 Mathias Vétillard
Mathias Vétillard Géraldine Schlecht-Louf
Géraldine Schlecht-Louf
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 04 June 2018
Sec. Antigen Presenting Cell Biology
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01232
Dendritic cells (DCs) are key antigen-presenting cells that control the induction of both tolerance and immunity. Understanding the molecular mechanisms regulating DCs commitment toward a regulatory- or effector-inducing profile is critical for better designing prophylactic and therapeutic approaches. Initially identified in dexamethasone-treated thymocytes, the glucocorticoid-induced leucine zipper (GILZ) protein has emerged as a critical factor mediating most, but not all, glucocorticoids effects in both non-immune and immune cells. This intracellular protein exerts pleiotropic effects through interactions with transcription factors and signaling proteins, thus modulating signal transduction and gene expression. GILZ has been reported to control the proliferation, survival, and differentiation of lymphocytes, while its expression confers anti-inflammatory phenotype to monocytes and macrophages. In the past twelve years, a growing set of data has also established that GILZ expression in DCs is a molecular switch controlling their T-cell-priming capacity. Here, after a brief presentation of GILZ isoforms and functions, we summarize current knowledge regarding GILZ expression and regulation in DCs, in both health and disease. We further present the functional consequences of GILZ expression on DCs capacity to prime effector or regulatory T-cell responses and highlight recent findings pointing to a broader role of GILZ in the fine tuning of antigen capture, processing, and presentation by DCs. Finally, we discuss future prospects regarding the possible roles for GILZ in the control of DCs function in the steady state and in the context of infections and chronic pathologies.
Dendritic cells (DCs) are the most potent antigen-presenting cells (APCs), with recognized ability to orchestrate tolerance and immunity. In parallel to antigen processing, they integrate antigen- and microenvironment-associated stimuli and translate them into membrane and cytokine signals for appropriate T-cell priming and polarization or T-cell tolerization (1). Thus, DCs are key regulators of immune homeostasis and gaining knowledge in the mechanisms controlling their polarization toward tolerogenic or immunogenic APCs is of critical importance for both prophylactic and therapeutic approaches in allergy, autoimmunity, inflammatory diseases, infections, and cancers.
Transcriptomic, phenotypic, and functional analyses have identified three steady-state DC subsets in human and mouse (2, 3). Conventional DCs (cDCs) excel in antigen presentation and encompass two subpopulations, namely cDC1 and cDC2, the former being functionally specialized in cross-presentation and type 1 T-helper (Th1) responses induction and the latter promoting Th2 and Th17 T-cell responses (4). The third subset corresponds to plasmacytoid DCs (pDCs) that are specialized in antiviral immunity (5). While each DC subset displays specialized immune-activating functions, all of them have also been shown to promote tolerance and favor regulatory T cells (Treg) differentiation, expansion, and/or activation in certain contexts. This points to mechanisms controlling DCs’ functional switch between tolerogenic and immunogenic APCs (6–8). In addition to these three well-defined DC subsets, inflammatory DCs can arise from monocytes in the course of inflammation, which contribute to innate immune responses and T-cell priming (9).
Among factors reported to skew DCs maturation toward a tolerogenic profile (10), glucocorticoids (GCs), rapamycine, interleukin (IL)-10, transforming growth factor-β (TGF-β), and vitamin D3 (vitD3) have been shown to promote the expression of the glucocorticoid-induced leucine zipper (GILZ) protein (11). GILZ was initially described in murine thymocytes treated with synthetic GCs (Dexamethasone, Dex) (12), but this intracellular protein is expressed in most tissues, including immune cells (12–19). GILZ has since been demonstrated to mediate GCs’ effects in human DCs (20, 21) and has more generally emerged as a regulator of DCs tolerogenic function in both mouse and human (20–27). More recently, we have unraveled that GILZ expression by DCs controls their efficiency at antigen capture and cross-presentation (27), suggesting that the extent of GILZ action in DCs may be broader than initially expected.
Herein, we provide a comprehensive review of recent insights into GILZ expression and functions in DCs and emphasize its implication as a regulator of DCs function, which modulates key processes ranging from antigen capture and presentation to functional maturation and T-cell priming.
Glucocorticoid-induced leucine zipper is encoded by the TGF-β-stimulated clone (TSC) 22 domain family protein 3 (Tsc22d3) gene located on the X-chromosome and is constitutively expressed in most tissues (12, 16, 28). GILZ is among the earliest and highest GC-induced genes. In addition to GC response elements, GILZ promoter harbors binding sites for several transcription factors, including Forkhead-Box O3, C-AMP Response Element-Binding protein, and Serum Responsive Factor. Two E-boxes, one GATA Box as well as putative-binding sites for signal transducer and activator of transcription 6, nuclear factor of activated T-cells and Octamer are also reported (29–32). Five GILZ isoforms exist in mouse, generated upon use of alternative initiation and splicing sites (13, 28). They differ in their N-terminal parts but four of them share conserved TSC and Leucine Zipper (LZ) domains and a C-terminal proline-rich region (PRR) (Figure 1). The term GILZ usually refers to the 137 amino acid (aa)-long GILZ1/Tsc22d3-2 protein in mouse and to the 134 aa-long isoform in human. As only this canonical isoform has been studied in DCs so far (20, 23, 24, 26, 27), we will adopt this nomenclature. Nevertheless, the other GILZ isoforms could be of functional relevance in DCs, as they may modulate GILZ1 function upon dimerization (33), competition, or ensure autonomous function owing to their unique N-terminal domain. GILZ has a short half-life [2–3 h (34–36)] and is quickly degraded upon ubiquitin-proteasome degradation (34). Several sites for posttranslational modification have been predicted in GILZ sequence (Figure 1), but only polyubiquitination by K48 ubiquitins has been confirmed so far (34).
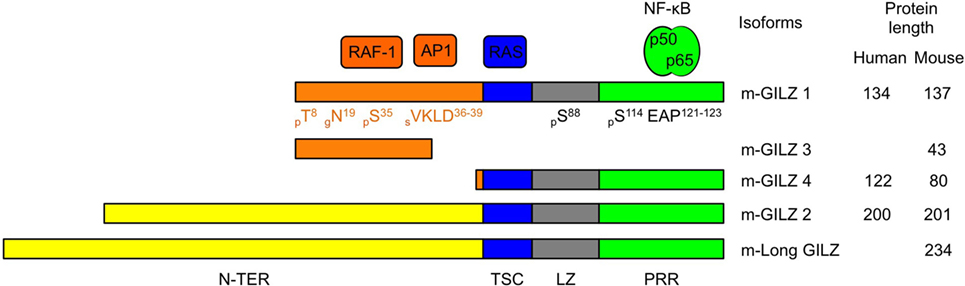
Figure 1. Glucocorticoid-induced leucine zipper (GILZ) isoforms and partners. Five murine GILZ (m-GILZ) isoforms, namely m-GILZ 1–4 and long GILZ (m-Long GILZ) have been identified in mouse (13, 28), with different N-TER domains while m-GILZ1, 2, 4 and m-Long GILZ encompass conserved TSC, LZ, and PRR domains. RAF1 (37) and AP1 (33) interact with GILZ1 and presumably GILZ3 N-TER domain. RAS interacts with the TSC (blue) domain (38). LZ (gray) domain allows dimerization (33). Nuclear factor Kappa B interacts with the PRR (green) domain and requires an EAP motif in positions 121–123 of GILZ1 (39). Predicted posttranslational modification sites are annotated with their positions (X) and the nature of the modification, i.e., phosphorylation, p; glycosylation, g; and sumoylation, s. Tsc22-d3 transcription starts either at a canonical AUG codon or at an upstream non-canonical AUG codon. GILZ isoforms derived from the use of the same codon display identical N-TER domain (orange or yellow, respectively). Protein sequences were aligned using BLAST. The scale is proportional to the real size of the protein. N-TER, N-terminal domain; TSC, TGF-β-stimulated clone; LZ, leucine zipper; PRR, proline-rich region.
In mouse lymphocytes, GILZ controls a wide range of processes including their activation, proliferation, survival, and differentiation (12, 40–43). GILZ also confers anti-inflammatory phenotype to innate immune cells, including human monocytes (25, 44, 45), mouse and human macrophages (35, 36, 46–48), human mastocytes (49), and mouse and human neutrophils (50, 51). Studies mostly done in T cells have reported that GILZ can exert its pleiotropic effects through direct interactions with transcription factors and signaling proteins, according to its cytoplasmic and nuclear distribution (12). GILZ directly binds to NF-κB-p65 (39, 46, 52), AP-1 (33), C/EBP (53), PU.1 (51) and prevents the nuclear translocation of FoxO3 (54). GILZ also interacts with Ras, Raf-1, and mTORC2, thereby inhibiting the MEK/ERK-1/2 and mTORC2/AKT pathways respectively (37, 38, 55). Finally, GILZ can also associate with nuclear DNA (19, 51, 56, 57) and modulate transcription upon competition with positive Th17-polarization regulators (56, 57) or relief of PU.1-mediated repression (51).
Glucocorticoid-induced leucine zipper expression by steady-state human and murine DCs was initially reported by our group, using quantitative RT-PCR, Western Blot, and flow cytometry (21, 23, 27). In mouse, we further documented heterogeneity in GILZ levels among splenic DC subsets, the cDC1 population displaying higher levels than the cDC2 and pDC subsets (27). As in monocytes (25, 44, 45), GILZ mRNA is quickly downregulated upon human blood DCs culture ex vivo (21), pointing to the requirement for an active mechanism to maintain GILZ expression in these cells. This tonic signal is most likely provided, at least in part, by endogenous GCs as in vivo glucocorticoid receptor (GR) blockade reduces GILZ expression in murine splenocytes (58). Accordingly, GILZ mRNA levels follow robust circadian rhythm in adipose tissue and muscle (59–61), and increase, upon restraint stress, in spleen and macrophages (62). However, daytime and stress-induced variations of GILZ levels in DCs have not been explored so far. As in other cell types, exogenous GCs promote GILZ expression in DCs, both in vitro and in vivo (11, 20, 21, 25, 58, 63). Thus, GILZ expression, which is quite low in mouse bone marrow-derived DCs (BMDCs) (23, 26, 27) and absent in human monocytes-derived DCs (Mo-DCs) (20, 45) is dose-dependently upregulated upon Dex treatment (11, 20, 45). GILZ transcription in Mo-DCs is modulated by DC-SCRIPT, a co-repressor of GR (64, 65) (Figure 2). Remarkably, DC-SCRIPT mRNA is selectively and highly expressed in cDCs in the steady state, but not in other immune cells [The immunological Genome Project (66)], suggesting a cDC-specific limitation of GILZ induction by GCs. While the contribution of DC-SCRIPT to GILZ level control in DC-subsets and its modulation upon cell-activation remain to be explored in vivo, the selective limitation of GR-induced GILZ expression in cDCs could potentiate the impact of other GILZ-inducing factors in these cells. Indeed, several molecules present in tissues at the steady state, such as vitD3 (11) and hepatocyte growth factor (HGF) (22), or produced in immunosuppressive microenvironments, as IL-10 and TGF-β, promote GILZ expression in DCs (20, 22, 25), supporting the hypothesis that GILZ is primarily induced in anti-inflammatory contexts (Figure 2). However, GILZ is also overexpressed in clinical grade Mo-DCs exposed to a maturation cocktail containing TNF-α, IL-1β, IL-6, and PGE2 (24), suggesting that pro-inflammatory factors can upregulate GILZ levels in certain conditions.
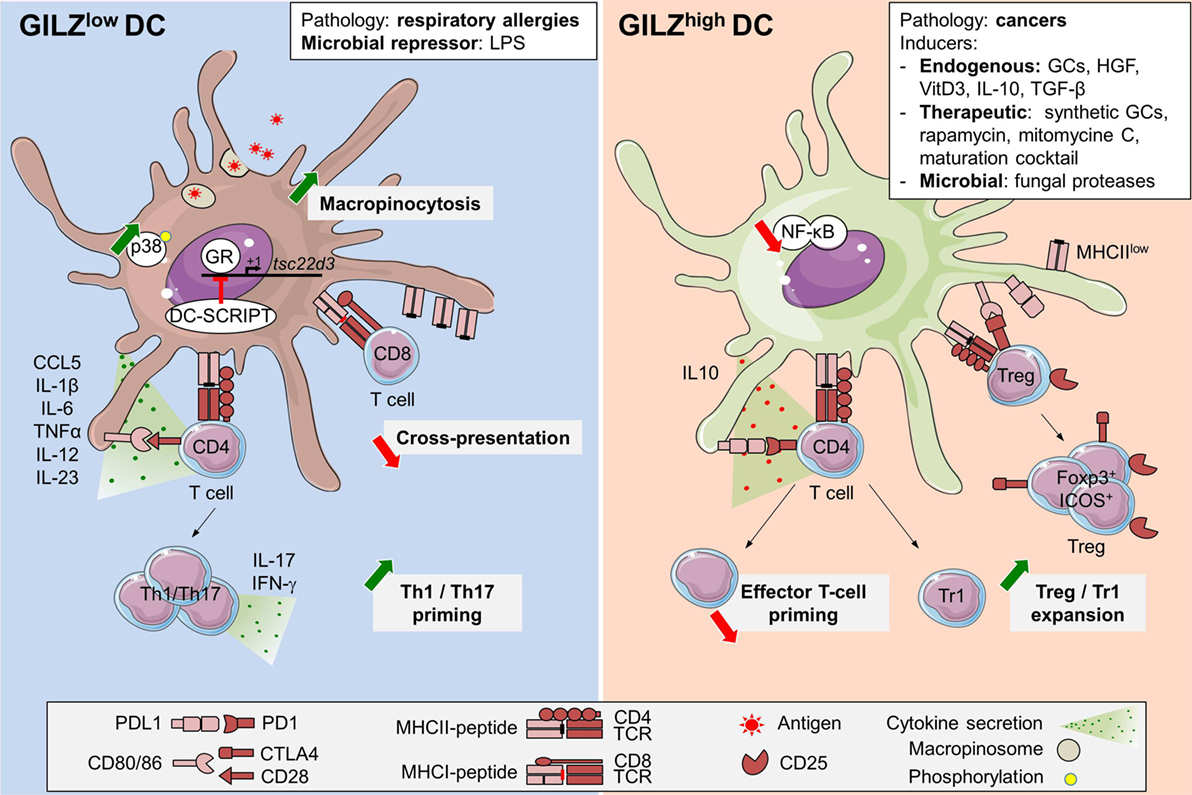
Figure 2. Glucocorticoid-induced leucine zipper (GILZ) regulation and functions in dendritic cells (DCs). GILZ expression in DCs can be induced by steady-state factors (11, 21, 22), immunosuppressive cytokines like TGF-β or interleukin (IL)-10 (20, 25), DC-maturation cocktail containing TNF-α, IL-1β, IL-6, and PGE2 (24), cell-derived factors (26, 45, 67), immunosuppressive drugs as synthetic GCs, rapamycin, and mitomycine C (11), fungal proteases (68), and cancer microenvironment (26). So far, the only exogenous GILZ repressor reported in DCs is LPS (26, 27). In addition, GILZ levels are reduced in blood DCs from respiratory allergic patients (21). DC-SCRIPT acts as an endogenous GR-repressor, thus limiting GILZ induction upon GCs exposure (64, 65). High GILZ levels promote PD-L1 expression and IL-10 production while limiting IL-12 and IL-23 secretion. Thus, GILZhi DCs are poor inducers of Th1 and Th17 T cells but efficient Treg and Tr1 activators (20, 23, 25). GILZ inhibits NF-κB functions upon interaction with p65 subunit (39, 46, 52). GILZ repression promotes macropinocytosis, likely upon increased p38-MAPK phosphorylation (27). GILZ deletion reduces antigen cross-presentation (27). GCs, glucocorticoids; TGF-β, transforming growth factor-β; IL-10, interleukin-10; HGF, hepatocyte growth factor; LPS, lipopolysaccharide; ICOS, inducible co-stimulator; Foxp3, Forkhead box p3; NF-κB, nuclear factor kappa B; CTLA4, cytotoxic T-lymphocyte-associated protein; Tr1, Type 1 regulatory T cells; Treg, regulatory T cells; p38-MAPK, p38 mitogen-activated protein kinase; GR, glucocorticoid receptor; cDC, conventional dendritic cell; tsc22d3, TGF-β-stimulated clone (TSC) 22 domain family protein 3.
So far, few microbial products were tested for their ability to control GILZ expression in DCs. Mouse DCs exposure to lipopolysaccharides downregulates GILZ at the transcript and protein levels, in vitro and in vivo (26, 27) (Figure 2). Conversely, fungal proteases from Aspergillus oryzae promote GILZ overexpression in human Mo-DCs, independently from the GR (68). Alteration of GILZ expression by microbial products has been reported in other cell types. In human epithelial cells, Yersinia enterocolitica YopT and Clostridium difficile Toxin B induce GILZ expression through USF-1 and -2 binding to TSC22D3 promoter (31). In microglia, Tsc22d3 was among the most downregulated genes from antibiotic-treated mice, suggesting a possible contribution of tonic signals from microbiota to GILZ levels control (69). Regarding viruses, Chikungunya and Respiratory Syncytial Virus infections downregulate GILZ expression in primary murine astrocytes (70) and in human epithelial cells (71), respectively. Conversely, Infectious Bursal Disease Virus protein VP4 suppresses GILZ degradation (34), thereby preventing type 1 IFN production in human fibroblast and keratinocyte cell lines (72). Whether GILZ modulation in the course of infections also occurs in DCs and the possible consequences and importance of such regulation remain to be addressed.
Altered GILZ expression in DCs has also been reported in the course of chronic pathologies. Blood DCs from respiratory allergic patients harbor reduced GILZ levels as compared to non-allergic healthy volunteers (21). Besides, GILZ overexpression has been found in tumor-associated DCs from A20 B-cell lymphoma-engrafted mice (26). While GILZ levels in tumor-infiltratring DCs have not been studied in humans so far, the hypothesis that GILZ could be induced in such pathologies is supported by the high GILZ expression detected in infiltrating macrophages in Burkitt’s lymphoma (46). Regarding the mechanisms that could contribute to such GILZ induction, Wang et al. found that hypoxia upregulated GILZ expression in macrophages and rats spleens (62). In addition, Lebson et al. reported GILZ induction in BMDCs exposed to A20- and B16-tumor cells conditioned medium, pointing to a role for soluble factors in this increased expression (26). These factors may include known GILZ inducers as reported in epithelial cell-conditioned medium-treated BMDCs (67). Additional studies have pointed to GILZ levels modulation in cells other than DCs during chronic inflammation. Thus, GILZ expression is reduced in skin lesions from atopic dermatitis (73) and psoriasis patients (63), as well as in macrophages from Crohn’s disease granuloma (46). Conversely, GILZ is overexpressed in the synovia of patients with active rheumatoid arthritis (RA) (58). Whether GILZ acts as an endogenous inhibitor of inflammation, as suggested by exacerbated imiquimod-induced psoriatic-like lesions in GILZ knock-out mice (63) and increased inflammation upon GILZ knock-down in synovial tissues in a murine model of RA (58), or whether it primarily mediates exogenous GCs effects (43) remains debated. The contribution of GILZ induction to GCs therapeutic effects is supported by several studies, two of them having assessed it in DCs. First, restoration of GILZ expression in DCs to normal levels was required for the increase of IL-10+CD4+ T cells, known to mediate oral GC-therapy beneficial effects in respiratory allergic patients (21). Second, in lupus patients, GILZ/prednisolone ratios in pDCs and myeloid DCs exhibited a negative correlation with disease activity (74). In addition to GCs, several therapeutic agents have been reported to induce GILZ in DCs, although the importance of this expression for their effects has not been explored. Thus, GILZ was identified among the most induced genes in Mo-DCs treated by Mitomycine C (75) and the Ca2+-targeting drug rapamycine (11) (Figure 2). Altogether, these data identify GILZ as a common intracellular marker upregulated upon treatment with several immunosuppressive therapeutic molecules. However, its importance in the drug’s effect on DCs function has been proven only for GCs so far.
Seminal work from our laboratory established that GILZ was required for Dex-, IL-10- and TGF-β-induced downregulation of CD80, CD86, and CD83 costimulatory molecules and the increase of immunoglobulin-like transcript 3, programmed death ligand 1 (PD-L1), and IL-10 in human DCs. These results were obtained in Mo-DCs and CD34+ cells-derived DCs, using both lentiviral transduction to overexpress GILZ and RNA-interference on Dex-treated cells to demonstrate GILZ requirement in the tolerogenic polarization of DCs (20, 25). Other groups established that GILZ was necessary for Dex-induced CD86 downregulation in BMDCs (26), PD-L1 induction, and IL-12 inhibition in clinical grade human DCs matured with a standard cocktail (24) and IL-10 secretion by splenic DCs isolated from HGF-treated mice in the course of experimental autoimmune encephalitis (22). GILZ overexpression was then further associated with regulatory phenotype in both human and mouse DCs (11, 23, 68). Of note, in certain studies, the GILZhiPD-L1hi DCs retained high expression of costimulatory molecules (22, 24). Using mice constitutively overexpressing GILZ in DCs (CD11c-GILZhi) and in line with the report of Cohen et al. (25), we could establish that the sole increase of GILZ levels did not alter DCs steady-state phenotype nor cytokine secretion, suggesting that the consequences of GILZ overexpression may depend on the context of GILZ induction. However, upon cognate interaction with T cells in vivo, GILZhi DCs produced higher amounts of IL-10 than control DCs and presented reduced MHC class II molecules levels (23), confirming their regulatory commitment. Additional insights into the importance of GILZ expression for DCs function came from total (63) and conditional (27) GILZKO mouse models. While these studies pointed to minor impact of GILZ deletion on DCs phenotype (27, 63), the cytokine response to TLR4 and TLR7 stimulations was markedly increased in GILZKO BMDCs, with higher IL-1α and β, IL-6, and IL-23 secretion (63), in line with GILZ inhibition promoting IL-12, TNF-α, and CCL5 production by Mo-DCs (20, 24, 45). In conclusion, GILZ expression in DCs promotes immature phenotype and IL-10 production while limiting secretion of Th1- and Th17-inducing cytokines (Figure 2), thereby favoring DCs polarization toward a tolerogenic profile (20–27).
The consequences of GILZ levels in DCs on their capacity to activate T-cell responses were first investigated in recall responses toward vaccine and cytomegalovirus antigens (20, 24). In these studies, Dex-induced GILZhi human Mo-DCs promoted poor autologous CD4+ T-cell proliferation and IFN-γ production, and this was reversed upon GILZ silencing. Along this line, clinical grade activated Mo-DCs were later shown to activate more efficiently CD8+ T-cell secondary responses when GILZ was knocked-down (24). Similar results were obtained for primary T-cell responses in vivo, the adoptive transfer of GILZhi DCs inducing poor effector T-cell (Teff) activation and IFN-γ secretion (23) while that of GILZKO (22) or GILZ-silenced (26) DCs led to enhanced CD4+ T cell proliferation, activation, as well as IFN-γ and IL-17 production (Figure 2). Remarkably, the poor Teff activation by GILZhi DCs was associated with the expansion of antigen-specific IL-10+CD4+ T cells in both human and mouse (20–23), which was abolished by GILZ silencing or genetic deletion in DCs (20–22). These IL-10+CD4+ T cells included Foxp3− cells, a phenotype consistent with Tr1 cells (20, 21), and Foxp3+ Treg (20–23), and inhibited Mo-DCs-induced autologous CD4+ and CD8+ T-cell proliferation through an IL-10-dependent antigen-specific mechanism (20, 21). We further established in vivo that Treg expansion by GILZhi DCs was abrogated upon depletion of pre-existing Treg (20, 23), supporting the conclusion that GILZhi DCs could expand thymus-derived Treg (tTreg) more efficiently than control DCs. Such an expansion might explain the spontaneous accumulation of Treg in CD11c-GILZhi mice, with an increased frequency of inducible co-stimulator (ICOS)hi Treg. Importantly, in the absence of transferred tTreg, GILZhi DCs still poorly induced conventional T cells activation, thus suggesting that GILZhi DCs were intrinsically inefficient in Teff priming, a characteristic of tolerogenic DCs (10). Altogether, the results from these studies identify GILZ expression levels in DCs as a critical switch controlling their ability to prime Teff versus Treg, mediating Dex- and HGF-induced effects on DCs phenotype, cytokine secretion, and T-cell priming capacities and required for their beneficial effects in respiratory allergic patients (21) and experimental autoimmune encephalitis (22).
Finally, GILZ expression in murine DCs has also been shown to control their antigen capture capacity. The initial report was made by Lebson et al., who observed that GILZ silencing led to increased ovalbumine (OVA) capture in BMDCs (26). Consistently, analysis of antigen uptake by DCs from CD11c-GILZKO mice revealed an increase in Dextran-FITC internalization by the splenic cDC1 subset in vivo. Using GILZKO and GILZhi BMDCs, we showed that GILZ selectively regulated Dextran, OVA, and Lucifer Yellow macropinocytosis, but not the phagocytosis of particulate zymosan. This control operated in immature and recently activated BMDCs, through a mechanism that may at least partly depend on the control of p38 mitogen-activated protein kinase phosphorylation (27). The fact that GILZ limits antigen internalization while Dex-treated DCs display enhanced antigen capture (76, 77) points to opposite effects of GILZ and GCs on certain DCs functions. Unexpectedly, the higher OVA macropinocytosis in GILZKO DCs did not result in increased peptide presentation to a CD4+ T-cell hybridoma and was even associated to reduced cross-presentation to a CD8+ T-cell hybridoma, despite efficient antigen degradation (27). These results suggested that the fine-tuning of antigen capture by GILZ in DCs might serve to regulate the quality rather that quantity of antigen available for presentation to T cells. It might also regulate antigen internalization for later transfer to B cells (78). Alternatively, GILZ might control additional steps in the antigen degradation and/or cross-presenting machinery of DCs. Further studies will be needed to address the physiological relevance of GILZ-mediated macropinocytosis modulation in DC subsets in adequate models in vivo. The link established between the macropinocytic process and the regulation of DC migration (79–81) suggests that GILZ expression might also control DCs trafficking in vivo, a question deserving dedicated analysis.
Glucocorticoid-induced leucine zipper has initially been identified in DCs as a key mediator of GCs’ effects, piloting DCs commitment toward a tolerogenic profile. Indeed, GILZ promotes immature phenotype and IL-10 production in DCs while limiting secretion of IL-12 and IL-23, thus favoring regulatory responses. In addition, GILZ also regulates DCs access to antigen by modulating macropinocytosis, thus pointing to its regulatory role at many steps of DCs function. Recent studies have revealed that GILZ expression in DCs is constitutive and can be modulated by their ontogeny and microenvironment, with cell-specific regulatory systems. Thus, GILZ may control DCs access to antigen and the issue of their interactions with T cells in the steady state as well as in certain pathologies. Furthermore, considering the wide effects reported for GILZ in other cell types, GILZ might impact DC biology in an even broader scale with possible effects on proliferation, cell survival, and migration. Deciphering the regulation of GILZ expression in DCs as well as its role in DC subsets function in vivo will deserve additional studies, which will provide insights into the fine mechanisms controlling DCs and might open new avenues for therapeutic approaches.
MV and GS-L jointly wrote the review and prepared the figures.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work benefited from data assembled by the Immgen Consortium. We thank Rim Makni-Bejar for correcting grammatical and syntax errors.
This work was supported by the Fondation ARC pour la Recherche sur le Cancer. MV is supported by a doctoral fellowship from the Doctoral School “Innovation Thérapeutique du fondamental à l’appliqué” (ED 569).
1. Pulendran B. The varieties of immunological experience: of pathogens, stress, and dendritic cells. Annu Rev Immunol (2015) 33:563–606. doi:10.1146/annurev-immunol-020711-075049
2. Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S, et al. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity (2016) 45(3):669–84. doi:10.1016/j.immuni.2016.08.015
3. See P, Dutertre CA, Chen J, Gunther P, McGovern N, Irac SE, et al. Mapping the human DC lineage through the integration of high-dimensional techniques. Science (2017) 356:6342. doi:10.1126/science.aag3009
4. Vu Manh TP, Bertho N, Hosmalin A, Schwartz-Cornil I, Dalod M. Investigating evolutionary conservation of dendritic cell subset identity and functions. Front Immunol (2015) 6:260. doi:10.3389/fimmu.2015.00260
5. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol (2015) 15(8):471–85. doi:10.1038/nri3865
6. Lutz MB. Induction of CD4(+) regulatory and polarized effector/helper T cells by dendritic cells. Immune Netw (2016) 16(1):13–25. doi:10.4110/in.2016.16.1.13
7. Domogalla MP, Rostan PV, Raker VK, Steinbrink K. Tolerance through education: how tolerogenic dendritic cells shape immunity. Front Immunol (2017) 8:1764. doi:10.3389/fimmu.2017.01764
8. Iberg CA, Jones A, Hawiger D. Dendritic cells as inducers of peripheral tolerance. Trends Immunol (2017) 38(11):793–804. doi:10.1016/j.it.2017.07.007
9. Segura E, Amigorena S. Inflammatory dendritic cells in mice and humans. Trends Immunol (2013) 34(9):440–5. doi:10.1016/j.it.2013.06.001
10. Gordon JR, Ma Y, Churchman L, Gordon SA, Dawicki W. Regulatory dendritic cells for immunotherapy in immunologic diseases. Front Immunol (2014) 5:7. doi:10.3389/fimmu.2014.00007
11. Zimmer A, Bouley J, Le Mignon M, Pliquet E, Horiot S, Turfkruyer M, et al. A regulatory dendritic cell signature correlates with the clinical efficacy of allergen-specific sublingual immunotherapy. J Allergy Clin Immunol (2012) 129(4):1020–30. doi:10.1016/j.jaci.2012.02.014
12. D’Adamio F, Zollo O, Moraca R, Ayroldi E, Bruscoli S, Bartoli A, et al. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity (1997) 7(6):803–12. doi:10.1016/S1074-7613(00)80398-2
13. Bruscoli S, Donato V, Velardi E, Di Sante M, Migliorati G, Donato R, et al. Glucocorticoid-induced leucine zipper (GILZ) and long GILZ inhibit myogenic differentiation and mediate anti-myogenic effects of glucocorticoids. J Biol Chem (2010) 285(14):10385–96. doi:10.1074/jbc.M109.070136
14. Bruscoli S, Velardi E, Di Sante M, Bereshchenko O, Venanzi A, Coppo M, et al. Long glucocorticoid-induced leucine zipper (L-GILZ) protein interacts with ras protein pathway and contributes to spermatogenesis control. J Biol Chem (2012) 287(2):1242–51. doi:10.1074/jbc.M111.316372
15. Cannarile L, Zollo O, D’Adamio F, Ayroldi E, Marchetti C, Tabilio A, et al. Cloning, chromosomal assignment and tissue distribution of human GILZ, a glucocorticoid hormone-induced gene. Cell Death Differ (2001) 8(2):201–3. doi:10.1038/sj.cdd.4400798
16. Cari L, Ricci E, Gentili M, Petrillo MG, Ayroldi E, Ronchetti S, et al. A focused real time PCR strategy to determine GILZ expression in mouse tissues. Results Immunol (2015) 5:37–42. doi:10.1016/j.rinim.2015.10.003
17. Eddleston J, Herschbach J, Wagelie-Steffen AL, Christiansen SC, Zuraw BL. The anti-inflammatory effect of glucocorticoids is mediated by glucocorticoid-induced leucine zipper in epithelial cells. J Allergy Clin Immunol (2007) 119(1):115–22. doi:10.1016/j.jaci.2006.12.532
18. Robert O, Boujedidi H, Bigorgne A, Ferrere G, Voican CS, Vettorazzi S, et al. Decreased expression of the glucocorticoid receptor-GILZ pathway in Kupffer cells promotes liver inflammation in obese mice. J Hepatol (2016) 64(4):916–24. doi:10.1016/j.jhep.2015.11.023
19. Shi X, Shi W, Li Q, Song B, Wan M, Bai S, et al. A glucocorticoid-induced leucine-zipper protein, GILZ, inhibits adipogenesis of mesenchymal cells. EMBO Rep (2003) 4(4):374–80. doi:10.1038/sj.embor.embor805
20. Hamdi H, Godot V, Maillot MC, Prejean MV, Cohen N, Krzysiek R, et al. Induction of antigen-specific regulatory T lymphocytes by human dendritic cells expressing the glucocorticoid-induced leucine zipper. Blood (2007) 110(1):211–9. doi:10.1182/blood-2006-10-052506
21. Karaki S, Garcia G, Tcherakian C, Capel F, Tran T, Pallardy M, et al. Enhanced glucocorticoid-induced leucine zipper in dendritic cells induces allergen-specific regulatory CD4(+) T-cells in respiratory allergies. Allergy (2014) 69(5):624–31. doi:10.1111/all.12379
22. Benkhoucha M, Molnarfi N, Dunand-Sauthier I, Merkler D, Schneiter G, Bruscoli S, et al. Hepatocyte growth factor limits autoimmune neuroinflammation via glucocorticoid-induced leucine zipper expression in dendritic cells. J Immunol (2014) 193(6):2743–52. doi:10.4049/jimmunol.1302338
23. Calmette J, Ellouze M, Tran T, Karaki S, Ronin E, Capel F, et al. Glucocorticoid-induced leucine zipper enhanced expression in dendritic cells is sufficient to drive regulatory T cells expansion in vivo. J Immunol (2014) 193(12):5863–72. doi:10.4049/jimmunol.1400758
24. Cathelin D, Met O, Svane IM. Silencing of the glucocorticoid-induced leucine zipper improves the immunogenicity of clinical-grade dendritic cells. Cytotherapy (2013) 15(6):740–9. doi:10.1016/j.jcyt.2013.02.005
25. Cohen N, Mouly E, Hamdi H, Maillot MC, Pallardy M, Godot V, et al. GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood (2006) 107(5):2037–44. doi:10.1182/blood-2005-07-2760
26. Lebson L, Wang T, Jiang Q, Whartenby KA. Induction of the glucocorticoid-induced leucine zipper gene limits the efficacy of dendritic cell vaccines. Cancer Gene Ther (2011) 18(8):563–70. doi:10.1038/cgt.2011.23 Epub 2011/05/07.,
27. Calmette J, Bertrand M, Vetillard M, Ellouze M, Flint S, Nicolas V, et al. Glucocorticoid-induced leucine zipper protein controls macropinocytosis in dendritic cells. J Immunol (2016) 197(11):4247–56. doi:10.4049/jimmunol.1600561
28. Soundararajan R, Wang J, Melters D, Pearce D. Differential activities of glucocorticoid-induced leucine zipper protein isoforms. J Biol Chem (2007) 282(50):36303–13. doi:10.1074/jbc.M707287200
29. Asselin-Labat ML, Biola-Vidamment A, Kerbrat S, Lombes M, Bertoglio J, Pallardy M. FoxO3 mediates antagonistic effects of glucocorticoids and interleukin-2 on glucocorticoid-induced leucine zipper expression. Mol Endocrinol (2005) 19(7):1752–64. doi:10.1210/me.2004-0206
30. Asselin-Labat ML, David M, Biola-Vidamment A, Lecoeuche D, Zennaro MC, Bertoglio J, et al. GILZ, a new target for the transcription factor FoxO3, protects T lymphocytes from interleukin-2 withdrawal-induced apoptosis. Blood (2004) 104(1):215–23. doi:10.1182/blood-2003-12-4295
31. Koberle M, Goppel D, Grandl T, Gaentzsch P, Manncke B, Berchtold S, et al. Yersinia enterocolitica YopT and Clostridium difficile toxin B induce expression of GILZ in epithelial cells. PLoS One (2012) 7(7):e40730. doi:10.1371/journal.pone.0040730
32. Ng HP, Jennings S, Wang J, Molina PE, Nelson S, Wang G. Non-canonical glucocorticoid receptor transactivation of gilz by alcohol suppresses cell inflammatory response. Front Immunol (2017) 8:661. doi:10.3389/fimmu.2017.00661
33. Mittelstadt PR, Ashwell JD. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J Biol Chem (2001) 276(31):29603–10. doi:10.1074/jbc.M101522200
34. He Z, Chen X, Fu M, Tang J, Li X, Cao H, et al. Infectious bursal disease virus protein VP4 suppresses type I interferon expression via inhibiting K48-linked ubiquitylation of glucocorticoid-induced leucine zipper (GILZ). Immunobiology (2017) 223(4–5):374–82. doi:10.1016/j.imbio.2017.10.048
35. Hoppstadter J, Diesel B, Eifler LK, Schmid T, Brune B, Kiemer AK. Glucocorticoid-induced leucine zipper is downregulated in human alveolar macrophages upon toll-like receptor activation. Eur J Immunol (2012) 42(5):1282–93. doi:10.1002/eji.201142081
36. Hoppstadter J, Kiemer AK. Glucocorticoid-induced leucine zipper (GILZ) in immuno suppression: master regulator or bystander? Oncotarget (2015) 6(36):38446–57. doi:10.18632/oncotarget.6197
37. Ayroldi E, Zollo O, Macchiarulo A, Di Marco B, Marchetti C, Riccardi C. Glucocorticoid-induced leucine zipper inhibits the Raf-extracellular signal-regulated kinase pathway by binding to Raf-1. Mol Cell Biol (2002) 22(22):7929–41. doi:10.1128/MCB.22.22.7929-7941.2002
38. Ayroldi E, Zollo O, Bastianelli A, Marchetti C, Agostini M, Di Virgilio R, et al. GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of Ras signaling. J Clin Invest (2007) 117(6):1605–15. doi:10.1172/JCI30724
39. Di Marco B, Massetti M, Bruscoli S, Macchiarulo A, Di Virgilio R, Velardi E, et al. Glucocorticoid-induced leucine zipper (GILZ)/NF-kappaB interaction: role of GILZ homo-dimerization and C-terminal domain. Nucleic Acids Res (2007) 35(2):517–28. doi:10.1093/nar/gkl1080
40. Bereshchenko O, Coppo M, Bruscoli S, Biagioli M, Cimino M, Frammartino T, et al. GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-beta signaling. Cell Rep (2014) 7(2):464–75. doi:10.1016/j.celrep.2014.03.004
41. Cannarile L, Cuzzocrea S, Santucci L, Agostini M, Mazzon E, Esposito E, et al. Glucocorticoid-induced leucine zipper is protective in Th1-mediated models of colitis. Gastroenterology (2009) 136(2):530–41. doi:10.1053/j.gastro.2008.09.024
42. Cannarile L, Fallarino F, Agostini M, Cuzzocrea S, Mazzon E, Vacca C, et al. Increased GILZ expression in transgenic mice up-regulates Th-2 lymphokines. Blood (2006) 107(3):1039–47. doi:10.1182/blood-2005-05-2183
43. Ngo D, Beaulieu E, Gu R, Leaney A, Santos L, Fan H, et al. Divergent effects of endogenous and exogenous glucocorticoid-induced leucine zipper in animal models of inflammation and arthritis. Arthritis Rheum (2013) 65(5):1203–12. doi:10.1002/art.37858
44. Ehrchen J, Steinmuller L, Barczyk K, Tenbrock K, Nacken W, Eisenacher M, et al. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood (2007) 109(3):1265–74. doi:10.1182/blood-2006-02-001115
45. Futterleib JS, Feng H, Tigelaar RE, Choi J, Edelson RL. Activation of GILZ gene by photoactivated 8-methoxypsoralen: potential role of immunoregulatory dendritic cells in extracorporeal photochemotherapy. Transfus Apher Sci (2014) 50(3):379–87. doi:10.1016/j.transci.2013.10.003
46. Berrebi D, Bruscoli S, Cohen N, Foussat A, Migliorati G, Bouchet-Delbos L, et al. Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: an anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood (2003) 101(2):729–38. doi:10.1182/blood-2002-02-0538
47. Pinheiro I, Dejager L, Petta I, Vandevyver S, Puimege L, Mahieu T, et al. LPS resistance of SPRET/Ei mice is mediated by Gilz, encoded by the Tsc22d3 gene on the X chromosome. EMBO Mol Med (2013) 5(3):456–70. doi:10.1002/emmm.201201683 Epub 2013/03/16.,
48. Yang YH, Aeberli D, Dacumos A, Xue JR, Morand EF. Annexin-1 regulates macrophage IL-6 and TNF via glucocorticoid-induced leucine zipper. J Immunol (2009) 183(2):1435–45. doi:10.4049/jimmunol.0804000
49. Godot V, Garcia G, Capel F, Arock M, Durand-Gasselin I, Asselin-Labat ML, et al. Dexamethasone and IL-10 stimulate glucocorticoid-induced leucine zipper synthesis by human mast cells. Allergy (2006) 61(7):886–90. doi:10.1111/j.1398-9995.2006.01065.x
50. Espinasse MA, Hajage D, Montravers P, Piednoir P, Dufour G, Tubach F, et al. Neutrophil expression of glucocorticoid-induced leucine zipper (GILZ) anti-inflammatory protein is associated with acute respiratory distress syndrome severity. Ann Intensive Care (2016) 6(1):105. doi:10.1186/s13613-016-0210-0
51. Ricci E, Ronchetti S, Pericolini E, Gabrielli E, Cari L, Gentili M, et al. Role of the glucocorticoid-induced leucine zipper gene in dexamethasone-induced inhibition of mouse neutrophil migration via control of annexin A1 expression. FASEB J (2017) 31(7):3054–65. doi:10.1096/fj.201601315R
52. Ayroldi E, Migliorati G, Bruscoli S, Marchetti C, Zollo O, Cannarile L, et al. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood (2001) 98(3):743–53. doi:10.1182/blood.V98.3.743
53. Pan G, Cao J, Yang N, Ding K, Fan C, Xiong WC, et al. Role of glucocorticoid-induced leucine zipper (GILZ) in bone acquisition. J Biol Chem (2014) 289(28):19373–82. doi:10.1074/jbc.M113.535237
54. Latre de Late P, Pepin A, Assaf-Vandecasteele H, Espinasse C, Nicolas V, Asselin-Labat ML, et al. Glucocorticoid-induced leucine zipper (GILZ) promotes the nuclear exclusion of FOXO3 in a Crm1-dependent manner. J Biol Chem (2009) 285(8):5594–605. doi:10.1074/jbc.M109.068346
55. Joha S, Nugues AL, Hetuin D, Berthon C, Dezitter X, Dauphin V, et al. GILZ inhibits the mTORC2/AKT pathway in BCR-ABL(+) cells. Oncogene (2012) 31(11):1419–30. doi:10.1038/onc.2011.328
56. Luz-Crawford P, Espinosa-Carrasco G, Ipseiz N, Contreras R, Tejedor G, Medina DA, et al. Gilz-activin A as a novel signaling axis orchestrating mesenchymal stem cell and Th17 cell interplay. Theranostics (2018) 8(3):846–59. doi:10.7150/thno.21793
57. Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature (2013) 496(7446):461–8. doi:10.1038/nature11981
58. Beaulieu E, Ngo D, Santos L, Yang YH, Smith M, Jorgensen C, et al. Glucocorticoid-induced leucine zipper is an endogenous antiinflammatory mediator in arthritis. Arthritis Rheum (2010) 62(9):2651–61. doi:10.1002/art.27566
59. Ayyar VS, DuBois DC, Almon RR, Jusko WJ. Mechanistic multi-tissue modeling of glucocorticoid-induced leucine zipper regulation: integrating circadian gene expression with receptor-mediated corticosteroid pharmacodynamics. J Pharmacol Exp Ther (2017) 363(1):45–57. doi:10.1124/jpet.117.242990
60. Gimble JM, Ptitsyn AA, Goh BC, Hebert T, Yu G, Wu X, et al. Delta sleep-inducing peptide and glucocorticoid-induced leucine zipper: potential links between circadian mechanisms and obesity? Obes Rev (2009) 10(Suppl 2):46–51. doi:10.1111/j.1467-789X.2009.00661.x
61. Miller BH, McDearmon EL, Panda S, Hayes KR, Zhang J, Andrews JL, et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci U S A (2007) 104(9):3342–7. doi:10.1073/pnas.0611724104
62. Wang Y, Ma YY, Song XL, Cai HY, Chen JC, Song LN, et al. Upregulations of glucocorticoid-induced leucine zipper by hypoxia and glucocorticoid inhibit proinflammatory cytokines under hypoxic conditions in macrophages. J Immunol (2012) 188(1):222–9. doi:10.4049/jimmunol.1002958
63. Jones SA, Perera DN, Fan H, Russ BE, Harris J, Morand EF. GILZ regulates Th17 responses and restrains IL-17-mediated skin inflammation. J Autoimmun (2015) 61:73–80. doi:10.1016/j.jaut.2015.05.010
64. Hontelez S, Karthaus N, Looman MW, Ansems M, Adema GJ. DC-SCRIPT regulates glucocorticoid receptor function and expression of its target GILZ in dendritic cells. J Immunol (2013) 190(7):3172–9. doi:10.4049/jimmunol.1201776
65. Sondergaard JN, Poghosyan S, Hontelez S, Louche P, Looman MW, Ansems M, et al. DC-SCRIPT regulates IL-10 production in human dendritic cells by modulating NF-kappaBp65 activation. J Immunol (2015) 195(4):1498–505. doi:10.4049/jimmunol.1402924
66. Heng TS, Painter MW. Immunological genome project C. The immunological genome project: networks of gene expression in immune cells. Nat Immunol (2008) 9(10):1091–4. doi:10.1038/ni1008-1091
67. Weitnauer M, Schmidt L, Ng Kuet Leong N, Muenchau S, Lasitschka F, Eckstein V, et al. Bronchial epithelial cells induce alternatively activated dendritic cells dependent on glucocorticoid receptor signaling. J Immunol (2014) 193(3):1475–84. doi:10.4049/jimmunol.1400446
68. Zimmer A, Luce S, Gaignier F, Nony E, Naveau M, Biola-Vidamment A, et al. Identification of a new phenotype of tolerogenic human dendritic cells induced by fungal proteases from Aspergillus oryzae. J Immunol (2011) 186(7):3966–76. doi:10.4049/jimmunol.1003184
69. Thion MS, Low D, Silvin A, Chen J, Grisel P, Schulte-Schrepping J, et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell (2017) 172(3):500–516.e16. doi:10.1016/j.cell.2017.11.042
70. Das T, Hoarau JJ, Jaffar Bandjee MC, Maquart M, Gasque P. Multifaceted innate immune responses engaged by astrocytes, microglia and resident dendritic cells against Chikungunya neuroinfection. J Gen Virol (2015) 96(Pt 2):294–310. doi:10.1099/vir.0.071175-0
71. Webster Marketon JI, Corry J, Teng MN. The respiratory syncytial virus (RSV) nonstructural proteins mediate RSV suppression of glucocorticoid receptor transactivation. Virology (2014) 449:62–9. doi:10.1016/j.virol.2013.11.014
72. Li Z, Wang Y, Li X, Li X, Cao H, Zheng SJ. Critical roles of glucocorticoid-induced leucine zipper in infectious bursal disease virus (IBDV)-induced suppression of type I Interferon expression and enhancement of IBDV growth in host cells via interaction with VP4. J Virol (2013) 87(2):1221–31. doi:10.1128/JVI.02421-12
73. Baek JO, Lee JR, Roh JY, Jung Y. Oral tolerance modulates the skin transcriptome in mice with induced atopic dermatitis. Allergy (2017) 73(4):962–6. doi:10.1111/all.13367
74. Jones SA, Toh AE, Odobasic D, Oudin MA, Cheng Q, Lee JP, et al. Glucocorticoid-induced leucine zipper (GILZ) inhibits B cell activation in systemic lupus erythematosus. Ann Rheum Dis (2016) 75(4):739–47. doi:10.1136/annrheumdis-2015-207744
75. Terness P, Oelert T, Ehser S, Chuang JJ, Lahdou I, Kleist C, et al. Mitomycin C-treated dendritic cells inactivate autoreactive T cells: toward the development of a tolerogenic vaccine in autoimmune diseases. Proc Natl Acad Sci U S A (2008) 105(47):18442–7. doi:10.1073/pnas.0807185105
76. Maggi J, Schinnerling K, Pesce B, Hilkens CM, Catalan D, Aguillon JC. Dexamethasone and monophosphoryl lipid A-modulated dendritic cells promote antigen-specific tolerogenic properties on naive and memory CD4(+) T cells. Front Immunol (2016) 7:359. doi:10.3389/fimmu.2016.00359
77. Piemonti L, Monti P, Allavena P, Leone BE, Caputo A, Di Carlo V. Glucocorticoids increase the endocytic activity of human dendritic cells. Int Immunol (1999) 11(9):1519–26. doi:10.1093/intimm/11.9.1519
78. Le Roux D, Le Bon A, Dumas A, Taleb K, Sachse M, Sikora R, et al. Antigen stored in dendritic cells after macropinocytosis is released unprocessed from late endosomes to target B cells. Blood (2012) 119(1):95–105. doi:10.1182/blood-2011-02-336123
79. Chabaud M, Heuze ML, Bretou M, Vargas P, Maiuri P, Solanes P, et al. Cell migration and antigen capture are antagonistic processes coupled by myosin II in dendritic cells. Nat Commun (2015) 6:7526. doi:10.1038/ncomms8526
80. Vargas P, Maiuri P, Bretou M, Saez PJ, Pierobon P, Maurin M, et al. Innate control of actin nucleation determines two distinct migration behaviours in dendritic cells. Nat Cell Biol (2016) 18(1):43–53. doi:10.1038/ncb3284
Keywords: dendritic cells, tolerance, TSC22D3, glucocorticoid-induced leucine zipper, antigen presentation, regulatory T cells
Citation: Vétillard M and Schlecht-Louf G (2018) Glucocorticoid-Induced Leucine Zipper: Fine-Tuning of Dendritic Cells Function. Front. Immunol. 9:1232. doi: 10.3389/fimmu.2018.01232
Received: 23 February 2018; Accepted: 16 May 2018;
Published: 04 June 2018
Edited by:
Pierre Guermonprez, King’s College London, United KingdomReviewed by:
Susan Kovats, Oklahoma Medical Research Foundation, United StatesCopyright: © 2018 Vétillard and Schlecht-Louf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Géraldine Schlecht-Louf, Z2VyYWxkaW5lLnNjaGxlY2h0LWxvdWZAdS1wc3VkLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.