 Mengjia Tang
Mengjia Tang Li Tian
Li Tian Guojing Luo
Guojing Luo Xijie Yu
Xijie Yu- Laboratory of Endocrinology and Metabolism, Department of Endocrinology and Metabolism, National Key Laboratory of Biotherapy/Collaborative Innovation Center of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Chengdu, Sichuan, China
Osteoimmunology is the interdiscipline that focuses on the relationship between the skeletal and immune systems. They are interconnected by shared signal pathways and cytokines. Interferon-gamma (IFN-γ) plays important roles in immune responses and bone metabolism. IFN-γ enhances macrophage activation and antigen presentation. It regulates antiviral and antibacterial immunity as well as signal transduction. IFN-γ can promote osteoblast differentiation and inhibit bone marrow adipocyte formation. IFN-γ plays dual role in osteoclasts depending on its stage. Furthermore, IFN-γ is an important pathogenetic factor in some immune-mediated bone diseases including rheumatoid arthritis, postmenopausal osteoporosis, and acquired immunodeficiency syndrome. This review will discuss the contradictory findings of IFN-γ in osteoimmunology and its clinical application potential.
Introduction
Osteoimmunology is the interdiscipline that focuses on the relationship between the skeletal and immune systems. In 2000, Arron and Choi first proposed the term “osteoimmunology” (1). Over the last 20 years, accumulating evidence has shown the involvement of the skeletal system in the process of hematoiesis and its influences in the immune system. Simultaneously, multiple immune cells and immune-derived cytokines are involved in bone homeostasis. The complex crossover between the skeletal and immune systems can also be found in various clinically relevant diseases such as osteoporosis, rheumatoid arthritis (RA), ankylosing spondylitis, human immunodeficiency virus (HIV), inflammatory bowel disease, and systemic lupus erythematosus (2–5). With a better understanding of osteoimmunology, there is higher possibility to prevent and treat immune-mediated bone diseases by targeting immune cells or immune-derived cytokines. Interferon-gamma (IFN-γ) is one of the immune-derived cytokines in the innate and adaptive immune responses. Recent studies have showed the active roles of IFN-γ in the differentiation of osteoclasts, osteoblasts, and bone marrow adipocytes. However, the effect of IFN-γ in osteoimmunology is less explored. This review will summarize current understanding in this field and discuss the clinical application value of IFN-γ in osteoimmunology.
Osteoimmunology: Interaction Between the Skeletal and Immune Systems
The immune system consists of immune organs, multiple immune cells, and immune factors, which participate in the process of immune defense, immune surveillance, and immune homeostasis. Bone cells including osteoclasts, osteoblasts, bone lining cells, and osteocytes are indispensable moderators for bone homeostasis. Bone provides physiological function for life by maintaining a balance between bone resorption and bone formation. The skeletal and immune systems are interconnected by shared signal pathways and intermediary cytokines (Figure 1).
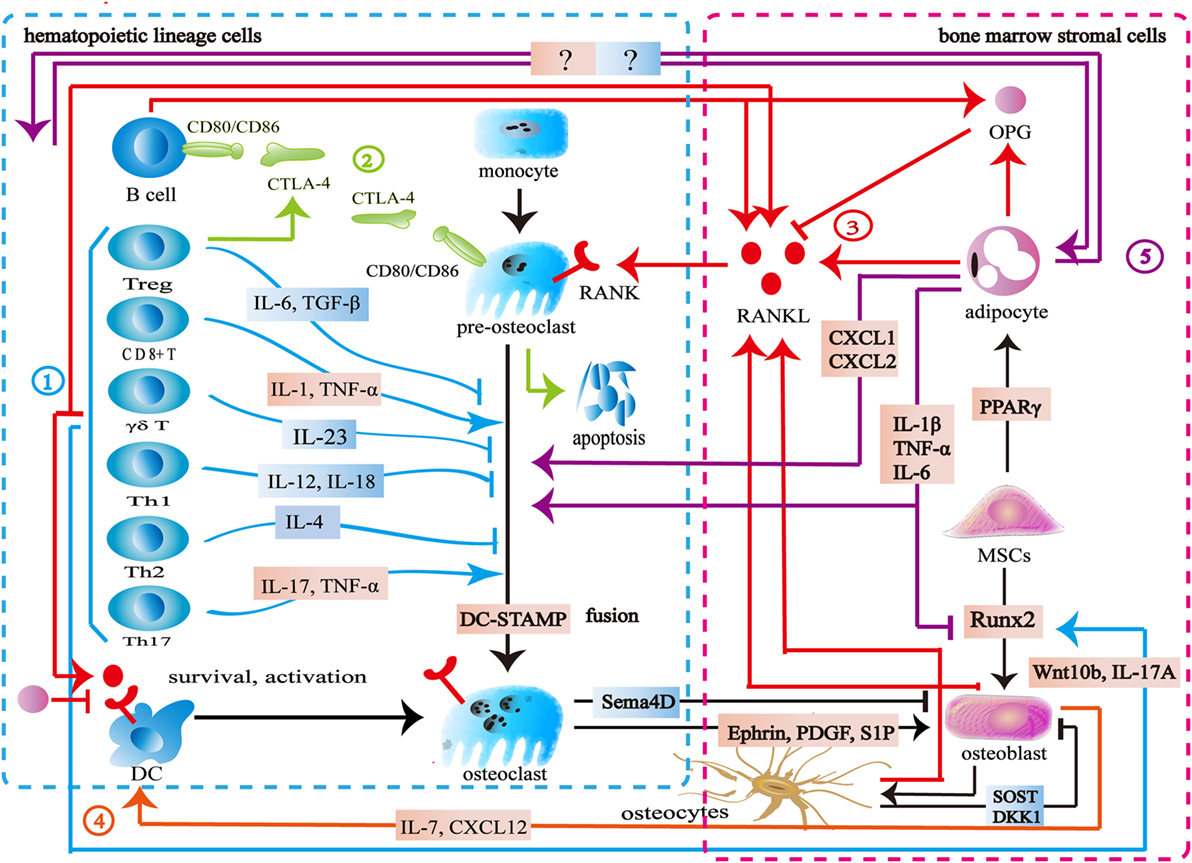
Figure 1. Interaction between the skeletal and immune systems. ① The immune cells produce various cytokines and molecules to regulate bone cells. IL-23, IL-6, IL-12, IL-18, IL-4, interferon-gamma (IFN-γ), TGF-β, and OPG inhibit osteoclastogenesis, while IL-1, IL-6, IL-17, TNF-α, TNF-β, RANKL, and macrophage colony stimulating factor promote osteoclast formation. Wnt10b, IL-17A, and BMP-2 enhance osteoblast formation. ② Cytotoxic T lymphocyte antigen 4 (CTLA-4) promotes the apoptosis of osteoclast precursors by binding to CD80/CD86 on the surface of osteoclasts to induce indoleamine 2,3-dioxygenase. CTLA-4 binds to CD80/CD86 on the surface of B cells to inhibit antigen presentation and the costimulatory signaling in T cells. ③ The skeletal and immune systems share RANK–RANKL–OPG pathways. ④ Osteoblasts produce CXCL12 and IL-7 to maintain lymphopoiesis. ⑤ Bone marrow adipocytes produce several factors to regulate osteoclasts, osteoblasts, and immune cells.
Bone marrow is both a hematopoietic organ and an immune organ. In the bone marrow microenvironment, hematopoietic stem cells differentiate into erythrocytes, granulocytes, platelets, and all kinds of immune cells including monocytes, dendritic cells, T cells, B cells, and natural killer (NK) cells (6, 7). Among them, the monocytes/macrophages from hematopoietic lineage are osteoclast precursors (8, 9). Furthermore, osteoblasts are derived from mesenchymal stem cells (MSCs) in the bone marrow (10). Bone marrow is located in relatively confined bone marrow cavity. Several diversified immune cells including CD4+ T cells, CD8+ T cells, Treg cells, B cells, dendritic cells, and NK cells reside in the bone marrow (11–21). Osteoclasts and osteoblasts attach to the bone surface. Osteocytes are embed in the bone matrix. Bone cells thus share the same microenvironment with immune cells in the bone marrow (Figure 1).
Similar to immune cells, osteoclasts originate from multipotent hematopoietic stem cells. Macrophage colony stimulating factor (M-CSF) binds to c-Fms to mediate the cytoskeletal rearrangement, survival, and proliferation of osteoclasts through the Grb2/extracellular signal-regulated kinase (ERK) and Src/PI3K/Akt pathways (22–25). Furthermore, M-CSF activates PU.1 and MITF to initiate early osteoclast differentiation (26). After that, the RANK–RANKL pathway regulates osteoclast differentiation. RANKL binds to RANK on osteoclast precursors and recruits tumor necrosis factor receptor-associated factors 6 (TRAF6) to activate the mitogen-activated protein kinase (MAPK) family and IkB kinase (27). The MAPK family includes ERK, c-Jun N-terminal kinase (JNK), and p38 MAPK (28, 29). The downstream signaling NF-κB and AP-1 regulate the expression of NFATc1 to promote osteoclast formation (30–33). TREM2 and OSCAR, immunoglobulin-like receptors on the surface of osteoclasts, are associated with immunoreceptor tyrosine-based activation motif (ITAM)-harboring adapters, FcRγ and DAP12, to initiate RANK–RANKL costimulatory signaling. The signaling pathways increase intracellular calcium levels and then regulate transcription of NFATc1 (34–36). Another important member of bone homeostasis is the osteoblasts derived from MSCs. Bone morphogenetic protein and Wnt signaling are responsible for the differentiating of MSCs toward the osteoprogenitor lineage (37, 38). Runt-related transcription factor 2 (Runx2) and osterix (Osx) play crucial roles in this process (39, 40). Finally, some osteoblasts can become osteocytes (41) (Figure 1).
As mentioned above, many signaling pathways are involved in regulating the formation of osteoblasts, osteocytes, and osteoclasts. Among them, the RANK–RANKL–OPG is the most important shared signaling pathway between the skeletal and immune systems. RANKL and RANK are expressed not only by the skeletal system but also by the immune system. In addition to osteoblasts and osteocytes, activated T cells, B cells, NK cells, fibroblasts, and thymus cells can also produce RANKL. Apart from osteoclasts and their progenitor cells, dendritic cells also express RANK (42). Previous studies suggested that osteoblasts were the main source of OPG. New findings show that B cells are also a source of OPG (43, 44). The RANK–RANKL pathway participates in the development of lymph nodes, the self-tolerance of T cells, the survival and apoptosis of dendritic cells, and antigen presentation (45, 46). As an antagonist of RANKL, OPG reduces T cell activation by antagonistic RANK–RANKL. It also directly inhibits the survival and function of dendritic cells (46, 47). In addition to its role as the downstream of the costimulatory signal of RANK–RANKL, ITAM also participates in various signaling cascade in the immune process. Adapter protein DAP12 and FcRγ with ITAM exist in a variety of immune cells including NK cells, neutrophils, macrophages, dendritic cells, basophils, eosinophilic cells, and mast cells to regulate immune response (48). Cytotoxic T lymphocyte antigen 4 (CTLA-4) binds to CD80/CD86 on the surface of the antigen-presenting cells to inhibit the costimulatory signaling of T cells. By binding to CD80/CD86 on the surface of osteoclasts and inducing indoleamine 2,3-dioxygenase (IDO), CTLA-4 promotes osteoclast precursor apoptosis (49). Moreover, Eph receptor, CD100, and osteopontin have the capacity of regulating both the skeletal and immune systems (50–56) (Figure 1).
A variety of immune cells including T cells, B cells, NK cells, and dendritic cells produce various kinds of cytokines to regulate bone metabolism. Activated CD4+T cells and CD8+ T cells secrete RANKL, TNF-α, IL-1, IL-6, and IL-17 to promote osteoclast formation. Th17 cells secrete IL-17, RANKL, TNF-α, IL-1, and IL-6 to induce osteoclast formation (57, 58). γδT cells secrete IL-23 to promote osteoclast formation (59, 60). Treg cells inhibit osteoclast formation directly through CTLA-4 and indirectly through TGF-β, IL-4, and IL-10 (57). FoxP3+ CD8 T-cells produce IFN-γ and CTLA-4 to inhibit osteoclastogenesis while RANKL to promote osteoclastogenesis (61). Both Th1 and Th2 cells secrete IFN-γ and IL-4 to inhibit osteoclast formation (62–64). Furthermore, IL-23 and IL-6 could inhibit osteoclast formation (65, 66). B cells produce TNF-α and TNF-β to promote osteoclast formation, while producing OPG, IFN-γ, and TGF-β to inhibit osteoclast formation (67–69). NK cells play an active role in osteoclast formation by producing RANKL and M-CSF (70). Apart from regulating osteoclasts, some immune cells produce cytokines to regulate osteoblast formation. CD8+ T cells produce Wnt10b to activate Wnt signaling while γδ T cells secrete IL-17A to promote bone formation (59, 71). Furthermore, multiple bone cells are capable of influencing the immune system. Osteoblasts regulate B cell development by expressing CXCL12 and IL-7 to influence the common lymphoid progenitors (72). DLL4 derived from osteoblasts promotes T-linage competent cell function (73). Osteocytes are involved in lymphopoiesis by maintaining primary lymphoid organ microenvironment (74) (Figure 1).
The interaction between bone cells and immune cells is acknowledged in osteoimmunology. However, the importance of bone marrow adipocytes in osteoimmunology seems to be ignored. Bone marrow adipocytes and osteoblasts share the same origin. Adipogenic transcription factors, PPARγ, and C/EBPs promote MSCs to differentiate into adipocytes in the bone marrow (75). As an important component of bone marrow, bone marrow adipocytes share a common microenvironment with bone cells and immune cells. Bone marrow adipocytes produce OPG and RANKL to regulate the RANK–RANKL–OPG pathway and osteoimmunology (76–78). In puberty, bone marrow adipocytes seem to be beneficial to the skeletal system, whereas the increase of bone marrow adipocytes is associated with bone loss in aging, starvation, osteoporosis, unloading, and type 1 diabetes (79). In these pathological states, bone marrow adipocytes produce several factors including M-CSF, IL-6, IL-1 β, TNF-α, CXCL1, and CXCL2 to disturb the balance between the skeletal and immune systems (77, 80). The exact mechanism remains unknown, however, this imbalance is hypothesized to increase osteoclastogenesis and reduce osteogenesis (81). Furthermore, CXCL1 and CXCL2 can activate osteoclasts (82). Further studies are needed to address the physiopathological roles among bone cells, immune cells, and bone marrow adipocytes in osteoimmunology (Figure 1).
Osteoimmunology is involved in immune-mediated bone diseases. Menopause and aging are pathogenesis factors for osteoporosis. An increasing number of studies have shown that immune activation and bone loss concomitantly initiate during aging and menopause (4, 52, 83). Glucocorticoids have inhibitory effects on a broad range of immune responses. Their clinical application may cause osteoporosis. The immunosuppression and bone loss may be correlated (84). In RA, activated immune cells (T cells and B cells) and upregulation of some immune factors, e.g., TNF-α and RANKL result in bone destruction and bone loss (52). Local bone erosion and hyperplasia coexist in ankylosing spondylitis. Th17, TNF-α, IL-23, and IL-17 are involved in the pathological process of ankylosing spondylitis (85). In addition, immune deficiency also has detrimental effects on bone health. Patients with acquired immunodeficiency syndrome have lower bone mineral density and a higher fracture risk. Activated B cells and T cells as well as various immune factors may be the reason behind this (86, 87). The imbalance of bone resorption and bone formation caused by immune dysfunction provides a clue to seek therapeutic targets through osteoimmunology.
Osteoimmunology emphasizes the interaction between the skeletal and the immune systems. The source of IFN-γ is not as widespread as TNF-α, IL-6 and more limited to the immune system (88). Several studies also showed that IFN-γ was involved in bone homeostasis. The involvement of IFN-γ in cancer, atherosclerosis, and hematopoiesis has already been summarized (89–92). However, the role of IFN-γ in osteoimmunology has not been reviewed in detail.
IFN-γ in Osteoimmunology
Signaling Pathway and Function of IFN-γ
Components of the interferon family include type I IFN, type II IFN, and type III IFN. Type I IFN and type III IFN contain several sub-types whereas IFN-γ is the only type II interferon. IFN-γ is produced by multiple types of lymphocyte cells including CD4+ and CD8+ T cells, Treg cells, FoxP3+ CD8 T-cells, γδ T cells, B cells, and NK cells (61, 93). Apart from lymphocyte cells, other immune cells are also capable of producing IFN-γ including monocyte/macrophage, dendritic cells, and neutrophile granulocytes (94–97). Abundant evidence suggests that T cells and NK cells are the major sources of IFN-γ (98). MSCs secrete low-level IFN-γ to regulate hematopoiesis (99). Early studies demonstrated that the monocyte/macrophage and dendritic cells produced IL-12 and IL-18 to upregulate IFN-γ in the monocyte/macrophage, dendritic cells, NK cells, and Th1 cells. IFN-γ also upregulates its own expression in the macrophages by autocrine (96). Through cell–cell contact and factors existing in the co-culture medium, MSCs facilitate IL-12/IL-18-stimulated NK cells to secrete IFN-γ (100). Furthermore, IL-4 and IL-10 negatively regulate its expression (96). In a word, a variety of immune cells produce IFN-γ and the activity and quantity of IFN-γ are dependent on certain inflammatory and immune states.
Interferon-gamma interacts with its receptors to trigger the cellular responses. IFN-γ receptors are ubiquitously expressed on the cellular surface of nearly all types of cells except for erythrocytes (101, 102). The functional receptors consist of receptor α chain (IFNGR1) and β chain (IFNGR2), both of which are vital for activating IFN-γ signaling (101–103). The expression of IFNGR1 seems to be constitutive. On the other hand, the expression of IFNGR2 is inducible because of its extremely low level at typical states. IFNGR1 is required for ligand binding with IFN-γ binding sets in the extracellular domain. Furthermore, IFNGR1 is critical for the downstream signal transduction with the tyrosine kinase JAK1 binding sets and STAT1 binding sets. IFNGR2 contains the tyrosine kinase JAK2 binding sets (101). The JAK–STAT1 pathway is the canonical signaling pathway of IFN-γ and commonly exists in many intracellular responses. By binding to receptors, IFN-γ activates JAK2 and JAK1. The formation of STAT1 binding sets leads to STAT1 recruitment, activation, and homodimer formation. STAT1 homodimers translocate to the nucleus and bind to the gamma-interferon activation site, the promoter regions of interferon-stimulated genes (ISGs), to initiate or turn off its expression (101, 104). ISGs play a vital role in the subsequent biological process (105). Apart from the canonical JAK–STAT1 pathway, IFN-γ also initiates the MAP kinase pathway, PI3K pathway, and NF-κB pathway. Meanwhile, IFN-γ could activate transcription factors STAT3, STAT5, and AP-1. The canonical and non-canonical signaling pathways form its complex signal network (106–128) (Figure 2).
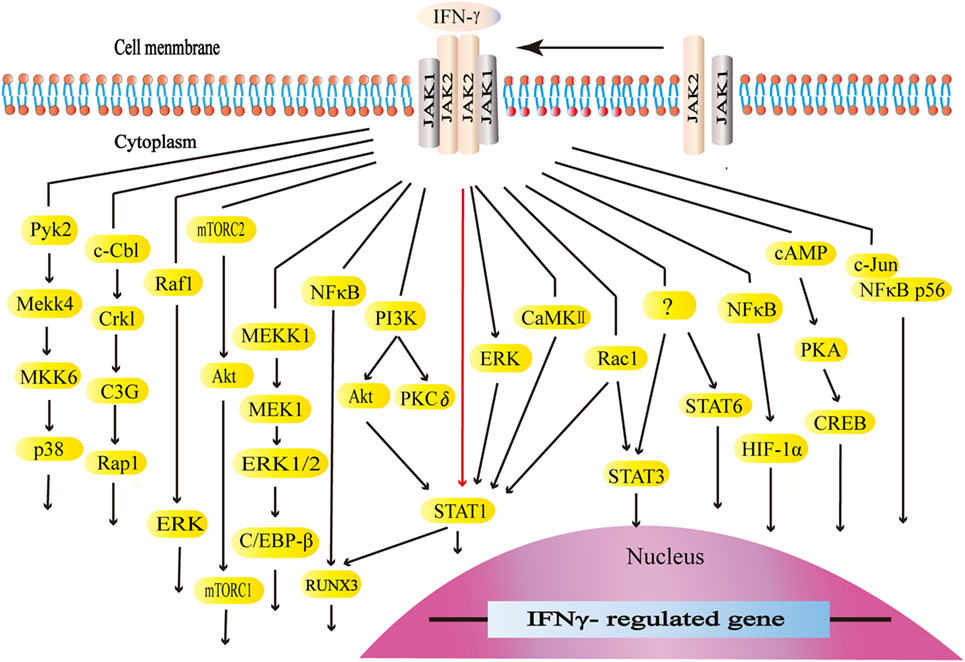
Figure 2. The canonical and non-canonical signaling pathways of interferon-gamma (IFN-γ). The JAK–STAT1 is the canonical signaling pathway. Apart from JAK–STAT1, IFN-γ activates other downstream factors. IFN-γ activates the kinase like PI3K, Pyk2, extracellular signal-regulated kinase (ERK), and CaMKII as well as GTPases like Raf1 and Rac1 to initiate multiple downstream signaling cascades. IFN-γ stimulates E3 ubiquitin-protein ligase c-Cbl, NF-κB, c-Jun, and cAMP, which are also involved in multiple pathways.
Interferon-gamma participates in many immunological functions (innate and adaptive immune responses) and inflammation. IFN-γ enhances macrophage activation and antigen presentation. It is broadly involved in antiviral and antibacterial immunity as well as signal transduction. Due to the complex role of IFN-γ in physiological and pathological conditions, it is difficult to label its effect as pro-inflammatory or anti-inflammatory (98). Many studies aim to explore the possibility of IFN-γ as a drug to treat certain types of cancer, idiopathic pulmonary fibrosis, invasive fungal infection, hepatic fibrosis, RA, and mycosis fungoides (129–134). The US Food and Drug Administration has approved IFN-γ-1b (marketed as Actimmune) to decrease the number and severity of infections in patients with chronic granulomatous disease and to delay the progression of severe, malignant osteopetrosis.
Effect of IFN-γ on the Skeletal System
Effects of IFN-γ on Osteoblasts
Ruiz et al. reported that human osteoblast-like cells express a slight level of IFN-γ (135). Early studies showed that IFN-γ increases alkaline phosphatase (ALP) activity in osteoblast-like cells derived from human femur (136). Meanwhile, IFN-γ promotes calvarial osteoblast formation by upregulating osteogenic factors including Runx2, osterix, Alp, and osteocalcin (137). Activated CD4+ T cells can promote human MSCs to differentiate into osteoblasts in the co-culture system. After blocking IFN-γ with antibodies, osteogenic activity is downregulated (138). Human MSCs also produce IFN-γ to promote its own osteogenic differentiation. The upregulated osteogenic transcription factors Runx2 is responsible for osteoblast formation (139). MSCs are able to differentiate into bone marrow adipocytes as well as osteoblasts. IFN-γ inhibits MSCs to differentiate into adipocytes through the downregulation of adipogenic transcription factor PPARγ and the upregulation of osteogenic transcription factor Runx2 (140). MSCs from IFN-γR−/− mice show a lower osteogenic differentiation due to lower Runx2 expression. Furthermore, IFN-γ siRNA inhibits osteoblast differentiation from MSCs (139, 141). However, the signaling pathway of IFN-γ in osteoblast differentiation is not clear yet.
Effects of IFN-γ on Osteoclasts
Previous studies demonstrated an inhibitory effect of IFN-γ on osteoclast differentiation (Figure 3). Various immune cells including B cells, pro-inflammatory M1 macrophages, dendritic cells, and NK cells produce IFN-γ to inhibit osteoclastogenesis (68, 70, 142). Furthermore, both activated and resting T cells secrete IFN-γ to inhibit osteoclast formation. The activated T-cell supernatant inhibits osteoclast differentiation from bone marrow macrophages. Anti-IFN-γ antibodies can block its effect in the culture system (143). The resting T cells also produce IFN-γ to reduce osteoclast formation when co-cultured with peripheral blood monocytes (144). IFN-γ downregulates c-Fms in monocyte-derived osteoclast precursors to reduce RANK. The M-CSF/c-Fms is critical for early osteoclast proliferation and differentiation. The decrease of RANK+ osteoclast precursors influences subsequent RANKL-dependent osteoclastogenesis (145). The recognized mechanism of IFN-γ in osteoclastogenesis is to promote TRAF6 degradation in RANKL-induced osteoclast differentiation (26, 146). IFN-γ activates the classical JAK–STAT1 pathway to initiate the ubiquitin-proteasome to increase the degradation of ubiquitin ligase TRAF6. Due to the downregulation of TRAF6, the downstream transcription factors of TRAF6 including NF-κB and JNK are inhibited, therefore, osteoclast formation is reduced (143, 147–149). However, two reports indicated that IFN-γ did not target on TRAF6 (147, 148). IFN-γ inhibits osteoclasts via downregulating NFATc1 (148, 149). IFN-γ inhibits osteoclast formation when IFN-γ and RANKL are simultaneously used in bone marrow macrophages or RAW264.7 cells. However, the inhibitory effect is almost completely neutralized after being pretreated with RANKL during a certain period of time (147). RANKL treatment with a short time did not resist the inhibitory effect of IFN-γ on osteoclastogenesis. Furthermore, high concentration RANKL might overly activate both the RANK–RANKL pathway and the RANK intracellular domain (ivvy535–538) to counteract the inhibitory effect (148). Apart from inhibiting osteoclast differentiation, IFN-γ also reduces osteoclast formation by promoting osteoclast apoptosis. IFN-γ inhibits TNF-α-induced osteoclastogenesis by inducing FasL expression in bone marrow cells. IFN-γ promotes osteoclast precursor apoptosis by Fas–FasL interaction (149). IFN-γ might increase FasL expression on osteoblasts by activating the NF-κB pathway to regulate osteoclast apoptosis (150). Furthermore, IFN-γ stimulates osteoblasts to produce NO to promote osteoclast apoptosis (151).
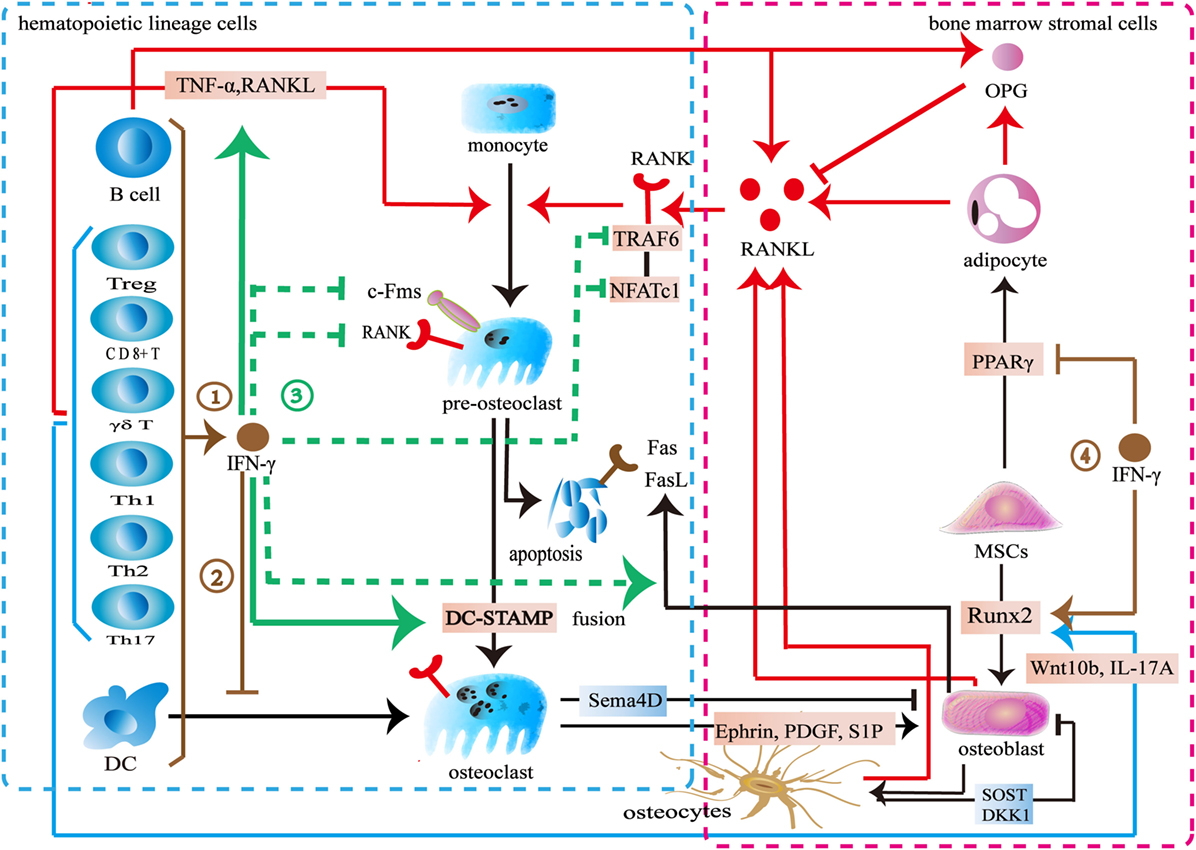
Figure 3. Effect of interferon-gamma (IFN-γ) in osteoimmunology. ① IFN-γ is produced by multiple immune cells including CD4+ T cells, CD8+ T cells, Treg cells, γδ T cells, B cells, and dendritic cells. ② IFN-γ inhibits the differentiation of DCs into osteoclasts. ③ IFN-γ reduces the expression of c-Fms on the surface of osteoclast precursors to inhibit its proliferation and differentiation. IFN-γ decreases the number of RANK+ osteoclast precursors to influence subsequent RANKL-dependent osteoclastogenesis. IFN-γ also increases the degradation of tumor necrosis factor receptor-associated factors 6 (TRAF6) or downregulates NFATc1 in the downstream of RANK–RANKL pathway to inhibit osteoclastogenesis in the early stage of osteoclast differentiation. IFN-γ enhances the apoptosis of osteoclast precursors through FasL. IFN-γ reduces osteoclast formation by directly inhibiting RANK–RANKL pathway, while stimulates the secretion of TNF-α and RANKL by T cells to promote osteoclast formation. IFN-γ promotes the fusion of mononucleate osteoclasts into functional osteoclasts through DC-STAMP. ④ IFN-γ upregulates the osteogenic transcription factor Runx2 and downregulates the adipogenic transcription factor PPARγ.
On the other hand, some studies showed that IFN-γ promotes osteoclast formation (Figure 3). IFN-γ promotes osteoclast maturation in the late stage of osteoclast formation. When monocytes/macrophages are differentiated into functional osteoclasts, they need to be matured into functional osteoclasts by fusion. A seven-transmembrane DC-STAMP protein is involved in osteoclast fusion. Miyamoto et al. (152) reported that NFATc1 and c-fos regulated the expression of DC-STAMP. IFN-γ upregulates c-fos and NFATc1 to ultimately increase DC-STAMP expression. Thus, the fusion of osteoclast is enhanced (153). IFN-γ induces osteoclasts to express IDO to inhibit T cells proliferation (146). However, IFN-γ increases MHC class II expression in the antigen-presenting cells to enhance T cells activation. T cells produce more TNF-α and RANKL to promote osteoclast formation (143, 154, 155). Furthermore, IFN-γ promotes multiple immune cells including monocytes and macrophages to secrete CXCL10. CXCL10 induces CD4+ T cells to secrete RANKL and TNF-α to promote the differentiation of osteoclasts (156, 157).
These contradictory results may result from the differences in the concentrations of IFN-γ, RANKL, and M-CSF, as well as the exposure time and the stage of osteoclast differentiation. IFN-γ inhibits osteoclast differentiation in a dose-dependent manner. However, low level of IFN-γ does not exert the inhibitory effect in osteoclast and treatment with high level of RANKL antagonizes its inhibitory effect in osteoclast differentiation. Also IFN-γ might potentially lose its inhibitory effect during a certain stage of osteoclasts. The inhibitory effect of IFN-γ seems to be limited to the early stage of osteoclast differentiation. The M-CSF/c-Fms signaling is important for early osteoclast proliferation and differentiation. The RANK–RANKL pathway is also vital for early osteoclast differentiation. IFN-γ downregulates c-Fms and RANK to inhibit osteoclastogenesis in the early stage of proliferation and differentiation. IFN-γ also increases the degradation of TRAF6 or downregulates the NFATc1 in the downstream of RANK–RANKL pathway to inhibit osteoclastogenesis in the early stage of osteoclast differentiation. However, IFN-γ promotes osteoclast maturation by increasing fusion in the late stage of osteoclast formation. Interestingly, there is an overlapped transcription factor. IFN-γ downregulates NFATc1 to inhibit osteoclastogenesis in the early stage of differentiation but upregulates NFATc1 to increase the fusion of osteoclast in the late stage of differentiation. In addition to its direct effect, IFN-γ also indirectly increases osteoclastic factors to promote osteoclast formation by activating T cells (26, 146). However, IFN-γ may reduce T cells proliferation by inducing the IDO in osteoclasts. IFN-γ activates immune response to promote osteoclast formation in the early stage whereas multinucleated osteoclasts inhibit the activation T cells in turn. In summary, the dual effect of IFN-γ in osteoclastogenesis may result from the different role of IFN-γ during different stages of osteoclast differentiation. Furthermore, the differences between direct and indirect actions also lead to the conflicting results.
Effects of IFN-γ on Bone Mass
IFN-γ−/− mice do not produce IFN-γ. IFN-γR−/− mice produce IFN-γ but lack the receptors. Both mice show lower bone mass (139, 141, 143, 155). Human T cell lymphotropic virus type 1-Tax transgenic mice (Tax+) have the potential to develop osteolytic bone tumors. Due to the absence of IFN-γ, Tax+IFN-γ−/− mice show more severe tumor-associated osteolytic destruction and lower bone mass with increased osteoclastogenesis. Administration of IFN-γ recused cancer-induced bone loss in Tax+IFN-γ−/− mice (158). Dcir−/− mice are easier to develop ankylosing enthesitis with slightly higher bone mass, while the knockout of IFN-γ counteracts the effect in Dcir−/− mice (137). Duque et al. found IFN-γ increased bone mass in wide-type mice (141).
However, some other studies demonstrated that systematic administration of IFN-γ can reduce bone mass. The clinic trial with recombinant human IFN-γ-1b revealed that IFN-γ promotes bone resorption by functional improvement of osteoclasts (159). Mann et al. found that systemic application of IFN-γ for 8 days led to cancellous bone loss in Sprague-Dawley rats (160). Gao et al. reported that IFN-γ treatment decreased spinal bone mass and increased serum level of C-terminal telopeptides in T cell–replete nude mice and wide-type mice, but not in T cell–nude mice, implicating the importance of T cells (155).
The knockout of IFN-γ or its receptors in animal models induces bone loss. It seems that IFN-γ is necessary to maintain normal bone mass. However, the administration of IFN-γ showed contradictory results. Under a certain medication schedule such as 2,000 or 10,000 IU three times a week for 6 weeks, IFN-γ may increase bone mass (141). However, IFN-γ may cause bone loss in some other schedule such as 1 × 106 IU/kg body weight twice a week for 3 weeks or 1 × 106 IU/kg body weight once a day for 8 days (155, 160). Long-term use of IFN-γ at a low dosage seems to promote bone formation and inhibit osteoclast differentiation at its early stage (141). However, short-term use of IFN-γ at a high dosage seems to act on the late stage of differentiation to promote osteoclast formation (160). Furthermore, short-term use of IFN-γ in a high dosage may activate immune responses especially T cell-mediated immune responses to promote osteoclastogenesis (155). In summary, IFN-γ may be vital to maintain normal bone mass but its dosage, frequency, usage, and course may be important factors. The balance of osteoblasts and osteoclasts, the predominant effect on osteoclasts differentiation, and the activation of immune cells may be the reason behind these contradictory results. Further studies are required to disclose its complex roles in different pathophysiological conditions.
IFN-γ Mediated Osteoimmunology in Diseases
Rheumatoid Arthritis
Rheumatoid arthritis is defined as inflammatory arthritis with periarticular and systemic bone loss (161–163). The etiology of RA is not completely clear but it is known that various adaptive immune responses and innate immune responses are involved in. Immune cells including B cells, T cells, NK cells, dendritic cells, macrophages, and immune factors play important roles in RA inflammatory and bone loss (164). Osteoclast activation and osteoblast inhibition lead to bone destruction and bone loss (161). Synovial fibroblasts, activated B cells, and T cells produce RANKL to influence the RANK–RANKL–OPG pathway and regulate osteoclast formation. Furthermore, synovial fibroblasts produce DKK-1 to block the Wnt signaling pathway and inhibit osteoblast formation. Inflammatory cytokines including TNF-α, IL-1, IL-6, IL-17, IL-18, and IL-23 also activate osteoclasts (165, 166).
Interferon-gamma also plays an important role in the development and progression of RA. NK-like T cells, monocytes, T cells, and B cells secret higher levels of IFN-γ in RA (167–171). The high levels of IFN-γ are found in RA plasma, synovial tissue, and synovial fluid (172, 173). With the progression of disease, IFN-γ levels show an upward trend in plasma (173). Cacciapaglia et al. found that the IFN-γ level in mononuclear cells from the peripheral blood was consistent with the serous of RA (174).
Several studies have reported a negative role of IFN-γ in RA. IFN-γ antibodies are a promising advantage to RA patients. Anti-IFN-γ and anti-TNF-α show similar efficacy in alleviating the synovial membrane thickness and clinical indices in RA (175, 176). In animal experiment, IFN-γ antibodies could reduce synovial proliferation, cell infiltration, bone erosion, and cartilage destruction in the joints of onset RA. Meanwhile, systemic administration of IFN-γ increases arthritic scores in the early stage of RA (177). IFN-γ seems to accelerate the onset and development of RA in the early stage. IFN-γ might initiate disease by activating inflammation and immune responses. IFN-γ mobilizes lymphocytes by activating the classic transcription factor STAT1 to promote RA inflammatory (171). Treg cells limit excessive immune responses, whereas IFN-γ derived from B cells inhibits the differentiation of Treg cells thus activating immune response in RA (170). Furthermore, T cells and macrophages activated by IFN-γ produce more RANKL and TNF-α to promote bone erosion. CXCL10 induced by IFN-γ promotes bone resorption by attracting immune cells such as T cells to be involved in RA (156).
Apart from promoting RA progression, some studies highlight a protective role of IFN-γ in RA. Some trials from 1980 to 1995 provided evidence of improvement and benefit. After the administration of IFN-γ, RA clinical symptoms were relieved (178–181). In animal experiment, IFN-γ−/− mice and IFN-γR−/− mice are easier to induce to be a model of RA (182). IFN-γ inhibited anti-CII antibody responses to inhibit type II collagen-induced arthritis (183). By contrast, IFN-γ antibody exacerbated the severity in the late stage of RA (177, 184). IFN-γ inhibits CII-induced T cell proliferation by limiting IgG1 and IgG2b antibody to the autoantigen in RA (185). The high temperature requirement A1 (HTRA1) is a protease and marker for the incidence and severity of RA. IFN-γ initiates p38 MAPK/STAT1 pathway to inhibit HTRA1 in RA joint tissues (166). As a destructive factor in the progression of RA, IL-17 produced by Th17 cells is involved in the RA pathogenesis. IFN-γ inhibits IL-17 production to alleviate RA (184, 186).
Interferon-gamma seems to exert different effects in different stages of RA. In animal experiment, IFN-γ accelerates the onset and development of RA in the early stage. IFN-γ may promote autoimmunity and activate immune response, increasing the level of osteoclastic factors and leading to the development of RA and bone destruction. The indirect stimulating effect of IFN-γ on osteoclasts may be predominant in the early stage of RA. However, IFN-γ seems to have a protective role in RA in the effector phase. The activation of immune systems in the onset of RA can attract more monocytes and macrophages from blood and peripheral tissue into joints to cause massive amplification of osteoclast formation. IFN-γ reduces c-Fms and RANK to inhibit osteoclast formation. IFN-γ also induces TRAF6 degradation or downregulates NFATc1 to inhibit the RANK–RANKL pathway to prevent osteoclast differentiation. In patients with RA, most of studies show administration of IFN-γ is an effective therapy, but some patients fail to display an adequate response. The difference in the genetic and epigenetic factors, the duel effect of IFN-γ in osteoclastogenesis, disease severity, and rate of progression may be the reason for these contradictory results.
Postmenopausal Osteoporosis
Menopause is a common factor of osteoporosis. Recent researches show that osteoimmunology is involved in the pathogenesis of postmenopausal osteoporosis (187). T-cell activity is increased but B-cells activity is decreased in postmenopausal women (188, 189). Immune cells produce some factors like IL-6, IL-7, IFN-γ, TNF-α, and RANKL to influence the skeletal and immune systems. The multiple cytokines like IL-6, IL-7, and TNF-α enhance the formation or activity of osteoclasts in estrogen deficiency. T cells and B cells produce RANKL to promote osteoclast formation (190, 191). Furthermore, IL-27 has a protective role on bone loss in estrogen deficiency (192). A clinical research showed CD4+ T cells secreted a significant lower IFN-γ in women with postmenopausal osteoporosis (187). Whereas two other studies showed no different in IFN-γ in patients with postmenopausal osteoporosis (193, 194). In animal experiment, IFN-γ level in bone marrow was increased after ovariectomy (150). The bone mass in the IFN-γ−/− mice did not significantly reduce after ovariectomy (155). Under estrogen deficiency, IFN-γ promoted the expression of class II transactivator in immune cells to enhance the antigen presentation between macrophages and T cells, which could upregulate TNF-α and RANKL and promote bone resorption (195). It seems at least in animal studies IFN-γ might enhance bone loss in estrogen deficiency through regulating osteoclastogenesis (26). Further study is definitely needed to clarify its pathogenesis.
Human Immunodeficiency Virus
Acquired immune deficiency syndrome (AIDS) is an acquired defect in cellular immunity associated with infection with the HIV. This syndrome leads to a large destruction of CD4+ T cells and immunodeficiency. The patients with AIDS are particularly vulnerable to bone loss and at higher risk for fracture due to HIV infection, antiretroviral therapy, hypogonadism, lower nutritional status, etc. (86). B cells produce more RANKL and less OPG to promote osteoclast formation in AIDS. After antiretroviral therapy, immune cells including T cells, B cells, and macrophages produce high levels of RANKL and TNF-α to activate osteoclasts (196). The serum level of IFN-γ is lower in exposed-uninfected individuals than that in HIV-unexposed individuals, but there is no data about the main source of IFN-γ. The lower level of IFN-γ might reduce antigen presentation and HLA class II expression to defend HIV infection (197). Some studies have shown IFN-γ inhibits HIV infection but the mechanism remains unknown (198, 199). Another study showed that IFN-γ reduced the expression of CXCR-4 and CCR-5 in monocyte surfaces to inhibit HIV replication (200). IFN-γ inhibits HIV-1 replication stimulated by RANKL (201). The gp120-stimulated T cells secrete RANKL to enhance osteoclast formation, which is an important reason for HIV-related bone loss. IFN-γ might inhibit RANKL-mediated osteoclast formation by promoting TRAF6 degradation in AIDS (201). The positive effect of IFN-γ on HIV infection and HIV-related bone loss make it a promising agent for clinical application.
Conclusion and Prospect
Over the past decade, osteoimmunology has been proposed as a new interdiscipline to understand the relationship between the skeletal and the immune systems. The effects of various immune factors on bone metabolism have led to the investigations of their clinical application. IFN-γ plays an important role in bone homeostasis via activation of complex signaling pathways. IFN-γ potentially promotes osteoblastogenesis but inhibits bone marrow adipocyte formation. However, IFN-γ plays conflicting roles in osteoclastogenesis. Its functions are dependent on the balance of direct or indirect effect, as well as the stage of osteoclast differentiation. IFN-γ inhibits the early differentiation of osteoclasts by targeting the RANK–RANKL pathway, whereas it promotes the fusion of mononucleated osteoclasts in the late stage of osteoclast formation. IFN-γ indirectly increases osteoclastic factors by activating immune responses. Under a condition of complete loss of IFN-γ signaling, normal bone mass cannot be maintained. However, the administration of IFN-γ may cause bone loss or inverse. It depends on the dosage, frequency, usage, and course. Furthermore, the pathogenesis, the disease severity, and rate of progression influence the effect of IFN-γ on immune-mediated bone diseases.
Future research should focus on the molecular pathways and conditions related to the different functions of IFN-γ in osteoimmunology. IFN-γ plays dual effect in osteoclastogenesis. Most studies showed that IFN-γ acts on multiple sites of RANK–RANKL pathway including RANK, TRAF6, and NFATc1. It is urgent to find out the key initial site. NFATc1 is an overlapped transcription factor. It is worth to investigate its role in the dual effect of IFN-γ in osteoclast formation. Second, we need to determine the appropriate dosage, frequency, usage, and course to apply IFN-γ in immune-mediated bone diseases. It is important to explore the effect of IFN-γ according to the pathological mechanisms of the disease. Regarding to RA, IFN-γ seems to exert different effects in different stages. Therefore, it may be important to find a suit appropriate time to use and explore the role of IFN-γ in the skeletal and immune systems in RA.
Author Contributions
XY provided the conception of the manuscript. MT and LT were contributed to writing of the manuscript. MT and GL provided the figure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 81770875, 81572639, 81370969) and the Science and Technology Department of Sichuan Province (2018SZ0142). The authors thank Xiao Yu from the University of Michigan Ann Arbor, for help with editing the language.
References
2. Horvathova M, Ilavska S, Stefikova K, Szabova M, Krivosikova Z, Jahnova E, et al. The cell surface markers expression in postmenopausal women and relation to obesity and bone status. Int J Environ Res Public Health (2017) 14(7):751. doi:10.3390/ijerph14070751
3. Titanji K. Beyond antibodies: B cells and the OPG/RANK-RANKL pathway in health, non-HIV disease and HIV-induced bone loss. Front Immunol (2017) 8:1851. doi:10.3389/fimmu.2017.01851
4. Iseme RA, McEvoy M, Kelly B, Agnew L, Walker FR, Attia J. Is osteoporosis an autoimmune mediated disorder? Bone Rep (2017) 7:121–31. doi:10.1016/j.bonr.2017.10.003
5. Sylvester FA. Inflammatory bowel disease: effects on bone and mechanisms. Adv Exp Med Biol (2017) 1033:133–50. doi:10.1007/978-3-319-66653-2_7
6. Gallagher JC. Advances in bone biology and new treatments for bone loss. Maturitas (2008) 60(1):65–9. doi:10.1016/j.maturitas.2008.04.005
7. Costa MHG, de Soure AM, Cabral JMS, Ferreira FC, da Silva CL. Hematopoietic niche – exploring biomimetic cues to improve the functionality of hematopoietic stem/progenitor cells. Biotechnol J (2018) 13(2). doi:10.1002/biot.201700088
8. Bar-Shavit Z. The osteoclast: a multinucleated, hematopoietic-origin, bone-resorbing osteoimmune cell. J Cell Biochem (2007) 102(5):1130–9. doi:10.1002/jcb.21553
9. Quinn JM, Neale S, Fujikawa Y, McGee JO, Athanasou NA. Human osteoclast formation from blood monocytes, peritoneal macrophages, and bone marrow cells. Calcif Tissue Int (1998) 62(6):527–31. doi:10.1007/s002239900473
10. Garg P, Mazur MM, Buck AC, Wandtke ME, Liu J, Ebraheim NA. Prospective review of mesenchymal stem cells differentiation into osteoblasts. Orthop Surg (2017) 9(1):13–9. doi:10.1111/os.12304
11. Chenery AL, Antignano F, Hughes MR, Burrows K, McNagny KM, Zaph C. Chronic Trichuris muris infection alters hematopoiesis and causes IFN-gamma-expressing T-cell accumulation in the mouse bone marrow. Eur J Immunol (2016) 46(11):2587–96. doi:10.1002/eji.201646326
12. Fu Y, Wang T, Xiu L, Shi X, Bian Z, Zhang Y, et al. Levamisole promotes murine bone marrow derived dendritic cell activation and drives Th1 immune response in vitro and in vivo. Int Immunopharmacol (2016) 31:57–65. doi:10.1016/j.intimp.2015.12.015
13. Siracusa F, Alp OS, Maschmeyer P, McGrath M, Mashreghi MF, Hojyo S, et al. Maintenance of CD8(+) memory T lymphocytes in the spleen but not in the bone marrow is dependent on proliferation. Eur J Immunol (2017) 47(11):1900–5. doi:10.1002/eji.201747063
14. Tokoyoda K, Zehentmeier S, Hegazy AN, Albrecht I, Grun JR, Lohning M, et al. Professional memory CD4+ T lymphocytes preferentially reside and rest in the bone marrow. Immunity (2009) 30(5):721–30. doi:10.1016/j.immuni.2009.03.015
15. Glatman Zaretsky A, Konradt C, Depis F, Wing JB, Goenka R, Atria DG, et al. T regulatory cells support plasma cell populations in the bone marrow. Cell Rep (2017) 18(8):1906–16. doi:10.1016/j.celrep.2017.01.067
16. Manko VM. B lymphocyte regulation of proliferation and differentiation of hematopoietic stem cells. Russ J Immunol (1996) 1(1):9–16.
17. Zhang XH, Wang GX, Zhu HH, Liu YR, Xu LP, Han W, et al. Recruitment of CD8(+) T cells into bone marrow might explain the suppression of megakaryocyte apoptosis through high expression of CX3CR1(+) in prolonged isolated thrombocytopenia after allogeneic hematopoietic stem cell transplantation. Ann Hematol (2015) 94(10):1689–98. doi:10.1007/s00277-015-2436-6
18. Okhrimenko A, Grun JR, Westendorf K, Fang Z, Reinke S, von Roth P, et al. Human memory T cells from the bone marrow are resting and maintain long-lasting systemic memory. Proc Natl Acad Sci U S A (2014) 111(25):9229–34. doi:10.1073/pnas.1318731111
19. Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature (2011) 474(7350):216–9. doi:10.1038/nature10160
20. Wang YX, Xu CG, Ran JL, Wu XC, Sun JH, Wang JD, et al. [In vitro activation of bone marrow natural killer T cells of aplastic anemia patients]. Zhonghua Xue Ye Xue Za Zhi (2010) 31(8):536–9. doi:10.3760/cma.j.issn.0253-2727.2010.08.009
21. Carlsten M, Baumann BC, Simonsson M, Jadersten M, Forsblom AM, Hammarstedt C, et al. Reduced DNAM-1 expression on bone marrow NK cells associated with impaired killing of CD34+ blasts in myelodysplastic syndrome. Leukemia (2010) 24(9):1607–16. doi:10.1038/leu.2010.149
22. Ross FP. M-CSF, c-Fms, and signaling in osteoclasts and their precursors. Ann N Y Acad Sci (2006) 1068:110–6. doi:10.1196/annals.1346.014
23. Sharma SM, Bronisz A, Hu R, Patel K, Mansky KC, Sif S, et al. MITF and PU.1 recruit p38 MAPK and NFATc1 to target genes during osteoclast differentiation. J Biol Chem (2007) 282(21):15921–9. doi:10.1074/jbc.M609723200
24. Kim K, Kim JH, Lee J, Jin HM, Lee SH, Fisher DE, et al. Nuclear factor of activated T cells c1 induces osteoclast-associated receptor gene expression during tumor necrosis factor-related activation-induced cytokine-mediated osteoclastogenesis. J Biol Chem (2005) 280(42):35209–16. doi:10.1074/jbc.M505815200
25. Matsumoto M, Kogawa M, Wada S, Takayanagi H, Tsujimoto M, Katayama S, et al. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J Biol Chem (2004) 279(44):45969–79. doi:10.1074/jbc.M408795200
26. Amarasekara DS, Yun H, Kim S, Lee N, Kim H, Rho J. Regulation of osteoclast differentiation by cytokine networks. Immune Netw (2018) 18(1):e8. doi:10.4110/in.2018.18.e8
27. Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Yano K, et al. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem Biophys Res Commun (1998) 253(2):395–400. doi:10.1006/bbrc.1998.9788
28. Matsumoto M, Sudo T, Saito T, Osada H, Tsujimoto M. Involvement of p38 mitogen-activated protein kinase signaling pathway in osteoclastogenesis mediated by receptor activator of NF-kappa B ligand (RANKL). J Biol Chem (2000) 275(40):31155–61. doi:10.1074/jbc.M001229200
29. Lee SE, Woo KM, Kim SY, Kim HM, Kwack K, Lee ZH, et al. The phosphatidylinositol 3-kinase, p38, and extracellular signal-regulated kinase pathways are involved in osteoclast differentiation. Bone (2002) 30(1):71–7. doi:10.1016/S8756-3282(01)00657-3
30. Inoue J, Ishida T, Tsukamoto N, Kobayashi N, Naito A, Azuma S, et al. Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp Cell Res (2000) 254(1):14–24. doi:10.1006/excr.1999.4733
31. Wong BR, Josien R, Lee SY, Vologodskaia M, Steinman RM, Choi Y. The TRAF family of signal transducers mediates NF-kappaB activation by the TRANCE receptor. J Biol Chem (1998) 273(43):28355–9. doi:10.1074/jbc.273.43.28355
32. Grigoriadis AE, Wang ZQ, Cecchini MG, Hofstetter W, Felix R, Fleisch HA, et al. c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science (1994) 266(5184):443–8. doi:10.1126/science.7939685
33. Ikeda F, Nishimura R, Matsubara T, Tanaka S, Inoue J, Reddy SV, et al. Critical roles of c-Jun signaling in regulation of NFAT family and RANKL-regulated osteoclast differentiation. J Clin Invest (2004) 114(4):475–84. doi:10.1172/JCI200419657
34. Fodor S, Jakus Z, Mocsai A. ITAM-based signaling beyond the adaptive immune response. Immunol Lett (2006) 104(1–2):29–37. doi:10.1016/j.imlet.2005.11.001
35. Negishi-Koga T, Takayanagi H. Ca2+-NFATc1 signaling is an essential axis of osteoclast differentiation. Immunol Rev (2009) 231(1):241–56. doi:10.1111/j.1600-065X.2009.00821.x
36. Zhao Q, Wang X, Liu Y, He A, Jia R. NFATc1: functions in osteoclasts. Int J Biochem Cell Biol (2010) 42(5):576–9. doi:10.1016/j.biocel.2009.12.018
37. Karner CM, Long F. Wnt signaling and cellular metabolism in osteoblasts. Cell Mol Life Sci (2017) 74(9):1649–57. doi:10.1007/s00018-016-2425-5
38. Sanchez-Duffhues G, Hiepen C, Knaus P, Ten Dijke P. Bone morphogenetic protein signaling in bone homeostasis. Bone (2015) 80:43–59. doi:10.1016/j.bone.2015.05.025
39. Komori T. Signaling networks in RUNX2-dependent bone development. J Cell Biochem (2011) 112(3):750–5. doi:10.1002/jcb.22994
40. Sinha KM, Zhou X. Genetic and molecular control of osterix in skeletal formation. J Cell Biochem (2013) 114(5):975–84. doi:10.1002/jcb.24439
41. Liu ZJ, Zhuge Y, Velazquez OC. Trafficking and differentiation of mesenchymal stem cells. J Cell Biochem (2009) 106(6):984–91. doi:10.1002/jcb.22091
42. Horwood NJ. Immune cells and bone: coupling goes both ways. Immunol Invest (2013) 42(7):532–43. doi:10.3109/08820139.2013.822762
43. Boyce AM. Denosumab: an emerging therapy in pediatric bone disorders. Curr Osteoporos Rep (2017) 15(4):283–92. doi:10.1007/s11914-017-0380-1
44. Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood (2007) 109(9):3839–48. doi:10.1182/blood-2006-07-037994
45. Theill LE, Boyle WJ, Penninger JM. RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu Rev Immunol (2002) 20:795–823. doi:10.1146/annurev.immunol.20.100301.064753
46. Seshasayee D, Wang H, Lee WP, Gribling P, Ross J, Van Bruggen N, et al. A novel in vivo role for osteoprotegerin ligand in activation of monocyte effector function and inflammatory response. J Biol Chem (2004) 279(29):30202–9. doi:10.1074/jbc.M403968200
47. Criscitiello C, Viale G, Gelao L, Esposito A, De Laurentiis M, De Placido S, et al. Crosstalk between bone niche and immune system: osteoimmunology signaling as a potential target for cancer treatment. Cancer Treat Rev (2015) 41(2):61–8. doi:10.1016/j.ctrv.2014.12.001
48. Hamerman JA, Ni M, Killebrew JR, Chu CL, Lowell CA. The expanding roles of ITAM adapters FcRgamma and DAP12 in myeloid cells. Immunol Rev (2009) 232(1):42–58. doi:10.1111/j.1600-065X.2009.00841.x
49. Takayanagi H. Osteoimmunology in 2014: two-faced immunology-from osteogenesis to bone resorption. Nat Rev Rheumatol (2015) 11(2):74–6. doi:10.1038/nrrheum.2014.219
50. Liu XL, Song J, Liu KJ, Wang WP, Xu C, Zhang YZ, et al. Role of inhibition of osteogenesis function by Sema4D/Plexin-B1 signaling pathway in skeletal fluorosis in vitro. J Huazhong Univ Sci Technolog Med Sci (2015) 35(5):712–5. doi:10.1007/s11596-015-1495-1
51. Ishida I, Kumanogoh A, Suzuki K, Akahani S, Noda K, Kikutani H. Involvement of CD100, a lymphocyte semaphorin, in the activation of the human immune system via CD72: implications for the regulation of immune and inflammatory responses. Int Immunol (2003) 15(8):1027–34. doi:10.1093/intimm/dxg098
52. Ginaldi L, De Martinis M. Osteoimmunology and beyond. Curr Med Chem (2016) 23(33):3754–74. doi:10.2174/0929867323666160907162546
53. Bryson JL, Bhandoola A. Editorial: ephs, ephrins, and early T cell development. J Leukoc Biol (2015) 98(6):877–9. doi:10.1189/jlb.1CE0315-132R
54. Shen LL, Zhang LX, Wang LM, Zhou RJ, Yang CZ, Zhang J, et al. Disturbed expression of EphB4, but not EphrinB2, inhibited bone regeneration in an in vivo inflammatory microenvironment. Mediators Inflamm (2016) 2016:6430407. doi:10.1155/2016/6430407
55. Santamaria MH, Corral RS. Osteopontin-dependent regulation of Th1 and Th17 cytokine responses in Trypanosoma cruzi-infected C57BL/6 mice. Cytokine (2013) 61(2):491–8. doi:10.1016/j.cyto.2012.10.027
56. Qi Y, Li X, Ma X, Xu L, Zhang X, Jiang X, et al. The role of osteopontin in the induction of the CD40 ligand in Graves’ disease. Clin Endocrinol (2014) 80(1):128–34. doi:10.1111/cen.12229
57. Pacifici R. T cells, osteoblasts, and osteocytes: interacting lineages key for the bone anabolic and catabolic activities of parathyroid hormone. Ann N Y Acad Sci (2016) 1364:11–24. doi:10.1111/nyas.12969
58. Hofbauer LC, Lacey DL, Dunstan CR, Spelsberg TC, Riggs BL, Khosla S. Interleukin-1beta and tumor necrosis factor-alpha, but not interleukin-6, stimulate osteoprotegerin ligand gene expression in human osteoblastic cells. Bone (1999) 25(3):255–9. doi:10.1016/S8756-3282(99)00162-3
59. Ono T, Okamoto K, Nakashima T, Nitta T, Hori S, Iwakura Y, et al. IL-17-producing gammadelta T cells enhance bone regeneration. Nat Commun (2016) 7:10928. doi:10.1038/ncomms10928
60. Quinn JM, Sims NA, Saleh H, Mirosa D, Thompson K, Bouralexis S, et al. IL-23 inhibits osteoclastogenesis indirectly through lymphocytes and is required for the maintenance of bone mass in mice. J Immunol (2008) 181(8):5720–9. doi:10.4049/jimmunol.181.8.5720
61. Buchwald ZS, Kiesel JR, DiPaolo R, Pagadala MS, Aurora R. Osteoclast activated FoxP3+ CD8+ T-cells suppress bone resorption in vitro. PLoS One (2012) 7(6):e38199. doi:10.1371/journal.pone.0038199
62. Horwood NJ, Elliott J, Martin TJ, Gillespie MT. IL-12 alone and in synergy with IL-18 inhibits osteoclast formation in vitro. J Immunol (2001) 166(8):4915–21. doi:10.4049/jimmunol.166.8.4915
63. Hong MH, Williams H, Jin CH, Pike JW. The inhibitory effect of interleukin-10 on mouse osteoclast formation involves novel tyrosine-phosphorylated proteins. J Bone Miner Res (2000) 15(5):911–8. doi:10.1359/jbmr.2000.15.5.911
64. Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med (2006) 203(12):2673–82. doi:10.1084/jem.20061775
65. Honda K. Interleukin-6 and soluble interleukin-6 receptor suppress osteoclastic differentiation by inducing PGE(2) production in chondrocytes. J Oral Sci (2011) 53(1):87–96. doi:10.2334/josnusd.53.87
66. Kamiya S, Nakamura C, Fukawa T, Ono K, Ohwaki T, Yoshimoto T, et al. Effects of IL-23 and IL-27 on osteoblasts and osteoclasts: inhibitory effects on osteoclast differentiation. J Bone Miner Metab (2007) 25(5):277–85. doi:10.1007/s00774-007-0766-8
67. Kozuka Y, Ozaki Y, Ukai T, Kaneko T, Hara Y. B cells play an important role in lipopolysaccharide-induced bone resorption. Calcif Tissue Int (2006) 78(3):125–32. doi:10.1007/s00223-005-0149-x
68. Choi Y, Kim JJ. B cells activated in the presence of Th1 cytokines inhibit osteoclastogenesis. Exp Mol Med (2003) 35(5):385–92. doi:10.1038/emm.2003.51
69. Weitzmann MN, Cenci S, Haug J, Brown C, DiPersio J, Pacifici R. B lymphocytes inhibit human osteoclastogenesis by secretion of TGFbeta. J Cell Biochem (2000) 78(2):318–24. doi:10.1002/(SICI)1097-4644(20000801)78:2<318::AID-JCB13>3.0.CO;2-N
70. Soderstrom K, Stein E, Colmenero P, Purath U, Muller-Ladner U, de Matos CT, et al. Natural killer cells trigger osteoclastogenesis and bone destruction in arthritis. Proc Natl Acad Sci U S A (2010) 107(29):13028–33. doi:10.1073/pnas.1000546107
71. Terauchi M, Li JY, Bedi B, Baek KH, Tawfeek H, Galley S, et al. T lymphocytes amplify the anabolic activity of parathyroid hormone through Wnt10b signaling. Cell Metab (2009) 10(3):229–40. doi:10.1016/j.cmet.2009.07.010
72. Okamoto K, Nakashima T, Shinohara M, Negishi-Koga T, Komatsu N, Terashima A, et al. Osteoimmunology: the conceptual framework unifying the immune and skeletal systems. Physiol Rev (2017) 97(4):1295–349. doi:10.1152/physrev.00036.2016
73. Yu VW, Saez B, Cook C, Lotinun S, Pardo-Saganta A, Wang YH, et al. Specific bone cells produce DLL4 to generate thymus-seeding progenitors from bone marrow. J Exp Med (2015) 212(5):759–74. doi:10.1084/jem.20141843
74. Sato M, Asada N, Kawano Y, Wakahashi K, Minagawa K, Kawano H, et al. Osteocytes regulate primary lymphoid organs and fat metabolism. Cell Metab (2013) 18(5):749–58. doi:10.1016/j.cmet.2013.09.014
75. Lecka-Czernik B, Rosen CJ, Kawai M. Skeletal aging and the adipocyte program: new insights from an “old” molecule. Cell cycle (2010) 9(18):3648–54. doi:10.4161/cc.9.18.13046
76. Goto H, Hozumi A, Osaki M, Fukushima T, Sakamoto K, Yonekura A, et al. Primary human bone marrow adipocytes support TNF-alpha-induced osteoclast differentiation and function through RANKL expression. Cytokine (2011) 56(3):662–8. doi:10.1016/j.cyto.2011.09.005
77. Hozumi A, Osaki M, Goto H, Sakamoto K, Inokuchi S, Shindo H. Bone marrow adipocytes support dexamethasone-induced osteoclast differentiation. Biochem Biophys Res Commun (2009) 382(4):780–4. doi:10.1016/j.bbrc.2009.03.111
78. Holt V, Caplan AI, Haynesworth SE. Identification of a subpopulation of marrow MSC-derived medullary adipocytes that express osteoclast-regulating molecules: marrow adipocytes express osteoclast mediators. PLoS One (2014) 9(10):e108920. doi:10.1371/journal.pone.0108920
79. Devlin MJ, Rosen CJ. The bone-fat interface: basic and clinical implications of marrow adiposity. Lancet Diabetes Endocrinol (2015) 3(2):141–7. doi:10.1016/S2213-8587(14)70007-5
80. Laharrague P, Fontanilles AM, Tkaczuk J, Corberand JX, Penicaud L, Casteilla L. Inflammatory/haematopoietic cytokine production by human bone marrow adipocytes. Eur Cytokine Netw (2000) 11(4):634–9.
81. Halade GV, El Jamali A, Williams PJ, Fajardo RJ, Fernandes G. Obesity-mediated inflammatory microenvironment stimulates osteoclastogenesis and bone loss in mice. Exp Gerontol (2011) 46(1):43–52. doi:10.1016/j.exger.2010.09.014
82. Hardaway AL, Herroon MK, Rajagurubandara E, Podgorski I. Marrow adipocyte-derived CXCL1 and CXCL2 contribute to osteolysis in metastatic prostate cancer. Clin Exp Metastasis (2015) 32(4):353–68. doi:10.1007/s10585-015-9714-5
83. Sapir-Koren R, Livshits G. Postmenopausal osteoporosis in rheumatoid arthritis: the estrogen deficiency-immune mechanisms link. Bone (2017) 103:102–15. doi:10.1016/j.bone.2017.06.020
84. Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther (2002) 96(1):23–43. doi:10.1016/S0163-7258(02)00297-8
85. Toussirot E. Biologics in spondyloarthritis: TNFalpha inhibitors and other agents. Immunotherapy (2015) 7(6):669–81. doi:10.2217/imt.15.28
86. Guerri-Fernandez R, Villar-Garcia J, Diez-Perez A, Prieto-Alhambra D. HIV infection, bone metabolism, and fractures. Arq Bras Endocrinol Metabol (2014) 58(5):478–83. doi:10.1590/0004-2730000003323
87. Weitzmann MN. Bone and the immune system. Toxicol Pathol (2017) 45(7):911–24. doi:10.1177/0192623317735316
88. Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol (2007) 96:41–101. doi:10.1016/S0065-2776(07)96002-2
89. Zaidi MR, Merlino G. The two faces of interferon-gamma in cancer. Clin Cancer Res (2011) 17(19):6118–24. doi:10.1158/1078-0432.CCR-11-0482
90. Miller CH, Maher SG, Young HA. Clinical use of interferon-gamma. Ann N Y Acad Sci (2009) 1182:69–79. doi:10.1111/j.1749-6632.2009.05069.x
91. de Bruin AM, Voermans C, Nolte MA. Impact of interferon-gamma on hematopoiesis. Blood (2014) 124(16):2479–86. doi:10.1182/blood-2014-04-568451
92. McLaren JE, Ramji DP. Interferon gamma: a master regulator of atherosclerosis. Cytokine Growth Factor Rev (2009) 20(2):125–35. doi:10.1016/j.cytogfr.2008.11.003
93. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol (2004) 75(2):163–89. doi:10.1189/jlb.0603252
94. Di Marzio P, Puddu P, Conti L, Belardelli F, Gessani S. Interferon gamma upregulates its own gene expression in mouse peritoneal macrophages. J Exp Med (1994) 179(5):1731–6. doi:10.1084/jem.179.5.1731
95. Neumann C, Sorg C. Immune interferon. I. Production by lymphokine-activated murine macrophages. Eur J Immunol (1977) 7(10):719–25. doi:10.1002/eji.1830071014
96. Frucht DM, Fukao T, Bogdan C, Schindler H, O’Shea JJ, Koyasu S. IFN-gamma production by antigen-presenting cells: mechanisms emerge. Trends Immunol (2001) 22(10):556–60. doi:10.1016/S1471-4906(01)02005-1
97. Sturge CR, Benson A, Raetz M, Wilhelm CL, Mirpuri J, Vitetta ES, et al. TLR-independent neutrophil-derived IFN-gamma is important for host resistance to intracellular pathogens. Proc Natl Acad Sci U S A (2013) 110(26):10711–6. doi:10.1073/pnas.1307868110
98. Lees JR. Interferon gamma in autoimmunity: a complicated player on a complex stage. Cytokine (2015) 74(1):18–26. doi:10.1016/j.cyto.2014.10.014
99. Dormady SP, Bashayan O, Dougherty R, Zhang XM, Basch RS. Immortalized multipotential mesenchymal cells and the hematopoietic microenvironment. J Hematother Stem Cell Res (2001) 10(1):125–40. doi:10.1089/152581601750098372
100. Thomas H, Jager M, Mauel K, Brandau S, Lask S, Flohe SB. Interaction with mesenchymal stem cells provokes natural killer cells for enhanced IL-12/IL-18-induced interferon-gamma secretion. Mediators Inflamm (2014) 2014:143463. doi:10.1155/2014/143463
101. Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol (1997) 15:563–91. doi:10.1146/annurev.immunol.15.1.563
102. Schreiber RD, Farrar MA, Hershey GK, Fernandez-Luna J. The structure and function of interferon-gamma receptors. Int J Immunopharmacol (1992) 14(3):413–9. doi:10.1016/0192-0561(92)90171-G
103. Marsters SA, Pennica D, Bach E, Schreiber RD, Ashkenazi A. Interferon gamma signals via a high-affinity multisubunit receptor complex that contains two types of polypeptide chain. Proc Natl Acad Sci U S A (1995) 92(12):5401–5. doi:10.1073/pnas.92.12.5401
104. Horvath CM. The Jak-STAT pathway stimulated by interferon gamma. Sci STKE (2004) 2004(260):tr8. doi:10.1126/stke.2602004tr8
105. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi:10.1146/annurev-immunol-032713-120231
106. Gough DJ, Levy DE, Johnstone RW, Clarke CJ. IFNgamma signaling-does it mean JAK-STAT? Cytokine Growth Factor Rev (2008) 19(5–6):383–94. doi:10.1016/j.cytogfr.2008.08.004
107. Sakatsume M, Stancato LF, David M, Silvennoinen O, Saharinen P, Pierce J, et al. Interferon gamma activation of Raf-1 is Jak1-dependent and p21ras-independent. J Biol Chem (1998) 273(5):3021–6. doi:10.1074/jbc.273.5.3021
108. Alsayed Y, Uddin S, Ahmad S, Majchrzak B, Druker BJ, Fish EN, et al. IFN-gamma activates the C3G/Rap1 signaling pathway. J Immunol (2000) 164(4):1800–6. doi:10.4049/jimmunol.164.4.1800
109. Hu J, Roy SK, Shapiro PS, Rodig SR, Reddy SP, Platanias LC, et al. ERK1 and ERK2 activate CCAAAT/enhancer-binding protein-beta-dependent gene transcription in response to interferon-gamma. J Biol Chem (2001) 276(1):287–97. doi:10.1074/jbc.M004885200
110. Behera AK, Kumar M, Lockey RF, Mohapatra SS. Adenovirus-mediated interferon gamma gene therapy for allergic asthma: involvement of interleukin 12 and STAT4 signaling. Hum Gene Ther (2002) 13(14):1697–709. doi:10.1089/104303402760293547
111. Nair JS, DaFonseca CJ, Tjernberg A, Sun W, Darnell JE Jr, Chait BT, et al. Requirement of Ca2+ and CaMKII for Stat1 Ser-727 phosphorylation in response to IFN-gamma. Proc Natl Acad Sci U S A (2002) 99(9):5971–6. doi:10.1073/pnas.052159099
112. Deb DK, Sassano A, Lekmine F, Majchrzak B, Verma A, Kambhampati S, et al. Activation of protein kinase C delta by IFN-gamma. J Immunol (2003) 171(1):267–73. doi:10.4049/jimmunol.171.1.267
113. Heller NM, Matsukura S, Georas SN, Boothby MR, Rothman PB, Stellato C, et al. Interferon-gamma inhibits STAT6 signal transduction and gene expression in human airway epithelial cells. Am J Respir Cell Mol Biol (2004) 31(5):573–82. doi:10.1165/rcmb.2004-0195OC
114. Liu L, Wang Y, Fan Y, Li CL, Chang ZL. IFN-gamma activates cAMP/PKA/CREB signaling pathway in murine peritoneal macrophages. J Interferon Cytokine Res (2004) 24(6):334–42. doi:10.1089/107999004323142196
115. Park EJ, Ji KA, Jeon SB, Choi WH, Han IO, You HJ, et al. Rac1 contributes to maximal activation of STAT1 and STAT3 in IFN-gamma-stimulated rat astrocytes. J Immunol (2004) 173(9):5697–703. doi:10.4049/jimmunol.173.9.5697
116. Zhang XF, Guo SZ, Lu KH, Li HY, Li XD, Zhang LX, et al. Different roles of PKC and PKA in effect of interferon-gamma on proliferation and collagen synthesis of fibroblasts. Acta Pharmacol Sin (2004) 25(10):1320–6.
117. Venkatesan BA, Mahimainathan L, Ghosh-Choudhury N, Gorin Y, Bhandari B, Valente AJ, et al. PI 3 kinase-dependent Akt kinase and PKCepsilon independently regulate interferon-gamma-induced STAT1alpha serine phosphorylation to induce monocyte chemotactic protein-1 expression. Cell Signal (2006) 18(4):508–18. doi:10.1016/j.cellsig.2005.05.022
118. Cho HJ, Kim SK, Jin SM, Hwang EM, Kim YS, Huh K, et al. IFN-gamma-induced BACE1 expression is mediated by activation of JAK2 and ERK1/2 signaling pathways and direct binding of STAT1 to BACE1 promoter in astrocytes. Glia (2007) 55(3):253–62. doi:10.1002/glia.20451
119. Wei B, Baker S, Wieckiewicz J, Wood KJ. IFN-gamma triggered STAT1-PKB/AKT signalling pathway influences the function of alloantigen reactive regulatory T cells. Am J Transplant (2010) 10(1):69–80. doi:10.1111/j.1600-6143.2009.02858.x
120. Deng J, Qian Y, Geng L, Xie H, Wang Y, Jiang G, et al. Involvement of ERK and JNK pathways in IFN-gamma-induced B7-DC expression on tumor cells. J Cancer Res Clin Oncol (2011) 137(2):243–50. doi:10.1007/s00432-010-0876-x
121. Li W, Henderson LJ, Major EO, Al-Harthi L. IFN-gamma mediates enhancement of HIV replication in astrocytes by inducing an antagonist of the beta-catenin pathway (DKK1) in a STAT 3-dependent manner. J Immunol (2011) 186(12):6771–8. doi:10.4049/jimmunol.1100099
122. Smyth D, McKay CM, Gulbransen BD, Phan VC, Wang A, McKay DM. Interferon-gamma signals via an ERK1/2-ARF6 pathway to promote bacterial internalization by gut epithelia. Cell Microbiol (2012) 14(8):1257–70. doi:10.1111/j.1462-5822.2012.01796.x
123. Lee WH, Chung MH, Tsai YH, Chang JL, Huang HM. Interferon-gamma suppresses activin A/NF-E2 induction of erythroid gene expression through the NF-kappaB/c-Jun pathway. Am J Physiol Cell Physiol (2014) 306(4):C407–14. doi:10.1152/ajpcell.00312.2013
124. Yang S, Yu M, Sun L, Xiao W, Yang X, Sun L, et al. Interferon-gamma-induced intestinal epithelial barrier dysfunction by NF-kappaB/HIF-1alpha pathway. J Interferon Cytokine Res (2014) 34(3):195–203. doi:10.1089/jir.2013.0044
125. Johnson S, Duncan J, Hussain SA, Chen G, Luo J, McLaurin C, et al. The IFNgamma-PKR pathway in the prefrontal cortex reactions to chronic excessive alcohol use. Alcohol Clin Exp Res (2015) 39(3):476–84. doi:10.1111/acer.12650
126. Gan H, Hao Q, Idell S, Tang H. Interferon-gamma promotes double-stranded RNA-induced TLR3-dependent apoptosis via upregulation of transcription factor Runx3 in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol (2016) 311(6):L1101–12. doi:10.1152/ajplung.00278.2016
127. Kroczynska B, Rafidi RL, Majchrzak-Kita B, Kosciuczuk EM, Blyth GT, Jemielity J, et al. Interferon gamma (IFNgamma) signaling via mechanistic target of rapamycin complex 2 (mTORC2) and regulatory effects in the generation of type II interferon biological responses. J Biol Chem (2016) 291(5):2389–96. doi:10.1074/jbc.M115.664995
128. Wang Y, Cao Y, Mangalam AK, Guo Y, LaFrance-Corey RG, Gamez JD, et al. Neuropilin-1 modulates interferon-gamma-stimulated signaling in brain microvascular endothelial cells. J Cell Sci (2016) 129(20):3911–21. doi:10.1242/jcs.190702
129. Skaria SD, Yang J, Condos R, Smaldone GC. Inhaled interferon and diffusion capacity in Idiopathic Pulmonary Fibrosis (IPF). Sarcoidosis Vasc Diffuse Lung Dis (2015) 32(1):37–42.
130. Delsing CE, Gresnigt MS, Leentjens J, Preijers F, Frager FA, Kox M, et al. Interferon-gamma as adjunctive immunotherapy for invasive fungal infections: a case series. BMC Infect Dis (2014) 14:166. doi:10.1186/1471-2334-14-166
131. Muir AJ, Sylvestre PB, Rockey DC. Interferon gamma-1b for the treatment of fibrosis in chronic hepatitis C infection. J Viral Hepat (2006) 13(5):322–8. doi:10.1111/j.1365-2893.2005.00689.x
132. Cai WM. Effect of interferon-gamma on hepatic fibrosis in chronic hepatitis B virus infection. Clin Gastroenterol Hepatol (2005) 3(12):1269. doi:10.1016/S1542-3565(05)00893-1
133. Veys EM, Menkes CJ, Emery P. A randomized, double-blind study comparing twenty-four-week treatment with recombinant interferon-gamma versus placebo in the treatment of rheumatoid arthritis. Arthritis Rheum (1997) 40(1):62–8. doi:10.1002/art.1780400110
134. Sugaya M, Tokura Y, Hamada T, Tsuboi R, Moroi Y, Nakahara T, et al. Phase II study of i.v. interferon-gamma in Japanese patients with mycosis fungoides. J Dermatol (2014) 41(1):50–6. doi:10.1111/1346-8138.12341
135. Ruiz C, Perez E, Garcia-Martinez O, Diaz-Rodriguez L, Arroyo-Morales M, Reyes-Botella C. Expression of cytokines IL-4, IL-12, IL-15, IL-18, and IFNgamma and modulation by different growth factors in cultured human osteoblast-like cells. J Bone Miner Metab (2007) 25(5):286–92. doi:10.1007/s00774-007-0767-7
136. Gowen M, MacDonald BR, Russell RG. Actions of recombinant human gamma-interferon and tumor necrosis factor alpha on the proliferation and osteoblastic characteristics of human trabecular bone cells in vitro. Arthritis Rheum (1988) 31(12):1500–7. doi:10.1002/art.1780311206
137. Maruhashi T, Kaifu T, Yabe R, Seno A, Chung SH, Fujikado N, et al. DCIR maintains bone homeostasis by regulating IFN-gamma production in T cells. J Immunol (2015) 194(12):5681–91. doi:10.4049/jimmunol.1500273
138. Croes M, Oner FC, van Neerven D, Sabir E, Kruyt MC, Blokhuis TJ, et al. Proinflammatory T cells and IL-17 stimulate osteoblast differentiation. Bone (2016) 84:262–70. doi:10.1016/j.bone.2016.01.010
139. Duque G, Huang DC, Macoritto M, Rivas D, Yang XF, Ste-Marie LG, et al. Autocrine regulation of interferon gamma in mesenchymal stem cells plays a role in early osteoblastogenesis. Stem Cells (2009) 27(3):550–8. doi:10.1634/stemcells.2008-0886
140. Vidal C, Bermeo S, Li W, Huang D, Kremer R, Duque G. Interferon gamma inhibits adipogenesis in vitro and prevents marrow fat infiltration in oophorectomized mice. Stem Cells (2012) 30(5):1042–8. doi:10.1002/stem.1063
141. Duque G, Huang DC, Dion N, Macoritto M, Rivas D, Li W, et al. Interferon-gamma plays a role in bone formation in vivo and rescues osteoporosis in ovariectomized mice. J Bone Miner Res (2011) 26(7):1472–83. doi:10.1002/jbmr.350
142. Yamaguchi T, Movila A, Kataoka S, Wisitrasameewong W, Ruiz Torruella M, Murakoshi M, et al. Proinflammatory M1 macrophages inhibit RANKL-induced osteoclastogenesis. Infect Immun (2016) 84(10):2802–12. doi:10.1128/IAI.00461-16
143. Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature (2000) 408(6812):600–5. doi:10.1038/35046102
144. Shinoda K, Sugiyama E, Taki H, Harada S, Mino T, Maruyama M, et al. Resting T cells negatively regulate osteoclast generation from peripheral blood monocytes. Bone (2003) 33(4):711–20. doi:10.1016/S8756-3282(03)00230-8
145. Ji JD, Park-Min KH, Shen Z, Fajardo RJ, Goldring SR, McHugh KP, et al. Inhibition of RANK expression and osteoclastogenesis by TLRs and IFN-gamma in human osteoclast precursors. J Immunol (2009) 183(11):7223–33. doi:10.4049/jimmunol.0900072
146. Xiong Q, Zhang L, Ge W, Tang P. The roles of interferons in osteoclasts and osteoclastogenesis. Joint Bone Spine (2016) 83(3):276–81. doi:10.1016/j.jbspin.2015.07.010
147. Huang W, O’Keefe RJ, Schwarz EM. Exposure to receptor-activator of NFkappaB ligand renders pre-osteoclasts resistant to IFN-gamma by inducing terminal differentiation. Arthritis Res Ther (2003) 5(1):R49–59. doi:10.1186/ar679
148. Cheng J, Liu J, Shi Z, Jules J, Xu D, Luo S, et al. Molecular mechanisms of the biphasic effects of interferon-gamma on osteoclastogenesis. J Interferon Cytokine Res (2012) 32(1):34–45. doi:10.1089/jir.2011.0019
149. Kohara H, Kitaura H, Fujimura Y, Yoshimatsu M, Morita Y, Eguchi T, et al. IFN-gamma directly inhibits TNF-alpha-induced osteoclastogenesis in vitro and in vivo and induces apoptosis mediated by Fas/Fas ligand interactions. Immunol Lett (2011) 137(1–2):53–61. doi:10.1016/j.imlet.2011.02.017
150. Wang L, Liu S, Zhao Y, Liu D, Liu Y, Chen C, et al. Osteoblast-induced osteoclast apoptosis by fas ligand/FAS pathway is required for maintenance of bone mass. Cell Death Differ (2015) 22(10):1654–64. doi:10.1038/cdd.2015.14
151. van’t Hof RJ, Ralston SH. Cytokine-induced nitric oxide inhibits bone resorption by inducing apoptosis of osteoclast progenitors and suppressing osteoclast activity. J Bone Miner Res (1997) 12(11):1797–804. doi:10.1359/jbmr.1997.12.11.1797
152. Miyamoto T. Regulators of osteoclast differentiation and cell-cell fusion. Keio J Med (2011) 60(4):101–5. doi:10.2302/kjm.60.101
153. Kim JW, Lee MS, Lee CH, Kim HY, Chae SU, Kwak HB, et al. Effect of interferon-gamma on the fusion of mononuclear osteoclasts into bone-resorbing osteoclasts. BMB Rep (2012) 45(5):281–6. doi:10.5483/BMBRep.2012.45.5.281
154. Cenci S, Toraldo G, Weitzmann MN, Roggia C, Gao Y, Qian WP, et al. Estrogen deficiency induces bone loss by increasing T cell proliferation and lifespan through IFN-gamma-induced class II transactivator. Proc Natl Acad Sci U S A (2003) 100(18):10405–10. doi:10.1073/pnas.1533207100
155. Gao Y, Grassi F, Ryan MR, Terauchi M, Page K, Yang X, et al. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest (2007) 117(1):122–32. doi:10.1172/JCI30074
156. Kwak HB, Ha H, Kim HN, Lee JH, Kim HS, Lee S, et al. Reciprocal cross-talk between RANKL and interferon-gamma-inducible protein 10 is responsible for bone-erosive experimental arthritis. Arthritis Rheum (2008) 58(5):1332–42. doi:10.1002/art.23372
157. Luster AD, Ravetch JV. Biochemical characterization of a gamma interferon-inducible cytokine (IP-10). J Exp Med (1987) 166(4):1084–97. doi:10.1084/jem.166.4.1084
158. Xu Z, Hurchla MA, Deng H, Uluckan O, Bu F, Berdy A, et al. Interferon-gamma targets cancer cells and osteoclasts to prevent tumor-associated bone loss and bone metastases. J Biol Chem (2009) 284(7):4658–66. doi:10.1074/jbc.M804812200
159. Shankar L, Gerritsen EJ, Key LL Jr. Osteopetrosis: pathogenesis and rationale for the use of interferon-gamma-1b. BioDrugs (1997) 7(1):23–9. doi:10.2165/00063030-199707010-00004
160. Mann GN, Jacobs TW, Buchinsky FJ, Armstrong EC, Li M, Ke HZ, et al. Interferon-gamma causes loss of bone volume in vivo and fails to ameliorate cyclosporin A-induced osteopenia. Endocrinology (1994) 135(3):1077–83. doi:10.1210/endo.135.3.8070349
161. Walsh NC, Gravallese EM. Bone remodeling in rheumatic disease: a question of balance. Immunol Rev (2010) 233(1):301–12. doi:10.1111/j.0105-2896.2009.00857.x
162. Baum R, Gravallese EM. Bone as a target organ in rheumatic disease: impact on osteoclasts and osteoblasts. Clin Rev Allergy Immunol (2016) 51(1):1–15. doi:10.1007/s12016-015-8515-6
163. Gough AK, Lilley J, Eyre S, Holder RL, Emery P. Generalised bone loss in patients with early rheumatoid arthritis. Lancet (1994) 344(8914):23–7. doi:10.1016/S0140-6736(94)91049-9
164. Angelotti F, Parma A, Cafaro G, Capecchi R, Alunno A, Puxeddu I. One year in review 2017: pathogenesis of rheumatoid arthritis. Clin Exp Rheumatol (2017) 35(3):368–78.
165. Shim JH, Stavre Z, Gravallese EM. Bone loss in rheumatoid arthritis: basic mechanisms and clinical implications. Calcif Tissue Int (2017) 102(5):533–46. doi:10.1007/s00223-017-0373-1
166. Hou Y, Lin H, Zhu L, Liu Z, Hu F, Shi J, et al. The inhibitory effect of IFN-gamma on protease HTRA1 expression in rheumatoid arthritis. J Immunol (2014) 193(1):130–8. doi:10.4049/jimmunol.1302700
167. Thanapati S, Ganu M, Giri P, Kulkarni S, Sharma M, Babar P, et al. Impaired NK cell functionality and increased TNF-alpha production as biomarkers of chronic chikungunya arthritis and rheumatoid arthritis. Hum Immunol (2017) 78(4):370–4. doi:10.1016/j.humimm.2017.02.006
168. Steiner G, Tohidast-Akrad M, Witzmann G, Vesely M, Studnicka-Benke A, Gal A, et al. Cytokine production by synovial T cells in rheumatoid arthritis. Rheumatology (Oxford) (1999) 38(3):202–13. doi:10.1093/rheumatology/38.3.202
169. Morita Y, Yamamura M, Kawashima M, Harada S, Tsuji K, Shibuya K, et al. Flow cytometric single-cell analysis of cytokine production by CD4+ T cells in synovial tissue and peripheral blood from patients with rheumatoid arthritis. Arthritis Rheum (1998) 41(9):1669–76. doi:10.1002/1529-0131(199809)41:9<1669::AID-ART19>3.0.CO;2-G
170. Olalekan SA, Cao Y, Hamel KM, Finnegan A. B cells expressing IFN-gamma suppress Treg-cell differentiation and promote autoimmune experimental arthritis. Eur J Immunol (2015) 45(4):988–98. doi:10.1002/eji.201445036
171. Karonitsch T, von Dalwigk K, Steiner CW, Bluml S, Steiner G, Kiener HP, et al. Interferon signals and monocytic sensitization of the interferon-gamma signaling pathway in the peripheral blood of patients with rheumatoid arthritis. Arthritis Rheum (2012) 64(2):400–8. doi:10.1002/art.33347
172. Xie YD, Jin L, Yu QW. [The role of IFN-gamma, IL-10, IL-12 and TRAIL in sera and synovium fluids from patients with rheumatoid arthritis]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2007) 23(6):536–7. doi:10.3321/j.issn:1007-8738.2007.06.014
173. Kokkonen H, Soderstrom I, Rocklov J, Hallmans G, Lejon K, Rantapaa Dahlqvist S. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum (2010) 62(2):383–91. doi:10.1002/art.27186
174. Cacciapaglia F, Buzzulini F, Arcarese L, Ferraro E, Afeltra A. The use of an interferon-gamma release assay as a biomarker of response to anti-TNF-alpha treatment. Drug Dev Res (2014) 75(Suppl 1):S50–3. doi:10.1002/ddr.21195
175. Nasonova VA, Lukina GV, Sigidin Ia A. [Neutralisation of interferon gamma – a new trend in therapy of rheumatoid arthritis]. Ter Arkh (2008) 80(5):30–7.
176. Sigidin YA, Loukina GV, Skurkovich B, Skurkovich S. Randomized, double-blind trial of anti-interferon-gamma antibodies in rheumatoid arthritis. Scand J Rheumatol (2001) 30(4):203–7. doi:10.1080/030097401316909530
177. Boissier MC, Chiocchia G, Bessis N, Hajnal J, Garotta G, Nicoletti F, et al. Biphasic effect of interferon-gamma in murine collagen-induced arthritis. Eur J Immunol (1995) 25(5):1184–90. doi:10.1002/eji.1830250508
178. Lemmel EM, Brackertz D, Franke M, Gaus W, Hartl PW, Machalke K, et al. Results of a multicenter placebo-controlled double-blind randomized phase III clinical study of treatment of rheumatoid arthritis with recombinant interferon-gamma. Rheumatol Int (1988) 8(2):87–93. doi:10.1007/BF00271840
179. Cannon GW, Emkey RD, Denes A, Cohen SA, Saway PA, Wolfe F, et al. Prospective 5-year followup of recombinant interferon-gamma in rheumatoid arthritis. J Rheumatol (1993) 20(11):1867–73.
180. Hofschneider P, Winter U, Lemmel E, Brölz B, Gaus W, Schmid S, et al. Double blind controlled phase III multicenter clinical trial with interferon gamma in rheumatoid arthritis. German Lymphokine Study Group. Rheumatol Int (1992) 12(5):175–85. doi:10.1007/BF00302149
181. Lemmel EM, Franke M, Gaus W, Hartl PW, Hofschneider PH, Miehlke K, et al. Results of a phase-II clinical trial on treatment of rheumatoid arthritis with recombinant interferon-gamma. Rheumatol Int (1987) 7(3):127–32. doi:10.1007/BF00270465
182. Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P. Accelerated collagen-induced arthritis in IFN-gamma receptor-deficient mice. J Immunol (1997) 158(11):5507–13.
183. Nakajima H, Takamori H, Hiyama Y, Tsukada W. The effect of treatment with interferon-gamma on type II collagen-induced arthritis. Clin Exp Immunol (1990) 81(3):441–5. doi:10.1111/j.1365-2249.1990.tb05353.x
184. Sarkar S, Cooney LA, White P, Dunlop DB, Endres J, Jorns JM, et al. Regulation of pathogenic IL-17 responses in collagen-induced arthritis: roles of endogenous interferon-gamma and IL-4. Arthritis Res Ther (2009) 11(5):R158. doi:10.1186/ar2838
185. Guedez YB, Whittington KB, Clayton JL, Joosten LA, van de Loo FA, van den Berg WB, et al. Genetic ablation of interferon-gamma up-regulates interleukin-1beta expression and enables the elicitation of collagen-induced arthritis in a nonsusceptible mouse strain. Arthritis Rheum (2001) 44(10):2413–24. doi:10.1002/1529-0131(200110)44:10<2413::AID-ART406>3.0.CO;2-E
186. Chu CQ, Swart D, Alcorn D, Tocker J, Elkon KB. Interferon-gamma regulates susceptibility to collagen-induced arthritis through suppression of interleukin-17. Arthritis Rheum (2007) 56(4):1145–51. doi:10.1002/art.22453
187. Breuil V, Ticchioni M, Testa J, Roux CH, Ferrari P, Breittmayer JP, et al. Immune changes in post-menopausal osteoporosis: the Immunos study. Osteoporos Int (2010) 21(5):805–14. doi:10.1007/s00198-009-1018-7
188. Faienza MF, Ventura A, Marzano F, Cavallo L. Postmenopausal osteoporosis: the role of immune system cells. Clin Dev Immunol (2013) 2013:575936. doi:10.1155/2013/575936
189. D’Amelio P, Grimaldi A, Di Bella S, Brianza SZ, Cristofaro MA, Tamone C, et al. Estrogen deficiency increases osteoclastogenesis up-regulating T cells activity: a key mechanism in osteoporosis. Bone (2008) 43(1):92–100. doi:10.1016/j.bone.2008.02.017
190. Zhao R. Immune regulation of osteoclast function in postmenopausal osteoporosis: a critical interdisciplinary perspective. Int J Med Sci (2012) 9(9):825–32. doi:10.7150/ijms.5180
191. Molnar I, Bohaty I, Somogyine-Vari E. IL-17A-mediated sRANK ligand elevation involved in postmenopausal osteoporosis. Osteoporos Int (2014) 25(2):783–6. doi:10.1007/s00198-013-2548-6
192. Shukla P, Mansoori MN, Kakaji M, Shukla M, Gupta SK, Singh D. Interleukin 27 (IL-27) alleviates bone loss in estrogen-deficient conditions by induction of early growth response-2 gene. J Biol Chem (2017) 292(11):4686–99. doi:10.1074/jbc.M116.764779
193. Zheng SX, Vrindts Y, Lopez M, De Groote D, Zangerle PF, Collette J, et al. Increase in cytokine production (IL-1 beta, IL-6, TNF-alpha but not IFN-gamma, GM-CSF or LIF) by stimulated whole blood cells in postmenopausal osteoporosis. Maturitas (1997) 26(1):63–71. doi:10.1016/S0378-5122(96)01080-8
194. Hustmyer FG, Walker E, Yu XP, Girasole G, Sakagami Y, Peacock M, et al. Cytokine production and surface antigen expression by peripheral blood mononuclear cells in postmenopausal osteoporosis. J Bone Miner Res (1993) 8(1):51–9. doi:10.1002/jbmr.5650080108
195. Roggia C, Tamone C, Cenci S, Pacifici R, Isaia GC. Role of TNF-alpha producing T-cells in bone loss induced by estrogen deficiency. Minerva Med (2004) 95(2):125–32.
196. Weitzmann MN, Ofotokun I. Physiological and pathophysiological bone turnover – role of the immune system. Nat Rev Endocrinol (2016) 12(9):518–32. doi:10.1038/nrendo.2016.91
197. Jaumdally SZ, Picton A, Tiemessen CT, Paximadis M, Jaspan HB, Gamieldien H, et al. CCR5 expression, haplotype and immune activation in protection from infection in HIV-exposed uninfected individuals in HIV-serodiscordant relationships. Immunology (2017) 151(4):464–73. doi:10.1111/imm.12743
198. Rihn SJ, Foster TL, Busnadiego I, Aziz MA, Hughes J, Neil SJ, et al. The envelope gene of transmitted HIV-1 resists a late interferon gamma-induced block. J Virol (2017) 91(7):e02254–16. doi:10.1128/JVI.02254-16
199. Cardone M, Ikeda KN, Varano B, Gessani S, Conti L. HIV-1-induced impairment of dendritic cell cross talk with gammadelta T lymphocytes. J Virol (2015) 89(9):4798–808. doi:10.1128/JVI.03681-14
200. Creery D, Weiss W, Lim WT, Aziz Z, Angel JB, Kumar A. Down-regulation of CXCR-4 and CCR-5 expression by interferon-gamma is associated with inhibition of chemotaxis and human immunodeficiency virus (HIV) replication but not HIV entry into human monocytes. Clin Exp Immunol (2004) 137(1):156–65. doi:10.1111/j.1365-2249.2004.02495.x
201. Fakruddin JM, Laurence J. HIV envelope gp120-mediated regulation of osteoclastogenesis via receptor activator of nuclear factor kappa B ligand (RANKL) secretion and its modulation by certain HIV protease inhibitors through interferon-gamma/RANKL cross-talk. J Biol Chem (2003) 278(48):48251–8. doi:10.1074/jbc.M304676200
Keywords: osteoimmunology, interferon-gamma, osteoclasts, osteoblasts, rheumatoid arthritis, postmenopausal osteoporosis, human immunodeficiency virus
Citation: Tang MJ, Tian L, Luo GJ and Yu XJ (2018) Interferon-Gamma-Mediated Osteoimmunology. Front. Immunol. 9:1508. doi: 10.3389/fimmu.2018.01508
Received: 27 February 2018; Accepted: 18 June 2018;
Published: 29 June 2018
Edited by:
Fabrice Cognasse, Groupe Sur L’immunité Des Muqueuses Et Agents Pathogènes (GIMAP), FranceReviewed by:
Udo S. Gaipl, Universitätsklinikum Erlangen, GermanyRajeev Aurora, Saint Louis University School of Medicine, United States
Copyright: © 2018 Tang, Tian, Luo and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xijie Yu, eGlqaWV5dUBzY3UuZWR1LmNu, eGlqaWV5dUBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work.