 Maria Pia Cicalese1,2,3†
Maria Pia Cicalese1,2,3† Jolanda Gerosa4†
Jolanda Gerosa4† Manuela Baronio5
Manuela Baronio5 Davide Montin6Francesco Licciardi6Annarosa Soresina7
Davide Montin6Francesco Licciardi6Annarosa Soresina7 Rosa Maria Dellepiane8Maurizio Miano9Lucia Augusta Baselli8Stefano Volpi10Carlo Dufour9,10Alessandro Plebani5Alessandro Aiuti1,2,3
Rosa Maria Dellepiane8Maurizio Miano9Lucia Augusta Baselli8Stefano Volpi10Carlo Dufour9,10Alessandro Plebani5Alessandro Aiuti1,2,3 Vassilios Lougaris5†
Vassilios Lougaris5† Georgia Fousteri4*†
Georgia Fousteri4*†
- 1San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET), IRCCS San Raffaele Scientific Institute, Milan, Italy
- 2Pediatric Immunohematology and Bone Marrow Transplantation Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 3Vita-Salute San Raffaele University, Milan, Italy
- 4Division of Immunology Transplantation and Infectious Diseases (DITID), Diabetes Research Institute (DRI) IRCCS San Raffaele Scientific Institute, Milan, Italy
- 5Department of Clinical and Experimental Sciences, Pediatrics Clinic and Institute of Molecular Medicine A. Novicelli, University of Brescia, ASST-Spedali Civili of Brescia, Brescia, Italy
- 6Immuno-Rheumatology, Department of Paediatrics II, Regina Margherita Hospital, Città della Salute e della Scienza di Torino, Torino, Italy
- 7Pediatrics Clinic, ASST-Spedali Civili di Brescia, Brescia, Italy
- 8Department of Pediatrics, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico, University of Milan, Milan, Italy
- 9Department of Emato-Oncology, IRCCS Instituto Giannina Gaslini, Genoa, Italy
- 10Department of Pediatrics, IRCCS Instituto Giannina Gaslini, Genoa, Italy
Mutations in genes that control class switch recombination and somatic hypermutation during the germinal center (GC) response can cause diverse immune dysfunctions. In particular, mutations in CD40LG, CD40, AICDA, or UNG cause hyper-IgM (HIGM) syndrome, a heterogeneous group of primary immunodeficiencies. Follicular helper (Tfh) and follicular regulatory (Tfr) T cells play a key role in the formation and regulation of GCs, but their role in HIGM pathogenesis is still limited. Here, we found that compared to CD40 ligand (CD40L)- and activation-induced cytidine deaminase (AICDA)-deficient patients, circulating Tfh and Tfr cells were severely compromised in terms of frequency and activation phenotype in a child with CD40 deficiency. These findings offer useful insight for human Tfh biology, with potential implications for understanding the molecular basis of HIGM syndrome caused by mutations in CD40.
Background
Hyper-IgM (HIGM) syndromes comprise a group of rare primary immunodeficiencies characterized by low or absent IgG and IgA and normal to elevated levels of IgM (1, 2). CD40 ligand (CD40L) deficiency is X-linked and the estimated frequency is 2:1,000,000 males (3). Although no data are available on the frequency of activation-induced cytidine deaminase (AID) deficiency, this disorder is estimated to affect less than 1:1,000,000 individuals (4). In contrast, there are only a few reported cases of CD40 and uracil N-glycosylase (UNG) deficiencies (5–9).
Patients affected by HIGM present defects in class switch recombination and somatic hypermutation, two events that take place in germinal centers (GCs) (2–6). These processes are compromised in HIGM patients due to impaired cross-talk between T and B cells caused by mutations in CD40LG and CD40 or intrinsic B cell defects due to mutations in AICDA and UNG (10, 11). Clinically, affected patients present recurrent respiratory and gastrointestinal infections, and in some cases autoimmune manifestations (1–3). Patients with mutations in CD40L or CD40 are particularly susceptible to Pneumocystis carinii pneumonia (PCP) and Cryptosporidium infection (3, 7). Autoimmune manifestations may occur in all forms of HIGM, although they seem more frequent in AID deficiency (12, 13). Two recent studies reported a reduction in the percentage of CD4+CD25+FOXP3+ T regulatory cells (Tregs), accompanied by an increase in the Th17/Treg cell ratio and Th1/Treg cell ratio in CD40L-deficient patients (14, 15). Similarly, patients with mutations in AICDA, but not UNG-deficient patients, displayed Treg cells with defective suppressive function (16). These results may explain the increased susceptibility to autoimmunity in these disorders.
Follicular helper T cells (CXCR5+FOXP3−) (Tfh) and follicular regulatory T cells (CXCR5+FOXP3+) (Tfr) are considered key players for the formation and regulation of GCs, respectively (17–19). To date, available data regarding these T cell subsets in HIGM syndromes are limited and in some cases discordant (16, 20, 21). Furthermore, studies on Tfh and Tfr cells in humans with an extremely rare form of HIGM caused by CD40 deficiency are completely lacking. Here, we report for the first time the distribution and phenotype of circulating Tfh and Tfr cells in a patient with CD40 deficiency and compare them to CD40L and AID-deficient patients.
Methods
Clinical Cases and Controls
Ten patients with diagnosis of monogenic HIGM syndromes referred from the Pediatrics Clinic, University of Brescia and ASST-Spedali Civili of Brescia, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico, Milan, IRCCS Instituto Giannina Gaslini, Genoa and Immuno-rheumatology, II, Regina Margherita Hospital, Turin, Italy, were enrolled in the study. All the subjects, their parents or guardians provided a written informed consent. The study was approved by the local ethics committee (Comitato Etico Ospedale San Raffaele, Milano) and performed in accordance with the Declaration of Helsinki.
The study group included one pediatric patient with CD40 deficiency, three patients with AID deficiency (one adult and two pediatric) and six patients with CD40L deficiency [two adult and four pediatric (one patient was analyzed in pediatric and adult age)]. Clinical characteristics of the study participants are shown in Table 1. Seven pediatric (6 months–10 years) and nine adult (23–40 years) healthy donors were included as controls (HC).
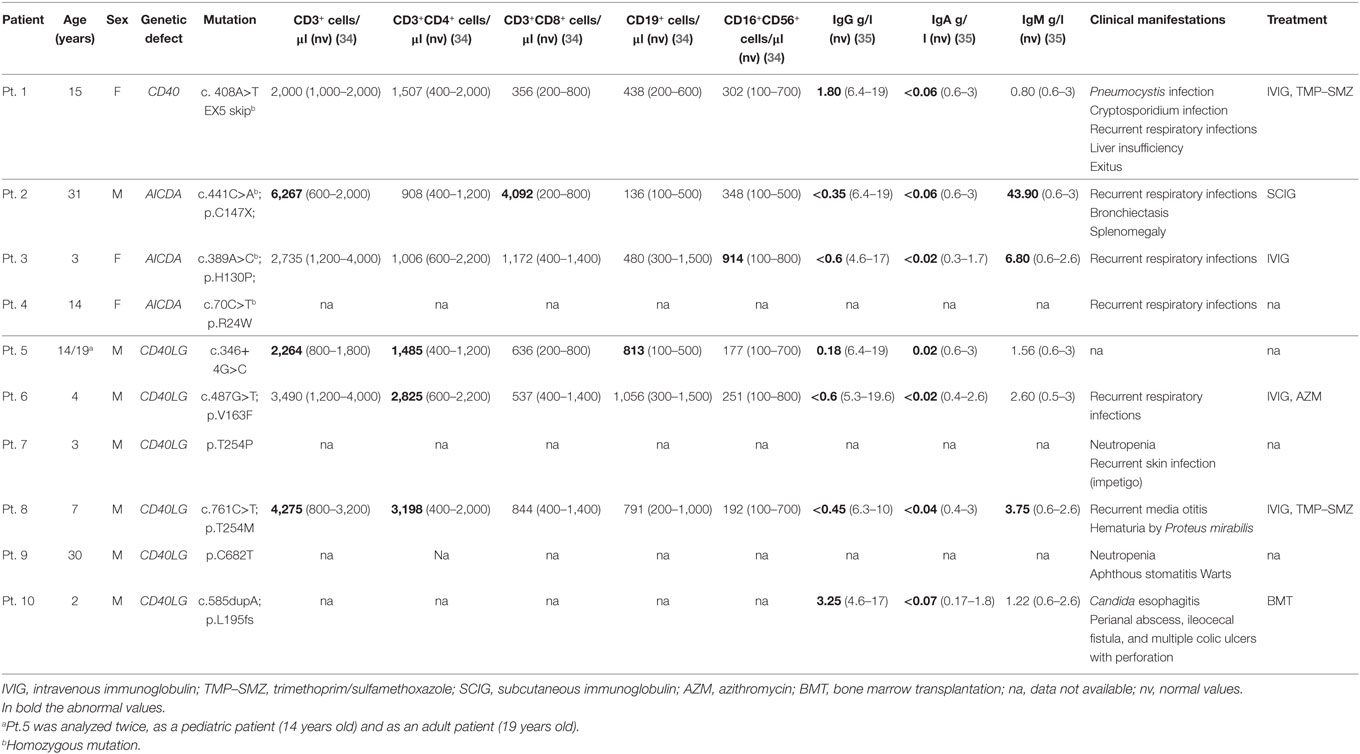
Table 1. Patients’ mutations, clinical characteristics and immunological profile.
Sample Collection and Analysis
Peripheral blood mononuclear cells (PBMCs) were isolated by density-gradient centrifugation over Lymphoprep (Stemcell) from heparinized venous blood and frozen in RPMI (Lonza) with addition of 10% DMSO and 25% FBS. PBMCs were then thawed in water bath at 37° and stained for flow cytometry with superficial antibodies against CD4 (SK3), CD3 (SK7), CD45RA (H100), CD19 (4G7), CD14 (TUK4), CD8 (BW135/80), CXCR5 (RF8B2), PD-1 (eBioJ105), and ICOS (ISA-3) (Ab clones are indicated in the parentheses). Cells were fixed and permeabilized for intracellular staining with the FOXP3 Transcription Factor Staining Buffer Set (eBioscience) and stained with antibodies against FOXP3 (259D). Cells were acquired on FACSCantoII (BD) and analyzed with FlowJo (Tree Star) software. Mann–Whitney statistical analysis was performed between HC and patient groups with the prism software (San Diego).
Results
Summary of Case Report
The CD40 deficient patient was born from first-degree consanguineous Italian parents. The patient was admitted at the age of 4 months for severe respiratory distress due to PCP, and subsequently re-admitted at the age of 2 years for another pneumonia event. During the second infectious episode, immunological work-up showed low IgG and IgA with normal IgM serum levels and received immunoglobulin replacement treatment and trimethoprim/sulfamethoxazole prophylaxis (Table 1). During follow-up, the patient presented recurrent respiratory infections. The clinical history was complicated by Cryptosporidium infection at the age of 12 years that led to progressive end-stage liver dysfunction. The patient over time developed esophageal varices, diffuse abdominal spider naevi with ascitis and severe mucositis and became transfusion dependent. Considering the patient’s severe clinical course, the parents decided against hematopoietic stem cell transplantation. The severity of her clinical conditions led to exitus at the age of 16 years (5).
Tfh and Tfr Cell Frequencies in Human CD40 Deficiency
Comparison between pediatric and adult HCs showed differences in the percentage of Tfh cells: pediatric HCs had lower frequency of Tfh cells within CD4+ T cells when compared with adult HCs (Figures 1A,B), reflecting the expansion of this memory T cell subset with age. Instead, frequencies of Tfr and Treg (CXCR5−FOXP3+) cells did not change with age (not shown). Since Tfh cells differed significantly between pediatric and adult HCs, patients were compared with age-matched HCs. Tfh, Tfr, and Treg cells were severely reduced in the child with CD40 deficiency as compared with healthy children (Figures 1B–E). Reduction in Tfh and Treg but not in Tfr cell frequencies were also observed in children with CD40L deficiency as compared with age-matched HCs, while children with AID deficiency had normal distribution of all cell subsets (Figures 1B–E).
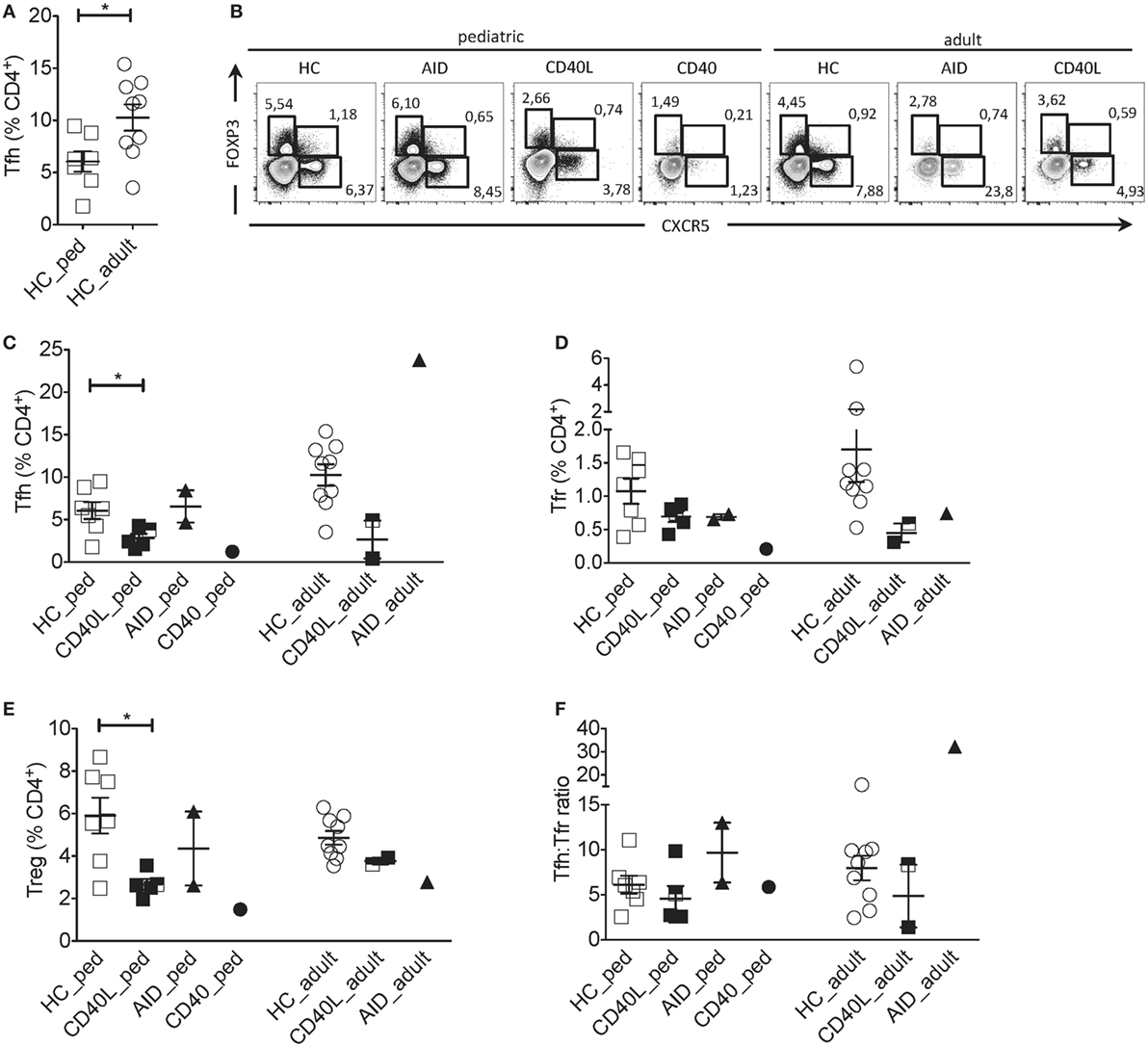
Figure 1. Circulating Tfh and Tfr cells in an individual with CD40 deficiency compared to patients with CD40L and AICDA mutations. (A) Percentage of Tfh (gated on singlets → lymphocytes → CD3+CD19−CD14−CD8− → CD4+CD3+) in healthy pediatric (3–15 years, n = 7, HC_ped) and healthy adult (19–31 years, n = 9; HC_adult) healthy controls (HCs). (B) Representative flow cytometry plots for Tfh (CXCR5+FOXP3−), Tfr (CXCR5+FOXP3+), and T regulatory (Treg) (CXCR5−FOXP3+) cells, gated on singlets → lymphocytes → CD3+CD19−CD14−CD8− → CD4+CD3+ in hyper-IgM (HIGM) patients and age-matched HCs. (C–E) Percentage of Tfh (C), Tfr (D), and Treg (E) cells in pediatric [CD40 ligand (CD40L), n = 5; activation-induced cytidine deaminase (AID), n = 2; CD40, n = 1] and adult (CD40L, n = 2; AID, n = 1) HIGM patients, compared with age-matched HCs [same as in panel (A)]. (F) Tfh:Tfr ratio cells in pediatric and adult HIGM patients (AID, CD40L, and CD40 deficiency, as indicated) compared with age-matched HCs. Bars: mean ± SEM. *p < 0.05 (Mann–Whitney test). Each dot represents one patient. Black & white squares represent longitudinal measurements of the same CD40L-deficient patient, collected at 14 and 19 years of age.
The circulating Tfh:Tfr ratio provides important insights into the function and regulation of GC responses when evaluated alongside the frequency of Tfh and Tfr. For example, an increased ratio of Tfh:Tfr was found in patients with Sjogren’s syndrome (22), suggesting the persistence of ongoing GC reactions. We observed that despite the alterations in Tfh and Tfr cells in CD40, CD40L and AID deficient pediatric patients, the ratio of Tfh:Tfr was similar to that observed in age-matched HCs (Figure 1F).
Circulating Tfh, Tfr and Treg cells were also reduced in adult CD40L-deficient patients compared with age-matched HCs (Figures 1B–E). While the Tfh:Tfr ratio was conserved in one adult CD40L-deficient patient, it was reduced in the other one (Figure 1F). PBMCs from one CD40L-deficient patient were examined at a pediatric and an adult age (marked with bicolor). The percentage of Tfh and Tfr cells remained relatively stable during the follow-up (Figure 1C). On the contrary, Tfh cell frequency was increased in the single adult AID-deficient patient (Figure 1C), while Tfr and Treg cells were not affected (Figures 1D,E).
Activation Phenotype of Tfh and Tfr Cells in Human CD40 Deficiency
Next, we addressed the expression of PD-1 and ICOS, two molecules typically expressed by activated Tfh cells that correlate with Tfh cell function (17–19). Given that the proportion of Tfh cells expressing PD-1 and ICOS was higher in pediatric as compared with adult HCs (Figures 2A,B), clinical samples were compared with age-matched HCs. The proportion of PD-1+ Tfh cells but not the PD-1 geometric mean fluorescence intensity (gMFI) was severely reduced in the CD40-deficient child but not the CD40L and AID-deficient children when compared with HCs (Figures 2B,C; Figure S1A in Supplementary Material). The proportions of ICOS+ and PD-1+ICOS+ Tfh cells but not ICOS gMFI were reduced in several patients affected by HIGM when compared with HCs, irrespective the genetic defect. However, the reduction in ICOS was more pronounced in the CD40-deficient child and some patients with CD40L deficiency (Figures 2B,D,E and data not shown).
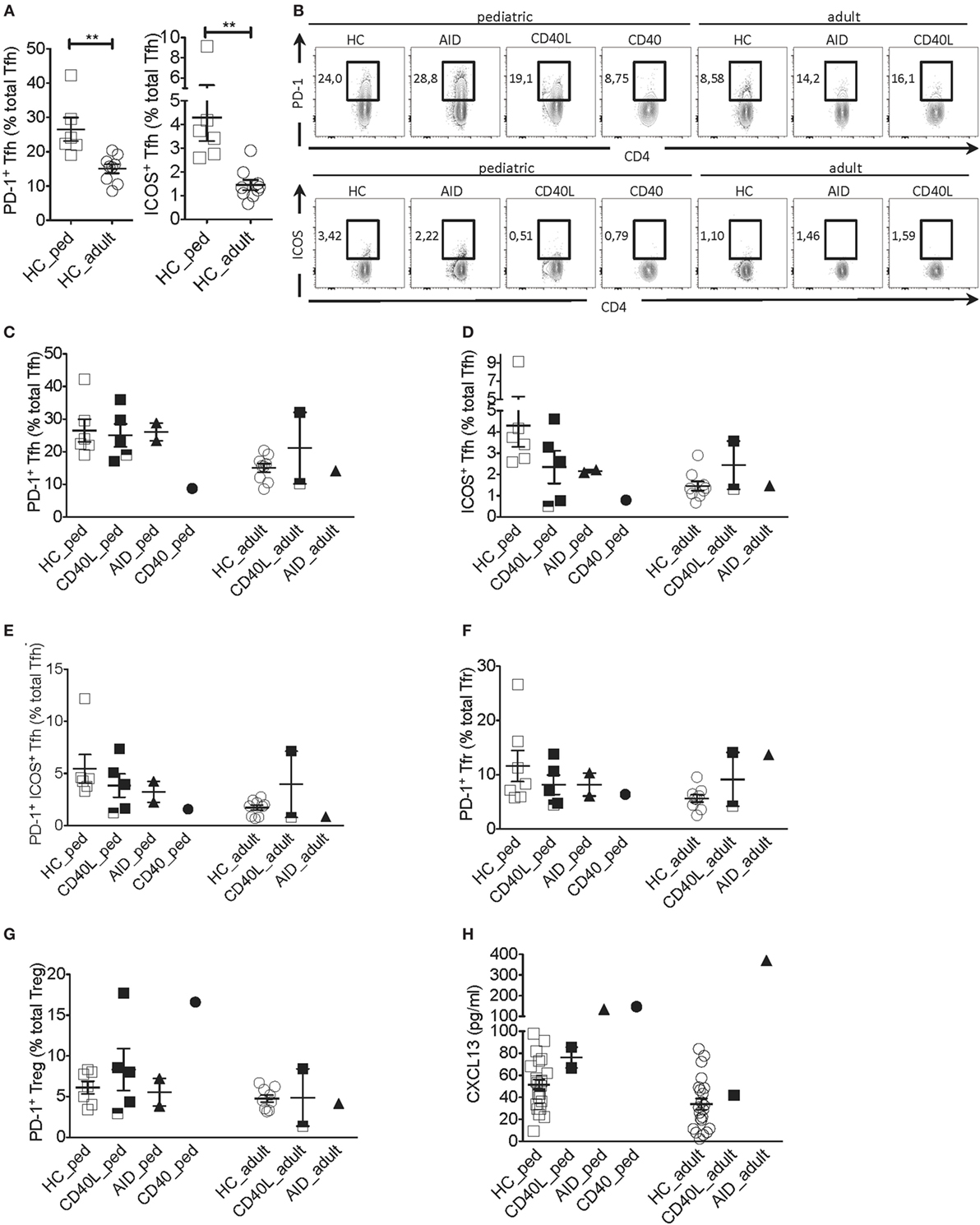
Figure 2. Tfh cell activation phenotype in a patient with CD40 deficiency compared with patients with CD40L and AICDA mutations. (A) Percentage of PD-1+ and ICOS+ Tfh cells in peripheral blood of a pediatric (HC_ped) and adult (HC_adult) healthy controls (HCs) as described in Figure 1 (gated on singlets → lymphocytes → CD3+CD19−CD14−CD8− → CD4+CD3+ → CD4+CXCR5+). (B) Representative plots for PD-1+ and ICOS+ within Tfh cells in hyper-IgM (HIGM) patients and age-matched HCs. (C,D) Percentage of PD-1+ (C) and ICOS+ (D) Tfh cells in pediatric and adult HIGM patients and age-matched HCs (same as in Figure 1). (E) Percentage of PD-1+ICOS+ Tfh cells in pediatric and adult HIGM patients compared with age-matched HCs. (F,G) The proportion of Tfr (F) and T regulatory (Treg) (G) cells expressing PD-1 in pediatric and adult HIGM patients compared with age-matched HCs. (H) CXCL13 levels in the plasma of pediatric [CD40 ligand (CD40L), n = 2; activation-induced cytidine deaminase (AID), n = 1; CD40, n = 1] and adult HIGM patients (CD40L, n = 1; AID, n = 1) and age-matched HCs (pediatric HC, n = 25, adult HC n = 23). Bars: mean ± SEM. **p < 0.01 (Mann–Whitney test). Each dot represents one patient. Black & white squares represent longitudinal measurements of the same CD40L-deficient patient, collected at 14 and 19 years of age.
Interestingly, the proportion Tfr and Treg cells expressing PD-1 was elevated in the pediatric CD40-deficient patient (Figure 2G). Treg cells also expressed higher levels of PD-1 in this patient (Figure S1C in Supplementary Material). Instead, the proportion and expression levels of PD-1 on Tfr and Treg cells in children with CD40L and AID deficiency were similar to those seen in HCs (Figures 2F,G; Figures S1B,C in Supplementary Material). The percentage of PD-1+ Tfr cells was slightly elevated in one adult CD40L-deficient and the adult AID-deficient patient as compared with HCs (Figures 2F,G).
Unexpectedly, plasma levels of CXCL13, a chemokine considered to be indicative of GC activity (23), were found elevated in the patient with CD40-deficiency, but were within the normal range in the CD40L-deficient patients (Figure 2H). Patients with AID deficiency had also elevated plasma levels of CXCL13 compared with HCs, in line with the hyper-reactive GCs described in this disorder (17).
Discussion
Our findings report the first phenotypic characterization of Tfh and Tfr in human CD40 deficiency and describe differences to other HIGM syndromes caused by mutations in CD40L and AICDA. The single patient with CD40 deficiency analyzed, showed a marked reduction in circulating Tfh and Tfr cells suggesting that lack of CD40 signals contribute to an early demise of GCs in humans, similarly to mice (24). While the precise mechanism of CD40 signaling is poorly understood, CD40 ligands on T cells and follicular dendritic cells (FDCs) were shown to be essential for T-dependent and T-independent GC responses, respectively (25). In contrast to other forms of HIGM and particularly the CD40L deficiency, patients with CD40 deficiency display a more severe clinical phenotype. It has been previously shown that CD40L is not the only ligand that can bind to CD40. A protein that binds activated complement 3b (C3b) and C4b, C4BP, on FDCs can also deliver signals to B cells via CD40 during T-cell independent responses (26). Possibly, the lack of these T-independent signals in human CD40 deficiency might have contributed in the aggravated clinical phenotype of this form of HIGM.
Antigen-specific interactions between Tfh cells and B cells are required from the very first stage of their interaction to allow B-cell differentiation toward the GC fate and the maintenance of the GC response (27). B cells in return, provide signals for GC Tfh formation to Tfh cells suggesting that B cells and Tfh cells are mutually dependent on each other for their differentiation into GC B cells and GC Tfh cells, respectively (17–19, 28, 29). CD40L–CD40 interactions along with integrin and SAP-dependent contacts between B and T cells were shown to be essential for this process (30–32). In agreement to previous reports, we found that patients with CD40L deficiency generated a small number of Tfh cells (21). Instead, circulating Tfh cells in a patient with CD40 deficiency were heavily compromised. Although the blood is not the most suitable tissue to address the effect of CD40:CD40L signaling in Tfh development, our results suggest that other mechanisms might be able to compensate for the lack of CD40L. AICDA mutations on the other hand, increased the number of circulating Tfh cells in line with previous results supporting the notion that the reduced efficiency in generating highly mutated antibodies leads to enhanced GC reactions and increased Tfh cell development (16).
Both CD40L and CD40 deficiencies were characterized by normal or even elevated concentration of CXCL13 in the plasma. Tfh along with FDCs are the main source of CXCL13 in the B cell follicles (23). Recently, a CXCR5−PD-1++ Tfh population was described in the tumor infiltrate of patients with breast cancer and the synovial fluid of patients with rheumatoid arthritis as an important source of CXCL13 (33). The percentage of CXCR5−PD-1++ cells was not increased in the blood of CD40- nor CD40L-deficient patients (data not included), suggesting the FDCs were the main source of CXCL13. Alternatively, the lack of “CXCL3 consumption” by GC Tfh and B cells might have led to an increase in the plasma CXCL13 levels in human CD40 and CD40L deficiency.
Concluding Remarks
Although studies on additional CD40-deficient patients are necessary, analysis of an individual with CD40 deficiency suggests that CD40 is possibly required for Tfh and Tfr development in humans. Compared with other genetically characterized forms of HIGM, i.e., due to mutations in CD40L and AICDA, patients with CD40 deficiency show a more severe clinical phenotype. According to our analyses, this could be partially explained by the more severe impairment of Tfh and Tfr cells. Longitudinal studies on a larger number of samples, i.e. blood and secondary lymphoid organs from patients with CD40 deficiency may offer useful insights in human Tfh biology, with important implications for understanding human GC development and potentially for the management of patients affected with HIGM or other primary immunodeficiencies.
Ethics Statement
All the subjects, their parents, or tutors for minors gave their written informed consent. The study was approved by the local ethic committee (Comitato Etico Ospedale San Raffaele, Milano) and performed in accordance with the Declaration of Helsinki.
Author Contributions
MC: contributed to scientific discussion and wrote the manuscript. JG: performed the experiments and data analysis and contributed to manuscript writing. MB: provided samples and contributed to manuscript writing. DM, FL, AS, RD, MM, LB, SV, CD, AP, and AA: provided samples and read and approved the manuscript. VL: provided samples, contributed to scientific discussion, and wrote the manuscript. GF: designed and supervised the study, coordinated scientific discussion and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge 5x1000 OSR PILOT & SEED GRANT by Ospedale San Raffaele and C. Golgi Foundation of Brescia for funding this study. Moreover, we acknowledge the nurses, the patients and their families.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01761/full#supplementary-material.
Figure S1. Geometric mean fluorescence intensity (gMFI) of PD-1 in a patient with CD40 deficiency compared with patients with CD40L and AICDA mutations. (A–C) gMFI of PD-1 on Tfh (A), Tfr (B) and Treg cells (C) in pediatric and adult hyper-IgM (HIGM) patients compared with age-matched health controls (HCs) (same as in Figure 1). Bars: mean ± SEM. Each dot represents one patient. Black & white squares represent longitudinal measurements of the same CD40 ligand (CD40L)-deficient patient, collected at 14 and 19 years of age.
Abbreviations
HIGM, hyper-IgM; CSR, class switch recombination; SHM, somatic hypermutation; GC, germinal center; AID, activation-induced cytidine deaminase; CD40L, CD40 ligand; UNG, uracil N-glycosylase; HC, healthy control; Tfh, T follicular helper; Treg, T regulatory; Tfr, T follicular regulatory; FDCs, follicular dendritic cells; C3b, complement 3b; TMP–SMZ, trimethoprim/sulfamethoxazole; PCP, Pneumocystis carinii pneumonia; HSCT, hematopoietic stem cell transplantation.
References
1. de la Morena MT. Clinical phenotypes of hyper-IgM syndromes. J Allergy Clin Immunol Pract (2016) 4(6):1023–36. doi:10.1016/j.jaip.2016.09.013
2. Davies EG, Thrasher AJ. Update on the hyper immunoglobulin M syndromes. Br J Haematol (2010) 149(2):167–80. doi:10.1111/j.1365-2141.2010.08077.x
3. Winkelstein JA, Marino MC, Ochs H, Fuleihan R, Scholl PR, Geha R, et al. The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine (Baltimore) (2003) 82(6):373–84. doi:10.1097/01.md.0000100046.06009.b0
4. Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell (2000) 102(5):565–75. doi:10.1016/S0092-8674(00)00079-9
5. Ferrari S, Giliani S, Insalaco A, Al-Ghonaium A, Soresina AR, Loubser M, et al. Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc Natl Acad Sci U S A (2001) 98(22):12614–9. doi:10.1073/pnas.221456898
6. Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat Immunol (2003) 4(10):1023–8. doi:10.1038/ni974
7. Kutukculer N, Moratto D, Aydinok Y, Lougaris V, Aksoylar S, Plebani A, et al. Disseminated cryptosporidium infection in an infant with hyper-IgM syndrome caused by CD40 deficiency. J Pediatr (2003) 142(2):194–6. doi:10.1067/mpd.2003.41
8. Lanzi G, Ferrari S, Vihinen M, Caraffi S, Kutukculer N, Schiaffonati L, et al. Different molecular behavior of CD40 mutants causing hyper-IgM syndrome. Blood (2010) 116(26):5867–74. doi:10.1182/blood-2010-03-274241
9. Mazzolari E, Lanzi G, Forino C, Lanfranchi A, Aksu G, Ozturk C, et al. First report of successful stem cell transplantation in a child with CD40 deficiency. Bone Marrow Transplant (2007) 40(3):279–81. doi:10.1038/sj.bmt.1705713
10. Lee WI, Torgerson TR, Schumacher MJ, Yel L, Zhu Q, Ochs HD. Molecular analysis of a large cohort of patients with the hyper immunoglobulin M (IgM) syndrome. Blood (2005) 105(5):1881–90. doi:10.1182/blood-2003-12-4420
11. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell (2000) 102(5):553–63. doi:10.1016/S0092-8674(00)00078-7
12. Durandy A, Cantaert T, Kracker S, Meffre E. Potential roles of activation-induced cytidine deaminase in promotion or prevention of autoimmunity in humans. Autoimmunity (2013) 46(2):148–56. doi:10.3109/08916934.2012.750299
13. Meyers G, Ng YS, Bannock JM, Lavoie A, Walter JE, Notarangelo LD, et al. Activation-induced cytidine deaminase (AID) is required for B-cell tolerance in humans. Proc Natl Acad Sci U S A (2011) 108(28):11554–9. doi:10.1073/pnas.1102600108
14. Tang WJ, An YF, Dai RX, Wang QH, Jiang LP, Tang XM, et al. Clinical, molecular, and T cell subset analyses in a small cohort of Chinese patients with hyper-IgM syndrome type 1. Hum Immunol (2014) 75(7):633–40. doi:10.1016/j.humimm.2014.04.014
15. Herve M, Isnardi I, Ng YS, Bussel JB, Ochs HD, Cunningham-Rundles C, et al. CD40 ligand and MHC class II expression are essential for human peripheral B cell tolerance. J Exp Med (2007) 204(7):1583–93. doi:10.1084/jem.20062287
16. Cantaert T, Schickel JN, Bannock JM, Ng YS, Massad C, Delmotte FR, et al. Decreased somatic hypermutation induces an impaired peripheral B cell tolerance checkpoint. J Clin Invest (2016) 126(11):4289–302. doi:10.1172/JCI84645
17. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41(4):529–42. doi:10.1016/j.immuni.2014.10.004
18. Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol (2015) 16(2):142–52. doi:10.1038/ni.3054
19. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Annu Rev Immunol (2016) 34:335–68. doi:10.1146/annurev-immunol-041015-055605
20. Bossaller L, Burger J, Draeger R, Grimbacher B, Knoth R, Plebani A, et al. ICOS deficiency is associated with a severe reduction of CXCR5+CD4 germinal center Th cells. J Immunol (2006) 177(7):4927–32. doi:10.4049/jimmunol.177.7.4927
21. Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, et al. Monogenic mutations differentially affect the quantity and quality of T follicular helper cells in patients with human primary immunodeficiencies. J Allergy Clin Immunol (2015) 136(4):993–1006.e1. doi:10.1016/j.jaci.2015.05.036
22. Fonseca VR, Agua-Doce A, Maceiras AR, Pierson W, Ribeiro F, Romao VC, et al. Human blood Tfr cells are indicators of ongoing humoral activity not fully licensed with suppressive function. Sci Immunol (2017) 2:eaan1487. doi:10.1126/sciimmunol.aan1487
23. Havenar-Daughton C, Lindqvist M, Heit A, Wu JE, Reiss SM, Kendric K, et al. CXCL13 is a plasma biomarker of germinal center activity. Proc Natl Acad Sci U S A (2016) 113(10):2702–7. doi:10.1073/pnas.1520112113
24. Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, et al. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity (1994) 1(3):167–78. doi:10.1016/1074-7613(94)90095-7
25. Vinuesa CG, Linterman MA, Goodnow CC, Randall KL. T cells and follicular dendritic cells in germinal center B-cell formation and selection. Immunol Rev (2010) 237(1):72–89. doi:10.1111/j.1600-065X.2010.00937.x
26. Brodeur SR, Angelini F, Bacharier LB, Blom AM, Mizoguchi E, Fujiwara H, et al. C4b-binding protein (C4BP) activates B cells through the CD40 receptor. Immunity (2003) 18(6):837–48. doi:10.1016/S1074-7613(03)00149-3
27. Lanzavecchia A. Antigen-specific interaction between T and B cells. Nature (1985) 314(6011):537–9. doi:10.1038/314537a0
28. Ansel KM, McHeyzer-Williams LJ, Ngo VN, McHeyzer-Williams MG, Cyster JG. In vivo-activated CD4 T cells upregulate CXC chemokine receptor 5 and reprogram their response to lymphoid chemokines. J Exp Med (1999) 190(8):1123–34. doi:10.1084/jem.190.8.1123
29. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med (2000) 192(11):1545–52. doi:10.1084/jem.192.11.1545
30. Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, Aruffo A. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci U S A (1992) 89(14):6550–4. doi:10.1073/pnas.89.14.6550
31. Van den Eertwegh AJ, Noelle RJ, Roy M, Shepherd DM, Aruffo A, Ledbetter JA, et al. In vivo CD40-gp39 interactions are essential for thymus-dependent humoral immunity. I. In vivo expression of CD40 ligand, cytokines, and antibody production delineates sites of cognate T-B cell interactions. J Exp Med (1993) 178(5):1555–65. doi:10.1084/jem.178.5.1555
32. Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature (2008) 455(7214):764–9. doi:10.1038/nature07345
33. Gu-Trantien C, Willard-Gallo K. PD-1(hi)CXCR5(-)CD4(+) TFH cells play defense in cancer and offense in arthritis. Trends Immunol (2017) 38(12):875–8. doi:10.1016/j.it.2017.10.003
34. Comans-Bitter WM, de Groot R, van den Beemd R, Neijens HJ, Hop WC, Groeneveld K, et al. Immunophenotyping of blood lymphocytes in childhood. Reference values for lymphocyte subpopulations. J Pediatr (1997) 130(3):388–93. doi:10.1016/S0022-3476(97)70200-2
Keywords: hyper-IgM syndrome, follicular helper T cells, follicular regulatory T cells, class switch recombination, somatic hypermutation, AICDA, CD40LG, CD40
Citation: Cicalese MP, Gerosa J, Baronio M, Montin D, Licciardi F, Soresina A, Dellepiane RM, Miano M, Baselli LA, Volpi S, Dufour C, Plebani A, Aiuti A, Lougaris V and Fousteri G (2018) Circulating Follicular Helper and Follicular Regulatory T Cells Are Severely Compromised in Human CD40 Deficiency: A Case Report. Front. Immunol. 9:1761. doi: 10.3389/fimmu.2018.01761
Received: 30 January 2018; Accepted: 16 July 2018;
Published: 06 August 2018
Edited by:
Satoshi Okada, Hiroshima University, JapanReviewed by:
Kohsuke Imai, Tokyo Medical and Dental University, JapanVanessa L. Bryant, Walter and Eliza Hall Institute of Medical Research, Australia
Cindy S. Ma, Garvan Institute of Medical Research, Australia
Copyright: © 2018 Cicalese, Gerosa, Baronio, Montin, Licciardi, Soresina, Dellepiane, Miano, Baselli, Volpi, Dufour, Plebani, Aiuti, Lougaris and Fousteri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Georgia Fousteri, Zm91c3RlcmkuZ2VvcmdpYSYjeDAwMDQwO2hzci5pdA==
†These authors have contributed equally to this work.