
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 24 July 2018
Sec. Autoimmune and Autoinflammatory Disorders
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01715
 Carole Le Coz1
Carole Le Coz1 Brian E. Nolan2
Brian E. Nolan2 Melissa Trofa1
Melissa Trofa1 Alicia M. Kamsheh1Mustafa K. Khokha3,4,5Saquib A. Lakhani4,5
Alicia M. Kamsheh1Mustafa K. Khokha3,4,5Saquib A. Lakhani4,5 Antonio Novelli6Elaine H. Zackai7,8
Antonio Novelli6Elaine H. Zackai7,8 Kathleen E. Sullivan1,8Silvana Briuglia9Tricia R. Bhatti8,10
Kathleen E. Sullivan1,8Silvana Briuglia9Tricia R. Bhatti8,10 Neil Romberg1,8*
Neil Romberg1,8*
Located contiguously on the long arm of the second chromosome are gene paralogs encoding the immunoglobulin-family co-activation receptors CD28 and cytotoxic T-lymphocyte-associated protein 4 (CTLA4). CD28 and CTLA4 share the same B7 ligands yet each provides opposing proliferative signals to T cells. Herein, we describe for the first time two unrelated subjects with coexisting CD28 and CTLA4 haploinsufficiency due to heterozygous microdeletions of chromosome 2q. Although their clinical phenotype, multi-organ inflammatory disease, is superficially similar to that of CTLA4 haploinsufficient autoimmune lymphoproliferative syndrome type V (ALPS5) patients, we demonstrate our subjects’ underlying immunopathology to be distinct. Unlike ALPS5 T cells which hyperproliferate to T-cell receptor-mediated activation and infiltrate organs, T cells from our subjects are hypoproliferative and do not. Instead of T cell infiltrates, biopsies of affected subject tissues demonstrated infiltrates of lineage negative lymphoid cells. This histologic feature correlated with significant increases in circulating type 3 innate lymphoid cells (ILC3s) and ILC3 cytokines, interleukin 22, and interleukin-17A. CTLA4-Ig monotherapy, which we trialed in one subject, was remarkably effective in controlling inflammatory diseases, normalizing ILC3 frequencies, and reducing ILC3 cytokine concentrations.
Located contiguously on the long arm of the second chromosome are gene paralogs encoding CD28 and cytotoxic T-lymphocyte-associated protein 4 (CTLA4). Both are immunoglobulin-family co-activation receptors that share the same B7 ligands yet each provides opposing proliferative signals to T cells (1). CD28 is a potent T-cell receptor (TCR) costimulatory molecule, whereas CTLA4 can either directly inhibit TCR signaling in cis or out-compete CD28 for B7 ligands in trans (1–4). CD28-deficient humans have not yet been described, but CD28-deficient mice exhibit impaired T-cell proliferation (1, 5). Recently, heterozygous CTLA4 loss-of-function mutations (CTLA4wt/mut) were identified in autoimmune lymphoproliferative syndrome type V patients (ALPS5; OMIM #616100) (6, 7). CTLA4wt/mut T cells hyperproliferate in vitro, and inflamed ALPS5 patient organs exhibit gross lymphocytic infiltrates.
Herein, we describe for the first time the immunologic consequences of coexisting CD28 and CTLA4 haploinsufficiency in two unrelated human subjects. Their condition, a multi-organ inflammatory disease, appears superficially similar to ALPS5 but lacks distinctive lymphoproliferative features. Although our subjects’ CD28-haploinsufficient T cells display low in vivo proliferative histories and divide poorly in vitro to TCR-mediated activation, we demonstrate B7 blockade with CTLA4-Ig to be a highly effective anti-inflammatory monotherapy in this clinical context.
Peripheral blood mononuclear cell (PBMCs) were separated from peripheral blood samples of subjects and healthy donor (HD) controls via Ficoll-Paque centrifugation. The mean age of HDs was 26.3 years (range 14–50 years), and 40% were male. Demographic and clinical information on subjects are listed in Table S1 in Supplementary Material. Study protocols were approved by the institutional review boards of the Children’s Hospital of Philadelphia or the University of Messina. Written informed consent has been obtained from subjects for their cases to be published in this report.
The following antibodies were used for flow cytometric staining: anti-CD19 Pacific Blue, anti-CD14 Pacific Blue, anti-CD117 APC-Cy7, anti-CD4 APC-Cy7, anti-CD25 PE, anti-CD127 APC, anti-CD45RO AF-700, anti-CXCR5 Pacific Blue, anti-PD-1 PE-Cy7, anti-inducible T-cell costimulator (ICOS) BV510, anti-Ki67 Pe, and anti-CD28 PeDazzle (all from BioLegend) and anti-CD3 eFluor 605NC (eBioscience). Intracellular staining with anti-Foxp3 Alexa Fluor 488 (BioLegend), anti-CTLA4 BV786 (BD Bioscience), and anti-pAKT APC (eBioscience) was performed using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) in accordance with the manufacturer. Stained cells were then characterized by flow cytometry using a LSRFortessa (BD Bioscience).
Interleukin-17A (IL-17A) and interleukin 22 (IL-22) concentrations in HD plasma and subject plasma obtained before and after 20 months of CTLA4-Ig therapy were determined using a LEGENDplex Th panel kit (BioLegend) in accordance with the manufacturer’s instructions.
CD4+CD45RO− T cells were sorted using the MoFlo Astrios EQ (Beckman Coulter), carboxyfluorescein diacetate succinimidyl ester (CFSE) (Thermo Fisher Scientific) labeled and plated at 100,000 cells/well in a 96-well plate in RPMI 10% FBS either with 1 µg/ml phytohemagglutinin (PHA, Sigma) and 1 ng/ml rIL2 (R&D systems) or anti-CD2/CD3/CD28 coated beads [T regulatory cell (Treg) suppression inspector human, Miltenyi]. At 4 days, cellular proliferation was determined by flow cytometry. Anti-CD28 dose responses were determined by culturing PBMC or CFSE-labeled CD4+CD45RO− T cells at 100,000 cells/well in a 96-well plate pre-coated with 1 µg/ml anti-CD3 (eBioscience) in the presence of various concentrations of anti-human CD28 (from 0.2 to 20 µg/ml; BD Biosciences). Phospho-AKT staining of CD4+ T cells was determined at 24 h by flow cytometry and CFSE dilution was assessed at 4 days.
CD4+ T cells were pre-enriched using the MojoSort™ Human CD4 T Cell Isolation Kit (BioLegend). CD4+CD45RO+CD25hiCD127lo/− Tregs and CD4+CD45RO−CD25− Tresp cells were sorted and labeled with CFSE. Treg and Tresp cells were co-cultured at a 1:1 ratio in the presence of anti-CD2/CD3/CD28 coated beads. At 3.5–4.5 days, co-cultures were stained for viability with the LIVE/DEAD kit (Thermo Fisher Scientific), and the proliferation of viable Tresp cells was assessed using CFSE dilution.
Tissue specimens were embedded in paraffin and fixed in formalin at the time of biopsy and held in storage. Prior to antibody staining, samples were deparaffinized and pre-treated with target retrieval solution (Dako, Santa Clara, CA, USA) for 15 min at boiling point. Slides were then cooled for 20 min before tissues were rinsed with buffer and subjected to antibody staining. Anti-CD3 (polyclonal, Dako) and anti-CD19 (Clone LE-CD19, Dako) both at a dilution of 1:200 were incubated with tissue sections at room temperature for 30 min. Sections were then mounted onto slides for visualization with bright field microscopy. For colonic biopsies, lymphoid cells infiltrating the glandular structures were enumerated across four representative regions and normalized to the number of gut epithelia in those same regions.
Our report focuses on two female subjects with intellectual disability and multi-organ inflammatory disease. Subject 1, now 20 years old, developed vitiligo universalis, thyroiditis, and lipodystrophy in her seventh year. At 12 years, she was diagnosed with anti-smooth muscle antibody-positive autoimmune hepatitis that was poorly responsive to azathioprine. Four years later, anti-GAD65 and anti-insulin antibody-positive insulin-dependent diabetes mellitus was recognized. Onset of chronic diarrhea at 17 years was associated with pan-enteropathic autoimmune enterocolitis. In her 18th year, she developed heart failure secondary to chronic pericarditis that progressed to tamponade physiology requiring a pericardiotomy (Figure S1 in Supplementary Material). Despite the unsuccessful application of various immunosuppressant therapies (azathioprine, rapamycin, colchicine, and corticosteroids), subject 1 exhibited an unremarkable infectious history. Laboratory assessments of protective immunity were generally unrevealing. She possessed normal serum IgG (1,320 mg/dl, nml 635–1,775) and IgM concentrations (110 mg/dl, nml 71–237) but a lower IgA concentration (44 mg/dl, nml 70–486). She generated protective vaccine titers to tetanus, diphtheria, and pneumococcus (Table S1 in Supplementary Material).
Subject 2, now 6 years old, also presented in infancy with neurodevelopmental delay and feeding difficulties. At that time, her skin was affected by a severe form of atopic dermatitis that evolved over her childhood into lichen sclerosis. At 15 months of age, subject 2 developed autoimmune urticaria and chronic diarrhea-associated enterocolitis with an associated recto-perineal fistula requiring surgical correction. At 6 years of age, subject 2 was diagnosed with anti-thyroperoxidase antibody-positive autoimmune thyroiditis. Despite a history of respiratory tract infections, assessments of subject 2 humoral immune function revealed normal IgG (710 mg/dl, nL 633–1,280) and IgM serum levels (49 mg/dl, 48–207) but a low IgA level (5 mg/dl, 33–202, Table S1 in Supplementary Material).
To determine if there was a genetic basis for their inflammatory diseases, we analyzed subject 1’s DNA using whole exome sequencing, which was unrevealing, and using chromosomal microarray, which identified a de novo heterozygous 11.6 Mb chromosome 2q33.1-q34 deletion (197,942,576–209,522,220; GRCh37/hg19). Several potentially pathologic genes within the deleted region were of interest including the intellectual disability gene SATB2 and the CD28/CTLA4/ICOS immune gene cluster (Figure 1A). Using a chromosomal microarray, we identified a more discrete de novo 8.3 Mb chromosome 2q33.1-q34 deletion (200,742,119–209,062,408), also encompassing SATB2 and the CD28/CTLA4/ICOS gene cluster, in subject 2 (Figure 1A).
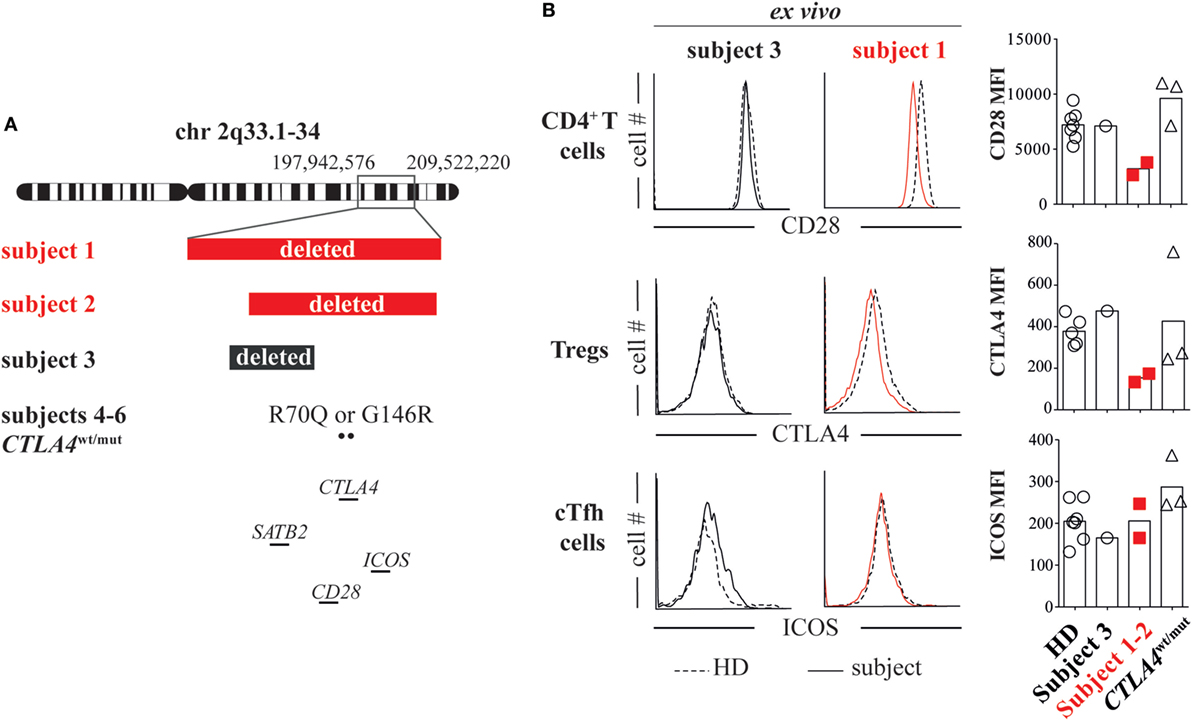
Figure 1. Heterozygous loss of the CD28/CTLA4/ICOS gene cluster decreases CD28 and cytotoxic T-lymphocyte-associated protein 4 (CTLA4) but not inducible T-cell costimulator (ICOS) expression. (A) Chromosome 2 breakpoints and mutation locations for subjects 1 through 6 are depicted. (B) Ex vivo CD4+ T cell CD28 expression, T regulatory cell (Treg) (CD127lowCD25high) CTLA4 expression and ICOS expression on cTfh cells (CXCR5+PD1+) were assessed in subject 1 (red line), subject 3 (black line), and healthy donor (HD, dashed line) blood samples by flow cytometry. CD28, ICOS, and CTLA4 average mean fluorescence intensities (MFIs) from subjects and HDs are summarized (right).
To investigate if heterozygous loss of CD28/CTLA4/ICOS alters protein expression, we compared CD28, CTLA4, and ICOS mean fluorescence intensities (MFIs) on subject 1/subject 2 T-cell subsets to MFIs on HD cells and also cells from a separate control subject (subject 3). Subject 3 is an intellectually disabled 9-year-old female with a de novo heterozygous 5 Mb 2q33.1 deletion encompassing SATB2 but not CD28/CTLA4/ICOS (199,631,959–204,602,266; GRCh37/hg19, Figure 1A; Table S1 in Supplementary Material) (8). She is immune competent and has not experienced inflammatory diseases. In agreement with previously published T-cell phenotypes from unaffected parents of ICOS-deficient patients (9) and consistent normal circulating CXCR5+PD-1+ T follicular helper (cTfh) cell frequencies (Table S1 in Supplementary Material), we found ICOS expression on subject 1/subject 2 cTfh cells, Tregs and naïve T cells to be indistinguishable from HD/subject 3 T-cell subsets (Figure 1B; Figure S2 in Supplementary Material). By contrast, CD28 and CTLA4 MFIs on subject 1/subject 2 CD4+ T cells and CD25hiCD127− Tregs were, respectively, half of HD/subject 3 counterpart cell MFIs (Figure 1B). Hence, heterozygous deletion of the CD28/CTLA4/ICOS gene cluster diminishes expression of CD28 and CTLA4 but not ICOS.
To identify the functional consequences of reduced CD28 expression, we compared subject 1/subject 2 T-cell proliferative responses against HD/subject 3 cells and also against cells from three additional subjects (subjects 4 through 6) with heterozygous ALPS5-associated CTLA4 mutations (CTLA4wt/mut, Table S1 in Supplementary Material). Although CTLA4 expression varied significantly on CTLA4wt/mut cells, CD28 and ICOS expression did not and was indistinguishable from HD control cells (Figure 1B). As previously reported (6, 10), CTLA4wt/mut CD4+ T cells skewed toward a memory phenotype and an increased frequency expressed the cell-cycle marker Ki67 (5–9%) compared with HD/subject 3 counterpart cells (2–3%). By contrast, less than 1% of subject 1 and subject 2 CD4+ T cells were Ki67+, suggesting a recent history of hypoproliferation (Figure 2A). To determine if subject 1/subject 2 proliferative responses were also reduced in vitro, we sorted their naïve T cells and stimulated them with anti-CD3 and anti-CD28 antibodies. After 4 days, only 24% of subject 1 and 32% of subject 2 T cells had divided, a proportion roughly half that of identically stimulated HD (58%) and subject 3 T cells (48%). By contrast, 80% of CTLA4wt/mut naïve T cells divided by culture day 4, a finding that agreed with previously published reports (Figure 2B) (6, 7). Increasing anti-CD28 concentrations improved subject 1 T-cell division somewhat but did not normalize AKT phosphorylation, a downstream indicator of CD28/TCR activity (Figure S3 in Supplementary Material). Notably, when stimulated with PHA and IL-2, subject 1/subject 2 T cells proliferated similar to HD controls (Figure 2C). Hence, CD28 haploinsufficiency imparts a pathway-specific T-cell proliferation defect.
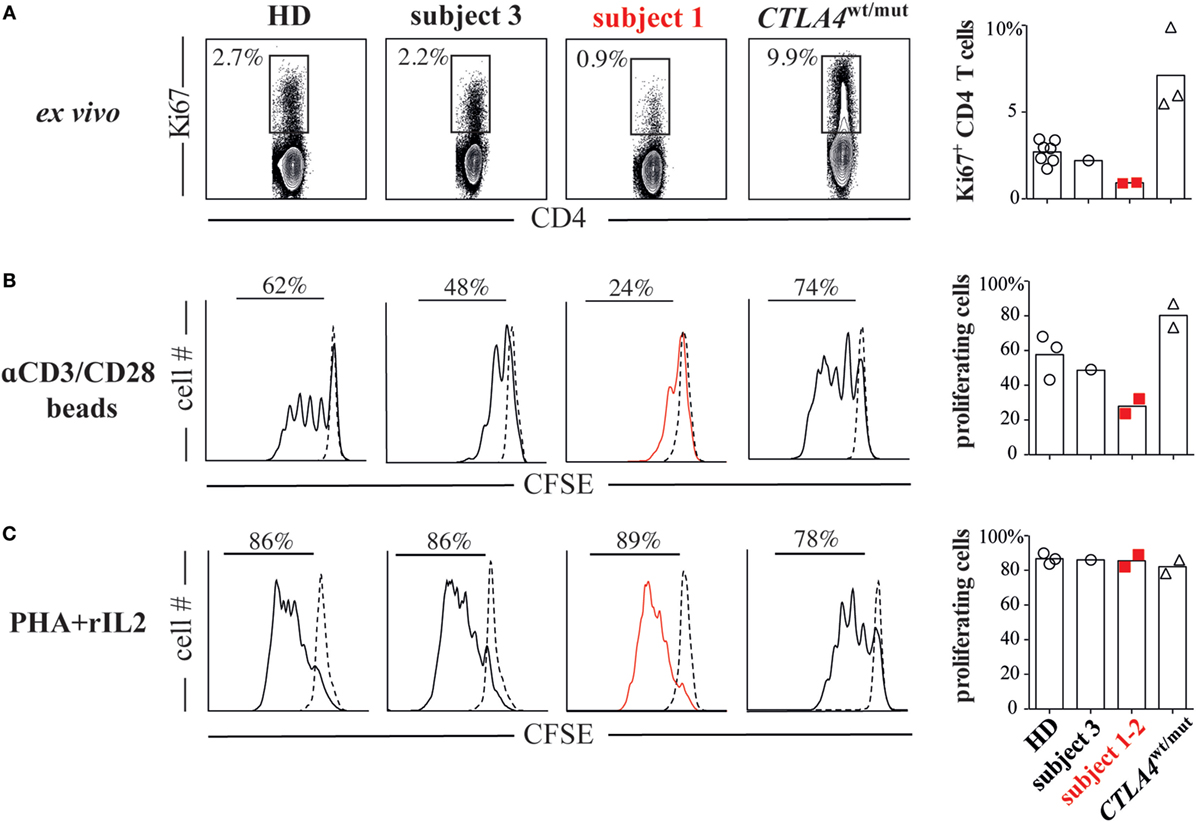
Figure 2. CD28 haploinsufficiency is associated with T-cell hypoproliferation. (A) Subject 1/subject 2 display decreased frequencies of Ki67+ CD4+ T cells. (B) CFSE dilutions in sorted naïve T cells (CD4+CD45RO−) from subjects and healthy donor (HDs) cultured for 4 days with anti-CD2/CD3/CD28 coated beads or (C) phytohemagglutinin (PHA, 1 µg/ml) and recombinant interleukin 2 (rIL-2, 1 ng/ml) are displayed.
To assess the suppressive potential of subject 1/subject 2 FOXP3+ Tregs, we first determined their frequencies in peripheral blood. Tregs comprised only 1% of subject 1 CD4+ T cells and 0.1% of subject 2 CD4+ T cells, considerably lower values than HDs (2.8–7.8%) and CTLA4wt/mut subjects (0.9–3.2%, Figure 3A). We then assessed the ability of subject 1 Tregs to suppress proliferation of naïve T responder cells (Tresp) when cultured in a 1:1 ratio. At culture day 4, subject 1 Tregs suppressed heterologous HD Tresp proliferation as poorly as CTLA4wt/mut Tregs, which were previously reported to exhibit this defect (Figure 3B) (6, 7). Thus, CTLA4 haploinsufficiency-associated Treg defects are balanced in vitro by CD28 haploinsufficiency-associated Tresp cell hypoproliferation.
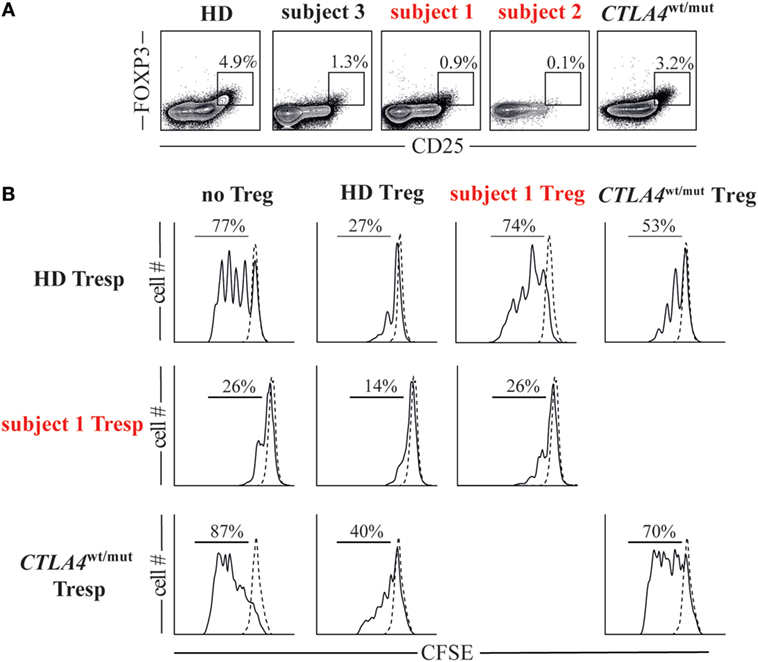
Figure 3. Cytotoxic T-lymphocyte-associated protein 4 (CTLA4)/CD28 haploinsufficiency is associated with quantitative and qualitative T regulatory cell (Treg) defects. (A) Frequencies of Foxp3+CD25hi Tregs in subjects and a representative healthy donor (HD) are displayed. (B) Histograms display day 4 cell proliferation of unstimulated (dotted line) and anti-CD2/CD3/CD28 stimulated (black line) T responder (Tresp) cells cultured with or without autologous or heterologous CD25hiCD127lo Tregs in a 1:1 Tresp to Treg ratio.
During the course of her many inflammatory diseases, subject 1 was multiply biopsied. To determine how T-cell defects we observed in vitro contributed to inflammatory pathology in vivo, we analyzed subject 1 colon, pericardium, liver, and duodenal tissues obtained during periods of active disease. Tissue damage was prominent in each sample, but lymphoid infiltrates were scant (Figure 4A; Figures S1, S4, and S5 in Supplementary Material). This pattern was especially striking in contrast to tissues from CTLA4wt/mut patients like subject 6 (6, 7). Although subject 1 and subject 6 gastrointestinal biopsies both exhibited pathologic surface epithelium attenuation, goblet cell loss, and increased crypt epithelium apoptosis, only subject 6 tissues exhibited prominent intraepithelial lymphoid cell (IELC) infiltrates, a common feature of autoimmune enteropathies (11) (Figure 4A, gold arrowheads). In fact, we found IELC frequencies in subject 6 duodenum, left colon and right colon to be 2.5×, 4×, and 20× greater than subject 1 biopsies from the same sites (P < 0.029, P < 0.029, and P < 0.04, Figure 4B). Immunohistochemical staining revealed that while most IELCs in subject 6 gastrointestinal biopsies were T cells (93%) (Figure 4C, black arrowheads), most (64%) subject 1 IELCs stained negative for CD3 and CD19 lineage markers (Figure 4C, white arrowheads and Figure S6 in Supplementary Material). Inflamed subject 2 tissues were not biopsied and therefore were unavailable for analysis.
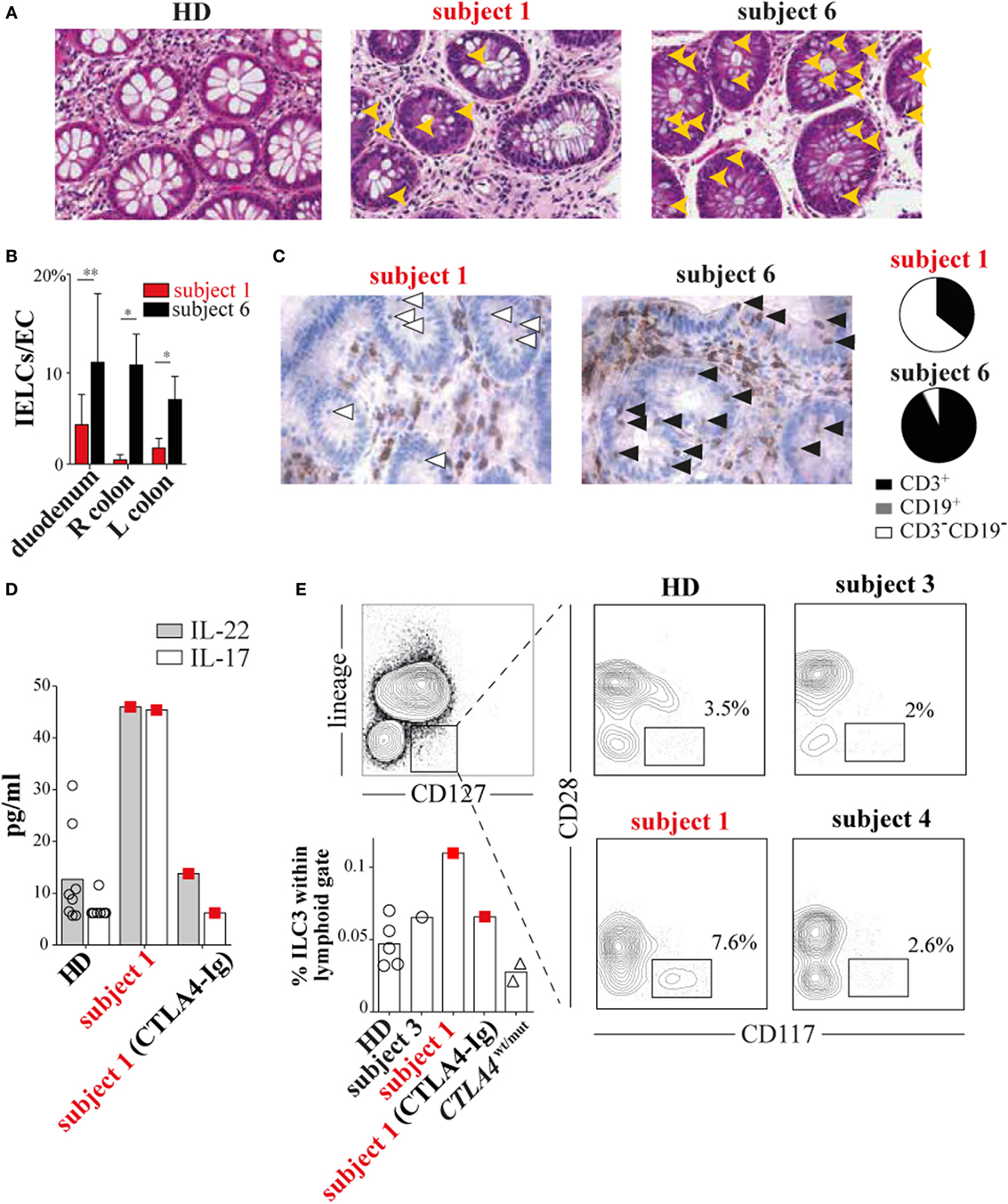
Figure 4. Subject 1 displays scant T-cell gut infiltrates with increased circulating type 3 innate lymphoid cells (ILC3s) and ILC3 cytokines. (A) Hematoxylin and eosin staining of healthy donor (HD), subject 1 and subject 6 colonic biopsies with intraepithelial lymphoid cells (IELCs) indicated (gold arrowheads). 40× original magnification for all images. (B) Average frequencies of IELCs per gland epithelial cell from subjects’ duodenum, right (R) colon, and left (L) colon biopsies are displayed (*P < 0.029, **P < 0.04, Mann–Whitney U test). (C) Anti-CD3 immunohistochemical staining (left) identifies T cells (black arrowheads) and non-T cells (white arrowheads). Pie charts (right) depict the proportions of different infiltrating lymphoid cell populations in subject 1 and subject 6 colonic biopsies. Total lymphoid cells counted per biopsy are n = 57 and n = 72, respectively. (D) Subject 1 plasma interleukin 22 (IL-22) and interleukin-17A (IL-17A) concentrations and (E) circulating ILC3 (CD3−CD19−CD14−CD11c−CD127+CD117+CD28−) frequencies are elevated and decrease after 20 months of cytotoxic T-lymphocyte-associated protein 4 (CTLA4)-Ig therapy.
Based upon encouraging reports describing improvement of ALPS5 enteropathy with CTLA4-Ig treatment (12, 13) and since other therapies had been ineffective, we began subject 1 on a trial of CTLA4-Ig. CTLA4-Ig monotherapy proved remarkably effective in treating subject 1 enterocolitis, hepatitis, and pericarditis, although its use was associated with progressive antibody deficiency. When comparing multiplex cytokine profiles of subject 1 plasma from before and during CTLA4-Ig therapy, the most dramatic differences were decreases in IL-22/IL-17A concentrations (Figure 4D).
Recently, ILCs, which lack antigen-specific receptors and traditional lineage markers (CD3, CD19, CD14, etc.), have emerged as potent mucosal cytokines producers, including IL-22 and IL17 (14). To determine if changes in circulating ILCs correlated with her inflammatory symptoms, we compared ILC frequencies in subject 1’s blood before CTLA4-Ig to frequencies in blood from HDs and subjects 3 through 6. All were similar (Figure S7 in Supplementary Material). ILCs can be further divided into types 1 through 3 on the basis of cytokine secretion signatures or by CD28 expression; CD28 is highly expressed by ILC1s and ILC2s but not ILC3s (15). Using a CD28 and CD117 gating strategy, we determined that ILC3s, the primary secretors of IL-22 and IL-17, were present at twice the frequency in subject 1 blood samples taken before CTLA4-Ig therapy than were present in HD samples (Figure 4E). With CTLA4-Ig, subject 1 ILC3 frequencies dropped precipitously and became indistinguishable from controls.
Herein, we describe two phenotypically similar subjects each with heterozygous loss of the CD28/CTLA4/ICOS gene cluster. Like CTLA4wt/mut ALPS5 patients, our subjects experienced multi-organ inflammation and exhibited a Treg suppressive defect. Yet, while ALPS5 patients typically display normal or only slightly reduced Treg numbers (6, 7), we found subject 1 Treg frequencies to be significantly reduced and subject 2’s Tregs to be nearly absent. Such scarcity parallels Treg-specific CD28 conditional knockout mice that experience multi-organ inflammatory disease because they fail to numerically balance suppressor and effector T cells during immune responses (16). Yet, unlike this mouse model, our subjects’ Tregs intrinsically lack suppressive capacity due to CTLA4 haploinsufficiency and our subjects’ effector T cells hypoproliferated to traditional TCR-focused activation due to CD28 haploinsufficiency. As T-cell hyperproliferation and related lymphocytic organ infiltration are the sine qua non of ALPS5 pathology, we conclude the inflammatory process affecting subject 1 and subject 2 is distinct from those affecting CTLA4wt/mut patients.
Instead of T-cell expansion in subject 1, we found that coexisting CD28 and CTLA4-haploinsufficiency correlated with a dramatic increase in circulating ILC3s and ILC3 cytokines. We speculate that because ILC3s do not express CD28, they may enjoy, in our subjects, a competitive advantage over ILC1s and ILC2s which potentially rely upon CD28 for homeostasis. As ILC3s have been previously shown to mediate mouse colitis and to be increased in the inflamed intestines of Crohn’s disease patients (14), expansion of these cells may explain subject 1’s enterocolitis and their contraction, its resolution. Currently, the prevailing view is that CTLA4-Ig dampens inflammation by out-competing CD28 expressed on T cells for B7 molecules (17), but given low CD28 expression and reduced Tresp cell proliferation, this mechanism does not adequately explain subject 1’s dramatic CTAL4-Ig response. One clue, in our specific context, may come from previous work demonstrating that CTLA4-Ig prolongs allograft survival in CD28/CTLA4 double knockout mice (18) and experiments that reveal CTLA4-Ig, but not CD28 blockade, inhibits T-dependent alloantibody production in rats (19). These related phenomena, first observed in animals and now in our CTLA4/CD28 haploinsufficient subjects, predict that a stimulatory B7 receptor(s) other than CD28 exists and that it is expressed by ILC3s.
This study was carried out in accordance with the recommendations and approved by the institutional review boards of the Children’s Hospital of Philadelphia and the University of Messina. All subjects gave written informed consent in accordance with the Declaration of Helsinki.
CC, MT, BN, AK, and NR performed experiments and analyzed data. BN and TB performed histological analyses. KS, SB, EZ, and NR provided patient samples. MK, SL, AN, and EZ genotyped subjects. CC and NR were responsible for study design and wrote the manuscript. All the authors reviewed the manuscript and provided scientific input.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the subjects and families for their participation.
This work was supported by grant numbers K23 AI115001 (to NR) from the National Institutes of Health-National Institute of Allergy and Infectious Diseases, T32-HD043021 (to BN), from the National Institutes of Health-Eunice Kennedy Shriver National Institute of Child Health and Human Development, and the Jeffrey Modell Foundation (to NR). Our study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of DECIPHER data is available from http://decipher.sanger.ac.uk and via email fromZGVjaXBoZXJAc2FuZ2VyLmFjLnVr. Funding for the DECIPHER project was provided by Wellcome Trust.
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01715/full#supplementary-material.
1. Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 costimulation: from mechanism to therapy. Immunity (2016) 44(5):973–88. doi:10.1016/j.immuni.2016.04.020
2. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med (1995) 182(2):459–65. doi:10.1084/jem.182.2.459
3. Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, et al. Molecular basis of T cell inactivation by CTLA-4. Science (1998) 282(5397):2263–6. doi:10.1126/science.282.5397.2263
4. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332(6029):600–3. doi:10.1126/science.1202947
5. Green JM, Noel PJ, Sperling AI, Walunas TL, Gray GS, Bluestone JA, et al. Absence of B7-dependent responses in CD28-deficient mice. Immunity (1994) 1(6):501–8. doi:10.1016/1074-7613(94)90092-2
6. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science (2014) 345(6204):1623–7. doi:10.1126/science.1255904
7. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med (2014) 20(12):1410. doi:10.1038/nm.3746
8. Balasubramanian M, Smith K, Basel-Vanagaite L, Feingold MF, Brock P, Gowans GC, et al. Case series: 2q33.1 microdeletion syndrome—further delineation of the phenotype. J Med Genet (2011) 48(5):290–8. doi:10.1136/jmg.2010.084491
9. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Dräger R, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol (2003) 4(3):261. doi:10.1038/ni902
10. Hou TZ, Verma N, Wanders J, Kennedy A, Soskic B, Janman D, et al. Identifying functional defects in patients with immune dysregulation due to LRBA and CTLA-4 mutations. Blood (2017) 129(11):1458–68. doi:10.1182/blood-2016-10-745174
11. Najarian RM, Hait EJ, Leichtner AM, Glickman JN, Antonioli DA, Goldsmith JD. Clinical significance of colonic intraepithelial lymphocytosis in a pediatric population. Mod Pathol (2009) 22(1):13. doi:10.1038/modpathol.2008.139
12. Lee S, Moon JS, Lee C-R, Kim H-E, et al. Abatacept alleviates severe autoimmune symptoms in a patient carrying a de novo variant in CTLA-4. J Allergy ClinImmunol (2016) 137(1):327–30. doi:10.1016/j.jaci.2015.08.036
13. Gupta NK, Yilmaz O, Fisher M, Yajnik V. Abatacept: a new treatment option for refractory adult autoimmune enteropathy. J Clin Gastroenterol (2014) 48(1):55–8. doi:10.1097/MCG.0b013e3182a4e0ec
14. Sonnenberg GF, Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med (2015) 21(7):698. doi:10.1038/nm.3892
15. Roan F, Stoklasek TA, Whalen E, Molitor JA, Bluestone JA, Buckner JH, et al. CD4+ group 1 innate lymphoid cells (ILC) form a functionally distinct ILC subset that is increased in systemic sclerosis. J Immunol (2016) 196(5):2051–62. doi:10.4049/jimmunol.1600364
16. Zhang R, Huynh A, Whitcher G, Chang J, Maltzman JS, Turka LA. An obligate cell-intrinsic function for CD28 in Tregs. J Clin Invest (2013) 123(2):580–93. doi:10.1172/JCI65013
17. Bluestone JA, St Clair EW, Turka LA. CTLA4Ig: bridging the basic immunology with clinical application. Immunity (2006) 24(3):233–8. doi:10.1016/j.immuni.2006.03.001
18. Mandelbrot DA, Oosterwegel MA, Shimizu K, Yamada A, Freeman GJ, Mitchell RN, et al. B7-dependent T-cell costimulation in mice lacking CD28 and CTLA4. J Clin Invest (2001) 107(7):881–7. doi:10.1172/JCI11710
Keywords: cytotoxic T-lymphocyte-associated protein 4, CD28, type 3 innate lymphoid cell, inflammation, regulatory T cell
Citation: Le Coz C, Nolan BE, Trofa M, Kamsheh AM, Khokha MK, Lakhani SA, Novelli A, Zackai EH, Sullivan KE, Briuglia S, Bhatti TR and Romberg N (2018) Cytotoxic T-Lymphocyte-Associated Protein 4 Haploinsufficiency-Associated Inflammation Can Occur Independently of T-Cell Hyperproliferation. Front. Immunol. 9:1715. doi: 10.3389/fimmu.2018.01715
Received: 12 April 2018; Accepted: 12 July 2018;
Published: 24 July 2018
Edited by:
Betty Diamond, Feinstein Institute for Medical Research, United StatesReviewed by:
Ueno Hideki, Icahn School of Medicine at Mount Sinai, United StatesCopyright: © 2018 Le Coz, Nolan, Trofa, Kamsheh, Khokha, Lakhani, Novelli, Zackai, Sullivan, Briuglia, Bhatti and Romberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neil Romberg, cm9tYmVyZ25AZW1haWwuY2hvcC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.