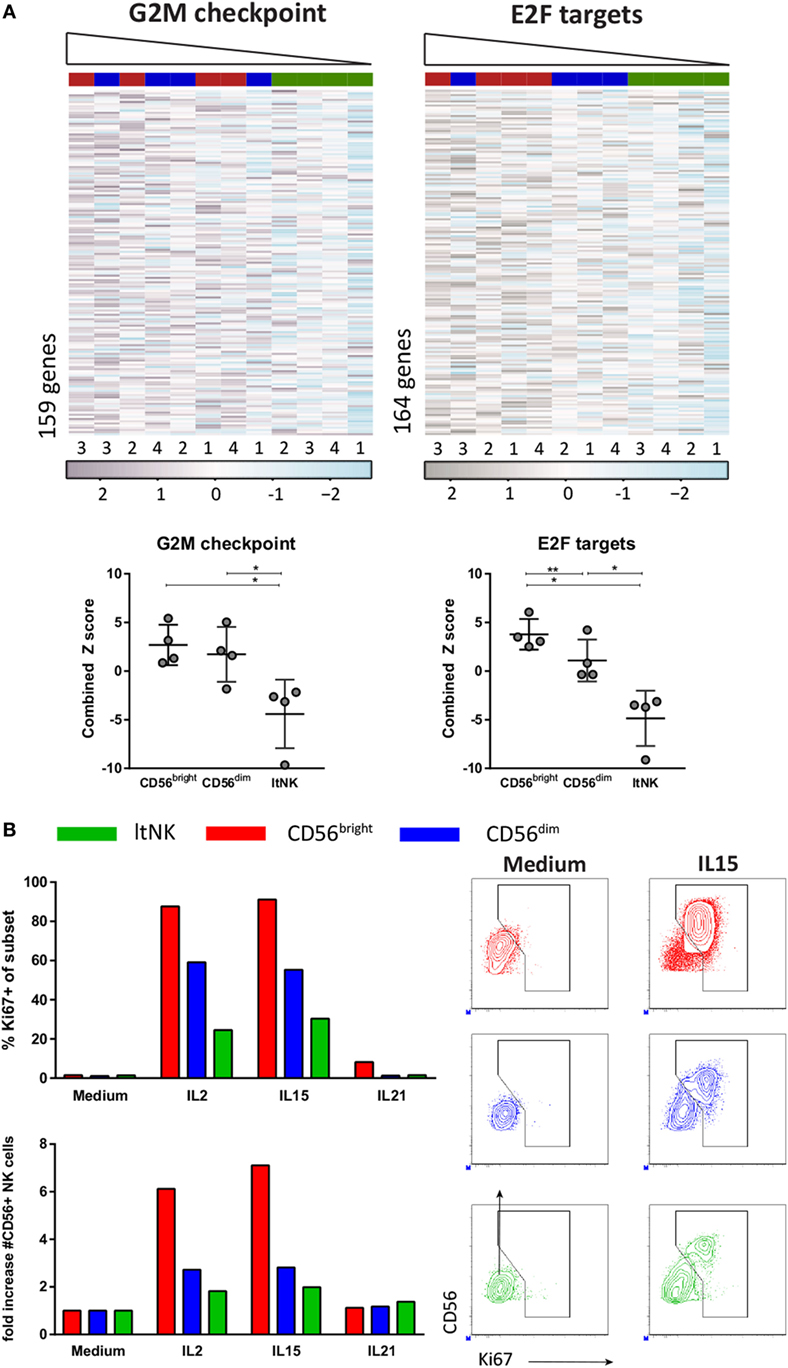
Figure 6. Proliferative capacity of ltNK cells is lower compared with CD56bright and CD56dim natural killer (NK) cells. (A) Gene set enrichment analysis using CAMERA and the Broad institute gene collections revealed that many cell-cycle related genes are downregulated by ltNK cells. Heatmaps show the normalized expression values of the hallmark gene sets G2M checkpoint (M5901) and E2F targets (M5925). The column order is based on the combined Z score of each donor NK cell population (high → low). The combined Z score is a quantification of the overall expression level of the genes per donor derived NK cell population. *P < 0.05, **P < 0.01, by one-way ANOVA. Shown is mean ± SD. (B) Proliferative capacity was assessed by stimulating sorted bone marrow-derived NK cell populations for 6 days with IL2, IL15, or IL21. Intracellular Ki67 expression was measured by flow cytometry. The number of CD56+ NK cells is based on flow cytometer counts. Shown are dot plots of medium control and IL15 stimulated NK cell populations.
LtNK and Tissue-Resident CD8+ Memory T Cells Share a Core Transcriptional and Phenotypic Profile
Many studies transcriptionally profiled CD8+ tissue-resident memory T (Trm) cells from various tissues. As earlier stated, DUSP6 and EGR2, identified in our GSEA, were reported to be higher expressed in human CD8+ Trm cells compared to circulating counterparts (23, 25). This led us to hypothesize about a transcriptional program, which is shared between ltNK and CD8+ Trm cells. We aimed to perform a comparative analysis of RNA sequence data from spleen derived CD69+CD8+ Trm and CD69−CD8+ effector memory T (Tem) cells (25), and our own NK cell dataset. We selected genes expressed in both T and NK cells and plotted the log2 FC of CD8+ Trm versus Tem cells against the log2 FC of ltNK versus CD56dim NK cells, and ltNK versus CD56bright NK cells (Figure 7A; Figure S5 in Supplementary Material). Importantly, the most differentially expressed genes were up- or downregulated in the same direction in all comparisons. Core-resident genes were selected based on a log2 FC ≥ 1 or ≤−1 in all three comparisons. In total, we revealed 83 genes, which have a universal differential expression pattern among resident CD8+ memory T and NK cells (Table S5 in Supplementary Material). Among the core genes, we identified S1PR1↓, CD62L↓, DUSP6↑, CXCR6↑, MKI67↓, and IFNG↑. This prompted us to investigate whether the surface molecule and transcription factor profile of ltNK cells (CD62L−, CD81high, CD11c−, CD49e−, CD29low, TIGIT+, DNAM1low, CXCR6+, Eomeshigh, Tbetlow) is similar to CD8+ Trm cells. Although CD69+ memory T cells comprise a highly diverse subset across tissues, CD69 was shown to be the major marker delineating circulating from resident T cells (25, 50). For this analysis, we compared resident CD69+ (Trm) and non-resident CD69− (Tem) CD8+ memory T cells from human BM (Figure S1 in Supplementary Material). Trm cells represented 28% (median) of BM CD8+ memory T cells (Figure 7B). In contrast to Trm cells from the often evaluated mucosal and epithelial tissues, CD103 was hardly expressed by CD8+ Trm cells in the BM (Figure S6 in Supplementary Material). Unlike NK cells, CD8+ memory T cells universally lacked CD11c (Figure S6 in Supplementary Material). Like ltNK cells, CD8+ Trm cells were CD62L−, CD81high, CD49e−, CD29low, TIGIT+, and DNAM−. Moreover, the majority of CD8+ Trm cells expressed CXCR6 (Figure 7C). In addition, Eomes and Tbet were higher and lower expressed in CD8+ Trm compared to CD8+ Tem cells in BM (Figure 7D). Collectively, we defined a core gene and molecular signature, which characterizes both lymphoid tissue-derived ltNK cells and CD8+ Trm cells.
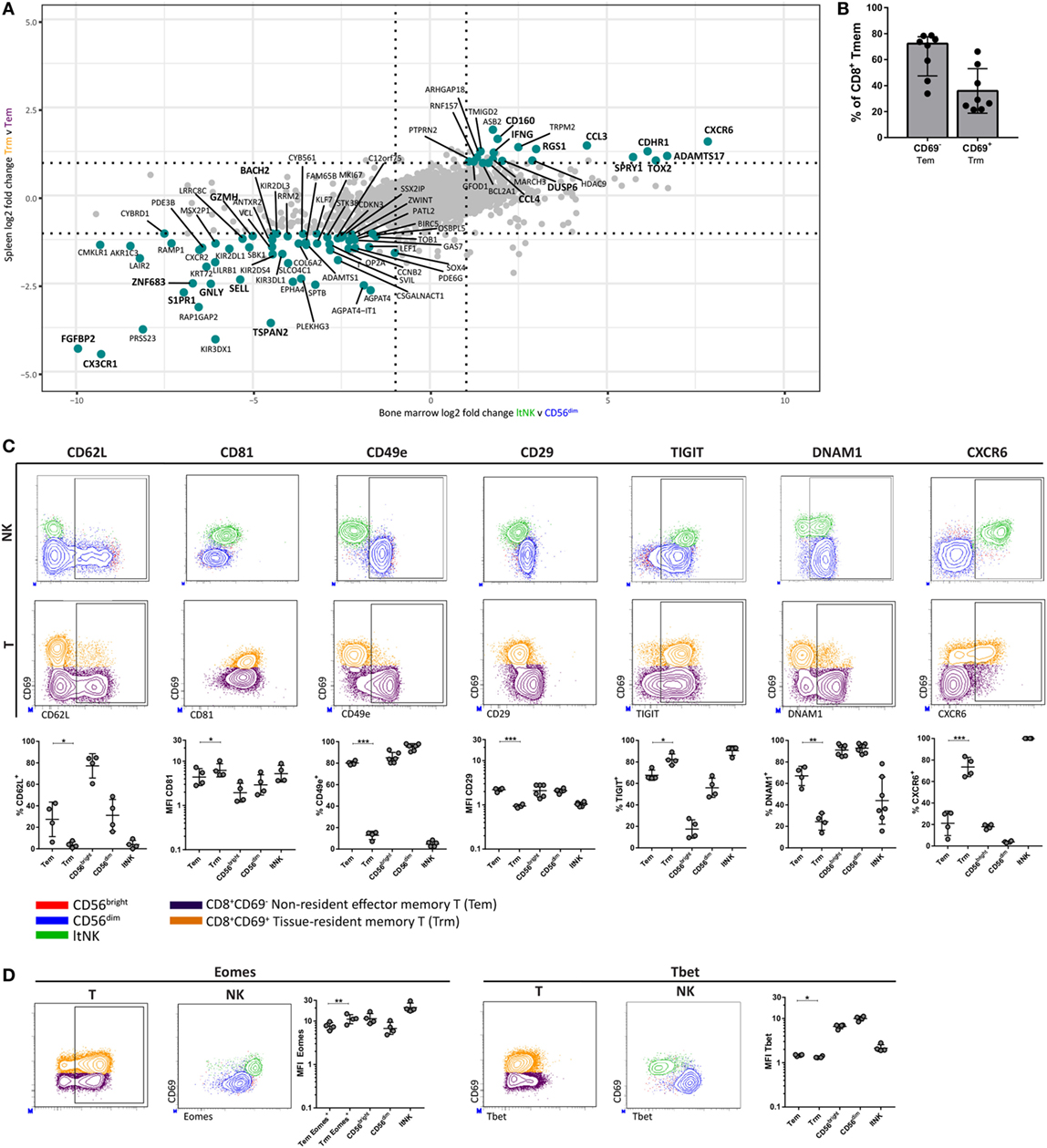
Figure 7. LtNK cells and tissue-resident CD8+ memory T cells share a core transcriptional and phenotypic profile. (A) The datasest GSE94964 contains RNAseq data of spleen-derived CD8+ effector memory T cells (CD3+CD8+CD45RA−CCR7−), which were subdivided into CD69+ tissue-resident memory (Trm) and CD69− effector memory T (Tem) cells. We compared this gene set to our RNAseq data to determine a core gene signature among Trm and ltNK cells in lymphoid tissues. The log2-fold change (FC) of 8,700 genes [which were both expressed in T and natural killer (NK) cells] was calculated. The log2 FC of ltNK versus CD56dim was plotted against the log2 FC of CD8+ Trm against Tem cells. Figure S5 in Supplementary Material depicts the plot, which contains the log2 FC of ltNK versus CD56bright NK cells. Core resident genes, which are depicted in turquoise were identified as genes with a log2 FC ≥ 1 or ≤−1 in all three comparisons. (B) Frequency of CD69− non-resident (Tem) and CD69+ resident CD8+ memory T (Trm) cells in bone marrow (BM). CD8+ memory T cells were defined as CD3+CD8+CCR7+ and included both effector memory and end-stage effector memory T cells. (C,D) Protein expression of surface molecules and transcription factors, which were differentially expressed by ltNK versus circulating NK cells were determined on CD8+ memory T cells (CD69+ versus CD69−). For comparison, plots visualizing the ltNK, CD56bright, and CD56dim NK cells are shown. The Eomes MFI on the CD8+ memory T cells was determined only on the Eomes+ CD8+ memory T cells, rather than on the whole population (as was done for the NK cells). Statistics of NK cell protein expression comparisons are shown in Figure 3. Dot plots depict a representative BM donor. MFI, mean fluorescence intensity. *P < 0.05, **P < 0.01, ***P < 0.001, by two-tailed paired t-test. Median and interquartile range are shown in (B). Mean ± SD are shown in (C,D).
Discussion
The majority of NK cell studies focused on circulating NK cells, which resulted in the classical subdivision of NK cells in the CD56bright and CD56dim NK cell subsets. We recently defined a non-circulating NK cell population, based on the combined expression of CD69 and CXCR6, which resides in lymphoid tissues. In this study, we further analyzed the CD69+CXCR6+ ltNK cells and performed RNA sequence analysis to provide more insight into the biology of ltNK cells. We demonstrated that the surface receptor, adhesion molecule, and transcription factor profiles of ltNK cells significantly differ from that of circulating NK cells. Thereby, we confirmed that ltNK cells constitute a unique subset and not reflect one of the two circulating populations trafficking through tissues, transiently acquiring a different phenotype. Additionally, we defined a core gene signature and surface molecule profile, which is shared by ltNK cells and CD8+ Trm cells, thereby further specifying tissue-residency.
The transcriptome and phenotype of ltNK cells support the notion that ltNK cells are tissue-resident. Molecules involved in cell trafficking are downregulated in both tissue-resident murine NK cells, human liver-resident NK cells, and ltNK cells: i.e., S1PR1, CCR7, and CD62L (8, 9, 12, 51, 52). S1PR1 enables tissue egress, while CCR7 and CD62L are required to enter lymphoid tissues via high endothelial venules (4, 5, 53). Several transcription factors are known to regulate expression of these trafficking molecules, mainly deduced from murine cell lineage specific knock-out models. The transcription factor (KLF2) promotes S1pr1 and Sell (CD62L) expression (42, 43). However, KLF2 was neither significantly downregulated by ltNK cells nor by human liver-resident NK cells (42, 43). Hobit represses S1pr1, Klf2, and Ccr7 and is preferentially expressed in murine-resident lymphocytes (26). However, ltNK cells show profoundly reduced HOBIT expression, whereas the circulating NK cells express high levels. Hence, despite the compatible surface profile of migration/adhesion molecules, the transcriptional network underlying tissue-residency in human ltNK cells seems to differ from that in mice.
The comparative gene expression analysis revealed several interesting differences in the integrin and tetraspanin repertoire between ltNK and circulating NK cells. By flow cytometry, we confirmed that these differences were also present at the protein level. Compared to circulating NK cells, CD81 (TSPAN28) and CD11c (ITGAX) were found to be higher and lower expressed by ltNK cells, respectively. Importantly, we identified the lack of CD49e (ITGA5) as a key marker to delineate ltNK cells from circulating NK cells. In line with this, ltNK cells had lower expression of the dimerizing partner CD29 (ITGB1). A recent paper demonstrated that human liver-resident NK cells are also negative for CD49e (10). Moreover, murine tissue-resident NK cells are marked by absence of CD49b, indicating that an altered integrin repertoire is characteristic for resident NK cells (51). Collectively, both the absence of CD49e and the presence of CD69 and CXCR6 can be used to define resident ltNK cells in humans.
In addition to the adhesion molecule profile, we observed that the inhibitory receptors TIGIT and CD96 were highly expressed by ltNK cells. TIGIT and CD96 share ligands with the activating receptor DNAM1 (CD226), which is expressed by the minority of ltNK cells (54). Engagement of TIGIT by PVR (CD155) or PVRL2 (CD112) can inhibit human NK cell cytotoxicity (55). Furthermore, the expression of TIGIT is inversely correlated with IFN-γ production (56). The functional profile of ltNK cells indeed revealed reduced expression of cytotoxic molecules (PRF1, GZMB, GZMH, GNLY), as well as reduced capacity to kill, to proliferate, and to produce IFN-γ, despite high levels of IFNG mRNA (12). This functional profile suggests that the effector function of ltNK cells is restrained. Alternatively, the appropriate stimuli to engage the functional potential of ltNK cells have not yet been identified.
By performing GSEA, we identified the transcription factor EGR2, the phosphatase DUSP6, the growth factor EGF, and “bone marrow stromal cells” as potential regulators of the ltNK cells transcriptome. EGR2 has been identified as transcriptional activator of Zbtb16 (PLZF) (57). PLZF represses Bach2, which on its turn represses Prdm1 (Blimp1) (45, 46). In line with this axis, ZBTB16 was the highest expressed in ltNK cells, while BACH2 was barely expressed at mRNA level in ltNK cells. LtNK cells express PRDM1 at levels comparable to CD56dim NK cells but higher than CD56bright NK cells. These findings are supportive for a regulatory axis with EGR2 as a central regulator. DUSP6 is an inhibitor of MAP kinase signaling. Although the function of DUSP6 in NK cells has not been reported, high levels of DUSP6 are associated with reduced TCR sensitivity (58). Moreover, DUSP6 inhibits TCR signaling and IFN-γ production in murine CD4 T cells, revealing another potential explanation for the difficulty we encountered in evoking IFN-γ production in ltNK cells (59, 60). Finally, genes that are upregulated by EGF and BM stromal cells were enriched in ltNK cells. Notably, EGF induces EGR2 as well (61). Therefore, it is tempting to speculate that growth factors secreted by stromal cells induce the ltNK cell transcriptome.
DUSP6 and EGR2 were reported to be differentially expressed by human CD8+ Trm cells versus their circulating counterparts (23, 25). This finding led us to speculate about a transcriptional program, which is shared among resident lymphocytes. We identified a core gene and surface molecule signature among ltNK cells and lymphoid tissue derived CD8+ Trm cells, which includes molecules associated with tissue-residency (S1PR1↓, CD62L↓), adhesion molecules (CD49e↓, CD29↓, CD81↑, TSPAN2↓), chemokine receptors (CXCR6↑, CX3CR1↓, CXCR2↓), activating/inhibitory receptors (TIGIT↑, DNAM1↓), transcription factors (Eomes↑, Tbet↓, HOBIT↓, BACH2↓), effector molecules (IFNG↑, CCL3↑, FGFBP2↓, GNLY↓, GZMH↓), and regulatory molecules (DUSP6↑, RGS1↑, Ki67↓). Notably, human liver-resident NK cells exhibit strong similarities to ltNK cells regarding phenotype (CD49e−CXCR6+EomeshighTbetlow) as well as the aforementioned core gene signature (except for CCL3) (7–10, 40). Moreover, human hepatic CD8+ Trm cells are CXCR6+EomeshighTbetlow and have reduced mRNA levels of HOBIT (24). These findings define a core gene signature of residency, which is shared by human tissue-resident NK cells and CD8+ Trm cells in lymphoid tissues and likely also in liver.
It is important to note that phenotypic heterogeneity does exist between tissue-resident lymphocytes in human non-mucosal tissue (e.g., spleen, BM, and liver) and in mucosal or epithelial tissues (e.g., tonsil, gut, skin, and lung), as has been elegantly demonstrated by mass cytometry (50). Although it is generally assumed that Trm cells are Eomes−/low (30, 62), we demonstrated that BM CD8+ Trm cells are Eomeshigh. Potentially, Eomes expression is dependent on the tissue where the lymphocytes reside. In contrast to the Eomeshigh BM derived ltNK cells and CD8+ Trm cells, human lung CD103+CD8+ Trm cells are Eomeslow and skin CD103+CD8+ Trm have reduced transcript levels of EOMES (21, 23). Moreover, CD49a and CD103 are both highly expressed by human tissue-resident NK cells in tonsil and CD8+ Trm cells in tonsil, lung, and skin, while those markers are nearly absent on ltNK cells and spleen and BM-derived CD8+ Trm cells (11, 12, 23, 50, 63). Eomes downregulation and CD103 and CD49a upregulation are hallmarks of TGF-β imprinting, indicating that local environmental cues govern the tissue-specific phenotype of tissue-resident lymphocytes (31, 64, 65). Therefore, it is important to distinguish non-mucosal lymphoid tissue from mucosal or epithelial tissue when discussing tissue-resident lymphocytes.
The function of ltNK cells still remains enigmatic. Taking all our observations into consideration, both CD8+ Trm cells and ltNK cells have low proliferative capacity (25), reduced expression of cytotoxic molecules, while expressing high IFN-γ mRNA. A key advantage of lymphocytes residing in tissues could be a rapid response to recurrent pathogens. For murine CD8+ Trm cells, it has been proven that pathogen-specific CD8+ Trm cells in skin can protect against and clear recurrent infections better than circulating memory CD8+ T cells (32, 66–70). Murine liver-resident NK cells confer hapten- and virus-specific memory responses (51, 71). These studies support a model in which ltNK cells and CD8+ Trm cells reside in lymphoid tissues awaiting to encounter a specific stimulus. As an alternative for the perforin/granzyme B-mediated pathway, cytolysis could involve death receptor ligands. FASL was upregulated in both ltNK cells and lung-resident CD8+ Trm cells (23). Still, the question remains which activating signals can release the functional brake or whether a distinct function is exerted.
The transcriptional signature of ltNK cells contributed to our understanding of the ltNK cell biology and yielded two important concepts. First, the ltNK cells are truly distinct from the two circulating NK cell subsets. Second, the transcriptional signature of the ltNK cells is very similar to that of (splenic) CD8+ Trm cells. Despite the lack of transcriptional data on simultaneously purified CD8+ Trm and Tem cells from BM in our study, we were able to provide further insight into tissue-resident features by comparing our own data with a publically available dataset. Validation of the BM ltNK and CD8+ Trm cell phenotype in other (lymphoid) tissues would be interesting, but is missing in this study due to the lack of healthy donor tissues. It remains presently unknown, which stimuli can trigger ltNK cells and more functional experiments are, therefore, required. Altogether, this study confirms the resident nature of ltNK cells and offers opportunities for further studies on the specific function of ltNK cells. Our comprehensive analysis facilitates new strategies to further study tissue-resident features, development of ltNK cells, and their role in immune regulation.
Data Availability Statement
The dataset generated for this study can be found in the GEO database under accession number GSE116178. To perform comparative analysis, the T cell dataset GSE94964 published by Kumar et al. was used.
Ethics Statement
This study was carried out in accordance with the recommendations of the Dutch national ethical and profession guidelines. The protocol was approved by the institutional review board. All subjects gave written informed consent in accordance with the Declaration of Helsinki.
Author Contributions
JM designed the study, performed experiments, analyzed data, and wrote the manuscript. GL designed and performed the RNA sequence experiment and wrote the paper. CV performed flow cytometry experiments. SK analyzed RNA sequence data. SZ analyzed RNA sequence data. HB designed and performed the RNA sequencing. MO contributed expertise and performed flow cytometry experiments. AL and MS supervised the study, designed experiments, and wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer FV and the handling Editor declared their shared affiliation.
Acknowledgments
We thank Dr. M. J. D. van Tol (Department of Pediatrics, Leiden University Medical Center) and Dr. J. van Bergen (Department of Immunohematology and Blood Transfusion, Leiden University Medical Center) for critical reading of the manuscript, Dr. S. Tsonaka (Department of Biomedical Data Sciences, Leiden University Medical Center) for contributing statistical expertise and the flow cytometry core facility of the Leiden University Medical Center for providing support for the FACS.
Funding
This work was financially supported with a grant from the Dutch Cancer Society (#UL 2011-5133). JM and GL were supported by fellowships from the Leiden University Medical Center and the graduate program of NWO.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01829/full#supplementary-material.
References
1. Bankovich AJ, Shiow LR, Cyster JG. CD69 suppresses sphingosine 1-phosophate receptor-1 (S1P1) function through interaction with membrane helix 4. J Biol Chem (2010) 285:22328–37. doi:10.1074/jbc.M110.123299
2. Shiow LR, Rosen DB, Brdicková N, Xu Y, An J, Lanier LL, et al. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature (2006) 440:540–4. doi:10.1038/nature04606
3. Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, et al. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol (2015) 194:2059–63. doi:10.4049/jimmunol.1402256
4. Pappu R, Schwab SR, Cornelissen I, Pereira JP, Regard JB, Xu Y, et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science (2007) 316:295–8. doi:10.1126/science.1139221
5. Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, Cyster JG. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science (2005) 309:1735–9. doi:10.1126/science.1113640
6. Montaldo E, Vacca P, Chiossone L, Croxatto D, Loiacono F, Martini S, et al. Unique Eomes+ NK cell subsets are present in uterus and decidua during early pregnancy. Front Immunol (2016) 6:646. doi:10.3389/fimmu.2015.00646
7. Stegmann KA, Robertson F, Hansi N, Gill U, Pallant C, Christophides T, et al. CXCR6 marks a novel subset of T-betloEomeshi natural killer cells residing in human liver. Sci Rep (2016) 6:26157. doi:10.1038/srep26157
8. Cuff AO, Robertson FP, Stegmann KA, Pallett LJ, Maini MK, Davidson BR, et al. Eomes hi NK cells in human liver are long-lived and do not recirculate but can be replenished from the circulation. J Immunol (2016) 197:4283–91. doi:10.4049/jimmunol.1601424
9. Hudspeth K, Donadon M, Cimino M, Pontarini E, Tentorio P, Preti M, et al. Human liver-resident CD56bright/CD16neg NK cells are retained within hepatic sinusoids via the engagement of CCR5 and CXCR6 pathways. J Autoimmun (2016) 66:40–50. doi:10.1016/j.jaut.2015.08.011
10. Aw Yeang HX, Piersma SJ, Lin Y, Yang L, Malkova ON, Miner C, et al. Cutting edge: human CD49e − NK cells are tissue resident in the liver. J Immunol (2017) 198:1417–22. doi:10.4049/jimmunol.1601818
11. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity (2013) 38:769–81. doi:10.1016/j.immuni.2013.02.010
12. Lugthart G, Melsen JE, Vervat C, van Ostaijen-ten Dam MM, Corver WE, Roelen DL, et al. Human lymphoid tissues harbor a distinct CD69 + CXCR6 + NK cell population. J Immunol (2016) 197:78–84. doi:10.4049/jimmunol.1502603
13. Melsen JE, Lugthart G, Lankester AC, Schilham MW. Human circulating and tissue-resident CD56(bright) natural killer cell populations. Front Immunol (2016) 7:262. doi:10.3389/fimmu.2016.00262
14. Björkström NK, Ljunggren H-G, Michaëlsson J. Emerging insights into natural killer cells in human peripheral tissues. Nat Rev Immunol (2016) 16:310–20. doi:10.1038/nri.2016.34
15. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol (2001) 22:633–40. doi:10.1016/S1471-4906(01)02060-9
16. Cooper M, Fehniger T, Turner SC, Chen KS, Ghaheri B, Ghayur T, et al. Human natural killer cells : a unique innate immunoregulatory role for the CD56 bright subset. Blood (2001) 97:3146–51. doi:10.1182/blood.V97.10.3146
17. Fauriat C, Long EEO, Ljunggren H-G, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood (2010) 115:2167–76. doi:10.1182/blood-2009-08-238469.A
18. Béziat V, Duffy D, Quoc SN, Le Garff-Tavernier M, Decocq J, Combadière B, et al. CD56brightCD16+ NK cells: a functional intermediate stage of NK cell differentiation. J Immunol (2011) 186:6753–61. doi:10.4049/jimmunol.1100330
19. Clark RA, Watanabe R, Teague JE, Schlapbach C, Tawa MC, Adams N, et al. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci Transl Med (2012) 4:1–11. doi:10.1126/scitranslmed.3003008
20. Zhu J, Peng T, Johnston C, Phasouk K, Kask AS, Klock A, et al. Immune surveillance by CD8αα+ skin-resident T cells in human herpes virus infection. Nature (2013) 497:494–7. doi:10.1038/nature12110
21. McCully ML, Ladell K, Andrews R, Jones RE, Miners KL, Roger L, et al. CCR8 expression defines tissue-resident memory T cells in human skin. J Immunol (2018) 200:1639–50. doi:10.4049/jimmunol.1701377
22. Woon HG, Braun A, Li J, Smith C, Edwards J, Sierro F, et al. Compartmentalization of total and virus-specific tissue-resident memory CD8+ T cells in human lymphoid organs. PLoS Pathog (2016) 12:e1005799. doi:10.1371/journal.ppat.1005799
23. Hombrink P, Helbig C, Backer RA, Piet B, Oja AE, Stark R, et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat Immunol (2016) 17:1467–78. doi:10.1038/ni.3589
24. Stelma F, de Niet A, Sinnige MJ, van Dort KA, van Gisbergen KPJM, Verheij J, et al. Human intrahepatic CD69 + CD8+ T cells have a tissue resident memory T cell phenotype with reduced cytolytic capacity. Sci Rep (2017) 7:6172. doi:10.1038/s41598-017-06352-3
25. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep (2017) 20:2921–34. doi:10.1016/j.celrep.2017.08.078
26. Mackay LK, Minnich M, Kragten NAM, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science (2016) 352:459–63. doi:10.1126/science.aad2035
27. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon M-L, et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat Immunol (2013) 14:1294–301. doi:10.1038/ni.2744
28. Wakim LM, Woodward-Davis A, Liu R, Hu Y, Villadangos J, Smyth G, et al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol (2012) 189:3462–71. doi:10.4049/jimmunol.1201305
29. Fernandez-Ruiz D, Ng WY, Holz LE, Ma JZ, Zaid A, Wong YC, et al. Liver-resident memory CD8 + T cells form a front-line defense against malaria liver-stage infection. Immunity (2016) 45:889–902. doi:10.1016/j.immuni.2016.08.011
30. Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, et al. T-box transcription factors combine with the cytokines TGF-β and IL-15 to control tissue-resident memory T cell fate. Immunity (2015) 43:1101–11. doi:10.1016/j.immuni.2015.11.008
31. Zhang N, Bevan M. Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity (2013) 39:687–96. doi:10.1016/j.immuni.2013.08.019
32. Sheridan BS, Pham Q-M, Lee Y-T, Cauley LS, Puddington L, Lefrançois L. Oral infection drives a distinct population of intestinal resident memory CD8+ T cells with enhanced protective function. Immunity (2014) 40:747–57. doi:10.1016/j.immuni.2014.03.007
33. Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol (2014) 15:R29. doi:10.1186/gb-2014-15-2-r29
34. Zhao S, Guo Y, Sheng Q, Shyr Y. Advanced heat map and clustering analysis using heatmap3. Biomed Res Int (2014) 2014:986048. doi:10.1155/2014/986048
35. Wickham H. Elegant graphics for data analysis. Media (2009) 35:211. doi:10.1007/978-0-387-98141-3
36. Heemskerk B, van Vreeswijk T, Veltrop-Duits LA, Sombroek CC, Franken K, Verhoosel RM, et al. Adenovirus-specific CD4+ T cell clones recognizing endogenous antigen inhibit viral replication in vitro through cognate interaction. J Immunol (2006) 177:8851–9. doi:10.4049/jimmunol.177.12.8851
37. Wu D, Smyth GK. Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res (2012) 40:1–12. doi:10.1093/nar/gks461
38. Lee E, Chuang H-Y, Kim J-W, Ideker T, Lee D. Inferring pathway activity toward precise disease classification. PLoS Comput Biol (2008) 4:e1000217. doi:10.1371/journal.pcbi.1000217
39. Serafini N, Vosshenrich CA, Di Santo JP. Transcriptional regulation of innate lymphoid cell fate. Nat Rev Immunol (2015) 15:415–28. doi:10.1038/nri3855
40. Collins A, Rothman N, Liu K, Reiner SL. Eomesodermin and T-bet mark developmentally distinct human natural killer cells. JCI Insight (2017) 2:e90063. doi:10.1172/jci.insight.90063
41. van Helden MJ, Goossens S, Daussy C, Mathieu A-L, Faure F, Marçais A, et al. Terminal NK cell maturation is controlled by concerted actions of T-bet and Zeb2 and is essential for melanoma rejection. J Exp Med (2015) 212:2015–25. doi:10.1084/jem.20150809
42. Carlson CM, Endrizzi BT, Wu J, Ding X, Weinreich MA, Walsh ER, et al. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature (2006) 442:299–302. doi:10.1038/nature04882
43. Bai A, Hu H, Yeung M, Chen J. Kruppel-like factor 2 controls T cell trafficking by activating L-selectin (CD62L) and sphingosine-1-phosphate receptor 1 transcription. J Immunol (2007) 178:7632–9. doi:10.4049/jimmunol.178.12.7632
44. Vieira Braga FA, Hertoghs KML, Kragten NAM, Doody GM, Barnes NA, Remmerswaal EBM, et al. Blimp-1 homolog Hobit identifies effector-type lymphocytes in humans. Eur J Immunol (2015) 45:2945–58. doi:10.1002/eji.201545650
45. Ochiai K, Katoh Y, Ikura T, Hoshikawa Y, Noda T, Karasuyama H, et al. Plasmacytic transcription factor Blimp-1 is repressed by Bach2 in B cells. J Biol Chem (2006) 281:38226–34. doi:10.1074/jbc.M607592200
46. Mao A-P, Constantinides MG, Mathew R, Zuo Z, Chen X, Weirauch MT, et al. Multiple layers of transcriptional regulation by PLZF in NKT-cell development. Proc Natl Acad Sci U S A (2016) 113:7602–7. doi:10.1073/pnas.1601504113
47. Amit I, Citri A, Shay T, Lu Y, Katz M, Zhang F, et al. A module of negative feedback regulators defines growth factor signaling. Nat Genet (2007) 39:503–12. doi:10.1038/ng1987
48. Hanafusa H, Torii S, Yasunaga T, Nishida E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat Cell Biol (2002) 4:850–8. doi:10.1038/ncb867
49. Baume DM, Robertson MJ, Levine H, Manley TJ, Schow PW, Ritz J. Differential responses to interleukin 2 define functionally distinct subsets of human natural killer cells. Eur J Immunol (1992) 22:1–6. doi:10.1002/eji.1830220102
50. Wong MT, Ong DEH, Lim FSH, Teng KWW, McGovern N, Narayanan S, et al. A high-dimensional atlas of human T cell diversity reveals tissue-specific trafficking and cytokine signatures. Immunity (2016) 45:442–56. doi:10.1016/j.immuni.2016.07.007
51. Peng H, Jiang X, Chen Y, Sojka DK, Wei H, Gao X, et al. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J Clin Invest (2013) 123:1444–56. doi:10.1172/JCI66381
52. Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, et al. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med (2014) 211:563–77. doi:10.1084/jem.20131560
53. Kunkel EJ, Butcher EC. Chemokines and the tissue-specific migration of lymphocytes. Immunity (2002) 16:1–4. doi:10.1016/S1074-7613(01)00261-8
54. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev (2017) 276:112–20. doi:10.1111/imr.12518
55. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A (2009) 106:17858–63. doi:10.1073/pnas.0903474106
56. Wang F, Hou H, Wu S, Tang Q, Liu W, Huang M, et al. TIGIT expression levels on human NK cells correlate with functional heterogeneity among healthy individuals. Eur J Immunol (2015) 45:2886–97. doi:10.1002/eji.201545480
57. Seiler MP, Mathew R, Liszewski MK, Spooner C, Barr K, Meng F, et al. Elevated and sustained expression of the transcription factors Egr1 and Egr2 controls NKT lineage differentiation in response to TCR signaling. Nat Immunol (2012) 13:264–71. doi:10.1038/ni.2230
58. Li G, Yu M, Lee WW, Tsang M, Krishnan E, Weyand CM, et al. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat Med (2012) 18:1518–24. doi:10.1038/nm.2963
59. González-Navajas JM, Fine S, Law J, Datta SK, Nguyen KP, Yu M, et al. TLR4 signaling in effector CD4 + T cells regulates TCR activation and experimental colitis in mice. J Clin Invest (2010) 120:570–81. doi:10.1172/JCI40055DS1
60. Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene (2007) 26:3203–13. doi:10.1038/sj.onc.1210412
61. Nagashima T, Shimodaira H, Ide K, Nakakuki T, Tani Y, Takahashi K, et al. Quantitative transcriptional control of ErbB receptor signaling undergoes graded to biphasic response for cell differentiation. J Biol Chem (2007) 282:4045–56. doi:10.1074/jbc.M608653200
62. Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol (2015) 16:1–11. doi:10.1038/nri.2015.3
63. Cheuk S, Schlums H, Gallais Sérézal I, Martini E, Chiang SC, Marquardt N, et al. CD49a expression defines tissue-resident CD8 + T cells poised for cytotoxic function in human skin. Immunity (2017) 46:287–300. doi:10.1016/j.immuni.2017.01.009
64. Cortez VS, Cervantes-Barragan L, Robinette ML, Bando JK, Wang Y, Geiger TL, et al. Transforming growth factor-β signaling guides the differentiation of innate lymphoid cells in salivary glands. Immunity (2016) 44:1–13. doi:10.1016/j.immuni.2016.03.007
65. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol (2017) 18:1004–15. doi:10.1038/ni.3800
66. Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8 + T RM cells providing global skin immunity. Nature (2012) 483:227–31. doi:10.1038/nature10851
67. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol (2009) 10:524–30. doi:10.1038/ni.1718
68. Wu T, Hu Y, Lee Y-T, Bouchard KR, Benechet A, Khanna K, et al. Lung-resident memory CD8 T cells (T RM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol (2014) 95:215–24. doi:10.1189/jlb.0313180
69. Ariotti S, Beltman JB, Chodaczek G, Hoekstra ME, van Beek AE, Gomez-Eerland R, et al. Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. Proc Natl Acad Sci U S A (2012) 109:19739–44. doi:10.1073/pnas.1208927109
70. Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science (2014) 346:98–101. doi:10.1126/science.1254536
Keywords: natural killer cells, tissue-resident, RNA sequence, CD8+ T cells, lymphoid tissue
Citation: Melsen JE, Lugthart G, Vervat C, Kielbasa SM, van der Zeeuw SAJ, Buermans HPJ, van Ostaijen-ten Dam MM, Lankester AC and Schilham MW (2018) Human Bone Marrow-Resident Natural Killer Cells Have a Unique Transcriptional Profile and Resemble Resident Memory CD8+ T Cells. Front. Immunol. 9:1829. doi: 10.3389/fimmu.2018.01829
Received: 11 June 2018; Accepted: 24 July 2018;
Published: 22 August 2018
Edited by:
Eric Vivier, INSERM U1104 Centre d’Immunologie de Marseille-Luminy, FranceReviewed by:
Amir Horowitz, Icahn School of Medicine at Mount Sinai, United StatesFrederic Vely, INSERM U1104 Centre d’Immunologie de Marseille-Luminy, France
Copyright: © 2018 Melsen, Lugthart, Vervat, Kielbasa, van der Zeeuw, Buermans, van Ostaijen-ten Dam, Lankester and Schilham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Janine E. Melsen, ai5lLm1lbHNlbkBsdW1jLm5s