 Katherine H. Restori
Katherine H. Restori Bharat T. Srinivasa
Bharat T. Srinivasa Brian J. Ward
Brian J. Ward Elizabeth D. Fixman
Elizabeth D. Fixman- 1Research Institute of the McGill University Health Centre, Montréal, QC, Canada
- 2Meakins-Christie Laboratories, Research Institute of the McGill University Health Centre, Montréal, QC, Canada
Infants are exposed to a wide range of potential pathogens in the first months of life. Although maternal antibodies acquired transplacentally protect full-term neonates from many systemic pathogens, infections at mucosal surfaces still occur with great frequency, causing significant morbidity and mortality. At least part of this elevated risk is attributable to the neonatal immune system that tends to favor T regulatory and Th2 type responses when microbes are first encountered. Early-life infection with respiratory viruses is of particular interest because such exposures can disrupt normal lung development and increase the risk of chronic respiratory conditions, such as asthma. The immunologic mechanisms that underlie neonatal host–virus interactions that contribute to the subsequent development of asthma have not yet been fully defined. The goals of this review are (1) to outline the differences between the neonatal and adult immune systems and (2) to present murine and human data that support the hypothesis that early-life interactions between the immune system and respiratory viruses can create a lung environment conducive to the development of asthma.
Introduction
The young of any species face microbial threats similar to those faced by adults. However, the developing mammalian immune system does not mount “adult-type” immune responses to most pathogens. Adaptive in many respects (see below), this immature pattern of response may also contribute to high levels of infection-related mortality and morbidity in young infants. Of the 5 million worldwide deaths in children <5 years of age in 2010, ~64% were due to infectious causes, 40% of which occurred between 1 and 29 days (1). Respiratory infections account for a majority of these neonatal deaths (1). Although bacteria contribute to this disease burden, viral infections are far more common. The annual economic burden of viral respiratory illnesses in the US alone is estimated to be ~$25 billion (2). Many of the respiratory viruses with the greatest impact in young children are well known and include, among others, respiratory syncytial virus (RSV), influenza A/B viruses, rhinoviruses (RVs), human metapneumovirus, parainfluenzaviruses 1–4 (PIV), bocaviruses, coronaviruses, and certain adenovirus strains.
While infant-pattern immune responses may enhance susceptibility to some infections, mounting adult-type responses may not be optimal for overall development either. The neonatal immune system must first and foremost distinguish between self and non-self, and then rapidly proceed to distinguish between benign or even “helpful” non-self (e.g., the commensal microbiome) and potential pathogens. During this period of immune education, the developing immune system has to strike a delicate and pathogen-specific balance between the induction of potentially damaging, pro-inflammatory responses mediated by T helper (Th) cell 1-type (Th1) and some Th17-type cells, less damaging inflammatory responses (e.g., Th2-type) or even suppressive responses mediated by regulatory T cells (Treg). Although the induction of suppressive responses may seem counterintuitive when dealing with potential pathogens, such responses appear to be essential to protect the fetus from reacting too vigorously to maternal antigens (3) or to colonization of the neonatal gut and other body surfaces with the normal microbial flora (4). The “cost” to the infant of mounting an aggressive, pro-inflammatory response can be very high. Early-life inflammation has been associated with a wide range of negative long-term consequences including overall development of the body and the brain, neuro-psychological reactions (e.g., to pain) and patterns of inflammatory response (5, 6) [reviewed in Ref. (7, 8)]. It is, therefore, entirely plausible that early-life experiences with respiratory pathogens can have long-lasting effects on the lung.
A number of epidemiological studies have linked respiratory viral infection during infancy with the later development of asthma [reviewed in Ref. (9–11)]. It is currently unknown whether this association is a direct effect of viral replication in respiratory tissues or results from a virus-induced exacerbation of an underlying predisposition to atopy. However, there is evidence from both human and animal studies to support both hypotheses. The purpose of this review is to examine the immunology of the host–virus interaction during the neonatal period in the context of asthma development. We will first briefly describe how the neonatal immune system differs from that of the adult. Because RSV, influenza, and RVs cause a large proportion of respiratory-tract infections in neonates, we will focus primarily on these three pathogens as models to better understand how early-life infection and antiviral immune responses might contribute to the subsequent development of asthma.
The Immune System of the Neonate
Adaptive Immunity in the Neonate
Prior to the 1980s, it was commonly thought that the neonatal immune system was in a state of tolerance (12), as demonstrated by mouse experiments in the 1950s showing the apparent lack of recall responses to antigens injected soon after birth (13). Indeed, neonates were considered by some to be “immunodeficient” (14). Upon discovery of different patterns of T cell response in the mid-1980s [e.g., Th1 vs. Th2 type: reviewed in Ref. (15)], it subsequently became clear that what appeared to be a deficiency in responding to recall antigens was instead a reflection of an intrinsic Th2/Treg bias in neonatal mice (16, 17).
A quarter of a century later, Th1- and Th2-type responses are two of the most well characterized aspects of cellular immunity. The classical Th1 response is characterized by production of the cytokines interferon (IFN)γ and interleukin (IL)-12, whereas Th2 responses are typified by production of IL-4 and IL-13. Th1-type responses tend to be pro-inflammatory in nature, with increases in CD8+ T cells, and are most often associated with challenges posed by intracellular organisms, including viruses. Th2 responses typically play a central role in defending against invasive helminths, but are also important in modulating potentially damaging inflammatory responses as well as in driving allergic pathologies (e.g., atopy, asthma). Other more recently recognized patterns of immune response have also been implicated in either promoting or preventing the development or evolution of asthma, including Th17-, Th9-, and Treg. In particular, Th17- and Th9-driven responses may promote exacerbations of severe asthma [reviewed in Ref. (18)] and/or promote allergic airways disease through interactions with Th2 cells (19, 20). Tregs, on the other hand, play a central role in limiting excessive reactivity to self and innocuous allergens (21).
While Th2 skewing may be a natural or default response pattern to microbial insults in the neonate, the neonatal immune system can induce adult-like, Th1/Th2 balanced or even Th1-predominant responses under certain conditions (17, 22, 23). Experimental situations in which such responses can be generated typically require potent Th1 inducers such as CpG motifs found in DNA vaccines (24) and oligonucleotide adjuvants (25, 26), mycobacterial cell wall antigens either in the form of live Bacille Calmette–Guerin (BCG) (27) or complete Freund’s adjuvant (17), or lipid-based adjuvants, such as liposomes (28) and oil-in-water emulsions (29). Several of these powerful stimuli have recently been shown to act via ligation of pattern recognition receptors (PRR) [e.g., toll-like receptor (TLR) 9 in the case of CpG motifs] (30). Even when an apparently “balanced” Th1/Th2 response has been generated in neonates, recall responses may still reveal an underlying Th2 bias (31).
The mechanisms by which subsequent antigen recall results in a Th2-biased response are still under active investigation. Neonatal mouse T cells produce significantly higher levels of IL-4 within 48 h of in vitro T cell receptor (TCR) stimulation (32) compared to adult T cells, which typically take several days to produce high levels of Th2 cytokines (33). This readiness to produce Th2 cytokines has been attributed to hypomethylation of the Th2 cytokine regulatory region on chromosome 11 in neonatal murine CD4+ T cells. A similar state of epigenetic control over Th2 cytokine production has also been observed in human neonatal CD4+ T cells (34). Yoshimoto and colleagues have recently shown that epigenetic modifications of the Th2 regulatory regions occur during fetal development in the mouse (35). The Th2 regulatory regions in the 14-day fetal thymus are hypomethylated and adult-like methylation patterns appear only 3–6 days after birth along with a decreased propensity to produce Th2 cytokines. Interestingly, this hypomethylation is restricted to the Th2 regulatory regions, and is not seen in the regulatory regions of the IFNγ or Foxp3 loci. Other factors that could potentially influence Th2-biased immune responses in neonates include relative antigen loads (36), lower total numbers of T cells in neonatal organs (37) and the presence of fetal-origin T cells that are strongly Th2-skewed (38).
Cross-regulation between Th subsets is well known and may be particularly effective in the very young. For example, there is evidence that Th1-type signaling is actively suppressed by Th2 cytokines in the context of both antigen challenge and recall in neonates. Using TCR transgenic mice, Li et al. showed not only that recall responses are primarily Th2-like but that these responses are accompanied by apoptosis of antigen-specific Th1 cells (39). This apoptosis of Th1 cells is driven predominantly by IL-4, is enhanced by IL-10, and occurs only in neonatal T cells (40). Neonatal (but not transplanted adult) Th1 cells co-express IL-13Rα1 and IL-4Rα. Upon exposure to IL-4 after antigen challenge, Th1 cells that carry this heteroreceptor undergo apoptosis (12), resulting in a Th2-biased antigen recall response. IL-13Rα1 upregulation on these Th2 cells is due to relatively low frequencies of IL-12-producing (CD8α+ CD4−) dendritic cells (DCs) in the neonate- a phenomenon that is reversed by day 6 of life when CD8α+ CD4− DCs mature and begin to secrete IL-12 (41). By ~8 days after birth, neonatal murine T cells respond to gradual increases in IL-12 by upregulating IL-12Rβ2 and suppressing IL-13Rα1, contributing to the sustained survival of antigen-specific Th1-type T cells (42).
Although the differences between neonatal and adult T cell responses are particularly striking, the capacity to mount competent humoral responses also varies considerably with age as different “waves” of B cell development occur through early-life (43). For example, antibody responses to T cell independent antigens such as polysaccharide antigens can be very weak during the first years of life. Compared to the adult humoral response to any given antigen, the neonatal response is characterized by generally lower levels of antibodies produced with a delayed onset, less efficient antibody affinity maturation (14) and higher levels of B cell apoptosis (44). Despite these clinical observations, major differences between neonatal and adult B cells have not yet been identified at the molecular level. As a result, the relative “defects” in antibody production for T cell independent antigens are likely attributable to other factors such as delayed appearance of B cells fully competent to handle such antigens or deficiencies in the organization of secondary response sites, such as lymphoid and follicular centers, which develop only a few weeks after birth in mice [reviewed in Ref. (14)].
Innate Immunity in the Neonate
Striking differences in innate immunity between neonates and adults have also been characterized, particularly with regard to IL-12, a key innate Th1 cytokine. The half-life of IL-12p40 mRNA is decreased in cord blood mononuclear cells and these cells produce far less IL-12 than adult peripheral blood mononuclear cells (PBMC) in response to LPS (45). The addition of IL-12 to cord blood cultures increases Th1-type responses to recall antigens in vitro (46), increases the activity of natural killer (NK) cells and enhances IFNγ production (45), and suppresses the induction of IL-13Rα1 (41). However, the administration of supplemental IL-12 to 1-week-old mice in vivo is not well tolerated, suppressing weight gain and increasing mortality compared to older mice (47). DCs are the main producers of IL-12 (12) and neonatal mice have fewer splenic DCs than adult animals (48), as well as weaker responses to antigen stimulation in vitro and in vivo (12). It is interesting that neonatal B cells can suppress both IL-12 production and signaling in neonatal DCs in an IL-10-dependent manner (49, 50). Because DCs are positioned at the nexus of the innate and adaptive immune responses, the characteristics of neonatal DCs are likely central to the relatively weak Th1 responses observed in neonates. Monocyte-derived DCs from human cord blood have decreased markers of activation, as well as lower IL-12p35 mRNA expression upon stimulation with ligands such as LPS, CD40 ligation, or Poly I:C, and are poor inducers of IFNγ from adult T cells (51). Neonatal DCs also produce less IFNα and IFNβ than adult cells (52, 53). Recently, it has been suggested that poor production of IFN by early-life DCs is due, at least in part, to posttranscriptional downregulation of TLR7/9 signaling and increases in the regulatory miRNAs, miR146 and miR155 (54). One important outcome of decreased type 1 IFN signaling would be diminished IFNγ responses in neonates.
Both cell surface and cytoplasmic PRRs, including TLRs, NOD-like receptors, and retinoic acid induced gene I (RIG-I)-like receptors, play important roles in the recognition of common microbial products (e.g., LPS, double stranded RNA) and in triggering immune responses in key innate effector cells such as monocytes and DCs. Responses of cord blood DCs to TLR ligation are both qualitatively and quantitatively different from adult DC responses, despite having similar levels of mRNA (53, 55). Using agonists specific to each TLR, Kollmann et al. (56) studied single cell responses of DCs and monocytes in human cord blood compared to neonatal or adult cells differentiated from PBMC. Overall, the neonatal monocytes and DCs produced more IL-6, IL-23, and IL-1β, which can induce Th17 differentiation/activity, as well as IL-10, a classic immunosuppressive cytokine, but less of the Th1-associated cytokines: IFNα, IFNγ, tumor necrosis factor (TNF)α, and IL-12p70. In another study of early-life responses, the ratio of TLR-induced IL-6/TNFα production by neonatal monocytes was high compared to adult cells. Newborn serum was found to have a similarly skewed ratio of these cytokines (57). Serial measurements in human infants suggest that the ratio of TLR-driven inflammatory (e.g., IFNα, IL-12p70, IFNγ) vs. suppressive/regulatory cytokines (e.g., IL-10) shifts slowly toward “adult” values over the first 2 years of life (58). Several factors likely contribute to the sluggish activity of TLRs in neonatal immune cells, including lower MyD88 levels (59), decreased DNA and co-activator binding of IFN regulatory factor 3 (52), and decreased nuclear translocation of IFN regulatory factor 7 (53). Together, these data suggest that stimulation of innate immune cells in neonates/infants favors Th2/Th17 and possibly Treg differentiation rather than Th1 development. This age-related bias in T cell education could plausibly play a pivotal role in the subsequent development of asthma.
There are still other differences between neonatal and adult innate response capabilities. For example, fetal and the neonatal invariant natural killer T cells (iNKT) cells produce equal amounts of IFNγ and IL-4 upon receptor stimulation in sharp contrast to adult iNKT cells that produce predominantly IFNγ (60). Although NK cells are present in higher numbers in the peripheral blood of neonates compared to adults, they express more inhibitory receptors, have truncated maturation and have lower overall functionality (61). The NK maturation defect in neonates has also recently been linked to transforming growth factor-beta (TGFβ), since murine NK cells engineered to lack TGFβ receptor were capable of fully maturing by 10 days after birth (62). Neonatal macrophages, upon stimulation with LPS and polysaccharide antigens, produce more IL-10 than adult macrophages, and secrete less IL-1β, IL-12, TNFα, probably due to weak TLR signaling (63). Neonatal macrophages are also less responsive to IFNγ due to a defect in STAT1 phosphorylation (64) despite having adult levels of phagocytic function (65). Walk and colleagues studied the expression of a number of inhibitory receptors on neonatal cells, and found increased expression of LAIR-1, CD31, and CD200 on a wide range of innate immune cells including neutrophils, monocytes, and NK cells as well as CD4+ and CD8+ T cells (66). At least some of the apparent “defects” in neonatal immune cell function can be overcome using specific stimuli either alone or in combination. As mentioned above, TLR8 signaling can drive a strong pro-inflammatory response from neonatal monocytes but can also induce neonatal NK cells to produce adult levels of IFNγ in an IL-12-dependent fashion (67).
Neonatal Lung Mucosal Immunity
During the postnatal period, the lung continues maturation begun in utero and undergoes alveolarization (mice, pnd4-21; humans, 36 weeks preterm to 1–2 years) and microvascular maturation (mice, pnd4-21 mice; humans, 0–3 years) (68, 69). A strong type-2 immune cell bias characterizes the lung mucosa during alveolarization in mice as populations of innate type 2 lymphoid cells (ILC2), mast cells, eosinophils, and basophils increase (69). Furthermore, a large proportion of tissue resident alveolar macrophages, unique populations that are self-maintained throughout life and which develop from fetal liver monocytes beginning at pnd3 in mice (70), express CD206, indicative of a type-2 alternatively activate phenotype (71). Conventional CD11b+ neonatal murine lung DCs, though few in number, process antigen more efficiently and more readily express CCR7 (69, 72, 73) than adult DCs and, compared to CD103+ DCs, preferentially migrate to the draining (mediastinal) lymph node and promote Th2 responses (69). In mice, lung delivery of house dust mite (HDM) extracts at this early stage of development promotes enhanced allergic airways disease [i.e., increased eosinophil recruitment to the lung, airway hyperresponsiveness (AHR)] (69, 73, 74), in a manner that is dependent upon IL-33 (discussed in detail below) and influenced by preferential differentiation of CD4+ T cells to a Th2-type phenotype expressing IL-4, IL-5, and IL-13. In fact, additional studies in mice have investigated canonical T lymphocyte responses in the neonatal vs. adult lung (72). In response to anti-CD3 treatment in vitro, CD3+, CD4/CD8 double negative T cells, which are present in greater frequency during the neonatal period than in adulthood, more readily express GATA-3 and produce more Th2 cytokines, IL-4 and IL-5 (72). Interestingly, priming of neonatal or adult lung DCs with BCG induced a Th2 response when cocultured with neonatal lymph node T cells, a response that switched to Th1 when cocultured with lymph node T cells from adult mice (72). Altogether, these data highlight the importance the temporal window of the alveolarization stage of lung development to promote rapid, detrimental Th2-type inflammatory responses in the lung.
Compelling new data implicate changes to the lung microbiome in differential regulation of maladaptive type-2 allergic responses between neonates and adults. The predominance of Firmicutes and Gammaproteobacteria in the lung is associated with allergic airways disease in both murine models of asthma (75) and human asthmatics (76) and are the major families that colonize the lung during the alveolar stage of development. These commensals change throughout ontogeny as the lung milieu adapts to harbor Bacteroidetes in adulthood (73). Interestingly, Helios+ Treg (CD4+Foxp3+CD25+) cells are abundant in the neonatal lung during alveolarization, but are non-tolerogenic as repeated HDM exposure causes robust eosinophilic airway inflammation, AHR, and mucus production associated with allergic airways disease. Later in life, when the lung microenvironment shifts to support Bacteroidetes, Helios− Tregs are abundant and successfully promote regulatory, antiinflammatory responses thereby providing protection to adult mice from allergic airways disease upon exposure to HDM (73).
The neonatal lung is comprised of type 2 innate immune cells and DCs, which rapidly home to the mediastinal LNs to educate naïve CD4+ T cells to develop Th2 responses. Furthermore, the postnatal lung supports a microbiome that promotes allergy and ineffectual Treg responses. Altogether, this type-2 biased neonatal mucosal lung environment has the potential to create a “perfect storm” for asthma development upon exposure to viruses or allergens. How this naturally biased state might synergize with early-life respiratory infections to create a lung microenvironment that favors the development of asthma is discussed in the next section.
Viral Respiratory Infections and Asthma
Allergic asthma is defined as a chronic inflammatory response to inhaled allergens characterized by intermittent airway obstruction, increased Th2 cytokine production, AHR and mucus production. At least 300 million people worldwide have asthma (77, 78). Despite intensive study, the etiology of asthma is still poorly understood, although a family history of atopy is consistently shown to be an important risk factor [reviewed in Ref. (79, 80)]. Genetics undoubtedly play a pivotal role in the development of asthma but genotype is not fully determinative. Environmental factors influence not only the overall risk of asthma but also the onset and severity of disease. As outlined above, respiratory infections are major causes of short-term morbidity and mortality in the first years of life (81) and viral infections are associated with up to 60% of all asthma exacerbations in the young (82). In addition to this apparent “direct” association with exacerbations (83–85), it is also possible that early-life exposure to specific viruses acts to prime some individuals for development of asthma later in life. Indeed, a growing body of epidemiological evidence suggests that early-life respiratory virus infections predispose infants to the development of asthma (see below). Murine models of both the induction of AHR and the exacerbation of existing airway disease in neonatal and infant animals are beginning to provide some mechanistic understanding of these phenomena. Because RSV, influenza viruses, and RVs are ubiquitous causes of respiratory infection in young infants, these agents will be our primary focus. Table 1 provides an overview of studies linking viral respiratory infections and wheeze or asthma in children.
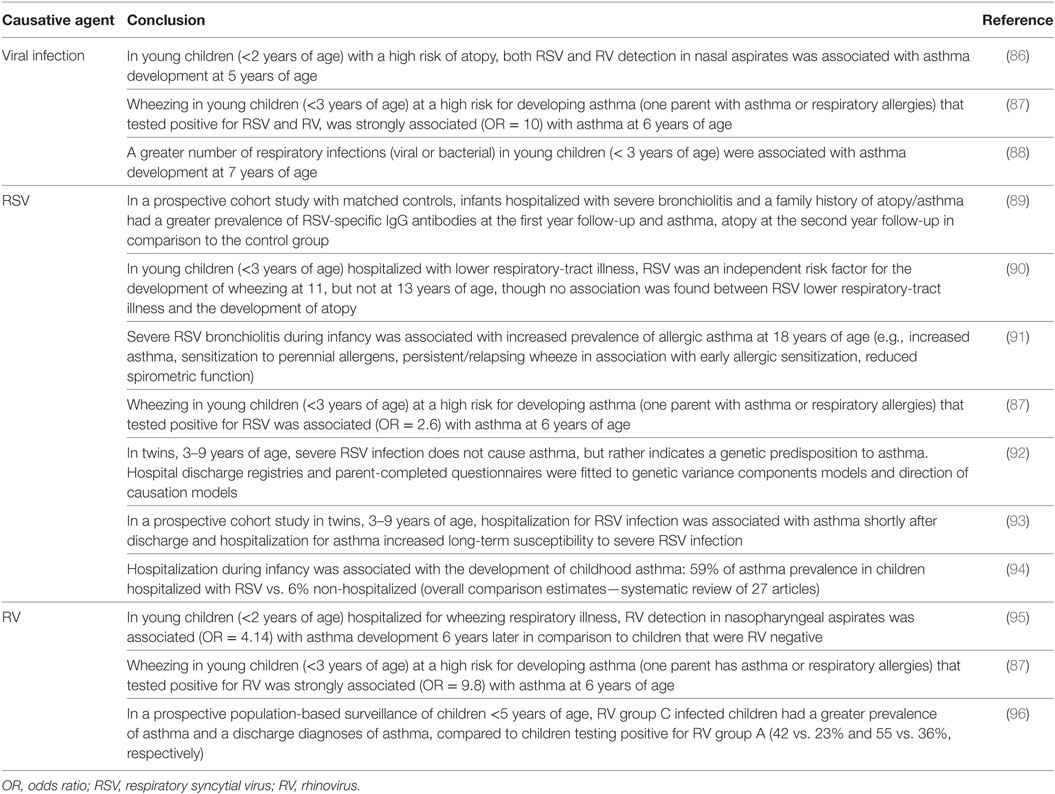
Table 1. Evidence linking respiratory viral infection in infancy and asthma development in childhood.
Respiratory Syncytial Virus
Background, Prevention, and Treatment
There are two principal antigenic sub-types of RSV (A and B), each with multiple genotypes based on their surface glycoprotein G (97). Although both sub-types can infect humans, most (>85%) symptomatic disease is attributable to type A viruses. These viruses are the leading global cause of serious viral respiratory illness in infants (98, 99). Based on seroepidemiology, nearly 90% of all infants are infected at least once by the age of two. There is no vaccine for RSV and treatment options are limited. Although aerosolized ribavirin may provide marginal benefit in severely ill children, this approach is cumbersome and used infrequently (100). The monthly prophylactic use of a monoclonal antibody that targets the surface fusion (F) glycoprotein (e.g., palivuzimab™ and others) can reduce hospitalization rates by about 50% (101) as well as the total number of wheezing days in the first year of life (102). However, the cost of prophylaxis is so high that this approach is generally restricted to infants at greatest risk of severe disease (preterm infants, infants with immunodeficiency, etc.) [reviewed in Ref. (103)].
Epidemiologic Link With Asthma
A broad range of epidemiological data strongly supports the association between early-life RSV-related hospitalization, and the subsequent development of asthma in childhood (90) with effects lasting into early adulthood (91). A recent meta-analysis of infants hospitalized with severe RSV during infancy found a 63% prevalence of asthma in children ≤5 years of age that increased to 92% in children between the ages 5 and 12. After 12 years of age, asthma prevalence in these children falls to 48% but all of these numbers are striking compared to background asthma rates of 1–7% in children with no history of early-life, RSV-associated hospitalization (94). The mechanisms that underlie this strong epidemiologic association are not yet fully understood and it has been argued that hospitalization with RSV is simply a “marker” for children genetically predisposed to asthma as opposed to an environmental risk factor for asthma development (104). In fact, it is likely that both are true to some degree. Given the frequency of RSV infection in the first years of life, hospitalization due to severe infection is relatively rare regardless of genetic background, occurring only when RSV moves into the lower respiratory tract (LRT), causing pneumonia and bronchiolitis (105). In most healthy, full-term babies and infants, RSV infection is limited to the upper respiratory tract and causes only mild-moderate symptoms. In a 10-year study in the US, the rates of RSV-related hospitalization ranged from 48.9 per 1,000 in infants less than 3 months of age to 26 per 1,000 in infants older than 1 year (106). Why some children progress to LRT complications is still unknown, but risk factors include a family history of atopy, preterm birth, congenital heart disease, and low levels of maternal anti-RSV antibodies [reviewed in Ref. (107)].
Immunology of RSV-Related Asthma Development
RSV Contains Pathogen-Associated Molecular Patterns That Induce Innate Type 2 Cytokines
The tissue tropism of RSV is narrow; restricted to cells of the airway epithelia both in vitro and in vivo. RSV entry on the apical side of these cells (108) is mediated by the viral attachment (G) and fusion (F) glycoproteins (109) that utilize surface glycosaminoglycans such as heparin and nucleolin (110), respectively, as receptors. As outlined above, a Th2-biased and/or immunosuppressive microenvironment in the lungs is associated with both the development and severity of asthma. There is now considerable evidence that the major RSV surface glycoproteins can directly impact the Th1/Th2 balance in the lung (111). Th2-biased airway inflammatory responses, including Th2 cytokine production and influx of eosinophils into the airways, are induced upon vaccination of BALB/c mice with vaccinia virus expressing the RSV G-protein (112–114). Compelling data implicate IL-9, a cytokine associated with the development of asthma (19) in RSV G-protein-dependent induction of Th2-biased airway inflammation in these mice (115). The RSV F protein may also contribute to the development of a Th2 microenvironment in the lung by signaling through TLR4 (116, 117) and there is strong recent data linking TLR4 genotype to severe RSV disease in humans (118). RSV infection of airway epithelial cells (both immortalized cell lines and primary human cells) in vitro increases expression of TLR4 (119) and epithelial cell-specific TLR4 signaling is required for Th2-type responses in the murine lung (120–122). For example, allergic airways disease induced by HDM depends upon airway epithelial cell-specific TLR4 expression and is associated with secretion of several innate cytokines associated with Th2-type responses [e.g., IL-25, IL-33, and thymic stromal lymphopoietin (TSLP)], as well as the classic Th2 cytokines IL-4 and IL-13 (122). RSV infection of bronchial epithelial cells also induces production of TSLP via activation of retinoic acid induced gene I (RIG-I) and downstream activation of nuclear factor-κB (123). Importantly, RSV infection of mice sensitized to the model allergen ovalbumin (OVA) leads to an increase in Th2 cytokine production in the lung following OVA challenge suggesting that the RSV effect on Th1/Th2 balance in the respiratory tract is not restricted to viral antigens (124, 125). More recently, prior RSV infection of young mice has been shown to enhance allergic airways disease induced by HDM (126).
Innate Type-2 Cytokines Activate Antigen-Presenting Cells
Recognition of the powerful effects of IL-25, IL-33, and TSLP produced by respiratory epithelial cells is an important new element in understanding the potential for respiratory viruses to cause and/or exacerbate asthma. These epithelial-origin cytokines play a pivotal role in both the initiation of Th2 type responses and the progression of allergic diseases (127).
Blockade of IL-25 or the absence of its receptor, IL-17RB, reduces Th2 cytokine production, mucus production, and/or AHR in murine RSV infection and RSV-induced asthma exacerbation models (128). Moreover, in the absence of NK cells, IL-25 secreted from airway epithelial cells upregulates the Notch ligand, Jagged1 on the surface of DCs, which induces the development of an RSV-specific Th2 response (129).
Treatment of murine bone marrow-derived macrophages with exogenous IL-25 or IL-33 can induce production of IL-5 and IL-13 in vitro (130). Intraperitoneal treatment of mice with IL-33 results in the differentiation of macrophages from the small intestinal lamina propria into an alternatively activated (M2 or AAM) phenotype in a STAT6-independent fashion. Such peritoneal macrophages are major producers of IL-13 in vivo. Furthermore, IL-33 links viral infection, macrophages, and ILC2-dependent production of IL-13 to AHR (131–134) and induces ILC2- and IL-13-dependent DC trafficking to the lymph nodes, promoting Th2 adaptive immunity (135). In the case of RSV infection models, IL-33 gene expression as well as the number of leukocytes expressing the IL-33 receptor (ST2) increase in adult BALB/c mice infected with RSV and interestingly, ST2 blockade decreases lung eosinophil recruitment, IL-13 levels and mucus production, without affecting IFNγ levels (136). ILC2s are an important source of IL-13 secretion post-RSV infection and evidence that they play a role in enhanced disease is provided by data showing that adoptive transfer of lung ILC2s purified from RSV-infected mice 2 h prior to RSV infection dramatically increases production of IL-13 and subsequent eosinophil infiltration (137). Similarly, IL-33 production in the lungs of RSV-infected neonatal mice contributes to ILC2 expansion as well as Th2-biased airway inflammatory responses following RSV re-infection in adults (138). Evidence that IL-33 may contribute to pathogenesis in human disease is provided by data showing that IL-33 levels are increased in nasal aspirates of infants hospitalized with RSV infection (138). Similarly, IL-33 mRNA levels are greater in nasal aspirates of infants hospitalized with RSV bronchiolitis with a family history of atopy compared to those with no such history (139).
Thymic stromal lymphopoietin likely also plays a role in the development of Th2 responses in RSV infection. In rat primary airway epithelial cell cultures, infection with RSV triggers an immediate increase in TSLP mRNA and protein, which induces myeloid DCs to express markers associated with Th2 polarization (140). Ex vivo RSV infection of human airway epithelial cells of healthy children and children with asthma results in TSLP induction and has been shown to contribute to Th2 inflammation (123). Infection of human primary bronchial airway epithelial cells also results in upregulation of functional TSLP receptor (TSLPR) on these cells, suggesting a feedback loop in which TSLP binding to its receptor increases production of more TSLP (141). TSLP also acts directly on DCs. Lung DCs, along with alveolar macrophages and ILC2s are among the “first responders” to viral infections and allergen exposure in the lung. The relationship between lung epithelial cells and DCs and the development of asthma has been the subject of several recent reviews (142–144) and provides a framework for understanding how RSV-infected epithelial cells may program lung DCs to promote Th2 immunity. TSLP secreted from epithelial cells infected with RSV (123, 140) induces OX40L expression on the surface of DCs (145) that, in turn, drives T cells toward a Th2 cell phenotype (146). A further link between TSLP and IL-25 in RSV infection is provided by evidence that IL-25 enhances the memory Th2 response induced by TSLP-activated DCs (147). IL-33 also upregulates OX40L surface expression on DCs, effectively promoting Th2 immunity in both allergy and RSV infection models (138, 148). TSLP signaling in ILC2s is also necessary for IL-13 production by these cells as TSLPR KO mice and blockade of TSLP signaling with anti-TSLP neutralizing antibody decrease IL-13 lung protein levels, mucus, AHR and weight loss during RSV infection (149).
In humans, when RSV infects primary myeloid and plasmacytoid-derived DCs (150) from healthy volunteers, the viral G-protein decreases DC activation (151). Suppression of DC maturation has also been observed by the viral non-structural (NS) proteins, NS1 and NS2 (152). Moreover, NS1-dependent activity in RSV-infected DCs suppresses the activation and proliferation of both migratory CD8+ T cells (CD103+CD8+) and Th17 cells while supporting the activation and proliferation of Th2 cytokine-producing CD4+ T cells (153). Together, these data suggest that RSV infection of epithelial cells and DCs may act synergistically to elicit a Th2-biased response in both mice and humans. This response pattern is likely accentuated by the already Th2-biased nature of the neonate.
Yet another piece of this puzzle may be the induction by RSV of long-lived, lung-resident macrophage populations. RSV infection in adult mice induces polarization of macrophages toward an AAM (M2) phenotype (126, 154). Similar polarization is observed upon RSV infection of peritoneal macrophages ex vivo (154). The classification of macrophages is largely based on cytokine production profiles upon activation and, as is the case with Th1 and Th2 lymphocytes, the cytokines IFNγ and IL-4 appear to play central roles in the development of M1 and M2 macrophages, respectively. Upon activation, M1 macrophages (also called classically activated macrophages or CAM) typically secrete inflammatory cytokines, such as IFNγ, IL-12, and IL-1, whereas M2 macrophages secrete IL-4, IL-13, and IL-10 (155). M2 macrophages play an important role in tissue repair (156, 157) [reviewed in Ref. (158)] mediated, at least in part, by TGFβ and platelet-derived growth factor. Studies in both human infants (159) and neonatal mice (160) have demonstrated that the production of TGFβ is increased upon RSV infection. On the one hand, this increase could reflect an adaptive M2 reparative response to the lung injury caused by RSV infection as shown by Shirey and colleagues (154). On the other hand, over-enthusiastic repair of virally damaged airways could be maladaptive with long-term changes in macrophage phenotype that promote pathologic changes in airway structure and function (131, 134, 161). It is, therefore, interesting that RSV infection of neonatal mice is associated with long-term increases in collagen deposition and airway remodeling (125, 162). Although the precise mechanisms by which RSV infection induces M2 polarization are still unknown, it seems likely that production of TSLP, IL-25, and/or IL-33 by RSV-infected or -exposed respiratory epithelial cells contributes to both M2 differentiation and long-term maintenance of a Th2-biased macrophage population in the lung (123, 130, 163, 164). Furthermore, as noted above, the NS1 and NS2 proteins of RSV inhibit the type-I IFN response in human macrophages (165), which could also contribute to the induction of M2 macrophages.
Limitations of These Studies
Much of the evidence outlined above in support of a Th2-biasing effect of RSV in the lung has been obtained using adult mice and human cells/cell lines. Neither of these strategies is ideal. The limitations of cell lines and even primary cells are obvious since these reductionist models cannot reproduce the complexity of in vivo host–virus interactions. The adult mouse model of RSV infection also has important drawbacks since adult mice are not susceptible to RSV. This model requires the instillation of high titers of virus (106–107 50% tissue culture infective dose/mL) directly into the LRT. Furthermore, RSV replication in adult mice is relatively poor and occurs primarily in alveolar pneumocytes rather than the bronchiolar epithelial cells as occurs in humans (166, 167). Perhaps most importantly, RSV infection of adult mice induces a potent Th1 response that rapidly clears the virus. The role of RSV-specific Th2 responses in this model may be restricted to the prevention of exaggerated Th1-mediated lung pathology (154). In the case of human clinical data, it is difficult to resolve the “chicken-and-egg” problem: do children with a genetic propensity to mount Th2 responses and develop asthma suffer more severe early-life RSV or does early and severe RSV infection cause the subsequent development of asthma (83, 168)?
Neonatal RSV Infection Model
In an attempt to address these limitations, several groups, including ours, have developed models of RSV infection in neonatal and young mice with re-infection or exposure to allergens later in life (126, 160, 162, 169, 170). These models demonstrate that early-life RSV infection (within 7 days of birth) elicits little IFNγ production in contrast to adult animals (169) and leads to a “pro-asthmatic” phenotype characterized by AHR, mucus production, airway remodeling, and severe disease upon subsequent allergen exposure or live RSV challenge (126, 160, 162, 170). Consistent with these Th2-biased responses, IL-4Rα is increased on pulmonary CD4+ T cells following RSV reinfection of adults (171). The exaggerated Th2-inflammatory response upon re-infection is dependent upon the age at initial infection and production of both IL-13 and IL-33 (138, 170) and is enhanced by the presence of anti-RSV IgE antibodies (172). Specifically targeting Th2 responses in the neonate can reduce these exaggerated responses upon adult RSV reinfection. For example, delivery of antisense oligonucleotides targeting the IL-4Rα (173) or a cell penetrating peptide targeting the STAT6 transcription factor (activated by both IL-4 and IL-13) (162) to neonatal mice at the time of RSV infection reduces enhanced disease upon RSV re-infection of adults. Interestingly, human cord blood CD4+ T cells also upregulate IL-4Rα upon ex vivo RSV stimulation (171). Consistent with these data, development of enhanced disease is T cell dependent in the neonatal challenge- adult rechallenge model. However, reduced inflammatory responses are seen only if CD4+ T cells are depleted at the time of adult re-infection but not during the neonatal exposure (174). On the other hand, depletion of CD8+ T cells during either the neonatal infection or adult re-infection significantly decreases enhanced disease in mice (174). How CD8+ T cells exposed to RSV during neonatal infection promote disease in adult re-infection is unclear, and somewhat paradoxical, given the protective role that CD8+ T cells are believed to play in recovery from viral infections. It is also possible that CD8+ T cells play slightly different roles in mouse vs. human disease (175). How CD4+ and CD8+ T cell populations influence the pathogenesis of RSV infection in reinfected adult mice is intriguing and deserves further evaluation. Finally, repeated RSV infection of weanling mice interferes with Treg-mediated tolerance and increases susceptibility to allergic asthma (176).
As described in the first section of this review, the neonatal immune system is characterized by a pre-existing Th2 bias. The addition of RSV infection of airway epithelial cells with production of the type-2 innate cytokines, IL-33 (138), TSLP (123), and IL-25 (128), would, therefore, be predicted to create an even more exaggerated type-2-biased microenvironment in the lung with activation of other immune cells (e.g., M2 macrophages, DCs, and/or ILC2 cells) and the development of AHR. When neonatal mice are treated with antibodies to neutralize TSLP, IL-33 or their target expressed on DCs, OX40L, the ability of early RSV infection to prime pathological Th2 responses upon re-infection of adults is reduced (138, 145). As discussed above, TSLP and the other innate type-2 cytokines, IL-25 and IL-33, all promote amplification, differentiation, and maintenance of M2 macrophages (130, 163, 164). Together, these data suggest that many different cells and cytokines, as well as other yet to be determined factors have the potential to contribute to the RSV-induced, Th2 microenvironment and the subsequent development of asthma (Figure 1).
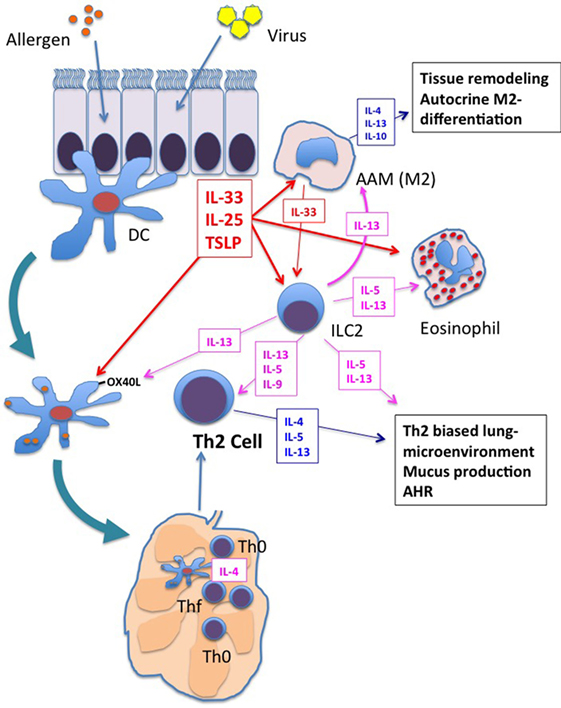
Figure 1. Shared responses in lung innate cells in response to respiratory virus infection and allergen exposure. Viral infection and/or allergen exposure of bronchial epithelial cells results in the secretion of the innate type-2 cytokines: thymic stromal lymphopoietin (TSLP), interleukin (IL)-25, and IL-33. Dendritic cells (DCs), macrophages, and innate type 2 lymphoid cell (ILC2s) are among the first responders in the lung. Each of these cytokines increases activation of DCs, including increased expression of OX40L. OX40L expressing DCs drive Th2 differentiation in the lung mediastinal lymph nodes. These innate type-2 cytokines also promote differentiation of M2 alternatively activated macrophages (AAM), which secrete type-2 cytokines IL-4, IL-13, and IL-10 and orchestrate type-2 inflammation and tissue repair in the lung. M2 macrophage cytokine/chemokine production contributes to enhanced type-2 biased inflammation and airway remodeling. IL-5 and IL-13 production from ILC2s promotes eosinophil influx and responses in DCs and macrophages that further enhance type-2 inflammation, Th2 differentiation, and airway hyperresponsiveness (AHR). Each of these responses is enhanced in neonates upon exposure to respiratory viruses, setting the stage for enhanced type-2 inflammation upon exposure to allergens.
An alternative to the “too much Th2” explanation for Th2-biased neonatal immune system would be “too little Th1”. IFNγ plays a key role in the induction and maintenance of Th1 responses and infection of neonatal mice with recombinant RSV expressing IFNγ prevents enhanced disease upon re-infection (177). A large proportion of the immune cells present in the neonatal mouse lung are macrophages, and these cells are particularly sensitive to stimulation by IFNγ. Indeed, the work of Empey and colleagues shows that IFNγ plays a central role in the balance between the inflammatory M1 macrophage phenotype and the immunosuppressive, M2 phenotype (71). While lung macrophages from adult mice effectively induce M1 macrophage markers in response to RSV, neonatal macrophages require exogenous IFNγ to express M1 markers, produce inflammatory cytokines and drive efficient viral clearance (71). Moreover, administration of intranasal clodronate liposomes that depletes macrophages significantly reduces the ability of IFNγ to promote viral clearance (178). Furthermore, Yamaguchi and colleagues (179) have shown that treatment of neonatal mice with the powerful Th1 adjuvant, CpG, prior to RSV infection prevents enhanced disease upon re-infection, possibly by promoting activation of antigen-presenting cells and production of IFNγ from NK cells. Remarkably, CpG treatment even 4 weeks after early-life infection provides protection (179). These observations raise the possibility that RSV vaccines incorporating CpG or other Th1-biasing adjuvants might be useful not only to prevent infection (180) but also to block the later development of asthma. Whether or not the effects of “more” IFNγ are antigen-specific or simply reflect an adjuvant-modified baseline “potential to respond” in the lung is also an important question. This last possibility is strongly suggested by the work of Remot and colleagues who observed that mucosal vaccination of neonatal mice with nanostructures formed by the RSV nucleoprotein (N) and a combination of Th1-biasing adjuvants (E. coli enterotoxin LT and CpG) provides greater protection from immunopathology upon reinfection by RSV later in life compared to nanostructures containing the N protein + LT (181). Together, these data suggest that preventing too much Th2 activity (driven by RSV or other stimuli) or promoting Th1-type activity (or both) may establish a long-term more “balanced” lung microenvironment that resists subsequent development of asthma.
As discussed above, RSV infection inhibits type I IFN production and plasmacytoid dendritic cell (pDC) responses (71, 150, 178).As a result, the maintenance of pDCs during RSV infection may promote beneficial Th1-type responses including the expansion of RSV-specific CD8+ T cells. In the challenge–rechallenge model, treatment of neonatal mice during the initial RSV infection with either IFNα or adoptively transferred adult pDCs leads to upregulation of IFNα expression and diminished Th2-biased lung inflammation when these mice are reinfected as adults (182). Further support for the role of pDCs comes from experiments in which adult mice are exposed to FMS-like tyrosine kinase 3 ligand (Flt3-L) prior to RSV infection. In response to Flt3-L, both conventional DC (CD11b+CD11c+) and pDC (CD11b−CD11c+B220+) populations expand in the lung and draining lymph nodes but only the pDCs protect against airway hyperreactivity, exaggerated Th2 cytokine expression, airway inflammation and mucus production (183). These Flt3-L-induced pDCs have upregulated type I IFN (IFN-α/β) expression and support the expansion of CD8+ T cell populations that decrease viral load. Depletion of these pDCs reduces the protective CD8+ Th1 response with greater AHR, mucus, viral titers, and Th2 cytokine expression (183, 184). Similarly, Flt3-L administration prior to neonatal RSV infection reduces airway mucus secretion and airway eosinophilia but promotes Th1 RSV-specific CD8+ T cells upon adult re-infection (185). Although these observations suggest that Flt3-L protects against RSV infection by inducing pDCs to produce type I IFN, alveolar macrophages (CD11c+SiglecF+) rather than pDCs (or epithelial cells), are thought to be the main producers of type I IFN in the adult mouse lung (186, 187). Regardless of whether pDCs or alveolar macrophages are the predominant source of type I IFN, these studies strongly suggest that promoting type I IFN production in the lung during early-life RSV infection is likely to lead to adaptive Th1 responses while suppression of type 1 IFN production may favor maladaptive Th2 responses.
Taken together, the data summarized above strongly suggest that RSV infection of the neonatal lung adds a further Th2 influence to an already Th2-biased respiratory microenvironment, with likely contributions from respiratory epithelial cells as well as multiple innate cell populations (e.g., macrophages, DCs, NK cells, ILC2s). The presence of multiple autocrine and paracrine amplification loops between these cells likely leads to the production of excessive IL-4 and IL-13 in the lung that increase both the short- and long-term risk that exposures to otherwise innocuous allergens will lead to immunopathologic responses including asthma (Figure 1).
Influenza A and B Viruses
Background, Prevention, and Treatment
Influenza viruses are far more diverse than RSV due, in large part, to their genetic organization (eight independently segregating genes in the A and B viruses that most commonly infect humans) and the enormous genetic reservoir of influenza viruses in aquatic birds and a number of mammalian species (e.g., most notably pigs but also cats, whales, elephants, skunks, among others) (188). The influenza viruses remain a leading cause of childhood pneumonia globally (189) and early-life infections are common despite the near universal availability of influenza vaccines in resource-rich settings (190). It has been estimated that without vaccination 10–40% of young children are infected by an influenza virus every year (191). Although a number of antivirals are available for influenza infections (e.g., M2 and neuraminidase inhibitors), these drugs have only modest efficacy and their use is further limited by resistance that is either pre-existing or is rapidly induced during treatment (192). Although influenza infections are clearly associated with exacerbations of asthma [reviewed in Ref. (193)], the putative link between early-life influenza and the subsequent development of asthma is much more tenuous. Compared to the large body of work implicating RSV (above), far less work has been done to address this question for influenza viruses.
Evidence for a Link Between Influenza Viruses and Asthma
At the current time, there is neither epidemiologic nor experimental evidence in humans suggesting a strong association between early-life influenza infection and asthma. Although work with murine models tends to support an association, these data are not consistent from one model or laboratory to another. Differences in the age of infection, both mouse and virus strain (i.e., human vs. mouse-adapted) and the infective dose may all contribute to these apparently contradictory results. For example, a study by Dahl et al. (194) showed that LRT infection of adult BALB/c mice with influenza A (HKx31 strain) at 6–8 weeks of age could predispose to AHR upon subsequent allergen sensitization (keyhole limpet). Surprisingly, AHR in this model was associated with both Th1-type (IFNγ, IgG2a) and Th2-type (IgG1) antigen-specific responses and the allergic phenotype could be adoptively transferred with pulmonary DCs. Using a similar protocol with OVA as a model allergen in BALB/c mice, Barends et al. found that infection with a mouse-adapted influenza A strain (A/PR/8/34) at the time of OVA challenge decreased Th2 cytokine production in the lungs but increased infiltration by eosinophils (195). Chang et al. have reported that infection of adult BALB/c mice with a reassortant H3N1 influenza A (Mem71) increases alveolar macrophage IL-33 production, driving natural helper cells (i.e., ILC2s) to produce IL-13 that promotes the development of AHR (132). Recently, elevated pleural IL-33 and ILC2s were found to mediate the induction of asthma-like responses (e.g., AHR, production of Th2 cytokines) following pdmH1N1 infection in Rag1−/− mice (on a C57Bl/6 background) that lack functional T and B lymphocytes (196).
Like the observations in adult mice, the reported outcomes after early-life influenza A infection are highly variable, sometimes protecting against, but sometimes promoting the development of AHR. When suckling BALB/c mice (2 weeks old) are infected with influenza H3N1 (again Mem71) and challenged with allergens as adults, AHR typically does not occur and the protective effect appears to be mediated by a subset of NKT cells that produce large amounts of IFNγ (197). At high inocula, many influenza viruses are lethal for young mice (198). Using low titer H1N1 virus (PR8), Lines and colleagues found a delayed T cell response with a pronounced influx of eosinophils into the lungs of C57Bl/6J neonates compared to adult mice (198). Ex vivo stimulation of lung cells with phorbol 12-myristate 13-acetate/ionomycin in these experiments showed delayed and lower levels of IFNγ in the neonatal immune T cells (198). Others have shown that infection of BALB/c mouse pups with PR8 on day 7 of life can result in long-term pulmonary dysfunction, AHR and an increase in inflammatory cytokines, neutrophils and alveolar macrophages in the lungs (199). In this model, IFNγ was not produced by the neonatal pulmonary CD8+ T cells, but adoptive transfer of adult CD8+ T cells prevented the long-term AHR (199). Although this body of work is much smaller than that dealing with RSV, the available data raise the possibility that deficient IFNγ production in the neonate during influenza virus infection may play a role in the induction of an asthmatic phenotype upon subsequent exposure to viruses or allergens. Further studies, including the development of a “standard” early-life mouse model of influenza infection are required to begin to understand the potential contribution of these viruses to asthma initiation.
Rhinoviruses
Background, Prevention, and Treatment
Rhinoviruses are the most important etiological agents of the “common cold”. Similar to RSV, RV can cause both upper and lower respiratory-tract illness at all ages. There are 100–150 strains of RV, divided into three groups: A, B, and C. Groups A and B were discovered in the 1990s, while RV-C, a new group with over 50 strains, was identified only in 2006 (200, 201). Most RV infections are thought to be minimally symptomatic or completely asymptomatic. Under 4 years of age, at least one RV can be found by nasal swab in 12–32% of asymptomatic children [reviewed in Ref. (202)]. Although both the scientific literature and the internet offer a wide variety of therapies to prevent or cure the common cold (203, 204), at the current time neither antivirals nor vaccines are available for RV infections.
Association of RV With Human Asthma
Similar to RSV, the RVs, and particularly the RV-C group, have been implicated in both the development of asthma and in exacerbation of wheezing illness (87, 96, 205–207). Interestingly, RV-associated wheezing tends to be more common in older infants (>1 year old) in contrast to RSV infection in which the most serious manifestations (i.e., bronchiolitis) occur primarily in those <1 year old (208, 209). However, both RSV and RV are frequently isolated in infants hospitalized with severe respiratory symptoms and wheezing (210). As noted above for RSV, it is very difficult to know if this observation means that children with a genetic propensity to wheeze are more likely to be hospitalized or if RV infections are a causal factor in the onset of wheezing illness (83, 168).
Immunology of RV and Asthma
Until recently, animal models suffered from the absence of a receptor for human RV on mouse cells. Most human RV strains bind to intracellular adhesion molecule-1 (ICAM-1) but not mouse ICAM-1 and only a minority (~10%) binds to low-density lipoprotein of both humans and mice (211). Bartlett et al. (212) have described two mouse models of RV infection, one in which BALB/c mice are infected with a low-density lipoprotein-binding isolate and a second based upon transgenic expression of human ICAM-1. Infection of adult mice in the latter model results in a strong Th1 response with abundant IFNγ production as well as exacerbation of allergic responses. To date, the transgenic model has not been used to study RV infection and asthma initiation in neonatal mice. A third model was recently described by Schneider et al. (213) in which 7-day-old BALB/c mice are infected with RV-1B, leading to the development of AHR and mucus production 4 weeks later. Surprisingly, this neonatal infection resulted in the production of IFNγ along with increases in inflammatory cytokines and chemokines, including TNFα, CXCL1, and CXCL2 in the lungs. These observations are very different from the findings in RSV-infected neonates. However, IL-13 was also strongly induced by RV-1B infection in neonatal mice but not in adult mice (214). Analysis of the lungs 35 days after infection showed that a majority of cells producing IL-13 were NKT cells, while CD4 T cells predominantly expressed IFNγ. There was also an increase in M2 macrophages in the lungs, cells that contribute to RV-1B-induced AHR in allergen-sensitized mice infected as adults (215). Neutralizing IL-13 or IL-4R decreased AHR in mice infected as neonates, suggesting that mechanisms similar to those operating in influenza (197) and RSV (138, 170, 173) may also play a role in RV infection. In HDM (Dermatophagoides farina)-sensitized BALB/c mice, RV infection decreases IL-10 and increases production of IL-13, RANTES and TNFα as well as eosinophil infiltration into the lungs (216). Th2 cytokines (IL-4, IL-13) increase the expression of the RV receptor (ICAM-1) on human respiratory epithelial (H292) cells (217). Antiviral responses (IFNβ/λ) appear to be decreased in primary bronchial epithelial cells isolated from children with atopy/asthma and are more permissive of RV16 replication (218). Serum IgE levels in these infants are positively correlated with viral RNA levels in ex vivo-infected epithelial cells and negatively correlated with RV-induced IFNβ/λ. Other investigators have found a negative correlation between IFN-induction by RV and airway Th2 responses, including eosinophilia and detectable IL-4, in children with a history of asthma both with and without atopy (218). In a human challenge study with RV14, both atopic and non-atopic subjects with low IFNγ/IL-5 ratios had more symptoms and prolonged viral shedding (219).
The dearth of studies on RV infection in neonatal mice makes it hard to draw conclusions about potential mechanisms of RV-mediated asthma initiation. Further work on neonatal models, particularly using the group C strains associated with clinical asthma in infants (87, 96, 205–207), would be useful. Like RSV, RV primarily targets respiratory epithelial cells, although RV infections are typically “patchy” and thus may not cause the same degree of damage as RSV.
Recent investigations of RV infection in both animal models and human studies have focused on the innate type 2 cytokines IL-25, TSLP, and IL-33. TSLP is secreted at a low concentration from primary human bronchial epithelial cells infected ex vivo with RV, but rise dramatically with the addition of IL-4 (220). TLSP and IL-33 are also upregulated in bronchial epithelial cells in response to in vitro RV infection but even greater increases in IL-25 are seen in cells from asthmatic patients post-RSV infection (221, 222). RV-induced production of IL-33 and/or TSLP blocks OVA-induced inhalational tolerance, with resultant lung eosinophilia and neutrophilia, as well as increased Th2 and decreased Treg cells. Delivery of neutralizing antibodies that target ST2 (the IL-33 receptor) or RV infection of TSLPR knock-out mice both disrupt the ability of RV to block tolerance in this model (222). These data suggest that RV may increase susceptibility to allergic airways disease, by increasing TSLP and/or IL-33, generating a lung environment conducive to immune recognition of normally harmless antigens/allergens. In a murine model of RV-induced asthma exacerbation using OVA, lung levels of IL-25, expressed by epithelial cells and infiltrating immune cells are greatest in mice exposed to both OVA and RV (221). OVA-RV exposed mice have increased levels of IL-4+ basophils, IL-4+ CD4+ T and ICOS+ ST2+ non-T (ILC2?) cells in the bronchoalveolar lavage (BAL) fluid, as well as enhanced airway eosinophilia and neutrophilia, and exacerbated Th2 cytokine production, all of which are reduced upon delivery of neutralizing IL-25 receptor antibody (α-IL-17RB) prior to RV infection. Interestingly, α-IL-17RB treatment also decreases lung tissue IL-33 and TSLP protein levels. These data suggest that targeting IL-25 receptor may hold therapeutic promise through mitigation of viral- and allergen-induced inflammation promoted by innate type 2 cytokines.
Interleukin-33 is produced by cultured bronchial epithelial and smooth muscle cells when infected with RV (223, 224). In response to in vivo infection with RV16, asthmatics have greater viral loads, increased BAL eosinophilia, and greater respiratory symptom scores compared to non-asthmatic controls. These parameters are also associated with reductions in both peak expiratory flow and FEV1. Moreover, elevated levels of the Th2 cytokines (IL-4, IL-5, IL-13) as well as IL-33 are present in the BAL fluid of RV16-infected asthmatics. Ex vivo incubation of activated (undifferentiated) CD4+ T cells (Th0) or ILC2 cells with supernatants from RV-infected bronchial epithelial cells leads to production of IL-5 and IL-13 by both cell types in an IL-33-dependent manner (223). Significantly, production of IL-5 and IL-13 is 100- to 200-fold greater in ILC2 cells compared to CD4+ T cells suggesting that IL-33-dependent activation of ILC2 cells during RV infection could play a major role in the development of asthma. Taken together, these data suggest that pronounced IL-33 secretion by smooth muscle cells and/or IL-25, TSLP, and IL-33 secretion by epithelial cells in response to RV infection may skew the lung microenvironment toward Th2-biased allergic airways disease through activation of both innate (e.g., macrophage, DCs, and ILC2 cells) and adaptive immune cells.
Conclusion
At the current time, a large body of RSV data in both humans and murine models strongly suggest that there is a maladaptive and “asthma-genic” interaction between the Th2-biased nature of the infant immune system and the Th2-promoting effects of the virus itself. It is also very likely that host genetics play an important role in determining both the short-term (e.g., hospitalization) and long-term (e.g., asthma) consequences of this interaction (90, 93). To date, the only factors known to increase susceptibility to RSV-induced asthma are family history of atopy, premature birth, and certain host genetic polymorphisms that are linked with severe disease (111). Whether or not any child, regardless of his/her genetic background can be “made” asthmatic by early-life RSV infection is an important question that cannot be answered at the current time. In favor of the hypothesis that asthmatics are “born” and not “made”, Gern and colleagues have reported bi-directional cytokine responses in cord blood mononuclear cells stimulated with mitogen or viral antigens (e.g., RSV, RV) from children who were later classified as either wheezing vs. non-wheezing at one year of age (225). Whether or not RSV is unique among early childhood respiratory virus exposures with regard to asthma development is also of great interest. The more limited data available suggest that the RV but not influenza virus infection may have effects similar to RSV. Bonnelykke et al. (88) reported that the number of viral or bacterial respiratory infection episodes within the first year of life is a greater predictor of asthma development than the particular viral or bacterial type(s) (88). In contrast to the contribution of respiratory viruses to asthma development, the role of several viruses, including RSV, RV, and influenza viruses, in asthma exacerbations is very clear (9, 84, 206, 226, 227). Of course, neonates and infants are not just exposed to respiratory viruses in the first weeks-months of life. Rather, they experience a wide range of immunologic challenges with both commensal and potentially pathogenic organisms at multiple epithelial sites throughout childhood. For example, Ege and colleagues have shown that children exposed to wide range of microbes early in life, including children that live on farms, have a reduced risk of developing asthma (the hygiene hypothesis) (228) and germ-free mice can more easily be made airway hyperresponsive than their non germ-free littermates (229). A great deal of work remains to be done to fully understand how the ubiquitous respiratory viruses discussed in this review, as well as other early-life microbial exposures, contribute to the development and perpetuation of asthma.
Such an improved understanding is essential to develop strategies to both prevent asthma development and to mitigate the symptoms of asthma once developed. This review has focused on how a small number of viruses may also contribute to the induction of asthma. In particular, we have described the growing evidence that innate immune effector cells may orchestrate early-life events in the lung to “set the stage” for the later development of asthma. Until very recently, the potential asthma-inducing role of respiratory epithelial cells, macrophages, DCs, ILC2s, and NK/NKT cells has been under-appreciated. Although vaccination to protect the very young from early-life respiratory viruses (including maternal immunization) is one possible strategy (230), these new data suggest that modulation of innate responses during early-life viral infections may also be successful. Of course, any strategy that seeks to alter either the antigen-specific or the “overall” immune response pattern of neonates/infants would have to be approached with great caution (e.g., risk of exaggerated Th1 immunopathology, less effective responses to other pathogens or vaccines). As outlined above, the Th2-biased nature of the neonatal immune system is a strategy that has been “proven” by evolution. Nonetheless, given that asthma has now reached essentially “pandemic” proportions, very few questions in clinical medicine are of greater importance. The concepts described in this review raise the possibility of completely novel strategies for dealing with this pandemic.
Author Contributions
KR, BS, BW, and EF contributed equally to the writing of the manuscript and production of the accompanying figure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported, in part, by operating grants from the Canadian Institutes of Health Research (CIHR; to BW; MOP 81254), as well as the United States Department of Defense through the Peer Reviewed Medical Research Program under Awards W81XWH-15-1-0697 and W81XWH-15-1-0698 (to EF and BW, respectively). The Meakins-Christie Laboratories/Research Institute of the McGill University Health Centre is supported, in part, by a center grant from Fonds de Recherche du Québec - Santé (FRQS).
Abbreviations
AAM, alternatively activated macrophage; AHR, airway hyperresponsiveness; CAM, classically activated macrophage; DC, dendritic cell; HDM, house dust mite; ICAM-1, intracellular adhesion molecule-1; IFN, interferon; IL, interleukin; ILC2, innate type 2 lymphoid cell; KLH, Keyhole limpet (antigen); M1, classically activated macrophage; M2, alternatively activated macrophages; NF-κB, nuclear factor-κB; NLR, NOD-like receptor; NK, natural killer; NS, non-structural (protein); OVA, ovalbumin; pDC, plasmacytoid dendritic cell; PRR, pattern recognition receptor; RLR, RIG-I-like receptor; RSV, respiratory syncytial virus; RV, rhinovirus; TGFβ, transforming growth factor β; Th, T helper; TLR, Toll-like receptor; TNFα, tumor necrosis factor α; Treg, T regulatory; TSLP, thymic stromal lymphopoietin.
References
1. Liu L, Johnson HL, Cousens S, Perin J, Scott S, Lawn JE, et al. Global, regional, and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet (2012) 379:2151–61. doi:10.1016/S0140-6736(12)60560-1
2. Bertino JS. Cost burden of viral respiratory infections: issues for formulary decision makers. Am J Med (2002) 112(Suppl 6A):42S–9S. doi:10.1016/S0002-9343(01)01063-4
3. Mold JE, Michaelsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science (2008) 322:1562–5. doi:10.1126/science.1164511
4. Chassin C, Kocur M, Pott J, Duerr CU, Gutle D, Lotz M, et al. miR-146a mediates protective innate immune tolerance in the neonate intestine. Cell Host Microbe (2010) 8:358–68. doi:10.1016/j.chom.2010.09.005
5. Spencer SJ, Hyland NP, Sharkey KA, Pittman QJ. Neonatal immune challenge exacerbates experimental colitis in adult rats: potential role for TNF-alpha. Am J Physiol Regul Integr Comp Physiol (2007) 292:R308–15. doi:10.1152/ajpregu.00398.2006
6. Benatti C, Alboni S, Capone G, Corsini D, Caggia F, Brunello N, et al. Early neonatal inflammation affects adult pain reactivity and anxiety related traits in mice: genetic background counts. Int J Dev Neurosci (2009) 27:661–8. doi:10.1016/j.ijdevneu.2009.07.009
7. Adams-Chapman I. Long-term impact of infection on the preterm neonate. Semin Perinatol (2012) 36:462–70. doi:10.1053/j.semperi.2012.06.009
8. Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol (2012) 71:444–57. doi:10.1002/ana.22620
9. Busse WW, Lemanske RF Jr, Gern JE. Role of viral respiratory infections in asthma and asthma exacerbations. Lancet (2010) 376:826–34. doi:10.1016/S0140-6736(10)61380-3
10. Sly PD, Kusel M, Holt PG. Do early-life viral infections cause asthma? J Allergy Clin Immunol (2010) 125:1202–5. doi:10.1016/j.jaci.2010.01.024
11. Tregoning JS, Schwarze J. Respiratory viral infections in infants: causes, clinical symptoms, virology, and immunology. Clin Microbiol Rev (2010) 23:74–98. doi:10.1128/CMR.00032-09
12. Zaghouani H, Hoeman CM, Adkins B. Neonatal immunity: faulty T-helpers and the shortcomings of dendritic cells. Trends Immunol (2009) 30:585–91. doi:10.1016/j.it.2009.09.002
13. Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature (1953) 172:603–6. doi:10.1038/172603a0
14. Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nat Rev Immunol (2004) 4:553–64. doi:10.1038/nri1394
15. Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol (1989) 7:145–73. doi:10.1146/annurev.iy.07.040189.001045
16. Chen N, Field EH. Enhanced type 2 and diminished type 1 cytokines in neonatal tolerance. Transplantation (1995) 59:933–41. doi:10.1097/00007890-199504150-00002
17. Forsthuber T, Yip HC, Lehmann PV. Induction of TH1 and TH2 immunity in neonatal mice. Science (1996) 271:1728–30. doi:10.1126/science.271.5256.1728
18. Oboki K, Ohno T, Saito H, Nakae S. Th17 and allergy. Allergol Int (2008) 57:121–34. doi:10.2332/allergolint.R-07-160
19. Soroosh P, Doherty TA. Th9 and allergic disease. Immunology (2009) 127:450–8. doi:10.1111/j.1365-2567.2009.03114.x
20. Cosmi L, Maggi L, Santarlasci V, Capone M, Cardilicchia E, Frosali F, et al. Identification of a novel subset of human circulating memory CD4(+) T cells that produce both IL-17A and IL-4. J Allergy Clin Immunol (2010) 125:222–30.e1–4. doi:10.1016/j.jaci.2009.10.012
21. Robinson DS. Regulatory T cells and asthma. Clin Exp Allergy (2009) 39:1314–23. doi:10.1111/j.1365-2222.2009.03301.x
22. Ridge JP, Fuchs EJ, Matzinger P. Neonatal tolerance revisited: turning on newborn T cells with dendritic cells. Science (1996) 271:1723–6. doi:10.1126/science.271.5256.1723
23. Pertmer TM, Oran AE, Madorin CA, Robinson HL. Th1 genetic adjuvants modulate immune responses in neonates. Vaccine (2001) 19:1764–71. doi:10.1016/S0264-410X(00)00388-1
24. Martinez X, Brandt C, Saddallah F, Tougne C, Barrios C, Wild F, et al. DNA immunization circumvents deficient induction of T helper type 1 and cytotoxic T lymphocyte responses in neonates and during early life. Proc Natl Acad Sci U S A (1997) 94:8726–31. doi:10.1073/pnas.94.16.8726
25. Kovarik J, Bozzotti P, Love-Homan L, Pihlgren M, Davis HL, Lambert PH, et al. CpG oligodeoxynucleotides can circumvent the Th2 polarization of neonatal responses to vaccines but may fail to fully redirect Th2 responses established by neonatal priming. J Immunol (1999) 162:1611–7.
26. Weeratna RD, Brazolot Millan CL, Mccluskie MJ, Davis HL. CpG ODN can re-direct the Th bias of established Th2 immune responses in adult and young mice. FEMS Immunol Med Microbiol (2001) 32:65–71. doi:10.1111/j.1574-695X.2001.tb00535.x
27. Marchant A, Goetghebuer T, Ota MO, Wolfe I, Ceesay SJ, De Groote D, et al. Newborns develop a Th1-type immune response to Mycobacterium bovis bacillus Calmette-Guerin vaccination. J Immunol (1999) 163:2249–55.
28. Kamath AT, Rochat AF, Christensen D, Agger EM, Andersen P, Lambert PH, et al. A liposome-based mycobacterial vaccine induces potent adult and neonatal multifunctional T cells through the exquisite targeting of dendritic cells. PLoS One (2009) 4:e5771. doi:10.1371/journal.pone.0005771
29. Barrios C, Brandt C, Berney M, Lambert PH, Siegrist CA. Partial correction of the TH2/TH1 imbalance in neonatal murine responses to vaccine antigens through selective adjuvant effects. Eur J Immunol (1996) 26:2666–70. doi:10.1002/eji.1830261118
30. Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol (2002) 20:709–60. doi:10.1146/annurev.immunol.20.100301.064842
31. Adkins B, Bu Y, Guevara P. The generation of Th memory in neonates versus adults: prolonged primary Th2 effector function and impaired development of Th1 memory effector function in murine neonates. J Immunol (2001) 166:918–25. doi:10.4049/jimmunol.166.2.918
32. Adkins B, Hamilton K. Freshly isolated, murine neonatal T cells produce IL-4 in response to anti-CD3 stimulation. J Immunol (1992) 149:3448–55.
33. Rose S, Lichtenheld M, Foote MR, Adkins B. Murine neonatal CD4+ cells are poised for rapid Th2 effector-like function. J Immunol (2007) 178:2667–78. doi:10.4049/jimmunol.178.5.2667
34. Webster RB, Rodriguez Y, Klimecki WT, Vercelli D. The human IL-13 locus in neonatal CD4+ T cells is refractory to the acquisition of a repressive chromatin architecture. J Biol Chem (2007) 282:700–9. doi:10.1074/jbc.M609501200
35. Yoshimoto M, Yoder MC, Guevara P, Adkins B. The murine Th2 locus undergoes epigenetic modification in the thymus during fetal and postnatal ontogeny. PLoS One (2013) 8:e51587. doi:10.1371/journal.pone.0051587
36. Sarzotti M, Robbins DS, Hoffman PM. Induction of protective CTL responses in newborn mice by a murine retrovirus. Science (1996) 271:1726–8. doi:10.1126/science.271.5256.1726
37. Garcia AM, Fadel SA, Cao S, Sarzotti M. T cell immunity in neonates. Immunol Res (2000) 22:177–90. doi:10.1385/IR:22:2-3:177
38. Prescott SL, Macaubas C, Holt BJ, Smallacombe TB, Loh R, Sly PD, et al. Transplacental priming of the human immune system to environmental allergens: universal skewing of initial T cell responses toward the Th2 cytokine profile. J Immunol (1998) 160:4730–7.
39. Li L, Legge KL, Min B, Bell JJ, Gregg R, Caprio J, et al. Neonatal immunity develops in a transgenic TCR transfer model and reveals a requirement for elevated cell input to achieve organ-specific responses. J Immunol (2001) 167:2585–94. doi:10.4049/jimmunol.167.5.2585
40. Li L, Lee HH, Bell JJ, Gregg RK, Ellis JS, Gessner A, et al. IL-4 utilizes an alternative receptor to drive apoptosis of Th1 cells and skews neonatal immunity toward Th2. Immunity (2004) 20:429–40. doi:10.1016/S1074-7613(04)00072-X
41. Lee HH, Hoeman CM, Hardaway JC, Guloglu FB, Ellis JS, Jain R, et al. Delayed maturation of an IL-12-producing dendritic cell subset explains the early Th2 bias in neonatal immunity. J Exp Med (2008) 205:2269–80. doi:10.1084/jem.20071371
42. Hoeman CM, Dhakal M, Zaghouani AA, Cascio JA, Wan X, Khairallah MT, et al. Developmental expression of IL-12Rbeta2 on murine naive neonatal T cells counters the upregulation of IL-13Ralpha1 on primary Th1 cells and balances immunity in the newborn. J Immunol (2013) 190:6155–63. doi:10.4049/jimmunol.1202207
43. Montecino-Rodriguez E, Dorshkind K. B-1 B cell development in the fetus and adult. Immunity (2012) 36:13–21. doi:10.1016/j.immuni.2011.11.017
44. Tian C, Kron GK, Dischert KM, Higginbotham JN, Crowe JE Jr. Low expression of the interleukin (IL)-4 receptor alpha chain and reduced signalling via the IL-4 receptor complex in human neonatal B cells. Immunology (2006) 119:54–62. doi:10.1111/j.1365-2567.2006.02405.x
45. Lee SM, Suen Y, Chang L, Bruner V, Qian J, Indes J, et al. Decreased interleukin-12 (IL-12) from activated cord versus adult peripheral blood mononuclear cells and upregulation of interferon-gamma, natural killer, and lymphokine-activated killer activity by IL-12 in cord blood mononuclear cells. Blood (1996) 88:945–54.
46. Min B, Legge KL, Li L, Caprio JC, Pack CD, Gregg R, et al. Neonatal tolerant immunity for vaccination against autoimmunity. Int Rev Immunol (2000) 19:247–64. doi:10.3109/08830180009088507
47. Kovarik J, Martinez X, Pihlgren M, Bozzotti P, Tao MH, Kipps TJ, et al. Limitations of in vivo IL-12 supplementation strategies to induce Th1 early life responses to model viral and bacterial vaccine antigens. Virology (2000) 268:122–31. doi:10.1006/viro.1999.0159
48. Willems F, Vollstedt S, Suter M. Phenotype and function of neonatal DC. Eur J Immunol (2009) 39:26–35. doi:10.1002/eji.200838391
49. Sun CM, Deriaud E, Leclerc C, Lo-Man R. Upon TLR9 signaling, CD5+ B cells control the IL-12-dependent Th1-priming capacity of neonatal DCs. Immunity (2005) 22:467–77. doi:10.1016/j.immuni.2005.02.008
50. Walker WE, Goldstein DR. Neonatal B cells suppress innate toll-like receptor immune responses and modulate alloimmunity. J Immunol (2007) 179:1700–10. doi:10.4049/jimmunol.179.3.1700
51. Goriely S, Vincart B, Stordeur P, Vekemans J, Willems F, Goldman M, et al. Deficient IL-12(p35) gene expression by dendritic cells derived from neonatal monocytes. J Immunol (2001) 166:2141–6. doi:10.4049/jimmunol.166.3.2141
52. Aksoy E, Albarani V, Nguyen M, Laes JF, Ruelle JL, De Wit D, et al. Interferon regulatory factor 3-dependent responses to lipopolysaccharide are selectively blunted in cord blood cells. Blood (2007) 109:2887–93. doi:10.1182/blood-2006-06-027862
53. Danis B, George TC, Goriely S, Dutta B, Renneson J, Gatto L, et al. Interferon regulatory factor 7-mediated responses are defective in cord blood plasmacytoid dendritic cells. Eur J Immunol (2008) 38:507–17. doi:10.1002/eji.200737760
54. Charrier E, Cordeiro P, Cordeau M, Dardari R, Michaud A, Harnois M, et al. Post-transcriptional down-regulation of toll-like receptor signaling pathway in umbilical cord blood plasmacytoid dendritic cells. Cell Immunol (2012) 276:114–21. doi:10.1016/j.cellimm.2012.04.010
55. Levy O, Zarember KA, Roy RM, Cywes C, Godowski PJ, Wessels MR. Selective impairment of TLR-mediated innate immunity in human newborns: neonatal blood plasma reduces monocyte TNF-alpha induction by bacterial lipopeptides, lipopolysaccharide, and imiquimod, but preserves the response to R-848. J Immunol (2004) 173:4627–34. doi:10.4049/jimmunol.173.7.4627
56. Kollmann TR, Crabtree J, Rein-Weston A, Blimkie D, Thommai F, Wang XY, et al. Neonatal innate TLR-mediated responses are distinct from those of adults. J Immunol (2009) 183:7150–60. doi:10.4049/jimmunol.0901481
57. Angelone DF, Wessels MR, Coughlin M, Suter EE, Valentini P, Kalish LA, et al. Innate immunity of the human newborn is polarized toward a high ratio of IL-6/TNF-alpha production in vitro and in vivo. Pediatr Res (2006) 60:205–9. doi:10.1203/01.pdr.0000228319.10481.ea
58. Corbett NP, Blimkie D, Ho KC, Cai B, Sutherland DP, Kallos A, et al. Ontogeny of toll-like receptor mediated cytokine responses of human blood mononuclear cells. PLoS One (2010) 5:e15041. doi:10.1371/journal.pone.0015041
59. Sadeghi K, Berger A, Langgartner M, Prusa AR, Hayde M, Herkner K, et al. Immaturity of infection control in preterm and term newborns is associated with impaired toll-like receptor signaling. J Infect Dis (2007) 195:296–302. doi:10.1086/509892
60. de Lalla C, Festuccia N, Albrecht I, Chang HD, Andolfi G, Benninghoff U, et al. Innate-like effector differentiation of human invariant NKT cells driven by IL-7. J Immunol (2008) 180:4415–24. doi:10.4049/jimmunol.180.7.4415
61. Guilmot A, Hermann E, Braud VM, Carlier Y, Truyens C. Natural killer cell responses to infections in early life. J Innate Immun (2011) 3:280–8. doi:10.1159/000323934
62. Marcoe JP, Lim JR, Schaubert KL, Fodil-Cornu N, Matka M, Mccubbrey AL, et al. TGF-beta is responsible for NK cell immaturity during ontogeny and increased susceptibility to infection during mouse infancy. Nat Immunol (2012) 13:843–50. doi:10.1038/ni.2388
63. Chelvarajan RL, Collins SM, Doubinskaia IE, Goes S, Van Willigen J, Flanagan D, et al. Defective macrophage function in neonates and its impact on unresponsiveness of neonates to polysaccharide antigens. J Leukoc Biol (2004) 75:982–94. doi:10.1189/jlb.0403179
64. Marodi L, Goda K, Palicz A, Szabo G. Cytokine receptor signalling in neonatal macrophages: defective STAT-1 phosphorylation in response to stimulation with IFN-gamma. Clin Exp Immunol (2001) 126:456–60. doi:10.1046/j.1365-2249.2001.01693.x
65. Speer CP, Gahr M, Wieland M, Eber S. Phagocytosis-associated functions in neonatal monocyte-derived macrophages. Pediatr Res (1988) 24:213–6. doi:10.1203/00006450-198808000-00015
66. Walk J, Westerlaken GH, Van Uden NO, Belderbos ME, Meyaard L, Bont LJ. Inhibitory receptor expression on neonatal immune cells. Clin Exp Immunol (2012) 169:164–71. doi:10.1111/j.1365-2249.2012.04599.x
67. Eljaafari FM, Takada H, Tanaka T, Doi T, Ohga S, Hara T. Potent induction of IFN-gamma production from cord blood NK cells by the stimulation with single-stranded RNA. J Clin Immunol (2011) 31:728–35. doi:10.1007/s10875-011-9528-4
68. Schittny JC, Mund SI, Stampanoni M. Evidence and structural mechanism for late lung alveolarization. Am J Physiol Lung Cell Mol Physiol (2008) 294:L246–54. doi:10.1152/ajplung.00296.2007
69. de Kleer IM, Kool M, De Bruijn MJ, Willart M, Van Moorleghem J, Schuijs MJ, et al. Perinatal activation of the interleukin-33 pathway promotes type 2 immunity in the developing lung. Immunity (2016) 45:1285–98. doi:10.1016/j.immuni.2016.10.031
70. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med (2013) 210:1977–92. doi:10.1084/jem.20131199
71. Empey KM, Orend JG, Peebles RS Jr, Egana L, Norris KA, Oury TD, et al. Stimulation of immature lung macrophages with intranasal interferon gamma in a novel neonatal mouse model of respiratory syncytial virus infection. PLoS One (2012) 7:e40499. doi:10.1371/journal.pone.0040499
72. Roux X, Remot A, Petit-Camurdan A, Nahori MA, Kiefer-Biasizzo H, Marchal G, et al. Neonatal lung immune responses show a shift of cytokines and transcription factors toward Th2 and a deficit in conventional and plasmacytoid dendritic cells. Eur J Immunol (2011) 41:2852–61. doi:10.1002/eji.201041224
73. Gollwitzer ES, Saglani S, Trompette A, Yadava K, Sherburn R, Mccoy KD, et al. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat Med (2014) 20:642–7. doi:10.1038/nm.3568
74. Saglani S, Mathie SA, Gregory LG, Bell MJ, Bush A, Lloyd CM. Pathophysiological features of asthma develop in parallel in house dust mite-exposed neonatal mice. Am J Respir Cell Mol Biol (2009) 41:281–9. doi:10.1165/rcmb.2008-0396OC
75. Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med (2014) 20:159–66. doi:10.1038/nm.3444
76. Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLoS One (2010) 5:e8578. doi:10.1371/journal.pone.0008578
77. Braman SS. The global burden of asthma. Chest (2006) 130:4S–12S. doi:10.1378/chest.130.1_suppl.4S
78. Locksley RM. Asthma and allergic inflammation. Cell (2010) 140:777–83. doi:10.1016/j.cell.2010.03.004
79. Subbarao P, Mandhane PJ, Sears MR. Asthma: epidemiology, etiology and risk factors. CMAJ (2009) 181:E181–90. doi:10.1503/cmaj.080612
80. Beasley R, Semprini A, Mitchell EA. Risk factors for asthma: is prevention possible? Lancet (2015) 386:1075–85. doi:10.1016/S0140-6736(15)00156-7
81. Wardlaw T, Salama P, Johansson EW, Mason E. Pneumonia: the leading killer of children. Lancet (2006) 368:1048–50. doi:10.1016/S0140-6736(06)69334-3
82. Johnston NW, Johnston SL, Duncan JM, Greene JM, Kebadze T, Keith PK, et al. The September epidemic of asthma exacerbations in children: a search for etiology. J Allergy Clin Immunol (2005) 115:132–8. doi:10.1016/j.jaci.2004.09.025
83. Message SD, Johnston SL. Viruses in asthma. Br Med Bull (2002) 61:29–43. doi:10.1093/bmb/61.1.29
84. Jackson DJ, Sykes A, Mallia P, Johnston SL. Asthma exacerbations: origin, effect, and prevention. J Allergy Clin Immunol (2011) 128:1165–74. doi:10.1016/j.jaci.2011.10.024
85. James KM, Peebles RS Jr, Hartert TV. Response to infections in patients with asthma and atopic disease: an epiphenomenon or reflection of host susceptibility? J Allergy Clin Immunol (2012) 130:343–51. doi:10.1016/j.jaci.2012.05.056
86. Kusel MM, De Klerk NH, Kebadze T, Vohma V, Holt PG, Johnston SL, et al. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J Allergy Clin Immunol (2007) 119:1105–10. doi:10.1016/j.jaci.2006.12.669
87. Jackson DJ, Gangnon RE, Evans MD, Roberg KA, Anderson EL, Pappas TE, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med (2008) 178:667–72. doi:10.1164/rccm.200802-309OC
88. Bonnelykke K, Vissing NH, Sevelsted A, Johnston SL, Bisgaard H. Association between respiratory infections in early life and later asthma is independent of virus type. J Allergy Clin Immunol (2015) 136:81–6.e4. doi:10.1016/j.jaci.2015.02.024
89. Sigurs N, Bjarnason R, Sigurbergsson F, Kjellman B, Bjorksten B. Asthma and immunoglobulin E antibodies after respiratory syncytial virus bronchiolitis: a prospective cohort study with matched controls. Pediatrics (1995) 95:500–5.
90. Stein RT, Sherrill D, Morgan WJ, Holberg CJ, Halonen M, Taussig LM, et al. Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years. Lancet (1999) 354:541–5. doi:10.1016/S0140-6736(98)10321-5
91. Sigurs N, Aljassim F, Kjellman B, Robinson PD, Sigurbergsson F, Bjarnason R, et al. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax (2010) 65:1045–52. doi:10.1136/thx.2009.121582
92. Thomsen SF, Van Der Sluis S, Stensballe LG, Posthuma D, Skytthe A, Kyvik KO, et al. Exploring the association between severe respiratory syncytial virus infection and asthma: a registry-based twin study. Am J Respir Crit Care Med (2009) 179:1091–7. doi:10.1164/rccm.200809-1471OC
93. Stensballe LG, Simonsen JB, Thomsen SF, Larsen AM, Lysdal SH, Aaby P, et al. The causal direction in the association between respiratory syncytial virus hospitalization and asthma. J Allergy Clin Immunol (2009) 123:131–7.e1. doi:10.1016/j.jaci.2008.10.042
94. Szabo SM, Levy AR, Gooch KL, Bradt P, Wijaya H, Mitchell I. Elevated risk of asthma after hospitalization for respiratory syncytial virus infection in infancy. Paediatr Respir Rev (2013) 13(Suppl 2):S9–15. doi:10.1016/S1526-0542(12)70161-6
95. Kotaniemi-Syrjanen A, Vainionpaa R, Reijonen TM, Waris M, Korhonen K, Korppi M. Rhinovirus-induced wheezing in infancy – the first sign of childhood asthma? J Allergy Clin Immunol (2003) 111:66–71. doi:10.1067/mai.2003.33
96. Miller EK, Edwards KM, Weinberg GA, Iwane MK, Griffin MR, Hall CB, et al. A novel group of rhinoviruses is associated with asthma hospitalizations. J Allergy Clin Immunol (2009) 123:98–104.e1. doi:10.1016/j.jaci.2008.10.007
97. Vandini S, Biagi C, Lanari M. Respiratory syncytial virus: the influence of serotype and genotype variability on clinical course of infection. Int J Mol Sci (2017) 18(8):E1717. doi:10.3390/ijms18081717
98. Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet (2010) 375:1545–55. doi:10.1016/S0140-6736(10)60206-1
99. Nair H, Simoes EA, Rudan I, Gessner BD, Azziz-Baumgartner E, Zhang JS, et al. Global and regional burden of hospital admissions for severe acute lower respiratory infections in young children in 2010: a systematic analysis. Lancet (2013) 381:1380–90. doi:10.1016/S0140-6736(12)61901-1
100. Krilov LR. Respiratory syncytial virus disease: update on treatment and prevention. Expert Rev Anti Infect Ther (2011) 9:27–32. doi:10.1586/eri.10.140
101. Mejias A, Ramilo O. Review of palivizumab in the prophylaxis of respiratory syncytial virus (RSV) in high-risk infants. Biologics (2008) 2:433–9.
102. Blanken MO, Rovers MM, Molenaar JM, Winkler-Seinstra PL, Meijer A, Kimpen JL, et al. Respiratory syncytial virus and recurrent wheeze in healthy preterm infants. N Engl J Med (2013) 368:1791–9. doi:10.1056/NEJMoa1211917
103. Shadman KA, Wald ER. A review of palivizumab and emerging therapies for respiratory syncytial virus. Expert Opin Biol Ther (2011) 11:1455–67. doi:10.1517/14712598.2011.608062
104. Feldman AS, He Y, Moore ML, Hershenson MB, Hartert TV. Toward primary prevention of asthma. Reviewing the evidence for early-life respiratory viral infections as modifiable risk factors to prevent childhood asthma. Am J Respir Crit Care Med (2015) 191:34–44. doi:10.1164/rccm.201405-0901PP
105. McNamara PS, Smyth RL. The pathogenesis of respiratory syncytial virus disease in childhood. Br Med Bull (2002) 61:13–28. doi:10.1093/bmb/61.1.13
106. Stockman LJ, Curns AT, Anderson LJ, Fischer-Langley G. Respiratory syncytial virus-associated hospitalizations among infants and young children in the United States, 1997–2006. Pediatr Infect Dis J (2012) 31:5–9. doi:10.1097/INF.0b013e31822e68e6
107. Szabo SM, Gooch KL, Bibby MM, Vo PG, Mitchell I, Bradt P, et al. The risk of mortality among young children hospitalized for severe respiratory syncytial virus infection. Paediatr Respir Rev (2013) 13(Suppl 2):S1–8. doi:10.1016/S1526-0542(12)00095-4
108. Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J Virol (2002) 76:5654–66. doi:10.1128/JVI.76.11.5654-5666.2002
109. Feldman SA, Audet S, Beeler JA. The fusion glycoprotein of human respiratory syncytial virus facilitates virus attachment and infectivity via an interaction with cellular heparan sulfate. J Virol (2000) 74:6442–7. doi:10.1128/JVI.74.14.6442-6447.2000
110. Tayyari F, Marchant D, Moraes TJ, Duan W, Mastrangelo P, Hegele RG. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat Med (2011) 17:1132–5. doi:10.1038/nm.2444
111. Lambert L, Sagfors AM, Openshaw PJ, Culley FJ. Immunity to RSV in early-life. Front Immunol (2014) 5:466. doi:10.3389/fimmu.2014.00466
112. Openshaw PJ, Clarke SL, Record FM. Pulmonary eosinophilic response to respiratory syncytial virus infection in mice sensitized to the major surface glycoprotein G. Int Immunol (1992) 4:493–500. doi:10.1093/intimm/4.4.493
113. Castilow EM, Meyerholz DK, Varga SM. IL-13 is required for eosinophil entry into the lung during respiratory syncytial virus vaccine-enhanced disease. J Immunol (2008) 180:2376–84. doi:10.4049/jimmunol.180.4.2376
114. Fuentes S, Coyle EM, Golding H, Khurana S. Nonglycosylated G-protein vaccine protects against homologous and heterologous respiratory syncytial virus (RSV) challenge, while glycosylated G enhances RSV lung pathology and cytokine levels. J Virol (2015) 89:8193–205. doi:10.1128/JVI.00133-15
115. Dodd JS, Lum E, Goulding J, Muir R, Van Snick J, Openshaw PJ. IL-9 regulates pathology during primary and memory responses to respiratory syncytial virus infection. J Immunol (2009) 183:7006–13. doi:10.4049/jimmunol.0900085
116. Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol (2000) 1:398–401. doi:10.1038/80833
117. Marr N, Turvey SE. Role of human TLR4 in respiratory syncytial virus-induced NF-kappaB activation, viral entry and replication. Innate Immun (2012) 18:856–65. doi:10.1177/1753425912444479
118. Caballero MT, Serra ME, Acosta PL, Marzec J, Gibbons L, Salim M, et al. TLR4 genotype and environmental LPS mediate RSV bronchiolitis through Th2 polarization. J Clin Invest (2015) 125:571–82. doi:10.1172/JCI75183
119. Monick MM, Yarovinsky TO, Powers LS, Butler NS, Carter AB, Gudmundsson G, et al. Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J Biol Chem (2003) 278:53035–44. doi:10.1074/jbc.M308093200
120. Dabbagh K, Dahl ME, Stepick-Biek P, Lewis DB. Toll-like receptor 4 is required for optimal development of Th2 immune responses: role of dendritic cells. J Immunol (2002) 168:4524–30. doi:10.4049/jimmunol.168.9.4524
121. Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med (2002) 196:1645–51. doi:10.1084/jem.20021340
122. Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via toll-like receptor 4 triggering of airway structural cells. Nat Med (2009) 15:410–6. doi:10.1038/nm.1946
123. Lee HC, Headley MB, Loo YM, Berlin A, Gale M Jr, Debley JS, et al. Thymic stromal lymphopoietin is induced by respiratory syncytial virus-infected airway epithelial cells and promotes a type 2 response to infection. J Allergy Clin Immunol (2012) 130:1187–96.e5. doi:10.1016/j.jaci.2012.07.031
124. Barends M, Van Oosten M, De Rond CG, Dormans JA, Osterhaus AD, Neijens HJ, et al. Timing of infection and prior immunization with respiratory syncytial virus (RSV) in RSV-enhanced allergic inflammation. J Infect Dis (2004) 189:1866–72. doi:10.1086/386341
125. Becnel D, You D, Erskin J, Dimina DM, Cormier SA. A role for airway remodeling during respiratory syncytial virus infection. Respir Res (2005) 6:122. doi:10.1186/1465-9921-6-122
126. Keegan AD, Shirey KA, Bagdure D, Blanco J, Viscardi RM, Vogel SN. Enhanced allergic responsiveness after early childhood infection with respiratory viruses: are long-lived alternatively activated macrophages the missing link? Pathog Dis (2016) 74:1–11. doi:10.1093/femspd/ftw047
127. Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity (2015) 43:29–40. doi:10.1016/j.immuni.2015.07.007
128. Petersen BC, Dolgachev V, Rasky A, Lukacs NW. IL-17E (IL-25) and IL-17RB promote respiratory syncytial virus-induced pulmonary disease. J Leukoc Biol (2014) 95(5):809–15. doi:10.1189/jlb.0913482
129. Kaiko GE, Phipps S, Angkasekwinai P, Dong C, Foster PS. NK cell deficiency predisposes to viral-induced Th2-type allergic inflammation via epithelial-derived IL-25. J Immunol (2010) 185:4681–90. doi:10.4049/jimmunol.1001758
130. Yang Z, Grinchuk V, Urban JF Jr, Bohl J, Sun R, Notari L, et al. Macrophages as IL-25/IL-33-responsive cells play an important role in the induction of type 2 immunity. PLoS One (2013) 8:e59441. doi:10.1371/journal.pone.0059441
131. Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med (2008) 14:633–40. doi:10.1038/nm1770
132. Chang YJ, Kim HY, Albacker LA, Baumgarth N, Mckenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol (2011) 12:631–8. doi:10.1038/ni.2045
133. Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, et al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol (2012) 129:216–27.e1–6. doi:10.1016/j.jaci.2011.10.036
134. Byers DE, Alexander-Brett J, Patel AC, Agapov E, Dang-Vu G, Jin X, et al. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest (2013) 123:3967–82. doi:10.1172/JCI65570
135. Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, Mcnagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity (2014) 40:425–35. doi:10.1016/j.immuni.2014.01.011
136. Zeng S, Wu J, Liu J, Qi F, Liu B. IL-33 receptor (ST2) signaling is important for regulation of Th2-mediated airway inflammation in a murine model of acute respiratory syncytial virus infection. Scand J Immunol (2015) 81:494–501. doi:10.1111/sji.12284
137. Liu J, Wu J, Qi F, Zeng S, Xu L, Hu H, et al. Natural helper cells contribute to pulmonary eosinophilia by producing IL-13 via IL-33/ST2 pathway in a murine model of respiratory syncytial virus infection. Int Immunopharmacol (2015) 28:337–43. doi:10.1016/j.intimp.2015.05.035
138. Saravia J, You D, Shrestha B, Jaligama S, Siefker D, Lee GI, et al. Respiratory syncytial virus disease is mediated by age-variable IL-33. PLoS Pathog (2015) 11:e1005217. doi:10.1371/journal.ppat.1005217
139. Bertrand P, Lay MK, Piedimonte G, Brockmann PE, Palavecino CE, Hernandez J, et al. Elevated IL-3 and IL-12p40 levels in the lower airway of infants with RSV-induced bronchiolitis correlate with recurrent wheezing. Cytokine (2015) 76:417–23. doi:10.1016/j.cyto.2015.07.017
140. Qiao J, Li A, Jin X. TSLP from RSV-stimulated rat airway epithelial cells activates myeloid dendritic cells. Immunol Cell Biol (2011) 89:231–8. doi:10.1038/icb.2010.85
141. Miazgowicz MM, Elliott MS, Debley JS, Ziegler SF. Respiratory syncytial virus induces functional thymic stromal lymphopoietin receptor in airway epithelial cells. J Inflamm Res (2013) 6:53–61. doi:10.2147/JIR.S42381
142. Hammad H, Lambrecht BN. Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat Rev Immunol (2008) 8:193–204. doi:10.1038/nri2275
143. Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med (2012) 18:684–92. doi:10.1038/nm.2737
144. Lambrecht BN, Hammad H. Lung dendritic cells in respiratory viral infection and asthma: from protection to immunopathology. Annu Rev Immunol (2012) 30:243–70. doi:10.1146/annurev-immunol-020711-075021
145. Han J, Dakhama A, Jia Y, Wang M, Zeng W, Takeda K, et al. Responsiveness to respiratory syncytial virus in neonates is mediated through thymic stromal lymphopoietin and OX40 ligand. J Allergy Clin Immunol (2012) 130:1175–86.e9. doi:10.1016/j.jaci.2012.08.033
146. Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, et al. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med (2005) 202:1213–23. doi:10.1084/jem.20051135
147. Wang YH, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, et al. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J Exp Med (2007) 204:1837–47. doi:10.1084/jem.20070406
148. Chu DK, Llop-Guevara A, Walker TD, Flader K, Goncharova S, Boudreau JE, et al. IL-33, but not thymic stromal lymphopoietin or IL-25, is central to mite and peanut allergic sensitization. J Allergy Clin Immunol (2013) 131:187–200.e1–8. doi:10.1016/j.jaci.2012.08.002
149. Stier MT, Bloodworth MH, Toki S, Newcomb DC, Goleniewska K, Boyd KL, et al. Respiratory syncytial virus infection activates IL-13-producing group 2 innate lymphoid cells through thymic stromal lymphopoietin. J Allergy Clin Immunol (2016) 138:814–24.e11. doi:10.1016/j.jaci.2016.01.050
150. Johnson TR, Johnson CN, Corbett KS, Edwards GC, Graham BS. Primary human mDC1, mDC2, and pDC dendritic cells are differentially infected and activated by respiratory syncytial virus. PLoS One (2011) 6:e16458. doi:10.1371/journal.pone.0016458
151. Johnson TR, Mclellan JS, Graham BS. Respiratory syncytial virus glycoprotein G interacts with DC-SIGN and L-SIGN to activate ERK1 and ERK2. J Virol (2012) 86:1339–47. doi:10.1128/JVI.06096-11
152. Munir S, Le Nouen C, Luongo C, Buchholz UJ, Collins PL, Bukreyev A. Nonstructural proteins 1 and 2 of respiratory syncytial virus suppress maturation of human dendritic cells. J Virol (2008) 82:8780–96. doi:10.1128/JVI.00630-08
153. Munir S, Hillyer P, Le Nouen C, Buchholz UJ, Rabin RL, Collins PL, et al. Respiratory syncytial virus interferon antagonist NS1 protein suppresses and skews the human T lymphocyte response. PLoS Pathog (2011) 7:e1001336. doi:10.1371/journal.ppat.1001336
154. Shirey KA, Pletneva LM, Puche AC, Keegan AD, Prince GA, Blanco JC, et al. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent. Mucosal Immunol (2010) 3:291–300. doi:10.1038/mi.2010.6
155. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci (2008) 13:453–61. doi:10.2741/2692
156. Ricardo SD, Van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest (2008) 118:3522–30. doi:10.1172/JCI36150
157. Marchetti V, Yanes O, Aguilar E, Wang M, Friedlander D, Moreno S, et al. Differential macrophage polarization promotes tissue remodeling and repair in a model of ischemic retinopathy. Sci Rep (2011) 1:76. doi:10.1038/srep00076
158. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol (2011) 11:723–37. doi:10.1038/nri3073
159. Thornburg NJ, Shepherd B, Crowe JE Jr. Transforming growth factor beta is a major regulator of human neonatal immune responses following respiratory syncytial virus infection. J Virol (2010) 84:12895–902. doi:10.1128/JVI.01273-10
160. You D, Becnel D, Wang K, Ripple M, Daly M, Cormier SA. Exposure of neonates to respiratory syncytial virus is critical in determining subsequent airway response in adults. Respir Res (2006) 7:107. doi:10.1186/1465-9921-7-107
161. Didierlaurent A, Goulding J, Patel S, Snelgrove R, Low L, Bebien M, et al. Sustained desensitization to bacterial toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med (2008) 205:323–9. doi:10.1084/jem.20070891
162. Srinivasa BT, Restori KH, Shan J, Cyr L, Xing L, Lee S, et al. STAT6 inhibitory peptide given during RSV infection of neonatal mice reduces exacerbated airway responses upon adult reinfection. J Leukoc Biol (2017) 101:519–29. doi:10.1189/jlb.4A0215-062RR
163. Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol (2009) 183:6469–77. doi:10.4049/jimmunol.0901575
164. Han H, Headley MB, Xu W, Comeau MR, Zhou B, Ziegler SF. Thymic stromal lymphopoietin amplifies the differentiation of alternatively activated macrophages. J Immunol (2013) 190:904–12. doi:10.4049/jimmunol.1201808
165. Spann KM, Tran KC, Chi B, Rabin RL, Collins PL. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages [corrected]. J Virol (2004) 78:4363–9. doi:10.1128/JVI.78.8.4363-4369.2004
166. Peebles RS Jr, Graham BS. Pathogenesis of respiratory syncytial virus infection in the murine model. Proc Am Thorac Soc (2005) 2:110–5. doi:10.1513/pats.200501-002AW
167. Johnson JE, Gonzales RA, Olson SJ, Wright PF, Graham BS. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod Pathol (2007) 20:108–19. doi:10.1038/modpathol.3800725
168. Martinez FD. Viruses and atopic sensitization in the first years of life. Am J Respir Crit Care Med (2000) 162:S95–9. doi:10.1164/ajrccm.162.supplement_2.ras-8
169. Culley FJ, Pollott J, Openshaw PJ. Age at first viral infection determines the pattern of T cell-mediated disease during reinfection in adulthood. J Exp Med (2002) 196:1381–6. doi:10.1084/jem.20020943
170. Dakhama A, Park JW, Taube C, Joetham A, Balhorn A, Miyahara N, et al. The enhancement or prevention of airway hyperresponsiveness during reinfection with respiratory syncytial virus is critically dependent on the age at first infection and IL-13 production. J Immunol (2005) 175:1876–83. doi:10.4049/jimmunol.175.3.1876
171. You D, Marr N, Saravia J, Shrestha B, Lee GI, Turvey SE, et al. IL-4Ralpha on CD4+ T cells plays a pathogenic role in respiratory syncytial virus reinfection in mice infected initially as neonates. J Leukoc Biol (2013) 93:933–42. doi:10.1189/jlb.1012498
172. Dakhama A, Lee YM, Ohnishi H, Jing X, Balhorn A, Takeda K, et al. Virus-specific IgE enhances airway responsiveness on reinfection with respiratory syncytial virus in newborn mice. J Allergy Clin Immunol (2009) 123:138–45.e5. doi:10.1016/j.jaci.2008.10.012
173. Ripple MJ, You D, Honnegowda S, Giaimo JD, Sewell AB, Becnel DM, et al. Immunomodulation with IL-4R alpha antisense oligonucleotide prevents respiratory syncytial virus-mediated pulmonary disease. J Immunol (2010) 185:4804–11. doi:10.4049/jimmunol.1000484
174. Tregoning JS, Yamaguchi Y, Harker J, Wang B, Openshaw PJ. The role of T cells in the enhancement of respiratory syncytial virus infection severity during adult reinfection of neonatally sensitized mice. J Virol (2008) 82:4115–24. doi:10.1128/JVI.02313-07
175. Arruvito L, Raiden S, Geffner J. Host response to respiratory syncytial virus infection. Curr Opin Infect Dis (2015) 28:259–66. doi:10.1097/QCO.0000000000000159
176. Krishnamoorthy N, Khare A, Oriss TB, Raundhal M, Morse C, Yarlagadda M, et al. Early infection with respiratory syncytial virus impairs regulatory T cell function and increases susceptibility to allergic asthma. Nat Med (2012) 18:1525–30. doi:10.1038/nm.2896
177. Harker JA, Lee DC, Yamaguchi Y, Wang B, Bukreyev A, Collins PL, et al. Delivery of cytokines by recombinant virus in early life alters the immune response to adult lung infection. J Virol (2010) 84:5294–302. doi:10.1128/JVI.02503-09
178. Eichinger KM, Egana L, Orend JG, Resetar E, Anderson KB, Patel R, et al. Alveolar macrophages support interferon gamma-mediated viral clearance in RSV-infected neonatal mice. Respir Res (2015) 16:122. doi:10.1186/s12931-015-0282-7
179. Yamaguchi Y, Harker JA, Wang B, Openshaw PJ, Tregoning JS, Culley FJ. Preexposure to CpG protects against the delayed effects of neonatal respiratory syncytial virus infection. J Virol (2012) 86:10456–61. doi:10.1128/JVI.01082-12
180. Cyr SL, Jones T, Stoica-Popescu I, Brewer A, Chabot S, Lussier M, et al. Intranasal proteosome-based respiratory syncytial virus (RSV) vaccines protect BALB/c mice against challenge without eosinophilia or enhanced pathology. Vaccine (2007) 25:5378–89. doi:10.1016/j.vaccine.2007.01.037
181. Remot A, Roux X, Dubuquoy C, Fix J, Bouet S, Moudjou M, et al. Nucleoprotein nanostructures combined with adjuvants adapted to the neonatal immune context: a candidate mucosal RSV vaccine. PLoS One (2012) 7:e37722. doi:10.1371/journal.pone.0037722
182. Cormier SA, Shrestha B, Saravia J, Lee GI, Shen L, Devincenzo JP, et al. Limited type I interferons and plasmacytoid dendritic cells during neonatal respiratory syncytial virus infection permit immunopathogenesis upon reinfection. J Virol (2014) 88:9350–60. doi:10.1128/JVI.00818-14
183. Smit JJ, Lindell DM, Boon L, Kool M, Lambrecht BN, Lukacs NW. The balance between plasmacytoid DC versus conventional DC determines pulmonary immunity to virus infections. PLoS One (2008) 3:e1720. doi:10.1371/journal.pone.0001720
184. Smit JJ, Rudd BD, Lukacs NW. Plasmacytoid dendritic cells inhibit pulmonary immunopathology and promote clearance of respiratory syncytial virus. J Exp Med (2006) 203:1153–9. doi:10.1084/jem.20052359
185. Remot A, Descamps D, Jouneau L, Laubreton D, Dubuquoy C, Bouet S, et al. Flt3 ligand improves the innate response to respiratory syncytial virus and limits lung disease upon RSV reexposure in neonate mice. Eur J Immunol (2016) 46:874–84. doi:10.1002/eji.201545929
186. Kolli D, Gupta MR, Sbrana E, Velayutham TS, Chao H, Casola A, et al. Alveolar macrophages contribute to the pathogenesis of human metapneumovirus infection while protecting against respiratory syncytial virus infection. Am J Respir Cell Mol Biol (2014) 51:502–15. doi:10.1165/rcmb.2013-0414OC
187. Goritzka M, Makris S, Kausar F, Durant LR, Pereira C, Kumagai Y, et al. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J Exp Med (2015) 212:699–714. doi:10.1084/jem.20140825
188. Parrish CR, Murcia PR, Holmes EC. Influenza virus reservoirs and intermediate hosts: dogs, horses, and new possibilities for influenza virus exposure of humans. J Virol (2015) 89:2990–4. doi:10.1128/JVI.03146-14
189. Rudan I, O’brien KL, Nair H, Liu L, Theodoratou E, Qazi S, et al. Epidemiology and etiology of childhood pneumonia in 2010: estimates of incidence, severe morbidity, mortality, underlying risk factors and causative pathogens for 192 countries. J Glob Health (2013) 3:010401. doi:10.7189/jogh.03.010401
190. Jefferson T, Rivetti A, Di Pietrantonj C, Demicheli V, Ferroni E. Vaccines for preventing influenza in healthy children. Cochrane Database Syst Rev (2012) 8:CD004879. doi:10.1002/14651858.CD004879.pub5
191. Effler PV. Every year is an influenza pandemic for children: can we stop them? Pediatrics (2012) 130:554–6. doi:10.1542/peds.2012-1171
193. Dulek DE, Peebles RS Jr. Viruses and asthma. Biochim Biophys Acta (2011) 1810:1080–90. doi:10.1016/j.bbagen.2011.01.012
194. Dahl ME, Dabbagh K, Liggitt D, Kim S, Lewis DB. Viral-induced T helper type 1 responses enhance allergic disease by effects on lung dendritic cells. Nat Immunol (2004) 5:337–43. doi:10.1038/ni1041
195. Barends M, De Rond LG, Dormans J, Van Oosten M, Boelen A, Neijens HJ, et al. Respiratory syncytial virus, pneumonia virus of mice, and influenza A virus differently affect respiratory allergy in mice. Clin Exp Allergy (2004) 34:488–96. doi:10.1111/j.1365-2222.2004.01906.x
196. Shim DH, Park YA, Kim MJ, Hong JY, Baek JY, Kim KW, et al. Pandemic influenza virus, pH1N1, induces asthmatic symptoms via activation of innate lymphoid cells. Pediatr Allergy Immunol (2015) 26:780–8. doi:10.1111/pai.12462
197. Chang YJ, Kim HY, Albacker LA, Lee HH, Baumgarth N, Akira S, et al. Influenza infection in suckling mice expands an NKT cell subset that protects against airway hyperreactivity. J Clin Invest (2011) 121:57–69. doi:10.1172/JCI44845
198. Lines JL, Hoskins S, Hollifield M, Cauley LS, Garvy BA. The migration of T cells in response to influenza virus is altered in neonatal mice. J Immunol (2010) 185:2980–8. doi:10.4049/jimmunol.0903075
199. You D, Ripple M, Balakrishna S, Troxclair D, Sandquist D, Ding L, et al. Inchoate CD8+ T cell responses in neonatal mice permit influenza-induced persistent pulmonary dysfunction. J Immunol (2008) 181:3486–94. doi:10.4049/jimmunol.181.5.3486
200. Lamson D, Renwick N, Kapoor V, Liu Z, Palacios G, Ju J, et al. MassTag polymerase-chain-reaction detection of respiratory pathogens, including a new rhinovirus genotype, that caused influenza-like illness in New York State during 2004–2005. J Infect Dis (2006) 194:1398–402. doi:10.1086/508551
201. Gern JE. The ABCs of rhinoviruses, wheezing, and asthma. J Virol (2010) 84:7418–26. doi:10.1128/JVI.02290-09
202. Jacobs SE, Lamson DM, St George K, Walsh TJ. Human rhinoviruses. Clin Microbiol Rev (2013) 26:135–62. doi:10.1128/CMR.00077-12
204. Nahas R, Balla A. Complementary and alternative medicine for prevention and treatment of the common cold. Can Fam Physician (2011) 57:31–6.
205. Renwick N, Schweiger B, Kapoor V, Liu Z, Villari J, Bullmann R, et al. A recently identified rhinovirus genotype is associated with severe respiratory-tract infection in children in Germany. J Infect Dis (2007) 196:1754–60. doi:10.1086/524312
206. Jackson DJ, Johnston SL. The role of viruses in acute exacerbations of asthma. J Allergy Clin Immunol (2010) 125:1178–87. doi:10.1016/j.jaci.2010.04.021
207. Hammond C, Kurten M, Kennedy JL. Rhinovirus and asthma: a storied history of incompatibility. Curr Allergy Asthma Rep (2015) 15:502. doi:10.1007/s11882-014-0502-0
208. Korppi M, Kotaniemi-Syrjanen A, Waris M, Vainionpaa R, Reijonen TM. Rhinovirus-associated wheezing in infancy: comparison with respiratory syncytial virus bronchiolitis. Pediatr Infect Dis J (2004) 23:995–9. doi:10.1097/01.inf.0000143642.72480.53
209. Jartti T, Lehtinen P, Vuorinen T, Ruuskanen O. Bronchiolitis: age and previous wheezing episodes are linked to viral etiology and atopic characteristics. Pediatr Infect Dis J (2009) 28:311–7. doi:10.1097/INF.0b013e31818ee0c1
210. Van Leeuwen JC, Goossens LK, Hendrix RM, Van Der Palen J, Lusthusz A, Thio BJ. Equal virulence of rhinovirus and respiratory syncytial virus in infants hospitalized for lower respiratory tract infection. Pediatr Infect Dis J (2012) 31:84–6. doi:10.1097/INF.0b013e31823345bf
211. Tuthill TJ, Papadopoulos NG, Jourdan P, Challinor LJ, Sharp NA, Plumpton C, et al. Mouse respiratory epithelial cells support efficient replication of human rhinovirus. J Gen Virol (2003) 84:2829–36. doi:10.1099/vir.0.19109-0
212. Bartlett NW, Walton RP, Edwards MR, Aniscenko J, Caramori G, Zhu J, et al. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat Med (2008) 14:199–204. doi:10.1038/nm1713
213. Schneider D, Hong JY, Popova AP, Bowman ER, Linn MJ, Mclean AM, et al. Neonatal rhinovirus infection induces mucous metaplasia and airways hyperresponsiveness. J Immunol (2012) 188:2894–904. doi:10.4049/jimmunol.1101391
214. Newcomb DC, Sajjan US, Nagarkar DR, Wang Q, Nanua S, Zhou Y, et al. Human rhinovirus 1B exposure induces phosphatidylinositol 3-kinase-dependent airway inflammation in mice. Am J Respir Crit Care Med (2008) 177:1111–21. doi:10.1164/rccm.200708-1243OC
215. Nagarkar DR, Bowman ER, Schneider D, Wang Q, Shim J, Zhao Y, et al. Rhinovirus infection of allergen-sensitized and -challenged mice induces eotaxin release from functionally polarized macrophages. J Immunol (2010) 185:2525–35. doi:10.4049/jimmunol.1000286
216. Kim E, Lee H, Kim HS, Won S, Lee EK, Kim HS, et al. The effect of rhinovirus on airway inflammation in a murine asthma model. Korean J Pediatr (2013) 56:482–9. doi:10.3345/kjp.2013.56.11.482
217. Bianco A, Sethi SK, Allen JT, Knight RA, Spiteri MA. Th2 cytokines exert a dominant influence on epithelial cell expression of the major group human rhinovirus receptor, ICAM-1. Eur Respir J (1998) 12:619–26. doi:10.1183/09031936.98.12030619
218. Baraldo S, Contoli M, Bazzan E, Turato G, Padovani A, Marku B, et al. Deficient antiviral immune responses in childhood: distinct roles of atopy and asthma. J Allergy Clin Immunol (2012) 130:1307–14. doi:10.1016/j.jaci.2012.08.005
219. Gern JE, Vrtis R, Grindle KA, Swenson C, Busse WW. Relationship of upper and lower airway cytokines to outcome of experimental rhinovirus infection. Am J Respir Crit Care Med (2000) 162:2226–31. doi:10.1164/ajrccm.162.6.2003019
220. Kato A, Favoreto S Jr, Avila PC, Schleimer RP. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol (2007) 179:1080–7. doi:10.4049/jimmunol.179.2.1080
221. Beale J, Jayaraman A, Jackson DJ, Macintyre JD, Edwards MR, Walton RP, et al. Rhinovirus-induced IL-25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci Transl Med (2014) 6:256ra134. doi:10.1126/scitranslmed.3009124
222. Mehta AK, Duan W, Doerner AM, Traves SL, Broide DH, Proud D, et al. Rhinovirus infection interferes with induction of tolerance to aeroantigens through OX40 ligand, thymic stromal lymphopoietin, and IL-33. J Allergy Clin Immunol (2016) 137:278–88.e6. doi:10.1016/j.jaci.2015.05.007
223. Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med (2014) 190:1373–82. doi:10.1164/rccm.201406-1039OC
224. Calven J, Akbarshahi H, Menzel M, Ayata CK, Idzko M, Bjermer L, et al. Rhinoviral stimuli, epithelial factors and ATP signalling contribute to bronchial smooth muscle production of IL-33. J Transl Med (2015) 13:281. doi:10.1186/s12967-015-0645-3
225. Gern JE, Brooks GD, Meyer P, Chang A, Shen K, Evans MD, et al. Bidirectional interactions between viral respiratory illnesses and cytokine responses in the first year of life. J Allergy Clin Immunol (2006) 117:72–8. doi:10.1016/j.jaci.2005.10.002
226. Beasley R, Coleman ED, Hermon Y, Holst PE, O’donnell TV, Tobias M. Viral respiratory tract infection and exacerbations of asthma in adult patients. Thorax (1988) 43:679–83. doi:10.1136/thx.43.9.679
227. Wark PA, Gibson PG. Asthma exacerbations. 3: pathogenesis. Thorax (2006) 61:909–15. doi:10.1136/thx.2005.045187
228. Ege MJ, Mayer M, Normand AC, Genuneit J, Cookson WO, Braun-Fahrlander C, et al. Exposure to environmental microorganisms and childhood asthma. N Engl J Med (2011) 364:701–9. doi:10.1056/NEJMoa1007302
229. Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science (2012) 336:489–93. doi:10.1126/science.1219328
Keywords: asthma, neonatal immune system, respiratory infections, respiratory syncytial virus, influenza, rhinovirus
Citation: Restori KH, Srinivasa BT, Ward BJ and Fixman ED (2018) Neonatal Immunity, Respiratory Virus Infections, and the Development of Asthma. Front. Immunol. 9:1249. doi: 10.3389/fimmu.2018.01249
Received: 09 November 2017; Accepted: 18 May 2018;
Published: 04 June 2018
Edited by:
Cecil Czerkinsky, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Rita Carsetti, Bambino Gesù Ospedale Pediatrico (IRCCS), ItalyValerie Verhasselt, University of Western Australia, Australia
Copyright: © 2018 Restori, Srinivasa, Ward and Fixman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian J. Ward, YnJpYW4ud2FyZCYjeDAwMDQwO21jZ2lsbC5jYQ==;
Elizabeth D. Fixman, ZWxpemFiZXRoLmZpeG1hbiYjeDAwMDQwO21jZ2lsbC5jYQ==