 Geison Cambri
Geison Cambri Marcelo Távora Mira
Marcelo Távora Mira- Graduate Program in Health Sciences, School of Medicine, Pontifícia Universidade Católica do Paraná, Curitiba, Paraná, Brazil
Genetics plays a crucial role in controlling susceptibility to infectious diseases by modulating the interplay between humans and pathogens. This is particularly evident in leprosy, since the etiological agent, Mycobacterium leprae, displays semiclonal characteristics not compatible with the wide spectrum of disease phenotypes. Over the past decades, genetic studies have unraveled several gene variants as risk factors for leprosy per se, disease clinical forms and the occurrence of leprosy reactions. As expected, several of these genes are immune-related; yet, hypothesis-free approaches have led to genes not classically linked to immune response. The PARK2, originally described as a Parkinson’s disease gene, illustrates the case: Parkin—the protein coded by PARK2—was defined as an important player regulating innate and adaptive immune responses only years after its description as a leprosy susceptibility gene. Interestingly, even with the use of powerful hypothesis-free study designs such as genome-wide association studies, most of the major gene effect controlling leprosy susceptibility remains elusive. One hypothesis to explain this “hidden heritability” is that rare variants not captured by classic association studies are of critical importance. To address this question, massively parallel sequencing of large segments of the human genome—even whole exomes/genomes—is an alternative to properly identify rare, disease-causing mutations. These mutations may then be investigated through sophisticated approaches such as cell reprogramming and genome editing applied to create in vitro models for functional leprosy studies.
Introduction
Infectious diseases are essentially caused by pathogens capable to transpose the immunological barrier and colonize the host organism. Exposure to an infectious agent is necessary but not sufficient to determine disease; exposed organisms need to be naturally susceptible and even then, clinical disease outcomes often display marked interindividual variation (1). The explanation for such variability can be addressed to different reasons, including environmental factors, divergence in virulence of pathogen strains and particularly, to the complex interplay between host and pathogens. A remarkable demonstration of this variability was observed in the Lübeck disaster occurred in the late 1920s: 251 neonates were accidentally infected by virulent Mycobacterium tuberculosis contaminating a batch of Bacille Calmette-Guérin (BCG) vaccine. Twenty-three infants (9.2%) did not show any clinical signs of tuberculosis and the mortality rate was 29%; 68% of neonates who presented clinical disease spontaneously progressed to cure (2).
Robust evidence that the host–pathogen interplay is largely influenced by the genetic make-up of the host has been brilliantly demonstrated in an adoptee study: predisposition to infectious disease was predominantly inherited, in an interesting contrast with cancer that was found to be much more dependent of non-genetic factors (3). Innate predisposition to infection seems to be particularly crucial for leprosy: it is estimated that only a small fraction (from 5 to 12%) of individuals exposed to Mycobacterium leprae are successfully infected (4, 5). Although leprosy is treatable by an efficient multidrug regimen available for free around the world, latest reports from 143 countries show 214,783 new cases, with India (63.08%), Brazil (11.74%), and Indonesia (7.83%) presenting the highest percentage of registered cases (6). Patients display a wide spectrum of clinical phenotypes that are related to individual differences in immune response and distributes between two poles: in one extreme, tuberculoid patients presents a strong cellular (Th1) immune response with increased production of pro-inflammatory cytokines such as interleukin-2 and interferon-γ and a low or inexistent bacillary load in lesions; on the other extreme of the spectrum, lepromatous leprosy is characterized by a predominantly antibody-based (Th2) immune response with predominant expression of interleukin-10 and interleukin-4 and a high number of M. leprae in skin smears. Borderline disease displays a gradient of immune features depending on the proximity to one of the poles (7, 8). During the course of the disease, treatment or even after cure, up to 50% of patients develop one of the two types of an aggressive, sudden inflammatory response known as leprosy reaction, the major cause of permanent neural damage with consequent disabilities today (9, 10).
The M. leprae is an acid-fast, Gram-positive bacillus incapable of growing in axenic media, thus strongly dependent on the host cellular environment. The bacterium presents a reduced genome and semi-clonal characteristics across strains distributed worldwide (11), reinforcing the impact of host genetics over control of disease per se, its clinical forms, and the occurrence of leprosy reactions. Decades of extensive research positions host genetics as a major player controlling susceptibility to leprosy (12, 13). Early evidence comes from genetic descriptive, DNA-free studies: leprosy occurrence displays strong familial aggregation (14) and concordance of infection is higher in monozygotic (59.7%) as compared to dizygotic twins (20%) (15). Several complex segregation analysis (CSA) consistently revealed the presence of a major gene effect controlling susceptibility to leprosy per se in different population samples of distinct genetic backgrounds, although no consensus on the exact model of inheritance has been achieved (16, 17). Later, hypothesis-free genome-wide linkage scans have identified chromosomal regions such as 10p13, 6q25-27, and 6p21 as positional candidates to harbor leprosy susceptibility genes (18, 19), and the first Genome-Wide Association Study (GWAS) on leprosy has been performed using a large Han Chinese sample set: a total of 491,883 single nucleotide polymorphisms (SNPs) spanned over the genome were first genotyped in 706 patients and 1,225 controls; the 93 markers associated with the smallest p-values were later tested for replication in two additional independent population samples (20). Combined, these molecular strategies have led to the description of a multitude of genes associated to leprosy (Figure 1; Table 1), several of them participating in host immune response and/or bacterial routes of infection and evasion from the immunological barrier.

Figure 1. Pathogenesis of leprosy with selected genes impacting on its phenotypes. Adapted from Mira (12) and Sauer et al. (13). For a more detailed list of genes, please refer to Table 1. Abbreviations: I, indeterminate; LL, lepromatous leprosy; BL, borderline-lepromatous; BB, borderline–borderline; BT, borderline-tuberculoid; TT, tuberculoid–tuberculoid; T1R, type-1 reaction; T2R, type-2 reaction.
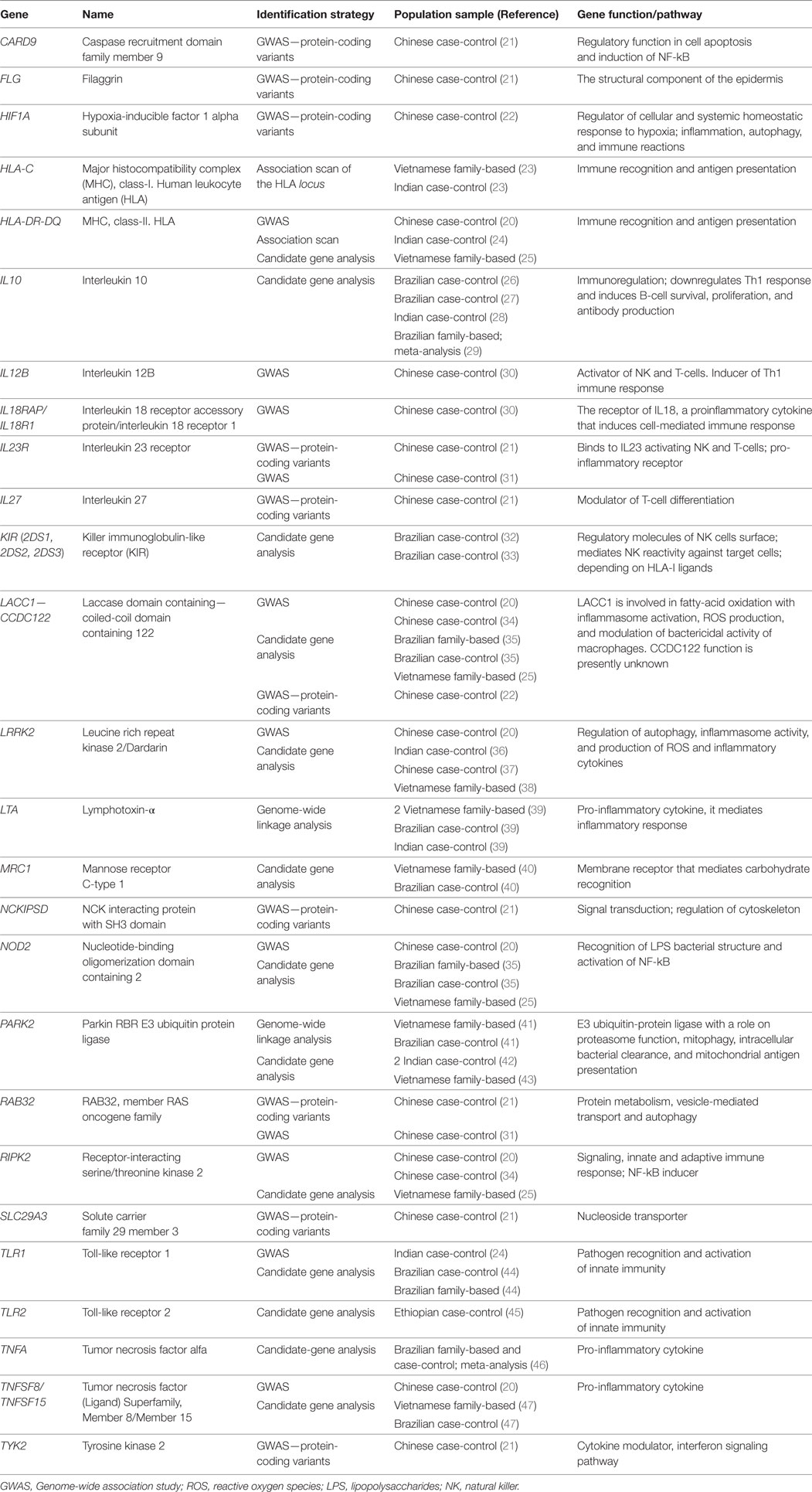
Table 1. Leprosy-associated genes with functional evidence or replicated status.
A natural functional and positional candidate genomic region has been the major histocompatibility complex (MHC)/human leukocyte antigen (HLA) located in the highly polymorphic 4 Mb interval at chromosome 6p21. The complex is essential for recognition, processing, and presentation of antigens during immune response. Genes located in all three MHC/HLA classes have been exhaustively studied in leprosy and haplotypes have been associated with both susceptibility and protection against the disease in distinct populations (13, 48). Killer immunoglobin-like receptors genes—KIR2DS1, 2DS2, and 3DS1—and their HLA ligands were associated with leprosy in a Brazilian population (32, 33) and HLA-C, a classical ligand for KIRs, was observed as a risk factor for leprosy in Vietnamese family based and Indian case-control populations (23). Genetic variants in the class-II HLA-DR–DQ locus have been consistently associated with protection against leprosy (20, 24). In the MHC class-III region, linkage disequilibrium mapping of the 6p21 region identified the low-producing A allele of the variant + 80 of Lymphotoxin-α (LTA + 80) as a risk factor for infection: association was reported in a Vietnamese, family-based sample and validated in a Brazilian case-control sample (39).
Receptors for pathogen-associated molecular patterns, classic molecules of the innate immune response, have been also consistently associated with leprosy. The non-synonymous single-nucleotide polymorphism G396S located at the Mannose Receptor C-type lectin (MRC1) gene located in region 10p13 was described as a risk factor for leprosy susceptibility in different populations (40). Polymorphisms in the toll-like receptor (TLR) family were repeatedly associated with leprosy and its phenotypes. Amino acid substitutions N248S and I602S in the TLR1 gene have been associated with susceptibility (44) and protection (24) against leprosy, respectively. SNP markers 597 C/T (rs3804099) and a 280 bp-length microsatellite of TLR2 have been associated with protection and increased risk of leprosy reactions, respectively (45) Another sensing molecule consistently associated with leprosy is the nucleotide-binding oligomerization domain 2 (NOD2), a cytoplasmic receptor responsible for recognizing intracellular pathogens via their peptidoglycan components of the bacterial cell wall. NOD2 involvement in leprosy was first identified in a GWAS (20) and later replicated (35); in addition, association of NOD2 variants with leprosy reaction has been detected (49). Functionally analysis has demonstrated that a structurally unique muramyl dipeptide of M. leprae is recognized by NOD2, triggering expression of interleukin-32 and monocytes differentiation into dendritic cells (50).
Cytokines regulating the Th1/Th2 immune responses have also been described associated with leprosy phenotypes. TNFA and IL10 genetic variants are classic risk factors for leprosy (29, 46); gene products TNF-α and IL-10 are major signature cytokines for the tuberculoid and lepromatous pole, respectively (51). More recently, GWAS have suggested a role in leprosy susceptibility control of IL12B, IL27, and pro-inflammatory receptors IL23R and IL18RAP/IL18R1 that regulates the adaptive immune response (21, 30). Functional assays indicate regulation of IL10 expression by IL27, inhibiting host defense through IFN-γ-induced antimicrobial activity (52).
A more comprehensive analysis of leprosy genetic studies reveals a complex network of interactions among associated genes. This is well exemplified by LRRK2, initially identified in the first leprosy GWAS (20) and later replicated in an Indian population (36); LRRK2 participates in the control of autophagy with involvement of the small GTPase RAB32 (53), which gene is associated with leprosy in two unrelated GWAS (21, 31). Interestingly, LRRK2 is also correlated to bacterial survival and co-localization as observed in RAW 264.7 cells infected by Salmonella typhimurium (54); although, the increase of LRRK2-kinase activity increases M. tuberculosis survival through reduction of phagosome maturation (55). Recently, LRRK2 has been associated with leprosy type-1-reaction, a pathological inflammatory response event (38). Finally, LRRK2 is a negative regulator of inflammasome activation (56, 57) and an inducer of ROS production (54, 57), two known mechanisms of immune defense against bacterial infections also modified by variants of LACC1, a gene consistently associated with leprosy (20, 25, 34, 35). Recently, the LACC1 contribution to leprosy risk has been reinforced: a GWAS-based analysis focusing on functional variants detected association between leprosy and a LACC1 missense variant (rs3764147, c.760A > G, p.Ile254Val) (22).
Several other genes enrolled in immune-response pathways have been associated to leprosy and its phenotypes; however, a full description of these studies goes beyond the scope of this paper, for more detailed data, please refer to Table 1 and Ref. (12, 13, 58, 59).
An intriguing aspect revealed by leprosy hypothesis-free genetic studies is the often identification of genes not classically related to immune response pathways—genetics studies on leprosy have contributed to the description of unsuspected immune-related roles for these genes; Parkin, the protein coded by PARK2, illustrates the case.
The PARK2/Parkin Case
The PARK2 gene was originally described in 1998 as a result of an investigation of microdeletions in patients carrying autosomal recessive juvenile parkinsonism (AR-JP): the authors isolated a 2,960 bp DNA sequence containing an open reading frame coding for a 465 amino acid protein. Characterization of the sequence by alignment and screening of DNA libraries led to the discovery of a ubiquitin-like protein, named Parkin due to its impact on Parkinson disease (60). Two years later, the PARK2 gene product was defined as a ubiquitin-protein ligase and its loss of function reputed as causal of AR-JP (61).
First evidence of a role for PARK2 in leprosy control came from a genome-wide linkage analysis. Genotyping of 388 microsatellite markers covering the whole genome (10 cM interval) was conducted in 86 Vietnamese families displaying 205 affect siblings; 11 chromosome regions were initially linked to leprosy. In a second-round of genotyping, all 11 regions were saturated with additional 89 markers and results evidenced strong co-segregation of the 6q25-q27 region and leprosy (maximum likelihood binomial LOD score 4.31; P = 5 × 10−6) (19). In a follow-up study, association fine mapping of the 6q25-27 genomic region using 208 independent simplex Vietnamese families lead to the discovery of SNPs clustered in the shared promoter region of PARK2 and PACRG genes, associated with increased risk of leprosy in two ethnically independent population, Vietnamese and Brazilian. Linkage-disequilibrium analysis evidenced two tag-SNPs—common allele “T” from PARK2_e01(-2599) and rare allele “C” from rs1040079—capturing the complete association information (41). Interestingly, PARK2, a non-immune related gene by the time of the study, was the first gene described and validated as having an impact on susceptibility to leprosy by a hypothesis-free, positional cloning strategy.
PARK2 association with leprosy was further replicated in an Indian population; however, association signal did not pass a conservative Bonferroni correction for multiple testing (62). Polymorphisms in PARK2/PACRG co-regulatory region were also found associated with leprosy risk in Croatian (63) and two unrelated Indians population samples (42). Moreover, the association was confirmed in independent Vietnamese and Indian samples with a remarkable contribution of age-at-diagnosis to the association signal (43). PARK2’s impact over susceptibility to infection was also demonstrated by association of the T allele-2599 to typhi and paratyphoid fever, diseases caused by Salmonella, an intracellular pathogen (64).
Parkin is an E3 ubiquitin-ligase involved in the proteasome pathway, in particular, the autophagy cellular mechanism of turnover of damaged biomolecules (lipids and proteins) and organelles. Parkin targets are marked and delivered to autophagosomes that are fused with lysosomes and consequently degraded. Of particular importance is the role of Parkin in the mitophagy pathway of mitochondrial recycling: along with PTEN-induced putative kinase protein 1 (PINK1), a mitochondrial kinase, Parkin modulates mitochondrial quality control by mediating the ubiquitination of mitochondrial proteins when the organelle is depolarized (65).
Autophagy has been described as an important defense mechanism aiming to destroy intracellular pathogens, an innate immune response process named xenophagy (66). Through this mechanism, invading microbes are labeled with ubiquitin and adaptor proteins (e.g., p62, NDP52, and optineurin) to be presented to autophagy protein LC3 and initialize assembly of the autophagosome (67). Bacterial degradation by xenophagy has been described against mycobacteria, including M. tuberculosis (68). More recently, Parkin has been described to participate in this pathway mediating resistance against M. tuberculosis and Salmonella enterica serovar Typhi. Parkin is essential for colocalization of ubiquitin along phagosomes markers within M. tuberculosis; murine bone-marrow-derived-macrophages bearing double knockouts for PARK2 are more susceptible to M. tuberculosis or S. enterica growth and present a decrease in survival rate after infection (69). Parkin role in the clearance of intracellular bacteria is corroborated by functional assays performed using dendritic cells infected by Chlamydia: autophagosome degradation of chlamydial infections and MHC-I antigen presentation are increased in presence of Parkin (70).
The influence of Parkin in T-cell stimulation has been also demonstrated in the mitochondrial antigen presentation (MitAP) pathway, based on the generation and trafficking of mitochondrial-derived vesicles (MDV) and mediated by Parkin and PINK1. Under stress, Sorting Nexin-9 (Snx9) and the GTPase Rab9 are recruited to mitochondria and triggers MDV formation; Parkin modulates this process by regulating the level of Snx9 in the cytosol in a proteasome-dependent manner, consequently repressing MitAP in antigen-presenting cells and impacting over immune tolerance (71). It is worth to note that MitAP is not mediated by mitophagy; also, Parkin has an effect upon the production of interleukin-6 and monocyte chemoattractant protein-1 (MCP-1/CCL2) (72), suggesting an impact of Parkin in multiple pathways related to immunity. Interestingly, this impact seems to be conserved among species since impairment of autophagic activity and lifespan after infection is observed in Drosophila melanogaster in the absence of PARK2 expression (69, 73).
In summary, genetic and functional studies have provided strong evidence of PARK2 as a key player in the pathogenesis of leprosy and other infectious diseases. However, these exciting findings are not enough to explain the strong genetic effect observed and estimated through CSA and twin studies—causal variants with high penetrance, able to explain the major gene effect, are yet to be evidenced. The strategies presented next might be powerful to contribute to the advance of the complete dissection of the molecular basis of susceptibility to infection.
Strategies and Future Perspectives
A main genetic assumption underlying classic genetic epidemiology studies is that common diseases are caused by common variants [i.e., nucleotide changes with Minor Allelic Frequency (MAF) > 1%]. Thus, genetic study designs, including GWAS, have been focusing on identifying these common variants and several have been associated to leprosy, some with consistent replication/validation across populations of distinct genetic backgrounds. This positive scenario led to the expectation that these powerful studies would reveal most—if not all—of the genetic variation impacting on susceptibility to common diseases in general and leprosy in particular. However, the picture that emerges today is distinct: GWAS have been revealing a large number of common variants associated with complex traits with very low odds ratios, and the combined effects explain ~5% or less of genetic variance to a given trait (74). These observations led to the term “missing heritability,” which can be at least partially explained by rare variants (MAF < 1%) with larger effects on phenotype or variants other than SNPs, such as copy number variants, both poorly represented in typical genotyping arrays (75). To address this hypothesis, massive deep sequencing technology has been proved to be a powerful tool. In recent years, advances in DNA sequencing chemistry and platforms have allowed an enormous improvement in data generation with a reduced cost (76). Therefore, human whole-genome sequencing (WGS) or whole-exome sequencing (WES) have become feasible and these approaches, especially WES has been proven effective to the identification of genes underlying several Mendelian diseases (77–79). Alternative designs might be used to reduce costs, improve power of detection, and increase individual sample sizes for sequencing; an insightful approach for leprosy might be to submit genes consistently associated with the disease to deep sequencing and search for new/rare variants as causal candidates. Moreover, exons can be preferentially targeted, as variants that cause amino acid change are more likely to have an impact on the phenotypes (80). This rationale was recently used to identify a common and a rare missense functional variant of LACC1 and HIF1A, respectively, as risk factors for leprosy, using WES and targeted second-generation sequencing (22). Another powerful approach has been the use of WGS/WES on the investigation of families or patients displaying extreme or atypical phenotypes; for example, individuals who do not present clinical disease albeit being exposed to an infectious microorganism, as it has been demonstrated for HIV (81, 82). As variants will be likely enriched in such cases, the discovery of causal mutations could be performed in smaller samples size (77).
A natural step further following genetic variant discovery is functional testing. Advances in genome edition technology and cell reprogramming have been allowing isogenic models ideal for functional studies on complex diseases. Such approaches have been proven useful to study neurological diseases such as Huntington’s (83), Parkinson’s (84), and Alzheimer’s disease (85). Genomic variants can be edited by CRISPR/Cas9 in the presence of a donor DNA harboring the nucleotide change; after DNA cleavage by Cas9 nuclease, the homology-directed repair machinery is activated and the donor-DNA is inserted (86), creating a feasible strategy to perform in vitro disease modeling with isogenic controls. In a potentially powerful combination, genome editing strategies could be applied to modify induced pluripotent stem cells (iPSC) (87) that could be differentiated into cell types, for example, targets of a specific pathogen. Although, Cas9 edition system might display off-targets, tools to reduce off-targets mutations have been developed, such as the use of Cas9 in a ribonucleoprotein complex (88) and nickases-Cas9 (89), which cleaves a single strand of DNA, thus a complementary pair of anti-sense gRNAs is necessary to induce mutation (90).
Conclusion
Genetics studies have significantly contributed to the understanding of the molecular basis of leprosy susceptibility and the pathophysiology of the disease. Interestingly, genome-wide, hypothesis-free studies led to the discovery of unsuspected immune-related genes such as PARK2 in the past and, more recently, LACC1 (22, 57, 91). Yet, the impact of rare variants upon disease mechanisms is largely unknown and causal variants that could explain the major gene effects are yet to be described. Advances in genome sequencing technology and functional studies approaches might contribute substantially to further advances in leprosy and other infectious/common diseases.
Author Contributions
GC designed the manuscript and performed major writing. MM contributed to the writing and provided senior supervision.
Conflict of Interest Statement
The authors declare no conflict of interest; the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
GC is supported by a post-doctoral scholarship by Brazilian Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
References
1. Chapman SJ, Hill AVS. Human genetic susceptibility to infectious disease. Nat Rev Genet (2012) 13(3):175–88. doi:10.1038/nrg3114
2. Fox GJ, Orlova M, Schurr E. Tuberculosis in newborns: the lessons of the “Lübeck Disaster” (1929–1933). PLoS Pathog (2016) 12(1):1–10. doi:10.1371/journal.ppat.1005271
3. Sorensen TIA, Nielsen GG, Andersen PK, Teasdale MA. Genetic and environmental influences on premature death in adult adoptees. N Engl J Med (1988) 318(12):727–32. doi:10.1056/NEJM198803243181202
4. Joyce MP. Historic aspects of human susceptibility to leprosy and the risk of conjugal transmission. Mem Inst Oswaldo Cruz (2012) 107(Suppl 1):17–21. doi:10.1590/S0074-02762012000900004
5. Sales AM, Ponce de Leon A, Düppre NC, Hacker MA, Nery JAC, Sarno EN, et al. Leprosy among patient contacts: a multilevel study of risk factors. PLoS Negl Trop Dis (2011) 5(3):1–6. doi:10.1371/journal.pntd.0001013
6. WHO. Global leprosy update, 2016: accelerating reduction of disease burden. Wkly Epidemiol Rec (2017) 92(35):501–20.
7. Ridley DS, Jopling WH. Classification of leprosy according to immunity. A five-group system. Int J Lepr Other Mycobact Dis (1966) 34(3):255–73.
8. Misch EA, Berrington WR, Vary JC Jr, Hawn TR. Leprosy and the human genome. Microbiol Mol Biol Rev (2010) 74(4):589–620. doi:10.1128/MMBR.00025-10
9. Fava V, Orlova M, Cobat A, Alcaïs A, Mira M, Schurr E. Genetics of leprosy reactions: an overview. Mem Inst Oswaldo Cruz (2012) 107(Suppl1):132–42. doi:10.1590/S0074-02762012000900020
10. Lastória JC, Abreu MA. Leprosy: review of epidemiological, clinical, and etiopathogenic aspects – part1. An Bras Dermatol (2014) 89(2):205–18. doi:10.1590/abd1806-4841.20142450
11. Monot M, Honoré N, Garnier T, Zidane N, Sherafi D, Paniz-mondolfi A, et al. Comparative genomic and phylogeographic analysis of Mycobacterium leprae. Nat Genet (2010) 41(12):1282–9. doi:10.1038/ng.477
12. Mira MT. Genetic host resistance and susceptibility to leprosy. Microbes Infect (2006) 8(4):1124–31. doi:10.1016/j.micinf.2005.10.024
13. Sauer MED, Salomão H, Ramos GB, D’Espindula HRS, Rodrigues RSA, Macedo WC, et al. Genetics of leprosy: expected-and unexpected-developments and perspectives. Clin Dermatol (2016) 34(1):96–104. doi:10.1016/j.clindermatol.2015.10.005
14. Shields ED, Russell DA, Pericak-Vance MA. Genetic epidemiology of the susceptibility to leprosy. J Clin Invest (1987) 79(4):1139–43. doi:10.1172/JCI112930
15. Chakravartti MR, Vogel F. A twin study on leprosy. In: Becker PE, Lenz W, Vogel F, Wendt GG, editors. Topics in Human Genetics. Stuttgart: Thieme (1973). p. 1–29.
16. Abel L, Demenais F. Detection of major genes for susceptibility to leprosy and its subtypes in a Caribbean island: Desirade island. Am J Hum Genet (1988) 42(2):256–66.
17. Lázaro FP, Werneck RI, Mackert CCO, Cobat A, Prevedello FC, Pimentel RP, et al. A major gene controls leprosy susceptibility in a hyperendemic isolated population from north of Brazil. J Infect Dis (2010) 201:1598–605. doi:10.1086/652007
18. Siddiqui MR, Meisner S, Tosh K, Balakrishnan K, Ghei S, Fisher SE, et al. A major susceptibility locus for leprosy in India maps to chromosome 10p13. Nat Genet (2001) 27(4):439–41. doi:10.1038/86958
19. Mira MT, Alcaïs A, Van Thuc N, Thai VH, Huong NT, Ba NN, et al. Chromosome 6q25 is linked to susceptibility to leprosy in a Vietnamese population. Nat Genet (2003) 33(3):412–5. doi:10.1038/ng1096
20. Zhang F-R, Huang W, Chen S-M, Sun L-D, Liu H, Li Y, et al. Genomewide association study of leprosy. N Engl J Med (2009) 361(27):2609–18. doi:10.1056/NEJMoa0903753
21. Liu H, Wang Z, Li Y, Yu G, Fu X, Wang C, et al. Genome-wide analysis of protein-coding variants in leprosy. J Invest Dermatol (2017) 137:2544–51. doi:10.1016/j.jid.2017.08.004
22. Wang D, Fan Y, Malhi M, Bi R, Wu Y, Xu M, et al. Missense variants in HIF1A and LACC1 contribute to leprosy risk in Han Chinese. Am J Hum Genet (2018) 102(5):794–805. doi:10.1016/j.ajhg.2018.03.006
23. Alter A, Huong NT, Singh M, Orlova M, Van Thuc N, Katoch K, et al. Human leukocyte antigen class I region single-nucleotide polymorphisms are associated with leprosy susceptibility in Vietnam and India. J Infect Dis (2011) 203(9):1274–81. doi:10.1093/infdis/jir024
24. Wong SH, Gochhait S, Malhotra D, Pettersson FH, Teo YY, Khor CC, et al. Leprosy and the adaptation of human toll-like receptor 1. PLoS Pathog (2010) 6(7):e1000979. doi:10.1371/journal.ppat.1000979
25. Grant AV, Alter A, Huong NT, Orlova M, Van Thuc N, Ba NN, et al. Crohn’s disease susceptibility genes are associated with leprosy in the Vietnamese population. J Infect Dis (2012) 206(11):1763–7. doi:10.1093/infdis/jis588
26. Santos AR, Suffys PN, Vanderborght PR, Moraes MO, Vieira LM, Cabello PH, et al. Role of tumor necrosis factor-alpha and interleukin-10 promoter gene polymorphisms in leprosy. J Infect Dis (2002) 186(11):1687–91. doi:10.1086/345366
27. Moraes MO, Pacheco AG, Schonkeren JJM, Vanderborght PR, Nery JAC, Santos AR, et al. Interleukin-10 promoter single-nucleotide polymorphisms as markers for disease susceptibility and disease severity in leprosy. Genes Immun (2004) 5(7):592–5. doi:10.1038/sj.gene.6364122
28. Malhotra D, Darvishi K, Sood S, Sharma S, Grover C, Relhan V, et al. IL-10 promoter single nucleotide polymorphisms are significantly associated with resistance to leprosy. Hum Genet (2005) 118(2):295–300. doi:10.1007/s00439-005-0042-8
29. Alvarado-Arnez LE, Amaral EP, Sales-Marques C, Durães SM, Cardoso CC, Nunes Sarno E, et al. Association of IL10 polymorphisms and leprosy: a meta-analysis. PLoS One (2015) 10(9):e0136282. doi:10.1371/journal.pone.0136282
30. Liu H, Irwanto A, Tian H, Fu X, Yu Y, Yu G, et al. Identification of IL18RAP/IL18R1 and IL12B as leprosy risk genes demonstrates shared pathogenesis between inflammation and infectious diseases. Am J Hum Genet (2012) 91(5):935–41. doi:10.1016/j.ajhg.2012.09.010
31. Zhang F, Liu H, Chen S, Low H, Sun L, Cui Y, et al. Identification of two new loci at IL23R and RAB32 that influence susceptibility to leprosy. Nat Genet (2011) 43(12):1247–51. doi:10.1038/ng.973
32. Jarduli L, Alves H, de Souza-Santana F, Marcos EV, Pereira A, Dias-Baptista IM, et al. Influence of KIR genes and their HLA ligands in the pathogenesis of leprosy in a hyperendemic population of Rondonópolis, Southern Brazil. BMC Infect Dis (2014) 14(1):438. doi:10.1186/1471-2334-14-438
33. Franceschi DSA, Mazini PS, Rudnick CCC, Sell AM, Tsuneto LT, Melo FC, et al. Association between killer-cell immunoglobulin-like receptor genotypes and leprosy in Brazil. Tissue Antigens (2008) 72:478–82. doi:10.1111/j.1399-0039.2008.01127.x
34. Liu H, Irwanto A, Fu X, Yu G, Yu Y, Sun Y, et al. Discovery of six new susceptibility loci and analysis of pleiotropic effects in leprosy. Nat Genet (2015) 47(3):267–71. doi:10.1038/ng.3212
35. Sales-Marques C, Salomão H, Fava VM, Alvarado-Arnez LE, Amaral EP, Cardoso CC, et al. NOD2 and CCDC122-LACC1 genes are associated with leprosy susceptibility in Brazilians. Hum Genet (2014) 133(12):1525–32. doi:10.1007/s00439-014-1502-9
36. Marcinek P, Jha AN, Shinde V, Sundaramoorthy A, Rajkumar R, Suryadevara NC, et al. LRRK2 and RIPK2 variants in the NOD 2-mediated signaling pathway are associated with susceptibility to Mycobacterium leprae in Indian populations. PLoS One (2013) 8(8):e73103. doi:10.1371/journal.pone.0073103
37. Wang D, Xu L, Lv L, Su LY, Fan Y, Zhang DF, et al. Association of the LRRK2 genetic polymorphisms with leprosy in Han Chinese from Southwest China. Genes Immun (2015) 16(2):112–9. doi:10.1038/gene.2014.72
38. Fava VM, Manry J, Cobat A, Orlova M, Van Thuc N, Ba NN, et al. A Missense LRRK2 variant is a risk factor for excessive inflammatory responses in leprosy. PLoS Negl Trop Dis (2016) 10(2):e0004412. doi:10.1371/journal.pntd.0004412
39. Alcaïs A, Alter A, Antoni G, Orlova M, Nguyen VT, Singh M, et al. Stepwise replication identifies a low-producing lymphotoxin-alpha allele as a major risk factor for early-onset leprosy. Nat Genet (2007) 39(4):517–22. doi:10.1038/ng2000
40. Alter A, De Léséleuc L, Van Thuc N, Thai VH, Huong NT, Ba NN, et al. Genetic and functional analysis of common MRC1 exon 7 polymorphisms in leprosy susceptibility. Hum Genet (2010) 127(3):337–48. doi:10.1007/s00439-009-0775-x
41. Mira MT, Alcaïs A, Nguyen VT, Moraes MO, Di Flumeri C, Vu HT, et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature (2004) 427(6975):636–40. doi:10.1038/nature02326
42. Chopra R, Ali S, Srivastava AK, Aggarwal S, Kumar B, Manvati S, et al. Mapping of PARK2 and PACRG overlapping regulatory region reveals LD structure and functional variants in association with leprosy in unrelated Indian population groups. PLoS Genet (2013) 9(7):e1003578. doi:10.1371/journal.pgen.1003578
43. Alter A, Fava VM, Huong NT, Singh M, Orlova M, Van Thuc N, et al. Linkage disequilibrium pattern and age-at-diagnosis are critical for replicating genetic associations across ethnic groups in leprosy. Hum Genet (2013) 132(1):107–16. doi:10.1007/s00439-012-1227-6
44. De Sales Marques C, Brito-De-Souza VN, Guerreiro LTA, Martins JH, Amaral EP, Cardoso CC, et al. Toll-like receptor 1 N248s single-nucleotide polymorphism is associated with leprosy risk and regulates immune activation during mycobacterial infection. J Infect Dis (2013) 208(1):120–9. doi:10.1093/infdis/jit133
45. Bochud P, Hawn TR, Siddiqui MR, Saunderson P, Abraham I, Argaw AT, et al. Toll-like receptor 2 (TLR2) polymorphisms are associated with reversal reaction in leprosy. J Infect Dis (2008) 197(2):253–61. doi:10.1086/524688
46. Cardoso CC, Pereira C, Brito-De-Souza VN, Duraes SMB, Ribeiro-alves M, Nery AC, et al. TNF-308G>A single nucleotide polymorphism is associated with leprosy among Brazilians: a genetic epidemiology assessment, meta-analysis, and functional study. J Infect Dis (2011) 204(8):1256–63. doi:10.1093/infdis/jir521
47. Fava VM, Cobat A, Van Thuc N, Latini ACP, Stefani MMA, Belone AF, et al. Association of TNFSF8 regulatory variants with excessive inflammatory responses but not leprosy per se. J Infect Dis (2015) 211(6):968–77. doi:10.1093/infdis/jiu566
48. Jarduli LR, Sell AM, Reis PG, Sippert EÂ, Ayo CM, Mazini PS, et al. Role of HLA, KIR, MICA, and cytokines genes in leprosy. Biomed Res Int (2013) 2013:98983. doi:10.1155/2013/989837
49. Sales-Marques C, Cardoso CC, Alvarado-Arnez LE, Illaramendi X, Sales AM, Hacker MDA, et al. Genetic polymorphisms of the IL6 and NOD2 genes are risk factors for inflammatory reactions in leprosy. PLoS Negl Trop Dis (2017) 11(7):e0005754. doi:10.1371/journal.pntd.0005754
50. Schenk M, Mahapatra S, Le P, Kim HJ, Choi AW, Brennan PJ, et al. Human NOD2 recognizes structurally unique muramyl dipeptides from Mycobacterium leprae. Infect Immun (2016) 84(9):2429–38. doi:10.1128/IAI.00334-16
51. Britton WJ, Lockwood DNJ. Leprosy. Lancet (2004) 363(9416):1209–19. doi:10.1016/S0140-6736(04)15952-7
52. Teles RMB, Kelly-scumpia KM, Sarno EN, Rea TH, Ochoa MT, Cheng G, et al. IL-27 suppresses antimicrobial activity in human leprosy. J Invest Dermatol (2015) 135(10):2410–7. doi:10.1038/jid.2015.195
53. Waschbüsch D, Michels H, Strassheim S, Ossendorf E, Kessler D, Gloeckner CJ, et al. LRRK2 transport is regulated by its novel interacting partner Rab32. PLoS One (2014) 9(10):e111632. doi:10.1371/journal.pone.0111632
54. Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens: commentary. J Immunol (2010) 185(9):5577–85. doi:10.4049/jimmunol.1000548
55. Härtlova A, Herbst S, Peltier J, Rodgers A, Bilkei-Gorzo O, Fearns A, et al. LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages. EMBO J (2018) 37(12):e98694. doi:10.15252/embj.201798694
56. Liu W, Liu X, Li Y, Zhao J, Liu Z, Hu Z, et al. LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella typhimurium infection. J Exp Med (2017) 214(10):3051–66. doi:10.1084/jem.20170014
57. Cader MZ, Boroviak K, Zhang Q, Assadi G, Kempster SL, Sewell GW, et al. C13orf31 (FAMIN) is a central regulator of immunometabolic function. Nat Immunol (2016) 17(9):1046–56. doi:10.1038/ni.3532
58. Fava VM, Schurr E. The complexity of the host genetic contribution to the human response to Mycobacterium leprae. In: Scollard D, Gillis T, editors. The International Textbook of Leprosy. Greenville: American Leprosy Missions (2016). p. 1–31.
59. Cardoso CC, Pereira AC, de Sales Marques C, Moraes MO. Leprosy susceptibility: genetic variations regulate innate and adaptive immunity, and and disease outcome. Future Microbiol (2011) 6(5):533–49. doi:10.2217/fmb.11.39
60. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature (1998) 392(6676):605–8. doi:10.1038/33416
61. Shimura H, Hattori N, Kubo SI, Mizuno Y, Asakawa S, Minoshima S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet (2000) 25(3):302–5. doi:10.1038/77060
62. Malhotra D, Darvishi K, Lohra M, Kumar H, Grover C, Sood S, et al. Association study of major risk single nucleotide polymorphisms in the common regulatory region of PARK2 and PACRG genes with leprosy in an Indian population. Eur J Hum Genet (2006) 14(4):438–42. doi:10.1038/sj.ejhg.5201563
63. Bakija-Konsuo A, Mulić R, Boraska V, Pehlic M, Huffman JE, Hayward C, et al. Leprosy epidemics during history increased protective allele frequency of PARK2/PACRG genes in the population of the Mljet Island, Croatia. Eur J Med Genet (2011) 54(6):e548–52. doi:10.1016/j.ejmg.2011.06.010
64. Ali S, Vollaard AM, Widjaja S, Surjadi C, Van De Vosse E, Van Dissel JT. PARK2/PACRG polymorphisms and susceptibility to typhoid and paratyphoid fever. Clin Exp Immunol (2006) 144(3):425–31. doi:10.1111/j.1365-2249.2006.03087.x
65. Seirafi M, Kozlov G, Gehring K. Parkin structure and function. FEBS J (2015) 282:2076–88. doi:10.1111/febs.13249
66. Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol (2014) 12(2):101–14. doi:10.1038/nrmicro3160
67. Sorbara MT, Girardin SE. Emerging themes in bacterial autophagy. Curr Opin Microbiol (2015) 23:163–70. doi:10.1016/j.mib.2014.11.020
68. Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell (2012) 150(4):803–15. doi:10.1016/j.cell.2012.06.040
69. Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature (2013) 501(7468):512–6. doi:10.1038/nature12566
70. Radomski N, Kagebein D, Liebler-Tenorio E, Karger A, Rufer E, Tews BA, et al. Mito-xenophagic killing of bacteria is coordinated by a metabolic switch in dendritic cells. Sci Rep (2017) 7(1):3923–41. doi:10.1038/s41598-017-04142-5
71. Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, et al. Parkinson’s disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell (2016) 166(2):314–27. doi:10.1016/j.cell.2016.05.039
72. de Léséleuc L, Orlova M, Cobat A, Girard M, Huong NT, Ba NN, et al. PARK2 mediates interleukin 6 and monocyte chemoattractant protein 1 production by human macrophages. PLoS Negl Trop Dis (2013) 7(1):e2015. doi:10.1371/journal.pntd.0002015
73. Cho JH, Park JH, Chung CG, Shim HJ, Jeon KH, Yu SW, et al. Parkin-mediated responses against infection and wound involve TSPO-VDAC complex in Drosophila. Biochem Biophys Res Commun (2015) 463(1–2):1–6. doi:10.1016/j.bbrc.2015.05.006
74. Orlova M, Di Pietrantonio T, Schurr E. Genetics of infectious diseases: hidden etiologies and common pathways. Clin Chem Lab Med (2011) 49(9):1427–37. doi:10.1515/CCLM.2011.620
75. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature (2009) 461(7265):747–53. doi:10.1038/nature08494
76. Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet (2016) 17(6):333–51. doi:10.1038/nrg.2016.49
77. Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet (2010) 11(6):415–25. doi:10.1038/nrg2779
78. Ku C, Naidoo N, Pawitan Y. Revisiting Mendelian disorders through exome sequencing. Hum Genet (2011) 129:351–70. doi:10.1007/s00439-011-0964-2
79. Manry J, Quintana-murci L. A genome-wide perspective of human diversity. Cold Spring Harb Perspect Med (2013) 3(1):1–16. doi:10.1101/cshperspect.a012450
80. Botstein D, Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet (2003) 33(3s):228–37. doi:10.1038/ng1090
81. Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell (1996) 86:367–77. doi:10.1016/S0092-8674(00)80110-5
82. Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene cohort study, multicenter hemophilia cohort study. Science (1996) 273(5283):1856–62. doi:10.1126/science.273.5283.1856
83. Xu X, Tay Y, Sim B, Yoon S-I, Huang Y, Ooi J, et al. Reversal of phenotypic abnormalities by CRISPR/Cas9-mediated gene correction in Huntington disease patient-derived induced pluripotent stem cells. Stem Cell Reports (2017) 8(3):619–33. doi:10.1016/j.stemcr.2017.01.022
84. Qing X, Walter J, Jarazo J, Arias-Fuenzalida J, Hillje A-L, Schwamborn JC. CRISPR/Cas9 and piggyBac-mediated footprint-free LRRK2-G2019S knock-in reveals neuronal complexity phenotypes and α-synuclein modulation in dopaminergic neurons. Stem Cell Res (2017) 24:44–50. doi:10.1016/j.scr.2017.08.013
85. Fong H, Wang C, Knoferle J, Walker D, Balestra ME, Tong LM, et al. Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Reports (2013) 1(3):226–34. doi:10.1016/j.stemcr.2013.08.001
86. Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature (2016) 533(7601):1–18. doi:10.1038/nature17664
87. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (2006) 126(4):663–76. doi:10.1016/j.cell.2006.07.024
88. Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, Sridharan M, et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol (2015) 208:44–53. doi:10.1016/j.jbiotec.2015.04.024
89. Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR cas9 for enhanced genome editing specificity. Cell (2013) 154(6):1380–9. doi:10.1016/j.cell.2013.08.021
90. Dianov GL, Hübscher U. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res (2013) 41(6):3483–90. doi:10.1093/nar/gkt076
Keywords: leprosy, genetics, association studies, PARK2, next-generation sequencing, disease modeling
Citation: Cambri G and Mira MT (2018) Genetic Susceptibility to Leprosy—From Classic Immune-Related Candidate Genes to Hypothesis-Free, Whole Genome Approaches. Front. Immunol. 9:1674. doi: 10.3389/fimmu.2018.01674
Received: 15 February 2018; Accepted: 06 July 2018;
Published: 20 July 2018
Edited by:
Pranab Kumar Das, Retired, Birmingham, United KingdomReviewed by:
Juan Li, Rockefeller University, United StatesNarinder K. Mehra, All India Institute of Medical Sciences, India
Neil Morgan, University of Birmingham, United Kingdom
Copyright: © 2018 Cambri and Mira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcelo Távora Mira, bS5taXJhQHB1Y3ByLmJy