 Sreya Bagchi
Sreya Bagchi Samantha Genardi
Samantha Genardi Chyung-Ru Wang
Chyung-Ru Wang- Department of Microbiology and Immunology, Northwestern University, Chicago, IL, United States
Dyslipidemia, or altered blood lipid content, is a risk factor for developing cardiovascular disease. Furthermore, several autoimmune diseases, including systemic lupus erythematosus, psoriasis, diabetes, and rheumatoid arthritis, are correlated highly with dyslipidemia. One common thread between both autoimmune diseases and altered lipid levels is the presence of inflammation, suggesting that the immune system might act as the link between these related pathologies. Deciphering the role of innate and adaptive immune responses in autoimmune diseases and, more recently, obesity-related inflammation, have been active areas of research. The broad picture suggests that antigen-presenting molecules, which present self-peptides to autoreactive T cells, can result in either aggravation or amelioration of inflammation. However, very little is known about the role of self-lipid reactive T cells in dyslipidemia-associated autoimmune events. Given that a range of autoimmune diseases are linked to aberrant lipid profiles and a majority of lipid-specific T cells are reactive to self-antigens, it is important to examine the role of these T cells in dyslipidemia-related autoimmune ailments and determine if dysregulation of these T cells can be drivers of autoimmune conditions. CD1 molecules present lipids to T cells and are divided into two groups based on sequence homology. To date, most of the information available on lipid-reactive T cells comes from the study of group 2 CD1d-restricted natural killer T (NKT) cells while T cells reactive to group 1 CD1 molecules remain understudied, despite their higher abundance in humans compared to NKT cells. This review evaluates the mechanisms by which CD1-reactive, self-lipid specific T cells contribute to dyslipidemia-associated autoimmune disease progression or amelioration by examining available literature on NKT cells and highlighting recent progress made on the study of group 1 CD1-restricted T cells.
Introduction
Over the years, it has become apparent that a range of rheumatological and dermatological autoimmune diseases like systemic lupus erythematosus (SLE) and psoriasis are associated with dyslipidemia (1, 2). In most cases, dyslipidemia in autoimmune diseases is characterized by altered serum cholesterol, triglyceride (TG), low-density lipoprotein (LDL), and high-density lipoprotein (HDL) levels (3–5). In this review, the words dyslipidemia and lipid abnormalities will be used interchangeably, while hyperlipidemia will be used to refer to cases in which an increase in cholesterol, TGs, and LDL was observed.
Atherogenic lipid profiles, characterized by increased serum cholesterol and TGs, have been observed up to 10 years prior to diagnosis of rheumatoid arthritis (6), while hypercholesterolemia in SLE patients has only been reported after onset of disease (7, 8). Overall, these data indicate that dyslipidemia might be linked to autoimmune disease. However, whether dyslipidemia acts as a potential trigger for the initiation of autoimmune diseases has not been investigated in-depth. It is known that inflammation plays a key role in autoimmune diseases (9–12). Several studies have shown that obesity gives rise to low-grade chronic inflammation and the incidence and severity of autoimmune diseases, particularly, rheumatoid arthritis and psoriasis, are increased in obese patients compared to population controls (13). These data suggest that metabolic inputs influence inflammatory outputs (14). In obese individuals, for example, adipocytes release pro-inflammatory cytokines and adipokines, like TNF-α, IL-1β, and leptin, which activate both innate and adaptive arms of the immune system (14, 15). Therefore, in addition to genetic and environmental stress, dyslipidemia-induced chronic inflammation could be a driver of autoimmune diseases, suggesting an intricate interplay between lipid metabolism, activation of the immune system, and subsequent development of autoimmune diseases.
One of the major players of the adaptive immune system involved in the pathophysiology of autoimmune diseases is self-antigen reactive T cells (16). Even though most self-reactive T cells are eliminated in the thymus by the process of negative selection, some can escape. These T cells can recognize self-antigens in the periphery (16), so the autoantigens present within the affected tissues are thought to activate autoreactive T cells. Antigen-presenting cells like macrophages, dendritic cells, and B cells present the antigens to T cells, which secrete pro-inflammatory cytokines, leading to tissue damage (17). In the case of SLE, activated Th1 and Th2 effector T cells help B cells to produce autoantibodies (18). These antibodies can form immune complexes, which will then damage the kidney and lead to nephritis (19). In psoriasis, Th1-related cytokines like IFN-γ secreted by effector T cells are known to play a pathogenic role (20). Additionally, IL-17A is considered to be pathogenic in several autoimmune diseases and IL-17A blocking antibodies are currently used for treating autoimmune diseases like rheumatoid arthritis and psoriasis (20–22). Thus far, most studies have looked in to the role of peptide-specific autoreactive T cells in autoimmune disease initiation, progression, and maintenance. However, given that inflammation forms the crux of most autoimmune diseases, and that dyslipidemia is a potential trigger of chronic inflammation, it is imperative to uncover the role of lipid-specific autoreactive T cells in dyslipidemia-associated autoimmune diseases. Since lipid antigen-presenting molecules are widely expressed on a range of antigen-presenting cells in different tissues, it is conceivable that they contribute to the activation of cognate T cells when presented with lipids in inflammatory environments (Figure 1). Work by our lab and others have demonstrated a role for lipid-autoreactive T cells in psoriasis (23, 24). Thus, understanding the role of self-lipid reactive T cells in dyslipidemia-associated autoimmune diseases would not only lead to better treatment options for a myriad of these diseases but also allow for development of preventive measures to either delay or eliminate their progression. Thus, the subsequent sections of this review will focus on detailed discussions of lipid reactive T cells and their role in major dyslipidemia-associated autoimmune diseases like SLE, psoriasis, and RA, as well as its associated comorbidities such as atherosclerosis and obesity.
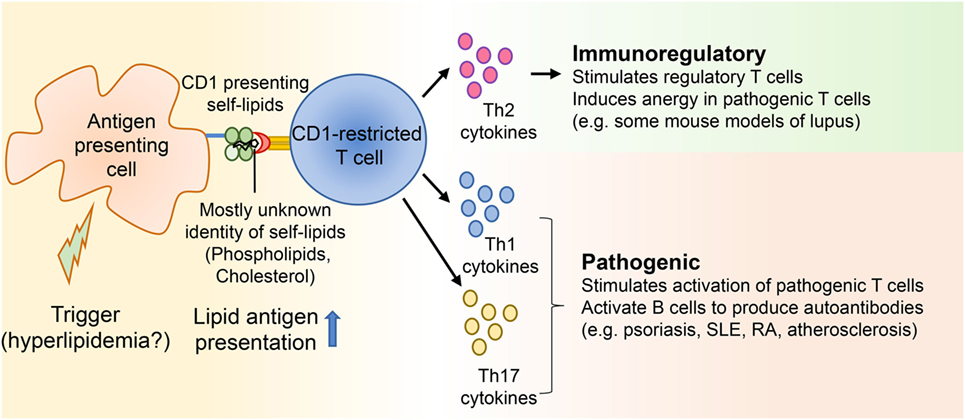
Figure 1. CD1-restricted T cells in hyperlipidemia associated autoimmune diseases. Autoimmune diseases develop as a result of genetic and environmental factors. Several studies suggest that some patients with diseases like systemic lupus erythematosus and psoriasis develop hyperlipidemia before onset of the autoimmune diseases. Yet, not many studies have directly demonstrated whether hyperlipidemia might be a trigger for certain autoimmune ailments. Since CD1 molecules present lipid antigens to T cells and a large portion of these T cells is autoreactive, self-lipid antigens can be presented to CD1-restricted T cells during autoimmune diseases, resulting in the secretion of Th1, Th2, and Th17 cytokines. These cytokines can then modulate either immunoregulatory or pathogenic immune responses by acting on both innate and adaptive immune players.
CD1 Molecules
CD1 are a subset of MHC I-like molecules capable of presenting lipid antigens to T cells (25). Unlike MHC molecules, which are highly polymorphic, CD1 molecules exhibit very limited polymorphism, suggesting that antigens presented by each CD1 molecule are similar from one individual to the other (26). CD1 molecules are found in placental mammals and also birds, which indicates their ancient lineage (27). However, there are varying numbers and types of CD1 isoforms present in different animal species (28–32). CD1 molecules are divided into groups 1, 2, and 3 based on sequence homology and patterns of expression. Group 1 CD1 consists of CD1a, CD1b, and CD1c while CD1d is the sole group 2 CD1 molecule (33). While the abovementioned CD1 isoforms are expressed on the cell surface and present lipid antigens to T cells, the group 3 CD1, CD1e, acts as a chaperone in intracellular compartments to aid with antigen loading onto other CD1 molecules (34–36).
CD1 Structure
CD1 genes are paralogs of MHC genes and are unlinked from the MHC locus; genes encoding all CD1 isoforms are located on the long arm of chromosome 1q22-23 in humans (37–39). Like MHC I molecules, CD1 molecules form heterodimers of heavy α chains with β2 microglobulin (β2m), held together by non-covalent interactions (40–43). The antigen-binding grooves of CD1 molecules are usually narrower, deeper, and more voluminous than MHC I molecules and are lined with hydrophobic/neutral residues that facilitate binding of lipid molecules (44–48). This structural diversity allows CD1 isoforms to bind a range of different lipids, thus suggesting that each isoform may play a non-redundant role in the immune system.
Antigens Presented by CD1 Molecules
Several studies have shown that CD1 molecules can present self-lipids to cognate T cells; yet, the physiological implication of self-lipid presentation under homeostatic and disease conditions remains unclear. We have recently shown that under conditions of hyperlipidemia, presentation of phospholipids and cholesterol by CD1b to cognate T cells drove the development of an inflammatory skin disease resembling psoriasis. In line with our findings, other groups have shown that CD1b can present phospholipids and GM1, a prototypic ganglioside, to T cells (49, 50). Apart from CD1b, CD1d is known to bind a range of glycosphingolipids and phospholipids (51–55). Interestingly, even though the antigen-binding groove of each CD1 molecule is unique, sulfatide, a sulfated glycolipid, is presented by all CD1 molecules, suggesting that each CD1 isoform is capable of presenting both shared and unique lipids (56). Additionally, CD1a can present the autoantigens phosphatidylcholine, lysophosphatidylcholine, and skin-derived apolar, headless oils (57, 58). CD1c can present a unique leukemia-associated methylated-lysophosphatidic acid and cholesteryl esters (59, 60). The ability of CD1 molecules to present such a diverse array of self lipids suggests their potential role in eliciting T cell responses under both steady state and pathological conditions.
CD1 Expression and Tissue Distribution
In humans, CD1 molecules are distributed on a myriad of cell types and tissues. Both group 1 (CD1a, CD1b, and CD1c) and group 2 CD1d molecules are expressed on double-positive (CD4+CD8+) thymocytes (61). In peripheral tissues, group 1 CD1 molecules are expressed exclusively by professional antigen-presenting cells. Dendritic cell subsets from lymph node and skin can express any of the group 1 CD1 isoforms, while B cells can express CD1c (61–63). In contrast to group 1 CD1, group 2 CD1d expression is more widely distributed, found on both hematopoietic and non-hematopoietic-derived cells. Examples of CD1d-expressing cells include epithelial cells of the small bowel and colon, keratinocytes in skin, and hepatocytes in liver (61). CD1 expression can be altered in various autoimmune and inflammatory conditions, thus dictating the flavor of lipid-specific T cell responses. For example, CD1d was upregulated in the psoriatic plaques from patients with active psoriasis, while patients with active psoriasis and dyslipidemia exhibited increased CD1b expression in skin (23, 64). In contrast, CD1d expression was lower in B cells isolated from SLE patients compared to healthy controls, resulting in reduced ability to stimulate CD1d-restricted T cell cytokine production ex vivo (65). CD1 a, b, c, and d were shown to be upregulated in human atherosclerotic plaques compared to non-atherosclerotic arteries, leading to the potential for increased CD1-restricted T cell activation and inflammation (66). Whether CD1 molecules become up or downregulated in the context of autoimmunity and inflammation seems to be dependent on their specific environments and much is still to be learned. Many researchers utilize mice for immunological studies; however, mice only express CD1d and do not express group 1 CD1 molecules, limiting the ability to study CD1-restricted T cells in the context of autoimmunity (67). As a result, little is known about group 1 CD1-restricted T cells under normal and pathologic conditions leading to autoimmunity.
Dysregulation in Antigen Presentation Under Dyslipidemia
Homeostatic presentation of lipid antigens by CD1 molecules can be disrupted by dyslipidemia. Cholesterol uptake and storage is a tightly regulated process that becomes dysregulated with genetic pre-dispositions and/or chronic overnutrition. Under steady-state conditions, cholesterol metabolism is regulated both intracellularly (SREBP2 pathway) and in circulation (LDL metabolism) (68). Dysregulated cholesterol metabolism within antigen-presenting cells has been linked to autoimmunity: Ito et al. reported that cholesterol accumulation within CD11c+ antigen-presenting cells drives autoreactive B cell and T cell expansion and promotes a lupus-like syndrome in mice (69). Our lab showed that serum from hyperlipidemic mice enhances IL-6 production by DCs, driving IL-17A production by autoreactive CD1b-restricted T cells in a model of psoriasis (23). Given that both group 1 and group 2 CD1 molecules are expressed by antigen-presenting cells and are upregulated in multiple autoimmune conditions, it will be important to further characterize the role that antigen-presenting cells play in autoimmunity and dyslipidemia in driving CD1-restricted T cell pathology.
CD1-Restricted T Cells
CD1d-Restricted Natural Killer T (NKT) Cells
CD1d-restricted NKT cells are divided into two main subsets, based on T cell receptor (TCR) usage. Type I NKT cells have an invariant TCR α chain (Vα14-Jα18 in mice and Vα24-Jα18 in humans) and thus are also referred to as invariant NKT (iNKT) cells (70–73). In mice, three β chains (Vβ7, Vβ8.2, and Vβ2) predominantly associate with the invariant α chain, while in humans, the invariant α chain pairs with Vβ11, an ortholog of the mouse Vβ8 (70–73). Unlike type I NKT cells, which have a semi-invariant TCR and recognize the marine sponge-derived lipid α-galactosylceramide (α-GalCer), type II NKT cells have diverse TCR usage and do not recognize α-GalCer (74–78). Rare populations of NKT cells (making up <1% of hematopoietic cells) have also been reported: NKT cells that use γδ chains for their TCR (primarily Vγ1.1 and Vδ6.3), and α-GalCer-reactive NKT cell population harboring a Vα10-Jα50 TCR were identified in mice (79, 80). Vα24− CD1d-α-GalCer-specific T cells expressing CD4 or CD8αβ and using diverse Vα/Vβ chains have been reported in humans (78). NKT cells are “educated” in the thymus where CD1d-expressing cortical thymocytes mediate their positive selection as opposed to thymic epithelial cells that select conventional T cells (81). The lipid(s) responsible for the selection of NKT cells are largely unknown, though a recent study suggested that ether-linked lysophospahtidylethanolamine and lysophosphatidic acids might play a role in the thymic selection of iNKT cells (82).
In the thymus, NKT cells are characterized by the expression of the transcription factor PLZF (promyelocytic leukemia zinc finger protein), which is thought to impart the “innate-like” features to these T cells (83, 84). As NKT cells exit the thymus, they exhibit a pre-activated phenotype and have the capacity to rapidly produce Th1, Th2, and Th17 related cytokines upon TCR stimulation (85). These cytokines are produced when NKT cells interact with either self or foreign lipid antigens presented by the CD1d molecule. It is known that a small proportion of conventional T cells are self-peptide reactive. In contrast, most NKT cells can recognize self-lipid antigens, although the ability of self-lipids to stimulate cytokine production is dependent on two inputs: (1) the strength of TCR signaling and (2) the presence of cytokine driven co-stimulation (e.g., IL-12/IL-18 secreted by TLR-activated DCs) (86, 87). The nature of self-lipids that activate NKT cells during steady state and/or during a specific pathogenic challenge remain largely unknown. Thus, by virtue of their pre-activated status and their ability to be activated by self-antigens in the presence of the correct cytokine milieu, it is conceivable that NKT cells play an important role, either pathogenic or protective, in a range of infectious and autoimmune diseases as well as tumor immunity.
Group 1 CD1-Restricted T Cells
In contrast to the copious amounts of information available on NKT cells, progress on group 1 CD1-restricted T cells is limited. Most studies have made use of long-term T cell clones isolated from patients infected with Mycobacterium tuberculosis and Mycobacterium leprae (88–94), though T cell clones derived from multiple sclerosis patients showing autoreactivity to several self glycosphingolipids have been described in the literature (49, 95). These T cell clones mostly have a diverse αβ TCR repertoire and can be CD4 or CD8 single positive or double negative, and capable of producing Th1, Th2, and/or Th17 related cytokines (88–94). While some of these T cell clones recognize lipid antigens from the mycobacterial cell wall, most CD1-restricted T cell clones described are autoreactive. In fact, the frequency of autoreactive group 1 CD1-restricted T cells makes up between 1 in 10 and 1 in 300 of all circulating T cells in humans, suggesting that they represent a substantial part of the T-cell repertoire in humans (96). Since most of the knowledge about these T cells comes from the study of T cell clones isolated from humans, the developmental program of group 1 CD1-restricted T cells and their physiologic responses during infection and autoimmunity are mostly unknown. Therefore, our lab generated a TCR transgenic mouse, expressing a CD1b-restricted self-lipid reactive TCR (HJ1Tg) and crossed it with mice co-expressing group 1 CD1b and CD1c (hCD1Tg) (97).
Characterization of HJ1Tg/hCD1Tg mice demonstrated that positive selection of autoreactive group 1 CD1-restricted T cells was mediated by thymocytes, similar to iNKT cells (97). CD1b-autoreactive HJ1 T cells were enriched in the liver and exhibited an activated/effector phenotype (CD44hi, CD69+, CD122+) in the naïve setting (97). Additionally, HJ1 T cells were shown to be protective against Listeria monocytogenes infection (97) and contribute to antitumor immunity against a CD1b+ T cell lymphoma (97, 98). In a recent study, we also demonstrated that under conditions of hyperlipidemia, HJ1 T cells contributed to the development of an inflammatory psoriasis-like skin inflammation (23). Although the nature of the lipid(s) recognized by HJ1 T cells in the context of Listeria infection remains unknown, HJ1 T cells in the context of hyperlipidemia were most likely activated by excess phospholipid and cholesterol species (23). These data suggest that while transient activation of group 1 CD1-autoreactive T cells may play a protective role in infections, chronic activation of autoreactive CD1-restricted T cells could lead to detrimental effects like initiation of inflammatory conditions and autoimmunity. A comparison of group 1 and group 2 CD1-restricted T cells is in Table 1.
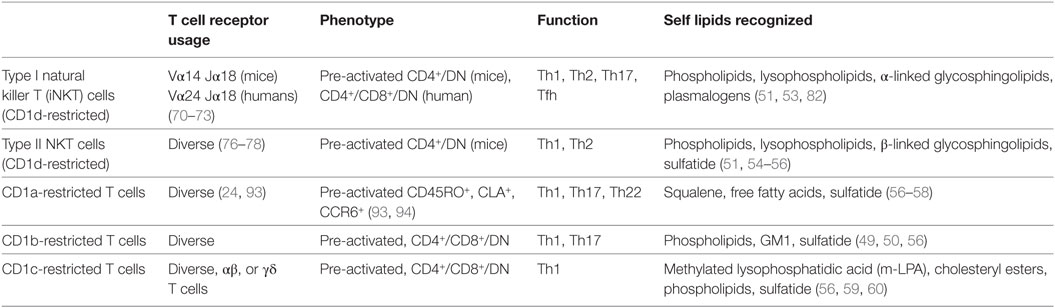
Table 1. Comparison of group 1 and group 2 CD1-restricted autoreactive T cells.
The Role of CD1-Restricted T Cells in Autoimmune Diseases Correlated with Dyslipidemia
Systemic Lupus Erythematosus
It has been well documented that atherosclerosis-related cardiovascular complications comprise the leading cause of death in SLE patients (2). Age, arterial hypertension, smoking habits, diabetes mellitus, obesity, and dyslipidemia are all known to be risk factors for the development of atherosclerosis-related cardiovascular diseases (CVD) among these patients (2). Dyslipidemia is specifically defined in SLE patients as increased total cholesterol, TG, LDL, and decreased levels of HDL (2). Studies have reported approximately 30% of SLE patients displaying signs of hyperlipidemia at the time of diagnosis, which increased to about 60% at 3 years post-diagnosis (99). Even though the mechanisms that cause hyperlipidemia in lupus patients is not well understood, some studies have suggested that the activity of enzyme lipoprotein lipase (LPL), which metabolizes lipids, is reduced and autoantibodies to LPL could be a reason for its reduced activity (100, 101). Like all autoimmune diseases, lupus is characterized by inflammation, often systemic. Under such conditions, LDL particles are more prone to oxidation (ox-LDL) (102). In fact, the process of atherosclerosis is initiated when macrophages ingest ox-LDL to become foam cells. Therefore, not surprisingly, anti-ox-LDL antibodies are present in lupus patients, contributing to the development of dyslipidemia-related cardiovascular ailments (103).
Since dyslipidemia and related pathologies are common in lupus patients and T cells are important players in lupus, exploring the role of lipid-reactive T cells is important. As iNKT cells are the most studied lipid-responsive T cell subset, copious amounts of information are available on these T cells in lupus, though some of the data are contradictory. When lupus was induced in C57BL/6 mice by injecting apoptotic cells, iNKT cells were activated and produced more IL-10 compared to IFN-γ; the absence of iNKT cells was associated with exacerbated disease as a result of increased autoantibody production and glomerular immune complex deposition (104). NKT cells also played a protective role when pristane, a hydrocarbon oil, was injected into BALB/c mice to stimulate a lupus-like disease. Absence of NKT cells (CD1d−/− mice) in this model led to increased nephritis and serum autoantibodies (105). iNKT cell-specific cytokine production, IL-4, and TNF-α, were also decreased, with an expansion of marginal zone B cells (105). Interestingly, activation of iNKT cells with α-GalCer, upon induction of lupus using pristane, protected BALB/c mice from disease, but aggravated disease in SJL mice (106). As noted in the SJL mice, several studies have reported that iNKT cells in BWF1 mice induced autoantibody production from B cells, which promotes lupus pathogenesis (107, 108). In humans, CD1d expression on B cells and iNKT cell frequency and proliferative capacity generally decreases in lupus patients (65, 109, 110), suggesting a protective role for iNKT cell during lupus. However, another study showed that iNKT cells from SLE patients could induce CD1d-dependent CD40/CD40L-dependent anti-dsDNA antibody production by B cells, demonstrating a pathogenic role for iNKT cells in SLE (111). Aside from NKT cells, the role of other lipid-reactive T cells in SLE remains largely unexplored. One study showed that CD1c-restricted T cells isolated from SLE patients can promote class-switched IgG autoantibodies, mediated by CD1c, IL-4, and CD40 (112). Given that dyslipidemia is very prevalent in lupus patients, it is not surprising that lipid-specific autoreactive T cells may contribute to disease progression. However, due to the conflicting role of iNKT cells and the dearth of knowledge about other CD1-restricted T cells in lupus, more research needs to be conducted before their potential can be harnessed in the clinic.
Psoriasis
Psoriasis, a primarily T cell driven autoimmune disease, which affects about 1–3% of the world’s population, is associated with hyperlipidemia (3, 113). Psoriatic patients also have a higher risk of developing cardiovascular disease such as atherosclerotic plaque formation (3). Additionally, psoriatic patients exhibit signs of systemic inflammation, which is largely mediated by neutrophils and T cells (20, 114). Although psoriatic patients are hyperlipidemic and psoriasis is a T cell driven disorder, the role of self-lipid reactive CD1-restricted T cells in psoriasis remains nebulous. Multiple studies have shown an increase in iNKT cells and CD1d expression within psoriatic lesions of mice and humans (64, 115, 116). Human skin engraftment of psoriatic and non-psoriatic skin onto SCID (severe combined immunodeficiency) mice has been the sole mouse model to study NKT cells in psoriasis; this model recapitulates a psoriasis-like disease in engrafted human skin when injected with psoriatic patient-derived lymphocytes (117, 118). An NKT cell line derived from a psoriatic patient was capable of producing IFN-γ when cocultured with CD1d-expressing keratinocytes and induced psoriasis plaque formation when injected into engrafted human skin SCID mice (64, 117). Since CD1d is expressed on keratinocytes and their expression is upregulated in psoriatic skin, it is thought that lipid antigens presented by these cells can activate NKT cells in vivo (61, 64).
Given the high frequency of autoreactive CD1-restricted T cells in humans and the presence of excess lipids under conditions of hyperlipidemia, it is surprising that the role of lipid-specific T cells remains understudied. Hyperlipidemia is induced in mice by either feeding them a high-fat diet, using mice that eat excessively due to genetic manipulations (obese mice) or knocking out genes important for lipid clearance from the blood. Recent work by our lab showed that upon induction of hyperlipidemia, CD1b-autoreactive T cells contributed to the development of psoriasis-like skin inflammation, characterized by a Th17 phenotype (23). Furthermore, there was preferential accumulation of phospholipids and cholesterol in the skin of these mice and the aforementioned lipids could be presented by CD1b to activate CD1b-autoreactive T cells. In humans, CD1b expression was increased in psoriatic compared to normal human skin and there were more CD1b-autoreactive T cells in the blood of affected individuals (23). Additionally, circulating CD1a-autoreactive T cell frequency increases in psoriasis patients compared to healthy controls (119). Presentation of self-lipids on CD1a drives the activation of cognate T cells with a Th17 effector phenotype. This results in the development of psoriatic-like skin inflammation (24, 119). Finally, headless apolar skin lipids can be presented by CD1a to CD1a-autoreactive T cells (58). It can be anticipated that some of these T cells might play a role in psoriasis, especially because they produce IL-17A and IL-22 in response to antigenic stimulation (58). A more in-depth examination of these T cells in psoriasis is warranted to determine the necessity of autoreactive CD1-restricted T cells in driving disease progression.
Rheumatoid Arthritis
Rheumatoid arthritis is an autoimmune disease that affects the connective tissue of the synovial joints. It affects about 0.3–1% of the world’s population, with women being more susceptible than men (120). Interestingly, it is now clear that affected individuals have a 50% increased risk of developing CVD, even though levels of lipids associated with an increased risk of CVD, like cholesterol and LDL, do not always show an association with the development of RA (121–124). The mechanisms of RA disease development and progression are not fully understood; it is speculated that RA-associated inflammation consists of a proinflammatory milieu with self-reactive T and B cells potentially contributing to disease pathogenesis. Thus, it is conceivable that self-lipid reactive T cells might play a role in disease development. It has been demonstrated that iNKT cell number and function is altered in the peripheral blood of RA patients (125). Along with decreases in iNKT cell frequency, their capacity to secrete Th2-related cytokine secretions is also diminished. These findings suggest that iNKT cells may be involved in the disease process of RA, although further studies need to be completed to link these findings with physiological relevance (126). In collagen-induced arthritis (CIA) mouse models, which are commonly used as a model for RA, the absence of iNKT ameliorates disease severity (127, 128). Similar results were obtained when the interaction of CD1d with NKT cells was blocked by administration of anti-CD1d antibody (128). Interestingly, administration of OCH, a ligand known to skew iNKT cells to a Th2 type response, ameliorated disease in wildtype but not iNKT cell-deficient mice (129). Further, neutralization of IL-4 and IL-10 abrogated the OCH-mediated therapeutic benefit (129). Another mouse model of RA uses serum or immunoglobulins from K/BxN mice transferred to wild-type mice to promote joint inflammation. RA disease pathology score was reduced in NKT cell-deficient mice transferred with serum compared to wild-type mice (130). Additionally, iNKT cells were shown to infiltrate the joints of mice given K/BxN serum, secreting IFN-γ and IL-4, which inhibited anti-inflammatory TGF-β secretion in joint fluid (130). These studies show that CD1d-restricted NKT cells contribute to the pathogenesis in multiple mouse models of RA. However, the role of group 1 CD1-restricted T cells in RA remains unknown. Further, whether the role of self-lipid reactive T cells changes in this disease model under conditions of dyslipidemia needs to be elucidated.
Obesity and Atherosclerosis
Due to the ease of tracking iNKT cells in vivo, it is no surprise that studies have examined the role of these T cells in high-fat diet fed, obese mice. In general, iNKT cells in adipose tissues have been implicated in maintaining immune homeostasis by producing IL-10, inducing an anti-inflammatory phenotype in macrophages (mediated by IL-4/STAT-6) and controlling the number and function of regulatory T cells (131). Recent studies have shown that CD1d expression and iNKT cell numbers decrease in obese humans and mice, leading to increased recruitment of proinflammatory macrophages in adipose tissues and insulin resistance (132–135). The cause of this decrease in iNKT cells is unclear, but insufficient stimulation of these T cells due to decreased CD1d expression could be a possibility. In contrast to the findings of the aforementioned studies, Wu et al. demonstrated that iNKT cells cause tissue inflammation in both high fat diet fed and obese mice (136). Further, adipocyte-specific CD1d deficiency ameliorated high fat diet-induced obesity and insulin resistance (137). A pathogenic contribution of iNKT cells under conditions of hyperlipidemia was also reported using β2m knockout mice injected with α-GalCer and fed a high fat diet. These mice had decreased inflammatory macrophage infiltration into adipose tissue compared to control C57BL/6 mice injected with α-GalCer and fed with a high fat diet (138). Since β2M knockout mice lack both CD8+ and NKT cells, the contribution of NKT cells alone to macrophage recruitment was not examined. Lastly, while one study has demonstrated a pathogenic role for type II NKT cells in high fat diet fed mice (139), no studies to date have explored group 1 CD1 expression and the role of group 1 CD1-restricted T cells in obesity.
Aside from diet-induced hyperlipidemia, knocking out genes important for lipid clearance like Apolipoprotein E (Apoe) and low density lipoprotein receptor (Ldlr) results in accumulation of lipids, characterized by increased serum cholesterol, TG and LDL with decreased HDL (140, 141). This lipid profile is similar to dyslipidemia observed in patients with autoimmune diseases. Traditionally these knockout mouse models have been used to study the formation of atherosclerotic plaques in murine blood vessels; unlike humans, mice do not naturally develop atherosclerotic plaques, even when put on high fat diet. Interestingly, in both Apoe and Ldlr knockout mice, a pathogenic role for iNKT cells has been reported. For example, CD1d deficiency significantly reduced atherosclerotic burden in atherosclerosis-prone mice (142–144). When mice were injected with α-GalCer, the process of atherosclerosis was accelerated and secretion of proatherogenic cytokines increased (142–144). Finally, it has been reported that iNKT cells play a pathogenic role during the initial phases of atherosclerotic plaque formation, but not during the later stages (143). Since atherosclerotic plaques harbor different species of oxidized and modified lipids, it is thought that CD1d-expressing DCs present these lipids to NKT cells, leading to their activation. This causes secretion of proinflammatory cytokines like IFN-γ, aggravating disease. A recent study showed that a monoclonal CD1b-autoreactive T cell did not significantly contribute to plaque formation in ApoE−/− mice at later stages of disease (30-week-old mice) (23). Since it is now appreciated that immune cells are instrumental in the maintenance of atherosclerotic plaques, it is of interest to further explore the role of group 1 CD1-autoreactive T cells in this disease, especially because autoreactive group 1 CD1-restricted T cells form a large proportion of CD1-reactive T cells in humans, and group 1 CD1 molecules are expressed in human atherosclerotic plaques but not in normal arterial walls (66).
The Effect of Statins, a Class of Lipid-Lowering Drugs, on Autoimmune Diseases
Statins, which are prescribed for lowering lipid levels in individuals with atherosclerosis, have favorable effects on autoimmune disease activity. In SLE patients, statin treatment was shown to lower proteinuria (145, 146). Additionally, statins can also have immunomodulatory functions such as increasing the presence of regulatory T cells, which are crucial for keeping autoimmune diseases at bay (147–149). Several studies have also noted a decrease in psoriasis area severity index scores in patients with psoriasis upon statin treatment (150, 151). However, another study did not find any statistically significant differences in psoriasis activity upon statin treatment (152). Therefore, it is too early to determine whether statins indeed ameliorate psoriasis. Finally, statin treatment has been shown to reduce disease activity scores of RA patients (153). These studies suggest a close link between lipid abnormalities and autoimmune diseases.
Conclusion
Several autoimmune diseases have been strongly linked to lipid abnormalities (2). Whether dyslipidemia acts as a trigger for disease development or whether dyslipidemia is an outcome of autoimmune diseases is not well understood. In diseases like atherosclerosis and SLE, hyperlipidemia is known to develop before disease onset (2) and cause chronic inflammation (154). T cells play an important role in autoimmune disease progression; therefore, it is essential to decipher the role of T cells, specifically self-lipid reactive T cells, in dyslipidemia-associated autoimmune diseases. Current information on the role of these T cells in autoimmune diseases is shown in Table 2. As mentioned above, most of the knowledge comes from the study of iNKT cells. Even so, a slew of contradictory results has made it difficult to reach definitive conclusions about their role under conditions of dyslipidemia-associated autoimmunity. The conflicting results regarding the role of iNKT cells in various diseases may arise from a range of factors. For example, it is known that different subsets of iNKT cells have different functions (155). A majority of iNKT cells are CD4+ and can secrete IFN-γ and IL-4 under different stimuli (156). However, NK1.1− iNKT cells are known to secrete more IL-17A (157). Additionally, the localization and microenvironment affect the function of iNKT cells. For example, adipose tissue-derived iNKT cells are known to play a regulatory role by not only producing IL-10 but also stimulating the activity of regulatory T cells (131). Furthermore, the genetic background of the mice can also affect iNKT function and thus their role in various disease conditions. It is known that iNKT cells from BALB/c mice secrete more IL-4 compared to IFN-γ than iNKT cells from C57BL/6 mice (155). Finally, microbiome compositions, which differ among animal housing facilities, may lead to discrepancies in results. Therefore, it is important to carefully evaluate these parameters before comparing results about iNKT cells in scientific studies. An additional parameter to note is that mouse models often do not fully mimic human autoimmunity. For example, mouse models of RA share autoreactive T cells, citrullinated autoantibodies, and macrophage/neutrophil infiltrate, but based on the method of disease induction, do not recapitulate the distribution of rheumatoid pannus and necessity of T cells as drivers of disease seen in humans (158). These reasons above highlight the importance of corroborating data from mouse models with human studies.
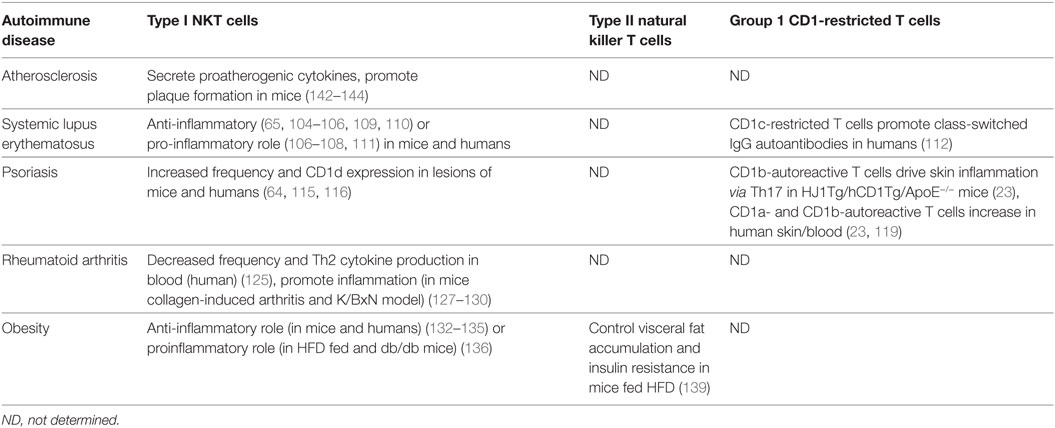
Table 2. Lipid-specific T cell involvement in autoimmune diseases and dyslipidemia.
Aside from CD1d, the study of group 1 CD1-restricted T cells under conditions of dyslipidemia and autoimmunity remains obscure. However, recent studies from our lab demonstrated a pathogenic role of CD1b-autoreactive T cells in hyperlipidemia-associated psoriasis-like skin inflammation (23). Additionally, very little is known about CD1d-restricted type II NKT cells, owing to the lack of markers to track them in vivo. The presence of type II NKT cells in mice makes them more amenable than group 1 CD1-restricted T cells to study in murine mouse models; however, the differences in lipid metabolism between mice and humans makes it critical to corroborate any murine findings of type II NKT cells with human studies. It is important to study these T cell subsets in more depth not only because they form a substantial proportion of the total human T cell population but also because a majority of these T cells are known to be autoreactive.
Though the role of iNKT cells has been deciphered in some autoimmune diseases, the identity of self-lipids recognized by these T cells remain largely unknown. The general thought is that antigen-presenting cells expressing CD1-self-lipid complexes activate CD1-restricted T cells. Activated T cells produce cytokines that are either immunomodulatory or inflammatory. The nature of the cytokines secreted is influenced by the functional state of the antigen-presenting cells, the type of self-lipid being presented, and the microenvironment. It is crucial to identify these lipids not only for tracking these T cells in vivo, but also for harnessing their potential in the clinic. Additionally, very few studies have examined the role of CD1-restricted T cells under dyslipidemia and autoimmune disorders. Thus, the study of self-lipid reactive T cells in this context is still in its infancy; more research investigating the links between autoimmunity, inflammation, and dyslipidemia could inform both diagnosis and treatment of autoimmune diseases in the future.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We sincerely apologize to colleagues whose work we have not cited due to space constraints or oversight. We thank Lavanya Visvabharathy and Eva Morgun for critical reading and helpful discussions. This work was supported by the National Institutes of Health R01 grants AI057460 and AI43407.
References
1. Ma C, Harskamp CT, Armstrong EJ, Armstrong AW. The association between psoriasis and dyslipidaemia: a systematic review. Br J Dermatol (2013) 168(3):486–95. doi:10.1111/bjd.12101
2. Toms TE, Panoulas VF, Kitas GD. Dyslipidaemia in rheumatological autoimmune diseases. Open Cardiovasc Med J (2011) 5:64–75. doi:10.2174/1874192401105010064
3. Pietrzak A, Michalak-Stoma A, Chodorowska G, Szepietowski JC. Lipid disturbances in psoriasis: an update. Mediators Inflamm (2010) 2010:1–13. doi:10.1155/2010/535612
4. Szabo MZ, Szodoray P, Kiss E. Dyslipidemia in systemic lupus erythematosus. Immunol Res (2017) 65(2):543–50. doi:10.1007/s12026-016-8892-9
5. Choy E, Ganeshalingam K, Semb AG, Szekanecz Z, Nurmohamed M. Cardiovascular risk in rheumatoid arthritis: recent advances in the understanding of the pivotal role of inflammation, risk predictors and the impact of treatment. Rheumatology (Oxford) (2014) 53(12):2143–54. doi:10.1093/rheumatology/keu224
6. van Halm VP, Nielen MM, Nurmohamed MT, van Schaardenburg D, Reesink HW, Voskuyl AE, et al. Lipids and inflammation: serial measurements of the lipid profile of blood donors who later developed rheumatoid arthritis. Ann Rheum Dis (2007) 66(2):184–8. doi:10.1136/ard.2006.051672
7. Bruce IN, Urowitz MB, Gladman DD, Hallett DC. Natural history of hypercholesterolemia in systemic lupus erythematosus. J Rheumatol (1999) 26(10):2137–43.
8. Urquizu-Padilla M, Balada E, Chacon P, Perez EH, Vilardell-Tarres M, Ordi-Ros J. Changes in lipid profile between flare and remission of patients with systemic lupus erythematosus: a prospective study. J Rheumatol (2009) 36(8):1639–45. doi:10.3899/jrheum.081097
9. O’Shea JJ, Ma A, Lipsky P. Cytokines and autoimmunity. Nat Rev Immunol (2002) 2(1):37–45. doi:10.1038/nri702
10. Cho JH, Feldman M. Heterogeneity of autoimmune diseases: pathophysiologic insights from genetics and implications for new therapies. Nat Med (2015) 21(7):730–8. doi:10.1038/nm.3897
11. Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol (2012) 32(9):2045–51. doi:10.1161/ATVBAHA.108.179705
12. Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med (1999) 340(2):115–26. doi:10.1056/NEJM199901143400207
13. Versini M, Jeandel PY, Rosenthal E, Shoenfeld Y. Obesity in autoimmune diseases: not a passive bystander. Autoimmun Rev (2014) 13(9):981–1000. doi:10.1016/j.autrev.2014.07.001
14. Greenberg AS, Obin MS. Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr (2006) 83(2):461S–5S. doi:10.1093/ajcn/83.2.461S
15. Monteiro R, Azevedo I. Chronic inflammation in obesity and the metabolic syndrome. Mediators Inflamm (2010) 2010. doi:10.1155/2010/289645
16. Bluestone JA, Bour-Jordan H, Cheng M, Anderson M. T cells in the control of organ-specific autoimmunity. J Clin Invest (2015) 125(6):2250–60. doi:10.1172/JCI78089
17. Riedhammer C, Weissert R. Antigen presentation, autoantigens, and immune regulation in multiple sclerosis and other autoimmune diseases. Front Immunol (2015) 6:322. doi:10.3389/fimmu.2015.00322
18. Fields ML, Nish SA, Hondowicz BD, Metzgar MH, Wharton GN, Caton AJ, et al. The influence of effector T cells and Fas ligand on lupus-associated B cells. J Immunol (2005) 175(1):104–11. doi:10.4049/jimmunol.175.1.104
19. Lech M, Anders HJ. The pathogenesis of lupus nephritis. J Am Soc Nephrol (2013) 24(9):1357–66. doi:10.1681/ASN.2013010026
20. Cai Y, Fleming C, Yan J. New insights of T cells in the pathogenesis of psoriasis. Cell Mol Immunol (2012) 9(4):302–9. doi:10.1038/cmi.2012.15
21. Honkanen J, Nieminen JK, Gao R, Luopajarvi K, Salo HM, Ilonen J, et al. IL-17 immunity in human type 1 diabetes. J Immunol (2010) 185(3):1959–67. doi:10.4049/jimmunol.1000788
22. Bellemore SM, Nikoopour E, Schwartz JA, Krougly O, Lee-Chan E, Singh B. Preventative role of interleukin-17 producing regulatory T helper type 17 (Treg 17) cells in type 1 diabetes in non-obese diabetic mice. Clin Exp Immunol (2015) 182(3):261–9. doi:10.1111/cei.12691
23. Bagchi S, He Y, Zhang H, Cao L, Van Rhijn I, Moody DB, et al. CD1b-autoreactive T cells contribute to hyperlipidemia-induced skin inflammation in mice. J Clin Invest (2017) 127(6):2339–52. doi:10.1172/JCI92217
24. Kim JH, Hu Y, Yongqing T, Kim J, Hughes VA, Le Nours J, et al. CD1a on Langerhans cells controls inflammatory skin disease. Nat Immunol (2016) 17(10):1159–66. doi:10.1038/ni.3523
25. Barral DC, Brenner MB. CD1 antigen presentation: how it works. Nat Rev Immunol (2007) 7(12):929–41. doi:10.1038/nri2191
26. Han M, Hannick LI, DiBrino M, Robinson MA. Polymorphism of human CD1 genes. Tissue Antigens (1999) 54(2):122–7. doi:10.1034/j.1399-0039.1999.540202.x
27. Dascher CC. Evolutionary biology of CD1. In: Moody DB, editor. T Cell Activation by CD1 and Lipid Antigens. Current Topics in Microbiology and Immunology. Berlin, Heidelberg: Springer (2007). p. 3–26.
28. Calabi F, Belt KT, Yu CY, Bradbury A, Mandy WJ, Milstein C. The rabbit CD1 and the evolutionary conservation of the CD1 gene family. Immunogenetics (1989) 30(5):370–7. doi:10.1007/BF02425277
29. Hayes SM, Knight KL. Group 1 CD1 genes in rabbit. J Immunol (2001) 166(1):403–10. doi:10.4049/jimmunol.166.1.403
30. Dascher CC, Hiromatsu K, Naylor JW, Brauer PP, Brown KA, Storey JR, et al. Conservation of a CD1 multigene family in the guinea pig. J Immunol (1999) 163(10):5478–88.
31. Bradbury A, Belt KT, Neri TM, Milstein C, Calabi F. Mouse CD1 is distinct from and co-exists with TL in the same thymus. EMBO J (1988) 7(10):3081–6.
32. Matsuura A, Hashimoto Y, Kinebuchi M, Kasai K, Ichimiya S, Katabami S, et al. Rat CD1 antigen: structure, expression and function. Transplant Proc (1997) 29(3):1705–6. doi:10.1016/S0041-1345(97)00023-7
33. Calabi F, Jarvis JM, Martin L, Milstein C. Two classes of CD1 genes. Eur J Immunol (1989) 19(2):285–92. doi:10.1002/eji.1830190211
34. Facciotti F, Cavallari M, Angenieux C, Garcia-Alles LF, Signorino-Gelo F, Angman L, et al. Fine tuning by human CD1e of lipid-specific immune responses. Proc Natl Acad Sci U S A (2011) 108(34):14228–33. doi:10.1073/pnas.1108809108
35. Cala-De Paepe D, Layre E, Giacometti G, Garcia-Alles LF, Mori L, Hanau D, et al. Deciphering the role of CD1e protein in mycobacterial phosphatidyl-myo-inositol mannosides (PIM) processing for presentation by CD1b to T lymphocytes. J Biol Chem (2012) 287(37):31494–502. doi:10.1074/jbc.M112.386300
36. de la Salle H, Mariotti S, Angenieux C, Gilleron M, Garcia-Alles LF, Malm D, et al. Assistance of microbial glycolipid antigen processing by CD1e. Science (2005) 310(5752):1321–4. doi:10.1126/science.1115301
37. Calabi F, Milstein C. A novel family of human major histocompatibility complex-related genes not mapping to chromosome 6. Nature (1986) 323(6088):540–3. doi:10.1038/323540a0
38. Albertson DG, Fishpool R, Sherrington P, Nacheva E, Milstein C. Sensitive and high resolution in situ hybridization to human chromosomes using biotin labelled probes: assignment of the human thymocyte CD1 antigen genes to chromosome 1. EMBO J (1988) 7(9):2801–5.
39. Yu CY, Milstein C. A physical map linking the five CD1 human thymocyte differentiation antigen genes. EMBO J (1989) 8(12):3727–32.
40. Martin LH, Calabi F, Lefebvre FA, Bilsland CA, Milstein C. Structure and expression of the human thymocyte antigens CD1a, CD1b, and CD1c. Proc Natl Acad Sci U S A (1987) 84(24):9189–93. doi:10.1073/pnas.84.24.9189
41. Martin LH, Calabi F, Milstein C. Isolation of CD1 genes: a family of major histocompatibility complex-related differentiation antigens. Proc Natl Acad Sci U S A (1986) 83(23):9154–8. doi:10.1073/pnas.83.23.9154
42. Knowles RW, Bodmer WF. A monoclonal antibody recognizing a human thymus leukemia-like antigen associated with beta 2-microglobulin. Eur J Immunol (1982) 12(8):676–81. doi:10.1002/eji.1830120810
43. Bauer A, Huttinger R, Staffler G, Hansmann C, Schmidt W, Majdic O, et al. Analysis of the requirement for beta 2-microglobulin for expression and formation of human CD1 antigens. Eur J Immunol (1997) 27(6):1366–73. doi:10.1002/eji.1830270611
44. Zajonc DM, Elsliger MA, Teyton L, Wilson IA. Crystal structure of CD1a in complex with a sulfatide self antigen at a resolution of 2.15 A. Nat Immunol (2003) 4(8):808–15. doi:10.1038/ni948
45. Zajonc DM, Crispin MD, Bowden TA, Young DC, Cheng TY, Hu J, et al. Molecular mechanism of lipopeptide presentation by CD1a. Immunity (2005) 22(2):209–19. doi:10.1016/j.immuni.2004.12.009
46. Gadola SD, Zaccai NR, Harlos K, Shepherd D, Castro-Palomino JC, Ritter G, et al. Structure of human CD1b with bound ligands at 2.3 A, a maze for alkyl chains. Nat Immunol (2002) 3(8):721–6. doi:10.1038/ni821
47. Zeng Z, Castano AR, Segelke BW, Stura EA, Peterson PA, Wilson IA. Crystal structure of mouse CD1: an MHC-like fold with a large hydrophobic binding groove. Science (1997) 277(5324):339–45. doi:10.1126/science.277.5324.339
48. Koch M, Stronge VS, Shepherd D, Gadola SD, Mathew B, Ritter G, et al. The crystal structure of human CD1d with and without alpha-galactosylceramide. Nat Immunol (2005) 6(8):819–26. doi:10.1038/ni1225
49. Shamshiev A, Donda A, Carena I, Mori L, Kappos L, De Libero G. Self glycolipids as T-cell autoantigens. Eur J Immunol (1999) 29(5):1667–75. doi:10.1002/(SICI)1521-4141(199905)29:05<1667::AID-IMMU1667>3.0.CO;2-U
50. Van Rhijn I, van Berlo T, Hilmenyuk T, Cheng TY, Wolf BJ, Tatituri RV, et al. Human autoreactive T cells recognize CD1b and phospholipids. Proc Natl Acad Sci U S A (2016) 113(2):380–5. doi:10.1073/pnas.1520947112
51. Gumperz JE, Roy C, Makowska A, Lum D, Sugita M, Podrebarac T, et al. Murine CD1d-restricted T cell recognition of cellular lipids. Immunity (2000) 12(2):211–21. doi:10.1016/S1074-7613(00)80174-0
52. Joyce S, Woods AS, Yewdell JW, Bennink JR, De Silva AD, Boesteanu A, et al. Natural ligand of mouse CD1d1: cellular glycosylphosphatidylinositol. Science (1998) 279(5356):1541–4. doi:10.1126/science.279.5356.1541
53. De Silva AD, Park JJ, Matsuki N, Stanic AK, Brutkiewicz RR, Medof ME, et al. Lipid protein interactions: the assembly of CD1d1 with cellular phospholipids occurs in the endoplasmic reticulum. J Immunol (2002) 168(2):723–33. doi:10.4049/jimmunol.168.2.723
54. Nair S, Boddupalli CS, Verma R, Liu J, Yang R, Pastores GM, et al. Type II NKT-TFH cells against Gaucher lipids regulate B-cell immunity and inflammation. Blood (2015) 125(8):1256–71. doi:10.1182/blood-2014-09-600270
55. Zhao J, Weng X, Bagchi S, Wang CR. Polyclonal type II natural killer T cells require PLZF and SAP for their development and contribute to CpG-mediated antitumor response. Proc Natl Acad Sci U S A (2014) 111(7):2674–9. doi:10.1073/pnas.1323845111
56. Shamshiev A, Gober HJ, Donda A, Mazorra Z, Mori L, De Libero G. Presentation of the same glycolipid by different CD1 molecules. J Exp Med (2002) 195(8):1013–21. doi:10.1084/jem.20011963
57. Bourgeois EA, Subramaniam S, Cheng TY, De Jong A, Layre E, Ly D, et al. Bee venom processes human skin lipids for presentation by CD1a. J Exp Med (2015) 212(2):149–63. doi:10.1084/jem.20141505
58. de Jong A, Cheng TY, Huang S, Gras S, Birkinshaw RW, Kasmar AG, et al. CD1a-autoreactive T cells recognize natural skin oils that function as headless antigens. Nat Immunol (2014) 15(2):177–85. doi:10.1038/ni.2790
59. Lepore M, de Lalla C, Gundimeda SR, Gsellinger H, Consonni M, Garavaglia C, et al. A novel self-lipid antigen targets human T cells against CD1c(+) leukemias. J Exp Med (2014) 211(7):1363–77. doi:10.1084/jem.20140410
60. Mansour S, Tocheva AS, Cave-Ayland C, Machelett MM, Sander B, Lissin NM, et al. Cholesteryl esters stabilize human CD1c conformations for recognition by self-reactive T cells. Proc Natl Acad Sci U S A (2016) 113(9):E1266–75. doi:10.1073/pnas.1519246113
61. Dougan SK, Kaser A, Blumberg RS. CD1 expression on antigen-presenting cells. In:Moody DB, editor. T Cell Activation by CD1 and Lipid Antigens. Current Topics in Microbiology and Immunology. Berlin, Heidelberg: Springer (2007). p. 113–41.
62. Pena-Cruz V, Ito S, Dascher CC, Brenner MB, Sugita M. Epidermal Langerhans cells efficiently mediate CD1a-dependent presentation of microbial lipid antigens to T cells. J Invest Dermatol (2003) 121(3):517–21. doi:10.1046/j.1523-1747.2003.12429.x
63. Smith ME, Thomas JA, Bodmer WF. CD1c antigens are present in normal and neoplastic B-cells. J Pathol (1988) 156(2):169–77. doi:10.1002/path.1711560212
64. Bonish B, Jullien D, Dutronc Y, Huang BB, Modlin R, Spada FM, et al. Overexpression of CD1d by keratinocytes in psoriasis and CD1d-dependent IFN-gamma production by NK-T cells. J Immunol (2000) 165(7):4076–85. doi:10.4049/jimmunol.165.7.4076
65. Bosma A, Abdel-Gadir A, Isenberg DA, Jury EC, Mauri C. Lipid-antigen presentation by CD1d(+) B cells is essential for the maintenance of invariant natural killer T cells. Immunity (2012) 36(3):477–90. doi:10.1016/j.immuni.2012.02.008
66. Melian A, Geng YJ, Sukhova GK, Libby P, Porcelli SA. CD1 expression in human atherosclerosis. A potential mechanism for T cell activation by foam cells. Am J Pathol (1999) 155(3):775–86.
67. Balk SP, Bleicher PA, Terhorst C. Isolation and expression of cDNA encoding the murine homologues of CD1. J Immunol (1991) 146(2):768–74.
68. Widenmaier SB, Hotamisligil GS. Immune cell intolerance for excess cholesterol. Immunity (2016) 45(6):1186–8. doi:10.1016/j.immuni.2016.12.006
69. Ito A, Hong C, Oka K, Salazar JV, Diehl C, Witztum JL, et al. Cholesterol accumulation in CD11c(+) immune cells is a causal and targetable factor in autoimmune disease. Immunity (2016) 45(6):1311–26. doi:10.1016/j.immuni.2016.11.008
70. Koseki H, Imai K, Nakayama F, Sado T, Moriwaki K, Taniguchi M. Homogenous junctional sequence of the V14+ T-cell antigen receptor alpha chain expanded in unprimed mice. Proc Natl Acad Sci U S A (1990) 87(14):5248–52. doi:10.1073/pnas.87.14.5248
71. Lantz O, Bendelac A. An invariant T cell receptor alpha chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4-8- T cells in mice and humans. J Exp Med (1994) 180(3):1097–106. doi:10.1084/jem.180.3.1097
72. Porcelli S, Yockey CE, Brenner MB, Balk SP. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J Exp Med (1993) 178(1):1–16. doi:10.1084/jem.178.1.1
73. Dellabona P, Padovan E, Casorati G, Brockhaus M, Lanzavecchia A. An invariant V alpha 24-J alpha Q/V beta 11 T cell receptor is expressed in all individuals by clonally expanded CD4-8- T cells. J Exp Med (1994) 180(3):1171–6. doi:10.1084/jem.180.3.1171
74. Matsuda JL, Naidenko OV, Gapin L, Nakayama T, Taniguchi M, Wang CR, et al. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J Exp Med (2000) 192(5):741–54. doi:10.1084/jem.192.5.741
75. Benlagha K, Weiss A, Beavis A, Teyton L, Bendelac A. In Vivo identification of glycolipid antigen-specific T cells using fluorescent CD1d tetramers. J Exp Med (2000) 191(11):1895–903. doi:10.1084/jem.191.11.1895
76. Cardell S, Tangri S, Chan S, Kronenberg M, Benoist C, Mathis D. CD1-restricted CD4+ T cells in major histocompatibility complex class II-deficient mice. J Exp Med (1995) 182(4):993–1004. doi:10.1084/jem.182.4.993
77. Behar SM, Podrebarac TA, Roy CJ, Wang CR, Brenner MB. Diverse TCRs recognize murine CD1. J Immunol (1999) 162(1):161–7.
78. Gadola SD, Dulphy N, Salio M, Cerundolo V. Valpha24-JalphaQ-independent, CD1d-restricted recognition of alpha-galactosylceramide by human CD4(+) and CD8alphabeta(+) T lymphocytes. J Immunol (2002) 168(11):5514–20. doi:10.4049/jimmunol.168.11.5514
79. Lees RK, Ferrero I, MacDonald HR. Tissue-specific segregation of TCRgamma delta+ NKT cells according to phenotype TCR repertoire and activation status: parallels with TCR alphabeta+NKT cells. Eur J Immunol (2001) 31(10):2901–9. doi:10.1002/1521-4141(2001010)31:10<2901::AID-IMMU2901>3.0.CO;2-#
80. Uldrich AP, Patel O, Cameron G, Pellicci DG, Day EB, Sullivan LC, et al. A semi-invariant Valpha10+ T cell antigen receptor defines a population of natural killer T cells with distinct glycolipid antigen-recognition properties. Nat Immunol (2011) 12(7):616–23. doi:10.1038/ni.2051
81. Bendelac A. Positive selection of mouse NK1+ T cells by CD1-expressing cortical thymocytes. J Exp Med (1995) 182(6):2091–6. doi:10.1084/jem.182.6.2091
82. Facciotti F, Ramanjaneyulu GS, Lepore M, Sansano S, Cavallari M, Kistowska M, et al. Peroxisome-derived lipids are self antigens that stimulate invariant natural killer T cells in the thymus. Nat Immunol (2012) 13(5):474–80. doi:10.1038/ni.2245
83. Savage AK, Constantinides MG, Han J, Picard D, Martin E, Li B, et al. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity (2008) 29(3):391–403. doi:10.1016/j.immuni.2008.07.011
84. Kovalovsky D, Uche OU, Eladad S, Hobbs RM, Yi W, Alonzo E, et al. The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nat Immunol (2008) 9(9):1055–64. doi:10.1038/ni.1641
85. Das R, Sant’Angelo DB, Nichols KE. Transcriptional control of invariant NKT cell development. Immunol Rev (2010) 238(1):195–215. doi:10.1111/j.1600-065X.2010.00962.x
86. Hegde S, Fox L, Wang X, Gumperz JE. Autoreactive natural killer T cells: promoting immune protection and immune tolerance through varied interactions with myeloid antigen-presenting cells. Immunology (2010) 130(4):471–83. doi:10.1111/j.1365-2567.2010.03293.x
87. Mallevaey T, Clarke AJ, Scott-Browne JP, Young MH, Roisman LC, Pellicci DG, et al. A molecular basis for NKT cell recognition of CD1d-self-antigen. Immunity (2011) 34(3):315–26. doi:10.1016/j.immuni.2011.01.013
88. Beckman EM, Porcelli SA, Morita CT, Behar SM, Furlong ST, Brenner MB. Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature (1994) 372(6507):691–4. doi:10.1038/372691a0
89. Rosat JP, Grant EP, Beckman EM, Dascher CC, Sieling PA, Frederique D, et al. CD1-restricted microbial lipid antigen-specific recognition found in the CD8+ alpha beta T cell pool. J Immunol (1999) 162(1):366–71.
90. Sieling PA, Ochoa MT, Jullien D, Leslie DS, Sabet S, Rosat JP, et al. Evidence for human CD4+ T cells in the CD1-restricted repertoire: derivation of mycobacteria-reactive T cells from leprosy lesions. J Immunol (2000) 164(9):4790–6. doi:10.4049/jimmunol.164.9.4790
91. Ulrichs T, Moody DB, Grant E, Kaufmann SH, Porcelli SA. T-cell responses to CD1-presented lipid antigens in humans with Mycobacterium tuberculosis infection. Infect Immun (2003) 71(6):3076–87. doi:10.1128/IAI.71.6.3076-3087.2003
92. Porcelli S, Morita CT, Brenner MB. CD1b restricts the response of human CD4-8- T lymphocytes to a microbial antigen. Nature (1992) 360(6404):593–7. doi:10.1038/360593a0
93. de Jong A, Pena-Cruz V, Cheng TY, Clark RA, Van Rhijn I, Moody DB. CD1a-autoreactive T cells are a normal component of the human alphabeta T cell repertoire. Nat Immunol (2010) 11(12):1102–9. doi:10.1038/ni.1956
94. Van Rhijn I, Kasmar A, de Jong A, Gras S, Bhati M, Doorenspleet ME, et al. A conserved human T cell population targets mycobacterial antigens presented by CD1b. Nat Immunol (2013) 14(7):706–13. doi:10.1038/ni.2630
95. Shamshiev A, Donda A, Prigozy TI, Mori L, Chigorno V, Benedict CA, et al. The alphabeta T cell response to self-glycolipids shows a novel mechanism of CD1b loading and a requirement for complex oligosaccharides. Immunity (2000) 13(2):255–64. doi:10.1016/S1074-7613(00)00025-X
96. de Lalla C, Lepore M, Piccolo FM, Rinaldi A, Scelfo A, Garavaglia C, et al. High-frequency and adaptive-like dynamics of human CD1 self-reactive T cells. Eur J Immunol (2011) 41(3):602–10. doi:10.1002/eji.201041211
97. Li S, Choi HJ, Felio K, Wang CR. Autoreactive CD1b-restricted T cells: a new innate-like T-cell population that contributes to immunity against infection. Blood (2011) 118(14):3870–8. doi:10.1182/blood-2011-03-341941
98. Bagchi S, Li S, Wang CR. CD1b-autoreactive T cells recognize phospholipid antigens and contribute to antitumor immunity against a CD1b+ T cell lymphoma. Oncoimmunology (2016) 5(9):e1213932. doi:10.1080/2162402X.2016.1213932
99. Tselios K, Koumaras C, Gladman DD, Urowitz MB. Dyslipidemia in systemic lupus erythematosus: just another comorbidity? Semin Arthritis Rheum (2016) 45(5):604–10. doi:10.1016/j.semarthrit.2015.10.010
100. Reichlin M, Fesmire J, Quintero-Del-Rio AI, Wolfson-Reichlin M. Autoantibodies to lipoprotein lipase and dyslipidemia in systemic lupus erythematosus. Arthritis Rheum (2002) 46(11):2957–63. doi:10.1002/art.10624
101. de Carvalho JF, Borba EF, Viana VS, Bueno C, Leon EP, Bonfa E. Anti-lipoprotein lipase antibodies: a new player in the complex atherosclerotic process in systemic lupus erythematosus? Arthritis Rheum (2004) 50(11):3610–5. doi:10.1002/art.20630
102. McMahon M, Grossman J, Skaggs B, Fitzgerald J, Sahakian L, Ragavendra N, et al. Dysfunctional proinflammatory high-density lipoproteins confer increased risk of atherosclerosis in women with systemic lupus erythematosus. Arthritis Rheum (2009) 60(8):2428–37. doi:10.1002/art.24677
103. Vaarala O, Alfthan G, Jauhiainen M, Leirisalo-Repo M, Aho K, Palosuo T. Crossreaction between antibodies to oxidised low-density lipoprotein and to cardiolipin in systemic lupus erythematosus. Lancet (1993) 341(8850):923–5. doi:10.1016/0140-6736(93)91213-6
104. Wermeling F, Lind SM, Jordo ED, Cardell SL, Karlsson MC. Invariant NKT cells limit activation of autoreactive CD1d-positive B cells. J Exp Med (2010) 207(5):943–52. doi:10.1084/jem.20091314
105. Yang JQ, Singh AK, Wilson MT, Satoh M, Stanic AK, Park JJ, et al. Immunoregulatory role of CD1d in the hydrocarbon oil-induced model of lupus nephritis. J Immunol (2003) 171(4):2142–53. doi:10.4049/jimmunol.171.4.2142
106. Singh AK, Yang JQ, Parekh VV, Wei J, Wang CR, Joyce S, et al. The natural killer T cell ligand alpha-galactosylceramide prevents or promotes pristane-induced lupus in mice. Eur J Immunol (2005) 35(4):1143–54. doi:10.1002/eji.200425861
107. Takahashi T, Strober S. Natural killer T cells and innate immune B cells from lupus-prone NZB/W mice interact to generate IgM and IgG autoantibodies. Eur J Immunol (2008) 38(1):156–65. doi:10.1002/eji.200737656
108. Zeng D, Liu Y, Sidobre S, Kronenberg M, Strober S. Activation of natural killer T cells in NZB/W mice induces Th1-type immune responses exacerbating lupus. J Clin Invest (2003) 112(8):1211–22. doi:10.1172/JCI200317165
109. Kojo S, Adachi Y, Keino H, Taniguchi M, Sumida T. Dysfunction of T cell receptor AV24AJ18+, BV11+ double-negative regulatory natural killer T cells in autoimmune diseases. Arthritis Rheum (2001) 44(5):1127–38. doi:10.1002/1529-0131(200105)44:5<1127::AID-ANR194>3.0.CO;2-W
110. Oishi Y, Sumida T, Sakamoto A, Kita Y, Kurasawa K, Nawata Y, et al. Selective reduction and recovery of invariant Valpha24JalphaQ T cell receptor T cells in correlation with disease activity in patients with systemic lupus erythematosus. J Rheumatol (2001) 28(2):275–83.
111. Shen L, Zhang H, Caimol M, Benike CJ, Chakravarty EF, Strober S, et al. Invariant natural killer T cells in lupus patients promote IgG and IgG autoantibody production. Eur J Immunol (2015) 45(2):612–23. doi:10.1002/eji.201444760
112. Sieling PA, Porcelli SA, Duong BT, Spada F, Bloom BR, Diamond B, et al. Human double-negative T cells in systemic lupus erythematosus provide help for IgG and are restricted by CD1c. J Immunol (2000) 165(9):5338–44. doi:10.4049/jimmunol.165.9.5338
113. Azfar RS, Gelfand JM. Psoriasis and metabolic disease: epidemiology and pathophysiology. Curr Opin Rheumatol (2008) 20(4):416–22. doi:10.1097/BOR.0b013e3283031c99
114. Reich K. The concept of psoriasis as a systemic inflammation: implications for disease management. J Eur Acad Dermatol Venereol (2012) 26(Suppl 2):3–11. doi:10.1111/j.1468-3083.2011.04410.x
115. Nickoloff BJ, Wrone-Smith T, Bonish B, Porcelli SA. Response of murine and normal human skin to injection of allogeneic blood-derived psoriatic immunocytes: detection of T cells expressing receptors typically present on natural killer cells, including CD94, CD158, and CD161. Arch Dermatol (1999) 135(5):546–52. doi:10.1001/archderm.135.5.546
116. Zhao Y, Fishelevich R, Petrali JP, Zheng L, Anatolievna MA, Deng A, et al. Activation of keratinocyte protein kinase C zeta in psoriasis plaques. J Invest Dermatol (2008) 128(9):2190–7. doi:10.1038/jid.2008.81
117. Nickoloff BJ, Bonish B, Huang BB, Porcelli SA. Characterization of a T cell line bearing natural killer receptors and capable of creating psoriasis in a SCID mouse model system. J Dermatol Sci (2000) 24(3):212–25. doi:10.1016/S0923-1811(00)00120-1
118. Gilhar A, Ullmann Y, Kerner H, Assy B, Shalaginov R, Serafimovich S, et al. Psoriasis is mediated by a cutaneous defect triggered by activated immunocytes: induction of psoriasis by cells with natural killer receptors. J Invest Dermatol (2002) 119(2):384–91. doi:10.1046/j.1523-1747.2002.01812.x
119. Cheung KL, Jarrett R, Subramaniam S, Salimi M, Gutowska-Owsiak D, Chen YL, et al. Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a. J Exp Med (2016) 213(11):2399–412. doi:10.1084/jem.20160258
120. Chaudhari K, Rizvi S, Syed BA. Rheumatoid arthritis: current and future trends. Nat Rev Drug Discov (2016) 15(5):305–6. doi:10.1038/nrd.2016.21
121. Solomon DH, Goodson NJ, Katz JN, Weinblatt ME, Avorn J, Setoguchi S, et al. Patterns of cardiovascular risk in rheumatoid arthritis. Ann Rheum Dis (2006) 65(12):1608–12. doi:10.1136/ard.2005.050377
122. Maradit-Kremers H, Crowson CS, Nicola PJ, Ballman KV, Roger VL, Jacobsen SJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum (2005) 52(2):402–11. doi:10.1002/art.20878
123. Nicola PJ, Maradit-Kremers H, Roger VL, Jacobsen SJ, Crowson CS, Ballman KV, et al. The risk of congestive heart failure in rheumatoid arthritis: a population-based study over 46 years. Arthritis Rheum (2005) 52(2):412–20. doi:10.1002/art.20855
124. Crowson CS, Liao KP, Davis JM III, Solomon DH, Matteson EL, Knutson KL, et al. Rheumatoid arthritis and cardiovascular disease. Am Heart J (2013) 166(4):622–8.e1. doi:10.1016/j.ahj.2013.07.010
125. Yanagihara Y, Shiozawa K, Takai M, Kyogoku M, Shiozawa S. Natural killer (NK) T cells are significantly decreased in the peripheral blood of patients with rheumatoid arthritis (RA). Clin Exp Immunol (1999) 118(1):131–6. doi:10.1046/j.1365-2249.1999.01018.x
126. Linsen L, Thewissen M, Baeten K, Somers V, Geusens P, Raus J, et al. Peripheral blood but not synovial fluid natural killer T cells are biased towards a Th1-like phenotype in rheumatoid arthritis. Arthritis Res Ther (2005) 7(3):R493–502. doi:10.1186/ar1695
127. Ohnishi Y, Tsutsumi A, Goto D, Itoh S, Matsumoto I, Taniguchi M, et al. TCR Valpha14 natural killer T cells function as effector T cells in mice with collagen-induced arthritis. Clin Exp Immunol (2005) 141(1):47–53. doi:10.1111/j.1365-2249.2005.02817.x
128. Chiba A, Kaieda S, Oki S, Yamamura T, Miyake S. The involvement of V(alpha)14 natural killer T cells in the pathogenesis of arthritis in murine models. Arthritis Rheum (2005) 52(6):1941–8. doi:10.1002/art.21056
129. Chiba A, Oki S, Miyamoto K, Hashimoto H, Yamamura T, Miyake S. Suppression of collagen-induced arthritis by natural killer T cell activation with OCH, a sphingosine-truncated analog of alpha-galactosylceramide. Arthritis Rheum (2004) 50(1):305–13. doi:10.1002/art.11489
130. Kim HY, Kim HJ, Min HS, Kim S, Park WS, Park SH, et al. NKT cells promote antibody-induced joint inflammation by suppressing transforming growth factor beta1 production. J Exp Med (2005) 201(1):41–7. doi:10.1084/jem.20041400
131. Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat Immunol (2015) 16(1):85–95. doi:10.1038/ni.3047
132. Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V, et al. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity (2012) 37(3):574–87. doi:10.1016/j.immuni.2012.06.016
133. Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KS, et al. Activation of natural killer T cells promotes M2 Macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4 (IL-4)/STAT6 protein signaling axis in obesity. J Biol Chem (2012) 287(17):13561–71. doi:10.1074/jbc.M112.350066
134. Schipper HS, Rakhshandehroo M, van de Graaf SF, Venken K, Koppen A, Stienstra R, et al. Natural killer T cells in adipose tissue prevent insulin resistance. J Clin Invest (2012) 122(9):3343–54. doi:10.1172/JCI62739
135. Huh JY, Kim JI, Park YJ, Hwang IJ, Lee YS, Sohn JH, et al. A novel function of adipocytes in lipid antigen presentation to iNKT cells. Mol Cell Biol (2013) 33(2):328–39. doi:10.1128/MCB.00552-12
136. Wu L, Parekh VV, Gabriel CL, Bracy DP, Marks-Shulman PA, Tamboli RA, et al. Activation of invariant natural killer T cells by lipid excess promotes tissue inflammation, insulin resistance, and hepatic steatosis in obese mice. Proc Natl Acad Sci U S A (2012) 109(19):E1143–52. doi:10.1073/pnas.1200498109
137. Satoh M, Hoshino M, Fujita K, Iizuka M, Fujii S, Clingan CS, et al. Adipocyte-specific CD1d-deficiency mitigates diet-induced obesity and insulin resistance in mice. Sci Rep (2016) 6:28473. doi:10.1038/srep28473
138. Ohmura K, Ishimori N, Ohmura Y, Tokuhara S, Nozawa A, Horii S, et al. Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice. Arterioscler Thromb Vasc Biol (2010) 30(2):193–9. doi:10.1161/ATVBAHA.109.198614
139. Satoh M, Andoh Y, Clingan CS, Ogura H, Fujii S, Eshima K, et al. Type II NKT cells stimulate diet-induced obesity by mediating adipose tissue inflammation, steatohepatitis and insulin resistance. PLoS One (2012) 7(2):e30568. doi:10.1371/journal.pone.0030568
140. Meir KS, Leitersdorf E. Atherosclerosis in the apolipoprotein-E-deficient mouse: a decade of progress. Arterioscler Thromb Vasc Biol (2004) 24(6):1006–14. doi:10.1161/01.ATV.0000128849.12617.f4
141. Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest (1993) 92(2):883–93. doi:10.1172/JCI116663
142. Tupin E, Nicoletti A, Elhage R, Rudling M, Ljunggren HG, Hansson GK, et al. CD1d-dependent activation of NKT cells aggravates atherosclerosis. J Exp Med (2004) 199(3):417–22. doi:10.1084/jem.20030997
143. Aslanian AM, Chapman HA, Charo IF. Transient role for CD1d-restricted natural killer T cells in the formation of atherosclerotic lesions. Arterioscler Thromb Vasc Biol (2005) 25(3):628–32. doi:10.1161/01.ATV.0000153046.59370.13
144. Major AS, Wilson MT, McCaleb JL, Ru Su Y, Stanic AK, Joyce S, et al. Quantitative and qualitative differences in proatherogenic NKT cells in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol (2004) 24(12):2351–7. doi:10.1161/01.ATV.0000147112.84168.87
145. Abud-Mendoza C, de la Fuente H, Cuevas-Orta E, Baranda L, Cruz-Rizo J, Gonzalez-Amaro R. Therapy with statins in patients with refractory rheumatic diseases: a preliminary study. Lupus (2003) 12(8):607–11. doi:10.1191/0961203303lu429oa
146. Ruiz-Limon P, Barbarroja N, Perez-Sanchez C, Aguirre MA, Bertolaccini ML, Khamashta MA, et al. Atherosclerosis and cardiovascular disease in systemic lupus erythematosus: effects of in vivo statin treatment. Ann Rheum Dis (2015) 74(7):1450–8. doi:10.1136/annrheumdis-2013-204351
147. Meng X, Zhang K, Li J, Dong M, Yang J, An G, et al. Statins induce the accumulation of regulatory T cells in atherosclerotic plaque. Mol Med (2012) 18:598–605. doi:10.2119/molmed.2011.00471
148. Mira E, Leon B, Barber DF, Jimenez-Baranda S, Goya I, Almonacid L, et al. Statins induce regulatory T cell recruitment via a CCL1 dependent pathway. J Immunol (2008) 181(5):3524–34. doi:10.4049/jimmunol.181.5.3524
149. Mausner-Fainberg K, Luboshits G, Mor A, Maysel-Auslender S, Rubinstein A, Keren G, et al. The effect of HMG-CoA reductase inhibitors on naturally occurring CD4+CD25+ T cells. Atherosclerosis (2008) 197(2):829–39. doi:10.1016/j.atherosclerosis.2007.07.031
150. Shirinsky IV, Shirinsky VS. Efficacy of simvastatin in plaque psoriasis: a pilot study. J Am Acad Dermatol (2007) 57(3):529–31. doi:10.1016/j.jaad.2007.05.040
151. Naseri M, Hadipour A, Sepaskhah M, Namazi MR. The remarkable beneficial effect of adding oral simvastatin to topical betamethasone for treatment of psoriasis: a double-blind, randomized, placebo-controlled study. Niger J Med (2010) 19(1):58–61. doi:10.4314/njm.v19i1.54216
152. Faghihi T, Radfar M, Mehrabian Z, Ehsani AH, Rezaei Hemami M. Atorvastatin for the treatment of plaque-type psoriasis. Pharmacotherapy (2011) 31(11):1045–50. doi:10.1592/phco.31.11.1045
153. Khattri S, Zandman-Goddard G. Statins and autoimmunity. Immunol Res (2013) 56(2–3):348–57. doi:10.1007/s12026-013-8409-8
154. Ridker PM. Hyperlipidemia as an instigator of inflammation: inaugurating new approaches to vascular prevention. J Am Heart Assoc (2012) 1(1):3–5. doi:10.1161/JAHA.112.000497
155. Lee YJ, Holzapfel KL, Zhu J, Jameson SC, Hogquist KA. Steady-state production of IL-4 modulates immunity in mouse strains and is determined by lineage diversity of iNKT cells. Nat Immunol (2013) 14(11):1146–54. doi:10.1038/ni.2731
156. Godfrey DI, Stankovic S, Baxter AG. Raising the NKT cell family. Nat Immunol (2010) 11(3):197–206. doi:10.1038/ni.1841
157. McNab FW, Pellicci DG, Field K, Besra G, Smyth MJ, Godfrey DI, et al. Peripheral NK1.1 NKT cells are mature and functionally distinct from their thymic counterparts. J Immunol (2007) 179(10):6630–7. doi:10.4049/jimmunol.179.10.6630
Keywords: CD1, dyslipidemia, antigen presentation, autoreactive T cells, natural killer T cells, animal models
Citation: Bagchi S, Genardi S and Wang C-R (2018) Linking CD1-Restricted T Cells With Autoimmunity and Dyslipidemia: Lipid Levels Matter. Front. Immunol. 9:1616. doi: 10.3389/fimmu.2018.01616
Received: 03 April 2018; Accepted: 29 June 2018;
Published: 16 July 2018
Edited by:
Luc Van Kaer, Vanderbilt University, United StatesReviewed by:
Gennaro De Libero, Universität Basel, SwitzerlandS. Subramanian, University of Washington, United States
Copyright: © 2018 Bagchi, Genardi and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chyung-Ru Wang, Y2h5dW5nLXJ1LXdhbmcmI3gwMDA0MDtub3J0aHdlc3Rlcm4uZWR1