 Shuhei Hosomi
Shuhei Hosomi- 1Division of Gastroenterology, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, United States
- 2Department of Gastroenterology, Osaka City University Graduate School of Medicine, Osaka, Japan
- 3Department of Gastroenterology and Hepatology, Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 4Division of Gastroenterology and Hepatology, Department of Medicine, University of Cambridge, Addenbrooke’s Hospital, Cambridge, United Kingdom
Natural-killer group 2 member D (NKG2D) is a well-characterized activating receptor expressed by natural killer (NK) cells, NKT cells, activated CD8+ T cells, subsets of γδ+ T cells, and innate-like T cells. NKG2D recognizes multiple ligands (NKG2D-ligands) to mount an innate immune response against stressed, transformed, or infected cells. NKG2D-ligand surface expression is tightly restricted on healthy cells through transcriptional and post-transcriptional mechanisms, while transformed or infected cells express the ligands as a danger signal. Recent studies have revealed that unfolded protein response pathways during endoplasmic reticulum (ER) stress result in upregulation of ULBP-related protein via the protein kinase RNA-like ER kinase-activating factor 4-C/EBP homologous protein (PERK-ATF4-CHOP) pathway, which can be linked to the pathogenesis of autoimmune diseases. Transformed cells, however, possess mechanisms to escape NKG2D-mediated immune surveillance, such as upregulation of carcinoembryonic antigen (CEA)-related cell adhesion molecule 1 (CEACAM1), a negative regulator of NKG2D-ligands. In this review, we discuss mechanisms of NKG2D-ligand regulation, with a focus on newly discovered mechanisms that promote NKG2D-ligand expression on epithelial cells, including ER stress, and mechanisms that suppress NKG2D-ligand-mediated killing of cancer cells, namely by co-expression of CEACAM1.
Introduction
Natural killer (NK) cells were originally identified as lymphocytes with cytotoxic reactivity against several types of cancer cells (1, 2), and are considered to be part of the innate immune cell compartment/family/pool because of lack of rearranged antigen-specific receptors by somatic recombination. Both activating and inhibitory receptors can regulate NK cells activity and recognize target cells (3). Inhibitory receptors, including killer cell immunoglobulin (Ig)-like receptors (KIRs) and leukocyte Ig-like receptors in humans, Ly49s in mouse, and CD94-Natural-killer group 2 member A receptors in human and mouse, recognize major histocompatibility complex (MHC) class I. If NK cells encounter cells that express MHC class I, an immune response against these cells is prevented by the inhibitory signals through receptor–ligand interactions. In contrast, cells in which MHC class I is downregulated, for example, in virus-infected cells or cancer cells, NK cells are activated by the lack of inhibitory signals, which makes the “diseased” cells prone to NK cell-mediated killing (the missing-self hypothesis) (4). The inhibitory function is mediated through immunoreceptor tyrosine-based inhibitory motif (ITIM) in the cytoplasmic domain of these inhibitory receptors (5).
Besides inhibitory receptors, numerous activating NK receptors, such as CD94-Natural-killer group 2 member C, natural-killer group 2 member D (NKG2D), NKp44, NKp46, KIRs in humans, Ly49 receptors, in mice have been identified (5). The combination of the activation receptors can synergistically mediate natural cytotoxicity (6). NKG2D, a type II transmembrane-anchored C-type lectin-like activating receptor, is a well-characterized activation receptor expressed by NK cells, NKT cells, activated CD8+ T cells, subsets of γδ+ T cells, and innate-like T cells, which are TCR+ NK1.1+ CD49ahigh CD103+ tissue-resident T lymphocytes with innate cytolytic activities, transcriptionally related to ILC1 (7–11). NKG2D can recognize multiple ligands (NKG2D-ligands), which are homologous to MHC class I molecules; MHC class I chain-related proteins A (MICA), MICB, UL16 binding protein 1 (ULBP1)–ULBP6 in human; retinoic acid early inducible 1 (RAE-1) (isoforms α–ε), H60 (isoforms a–c), and murine UL16-binding protein like transcript 1 (MULT1) in mouse (12). Interaction of these ligands with NKG2D results in NK cell cytotoxicity via signal transducing adapter molecule DAP10 in human and both DAP10 and DAP12 in mouse (10). Surface expression of NKG2D-ligands on healthy cells is tightly restricted by regulation at transcriptional and posttranscriptional levels, to ensure that healthy cells are not recognized by the innate immune system. The mechanisms involved in NKG2D-ligand expression regulation have been studied extensively [reviewed in Ref. (12, 13)].
Emerging evidence shows that intracellular stress can also induce the NKG2D-ligand expression. In this review, we summarize the mechanisms of NKG2D-ligand regulation. We focus specifically on recent advances in our understanding of how endoplasmic reticulum (ER) stress leads to NKG2D-ligand surface expression, and eventually group 1 innate lymphoid cells (ILCs)-mediated inflammation, particularly inflammatory bowel diseases, which are associated with several ER stress-related genes. In addition, we discuss the mechanisms by which NKG2D-L are suppressed on the other hand and specifically through carcinoembryonic antigen (CEA)-related cell adhesion molecule 1 (CEACAM1).
Regulation of NKG2D-Ligands
Regulation of NKG2D-Ligands by Cellular Stress and ER Stress
As NKG2D-ligand expression signals the immune system to recognize infected or transformed cells, a variety of stress pathways have been demonstrated to regulate NKG2D-ligand expression via different mechanisms (Table 1). Oxidative stress leads to accumulation of H2O2, which induces NKG2D-ligand including MICA/B and ULBP1–4 via activation of the mitogen-activated protein kinases pathway (14, 15). In contrast, heat shock can transcriptionally regulate MICA/B, as the promoter regions of the MIC genes have heat shock elements that can be recognized by heat shock factor 1 (HSF1) (15–17). Knockdown of HSF1 has been shown to suppress MICB, but not MICA, membrane expression leading to a reduction in NK cell-mediated cytotoxicity (18). In mice, heat shock induces MULT1 protein expression in fibroblasts and transformed cells by altering protein stability (19). One of the mechanisms associated with regulation of MULT1 surface expression by heat shock could be the membrane-associated RING-CH (MARCH) family of E3 ubiquitin ligase. While MULT1 is post-transcriptionally regulated by ubiquitin-dependent degradation by the MARCH family in unstressed cells, MULT1 ubiquitination and degradation are reduced in response to heat shock stress (19, 20).
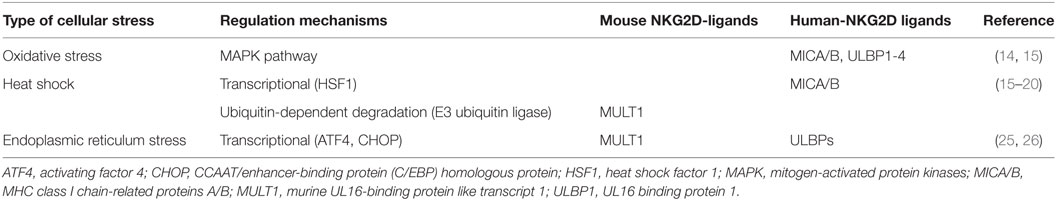
Table 1. Natural-killer group 2 member D (NKG2D)-ligands regulation by cellular stress.
Until recently, little was known about how cellular stress that leads to disturbances in proteostasis, and eventually ER stress, interact with regulation of NKG2D-ligand expression. ER stress is caused by the accumulation of unfolded and misfolded proteins in the ER arising from either primary (genetic) or secondary (environmental) factors (21). Highly secretory cells are highly susceptible to ER stress. These include Ig-producing plasma cells, insulin-secreting β-cells in the pancreas and intestinal epithelial cells, in particular Paneth cells and goblet cells (22). ER stress leads to the accumulation of unfolded or misfolded proteins within the ER lumen, which triggers three ER stress sensors to induce the so-called unfolded protein response (UPR). These include inositol-requiring transmembrane kinase-endonuclease 1 (IRE1), protein kinase RNA-like ER kinase (PERK), and activated transcription factor 6 (22). The main goal of UPR activation is to restore proteostasis and enhance the secretory capacity of the ER. In the IRE1 arm of the UPR, phosphorylated IRE1 possesses endoribonuclease activity that excises a 26-nucleotide sequence of X-box binding protein 1 (Xbp1) mRNA resulting in a frame shift and generation of a transcriptionally active isoform that functions as a transactivator of UPR target genes.
In the PERK arm, translation is suppressed by phosphorylation of elongation initiation factor 2α (eIF2α) to allow the cell to temporarily cope with excessive ER stress. Paradoxically, translation of some proteins, such as activating factor 4 (ATF4) is favored. Initially, ATF4 can induce several protective cellular pathways, among others autophagy (see below). However, if ER stress is excessive or prolonged, ATF4 induces apoptosis-related transcription factors, such as CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP) (23).
Besides cell death, ER stress elicits inflammatory responses, and hence it was hypothesized that UPR-related proteins can induce surface expression of NKG2D-ligands. Surprisingly, Xbp1 knockdown in the mouse immortalized small intestinal epithelial cell line MODE-K (24) (shXbp1 MODE-K by a short hairpin Xbp1 lentiviral vector), which causes ER stress (25), was shown to induce very strong induction of NKG2D-ligand MULT1 (both on mRNA level and surface protein expression), whereas inflammatory signals induced after stimulation with a variety of TLR ligands did not (25). Even more interesting was the fact that it appeared to be specific for MULT1, as both RAE-1 and H60 were not strongly induced. In contrast, expression of MHC class I, which is recognized by NK cell inhibitory receptors, was not affected by Xbp1 knockdown in vitro and knockout in vivo. The effect was not specific for Xbp1 deletion, as ER stress induction by administration of thapsigargin similarly induced strong upregulation of MULT1 surface expression. In addition, similar induction of ULBPs (the human ortholog of MULT1) was observed in a variety of human cell lines, including intestinal, gastric, esophageal, and hepatic cancer cell lines.
Intriguingly, ER stress protein ATF4 was found to be important in NKG2D-ligand upregulation using a completely different approach in a human cancer cell line HAP1 (26). This cancer cell line constitutively expresses ULBP1 and after treatment with a retroviral promoter trap vector, which randomly knocks out genes, the cell lines that had significant downregulation of ULBP1 surface expression were screened for gene enrichment. This screen revealed that ATF4 is important for the induction of ULBP1, which was confirmed by demonstrating that knockdown of ATF4 strongly decreased ULBP1 transcription. In addition, ATF4 was shown to have direct ULBP1 promotor binding sites and directly transactivates the ULBP1 promoter (26). In contrast to this study in human cancer cell lines, we have identified CHOP as a transcription factor that binds the promoter of the mouse ortholog of ULBP1, MULT1, using chromatin immunoprecipitation and luciferase assays. CHOP is downstream of PERK-ATF4, but can also be induced by other ER stress-associated pathway elements (27). Interestingly, MODE-K cells with silenced CHOP using shRNA, or primary intestinal epithelial cells from Chop−/− mice examined ex vivo show downregulation of ER stress-dependent induction of MULT1 on the surface of intestinal epithelial cells (Figure 1).
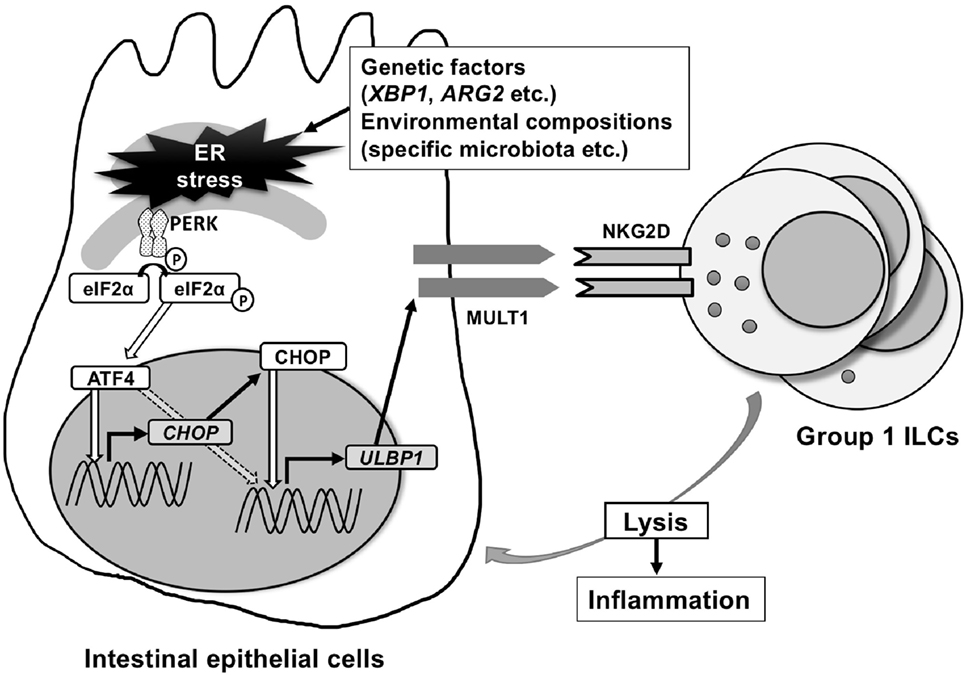
Figure 1. Endoplasmic reticulum (ER) stress-inducing murine UL16-binding protein like transcript 1 (MULT1) in mouse. MULT1 [encoded by UL16 binding protein 1 (Ulbp1)], but not other natural-killer group 2 member D (NKG2D)-ligands, mRNA, and surface expression are upregulated on ER-stressed mouse small intestinal epithelial cells. This occurs through activating factor 4 (ATF4) and/or C/EBP homologous protein (CHOP) presumably downstream of the protein kinase RNA-like ER kinase pathway. CHOP as a transcription factor binds directly to the promoter area of Ulbp1, which upon expression subsequently activates NKG2D-expressing intraepithelial group 1 innate lymphoid cells (ILCs) [natural killer (NK) cells and ILC1] that accumulate within the intestinal epithelium and promote small intestinal inflammation.
Together, these two recent studies indicate that the PERK–ATF4–CHOP pathway of the UPR is a highly conserved ER-stress-specific mechanism of regulation of NKG2D-ligands. Interestingly, ER stress is a common pathophysiological phenomenon in the two disease processes that have been mostly linked to NKG2D-ligand expression, namely cancer, discussed below, and (viral) infection. Viral infection can strongly induce NKG2D-ligands. For example, human immunodeficiency virus type 1 (HIV-1) induces DNA stress/damage checkpoint arrest initiated by the DNA damage-sensing protein kinase (ataxia telangiectasia-mutated and Rad3-related: ATR) (28), resulting in upregulation of NKG2D-ligands (29, 30). Recognition of viral products, such as double-stranded (dsRNA), by retinoic acid-inducible gene I and melanoma differentiation-associated gene 5, both cytoplasmic pattern recognition receptors, also upregulate MICA and ULBP2 expression (31). Since viral replication requires the host ER for the production of their structural and non-structural proteins, viral infection has been strongly associated with activation of the UPR as well, and further research is required to investigate how the UPR in viral infection affects NKG2D-ligand expression.
Consequences of ER Stress-Induced Regulation of NKG2D-Ligands in the Gastrointestinal Tract
In genome-wide association studies, MICs gene have been associated with susceptibility to a variety of autoimmune diseases, including systemic type 1 diabetes, rheumatoid arthritis, and lupus erythematosus (32–34). Consequently, NKG2D/NKG2D ligand interactions have been hypothesized to be involved in their pathogenesis (35–38).
In the intestine, NKG2D/NKG2D-ligand interactions have mainly been studied in celiac disease. Celiac disease is a gluten-driven innate and acquired immune cell-mediated enteropathy characterized by findings of intraepithelial lymphocytosis, crypt hyperplasia, and villous atrophy, and a positive response to a gluten free-diet (39). The fractions of NKG2D+ NK cells and NKT cells among the intraepithelial mononuclear cells in active celiac disease are significantly increased as compared to inactive celiac disease or control subjects (40). Increased MICA expression on the intestinal epithelium in active celiac disease via a pathway involving IL-15 induction by gliadin triggers activation of intraepithelial T cells, allowing for the killing of intestinal epithelial cells (41). Details of the MICA/B expression pattern showed that the expression was observed not only on epithelial cells but also in the cytoplasm of intraepithelial T lymphocytes in patients with active celiac disease, suggesting extensive stress conditions are present in active celiac disease (42). Interestingly, some studies have suggested that ER stress could be involved in NKG2D-ligand expression regulation in celiac disease (42).
In Crohn’s disease, several studies have also suggested involvement of the NKG2D/NKG2D-ligand pathways in the pathogenesis of this disorder (43–45). Specifically, MICA and MICB expression has been demonstrated to be increased on intestinal epithelial cells in patients with inflammatory bowel disease (46, 47). Moreover, a subset of CD4+ T cells expressing NKG2D was increased in patients with Crohn’s disease and functionally active through MICA–NKG2D interactions, leading to interferon-γ production. In pediatric Crohn’s disease and ulcerative colitis, ULBP1 and ULBP2 is upregulated on infiltrating immune cells in active inflammatory lesions (48). Interestingly, a recent clinical trial revealed efficacy of a blocking anti-NKG2D IgG4 antibody in patients with active Crohn’s disease (49), suggesting that NKG2D/NKG2D-ligand interactions are of importance in the pathogenesis of Crohn’s disease.
As ER stress has also been linked to the pathogenesis of Crohn’s disease (50), and as we demonstrated that ER stress regulates NKG2D-ligands on intestinal epithelial cells, we subsequently investigated the functional consequences of epithelial ER stress-induced NKG2D-ligand expression and its role in the development of spontaneous enteritis in a mouse model of this condition. We demonstrated that increased MULT1 expression on intestinal epithelial cells of Xbp1ΔIEC mice was accompanied by increased quantities of NKG2D-expressing intraepithelial group 1 ILCs (NK cells and ILC1) (25). The group 1 ILCs within the epithelium also exhibited evidence of increased activation as demonstrated by increased surface expression of NKG2D and CD25. Indeed, IELs from Xbp1ΔIEC mice exhibited increased cytotoxicity in comparison to the activity observed with IELs from wild-type mice, suggesting an involvement in inflammation, which was supported by amelioration of inflammation upon NKG2D blockade. Furthermore, ER stress-induced intestinal inflammation due to Xbp1 deletion in the intestinal epithelium was uniquely comprised of a significant component of innate immune activation as concomitant loss of the adaptive immune system in Rag1−/−Xbp1ΔIEC double mutant mice did not affect the severity or kinetics of the inflammation. However, depletion of NK cells significantly diminished inflammation that emerged from tamoxifen-driven, and thus temporally controlled induction of ER stress during adult life in Xbp1T-ΔIEC mice. Similarly, spontaneous enteritis was reversed by NKG2D blockade in Xbp1T-ΔIEC mice, in line with what has been shown for mouse colitis models (51, 52). Thus, our study demonstrates that ER stress in the small intestine leads to spontaneous enteritis that depends significantly on the presence of an innate immune system. Further, the development of ER-stress-mediated inflammation in this context involves the CHOP-dependent induction of NKG2D-ligand MULT1 and its recognition by NKG2D on group 1 ILC, which are increased in the intestinal epithelium. Future studies are required to investigate how ER stress is linked to NKG2D-ligand expression in the human gut, particularly in the setting of complex diseases including inflammatory bowel disease. Methods to resolve pathologic levels of ER stress could, therefore, be a potential target for therapies in this still uncurable disease.
Regulation of NKG2D-Ligand Expression in Cancer
NKG2D-ligand expression has been extensively studied in the setting of cancer. The expression of NKG2D-ligands in cancer can be hypothesized to serve in activating the immune system for elimination of the excessively proliferating cancer cells. E2F transcription factors, for example, regulate cell proliferation but at the same time can induce specific NKG2D-ligands such as RAE-1, but not MULT1 and H60 in mouse fibroblasts by directly binding the promoter region of Raet1 genes (53).
DNA damage responses to genotoxic stress coordinates activation of transcription, cell cycle control, apoptosis, and DNA repair processes mediated by a number of protein kinases, including ATM (ataxia telangiectasia mutated) and ATR (ATM and Rad3-related) protein kinases (54). In both mouse and human non-tumor cells, genotoxic stress and inhibition of DNA replication via the DNA damage pathway through ATM or ATR protein kinases, leads to increased surface expression of NKG2D-ligands (55). In tumor cell lines, constitutive ligand expression on the cell surface is suppressed by pharmacological or genetic inhibition of ATR, ATM, or Chk1 (55).
p53, induced in response to DNA damage, can also regulate human ULBP1 and ULBP2 by binding to p53-responsive elements in the promoter area of ULBP1 and ULBP2 gene (56–58). However, several microRNAs that are induced by p53 (miR-34a and miR-34c) have been shown to suppress ULBP2 expression by directly binding to the 3′-UTR of ULBP1 mRNA (59).
Other mechanisms of NKG2D-ligand induction in response to DNA damage response include the stimulator of interferon genes (STING)-dependent DNA sensor pathway (60). The accumulation of cytosolic DNA by DNA damage response activates STING-dependent DNA sensors, leading to the activation of TANK binding kinase 1 and interferon regulatory factor 3, which in turn are associated with RAE1 expression (60).
Inhibition of NKG2D-Ligand Expression on Tumor Cells by CEACAM1
As NKG2D-ligand expression on tumor cells is critical for the recognition and clearance of tumors, tumor cells have evolved by developing several mechanisms to escape from immune surveillance. Several studies have demonstrated that soluble forms of NKG2D-ligands that were derived from cancer cells by either proteolytic shedding (61–63), alternative splicing (64), or exosome secretion (65) can impair NKG2D-mediated cytotoxicity by negatively regulating NKG2D expression or recognition (66). In addition, metastasis-associated microRNA miR-10b (also known as metastamir), which promotes tumor invasion and metastasis by targeting multiple genes, downregulates MICB expression by binding directly to the 3′-UTR of MICB (67).
CEACAM1, a member of the CEA family of Ig like transmembrane glycoproteins (68), is involved in the negative regulation of NKG2D-ligands in cancer (69–71). CEACAM1 is expressed in mouse and humans and is characterized by numerous transmembrane isoforms that derived from alternative splicing mechanisms [reviewed in Ref. (68, 72)]. This mechanism generates CEACAM1 variants that share a membrane distal IgV-like domain (N-domain), which functions in homophilic or heterophilic interactions, that is coupled to variable numbers of IgC2 domains and linked to either a long (L) or short cytoplasmic domain. NK cells and T cells predominantly express CEACAM1-L isoforms that contain two immunoreceptor tyrosine-based inhibitory motifs in their cytoplasmic tail which serves to recruit Src homology phosphatase 1 (SHP1) and SHP2 after phosphorylation by Src-related kinases (68, 73). Ligation of CEACAM1-L isoforms on NK cells by CEACAM1 on tumor cells suppresses NK cytolytic function as CEACAM1 on the NK cells negatively regulates NKG2D signaling (74). Specifically, recruitment of SHP1 by CEACAM1 leads to dephosphorylation of the guanine nucleotide exchange factor Vav1, one of the most proximal elements associated with NKG2D-mediated cytolytic signaling (75). At the same time, CEACAM1 expression on the tumor cells has been shown to regulate NKG2D-ligand expression. Specifically, silencing of CEACAM1 in mouse and human tumor cells upregulates expression of NKG2D-ligands on the cell surface of the tumor cell and makes them more highly susceptible to NK cell-mediated cytotoxicity (Figure 2) (76). Silencing CEACAM1 did not alter the transcriptional levels of NKG2D-ligands. Indeed, cell surface expression of RAE-1 was increased in CEACAM-1 silenced cells, whereas intracellular RAE-1 protein was decreased in comparison to non-silenced cells. Furthermore, RAE-1 on the cell surface of CEACAM1 silenced cells possessed an increased quantity of carbohydrate side-chain modifications in comparison to CEACAM1 non-silenced cells, in which RAE-1 accumulates intracellularly as an incompletely glycosylated protein (76). Thus, CEACAM1 can regulate glycosylation of NKG2D-ligands resulting in the NKG2D-ligands retention in an intracellular compartment.
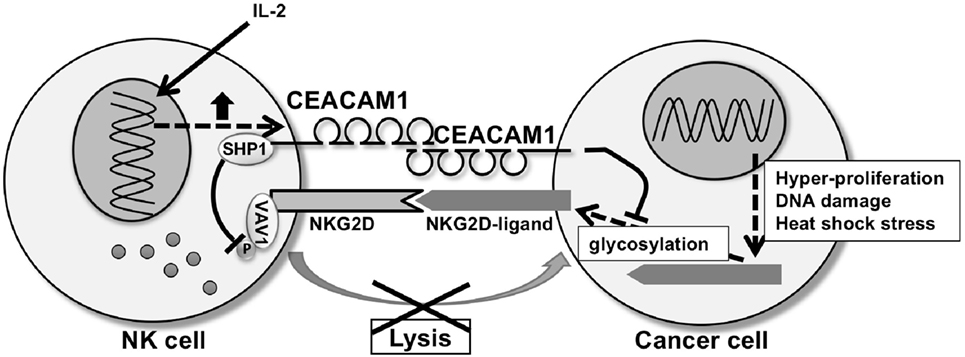
Figure 2. CEA-related cell adhesion molecule 1 (CEACAM1) regulating natural-killer group 2 member D (NKG2D)-ligands expression and NKG2D function. CEACAM1 regulates glycosylation of NKG2D-ligands, resulting in downregulation of the NKG2D-ligand expression on the cell surface of the tumor cell. CEACAM1 induced by interleukin-2 inhibits NKG2D-mediated cytotoxic function through recruitment of Src homology phosphatase 1 (SHP1) to phosphorylated CEACAM1 which leads to dephosphorylation of Vav1 on the natural killer (NK) cell. CEACAM1 on the NK cell and tumor cell interact homophilically through the N-domain of CEACAM1.
Conclusion
NKG2D/NKG2D-ligands interactions play a critical role in the immune surveillance of sick cells, such as those that are infected or have undergone neoplastic transformation. In such contexts, the expression and function of NKG2D and NKG2D-ligands are critical for removal of the altered cells and resolution of the condition. However, in certain circumstances cancer and infected cells have developed the ability to avoid immune surveillance that is mediated by NKG2D/NKG2D-ligands which allows progression of the infection or tumor. Our recent studies indicate that CEACAM1 plays an important role in NKG2D/NKG2D-ligand function and expression, respectively, suggesting that manipulation of this pathway may be beneficial in such disorders. On the other hand, NKG2D/NKG2D-ligand expression and function may inappropriately be associated with distressed organs or cells in autoimmune disease, suggesting that their blockade would be an important means of resolving inflammation consistent with their current evaluation in clinical trials in inflammatory bowel disease (49). Our recent studies thus suggest that enhancement of CEACAM1 function might be beneficial in such disorders by enhancing inhibition of NK and ILC1-mediated activation due to NKG2D/NKG2D ligand interactions. This is consistent with previous studies that application of CEACAM1 ligands to mouse models of colitis can suppress inflammation (77). This is particularly interesting as a key feature of inflammatory bowel disease which is the common induction of ER stress within the intestinal epithelium, which can serve as nidus for development of inflammation (22, 50). As such, the recent evidence that ER stress is linked to the upregulation of specific NKG2D-ligand and activation of intraepithelial NK cells and ILC1, together imply that blockade of NKG2D and NKG2D ligand interactions may have wide benefit in inflammatory bowel disease and potentially other autoimmune conditions.
Author Contributions
All authors wrote and edited the manuscript and gave final approval of the manuscript.
Conflict of Interest Statement
RB is a consultant to Syntalogic Pharmaceuticals which is developing therapies that target CEACAM1. The other authors have no conflict of interest.
Acknowledgments
The authors thank Aman Riar and Tara Traylor for administrative assistance in this manuscript.
Funding
This work was supported by JSPS KAKENHI Grant Number 2689323 and 16K19162, Japan Foundation for Applied Enzymology (SH); Rubicon grant 825.13.012, Netherlands Organization for Scientific Research (JG); Wellcome Trust Senior Investigator Award 106260/Z/14/Z, the European Research Council HORIZON2020/ERC grant no. 648889 (AK); NIH grants DK044319, DK051362, DK053056, DK088199, the Harvard Digestive Diseases Center (HDDC) DK034854 (RB).
References
1. Kiessling R, Klein E, Wigzell H. "Natural" killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol (1975) 5:112–7. doi:10.1002/eji.1830050208
2. Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer (1975) 16:216–29. doi:10.1002/ijc.2910160204
3. Lanier LL. NK cell recognition. Annu Rev Immunol (2005) 23:225–74. doi:10.1146/annurev.immunol.23.021704.115526
4. Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature (1986) 319:675–8. doi:10.1038/319675a0
5. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol (2008) 9:495–502. doi:10.1038/ni1581
6. Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood (2006) 107:159–66. doi:10.1182/blood-2005-04-1351
7. Champsaur M, Lanier LL. Effect of NKG2D ligand expression on host immune responses. Immunol Rev (2010) 235:267–85. doi:10.1111/j.0105-2896.2010.00893.x
8. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity (2013) 38:769–81. doi:10.1016/j.immuni.2013.02.010
9. Wencker M, Turchinovich G, Di Marco Barros R, Deban L, Jandke A, Cope A, et al. Innate-like T cells straddle innate and adaptive immunity by altering antigen-receptor responsiveness. Nat Immunol (2014) 15:80–7. doi:10.1038/ni.2773
10. Lanier LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res (2015) 3:575–82. doi:10.1158/2326-6066.CIR-15-0098
11. Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, et al. Cancer immunosurveillance by tissue-resident innate lymphoid cells and innate-like T cells. Cell (2016) 164:365–77. doi:10.1016/j.cell.2016.01.002
12. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol (2013) 31:413–41. doi:10.1146/annurev-immunol-032712-095951
13. Spies T. Regulation of NKG2D ligands: a purposeful but delicate affair. Nat Immunol (2008) 9:1013–5. doi:10.1038/ni0908-1013
14. Borchers MT, Harris NL, Wesselkamper SC, Vitucci M, Cosman D. NKG2D ligands are expressed on stressed human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol (2006) 291:L222–31. doi:10.1152/ajplung.00327.2005
15. Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T. Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J Immunol (2007) 178:961–9. doi:10.4049/jimmunol.178.2.961
16. Groh V, Steinle A, Bauer S, Spies T. Recognition of stress-induced MHC molecules by intestinal epithelial gammadelta T cells. Science (1998) 279:1737–40. doi:10.1126/science.279.5357.1737
17. Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci U S A (1996) 93:12445–50. doi:10.1073/pnas.93.22.12445
18. Schilling D, Kuhnel A, Tetzlaff F, Konrad S, Multhoff G. NZ28-induced inhibition of HSF1, SP1 and NF-kappaB triggers the loss of the natural killer cell-activating ligands MICA/B on human tumor cells. Cancer Immunol Immunother (2015) 64:599–608. doi:10.1007/s00262-015-1665-9
19. Nice TJ, Coscoy L, Raulet DH. Posttranslational regulation of the NKG2D ligand Mult1 in response to cell stress. J Exp Med (2009) 206:287–98. doi:10.1084/jem.20081335
20. Nice TJ, Deng W, Coscoy L, Raulet DH. Stress-regulated targeting of the NKG2D ligand Mult1 by a membrane-associated RING-CH family E3 ligase. J Immunol (2010) 185:5369–76. doi:10.4049/jimmunol.1000247
21. Kaser A, Adolph TE, Blumberg RS. The unfolded protein response and gastrointestinal disease. Semin Immunopathol (2013) 35:307–19. doi:10.1007/s00281-013-0377-5
22. Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol (2016) 16:469–84. doi:10.1038/nri.2016.62
23. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta (2013) 1833:3460–70. doi:10.1016/j.bbamcr.2013.06.028
24. Vidal K, Grosjean I, Evillard JP, Gespach C, Kaiserlian D. Immortalization of mouse intestinal epithelial cells by the SV40-large T gene. Phenotypic and immune characterization of the MODE-K cell line. J Immunol Methods (1993) 166:63–73. doi:10.1016/0022-1759(93)90329-6
25. Hosomi S, Grootjans J, Tschurtschenthaler M, Krupka N, Matute JD, Flak MB, et al. Intestinal epithelial cell endoplasmic reticulum stress promotes MULT1 up-regulation and NKG2D-mediated inflammation. J Exp Med (2017) 214:2985–97. doi:10.1084/jem.20162041
26. Gowen BG, Chim B, Marceau CD, Greene TT, Burr P, Gonzalez JR, et al. A forward genetic screen reveals novel independent regulators of ULBP1, an activating ligand for natural killer cells. Elife (2015) 4:e08474. doi:10.7554/eLife.08474
27. Li Y, Guo Y, Tang J, Jiang J, Chen Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim Biophys Sin (Shanghai) (2014) 46:629–40. doi:10.1093/abbs/gmu048
28. Roshal M, Kim B, Zhu Y, Nghiem P, Planelles V. Activation of the ATR-mediated DNA damage response by the HIV-1 viral protein R. J Biol Chem (2003) 278:25879–86. doi:10.1074/jbc.M303948200
29. Richard J, Sindhu S, Pham TN, Belzile JP, Cohen EA. HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood (2010) 115:1354–63. doi:10.1182/blood-2009-08-237370
30. LaThangue NB, Latchman DS. Nuclear accumulation of a heat-shock 70-like protein during herpes simplex virus replication. Biosci Rep (1987) 7:475–83. doi:10.1007/BF01116504
31. Esteso G, Guerra S, Vales-Gomez M, Reyburn HT. Innate immune recognition of double-stranded RNA triggers increased expression of NKG2D ligands after virus infection. J Biol Chem (2017) 292:20472–80. doi:10.1074/jbc.M117.818393
32. Yu P, Zhu Q, Chen C, Fu X, Li Y, Liu L, et al. Association between major histocompatibility complex class I chain-related gene polymorphisms and susceptibility of systemic lupus erythematosus. Am J Med Sci (2017) 354:430–5. doi:10.1016/j.amjms.2017.06.003
33. Martinez A, Fernandez-Arquero M, Balsa A, Rubio A, Alves H, Pascual-Salcedo D, et al. Primary association of a MICA allele with protection against rheumatoid arthritis. Arthritis Rheum (2001) 44:1261–5. doi:10.1002/1529-0131(200106)44:6<1261::AID-ART217>3.0.CO;2-L
34. Field SF, Nejentsev S, Walker NM, Howson JM, Godfrey LM, Jolley JD, et al. Sequencing-based genotyping and association analysis of the MICA and MICB genes in type 1 diabetes. Diabetes (2008) 57:1753–6. doi:10.2337/db07-1402
35. Flodstrom-Tullberg M, Bryceson YT, Shi FD, Hoglund P, Ljunggren HG. Natural killer cells in human autoimmunity. Curr Opin Immunol (2009) 21:634–40. doi:10.1016/j.coi.2009.09.012
36. Van Belle TL, von Herrath MG. The role of the activating receptor NKG2D in autoimmunity. Mol Immunol (2009) 47:8–11. doi:10.1016/j.molimm.2009.02.023
37. Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci U S A (2003) 100:9452–7. doi:10.1073/pnas.1632807100
38. Yang D, Tian Z, Zhang M, Yang W, Tang J, Wu Y, et al. NKG2D(+)CD4(+) T cells kill regulatory T cells in a NKG2D-NKG2D ligand-dependent manner in systemic lupus erythematosus. Sci Rep (2017) 7:1288. doi:10.1038/s41598-017-01379-y
40. Marafini I, Monteleone I, Di Fusco D, Sedda S, Cupi ML, Fina D, et al. Celiac disease-related inflammation is marked by reduction of Nkp44/Nkp46-double positive natural killer cells. PLoS One (2016) 11:e0155103. doi:10.1371/journal.pone.0155103
41. Hüe S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity (2004) 21:367–77. doi:10.1016/j.immuni.2004.06.018
42. Allegretti YL, Bondar C, Guzman L, Cueto Rua E, Chopita N, Fuertes M, et al. Broad MICA/B expression in the small bowel mucosa: a link between cellular stress and celiac disease. PLoS One (2013) 8:e73658. doi:10.1371/journal.pone.0073658
43. Ding Y, Xia B, Lü M, Zhang Y, Li J, Ye M, et al. MHC class I chain-related gene A-A5.1 allele is associated with ulcerative colitis in Chinese population. Clin Exp Immunol (2005) 142:193–8. doi:10.1111/j.1365-2249.2005.02907.x
44. Lü M, Xia B, Li J, Ye M, Zhang X, Tan Q. MICB microsatellite polymorphism is associated with ulcerative colitis in Chinese population. Clin Immunol (2006) 120:199–204. doi:10.1016/j.clim.2006.03.001
45. Martinez-Chamorro A, Moreno A, Gómez-García M, Cabello MJ, Martin J, Lopez-Nevot MÁ. MICA*A4 protects against ulcerative colitis, whereas MICA*A5.1 is associated with abscess formation and age of onset. Clin Exp Immunol (2016) 184:323–31. doi:10.1111/cei.12786
46. Perera L, Shao L, Patel A, Evans K, Meresse B, Blumberg R, et al. Expression of nonclassical class I molecules by intestinal epithelial cells. Inflamm Bowel Dis (2007) 13:298–307. doi:10.1002/ibd.20026
47. Allez M, Tieng V, Nakazawa A, Treton X, Pacault V, Dulphy N, et al. CD4+NKG2D+ T cells in Crohn’s disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology (2007) 132:2346–58. doi:10.1053/j.gastro.2007.03.025
48. La Scaleia R, Stoppacciaro A, Oliva S, Morrone S, Di Nardo G, Santoni A, et al. NKG2D/Ligand dysregulation and functional alteration of innate immunity cell populations in pediatric IBD. Inflamm Bowel Dis (2012) 18:1910–22. doi:10.1002/ibd.22899
49. Allez M, Skolnick BE, Wisniewska-Jarosinska M, Petryka R, Overgaard RV. Anti-NKG2D monoclonal antibody (NNC0142-0002) in active Crohn’s disease: a randomised controlled trial. Gut (2017) 66:1918–25. doi:10.1136/gutjnl-2016-311824
50. Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell (2008) 134:743–56. doi:10.1016/j.cell.2008.07.021
51. Kjellev S, Haase C, Lundsgaard D, Ursø B, Tornehave D, Markholst H. Inhibition of NKG2D receptor function by antibody therapy attenuates transfer-induced colitis in SCID mice. Eur J Immunol (2007) 37:1397–406. doi:10.1002/eji.200636473
52. Ito Y, Kanai T, Totsuka T, Okamoto R, Tsuchiya K, Nemoto Y, et al. Blockade of NKG2D signaling prevents the development of murine CD4+ T cell-mediated colitis. Am J Physiol Gastrointest Liver Physiol (2008) 294:G199–207. doi:10.1152/ajpgi.00286.2007
53. Jung H, Hsiung B, Pestal K, Procyk E, Raulet DH. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. J Exp Med (2012) 209:2409–22. doi:10.1084/jem.20120565
54. Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature (2000) 408:433–9. doi:10.1038/35044005
55. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature (2005) 436:1186–90. doi:10.1038/nature03884
56. Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science (2000) 287:1824–7. doi:10.1126/science.287.5459.1824
57. Textor S, Fiegler N, Arnold A, Porgador A, Hofmann TG, Cerwenka A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res (2011) 71:5998–6009. doi:10.1158/0008-5472.CAN-10-3211
58. Li H, Lakshmikanth T, Garofalo C, Enge M, Spinnler C, Anichini A, et al. Pharmacological activation of p53 triggers anticancer innate immune response through induction of ULBP2. Cell Cycle (2011) 10:3346–58. doi:10.4161/cc.10.19.17630
59. Heinemann A, Zhao F, Pechlivanis S, Eberle J, Steinle A, Diederichs S, et al. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res (2012) 72:460–71. doi:10.1158/0008-5472.CAN-11-1977
60. Lam AR, Bert NL, Ho SS, Shen YJ, Tang LF, Xiong GM, et al. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res (2014) 74:2193–203. doi:10.1158/0008-5472.CAN-13-1703
61. Salih HR, Rammensee HG, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol (2002) 169:4098–102. doi:10.4049/jimmunol.169.8.4098
62. Wu JD, Atteridge CL, Wang X, Seya T, Plymate SR. Obstructing shedding of the immunostimulatory MHC class I chain-related gene B prevents tumor formation. Clin Cancer Res (2009) 15:632–40. doi:10.1158/1078-0432.CCR-08-1305
63. Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, Mann HH, et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature (2007) 447:482–6. doi:10.1038/nature05768
64. Cao W, Xi X, Hao Z, Li W, Kong Y, Cui L, et al. RAET1E2, a soluble isoform of the UL16-binding protein RAET1E produced by tumor cells, inhibits NKG2D-mediated NK cytotoxicity. J Biol Chem (2007) 282:18922–8. doi:10.1074/jbc.M702504200
65. Fernández-Messina L, Ashiru O, Boutet P, Agüera-González S, Skepper JN, Reyburn HT, et al. Differential mechanisms of shedding of the glycosylphosphatidylinositol (GPI)-anchored NKG2D ligands. J Biol Chem (2010) 285:8543–51. doi:10.1074/jbc.M109.045906
66. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature (2002) 419:734–8. doi:10.1038/nature01112
67. Tsukerman P, Stern-Ginossar N, Gur C, Glasner A, Nachmani D, Bauman Y, et al. MiR-10b downregulates the stress-induced cell surface molecule MICB, a critical ligand for cancer cell recognition by natural killer cells. Cancer Res (2012) 72:5463–72. doi:10.1158/0008-5472.CAN-11-2671
68. Gray-Owen SD, Blumberg RS. CEACAM1: contact-dependent control of immunity. Nat Rev Immunol (2006) 6:433–46. doi:10.1038/nri1864
69. Markel G, Lieberman N, Katz G, Arnon TI, Lotem M, Drize O, et al. CD66a interactions between human melanoma and NK cells: a novel class I MHC-independent inhibitory mechanism of cytotoxicity. J Immunol (2002) 168:2803–10. doi:10.4049/jimmunol.168.6.2803
70. Stern N, Markel G, Arnon TI, Gruda R, Wong H, Gray-Owen SD, et al. Carcinoembryonic antigen (CEA) inhibits NK killing via interaction with CEA-related cell adhesion molecule 1. J Immunol (2005) 174:6692–701. doi:10.4049/jimmunol.174.11.6692
71. Markel G, Seidman R, Stern N, Cohen-Sinai T, Izhaki O, Katz G, et al. Inhibition of human tumor-infiltrating lymphocyte effector functions by the homophilic carcinoembryonic cell adhesion molecule 1 interactions. J Immunol (2006) 177:6062–71. doi:10.4049/jimmunol.177.9.6062
72. Dankner M, Gray-Owen SD, Huang YH, Blumberg RS, Beauchemin N. CEACAM1 as a multi-purpose target for cancer immunotherapy. Oncoimmunology (2017) 6:e1328336. doi:10.1080/2162402X.2017.1328336
73. Nagaishi T, Pao L, Lin SH, Iijima H, Kaser A, Qiao SW, et al. SHP1 phosphatase-dependent T cell inhibition by CEACAM1 adhesion molecule isoforms. Immunity (2006) 25:769–81. doi:10.1016/j.immuni.2006.08.026
74. Hosomi S, Chen Z, Baker K, Chen L, Huang YH, Olszak T, et al. CEACAM1 on activated NK cells inhibits NKG2D-mediated cytolytic function and signaling. Eur J Immunol (2013) 43:2473–83. doi:10.1002/eji.201242676
75. Upshaw JL, Arneson LN, Schoon RA, Dick CJ, Billadeau DD, Leibson PJ. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat Immunol (2006) 7:524–32. doi:10.1038/ni1325
76. Chen Z, Chen L, Baker K, Olszak T, Zeissig S, Huang YH, et al. CEACAM1 dampens antitumor immunity by down-regulating NKG2D ligand expression on tumor cells. J Exp Med (2011) 208:2633–40. doi:10.1084/jem.20102575
Keywords: natural-killer group 2 member D, natural-killer group 2 member D-ligand, murine UL16-binding protein like transcript 1, UL16 binding protein 1, endoplasmic reticulum stress, CEA-related cell adhesion molecule 1
Citation: Hosomi S, Grootjans J, Huang Y-H, Kaser A and Blumberg RS (2018) New Insights Into the Regulation of Natural-Killer Group 2 Member D (NKG2D) and NKG2D-Ligands: Endoplasmic Reticulum Stress and CEA-Related Cell Adhesion Molecule 1. Front. Immunol. 9:1324. doi: 10.3389/fimmu.2018.01324
Received: 17 February 2018; Accepted: 28 May 2018;
Published: 18 June 2018
Edited by:
Nadia Guerra, Imperial College London, United KingdomReviewed by:
Jianhua Yu, The Ohio State University, United StatesChiara Romagnani, Deutsches Rheuma-Forschungszentrum (DRFZ), Germany
Copyright: © 2018 Hosomi, Grootjans, Huang, Kaser and Blumberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuhei Hosomi, m1265271@med.osaka-cu.ac.jp