 Jonathan Fisher
Jonathan Fisher John Anderson
John Anderson- University College London, London, United Kingdom
Sharing both innate and adaptive immune properties, γδT cells are attractive candidates for cellular engineering. As the cancer immunotherapy field becomes increasingly busy, orthogonal approaches are required to drive advancement. Engineering of alternative effector cell types such as γδT cells represents one such approach. γδT cells can be modified using many of the techniques used in αβT cell engineering, with the added advantage of innate-like tumor recognition and killing. Progress has been made in T-cell receptor transfer to and from γδT cells as well as in a number of chimeric antigen receptor-based strategies. As the cancer immunotherapy field moves beyond repetitive iteration of established constructs to more creative solutions, γδT cells may offer an attractive chassis to drive anti-tumor responses that are not only broader, but also possess a more favorable safety profile.
Introduction
Cellular engineering has offered many options for redirecting immune responses against cancer. In some cases, the clinical responses have been remarkable (1–5) but there are still many challenges to overcome. Broadly speaking, redirection of T-cell responses against specific tumor-associated antigens (TAAs) has been achieved in two ways: T-cell receptor (TCR) gene transfer or chimeric antigen receptor (CAR) expression. TCR gene transfer involves expression of a TCR derived from a tumor antigen-reactive T-cell (6–8). The TCR is typically derived from a tumor infiltrating lymphocyte or from in vitro antigen-stimulated blood. Chimeric antigen receptors (CARs) are more synthetic in nature and comprise an ectodomain that directly binds a cell surface molecule specific for the tumor and endodomains, which provide T cell signaling. The ectodomain is most commonly a single-chain variable fragment derived from a monoclonal antibody, and the endodomains usually include CD3ζ in combination with one or more costimulatory domains derived from molecules such as CD28 or 4-1BB (9, 10).
The majority of cellular engineering approaches have been applied to αβT cells, which are easy to expand and purify from peripheral blood. Notable attention has been given to αβT cells engineered to express second- and third-generation CARs against targets such as CD19 (2, 11–14) and CAR-T cells targeting CD19 recently received FDA approval for sale in the United States for the treatment of diffuse large B-cell lymphoma and acute lymphoblastic leukemia (ALL).
Engineering approaches that redirect immune cells to target single antigens via a CAR or MHC-presented TAA epitopes have limitations. TCR transfer depends on the ability to isolate a HLA-matched TCR against a processed antigen presented by tumor cells (10), and is susceptible to tumor immune-evasion strategies such as downregulation of MHC (15) or loss of redundant neo-antigens (16). Transferred TCRs against TAAs can also lead to unexpected side-effects due to cross-reactivity with unrelated peptides. One study targeting MAGE-3A with a HLA-A*01 restricted TCR led to fatal cardiotoxicity due to cross-reactivity with epitopes derived from the striated-muscle protein, titin (17), though a later study targeting the same molecule but using a different TCR construct did not generate this toxicity and led to objective partial responses in 9/17 patients (18). This difference may be explicable due to recognition of different epitopes, but highlights the potential for unexpected toxicity.
Chimeric antigen receptors remove the need for HLA-matching and antigen presentation on tumor MHC by bypassing the αβTCR entirely, but antigen selection presents a challenge. CAR-T cells target both healthy and tumor cells expressing their cognate antigen (10); for example, anti-CD19 CARs kill CD19+ ALL as well as healthy CD19+ B-cells (19). In the context of CD19, B-cell aplasia is considered an acceptable cost, but targeting of other antigens such as carbonic anhydrase IX or ErbB2 has led to unexpected and sometimes fatal toxicity (albeit only at very high T cell dose in the case of ErbB2) (20, 21). Furthermore, the specificity of CAR-targeting provides a prime opportunity for immune-evasion through antigen loss, which has proven to be a particular issue in anti-CD19 CAR-T therapy (22).
Use of alternative cell types in cancer immunotherapy is not a novel concept. Adoptively transferred allogeneic NK cells or cytokine-induced killer cells have shown clinical efficacy against metastatic melanoma (23), renal cell carcinoma, acute myeloid leukemia, and Hodgkins lymphoma (24). While engineering of these cell types has lagged behind that of conventional αβT cells, CAR transduced NK cell lines have been successfully directed against CD19 (25), CD20 (26), the disialoganglioside GD2 (27), ErbB2 (28), and other TAAs (29). NK cell specificity to tumors has been enhanced using exogenous constructs such as bispecific antibodies that enhance or manipulate the synapse between NK cell and target (30). NKT cells expressing CARs have also been developed (31). Such modified NKT cells targeting the ganglioside GD2 are about to enter phase I trials in patients with neuroblastoma (clinical trial ID NCT03294954). This range of approaches demonstrates the feasibility of using effector cells with an innate immune phenotype, possessing broader tumor recognition potential.
Properties of γδT Cells
In vitro and in vivo, γδT cells exhibit potent anti-tumor responses suggesting natural roles in tumor control and potential for therapeutic exploitation (32–35). Of particular interest, a recent correlation between the molecular profile of the tumor immune microenvironment and prognosis in over 5,000 tumor samples indicated that the presence of infiltrating γδT cells was the strongest predictor of positive outcome (36).
γδT cells comprise only 1–10% of circulating T-cells (37), diverging from αβT cells in the thymus, with lineage commitment completed by the DN3 stage of thymic development (38). The dynamics of the γδT cell repertoire during fetal development and later adult life are complex, and while initial evidence suggested that the Vγ9Vδ2 subset, being small at birth (39), expanded purely in response to environmental pathogens, Dimova et al. showed that effector Vγ9Vδ2+ cells make up the bulk of the γδT cell repertoire in second trimester fetuses. This population contracts and loses its dominance toward full gestation, when Vγ9−Vδ1+ subsets predominate (40). These results were corroborated by Ravens et al. who used next-generation sequencing of the γδTCR repertoire in cord blood to reveal higher proportions of Vγ2–5 and Vδ1, 3 and 5 TCR chains compared to healthy adult circulation (41). Later in life, while adult human peripheral γδT cells expressing Vγ2–5, 8–9, and Vδ1–8 chains (42) can all be detected in peripheral blood of healthy donors and cancer patients (37), Vγ9Vδ2+ cells predominate in the circulation, and the age-related extrathymic increase in circulating Vγ9Vδ2+ proportions (39, 43) is well documented. Interestingly, this trend shows geographical variation; γδTCR repertoires of individuals from sub-Saharan Africa show greater enrichment of Vδ1+ cells compared to that of Caucasians living in Europe or America. This difference is not linked to malaria exposure and raises the possibility that the circulating γδTCR repertoire is shaped by environmental factors such as the endemic microbiome (43, 44).
Human Vγ9Vδ2+ T cells have been subjected to closer analysis than other subsets. They respond to targets with a high phosphoantigen burden, associated with malignant transformation and disordered EGFR signaling (45, 46). Importantly, this recognition is not dependent on peptide epitopes bound to MHC, distinguishing Vγ9Vδ2 T cells from αβT cells (47). Isopentenyl-5-pyrophosphate (IPP), a phosphoantigen by-product of the mevalonate pathway of cholesterol biosynthesis is the prototypic phosphoantigen in the context of human Vγ9Vδ2 T cell–tumor interactions (48, 49), though other phosphoantigens such as bromohydrin pyrophosphate and the microbially derived (E)-4-Hydroxy-3-methyl-but-2-enyl pyrophosphate have much lower EC50 values for Vγ9Vδ2 T cell activation (50). Because IPP production can be enhanced using aminobisphosphonates, Vγ9Vδ2+ T cells can be easily expanded from the blood of healthy donors and cancer patients using inexpensive and well-validated compounds in combination with low-dose IL-2 (50). Aminobisphosphonates inhibit the mevalonate pathway enzyme farnesyl pyrophosphate synthase, which is downstream of IPP and leads to its accumulation (51, 52). The approach allows production of large numbers of highly purified Vγ9Vδ2+ T cells using a relatively simple protocol (37, 53).
The precise mechanism of Vγ9Vδ2 TCR stimulus is still being clarified. There is a high degree of CDR3 sequence homology between TCR chains from fresh Vγ9Vδ2+ γδT cells, those expanded using aminobisphosphonates and those which expand in response to co-culture with microbes such as Escherichia coli (54). There is also homology in Vδ chain CDR3 regions between cells from unrelated individuals following phosphoantigen exposure (37). These factors reinforce the evidence that the Vγ9Vδ2 TCR responds to a ligand held in-common across donors. While previous reports have implicated F1-ATPase as the ligand (55, 56), strong recent evidence points to butyrophilin 3A1 (BTN3A1) (57, 58), which is stabilized in the membrane and undergoes a conformational change when its intracellular 30.2 domain is bound by IPP.
γδT cells also receive inputs from multiple co-stimulatory receptors and receptors usually associated with NK cells (59, 60), such as NKG2D (61), DNAM-1 (62), and Fcγ receptors, such as FcγRIII (CD16) (34, 63). Consequently, Vγ9Vδ2+ T cells exhibit NK-cell like properties of potent antibody-dependent and independent cytotoxicity (34, 53, 63). Less is known about the ligands of non-Vγ9Vδ2 γδTCRs, perhaps due to the diversity of targets and their MHC-independent activity. Numerous ligands have been identified but a clear pattern has not yet emerged, for example, Vδ1+ γδT cells have been shown to have against cells expressing lipids, such as CD1c (64), CD1d-sulphatide (65), CD1d-α-GalCer (66), but also against the MHC-associated molecules MICA (67) and MICB (68).
TCR Gene Transfer in the Context of γδTCR+ Cells
Transfer of specificity through the transfer of murine α and β TCR genes was first used to target the hapten molecule, fluorescein (69), an approach which has subsequently been used to redirect αβT cell immunity against antigens from viral (70) and tumor (71) targets, notably in highly immunogenic tumors, such as melanoma.
Transferring a new αβTCR gene construct into an αβT cells runs the risk of TCR chain mis-pairing unless the endogenous α and β chains are suppressed (72). Mis-pairing can lead to inefficient expression of the novel construct and may lead to the generation of self-reactive TCR clones leading to off-target toxicity (73). Using murine constant regions or altering arrangement of cysteines in the transferred TCRs prevents this (74). While there is a risk of the host mounting an anti-murine immune response with subsequent reduction in immunotherapeutic efficacy, this has not been seen in practice, and many leading groups favor murinized TCRs. One study in which 23% of patients developed anti-murine-TCR antibodies showed that these anti-murine responses had no effect on clinical outcome (75). Were an anti-murine response to be of concern, however, an alternative allowing use of entirely human TCRs is to use γδT cells as the substrate for gene transfer, as the γ and δ TCR chains do not mis-pair with transferred α or β chains (Figure 1A). Dorrie et al. (76) demonstrated that γδT cells could be induced to express a HLA-A*0101 restricted αβTCR targeting a peptide derived from an adenovirus hexon protein. Engineered γδT cells produced more IFNγ and TNFα than CD8+ αβT cells expressing the same TCR and had equivalent cytotoxicity against autologous adenovirus-infected dendritic cells. Similar antigen-specific cytokine release was demonstrated by Harrer et al. when γδT cells expressing a gp100/HLA-A2 restricted αβTCR were exposed to gp100+ melanoma cells (77).
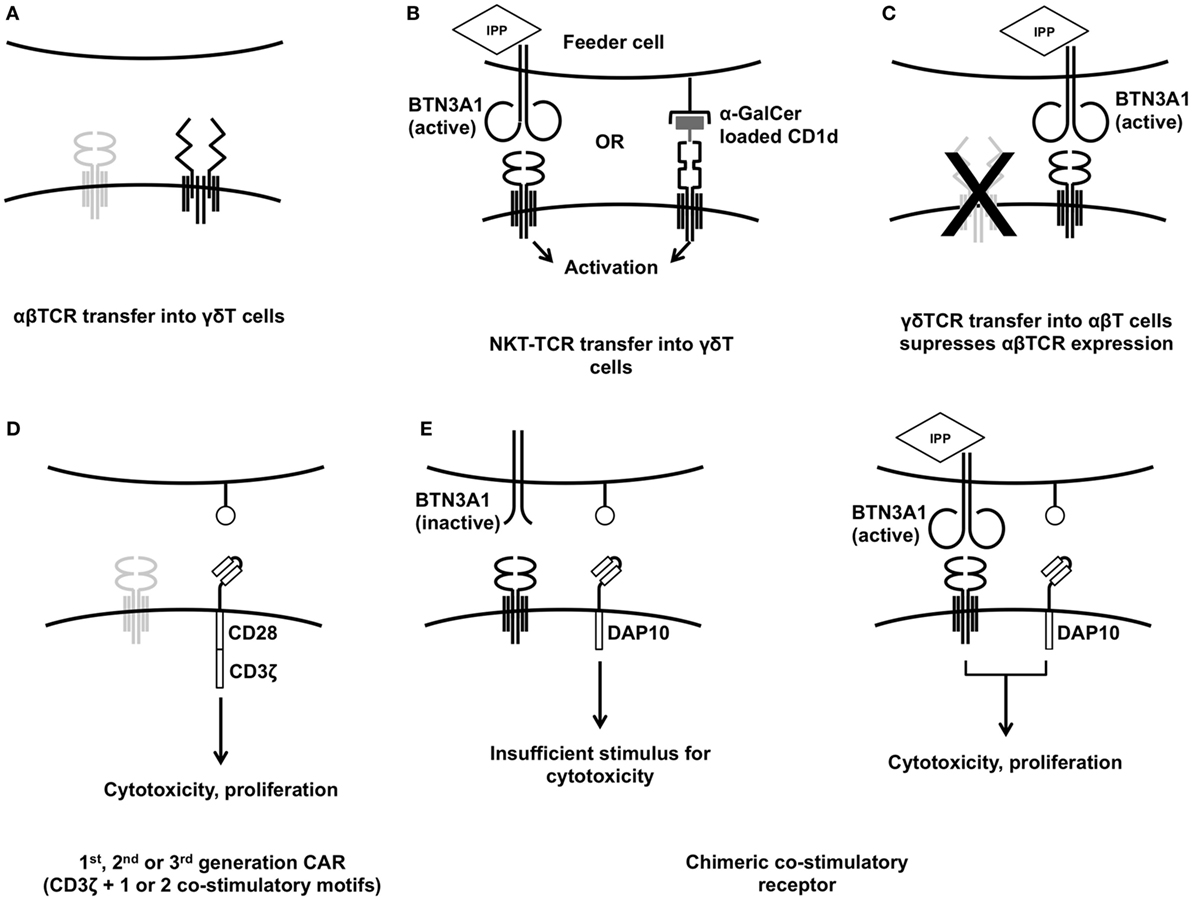
Figure 1. Established strategies for engineering γδT cells. Transferring specific αβT-cell receptor (TCR) into γδT cells (A) gives greater control of the final cell product and is one strategy to avoid TCR mis-pairing. Transferring NKT-TCRs into γδT cells (B) allows them to be activated for cytotoxicity using feeder cells treated with αGalCer or zoledronic acid. Transfer of γδTCRs into αβT cells has been used to impart broader anti-tumor reactivity (C). Anti-tumor cytotoxicity can be enhanced using conventional first-, second-, or third-generation chimeric antigen receptors (D) but the innate tumor recognition provided by the γδTCR can also be harnessed to deliver a more tumor-specific response (E).
Some researchers have highlighted the restrictions inherent in αβTCR gene transfer, in particular the restriction to particular HLA types and the possibility of antigen-negative escape variants (16). Transfer of TCRs derived from invariant natural killer T (iNKT) cells into γδT cells and transfer of γδTCRs into αβT cells (78) have both been used to overcome this. In humans, iNKT cells express the nearly invariant TCR encoded by Vα24Jα18 which responds to glycolipids presented on the HLA-class-I-like molecule CD1d. Like Vγ9Vδ2 TCR activation, this response is not MHC restricted (79). γδT cells expressing TCRs derived from iNKT cells can be stimulated by co-culture with either zoledronic acid treated HEK293T cells or HEK293T cells pulsed with the exogenous glycolipid α-galactosylceramide (α-GalCer, Figure 1B). Both stimulations led to enhanced cytotoxicity against the CD1d− leukemia cell line K562 (79). Continuing on the theme of using MHC-unrestricted TCRs, γδTCR gene transfer into αβT cells (Figure 1C) has yielded exciting results. The Vγ9Vδ2 TCR clone G115 (80) was expressed in αβT cells by Marcu-Malina et al. (78). They demonstrated that both the were required for either to be detected, indicating that mis-pairing with endogenous α or β chains was not occurring. The γδTCR-expressing αβT cells showed similar functional properties to “native” Vγ9Vδ2 cells including cytotoxicity against the Daudi cell line, release of TNFα and IFNγ, enhancement of cytotoxicity following target pre-treatment with aminobisphosphonates, and the ability to induce dendritic cell maturation. Vγ9Vδ2 transduced αβT cells showed a surprising lack of alloreactivity, linked to a downregulation of their endogenous αβTCRs (78), and were able to mount responses against a broad panel of tumor cell lines. This lack of γδT cell alloreactivity against non-transformed cells is corroborated by other in vitro data on both Vδ1+ and Vγ9Vδ2+ γδT cells (37).
Though the Vγ9Vδ2 TCRs derived from different T cell clones show varying anti-tumor responses, linked to small differences in the γ9 and δ2 CDR3 regions; no correlation was found between the expression of NKG2D, CD158a, NKAT-2, or NKB-1 and anti-tumor reactivity (81). This suggests that altering the functional avidity of interaction between BTN3A1 and the Vγ9Vδ2 TCR is a rich area for optimization. Using CD4+ αβT cells as the recipient cells, Gründer et al. performed alanine scanning between positions δ2-G115L109 and δ2-G115T113 and between γ9-G115E108 and γ9-G115E111, to demonstrate that the length and sequences in these CDR3 regions were critical for ligand interaction, with particular importance being placed on γ9-G115A109, in addition to the J-region residues δ109 and δ117 (81).
Such detailed knowledge of Vγ9Vδ2 avidity means that highly optimized γδTCRs can be expressed in more readily available αβT cells. γδTCR-engineered αβT cells prevented tumor growth in an immunodeficient (irradiated Rag−/−γc−/−) murine model of Burkitt lymphoma (Daudi) and multiple myeloma (OPM2) and also protected mice who had responded to initial treatment from re-challenge with OPM2 performed 120 days after the first tumor and T-cell injection (82). The downregulation of the αβTCR in the transduced cell population allows for facile selection of cells by αβTCR depletion, rather than positive selection of the transduced cells. This “untouched” cell product does not require co-expression of a marker gene and can be processed using pre-existing αβT cell depletion techniques currently used before some bone-marrow transplants, making it highly amenable to GMP-compliant manufacture.
γδT Cells Expressing CARs
While harnessing the innate potential of the γδTCR is a highly attractive option, manipulating cellular behavior in an antigen-specific manner using CARs (Figure 1D) remains one of the mainstays of modern immunotherapeutics. Compared to the substantial body of literature on αβT cells expressing CARs, there are relatively few reports of CAR-γδT cells. First described in 2004 (83), γδT cells expressing a first-generation CAR-targeting GD2 (14.G2aζ) which is expressed on the surface of neuroblastoma and Ewing sarcoma cells (84, 85) showed enhanced antigen-specific tumor reactivity. Following co-culture with the GD2+ neuroblastoma cell line LAN-1, 14.G2aζ+Vγ9+ cells showed greater production of the Th1 cytokine IFNγ compared to non-transduced zoledronate expanded 14.G2aζ−Vγ9+ γδT cells. This effect was mirrored in the expression of the T-cell activation marker CD69, which also upregulated the presence of the tumor cells. In the absence of GD2+ cells, 14.G2aζ+Vγ9+ γδT cells showed only 1.5 ± 0.5% IFNγ+CD69+ but following co-culture with GD2+ LAN-1 targets this rose to 33 ± 3%. Background production of IFNγ by non-transduced effectors exposed to LAN-1 was low (5.7 ± 1.2%). Similar results were seen when γδT cells expressing the CD19ζ CAR were co-cultured with CD19+ cell lines Daudi, Raji, and Reh (83), with substantial increases in target-dependent IFNγ production by mixed populations of CD19ζ+/− γδT cells. While Daudi is known to engage the γδTCR and is highly susceptible to γδT cell-mediated killing in its own right, Raji is usually considered to be a γδT cell resistant cell line (86), and it was in this model that the highest IFNγ production was seen, suggesting that CAR expression could overcome some of the immune-escape mechanisms shown by the target cells.
Since the publication of the work of Rischer et al. (83), progress in immunotherapy using adoptively transferred γδT cells has focused on the expansion of un-engineered γδT cells (32, 35). Whereas earlier studies used aminobisphosphonates to generate a predominantly Vδ2+ population (83, 87), a series of papers eventually demonstrated the possibility for expanding γδT cells with a broad range of γδTCR subsets using either plant-derived T-cell mitogens such as concanavalin A (88–90) or artificial antigen-presenting cells (aAPC) engineered to express co-stimulatory ligands and membrane-bound IL-15 (91). Two groups used a CD19+ aAPC system to expand Vδ2− γδT cells, demonstrating that the repertoire of γδT cells produced could be influenced by the loading of anti-γδTCR antibodies to the CD64 expressed on the aAPC (37, 92). Furthermore, this approach can be used to specifically propagate anti-CD19 CAR+ γδT cells (93). Deniger and colleagues generated a CAR+ γδTCR+ population containing a broad range of Vγ and Vδ chain combinations using negative selection following CAR gene transfer to the whole peripheral blood mononuclear cell (PBMC) population. γδT cells were isolated on the day after electroporation and propagated on CD19+CD64+CD86+CD137L+IL-15+ aAPCs in the presence of IL-2 and IL-21; the aAPCs were refreshed weekly. The resultant γδT cell population showed low expression of exhaustion markers such as CD57 and contained a heterogeneous mixture of memory phenotypes. This expansion technique has been shown to preserve the distribution of Vδ1+, Vδ2+, and Vδ1−/Vδ2− γδT cell subsets within a donor PBMC sample (37, 92). Singh et al. had previously demonstrated that culture using this aAPC system produced a selection pressure for CAR+ αβT cells (94) resulting in >90% CAR+ αβT cells after 28 days of co-culture, but this effect was muted when CAR+ γδT cells were expanded, presumably due to the inherent reactivity of non-transduced γδT cells against the aAPC leading to non-specific proliferation. aAPC based expansion may be particularly advantageous for γδT cells due to their expression of CD28 and CD137 which interact with CD86 and CD137L on the aAPC, and expression of CCR7 and CD62L by the CAR+γδTCR+ cells suggested that they had the capacity to home to the bone marrow and lymph nodes where CD19+ leukemia is known to reside. The CAR+ cells produced IFNγ, TNFα, MIP-1α, MIP1β, and RANTES following CAR activation through co-culture with a huCD19+ murine cell line which does not engage the γδTCR due to inter-species differences (95, 96), and killed human CD19+ cell lines with much greater efficacy than CAR−γδTCR+ cells (93). Immunodeficient mice xenografted with CD19+ffLuc+ NALM6 B-cell leukemia showed enhanced survival following CAR-γδT cell treatment compared to untreated, though a non-transduced or irrelevant CAR control was not included in the in vivo study so the in vivo activity is harder to dissect.
Engineering strategies which harness the innate properties of Vγ9Vδ2 T cells would seem to be the best justification for using them as an alternative “chassis” for CAR-T cell therapy. CARs were initially developed to bypass the αβTCR, limited as it is by MHC restriction and a requirement for specific TAA epitopes to be presented. The Vγ9Vδ2 TCR is not subject to these limitations; through its MHC-unrestricted detection of moieties associated with cellular stress. As such, there is an opportunity to “tune” the CAR-T cell response by modulating the level of stimulus delivered by the CAR. So far, this has been demonstrated in the context of neuroblastoma, against which Vγ9Vδ2+ T cells have minimal innate cytotoxicity (37), in part due to the tumor shedding soluble NKG2D ligands which block NKG2D activation (97, 98). If further stimulus is provided to the γδT cell, this cytotoxicity can be restored, either via a conventional second-generation CAR (90, 99) (Figure 1D), opsonization of the target cell (34, 37) or, as was recently shown, by restoring the NKG2D signal using a chimeric costimulatory receptor (CCR), that lacks CD3ζ, but contains the endodomain motif from the NKG2D adaptor, DAP10 (GD2-DAP10, Figure 1E). This approach enhanced killing of GD2+ neuroblastoma cells but did not induce cytotoxicity against GD2+ cells that did not engage the Vγ9Vδ2 TCR. Cytokine release was also controllable using this “AND gate” system; IL-2, IFNγ, and TNFα were only released from GD2-DAP10+Vδ2+ cells when they received both CD3 and CCR stimulus, whereas in GD2-28ζ+Vδ2+ cells, only CAR stimulus was required (99).
Chimeric costimulatory receptors have also been used in the context of αβT cells. They can deliver an isolated costimulatory signal to support antigen-specific proliferation (100), enhance tumor specificity by dividing CAR and CCR stimuli such that two antigens are required for activation (101), or reverse the suppressive effects of tumor PD-L1 through a PD1-CD28 chimeric receptor (102). In two of these studies, a separate CD3 signal was provided, either using OKT3 anti-CD3 (102), or a separate CD3ζ containing CAR (101). The earlier work by Krause et al. was particularly innovative; a chimeric anti-GD2 receptor with a CD28 endodomain supported antigen-specific proliferation of αβT cells in a TCR or CD3-dependent manner. When tested in the context of GD2+/− tumor cells, they also confirmed that signal 1 could be provided by the TCR (100) and that their CCR would function under these conditions. The promiscuous, MHC-independent reactivity of γδT cells to danger-associated molecular patterns rather than MHC-restricted peptide epitopes could offer an opportunity to broaden this approach.
In addition to the possibility of avoiding on-target off-tumor toxicity, CAR expressing γδT cells retain the ability to antigen to cross-present (53, 90, 103–105). A recent study indicated that γδT cells transduced with second-generation anti-GD2 CARs (GD2-28ζ) retain the ability to cross-present TAAs leading to a clonal expansion of αβT cells. Using a 25 amino acid fragment of the melanoma antigen MART-1 which encompasses a 10 amino acid epitope but is too long to be MHC-presented in its un-processed form, Capsomidis et al. demonstrated that HLA-A201+V2δ+GD2-28ζ+ cells pulsed with the long peptide were able to elicit secondary expansions in αβT cells expressing a HLA-A201-restricted MART-1 αβ TCR (90). Vδ2+GD2-28ζ+ cells also retained the ability to migrate toward tumor cell lines; GD2-28ζ expression had no effect on migration in either Vδ1+ or Vδ2+ subsets in an in vitro trans-well assay. The next step in these investigations would be to show that γδT cells can cross-present antigens derived from cells that they have themselves killed. If this were successful it would raise the possibility that the broad anti-tumor reactivity of γδT cells could be used to prime a diverse population of autologous αβT cells against many tumor-derived antigens simultaneously. Demonstration of enhanced anti-tumor activity by “γδT-cell primed” αβT cells would further validate this approach.
Transduction Strategies for γδT Cells
When engineering γδT cells it is important to select appropriate tools. In general, long-lasting transduction strategies which work well for αβT cells work well for γδT cells also. The predicted shorter lifespan of infused γδT cells offer the opportunity to use more transient engineering approaches as well, as the infused cells may not persist in the host long-term.
Many groups continue to use gammaretroviral vectors for transducing γδT cells. High transduction efficiencies are achievable using a Maloney murine leukemia virus-based vector, SFG (87), pseudotyped with the envelope of the feline endogenous retrovirus RD114 (99), or gibbon-ape leukemia virus envelopes. Gammaretroviral transduction has the advantage of allowing preparation of large, high titer batches of virus because of the availability of packaging cell lines which can be stably transduced to produce virus containing the construct of interest (106). Gammaretroviruses, lacking the machinery to penetrate the nucleus, require the cells to be actively cycling in order to achieve transduction as viral nucleic acids can enter through nuclear pores (107). This is not a restriction in the engineering of αβT cells, and the specific and rapid expansion of Vγ9Vδ2+ T cells in response to aminobisphosphonates allows for similar strategies to be applied. Transduction of other γδT cell subsets using gammaretroviruses following concanvaulin A driven expansion is less predictable, however, with variable yield (90). There has been some concern regarding the potential for insertion-site-mediated mutagenesis following gammaretroviral gene transfer (108), which has prompted some in the field to favor lentiviral vectors which have a safer insertional profile (107). There is little published data to compare lentiviral transduction techniques for γδT cells, though one group did find that the use of a vesicular stomatitis Indiana virus G-protein containing envelope in combination with a simian immunodeficiency virus transfer vector consistently provided higher transduction efficiency than a human immunodeficiency virus-based vector with the same envelope (transduction efficiency 65% vs 42%, p = 0.04) (109). New genome editing technologies, such as CRISPR-CAS, allow targeting integration of viral vectors with several potential advantages including avoidance of integration into oncogenic loci, and integration into loci that optimize CAR or TCR expression (11, 110).
Non-viral methods of transduction have provided particular advantages when engineering γδT cells. The Sleeping Beauty Transposon system (111, 112) uses enzymes originally derived from fish to insert new genetic material into host cells. It has not yet been established that the SB-transposon system is more efficacious than lentiviral transduction, and there is an unknown potential for insertional mutagenesis (113). Cells must be electroporated in order for gene transfer to occur, but do not require a specific proliferative stimulus. As described above, Deniger et al. used this to good effect to express an anti-CD19 CAR (CD19RCD28) in a polyclonal repertoire of γδT cells which were subsequently expanded using CD19+ aAPCs (91, 93). Unlike proliferation-driven transduction techniques, there was no “skewing” of the γδT cell population toward a particular Vγ/Vδ subset. This may be of particular interest if engineered γδT cells were to be directed against epithelial tumors, as non-Vδ2+ γδT cells are enriched in epithelial surfaces, a tropism which could be harnessed.
Viral or transposon-based gene transfer generates stable construct expression over time by integrating into the transduced cell genome. This has been considered important in the CAR-T cell field as it allows for persistence of CAR-T cells for weeks or months. More transient CAR expression strategies have been suggested as a means of reducing toxicity following CAR-T cell infusion. mRNA transfection using electroporation was used to generate γδT cells expressing NKT-cell derived TCRs (79) and, more recently, HLA-A2/gp100-specific TCR or CARs targeting melanoma-associated-chondroitin-sulfate-proteoglycan (MCSP). MCSP is a tumor-associated antigen expressed on melanoma (114), glioma (115), triple-negative breast cancer (116), and sarcomas (117). Expression peaked at around 24 h after transfection, returning to baseline by around 72 h (77). Similar techniques have been tested clinically in the context of CAR+ αβT cells, where they were used to transfect cells with a construct targeting mesothelin (118), though repeated infusions of CAR-T cells were required, presumably because of the need to “top-up” the reservoir of circulating CAR-T cells as expression was lost.
Concluding Comments
While there is promising data to suggest that gene-modified γδT cells may be an attractive candidate for clinical studies, the bulk of enthusiasm in the cellular immunotherapy field focuses on αβT cells. Undoubtedly, more data are available on αβT cell engineering, increasing the likelihood of introducing novel constructs to the clinic. γδT cells appear to cause less graft-versus-host disease than αβT cells while retaining graft-versus leukemia activity in the hematopoietic stem-cell transplant setting (119). In vivo data on the function of CCR expressing γδT cells and/or the antigen cross-presentation capacity of γδT cells are not yet available. Reduced toxicity and the potential for antigen cross-presentation are compelling arguments for the potential benefits of γδT cells over αβT cells as a substrate for CAR expression. The difficulty of modeling the more subtle aspects human γδT cell activity in a murine system makes this data particularly hard to generate. However, within the increasingly crowded field of cancer immunotherapy, orthogonal approaches to cellular engineering are required to move the field forward. The challenges of off-tumor toxicity, poor penetration of solid tumors and tumor immune evasion need to be addressed. It is no surprise that CD19 CAR-T therapies have been more successful than others, depletion of healthy CD19+ B-cells is considered an acceptable toxicity and the disease resides in the hematological compartment. In other cases, on-target off-tumor toxicity has been severe or fatal, experiences which have shaped the way that target antigens are chosen (20, 21). Investigating the potential of alternative CAR “chassis” to harness the innate characteristics of particular cell types factorially increases the number of options available. As cells sharing adaptive properties of conventional αβT-cells and innate properties of NK-cells, γδT cells are a highly attractive and potentially efficient candidate for this process of optimization.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
JA holds stock in Autolus PLC and JA and JF perform consulting work for TC Biopharm.
Funding
JA receives funding from the NIHR Great Ormond Street Biomedical Resaerch Centre, Research In Childhood Cancer, and Great Ormond Street Hospital Charity. JF is funded by a Wellcome Trust Post doctoral clinical fellowship (Grant no. 110022/Z/15/Z).
References
1. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood (2010) 116:4099–102. doi:10.1182/blood-2010-04-281931
2. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371:1507–17. doi:10.1056/NEJMoa1407222
3. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood (2015) 125:4017–23. doi:10.1182/blood-2014-12-580068
4. Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood (2012) 119:3940–50. doi:10.1182/blood-2011-10-387969
5. Brentjens RJ, Brentjens RJ, Davila ML, Davila ML, Rivière I, Rivière I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5:ra38–177. doi:10.1126/scitranslmed.3005930
6. Zhang L, Morgan RA. Genetic engineering with T cell receptors. Adv Drug Deliv Rev (2012) 64:756–62. doi:10.1016/j.addr.2011.11.009
7. Karpanen T, Olweus J. T-cell receptor gene therapy – ready to go viral? Mol Oncol (2015) 9:2019–42. doi:10.1016/j.molonc.2015.10.006
8. Morris EC, Stauss HJ. Optimizing T-cell receptor gene therapy for hematologic malignancies. Blood (2016) 127:3305–11. doi:10.1182/blood-2015-11-629071
9. Johnson LA, June CH. Driving gene-engineered T cell immunotherapy of cancer. Cell Res (2016) 27:38–58. doi:10.1038/cr.2016.154
10. Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature (2017) 545:423–31. doi:10.1038/nature22395
11. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature (2017) 543:113–7. doi:10.1038/nature21405
12. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med (2018) 378:449–59. doi:10.1056/NEJMoa1709919
13. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RPT, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol (2015) 33:540–9. doi:10.1200/JCO.2014.56.2025
14. Davila ML, Rivière I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med (2014) 6:ra25–224. doi:10.1126/scitranslmed.3008226
15. Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol (2016) 39:44–51. doi:10.1016/j.coi.2015.12.007
16. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res (2015) 21:687–92. doi:10.1158/1078-0432.CCR-14-1860
17. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood (2013) 122:863–71. doi:10.1182/blood-2013-03-490565
18. Lu Y-C, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, et al. Treatment of patients with metastatic cancer using a major histocompatibility complex class II-restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. J Clin Oncol (2017) 35:3322–9. doi:10.1200/JCO.2017.74.5463
19. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood (2012) 119:2709–20. doi:10.1182/blood-2011-10-384388
20. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18:843–51. doi:10.1038/mt.2010.24
21. Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol (2006) 24:e20–2. doi:10.1200/JCO.2006.05.9964
22. Ruella M, Maus MV. Catch me if you can: leukemia escape after CD19-directed T cell immunotherapies. Comput Struct Biotechnol J (2016) 14:357–62. doi:10.1016/j.csbj.2016.09.003
23. Gammaitoni L, Giraudo L, Macagno M, Leuci V, Mesiano G, Rotolo R, et al. Cytokine-induced killer cells kill chemo-surviving melanoma cancer stem cells. Clin Cancer Res (2017) 23:2277–88. doi:10.1158/1078-0432.CCR-16-1524
24. Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol (2013) 10:230–52. doi:10.1038/cmi.2013.10
25. Boissel L, Betancur M, Wels WS, Tuncer H, Klingemann H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk Res (2009) 33:1255–9. doi:10.1016/j.leukres.2008.11.024
26. Müller T, Uherek C, Maki G, Chow KU, Schimpf A, Klingemann H-G, et al. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol Immunother (2008) 57:411–23. doi:10.1007/s00262-007-0383-3
27. Esser R, Müller T, Stefes D, Kloess S, Seidel D, Gillies SD, et al. NK cells engineered to express a GD2-specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med (2012) 16:569–81. doi:10.1111/j.1582-4934.2011.01343.x
28. Schönfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther (2015) 23:330–8. doi:10.1038/mt.2014.219
29. Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer (2016) 16:7–19. doi:10.1038/nrc.2015.5
30. Davis ZB, Vallera DA, Miller JS, Felices M. Natural killer cells unleashed: checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin Immunol (2017) 31:64–75. doi:10.1016/j.smim.2017.07.011
31. Tian G, Courtney AN, Jena B, Heczey A, Liu D, Marinova E, et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J Clin Invest (2016) 126:2341–55. doi:10.1172/JCI83476
32. Fisher JPH, Heuijerjans J, Yan M, Gustafsson K, Anderson J. γδ T cells for cancer immunotherapy: a systematic review of clinical trials. Oncoimmunology (2014) 3:e27572. doi:10.4161/onci.27572
33. Braza MS, Klein B. Anti-tumour immunotherapy with Vγ9Vδ2 T lymphocytes: from the bench to the bedside. Br J Haematol (2012) 160:123–32. doi:10.1111/bjh.12090
34. Fisher JPH, Flutter B, Wesemann F, Frosch J, Rossig C, Gustafsson K, et al. Effective combination treatment of GD2-expressing neuroblastoma and Ewing’s sarcoma using anti-GD2 ch14.18/CHO antibody with Vγ9Vδ2+ γδT cells. Oncoimmunology (2016) 5:e1025194. doi:10.1080/2162402X.2015.1025194
35. Fournié J-J, Sicard H, Poupot M, Bezombes C, Blanc A, Romagné F, et al. What lessons can be learned from γδ T cell-based cancer immunotherapy trials? Cell Mol Immunol (2013) 10:35–41. doi:10.1038/cmi.2012.39
36. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med (2015) 21:938–45. doi:10.1038/nm.3909
37. Fisher J, Yan M, Heuijerjans J, Carter L, Abolhassani A, Frosch J, et al. Neuroblastoma killing properties of V-delta 2 and V-delta2 negative gamma delta T cells following expansion by artificial antigen presenting cells. Clin Cancer Res (2014) 20(22):5720–32. doi:10.1158/1078-0432.CCR-13-3464
38. Fahl SP, Coffey F, Wiest DL. Origins of γδ T cell effector subsets: a riddle wrapped in an enigma. J Immunol (2014) 193:4289–94. doi:10.4049/jimmunol.1401813
39. Parker CM, Groh V, Band H, Porcelli SA, Morita C, Fabbi M, et al. Evidence for extrathymic changes in the T cell receptor gamma/delta repertoire. J Exp Med (1990) 171:1597–612. doi:10.1084/jem.171.5.1597
40. Dimova T, Brouwer M, Gosselin F, Tassignon J, Leo O, Donner C, et al. Effector Vγ9Vδ2 T cells dominate the human fetal γδ T-cell repertoire. Proc Natl Acad Sci U S A (2015) 112:E556–65. doi:10.1073/pnas.1412058112
41. Ravens S, Schultze-Florey C, Raha S, Sandrock I, Drenker M, Oberdörfer L, et al. Human γδ T cells are quickly reconstituted after stem-cell transplantation and show adaptive clonal expansion in response to viral infection. Nat Immunol (2017) 18:393–401. doi:10.1038/ni.3686
42. Lefranc MP. Nomenclature of the human T cell receptor genes. Curr Protoc Immunol (2001) Appendix 1:Appendix 1O. doi:10.1002/0471142735.ima01os40
43. Pauza CD, Cairo C. Evolution and function of the TCR Vgamma9 chain repertoire: it’s good to be public. Cell Immunol (2015) 296(1):22–30. doi:10.1016/j.cellimm.2015.02.010
44. Hviid L, Akanmori BD, Loizon S, Kurtzhals JA, Ricke CH, Lim A, et al. High frequency of circulating gamma delta T cells with dominance of the v(delta)1 subset in a healthy population. Int Immunol (2000) 12:797–805. doi:10.1093/intimm/12.6.797
45. Poupot M, Fournié J-J. Non-peptide antigens activating human Vgamma9/Vdelta2 T lymphocytes. Immunol Lett (2004) 95:129–38. doi:10.1016/j.imlet.2004.06.013
46. Asslan R, Pradines A, Pratx C, Allal C, Favre G, Le Gaillard F. Epidermal growth factor stimulates 3-hydroxy-3-methylglutaryl-coenzyme A reductase expression via the ErbB-2 pathway in human breast adenocarcinoma cells. Biochem Biophys Res Commun (1999) 260:699–706. doi:10.1006/bbrc.1999.0945
47. Vantourout P, Hayday A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat Rev Immunol (2013) 13:88–100. doi:10.1038/nri3384
48. Benzaïd I, Mönkkönen H, Bonnelye E, Mönkkönen J, Clézardin P. In vivo phosphoantigen levels in bisphosphonate-treated human breast tumors trigger Vγ9Vδ2 T-cell antitumor cytotoxicity through ICAM-1 engagement. Clin Cancer Res (2012) 18:6249–59. doi:10.1158/1078-0432.CCR-12-0918
49. Cabillic F, Toutirais O, Lavoué V, La Pintière de CT, Daniel P, Rioux-Leclerc N, et al. Aminobisphosphonate-pretreated dendritic cells trigger successful Vgamma9Vdelta2 T cell amplification for immunotherapy in advanced cancer patients. Cancer Immunol Immunother (2010) 59:1611–9. doi:10.1007/s00262-010-0887-0
50. Belmant C, Decise D, Fournié J-J. Phosphoantigens and aminobisphosphonates: new leads targeting γδ T lymphocytes for cancer immunotherapy. Drug Discov Today Ther Strateg (2006) 3:17–23. doi:10.1016/j.ddstr.2006.02.001
51. Luckman SP, Hughes DE, Coxon FP, Graham R, Russell G, Rogers MJ. Nitrogen-containing bisphosphonates inhibit the mevalonate pathway and prevent post-translational prenylation of GTP-binding proteins, including Ras. J Bone Miner Res (1998) 13:581–9. doi:10.1359/jbmr.1998.13.4.581
52. Thompson K, Rogers MJ. Statins prevent bisphosphonate-induced gamma,delta-T-cell proliferation and activation in vitro. J Bone Miner Res (2004) 19:278–88. doi:10.1359/JBMR.0301230
53. Himoudi N, Morgenstern DA, Yan M, Vernay B, Saraiva L, Wu Y, et al. Human γδT lymphocytes are licensed for professional antigen presentation by interaction with opsonized target cells. J Immunol (2012) 188:1708–16. doi:10.4049/jimmunol.1102654
54. Barisa M, Kramer AM, Majani Y, Moulding D, Saraiva L, Bajaj-Elliott M, et al. E. coli promotes human Vγ9Vδ2 T cell transition from cytokine-producing bactericidal effectors to professional phagocytic killers in a TCR-dependent manner. Sci Rep (2017) 7:15. doi:10.1038/s41598-017-02886-8
55. Scotet E, Martinez LO, Grant E, Barbaras R, Jenö P, Guiraud M, et al. Tumor recognition following Vgamma9Vdelta2 T cell receptor interactions with a surface F1-ATPase-related structure and apolipoprotein A-I. Immunity (2005) 22:71–80. doi:10.1016/j.immuni.2004.11.012
56. Mookerjee-Basu J, Vantourout P, Martinez LO, Perret B, Collet X, Périgaud C, et al. F1-adenosine triphosphatase displays properties characteristic of an antigen presentation molecule for Vgamma9Vdelta2 T cells. J Immunol (2010) 184:6920–8. doi:10.4049/jimmunol.0904024
57. Vavassori S, Kumar A, Wan GS, Ramanjaneyulu GS, Cavallari M, Daker El S, et al. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human γδ T cells. Nat Immunol (2013) 14:908–16. doi:10.1038/ni.2665
58. Di Marco Barros R, Roberts NA, Dart RJ, Vantourout P, Jandke A, Nussbaumer O, et al. Epithelia use butyrophilin-like molecules to shape organ-specific γδ T cell compartments. Cell (2016) 167:203–218.e17. doi:10.1016/j.cell.2016.08.030
59. Hayday AC. γδ T cells and the lymphoid stress-surveillance response. Immunity (2009) 31:184–96. doi:10.1016/j.immuni.2009.08.006
60. Lawand M, Déchanet-Merville J, Dieu-Nosjean M-C. Key features of gamma-delta T-cell subsets in human diseases and their immunotherapeutic implications. Front Immunol (2017) 8:761. doi:10.3389/fimmu.2017.00761
61. Nedellec S, Sabourin C, Bonneville M, Scotet E. NKG2D costimulates human V gamma 9V delta 2 T cell antitumor cytotoxicity through protein kinase C theta-dependent modulation of early TCR-induced calcium and transduction signals. J Immunol (2010) 185:55–63. doi:10.4049/jimmunol.1000373
62. Gertner-Dardenne J, Castellano R, Mamessier E, Garbit S, Kochbati E, Etienne A, et al. Human Vγ9Vδ2 T cells specifically recognize and kill acute myeloid leukemic blasts. J Immunol (2012) 188:4701–8. doi:10.4049/jimmunol.1103710
63. Gertner-Dardenne J, Bonnafous C, Bezombes C, Capietto AH, Scaglione V, Ingoure S, et al. Bromohydrin pyrophosphate enhances antibody-dependent cell-mediated cytotoxicity induced by therapeutic antibodies. Blood (2009) 113:4875–84. doi:10.1182/blood-2008-08-172296
64. Spada FM, Grant EP, Peters PJ, Sugita M, Melián A, Leslie DS, et al. Self-recognition of CD1 by gamma/delta T cells: implications for innate immunity. J Exp Med (2000) 191:937–48. doi:10.1084/jem.191.6.937
65. Bai L, Picard D, Anderson B, Chaudhary V, Luoma A, Jabri B, et al. The majority of CD1d-sulfatide-specific T cells in human blood use a semiinvariant Vδ1 TCR. Eur J Immunol (2012) 42:2505–10. doi:10.1002/eji.201242531
66. Uldrich AP, Le Nours J, Pellicci DG, Gherardin NA, McPherson KG, Lim RT, et al. CD1d-lipid antigen recognition by the γδ TCR. Nat Immunol (2013) 14:1137–45. doi:10.1038/ni.2713
67. Xu B, Pizarro JC, Holmes MA, McBeth C, Groh V, Spies T, et al. Crystal structure of a gammadelta T-cell receptor specific for the human MHC class I homolog MICA. Proc Natl Acad Sci U S A (2011) 108:2414–9. doi:10.1073/pnas.1015433108
68. Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci U S A (1999) 96:6879–84. doi:10.1073/pnas.96.12.6879
69. Dembić Z, Haas W, Weiss S, McCubrey J, Kiefer H, Boehmer von H, et al. Transfer of specificity by murine alpha and beta T-cell receptor genes. Nature (1986) 320:232–8. doi:10.1038/320232a0
70. Cooper LJ, Kalos M, Lewinsohn DA, Riddell SR, Greenberg PD. Transfer of specificity for human immunodeficiency virus type 1 into primary human T lymphocytes by introduction of T-cell receptor genes. J Virol (2000) 74:8207–12. doi:10.1128/JVI.74.17.8207-8212.2000
71. Clay TM, Custer MC, Sachs J, Hwu P, Rosenberg SA, Nishimura MI. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol (1999) 163:507–13.
72. Berdien B, Mock U, Atanackovic D, Fehse B. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther (2014) 21:539–48. doi:10.1038/gt.2014.26
73. Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med (2010) 16:565–70, 1p following 570. doi:10.1038/nm.2128
74. Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res (2006) 66:8878–86. doi:10.1158/0008-5472.CAN-06-1450
75. Davis JL, Theoret MR, Zheng Z, Lamers CHJ, Rosenberg SA, Morgan RA. Development of human anti-murine T-cell receptor antibodies in both responding and nonresponding patients enrolled in TCR gene therapy trials. Clin Cancer Res (2010) 16:5852–61. doi:10.1158/1078-0432.CCR-10-1280
76. Dörrie J, Krug C, Hofmann C, Müller I, Wellner V, Knippertz I, et al. Human adenovirus-specific γ/δ and CD8+ T cells generated by T-cell receptor transfection to treat adenovirus infection after allogeneic stem cell transplantation. PLoS One (2014) 9:e109944. doi:10.1371/journal.pone.0109944
77. Harrer DC, Simon B, Fujii S-I, Shimizu K, Uslu U, Schuler G, et al. RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: a safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer (2017) 17:551. doi:10.1186/s12885-017-3539-3
78. Marcu-Malina V, Heijhuurs S, van Buuren M, Hartkamp L, Strand S, Sebestyen Z, et al. Redirecting αβ T cells against cancer cells by transfer of a broadly tumor-reactive γδT-cell receptor. Blood (2011) 118:50–9. doi:10.1182/blood-2010-12-325993
79. Shimizu K, Shinga J, Yamasaki S, Kawamura M, Dörrie J, Schaft N, et al. Transfer of mRNA encoding invariant NKT cell receptors imparts glycolipid specific responses to T cells and γδT cells. PLoS One (2015) 10:e0131477. doi:10.1371/journal.pone.0131477
80. Allison TJ, Winter CC, Fournie JJ, Bonneville M, Garboczi DN. Structure of a human gammadelta T-cell antigen receptor. Nature (2001) 411:820–4. doi:10.1038/35081115
81. Gründer C, van Dorp S, Hol S, Drent E, Straetemans T, Heijhuurs S, et al. γ9 and δ2CDR3 domains regulate functional avidity of T cells harboring γ9δ2TCRs. Blood (2012) 120:5153–62. doi:10.1182/blood-2012-05-432427
82. Straetemans T, Gründer C, Heijhuurs S, Hol S, Slaper-Cortenbach I, Bönig H, et al. Untouched GMP-ready purified engineered immune cells to treat cancer. Clin Cancer Res (2015) 21:3957–68. doi:10.1158/1078-0432.CCR-14-2860
83. Rischer M, Pscherer S, Duwe S, Vormoor J, Jurgens H, Rossig C. Human gammadelta T cells as mediators of chimaeric-receptor redirected anti-tumour immunity. Br J Haematol (2004) 126:583–92. doi:10.1111/j.1365-2141.2004.05077.x
84. Kailayangiri S, Altvater B, Meltzer J, Pscherer S, Luecke A, Dierkes C, et al. The ganglioside antigen GD2 is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br J Cancer (2012) 106:1123–33. doi:10.1038/bjc.2012.57
85. Schulz G, Cheresh DA, Varki NM, Yu A, Staffileno LK, Reisfeld RA. Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma patients. Cancer Res (1984) 44:5914–20.
86. L’Faqihi FE, Guiraud M, Dastugue N, Brousset P, Le Bouteiller P, Halary F, et al. Acquisition of a stimulatory activity for Vgamma9/Vdelta2 T cells by a Burkitt’s lymphoma cell line without loss of HLA class I expression. Hum Immunol (1999) 60:928–38. doi:10.1016/S0198-8859(99)00076-2
87. Rivière I, Brose K, Mulligan RC. Effects of retroviral vector design on expression of human adenosine deaminase in murine bone marrow transplant recipients engrafted with genetically modified cells. Proc Natl Acad Sci U S A (1995) 92:6733–7. doi:10.1073/pnas.92.15.6733
88. Siegers GM, Ribot EJ, Keating A, Foster PJ. Extensive expansion of primary human gamma delta T cells generates cytotoxic effector memory cells that can be labeled with Feraheme for cellular MRI. Cancer Immunol Immunother (2013) 62(3):571–83. doi:10.1007/s00262-012-1353-y
89. Siegers GM, Dhamko H, Wang X-H, Mathieson AM, Kosaka Y, Felizardo TC, et al. Human Vδ1 γδ T cells expanded from peripheral blood exhibit specific cytotoxicity against B-cell chronic lymphocytic leukemia-derived cells. Cytotherapy (2011) 13:753–64. doi:10.3109/14653249.2011.553595
90. Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, et al. Chimeric antigen receptor-engineered human gamma delta T cells: enhanced cytotoxicity with retention of cross presentation. Mol Ther (2018) 26:354–65. doi:10.1016/j.ymthe.2017.12.001
91. Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC, et al. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol Ther (2007) 15:981–8. doi:10.1038/mt.sj.6300134
92. Deniger DC, Maiti S, Mi T, Switzer K, Ramachandran V, Hurton LV, et al. Activating and propagating polyclonal gamma delta T cells with broad specificity for malignancies. Clin Cancer Res (2014) 20:5708–19. doi:10.1158/1078-0432.CCR-13-3451
93. Deniger DC, Switzer K, Mi T, Maiti S, Hurton L, Singh H, et al. Bispecific T-cells expressing polyclonal repertoire of endogenous γδ T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol Ther (2013) 21:638–47. doi:10.1038/mt.2012.267
94. Singh H, Figliola MJ, Dawson MJ, Huls H, Olivares S, Switzer K, et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res (2011) 71:3516–27. doi:10.1158/0008-5472.CAN-10-3843
95. Kato Y, Tanaka Y, Tanaka H, Yamashita S, Minato N. Requirement of species-specific interactions for the activation of human gamma delta T cells by pamidronate. J Immunol (2003) 170:3608–13. doi:10.4049/jimmunol.170.7.3608
96. Green AE, Lissina A, Hutchinson SL, Hewitt RE, Temple B, James D, et al. Recognition of nonpeptide antigens by human Vγ9Vδ2 T cells requires contact with cells of human origin. Clin Exp Immunol (2004) 136:472–82. doi:10.1111/j.1365-2249.2004.02472.x
97. Raffaghello L, Prigione I, Airoldi I, Camoriano M, Levreri I, Gambini C, et al. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia (2004) 6:558. doi:10.1593/neo.04316
98. Raffaghello L, Prigione I, Airoldi I, Camoriano M, Morandi F, Bocca P, et al. Mechanisms of immune evasion of human neuroblastoma. Cancer Lett (2005) 228:155–61. doi:10.1016/j.canlet.2004.11.064
99. Fisher J, Abramowski P, Wisidagamage Don ND, Flutter B, Capsomidis A, Cheung GW-K, et al. Avoidance of on-target off-tumor activation using a co-stimulation-only chimeric antigen receptor. Mol Ther (2017) 25(5):1234–47. doi:10.1016/j.ymthe.2017.03.002
100. Krause A, Guo H-F, Latouche J-B, Tan C, Cheung N-KV, Sadelain M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J Exp Med (1998) 188:619–26. doi:10.1084/jem.188.4.619
101. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol (2013) 31:71–5. doi:10.1038/nbt.2459
102. Prosser ME, Brown CE, Shami AF, Forman SJ, Jensen MC. Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol Immunol (2012) 51:263–72. doi:10.1016/j.molimm.2012.03.023
103. Anderson J, Gustafsson K, Himoudi N, Yan M, Heuijerjans J. Licensing of γδT cells for professional antigen presentation: a new role for antibodies in regulation of antitumor immune responses. Oncoimmunology (2012) 1:1652–4. doi:10.4161/onci.21971
104. Moser B, Eberl M. γδ T-APCs: a novel tool for immunotherapy? Cell Mol Life Sci (2011) 68:2443–52. doi:10.1007/s00018-011-0706-6
105. Brandes M, Willimann K, Moser B. Professional antigen-presentation function by human gammadelta T cells. Science (2005) 309:264–8. doi:10.1126/science.1110267
106. Danos O, Mulligan RC. Safe and efficient generation of recombinant retroviruses with amphotropic and ecotropic host ranges. Proc Natl Acad Sci U S A (1988) 85:6460–4. doi:10.1073/pnas.85.17.6460
107. Biasco L, Rothe M, Schott JW, Schambach A. Integrating vectors for gene therapy and clonal tracking of engineered hematopoiesis. Hematol Oncol Clin North Am (2017) 31:737–52. doi:10.1016/j.hoc.2017.06.009
108. Bosticardo M, Ghosh A, Du Y, Jenkins NA, Copeland NG, Candotti F. Self-inactivating retroviral vector-mediated gene transfer induces oncogene activation and immortalization of primary murine bone marrow cells. Mol Ther (2009) 17:1910–8. doi:10.1038/mt.2009.172
109. Lamb LS, Bowersock J, Dasgupta A, Gillespie GY, Su Y, Johnson A, et al. Engineered drug resistant γδ T cells kill glioblastoma cell lines during a chemotherapy challenge: a strategy for combining chemo- and immunotherapy. PLoS One (2013) 8:e51805. doi:10.1371/journal.pone.0051805
110. MacLeod DT, Antony J, Martin AJ, Moser RJ, Hekele A, Wetzel KJ, et al. Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T cells. Mol Ther (2017) 25:949–61. doi:10.1016/j.ymthe.2017.02.005
111. Ivics Z, Hackett PB, Plasterk RH, Izsvák Z. Molecular reconstruction of sleeping beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell (1997) 91:501–10. doi:10.1016/S0092-8674(00)80436-5
112. Aronovich EL, McIvor RS, Hackett PB. The sleeping beauty transposon system: a non-viral vector for gene therapy. Hum Mol Genet (2011) 20:R14–20. doi:10.1093/hmg/ddr140
113. Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev (2017) 4:92–101. doi:10.1016/j.omtm.2016.12.006
114. Campoli MR, Chang C-C, Kageshita T, Wang X, McCarthy JB, Ferrone S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): a melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit Rev Immunol (2004) 24:267–96. doi:10.1615/CritRevImmunol.v24.i4.40
115. Yadavilli S, Hwang EI, Packer RJ, Nazarian J. The role of NG2 proteoglycan in glioma. Transl Oncol (2016) 9:57–63. doi:10.1016/j.tranon.2015.12.005
116. Wang X, Osada T, Wang Y, Yu L, Sakakura K, Katayama A, et al. CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J Natl Cancer Inst (2010) 102:1496–512. doi:10.1093/jnci/djq343
117. Godal A, Bruland O, Haug E, Aas M, Fodstad O. Unexpected expression of the 250 kD melanoma-associated antigen in human sarcoma cells. Br J Cancer (1986) 53:839–41. doi:10.1038/bjc.1986.142
118. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res (2014) 2:112–20. doi:10.1158/2326-6066.CIR-13-0170
Keywords: gamma delta, chimeric antigen receptor, adoptive transfer, alpha beta T cells, cancer immunotherapy
Citation: Fisher J and Anderson J (2018) Engineering Approaches in Human Gamma Delta T Cells for Cancer Immunotherapy. Front. Immunol. 9:1409. doi: 10.3389/fimmu.2018.01409
Received: 09 March 2018; Accepted: 06 June 2018;
Published: 26 June 2018
Edited by:
Thomas Herrmann, Universität Würzburg, GermanyReviewed by:
Martin Wilhelm, Klinikum Nürnberg, GermanyDrew C. Deniger, National Institutes of Health (NIH), United States
Copyright: © 2018 Fisher and Anderson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Fisher, am9uYXRoYW4uZmlzaGVyJiN4MDAwNDA7dWNsLmFjLnVr