
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 21 June 2018
Sec. Inflammation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01418
 Tomohiro Fukaya1,2
Tomohiro Fukaya1,2 Takehito Fukui1,3
Takehito Fukui1,3 Tomofumi Uto1,2
Tomofumi Uto1,2 Hideaki Takagi1,2Junta Nasu1,3
Hideaki Takagi1,2Junta Nasu1,3 Noriaki Miyanaga1,4
Noriaki Miyanaga1,4 Keiichi Arimura1,3
Keiichi Arimura1,3 Takeshi Nakamura1,4Haruhiko Koseki5
Takeshi Nakamura1,4Haruhiko Koseki5 Narantsog Choijookhuu6
Narantsog Choijookhuu6 Yoshitaka Hishikawa6
Yoshitaka Hishikawa6 Katsuaki Sato1,2*
Katsuaki Sato1,2*
Disruption of skin homeostasis can lead to inflammatory cutaneous diseases resulting from the dysregulated interplay between epithelial keratinocytes and immune cells. Interleukin (IL)-22 signaling through membrane-bound IL-22 receptor 1 (IL-22R1) is crucial to maintain cutaneous epithelial integrity, and its malfunction mediates deleterious skin inflammation. While IL-22 binding protein (IL-22BP) binds IL-22 to suppress IL-22 signaling, how IL-22BP controls epithelial functionality to prevent skin inflammation remains unclear. Here, we describe the pivotal role of IL-22BP in mediating epithelial autoregulation of IL-22 signaling for the control of cutaneous pathogenesis. Unlike prominent expression of IL-22BP in dendritic cells in lymphoid tissues, epidermal keratinocytes predominantly expressed IL-22BP in the skin in the steady state, whereas its expression decreased during the development of psoriatic inflammation. Deficiency in IL-22BP aggravates psoriasiform dermatitis, accompanied by abnormal hyperproliferation of keratinocytes and excessive cutaneous inflammation as well as enhanced dermal infiltration of granulocytes and γδT cells. Furthermore, IL-22BP abrogates the functional alternations of keratinocytes upon stimulation with IL-22. On the other hand, treatment with IL-22BP alleviates the severity of cutaneous pathology and inflammation in psoriatic mice. Thus, the fine-tuning of IL-22 signaling through autocrine IL-22BP production in keratinocytes is instrumental in the maintenance of skin homeostasis.
The maintenance of structural and immunological barrier function in the skin is a prerequisite for limiting exposure to potentially harmful environmental substances and preventing systemic dissemination and entry of commensal and pathogenic microorganisms (1, 2). Therefore, tight regulation for crosstalk between epithelial, mesenchymal, and immune cells is essential to achieve effective host defense, maintain a healthy skin homeostasis, and allow normal wound repair after injury (1, 2). However, disruption of skin homeostasis is thought to cause chronic inflammatory skin diseases resulting from the dysregulated interplay between epithelial keratinocytes and infiltrating inflammatory leukocytes (1–3).
Interleukin (IL)-22 is a member of the IL-10 cytokine family with multiple functions in various inflammatory responses, depending on the environmental context. IL-22 is mainly produced by CD4+ effector T (Teff) cells, including T helper 1 (Th1) cells, Th17 cells, and Th22 cells, as well as innate lymphoid cells (ILCs) 3 (4–6). IL-22 signals through a heterodimeric transmembrane receptor complex composed of IL-22 receptor 1 (IL-22R1, also known as IL-22RA1) and IL-10 receptor 2 (IL-10R2), whereas IL-10 signals through IL-10R1 and IL-10R2 (4–6). In contrast to the ubiquitous expression of IL-10R2 on immune cells, the expression of IL-22R1 is restricted to non-hematopoietic cells at body barriers including epithelial cells of the lung and gastrointestinal tract and keratinocytes in the skin, thereby allowing IL-22 to be an important cytokine mediating the crosstalk between leukocytes and epithelia at barrier surfaces (4–6). Similar to other members of the IL-10 family, IL-22 mediates its effects through the IL-22R1/IL-10R2 complex and subsequent JAK–signal transducer and activator of transcription (STAT) signaling pathways, including Jak1, Tyk2, and STAT3 (6). As the main physiological function of IL-22 includes a reinforcement of epithelium barrier function through induction of antimicrobial peptides (AMPs) to promote antimicrobial immunity (7), and wound healing through induction of epithelial cell proliferation and survival following tissue damage in pathophysiological conditions (8–10), it appears to have an important role in the maintenance of epithelial integrity at barrier surfaces (11, 12). Nevertheless, continuous exacerbated or uncontrolled IL-22 signaling leads to undesirable tissue inflammation, accelerating certain immune pathologies such as psoriasis and rheumatoid arthritis (6, 13, 14), probably depending on the inflammatory milieu through synergistic action with other proinflammatory cytokines. Therefore, the fine-tuning of IL-22 signaling is implicated to be required to maintain epithelial integrity and control cutaneous tissue inflammation and immunosurveillance, as well as normal skin wound healing.
Unlike other members of the IL-10 cytokine family, IL-22 has a soluble-secreted receptor, known as IL-22 binding protein (IL-22BP, also known as IL-22RA2) (6, 15–17). Although IL-22BP shares the highest structural homology with the IL-22R1 chain, IL-22BP exhibits a much higher affinity for IL-22 than IL-22R1 and therefore prevents the binding of IL-22 to IL-22R1 (18–20). While the constitutive expression of IL-22BP has been reportedly observed in several tissues at various levels, subsets of CD11b+ conventional dendritic cells (cDCs) are a major source of IL-22BP in the gastrointestinal tract (21), and appear to be involved in regulation of intestinal tissue repair and tumorigenesis in the colon through the suppression of IL-22 function (22). Furthermore, IL-22BP expressed by CD11b+ cDCs has been shown to promote the bacterial uptake into Payer’s patch (PP) by inhibiting IL-22 signaling in the follicle-associated epithelium (23). On the other hand, IL-22BP has reportedly mediated a pathogenic role of intestinal CD4+ T cells in inflammatory bowel disease through impairment of the protective action of IL-22 (24). However, despite data linking the imbalance in IL-22–IL-22BP axis to psoriatic dermatitis (14, 25), intrinsic cutaneous cellular sources of IL-22BP and the role of endogenous IL-22BP in the functionality of keratinocytes that impact skin pathologic inflammation remain unclear.
Here, we address how IL-22BP controls epithelial functionality to prevent skin inflammation. We provide evidence that IL-22BP is predominantly expressed in keratinocytes in the skin that are different from other tissues, suppressing their IL-22-mediated functional alternations. We further demonstrate that deficiency of IL-22BP promotes the progression of psoriasiform dermatitis mediated through enhanced epidermal hyperplasia and inflammation. Collectively, these findings indicate that IL-22BP mediates epithelial autoregulation of IL-22 signaling to control cutaneous pathogenesis.
The following 8- to 12-week-old mice were used in this study. C57BL/6 mice (Japan Clea), B6.FVB-TgItgax-DTR/EGFP Lan/J (CD11c-DTR/EGFP) mice (26) (referred to as cDC-ablated mice), and B6. Il22ra2−/− mice as described below. All mice were bred and maintained in SPF and germ-free (GF) conditions in the animal facility at the University of Miyazaki. All experiments were performed in accordance with institutional guidelines of the Animal Experiment Committee and Gene Recombination Experiment Committee. For the systemic ablation of cDCs, B6.CD11c-DTR/EGFP mice were intraperitoneally (i.p.) injected with DT (100 ng/mouse; Sigma-Aldrich), and B6.WT mice were also treated with DT as controls.
Generation of Il22ra2−/− mice, tissue and cell isolation, flow cytometry, quantitative RT-PCR, generation of human immunoglobulin G Fc fusion protein, in vitro CD4+ T-cell differentiation assay, cell culture, skin inflammation, histopathological assessment, and immunohistochemical analysis are described in Supplementary Material.
Data are expressed as the mean ± SD from 3 to 10 individual samples in a single experiment, and we performed at least three independent experiments. The statistical significance of the differences between the values obtained was evaluated by ANOVA. A P value of <0.01 was considered significant.
Previous studies have shown that the Il22ra2 transcript is preferentially expressed in secondary lymphoid tissues and intestinal tissues compared with other tissues, and its expression is mainly detected in cDCs (21–23). However, the expression level and cellular source of IL-22BP in the skin relative to other lymphoid and non-lymphoid tissues remain unknown. To understand the role of endogenous IL-22BP in the control of skin homeostasis, we first compared the transcriptional expression levels of Il22ra2, as well as Il22, throughout lymphoid and non-lymphoid tissues, including epithelial tissues of the skin, in the steady-state condition. Similar to the published reports (5, 6), high transcriptional expressions of Il22 and Il22ra2 were found in peripheral lymph nodes (PLNs) and mesenteric lymph nodes (MLNs), as well as colon and PP (Figures S1A,B in Supplementary Material), and CD11chighI-A/I-E+ cDCs showed a higher expression of the Il22ra2 transcript than other leukocytes in both PLNs and MLNs, but not in the spleen (Spl) (Figures S1C–E in Supplementary Material). On the other hand, the marked expression level of the Il22ra2 transcript in the ears and back skin was comparable to that of PP, whereas little or no transcriptional expression of the Il22 was observed in the skin (Figures S1A,B in Supplementary Material). Furthermore, the prominent expression of Il22ra2 transcript was detected in CD45− epidermal keratinocytes and CD45− dermal mesenchymal cells rather than CD45+I-A/I-E− leukocytes and CD45+I-A/I-E+ leukocytes in the epidermis and dermis, respectively (Figures S1F,G in Supplementary Material).
To confirm that cutaneous epithelial cells, but not CD11chighI-A/I-E+ cDCs, act as the main producer of IL-22BP, we examined the transcriptional expression of Il22ra2 in the absence of cDCs using CD11c-diphtheria toxin (DT) receptor (DTR)/enhanced green fluorescent protein (EGFP) transgenic (Tg) mice (26), in which CD11chighEGFP+ cDCs were depleted after a single DT injection (referred to as cDC-ablated mice). The depletion of cDCs enhanced the expression levels of the Il22ra2 transcript in Spl possibly due to the massive splenic infiltration of CD11b+Gr-1+ neutrophils (27, 28) with their high expression after DT treatment (Figures S2A–C in Supplementary Material). By contrast, the elimination of cDCs led to a reduction in the transcriptional expression of Il22ra2 in PLNs, MLNs, and PP (Figure S2A in Supplementary Material), suggesting that cDCs mainly produce IL-22BP in these secondary lymphoid tissues. However, the cutaneous expression level of the Il22ra2 transcript was not affected the absence of cDCs (Figure S2A in Supplementary Material).
Collectively, these results indicate that epidermal keratinocytes are the major cellular source of IL-22BP in the skin in the steady-state condition.
To clarify the kinetics for the expression of IL-22BP during the skin inflammation, we accessed the expression level of the Il22ra2 transcript in the skin and skin-draining PLNs in psoriasiform dermatitis induced by topical application of imiquimod (IMQ), known as a synthetic Toll-like receptor 7 (TLR7) ligand on the ear skin (29, 30). The transcriptional expression of Il22ra2 was decreased during the development of psoriasiform dermatitis in ear skin and skin-draining PLNs (Figures S1H,I in Supplementary Material).
As retinoic acid (RA) enhanced the transcriptional expression of Il22ra2 on human monocyte-derived DCs (21), we examined the influence of topical application of RA on the expression of the Il22ra2 transcript in keratinocytes. In contrast to the enhanced transcriptional expression of Il22ra2 in bone marrow (BM)-derived cDCs upon stimulation with RA, reduced expression of the Il22ra2 transcript was observed in keratinocytes after treatment with RA (Figures S2D,E in Supplementary Material). Indeed, the topical application of RA reduced the transcriptional expression of Il22ra2 in ear skin (Figure S2F in Supplementary Material).
It has been shown that the production of IL-22 and bacterial colonization is reciprocally regulated in the mucosal surface of the intestine (23, 31, 32). To address the influence of bacterial colonization on the production of IL-22 and IL-22BP, we compared their transcriptional expressions in the skin between specific pathogen-free (SPF) and GF wild-type (WT) mice. While the cutaneous expression of the Il22 transcript was barely detected in GF mice when compared with that of SPF mice, comparable expression of the Il22ra2 transcript was observed in the ear skin in GF and SPF mice (Figure S2G in Supplementary Material).
Accumulating evidence indicates that IL-22 mediates keratinocyte proliferation and diffuse epidermal hyperplasia that cause thickening of the epidermis known as acanthosis, inhibits terminal differentiation of keratinocytes leading to incomplete keratinization that causes parakeratosis, and induces the secretion of AMPs (14, 25). To evaluate the pathophysiological role of IL-22BP in vivo, we generated mice lacking the Il22ra2 gene (Il22ra2−/−; Figure S3 in Supplementary Material). Il22ra2−/− mice were born at the expected frequencies, and homozygous mice were healthy with normal cellularity in primary and secondary lymphoid tissues (Figure S4 in Supplementary Material). Upon topical application of IMQ on the ear skin, Il22ra2−/− mice exhibited a more prominent psoriasiform inflammation, with thickening and scaling than WT mice (Figures 1A,B). Furthermore, the intradermal (i.d.) injection of IL-22 fusion protein, which consists of IL-22 and the Fc fragment of human immunoglobulin G (huIgG) (IL-22-huIgFc), caused more severe psoriatic inflammation in Il22ra2−/− mice than WT mice (Figure 1C), suggesting that the dysregulation of IL-22 signaling mediates the progression of psoriatic inflammation. Histological analyses confirmed that Il22ra2−/− mice displayed a more significant parakeratosis, acanthosis, and Munro’s microabscesses, as well as cutaneous infiltration of mononuclear cells, including Gr-1+ granulocytes and γδTCR+ T cells than WT mice following topical application of IMQ (Figures 1D–G).
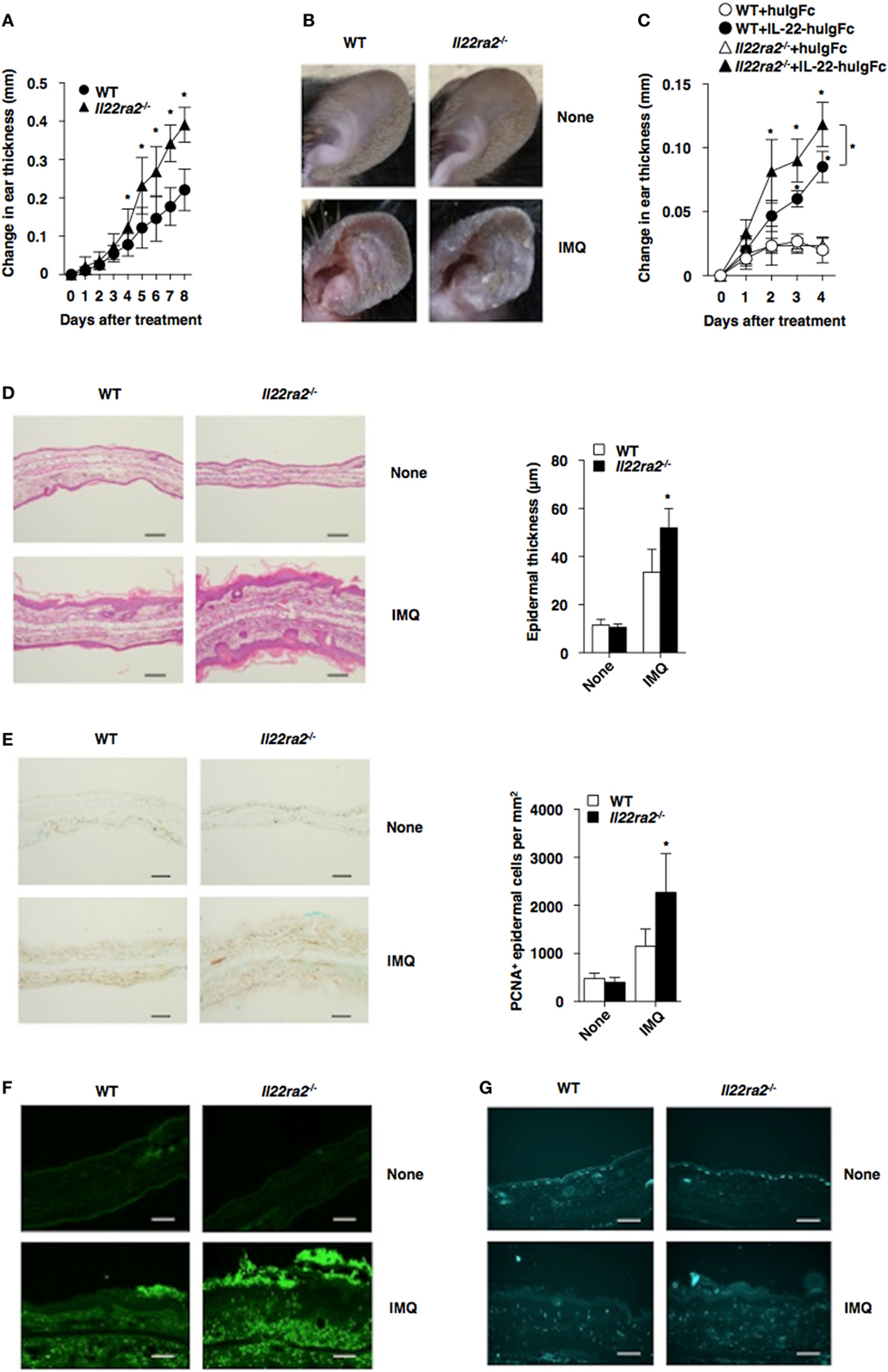
Figure 1. IL-22 binding protein deficiency exacerbates psoriatic dermatitis. (A,B) Wild-type (WT) mice (n = 5) and Il22ra2−/− mice (n = 5) were treated topically with imiquimod (IMQ) on the left ear skin every day for 8 days. (A) Ear thickness was evaluated for 8 days. Data were obtained from five individual samples in a single experiment. *P < 0.01 compared with WT mice. (B) Representative pictures of ear skin lesions at 8 days. (C) WT mice (n = 5) and Il22ra2−/− mice (n = 5) were treated with or without Fc fragment of human immunoglobulin (huIgFc) or interleukin (IL)-22-huIgFc, and ear thickness was evaluated for 4 days. Data were obtained from five individual samples in a single experiment. *P < 0.01 compared with WT mice. (D–G) Psoriatic dermatitis was induced by the topical application with IMQ on the left ear of WT mice (n = 5) and Il22ra2−/− mice (n = 5). (D) Representative hematoxylin and eosin sections (magnification; 20×) of ear skin at days 0 and 8 (left panel), and epidermal thickness was evaluated at 8 days (right panel). Data were obtained from five individual samples in a single experiment. *P < 0.01 compared with WT mice. (E) Representative immunohistochemical sections for detecting proliferating cell nuclear antigen (PCNA) (magnification; 20×) of ear skin at days 0 and 8 (left panel), and the quantification of PCNA+ epidermal cells of the epidermis (right panel) at 8 days. Data were obtained from five individual samples in a single experiment. *P < 0.01 compared with WT mice. (F,G) Representative immunohistochemical sections (magnification; 20×) for detecting Gr-1 (F) and γδTCR (G) of ear skin at days 0 and 8. All data are representative of at least three independent experiments.
To clarify how IL-22BP controls psoriasiform inflammation, we compared the transcriptional expressions of cytokines and chemokines, as well as epithelial inflammation-related molecules, in psoriatic lesions between WT mice and Il22ra2−/− mice (Figure 2; Figure S5A in Supplementary Material). Il22ra2−/− mice showed higher transcriptional expression of Il17a, Il22, Il1b, Csf3, Cxcl1, Mmp9, Il19, Il20, Il24, Reg3g (33), S100a7, and Saa1 (34–36) than WT mice. On the other hand, they exhibited lower transcriptional expressions of Cc120 (14), Lor, and Krt10 which are known as terminal differentiation marker genes (33), than WT mice.
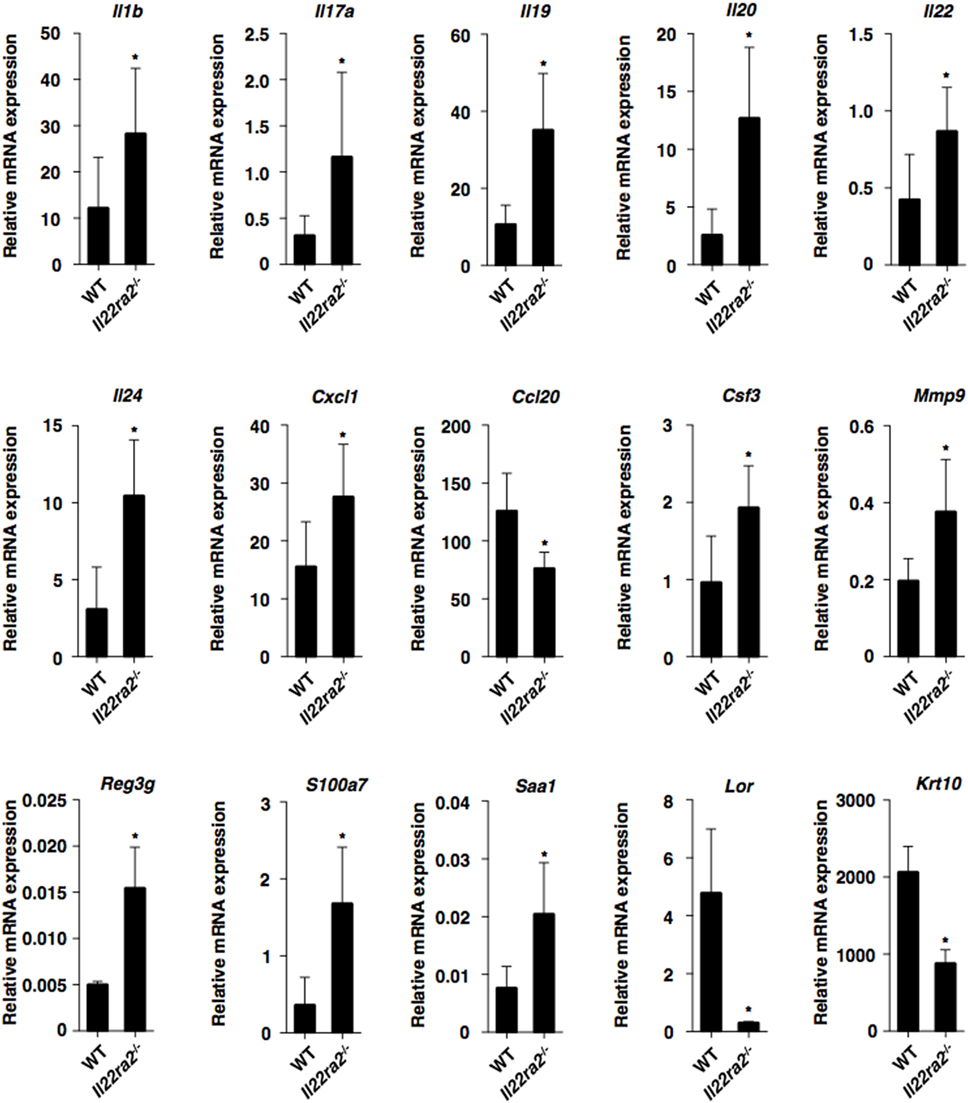
Figure 2. IL-22 binding protein deficiency promotes psoriatic inflammation. Transcriptional expression of cytokines, chemokines, and epithelial inflammation-related molecules in ear skin was analyzed by quantitative reverse transcription polymerase chain reaction at 8 days after topical application of imiquimod on the ear skin in wild-type (WT) mice (n = 5) and Il22ra2−/− mice (n = 5), and expression was normalized to the Actb transcript. Data are the mean ± SD from three individual samples in a single experiment. *P < 0.01 compared with WT mice. All data are representative of at least three independent experiments.
We also assessed the impact of IL-22BP on the infiltration of inflammatory leukocytes in the skin following initiation of psoriasiform dermatitis. Il22ra2−/− mice showed enhanced or reduced epidermal infiltration of neutrophils and γδTCRlow T cells, as well as CD4+ T cells or DCs, whereas they exhibited enhanced or reduced dermal infiltration of neutrophils or DCs when compared with WT mice (Figure 3). Furthermore, Il22ra2−/− mice showed higher accumulation of neutrophils, macrophages, cDCs, plasmacytoid DCs, and γδTCR+ T cells in the skin-draining PLNs than WT mice (Figure S5B in Supplementary Material).
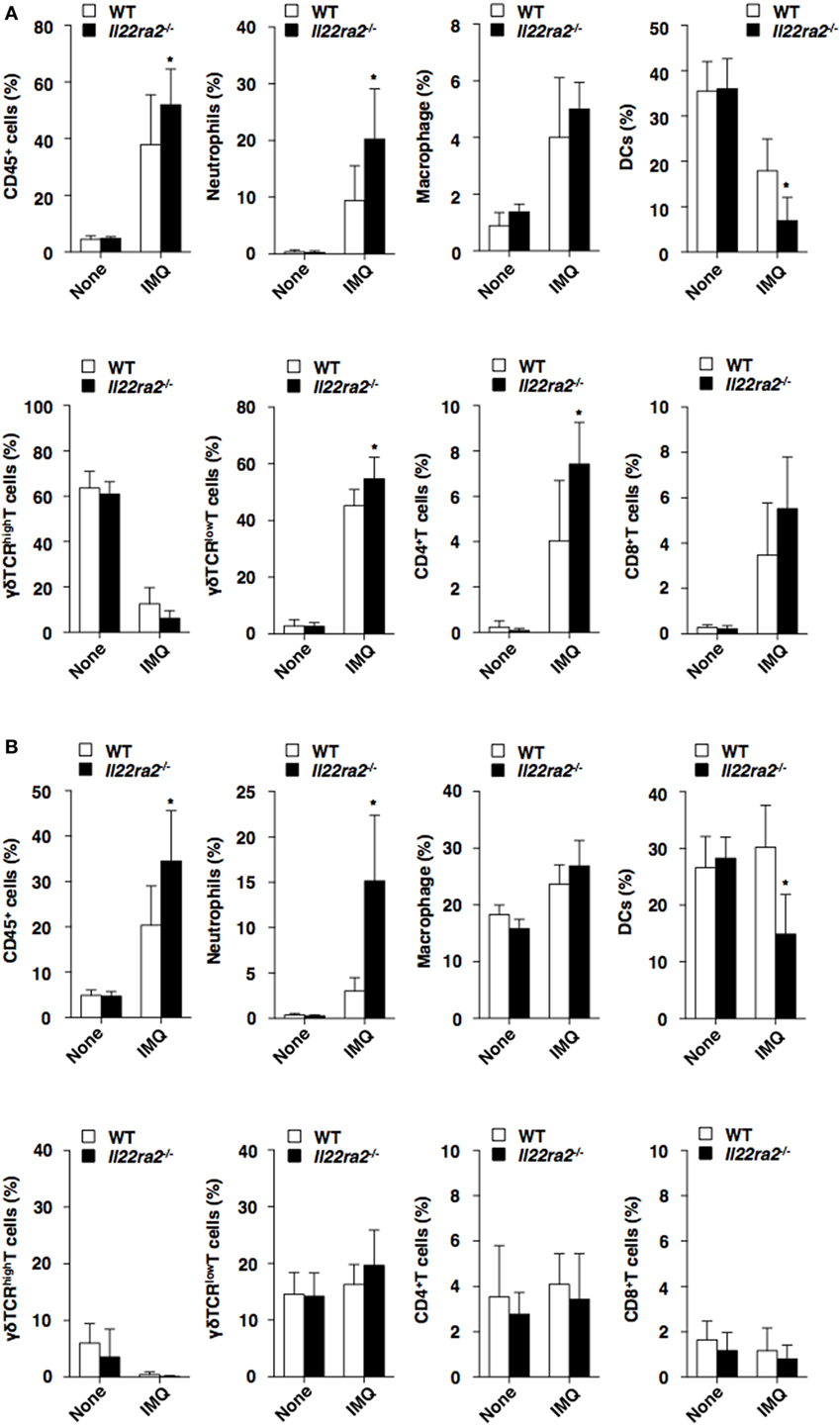
Figure 3. Absence of IL-22 binding protein enhances the accumulation of inflammatory leukocytes in psoriatic skin. The frequency of leukocytes in the epidermis (A) and dermis (B) was analyzed by flow cytometry at days 0 and 8 after topical application of imiquimod (IMQ) on the ear skin in wild-type (WT) mice (n = 5) and Il22ra2−/− mice (n = 5). Data are the mean ± SD from three individual samples in a single experiment. *P < 0.01 compared with WT mice. All data are representative of at least three independent experiments.
Taken together, these results indicate that a deficiency in IL-22BP exacerbates skin inflammation.
We further examined the impact of IL-22BP on the IL-22-mediated changes in the functionality of keratinocytes. While IL-22-huIgFc elicited the proliferation of keratinocytes in the presence or absence of extracellular calcium that triggered their differentiation, IL-22BP-huIgFc, but not huIgFc, almost completely inhibited their proliferation (Figures 4A,B). Furthermore, IL-22-huIgFc reduced the expressions of the transcripts of terminal differentiation marker genes (Lor and Krt10) of keratinocytes (Figures 4C,D). However, IL-22BP-huIgFc, but not huIgFc, inversely regulated the IL-22-mediated changes in the transcripts (Figures 4C,D). On the other hand, IL-22-huIgFc induced the translocation of phosphorylated STAT3 (pSTAT3), but not pSTAT1 and STAT5, into the nucleus in keratinocytes (37), whereas IL-22BP-huIgFc impaired the IL-22-mediated change in pSTAT3 (Figure 4E; Figure S6 in Supplementary Material).
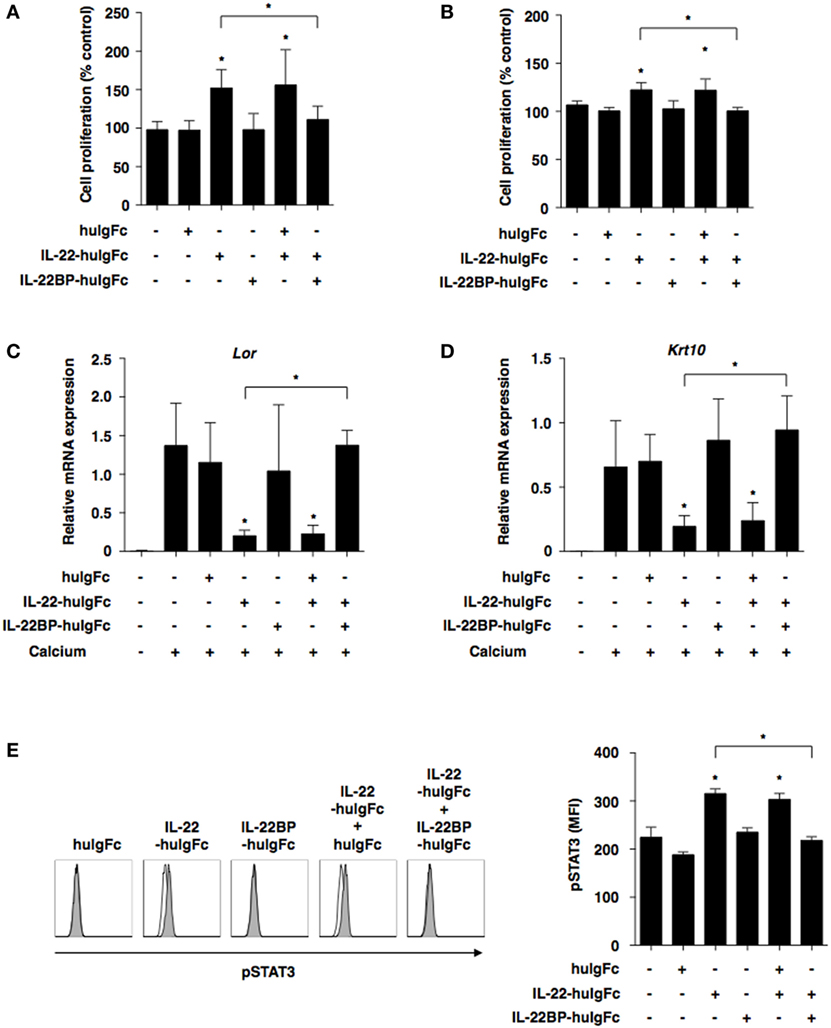
Figure 4. IL-22 binding protein (IL-22BP) suppresses the interleukin (IL)-22-mediated functional alternation of keratinocytes. (A–D) Keratinocytes were cultured with or without Fc fragment of human immunoglobulin (huIgFc), IL-22-huIgFc, and/or IL-22BP-huIgFc for 3 days. (A,B) Proliferative responses of keratinocytes in the absence (A) or presence (B) of extracellular calcium, with data expressed as percentage of untreated control (medium alone). Data are the mean ± SD from three individual samples in a single experiment. *P < 0.01 compared with untreated control or among groups. (C,D) Transcriptional expression of Lor (C) and Krt10 (D) in keratinocytes in the presence of extracellular calcium was analyzed by quantitative reverse transcription polymerase chain reaction at 3 days, and expression was normalized to the Actb transcript. Data are the mean ± SD from three individual samples in a single experiment. *P < 0.01 compared with untreated control (extracellular calcium) or among groups. (E) Keratinocytes were cultured with or without huIgFc, IL-22-huIgFc, and/or IL-22BP-huIgFc in the absence of extracellular calcium for 30 min, and the expression of phosphorylated STAT3 (pSTAT3) in nucleus was analyzed by flow cytometry. Data are presented as a histogram (left panel). Data are the mean fluorescence intensity (MFI) ± SD from three individual samples in a single experiment (right panel). *P < 0.01 compared with untreated control or among groups. All data are representative of at least three independent experiments.
Collectively, these results indicate that IL-22BP suppresses IL-22-mediated functional changes in keratinocytes.
The above study suggests that the augmentation of IL-22 signaling is implicated to directly mediate the alternation of epithelial functionality for the progression of skin inflammatory pathogenicity in the absence of IL-22BP without affecting the function of immune cells. This is because the expression of IL-22R1 is restricted to keratinocytes in the skin. On the other hand, IL-17-producing lymphocytes have been shown to play a critical role in the development of psoriasiform dermatitis (38, 39). To verify how IL-22 signaling in epithelial keratinocytes controls the IL-17 cytokine axis to elicit the development of psoriasiform inflammation, we addressed the influence of IL-22BP deficiency on the generation of lymphocytes producing IL-17A or IL-22 in the skin-draining PLNs in psoriatic mice. Importantly, the generation of Th17 cells and IL-17A-producing γδTCR+ T cells, but not IL-17A-producing CD8+ T cells, was significantly enhanced in the absence of IL-22BP (Figures 5A–D). Similarly, IL-22BP deficiency markedly promoted the generation of Th22 cells and IL-22-producing γδTCR+ T cells, while it had almost no effect on the induction of IL-22-producing CD8+ T cells (Figures 5E–H).

Figure 5. IL-22 binding protein deficiency increases the generation of lymphocytes producing interleukin (IL)-17A or IL-22 during the development of psoriatic inflammation. The frequency of IL-17A-producing cells (A–D) and IL-22-producing cells (E–H) among CD3+ T cells (A,E), γδTCR+ T cells (B,F), CD4+ T cells (C,G), and CD8+ T cells (D,H) in the skin-draining peripheral lymph nodes was analyzed by flow cytometry at days 0 and 8 after topical application of imiquimod (IMQ) on the ear skin in wild-type (WT) mice (n = 5) and Il22ra2−/− mice (n = 5). Data are presented as a contour plot, and numbers represent the proportion of the indicated cell populations in each gate (left panel). Data are the mean ± SD from five individual samples in a single experiment (right panel). *P < 0.01 compared with WT mice. All data are representative of at least three independent experiments.
It has been shown that serum amyloid A1 (SAA1) participates in the functional differentiation of Th17 cells in the intestines (34–36). As increased cutaneous expression of the Saa1 transcript was observed in the absence of IL-22BP (Figure 2), we examined the influence of SAA1 on the generation of Th17 cells and Th22 cells in vitro (Figure S7 in Supplementary Material). Stimulation of CD4+ T cells with SAA1 enhanced the generation of Th17 cells and Th22 cells in the presence of IL-6 or IL-6 plus TGF-β.
Taken together, these results indicate that the absence of IL-22BP promotes the formation of the pathogenic IL-17–IL-22 cytokine milieu to induce psoriatic inflammation.
To further demonstrate the protective effect of IL-22BP against skin inflammation, we examined the influence of IL-22BP-huIgFc on the progression of IMQ-induced psoriatic dermatitis in WT mice. Treatment with IL-22BP-huIgFc, but not huIgFc, alleviated the severity of psoriatic pathology as confirmed by histological analyses in psoriatic mice as compared with untreated controls (Figures 6A–C). Furthermore, IL-22BP-huIgFc, but not huIgFc, attenuated the changes in the transcriptional expression levels of Il17a, Il19, Il22, Il24, S100a7, and Saa1, as well as Krt10 and Il25 (40) in psoriatic mice (Figure 6D; Figure S8 in Supplementary Material).
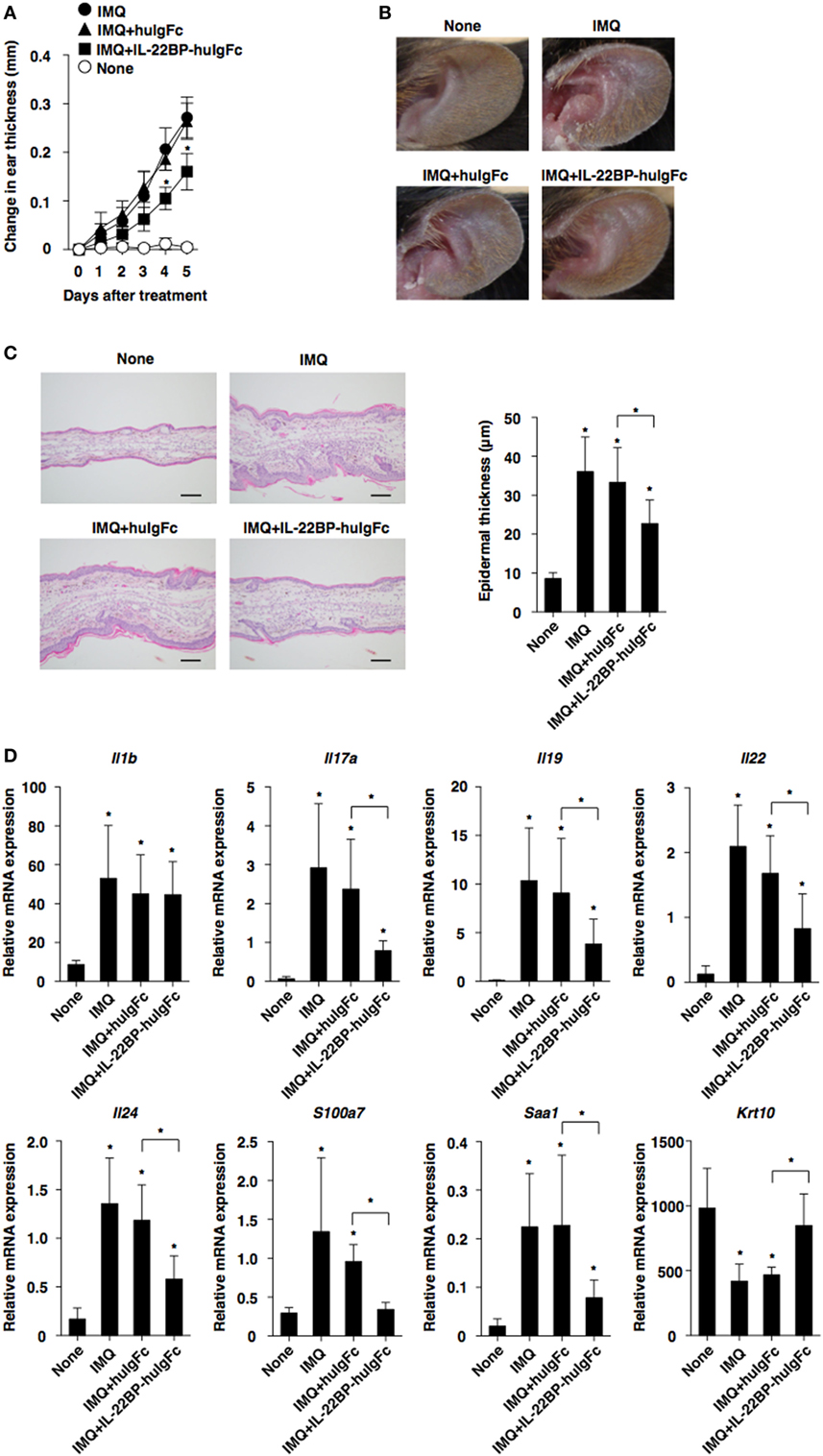
Figure 6. Protection of IL-22 binding protein (IL-22BP) against the development of imiquimod (IMQ)-induced psoriasiform dermatitis. Wild-type mice (each n = 5) that had received topical application of IMQ on the left ear skin every day for 5 days were treated with or without Fc fragment of human immunoglobulin (huIgFc) or IL-22BP-huIgFc every other day. (A) Ear thickness was evaluated at 5 days. Data were obtained from five individual samples in a single experiment. *P < 0.01 compared with untreated control. (B) Representative pictures of ear skin lesions at 5 days. (C) Representative hematoxylin and eosin sections (magnification; 20×) of ear skin at days 0 and 5 (left panel), and epidermal thickness was evaluated at 5 days (right panel). Data were obtained from five individual samples in a single experiment. *P < 0.01 compared with untreated control. (D) Transcriptional expression of cytokines and epithelial inflammation-related molecules in ear skin were analyzed by quantitative reverse transcription polymerase chain reaction at 5 days, and expression was normalized to the Actb transcript. Data are the mean ± SD from three individual samples in a single experiment. *P < 0.01 compared with untreated control. All data are representative of at least three independent experiments.
Collectively, these results indicate that treatment with IL22BP not only controls protection against inflammatory pathogenesis but also inhibits the malfunction of IL-22 signaling in keratinocytes during the development of psoriatic dermatitis.
Accumulating results suggest that IL-22 mediates epithelial integrity through membrane-bound IL-22R1 to control antimicrobial immunity, inflammation and tissue repair at barrier surfaces. Although dysregulated function of IL-22 is thought to be involved in the initiation and progression of epithelial pathogenesis, how epithelial IL-22 signaling is regulated in the skin remains unclear. In contrast to cDCs acting as main producers of IL-22BP to control epithelial functionality in the gastrointestinal tract, we showed the critical role of IL-22BP in mediating autoregulation of IL-22 signaling in keratinocytes to maintain skin homeostasis. Furthermore, IL-22BP protected against the development of psoriatic dermatitis mediated through abortive epidermal hyperplasia and inflammation.
Different from the intestinal mucosal tissues and secondary lymphoid tissues, in which cDCs are the major source of IL-22BP (21–23), epidermal keratinocytes and dermal mesenchymal cells exhibited its constitutive prominent production in the skin. Furthermore, the production of IL-22BP was reduced in the skin and skin-draining PLNs during the development of psoriasiform dermatitis. While stimulation with RA enhanced production of IL-22BP in cDCs (21), this stimulation somewhat diminished its production in keratinocytes. On the other hand, analysis of GF mice revealed that the steady-state production of IL-22 could require cutaneous microbial stimulation in the skin, whereas the cutaneous colonization of commensals could not affect the constitutive production of IL-22BP under the physiological condition. Collectively, these results suggest that the autocrine loop of IL-22BP constitutes a key element for regulating the responsiveness of epidermal keratinocytes to IL-22 signaling under the steady-state condition, whereas inflammatory stimulation abrogates their production of IL-22BP under the pathological condition. Although the precise mechanism responsible for the initiation of IL-22BP production in keratinocytes remains unclear, its transcript may be upregulated during the differentiation of keratinocytes from their progenitors. Further study will be needed to test this possibility.
While the pathogenesis of psoriasis involves several axes of immune dysregulation, the impact of IL-22BP on the epithelial functionality leading to the development of this cutaneous immune pathology remains unclear. Our results clearly showed that IL-22BP deficiency aggravated the incidence and severity of psoriatic dermatitis, accompanied by massive acanthosis and parakeratosis, as well as dermal leukocytic infiltration. Indeed, the absence of IL-22BP exacerbated IL-22-induced psoriatic dermatitis. Furthermore, IL-22BP deficiency led to enhanced or reduced expressions of AMPs (Reg3g and S100a7) and acute phase protein (Saa1) or terminal differentiation marker genes (Lor and Krt10) in keratinocytes. Taken together, these results suggest that IL-22BP works with epithelial autoregulation of IL-22 signaling in keratinocytes to control epithelial functionality to avoid undesirable skin inflammation.
Despite the restricted expression of IL-22R1 on non-hematopoietic cells allowing them to respond to IL-22 (12–14), IL-22BP deficiency caused drastic enhancement of cutaneous production of various inflammatory cytokines and chemokines, as well as the infiltration and accumulation of inflammatory leukocytes in the skin and skin-draining PLNs in psoriatic mice. Furthermore, the augmented transcriptional expression of several cytokines (Il19, Il20, and Il24) as sources of keratinocytes (5, 41) was observed in the pathological sites in the absence of IL-22BP. As the IL-20 subfamily of these cytokines secreted by keratinocytes has been implicated not only to act on various types of immune cells and non-hematopoietic cells but also to cause their further activation through a positive feedback loop, our finding suggests that IL-22BP impair the IL-22-mediated activation of keratinocytes to elicit the IL-19/IL-20/IL-24 axis, and this results in the amelioration of psoriatic dermatitis.
Although it has been shown that IL-22BP blocks the binding of IL-22 to IL-22R1 (18–20), how IL-22BP regulates the IL-22-mediated functional changes in keratinocytes remains unknown. We showed that IL-22BP almost completely inhibited the increased proliferation of keratinocytes and their aberrant differentiation upon stimulation with IL-22. Furthermore, IL-22BP impaired their IL-22-induced nuclear translocation of pSTAT3. Thus, IL-22BP could abrogate the IL-22-mediated alternation of keratinocyte functionality to alleviate acanthosis and parakeratosis, as well as skin inflammation during the progression of psoriatic dermatitis.
Growing evidence has shown that IL-23–IL-17 cytokine axis is a critical in the pathogenesis of psoriasis development (38, 39). While IL-23 produced by macrophages and cDCs reportedly participated in IL-17 secretion from Th17 cells and γδT cells as well as ILC3 (38, 39), IL-22BP deficiency had no effect on the expression of Il23a in psoriatic skin, implying that the IL-22BP-mediated impairment of IL-22 signaling does not contribute to control the ability of these phagocytes to produce IL-23. On the other hand, we showed that the generation of Th17 cells and IL-17A-producing γδTCR+ T cells, as well as Th22 cells and IL-22-producing γδTCR+ T cells were markedly enhanced in skin-draining PLNs under deficiency of IL-22BP. Previous studies have shown that IL-1β and SAA1 potentiated the production of IL-17A and IL-22 in Th17 cells and Th22 cells (34–36) as well as γδTCR+ T cells (42, 43) in mucosal tissues. Indeed, the absence of IL-22BP resulted in the enhanced transcriptional expression of Il1b and Saa1 in psoriatic skin, and stimulation with SAA1 enhanced the generation of Th17 cells and Th22 cells under Th17- and Th22-polarized culture conditions. Collectively, these results suggest that IL-22BP inhibits the formation of the IL-1β-SAA1-enriched milieu, which accelerates the generation of IL-17/IL-22-producing lymphocytes and their activation, mediated through the abortive IL-22-mediated activation of keratinocytes.
In accordance with the potent inhibitory function of IL-22BP on the IL-22-mediated functional alternation of keratinocytes in vitro, treatment of psoriatic mice with IL-22BP exerted marked protection against the development of skin pathogenesis, possibly due to the blockade of abnormal changes of keratinocytes. As the observed function of IL-22BP to alleviate psoriatic disease onset is comparable to that of anti-IL-22 monoclonal Ab (14), the use of IL-22BP could be a candidate treatment of this pathogenesis. Taken together, these results suggest that IL-22BP neutralizes the pathogenic actions of IL-22 signaling in keratinocytes to ameliorate psoriatic dermatitis.
In conclusion, we described the critical role of IL-22BP in mediating autoregulation of IL-22 signaling in keratinocytes to control cutaneous pathogenesis, and therefore fine tuning of the balance in the IL-22–IL-22R1–IL-22BP axis is essential to maintain epithelial homeostasis to evoke antimicrobial immunity, tissue repair at barrier surfaces, and prevent undesirable skin inflammatory disorders. Thus, IL-22BP may constitute an attractive target for the intervention and treatment of skin autoimmune and inflammatory disorders.
This study was carried out in accordance with the recommendations of Japanese national guidelines and institutional review board of University of Miyazaki. The protocol was approved by the Institutional Animal Care and Use Committee of University of Miyazaki.
KS designed all experiments, analyzed data, and wrote the manuscript. TFukaya, TFukui, TU, HT, JN, NM, KA, TN, and NC did experiments. HK and YH provided reagents and information.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all members of the animal facility at University of Miyazaki; Y. Sato and I. Goto for secretarial assistance; and Y. Kawagoe for technical help in cell sorting.
This work was supported by a Grant-in-Aid for Scientific Research (B) (KS;18H02670), challenging Exploratory Research (KS;16K15291) and for Young Scientists (B) (TU;17K15027, TFukaya;17K15732, and HT;26860337) from the Ministry of Education, Science and Culture of Japan, the Project for Cancer Research And Therapeutic Evolution (P-CREATE) from Japan Agency for Medical Research and development (AMED) (KS), the Uehara Memorial Foundation (HT), Takeda Science Foundation (TU and TFukaya), the NOVARTIS Foundation (Japan) for the Promotion of Science (HT), Kato Memorial Bioscience Foundation (HT), Daiichi Sankyo Foundation of Life Science (KS), the Naito Foundation (KS), GSK Japan Research Grant 2016 (TFukaya), GSK Japan Research Grant 2017 (HT), and Bristol-Myers Squibb Foundation Grants (KS).
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01418/full#supplementary-material.
Ab, antibody; BM, bone marrow; BM-cDCs, BM-derived conventional dendritic cells; DT, diphtheria toxin; DTR, DT receptor; EGFP, enhanced green fluorescent protein; FAE, follicle-associated epithelium; GF, germ-free; H&E, hematoxylin and eosin; huIgFc, Fc fragment of human immunoglobulin; IBD, inflammatory bowel diseases; i.d., intradermal; IFN, interferon; IFN-I, type I IFN; IgG, immunoglobulin G; IL, interleukin; ILCs, innate lymphoid cells; IL-10R, IL-10 receptor; IL-22BP, IL-22 binding protein; IL-22R, IL-22 receptor; IMQ, imiquimod; i.p., intraperitoneally; mAb, monoclonal Ab; MFI, mean fluorescence intensity; MLNs, mesenteric lymph nodes; PCNA, proliferating cell nuclear antigen; pDCs, plasmacytoid DCs; PLNs, peripheral lymph nodes; PP, Payer’s patch; pSTAT, phosphorylated signal transducer and activator of transcription; RA, retinoic acid; RT-qPCR, quantitative reverse transcription polymerase chain reaction; SAA, serum amyloid A; SPF, specific pathogen-free; Spleen, Spl; Teff, effector T; TGF, transforming growth factor; Th, T helper; TLR, toll-like receptors; TNF, tumor necrosis factor; WT, wild type.
1. Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol (2014) 14(5):289–301. doi:10.1038/nri3646
2. Lowes MA, Suárez-Fariñas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol (2014) 32:227–55. doi:10.1146/annurev-immunol-032713-120225
3. Gilliet M, Lande R. Antimicrobial peptides and self-DNA in autoimmune skin inflammation. Curr Opin Immunol (2008) 20(4):401–7. doi:10.1016/j.coi.2008.06.008
4. Eyerich S, Eyerich K, Cavani A, Schmidt-Weber C. IL-17 and IL-22: siblings, not twins. Trends Immunol (2010) 31(9):354–61. doi:10.1016/j.it.2010.06.004
5. Rutz S, Wang X, Ouyang W. The IL-20 subfamily of cytokines – from host defence to tissue homeostasis. Nat Rev Immunol (2014) 14(12):783–95. doi:10.1038/nri3766
6. Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R system. Nat Rev Drug Discov (2014) 3(1):21–38. doi:10.1038/nrd4176
7. Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med (2008) 14(3):275–81. doi:10.1038/nm1710
8. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity (2007) 27(4):647–59. doi:10.1016/j.immuni.2007.07.023
9. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity (2008) 29(6):947–57. doi:10.1016/j.immuni.2008.11.003
10. Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature (2015) 528(7583):560–4. doi:10.1038/nature16460
11. Sonnenberg GF, Fouser LA, Artis D. Functional biology of the IL-22-IL-22R pathway in regulating immunity and inflammation at barrier surfaces. Adv Immunol (2010) 107:1–29. doi:10.1016/B978-0-12-381300-8.00001-0
12. Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol (2011) 12(5):383–90. doi:10.1038/ni.2025
13. Ma HL, Liang S, Li J, Napierata L, Brown T, Benoit S, et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest (2008) 118(2):597–607. doi:10.1172/JCI33263
14. Van Belle AB, de Heusch M, Lemaire MM, Hendrickx E, Warnier G, Dunussi-Joannopoulos K, et al. IL-22 is required for imiquimod-induced psoriasiform skin inflammation in mice. J Immunol (2012) 188(1):462–9. doi:10.4049/jimmunol.1102224
15. Dumoutier L, Lejeune D, Colau D, Renauld JC. Cloning and characterization of IL-22 binding protein, a natural antagonist of IL-10-related T cell-derived inducible factor/IL-22. J Immunol (2001) 166(12):7090–5. doi:10.4049/jimmunol.166.12.7090
16. Kotenko SV, Izotova LS, Mirochnitchenko OV, Esterova E, Dickensheets H, Donnelly RP, et al. Identification, cloning, and characterization of a novel soluble receptor that binds IL-22 and neutralizes its activity. J Immunol (2001) 166(12):7096–103. doi:10.4049/jimmunol.166.12.7096
17. Xu W, Presnell SR, Parrish-Novak J, Kindsvogel W, Jaspers S, Chen Z, et al. A soluble class II cytokine receptor, IL-22RA2, is a naturally occurring IL-22 antagonist. Proc Natl Acad Sci U S A (2002) 98(17):9511–6. doi:10.1073/pnas.171303198
18. Logsdon NJ, Jones BC, Josephson K, Cook J, Walter MR. Comparison of interleukin-22 and interleukin-10 soluble receptor complexes. J Interferon Cytokine Res (2002) 22(11):1099–112. doi:10.1089/10799900260442520
19. Wolk K, Witte E, Hoffmann U, Doecke WD, Endesfelder S, Asadullah K, et al. IL-22 induces lipopolysaccharide-binding protein in hepatocytes: a potential systemic role of IL-22 in Crohn’s disease. J Immunol (2007) 78(9):5973–81. doi:10.4049/jimmunol.178.9.5973
20. Jones BC, Logsdon NJ, Walter MR. Structure of IL-22 bound to its high-affinity IL-22R1 chain. Structure (2008) 16(9):1333–44. doi:10.1016/j.str.2008.06.005
21. Martin JC, Bériou G, Heslan M, Chauvin C, Utriainen L, Aumeunier A, et al. Interleukin-22 binding protein (IL-22BP) is constitutively expressed by a subset of conventional dendritic cells and is strongly induced by retinoic acid. Mucosal Immunol (2014) 7(1):101–13. doi:10.1038/mi.2013.28
22. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature (2012) 491(7423):259–63. doi:10.1038/nature11535
23. Jinnohara T, Kanaya T, Hase K, Sakakibara S, Kato T, Tachibana N, et al. IL-22BP dictates characteristics of Peyer’s patch follicle-associated epithelium for antigen uptake. J Exp Med (2017) 214(6):1607–18. doi:10.1084/jem.20160770
24. Pelczar P, Witkowski M, Perez LG, Kempski J, Hammel AG, Brockmann L, et al. A pathogenic role for T cell-derived IL-22BP in inflammatory bowel disease. Science (2016) 354(6310):358–62. doi:10.1126/science.aah5903
25. Martin JC, Wolk K, Bériou G, Abidi A, Witte-Händel E, Louvet C, et al. Limited presence of IL-22 binding protein, a natural IL-22 inhibitor, strengthens psoriatic skin inflammation. J Immunol (2017) 198(9):3671–8. doi:10.4049/jimmunol.1700021
26. Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity (2002) 17(2):211–20. doi:10.1016/S1074-7613(02)00365-5
27. Tittel AP, Heuser C, Ohliger C, Llanto C, Yona S, Hämmerling GJ, et al. Functionally relevant neutrophilia in CD11c diphtheria toxin receptor transgenic mice. Nat Methods (2012) 9(4):385–90. doi:10.1038/nmeth.1905
28. Jiao J, Dragomir AC, Kocabayoglu P, Rahman AH, Chow A, Hashimoto D, et al. Central role of conventional dendritic cells in regulation of bone marrow release and survival of neutrophils. J Immunol (2014) 192(7):3374–82. doi:10.4049/jimmunol.1300237
29. Flutter B, Nestle FO. TLRs to cytokines: mechanistic insights from the imiquimod mouse model of psoriasis. Eur J Immunol (2013) 43(12):3138–46. doi:10.1002/eji.201343801
30. Takagi H, Arimura K, Uto T, Fukaya T, Nakamura T, Choijookhuu N, et al. Plasmacytoid dendritic cells orchestrate TLR7-mediated innate and adaptive immunity for the initiation of autoimmune inflammation. Sci Rep (2016) 6:24477. doi:10.1038/srep24477
31. Behnsen J, Jellbauer S, Wong CP, Edwards RA, George MD, Ouyang W, et al. The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity (2014) 40(2):262–73. doi:10.1016/j.immuni.2014.01.003
32. Schreiber F, Arasteh JM, Lawley TD. Pathogen resistance mediated by IL-22 signaling at the epithelial-microbiota interface. J Mol Biol (2015) 427(23):3676–82. doi:10.1016/j.jmb.2015.10.013
33. Lai Y, Li D, Li C, Muehleisen B, Radek KA, Park HJ, et al. The antimicrobial protein REG3A regulates keratinocyte proliferation and differentiation after skin injury. Immunity (2012) 37(1):74–84. doi:10.1016/j.immuni.2012.04.010
34. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139(3):485–98. doi:10.1016/j.cell.2009.09.033
35. Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell (2015) 163(2):381–93. doi:10.1016/j.cell.2015.08.061
36. Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell (2015) 163(2):367–80. doi:10.1016/j.cell.2015.08.058
37. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innateimmunity of tissues. Immunity (2004) 21(2):241–54. doi:10.1016/j.immuni.2004.07.007
38. Cai Y, Shen X, Ding C, Qi C, Li K, Li X, et al. Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity (2011) 35(4):596–610. doi:10.1016/j.immuni.2011.08.001
39. Lowes MA, Russell CB, Martin DA, Towne JE, Krueger JG. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol (2013) 34(4):174–81. doi:10.1016/j.it.2012.11.005
40. Takahashi K, Hirose K, Kawashima S, Niwa Y, Wakashin H, Iwata A, et al. IL-22 attenuates IL-25 production by lung epithelial cells and inhibits antigen-induced eosinophilic airway inflammation. J Allergy Clin Immunol (2011) 128(5):1067–76. doi:10.1016/j.jaci.2011.06.018
41. Kumari S, Bonnet MC, Ulvmar MH, Wolk K, Karagianni N, Witte E, et al. Tumor necrosis factor receptor signaling in keratinocytes triggers interleukin- 24-dependent psoriasis-like skin inflammation in mice. Immunity (2013) 39(5):899–911. doi:10.1016/j.immuni.2013.10.009
42. Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, et al. Retinoic acid expression associates with enhanced IL-22 production by γδ T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med (2013) 210(6):1117–24. doi:10.1084/jem.20121588
Keywords: interleukin-22, interleukin-22 binding protein, autoregulation, epidermal keratinocytes, skin inflammation, psoriasiform dermatitis, effector T cells
Citation: Fukaya T, Fukui T, Uto T, Takagi H, Nasu J, Miyanaga N, Arimura K, Nakamura T, Koseki H, Choijookhuu N, Hishikawa Y and Sato K (2018) Pivotal Role of IL-22 Binding Protein in the Epithelial Autoregulation of Interleukin-22 Signaling in the Control of Skin Inflammation. Front. Immunol. 9:1418. doi: 10.3389/fimmu.2018.01418
Received: 01 May 2018; Accepted: 06 June 2018;
Published: 21 June 2018
Edited by:
Akihiko Yoshimura, Keio University, JapanReviewed by:
Kiyoshi Hirahara, Chiba University, JapanCopyright: © 2018 Fukaya, Fukui, Uto, Takagi, Nasu, Miyanaga, Arimura, Nakamura, Koseki, Choijookhuu, Hishikawa and Sato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katsuaki Sato, a2F0c3Vha2lfc2F0b0BtZWQubWl5YXpha2ktdS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.