 Ute Greczmiel
Ute Greczmiel Annette Oxenius
Annette Oxenius- Institute of Microbiology, ETH Zürich, Zürich, Switzerland
Chronic infections with non-cytopathic viruses constitutively expose virus-specific adaptive immune cells to cognate antigen, requiring their numeric and functional adaptation. Virus-specific CD8 T cells are compromised by various means in their effector functions, collectively termed T cell exhaustion. Alike CD8 T cells, virus-specific CD4 Th1 cell responses are gradually downregulated but instead, follicular T helper (TFH) cell differentiation and maintenance is strongly promoted during chronic infection. Thereby, the immune system promotes antibody responses, which bear less immune-pathological risk compared to cytotoxic and pro-inflammatory T cell responses. This emphasis on TFH cells contributes to tolerance of the chronic infection and is pivotal for the continued maturation and adaptation of the antibody response, leading eventually to the emergence of virus-neutralizing antibodies, which possess the potential to control the established chronic infection. However, sustained high levels of TFH cells can also result in a less stringent B cell selection process in active germinal center reactions, leading to the activation of virus-unspecific B cells, including self-reactive B cells, and to hypergammaglobulinemia. This dispersal of B cell help comes at the expense of a stringently selected virus-specific antibody response, thereby contributing to its delayed maturation. Here, we discuss these opposing facets of TFH cells in chronic viral infections.
Introduction
Non-or poorly cytopathic viruses like human immunodeficiency virus 1 (HIV-1), hepatitis B virus (HBV), and hepatitis C virus (HCV) in humans or lymphocytic choriomeningitis virus (LCMV) in mice can induce persistent infections employing several mechanisms to evade control by the immune system. Continuous high-level viral replication and therefore high viral burden in the host is a major factor leading to numeric reduction and functional impairment of virus-specific cytotoxic CD8 T cells and Th1 CD4 T cells, collectively termed T cell exhaustion [reviewed in Ref. (1–3)]. In this setting, immune effector functions being less prone to induce immunopathology, like the humoral arm of immunity, are beneficial to contain viral spread (4–10). Especially, virus-neutralizing antibodies can inhibit new infection of host cells and thereby effectively limit viral spread. However, isotype-switched neutralizing antibodies often appear very late after the onset of persistent viral infections, being frequently delayed for several weeks to months (5, 11, 12). B cell dysregulation, including hypergammaglobulinemia and polyclonal B cell activation, contributes to the late emergence of virus-neutralizing antibodies (13, 14). Furthermore, mutational viral evolution results in selection of variants that escape the neutralizing antibody response, promoting persistence of the infection (12, 15–22).
Isotype-switched antibody responses are elicited in a T-help-dependent manner, being regulated by the interaction between follicular T helper (TFH) cells and cognate B cells (23). Activity of TFH cells is regulated by the transcriptional repressor B cell lymphoma (Bcl)-6 (24–26) which sustains, among other functions, upregulation of the chemokine receptor CX-chemokine receptor (CXCR) 5 that in turn mediates localization of TFH cells to the B cell follicle (27–29). There, TFH cells initiate B cell differentiation into either short-lived plasmablasts or germinal center (GC) B cells (30–33). Conversely, contact between TFH cells and cognate B cells is necessary to induce differentiation of TFH cells into GC TFH cells and to sustain their TFH phenotype (34–37), albeit this is disputed to also hold in case of persistent viral infections (38). TFH cells mediate affinity selection of B cells that have undergone proliferation and somatic hypermutation (SHM) by delivering survival signals via ICOS, CD40 ligand (CD40L), and the cytokine IL-21, depending on the affinity of the B cell for a given antigen (39–41). Therefore, TFH cells are essential for the induction and maintenance of the GC response.
Interestingly, TFH cells accumulate during the persistent phase of viral infections with non- or poorly cytopathic viruses (8, 38, 42, 43) while differentiation of naïve CD4 T cells into Th1 CD4 T cells is largely abrogated in this phase due to a sustained IFN-I environment (44). The expansion of the TFH population is most likely driven by follicular dendritic cell (FDC)-derived IL-6 signaling via signal transducer and activator of transcription (STAT)-3 (8, 43, 45), and the prolonged persistence of viral antigen in the host environment (46). It would be intriguing to conjecture an essential role of the sustained expansion of the TFH cell population for the eventual induction of the virus-neutralizing antibody response and also adaptation of the protective response to an evolving virus. However, accumulation of TFH cells might also contribute to the observed B cell dysregulation and thereby delay of the neutralizing antibody response (Figure 1). Here, we discuss evidence for both, promotion of late emergence of virus-neutralizing antibodies and dysregulated B cell responses in the context of chronic viral infections, focusing on experimental LCMV infection in mice and HIV-1, HCV, and HBV infection in humans (Table 1).
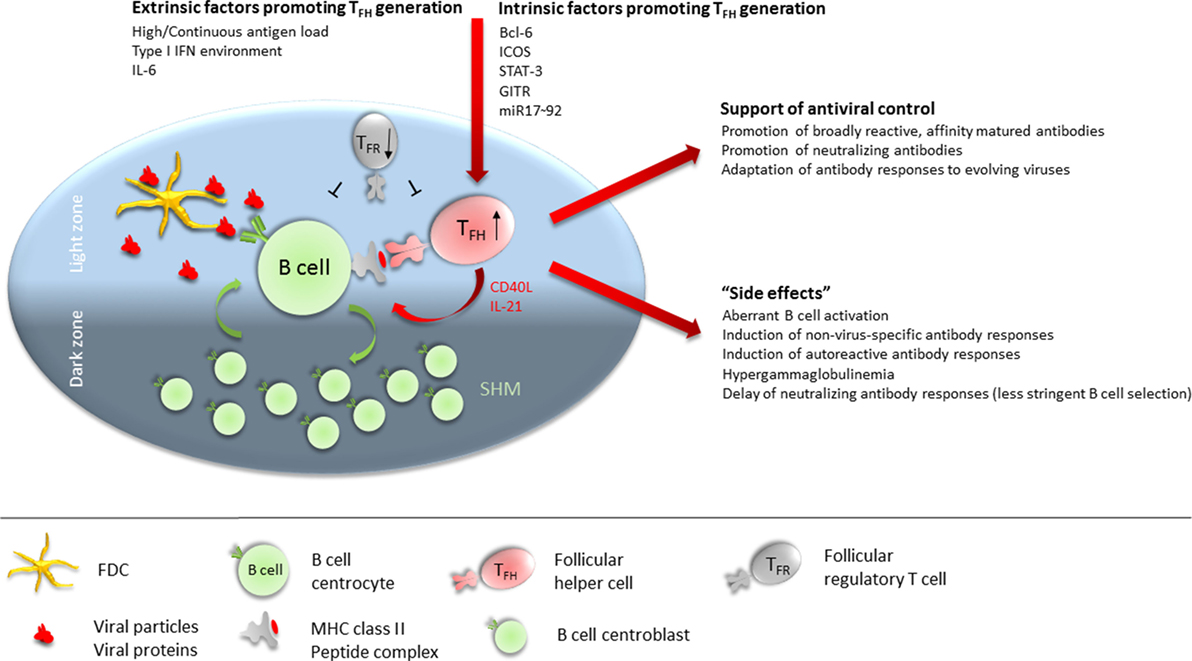
Figure 1. Follicular T helper (TFH) cells at the cross-road of helping versus inhibiting. TFH numbers are numerically increased in many chronic viral infections. Extrinsic factors contributing to promote TFH differentiation during chronic viral infections include continuous high antigen load, sustained type 1 IFN environment, and IL-6 availability. Intrinsically, Bcl-6, ICOS, signal transducer and activator of transcription (STAT)-3, GITR, and miR17–92 expression in CD4 T cells is required for (efficient) TFH differentiation. In the germinal center (GC), TFH cells preferentially localize to the light zone (LZ) where they interact via their TCR with B cells presenting antigenic peptides on MHC class II. B cells acquire antigen from follicular dendritic cells (FDCs) in the LZ which serve as antigen depot. FDCs retain antigen in form of antibody–antigen complexes or opsonized antigen via Fc and complement receptors. Cognate interaction between B cells and TFH provides survival, proliferation, and differentiation signals to the B cell in form of CD40 engagement and IL-21 supply. B cells will then either differentiate into antibody-secreting plasmablasts and long-lived plasma cells, into memory B cells, or enter the GC dark zone where the proliferate and undergo somatic hypermutation of their antibody variable regions before re-entering the LZ for selection of high-affinity B cells clones. Sustained activity of TFH cells is required throughout chronic viral infection to promote broadly reactive, affinity matured, and neutralizing antibodies and to adapt antibody specificity to emerging viral variants. Conversely, the high numbers of TFH cells present during many chronic viral infections render the GC LZ B cell activation and selection process less stringent, leading to aberrant B cell activation, induction of non virus-specific antibodies (including autoantibodies), hypergammaglobulinemia, and delayed generation of neutralizing antibody responses. Further contributing to a dysregulated TFH/B cell interaction in GCs is a dysbalanced ratio of TFH:TFR cells, often with reduced numbers of follicular regulatory T (TFR) cells in chronic viral infections.
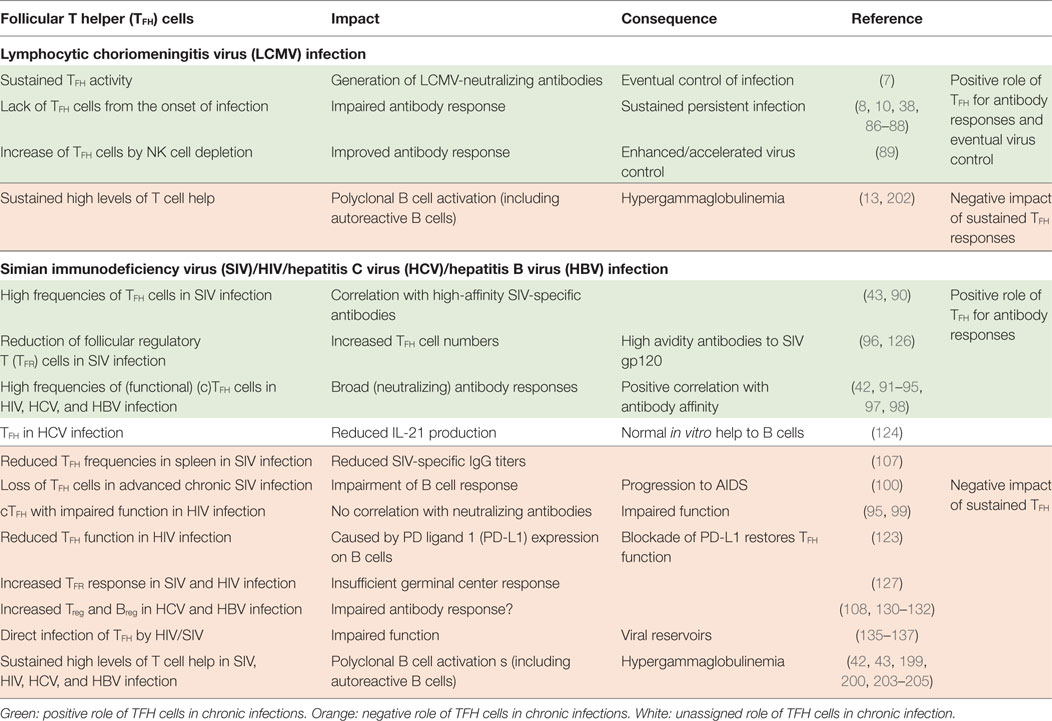
Table 1. Regulation and role of TFH cells in chronic viral infections.
TFH Cells
Follicular T helper cells are the main regulators of T-help-dependent antibody responses (23). Instruction of TFH cell differentiation is mediated in two steps. Priming of CD4 T cells that commit to the TFH cell lineage takes place in the T cell zone and is mediated by conventional DCs or monocyte-derived DCs (47, 48). In a second step, differentiation to TFH cells is further instructed and the TFH phenotype stably established by interactions between primed TFH cells and B cells at the border between T cell and B cell zone.
Which factors/cytokines instruct TFH differentiation is not entirely resolved, but both IL-6 and IL-21 can induce TFH differentiation via signaling through the transcription factor STAT-3 (49, 50). In the context of a persistent LCMV infection, it has furthermore been shown that late FDC-derived IL-6 is essential for TFH cell maintenance and eventual control of the infection (8).
CD4 T cells differentiating to TFH cells upregulate the hallmark transcriptional repressor Bcl-6 (24–26). Bcl-6 promotes commitment to the TFH cell lineage by repression of Blimp-1, which mediates expression of genes that are involved in the differentiation into other CD4 T cell lineages (24). Furthermore, Bcl-6 promotes localization of TFH cells toward the B cell follicle where T-help-dependent antibody responses take place. This is achieved in two different ways. For one, Bcl-6 represses the expression of molecules promoting localization in the T cell zone or egress from secondary lymphoid organs, i.e., CC chemokine receptor-7, Epstein–Barr virus-induced G-protein-coupled receptor (EBI)-2, or P-selectin glycoprotein-1 (51–53). Furthermore, Bcl-6 stabilizes the expression of CXCR5 on TFH cells, which is upregulated by the transcription factor achaete-scute homolog-2 (ASCL-2) upon priming (54). CXCR5 is essential for the localization of TFH cells toward the CXCL13-rich B cell follicles (27–29). TFH cells can further be distinguished by expression of other typical markers, which have important functions in mediating cognate interactions with B cells and thus sustaining antibody responses. Among these markers are the costimulatory molecules inducible T-cell costimulator (ICOS) and CD40L, the immunoregulatory molecule PD-1, their hallmark cytokine IL-21, and the T cell adaptor protein SAP (23, 55–58). Expression of these markers is moderate after priming and needs to be sustained and increased by interaction of TFH cells with cognate B cells and by ICOS signals delivered by ICOS ligand expressing bystander B cells in the interfollicular zone (34–37). These first interactions between TFH cells and B cells also determine the differentiation of TFH cells into GC TFH cells that induce and maintain the GC response and play an important role in the positive selection of affinity matured B cell clones. Expression of TFH markers is highest in GC TFH cells (37, 59, 60). After an immune response, some TFH cells have been shown to differentiate into long-lived memory cells, which downregulate some of their typical TFH markers like CXCR5, Bcl-6, and PD-1 (61, 62).
In humans, there have been further reports about circulating TFH-like cells that express CXCR5 and display a memory phenotype. Their expression of ICOS, PD-1, and Bcl-6 is reduced as well. However, these cells are efficient producers of IL-21 and IL-10 in in vitro coculture and effective inducers of B cell differentiation (23).
Closely related to TFH cells and equally important for the regulation of the GC responses are the so-called follicular regulatory T (TFR) cells. These are derived from thymus-derived T regulatory (Treg) cells, which adopt some TFH cell characteristics, like CXCR5 and Bcl-6 expression, to be able to migrate into B cell follicles. However, TFR cells lack expression of CD40L, IL-4, or IL-21 and have a higher expression of PD-1 and negatively regulate the GC response (63–65).
GC Response
The first encounter between TFH cells and activated B cells occurs in the interfollicular zone which lies at the border between T cell zone and B cell follicle (66–68). Here, interaction between TFH cells and cognate B cells induces a first round of B cell proliferation and instructs them to undergo one of three possible differentiation pathways. Either B cells undergo differentiation into short-lived extra-follicular plasmablasts, which produce a first wave of low affinity antibodies, or into GC-independent memory B cells or into GC B cells (30–33, 69). B cells destined to induce the GC response migrate with a subset of TFH cells, GC TFH cells, further into the B cell follicle. This migration is mediated by downregulation of EBI2 and upregulation of Sphingosine-1-phosphate receptor 2 on both B and TFH cells (66, 69–71).
The GC is partitioned into two distinct zones, the dark zone (DZ) and the light zone (LZ). In the DZ the cytokine CXCL12 is predominantly produced while the cytokine CXCL13 is predominantly produced in the LZ. Thereby, localization of B cells in DZ and LZ is controlled by differential expression of the chemokine receptors CXCR4 (migration into DZ) and CXCR5 (migration into LZ) (72). In the DZ, B cells undergo sequential rounds of proliferation (73–76). During this process, B cells upregulate the activation-induced cytidine deaminase (AID) which introduces point mutations into the variable regions of the B cell expressed BCRs/antibodies, a process termed SHM (77–79). Thereby, clonal B cell variants with different affinities toward one given antigen are generated. The activity of AID is also essential for class-switch reactions, which change the isotype of the antibodies (77, 79).
As SHM is a random process, it is necessary for B cells to undergo a selection process to ensure affinity maturation of the antibody repertoire and to exclude B cells that lost affinity for one antigen, decreased their affinity or even developed into autoreactive B cells. This selection process takes place in the LZ of the GC where also most of the GC TFH cells and FDCs are located (73, 75, 76). Upon entry into the LZ, B cells take up antigen which is stored on/presented by FDCs via their mutated BCR according to their affinity toward the antigen. Afterward, B cells present processed antigen to cognate GC TFH cells via their surface MHC II molecules. Higher affinity B cells are believed to have a competitive advantage in taking up FDC-stored antigen and thus are able to present more antigen on their surface MHC II molecules (80). The amount of presented antigen determines the amount of survival signals via ICOS, CD40L, and IL-21 a B cell clone receives from cognate GC TFH cells (39, 40, 76).
B cell clones that do not receive sufficient survival signals and therefore are negatively selected undergo apoptosis mediated by binding of Fas, expressed by the B cell, to FasL, expressed by the GC TFH cell (41, 81). Positively selected B cell clones either undergo another round of proliferation and SHM in the DZ or leave the GC reaction as long-lived plasma cells or memory B cells (74, 75, 82). B cells with the highest affinity may preferentially differentiate into plasma cells (69, 83, 84). B cell clones with a lower affinity, however, rather differentiate into memory B cells (82).
TFH Cells are Essential for the Emergence of Virus-Neutralizing Antibodies and Control of Persistent Viral Infection
The role of TFH cells for viral control during persistent viral infections, which is assumed to be dependent on development of neutralizing antibodies during the GC response (13, 16, 85), has been widely studied in the setting of persistent LCMV infection. For example, mice harboring a constitutive CXCR5 deficiency, and therefore being unable to develop TFH cells (and B cell follicles), exhibit an abrogated antibody response and prolonged viral persistence (38). Likewise, IL-6−/− (8), IL-6 signaling-deficient (10), STAT3−/− (86), glucocorticoid-induced tumor necrosis factor receptor related protein (GITR)-deficient mice (87), and mice with a T cell-specific deletion of the miR17–92 family of microRNAs (88) fail to elicit or maintain a TFH cell response upon (persistent) LCMV infection and are unable to eventually control the infection. Conversely, increasing the number of TFH cells by NK cell depletion accelerated viral clearance by improving the virus-specific antibody response (89).
Similar correlations between TFH cells and the appearance of protective antibody responses were observed in other persistent viral infections, e.g., with simian immunodeficiency virus (SIV), where the frequency of TFH cells positively correlated with the appearance of high-affinity SIV-specific antibodies in infected rhesus macaques (RM) (43). These TFH cells adopted a Th1-like profile regarding their chemokine receptor and cytokine expression (90). Furthermore, the quantity of TFH cells was higher in slow/non-progressor SIV-infected RMs, in which the virus was better contained, as compared with progressor SIV-infected RMs. The increase in TFH cell numbers in slow progressor correlated with higher titers of SIV-specific IgG antibodies in serum of infected RMs (43). Also, in chronically infected HIV-1, HCV and HBV patients, increased frequencies of a circulating population of cells with TFH characteristics (cTFH) (CXCR5+CXCR3−PD-1+) were observed (42, 91–98), which seemed closely related to GC TFH cells, based on their gene expression and cytokine profile. These were able to induce B cell differentiation in vitro and correlated with the appearance of broadly HIV-neutralizing antibodies (91, 92). In HIV controllers, an expanded population of functional gp120-specific TFH cells in blood correlated with gp120-specific B cell frequencies (93). However, other studies reported a reduced capacity of cTFH cells to provide help to B cells in (advanced) HIV-1 infected individuals (95, 99) or even loss of TFH cells in SIV-infected RMs (100).
Another indication implicating TFH cells in the eventual emergence of virus-neutralizing antibodies during persistent viral infection is the high frequency of somatic mutations in the variable regions of these antibodies (11, 19, 101, 102). SHM predominantly takes place in the GC and selection of high-affinity clones is supported by GC TFH cells (76). It is therefore tempting to speculate that continued activity of TFH cells during chronic viral infections is required for a continuous selection process of virus-specific B cell clones. This results not only in a continuous increase of their affinity toward viral antigens but also allows them to evolve to bind (and neutralize) to viral quasi species that emerge in vivo under selection pressures.
Indeed, we recently presented experimental evidence that sustained presence of CXCR5+/+ or Bcl6+/+ TFH cells is strictly required for the (late) emergence of LCMV-neutralizing antibodies. Using a novel in vivo experimental system allowed conditional depletion of specifically TFH cells or all LCMV-specific CD4 T cells during established persistent LCMV infection, after the initial establishment of the virus-specific IgG antibody response (7). This permitted, in contrast to previous studies (8, 10, 38, 86), to examine the function of TFH cells and LCMV-specific CD4 T cells during persistent viral infection beyond the mere induction of the virus-specific antibody response. This study revealed that LCMV-specific TFH cells (i.e., CXCR5+/+ or Bcl6+/+ CD4 T cells) were dispensable for maintaining overall LCMV-specific IgG titers and LCMV-specific IgG secreting plasma cells in spleen and bone marrow. By contrast, continued presence of LCMV-specific CD4 T cells was required to maintain overall LCMV-specific IgG titers as well as LCMV-specific IgG secreting plasma cells in bone marrow, suggesting that non-TFH LCMV-specific CD4 T cells are able to support an extra-follicular response to maintain the pool of LCMV-specific antibody-secreting cells and hence LCMV-specific IgG titers. However, sustained activity of TFH cells was strictly required for the development of LCMV-neutralizing antibodies by GC B cells (7), as conditional depletion of TFH cells reduced GC B cell numbers and abrogated emergence of antibodies with neutralizing capacity. Moreover, TFH cells seemed to be essential in driving the adaptation of the IgG response toward the contemporaneous circulating LCMV species, lending support to the notion that sustained TFH activity is important for continued selection of B cells. Importantly, the appearance of neutralizing antibodies was required for eventual control of an established persistent LCMV infection, demonstrating the importance of these antibodies and sustained presence and activity of TFH cells for control of a persistent infection in absence of overt immunopathology (7).
Nevertheless, the belated appearance of neutralizing antibodies in the setting of such persistent infections indicates possible restrictions of TFH cell function and/or their interactions with cognate B cells, which are discussed in the following sections.
Factors Curtailing TFH Cell Function upon Persistent Viral Infection
Optimal delivery of TFH cell help to cognate B cells as well as optimal TFH cell differentiation includes a chain of distinct steps at specific localizations in lymphoid tissue as well as a series of cell–cell interactions (23). With respect to localization-dependent processes, the structural integrity of secondary lymphoid organs is crucial allowing for initial encounter of activated CD4 and B cells as well as the establishment of GCs in the B cell follicle, including dark and LZ as designated compartments for proliferation, SHM and B cell selection. Cell–cell interactions that support B cell activation and production of (affinity-matured) antibodies comprise direct contact between activated CD4 T cell and cognate B cells initially at the T/B border, and later between TFH cells and cognate B cells in the GC LZ. Interference with any of these steps might lead to suboptimal antibody responses, which negatively affects control of persistent viral infections.
Destruction of Lymphoid Architecture
One possible influence on the establishment and the quality of TFH and GC B cell responses upon persistent viral infection is (immune-mediated) destruction of the lymphoid tissue architecture (11, 103–110). In chronic LCMV infection, this destruction is largely due to CD8 T cell-mediated cytotoxic activity directed against infected stromal cells as well as sustained type 1 IFN signaling and has been shown to hamper cognate interactions between T and B cells (11, 103–105, 111, 112). In SIV or HIV infection, immune activation-induced fibrosis of lymphoid tissues seems to play a major role in functional deterioration of secondary lymphoid organ structure and function, mediated by Treg-dependent transforming growth factor-beta 1 signaling and ensuing collagen deposition (109, 110).
Simian immunodeficiency virus-infected RMs with an expanded TFH cell population and increased SIV-specific antibody responses displayed a more intact lymph node (LN) structure as compared with fast progressing SIV-infected RMs with a less expanded TFH cell compartment (43, 107). This indicates that an intact lymphoid architecture is beneficial for virus-specific antibody responses and containment of the persistent infection.
Although destruction of lymphoid organ architecture is often attributed to cytotoxic CD8 T cells (104), an additional involvement of cytotoxic CD4 T cells during persistent LCMV infection has been shown (113). Cytotoxic CD4 T cells specifically targeted marginal zone (MZ) B cells, MZ macrophages, and metallophilic macrophages (113), subsets which have been implicated in the optimal induction of antibody responses (114–116). Analogous, depletion of MZ B cells was also reported in the context of persistent HIV infection (117) and a strong T helper response, possibly comprising cytotoxic CD4 T cells, is associated with low neutralizing antibody titers in persistent HCV infection (118).
During persistent LCMV infection, restoration of the lymphoid tissue architecture is closely associated with the onset of the neutralizing antibody response occurring between d40 and d80 post-infection (pi) (5, 103). During acute LCMV infection, lymphoid architecture is disrupted by day 8 pi and full reorganization, initiated by viral clearance and contraction of the CD8 T cell response, is only completed by d25 pi (103). During persistent LCMV infection, due to persistence of viral antigen and prolonged activity of CD8 and CD4 T cells before undergoing T cell exhaustion (1, 119–121), disruption of the lymphoid organ architecture is likely protracted as compared with acute LCMV infection. This underscores the relevance of lymphoid tissue reorganization and the onset of the LCMV-neutralizing antibody response, further emphasizing the beneficial effect of an intact lymphoid architecture and thereby optimal T and B cell interactions for the development of virus-neutralizing antibodies.
B Cell Dysfunction
In the context of HIV and SIV infection, B cell dysfunction was reported by a number of studies, characterized by loss of naïve and resting memory B cells, increases of activated B cells and tissue-like memory B cells, expansion of regulatory B cells, and altered functionality [reviewed in Ref. (122)].
In SIV or HIV infection, B cells were reported to actively render TFH cells ineffective in delivering help to B cells. GC B cells isolated from HIV or SIV-infected individuals/animals displayed a higher expression of PD ligand 1 (PD-L1) as compared with B cells isolated from healthy donors. Therefore, TFH cells received more signals via PD-1 during HIV/SIV infections, which mediated downregulation of IL-21 and IL-4 expression, and at the same time had a negative impact on TFH cell survival and proliferation (123). This impaired their B helper capacity, as observed in in vitro coculture experiments. Blocking of PD-L1 on B cells derived from HIV or SIV-infected donors, however, increased the ability of TFH cells to provide help to B cells as well as their cytokine expression (123). This also proved that TFH cells are in principle capable of providing sufficient help to B cells.
Furthermore, cTFH cells exhibiting reduced IL-21 expression as compared with healthy donors were identified in blood of persistently HCV infected patients (124). Surprisingly, however, in contrast to HIV and SIV infection, these cells proved to be capable of providing help to B cells in in vitro coculture experiments (124). These differences might be due to the different usage of B cell subsets in the coculture settings. Cocultures in the context of HIV/SIV infection were set up with GC-enriched B cells (123) while cocultures in the context of HCV infection used memory B cells (124). It is also conceivable that different non-or poorly cytopathic viruses use different mechanisms to render the antibody response ineffective upon persistent infection.
Altered Ratios of Regulatory Cells
In the setting of a recent HIV vaccination trial, it was established that the ratio of TFH cells to GC B cells is more important for the quality of the antibody response and eventual emergence of neutralizing antibodies than the total cell numbers. In this context, interaction of few GC B cells with one TFH cell was positively correlated with the occurrence of broadly neutralizing antibodies (bnab) (125). Furthermore, GC responses are subject to regulation by regulatory cells, in particular by TFR cells, which control the GC response to prevent aberrant production of antibodies (64). It has been shown in some studies that the frequency of TFR cells is reduced upon persistent infection with HIV and SIV (96, 126), albeit other studies have reported an increase of this population in HIV and SIV infection (127). Decreased levels of TFR cells favor the observed expansion of TFH cells and could indicate a less regulated GC response, hampering the induction of protective antibody responses for instance by a less stringent selection process and promoting unspecific B cell activation leading to hypergammaglobulinemia. Conversely, an expanded TFR population might contribute to inefficient GC responses [reviewed in Ref. (128)].
Also, in the context of persistent LCMV infection of lymphopenic mice lacking regulatory T cells, the induction of protective antibody responses was shown to be impaired (46, 129). Adoptive transfer of Treg improved the LCMV-specific antibody response and viral clearance drastically (129), proving the importance of balanced ratios between regulatory cells and TFH and GC B cells during the GC reaction.
Interestingly, in contrast to SIV, HIV and LCMV infection in lymphopenic mice, persistently HCV- or HBV-infected patients displayed an increase of regulatory B cells and Tregs as compared with healthy donors. This was associated with increases in IL-10 expression and increased PD-L1 expression on Treg cells (108, 130–132), which together might impair HCV- and HBV-specific antibody responses being associated with poor virus elimination and damage to lymphoid tissue (108).
Accumulation of TFH Cells Which Are Not Specific for Antigens Carrying Neutralizing Epitopes
Upon HIV infection, a predominant expansion of TFH cells that are specific for group-specific antigen is reported (42). However, induction of bnab seems to be associated with Env-specific TFH cells (91). Therefore, specific expansion of TFH cell populations, which are not recognizing the protein carrying neutralizing epitopes, could further contribute to the delayed emergence of neutralizing antibodies. TFH cells with other specificities would predominantly favor the survival of B cells expressing antibodies that are not specific for the neutralizing epitope. However, such intramolecular T cell help does not seem to be generally required and depends on the structure of the B cell activating viral antigen. While individual viral proteins engaging specific BCR would require intramolecular help, B cells interacting with intact or defective virions or virus-derived protein complexes could also be activated by TFH cells that are not necessarily specific for the protein containing the neutralizing epitopes (133, 134). Thus, it would be interesting to understand in more detail the structures of the selecting viral antigens/antigen complexes in the context of persistent viral infections to delineate more precisely the specificities of beneficial TFH responses.
Direct Infection of TFH and TFR Cells in HIV and SIV Infection
Upon HIV and SIV infection, TFH functionality is additionally compromised by their direct infection. CXCR5+ CD4 T cells are generally more permissive for HIV and SIV infection as compared with CXCR5− CD4 T cells, with GC TFH exhibiting the highest permissiveness (135–137). TFR cells are also highly permissive for HIV infection—even more so than TFH cells (138).
Surprisingly, infected TFH cells are not directly eliminated as compared with infected CXCR5− CD4 T cells. This might be due to the fact that only few CD8 T cells express CXCR5 and therefore cannot efficiently enter the B cell follicle where the infected TFH cells reside (136, 139, 140). In that way, TFH cells serve as viral reservoirs. However, during SIV and HIV infection, the CXCR5+ CD8 T cells that enter the GCs seem to contribute to control of infection (141), or alternatively negatively regulate T and B cells responses via IL-10 and Tim3-dependent processes (142). Infected GC TFH cells downregulate TFH markers during active viral replication (135) which might negatively affect their B cell helper functions, rendering the induction of antibody responses less effective.
Viral Evolution Can Mediate Evasion from the Neutralizing Antibody Response—Arms Race between Virus and the Humoral Immune Response
Besides immunological and secondary lymphoid organ topographical factors that might curtail effective TFH responses and thereby induction of neutralizing antibody responses, viral mutation can contribute to the establishment of persistence by escape from imposed immune pressure such as the humoral immune response. RNA viruses are known to evolve upon infection due to a high mutation rate during viral replication with their non-proofreading RNA-dependent RNA polymerase (or reverse transcriptase) and exist as a so-called quasi species in the infected host (143, 144). These high mutation rates allow the rapid adaptation of RNA viruses to changing environments and selective immune pressures (145). Viruses like HIV, HCV and LCMV take advantage of this viral evolution for the establishment of persistence, e.g., by sequential evasion from the adaptive immune response.
In persistent LCMV infection, especially in settings of reduced or absent CD8 T cell responses, escape variants from the neutralizing antibody response emerge that promote persistence of LCMV (15, 16, 146). This escape was mediated by only few amino acid substitutions in the neutralizing epitope contained in GP1 (15). However, LCMV generally has a rather low mutation rate, with 2.6 × 10−4 to 5.5 × 10−5 mutations per round of replication (147), compared with other RNA viruses with 10−3 to 10−5 miss-incorporations per copied nucleotide (15, 147, 148). Generally, selection of mutations was reduced or lacking in absence of neutralizing antibodies, indicating positive selection of escape viral variants upon antibody-imposed immune pressure (15). As recently published, escape of LCMV from the neutralizing antibody response also occurs in presence of a normal CD8 T cell response, meaning that neutralization of contemporaneous virus isolates lagged behind neutralization of the inoculating virus (7). This raises the question of how viral diversity is affected in absence of TFH cells. As animals with a conditional depletion of CXCR5+/+ TFH cells did not develop effective neutralizing antibodies against neither the inoculum or contemporaneous virus isolates, circulating antibodies most likely exhibited reduced immune pressure on the neutralizing epitopes (7). One would speculate that viral diversity is more restricted in absence of TFH cells as compared with control situations. Whether this prediction holds will have to be investigated in future studies.
Escape from the neutralizing antibody response and subsequent adaptation of the humoral immune response to new viral variants is more extensively investigated in persistent viral infection with HIV or HCV as compared with persistent LCMV infection. HIV and HCV infection share the common feature that the neutralizing antibody response is at first only directed against the autologous virus, while neutralization of heterologous viral variants by bnab is rather rare and only occurs later during infection (22, 149–152). Moreover, in HIV and HCV infection, the neutralizing antibody response toward the autologous virus usually lags behind the concurrent evolution of the viral quasi species, meaning that antibodies isolated from a given time point generally fail to neutralize contemporaneous virus isolates but are able to neutralize isolates from prior time points (22, 149, 152–154). Thus, during persistent viral infections a molecular arms race is taking place between the virus and the humoral immune response.
Mutations conferring escape are mostly accumulating in variable regions of the viral envelope (env), against which neutralizing antibodies are directed, e.g., the variable loops of HIV gp120 (19, 155) or the hypervariable region (HVR) of HCV (156, 157). Either these variable regions cover more conserved neutralizing epitopes or these regions contain the first neutralizing epitopes as in case of the HVR of HCV (20, 155–157). In addition, shielding of neutralizing epitopes by establishment of a glycan shield via mutational introduction of glycosylation sites is used by persistently infecting viruses to hamper binding of neutralizing antibodies by steric hindrance (22, 154, 158–162). Generally, neutralizing antibodies detect deglycosylated forms of the virion better than the glycosylated form as shown for HIV or Arenavirus infections (161, 163–165). Glycans reduce the on-rate of the neutralizing antibody and thereby limit their neutralizing capacity (161). In case of HIV infection, some glycans also increase the flexibility of the variable loops of the envelope protein, thereby increasing the binding entropy for neutralizing antibodies, which is unfavorable (166). Interestingly, however, in some HIV-infected patients, neutralizing antibodies that are able to penetrate the glycan shield by binding one or multiple conserved glycans (e.g., glycans at position N332 or N301) and simultaneously to gp120 protein residues (167–171) were elicited. This clearly shows that the humoral immune response is in principle able to develop antibodies that are able to bypass mechanisms conferring escape from the neutralizing antibody response.
Yet, in persistent HIV or HCV infections such bnab that bind to more conserved epitopes like glycan patches occur rather seldom (149, 150, 172–175). Most bnab are characterized by a high amount of somatic mutations, long CDRH3 regions and preferential usage of specific heavy and light chains (175–180). A high rate of somatic mutations can be observed in neutralizing antibodies against Arenaviruses like LCMV as well (11). Precursors of bnabs can be identified already early during the virus-specific humoral immune response upon persistent HIV infection (171, 181–186). The slow development of such precursors toward a bnab, together with the high quantity of somatic mutations, indicates that neutralizing antibodies mature over a prolonged period of time in the GC, including selection by TFH cells to develop the necessary neutralization breadth. Factors curtailing the GC response as described earlier might well contribute to the impaired or delayed emergence of such bnab. Furthermore, to allow the continued development of bnab in the GC response, their evolution/selection has to occur against viral variants that do not undergo complete viral escape from these bnab precursors (187).
Interestingly, diversity of the viral variants and the viral load influence the development of bnabs in HIV infection. Prolonged viremia and a higher diversity of the env are positively correlated with their induction (172, 173, 188–192). However, it is still a matter of discussion whether early diversity of the env (189, 190, 192), as, for example, achieved by superinfections (192) or a high diversity of the contemporaneous env genes is correlated with the emergence of bnab (193). Nevertheless, diversification of the viral variants is often observed before the onset of bnab responses (183, 184). Analogous, protracted viremia in persistent infections with Arenaviruses like LCMV is favorable for the induction of the neutralizing antibody response (11).
So far, little is known about the overall evolution of the LCMV-specific antibody response over the course of a chronic infection. Sustained TFH activity is crucial for the eventual emergence of neutralizing antibodies (7). However, how this sustained TFH activity supports the emergence of neutralizing antibodies is unclear. It could either be via continuous rounds of SHM and selection of B cells which would eventually give rise to antibodies with neutralizing capacity. Alternatively, TFH cells might be required for continued recruitment of new B cell clones into the GC response, thereby contributing to an overall broadening of the antibody repertoire. Interestingly, a recent vaccination study in humans repetitively exposed to the malaria parasite Plasmodium falciparum revealed that selection of potent B cell precursors from the naïve or memory pool contributed more efficiently to a potent antibody response to a complex antigen than the process of affinity maturation (194).
In the context of a chronic viral infection, it would be interesting to elucidate how viral diversity is reflected/presented in the GC response, leading to selection of the precursors of B cells producing neutralizing antibodies. Generally, GC B cells are dependent on taking up antigen from FDCs for affinity selection (76). However, whether this holds true in a setting with abundant free viral antigen during persistent viral infection still has to be determined. Interestingly, however, FDCs have been shown to be archives of viral quasi species upon HIV infection (195, 196), which would indicate constant binding of viral variants and their presentation. This would suggest that FDCs could also present the newest contemporaneous viral variants to B cells, which are then selected according to their affinity toward these variants. Yet, it remains unclear how fast the turnover rate of antigen presented by FDCs is in the setting of a chronic viral infection, which, in case of slow turnover, might lead to delays in the selection of B cells against the newest contemporaneous viral variants.
Moreover, emergence of bnab upon persistent HIV infection is also determined by the rate at which somatic mutations are acquired by B cell clones. For some bnab families it has been determined that the mutation rate was faster than that of the virus (181, 197), which enabled the host to “overtake” the viral evolution and develop an effective neutralizing antibody response. Concerning the role of TFH cells in the selection process of B cells producing such neutralizing antibodies, it has been established recently that the interaction intensity between TFH cells and GC B cells determines the quantity of proliferation rounds and therefore the quantity of somatic mutations a B cell can acquire (198). Therefore, it would be of interest to determine the influence of TFH cells on the mutation rate of such B cell clones. This could be achieved using the novel in vivo experimental model that allows conditional depletion of TFH cells upon persistent LCMV infection (7). Virus-specific plasma cells, developed in presence or absence of TFH cells, could be isolated at different time points pi, and the quantity of somatic mutations in the variable regions of heavy and light chains could be determined by NGS. Isolating contemporaneous virus isolates at the same time point and determining the sequences of their neutralizing epitopes by NGS could be used to relate the evolution kinetics of virus-specific B cells to the evolution of the virus. This approach could also be employed to determine whether the observed preferential usage of specific heavy and light chains by neutralizing antibodies is influenced by the absence of TFH cells. In absence of continuous TFH activity, one could conjecture that the overall diversity of B cell clones is increased, as the selection process is most likely much less stringent in absence of TFH cells, and the overall frequency of somatic mutations in B cells might be reduced due to insufficient selection and consecutive rounds of SHM.
Does Accumulation of TFH Cells Contribute to Dysregulated B Cell Responses upon Persistent Viral Infection?
During persistent viral infections with LCMV, HIV, SIV, or HCV, dysregulated B cell responses are observed. This includes the induction of hypergammaglobulinemia and polyclonal B cell activation, resulting in the emergence of seemingly virus-unspecific antibodies and in some cases even autoimmune reactive antibodies (199–205). However, in a recent study examining antibody responses toward Salmonella Typhimurium infection, it was shown that the seemingly predominantly Salmonella-unspecific antibody response was in fact of very low affinity toward Salmonella that increased due to affinity maturation in extra-follicular patches (206). Therefore, it would be interesting to investigate whether unspecific antibody responses elicited upon persistent viral infections might also display very low (undetectable in commonly used read-outs) affinities for the virus, which might improve upon affinity maturation and then allows recruitment into the virus-specific antibody response.
The described B cell dysfunctions have been further linked to the delayed appearance of neutralizing antibodies and in the context of persistent LCMV infection have been shown to be dependent on CD4 T cell help to cognate B cells via CD40:CD40L signals (13, 202, 207). It is believed that the virus-unspecific B cells acquire viral antigen from the environment and present it via their surface MHC II molecules to cognate CD4 T cells. How exactly virus-unspecific B cells acquire viral antigen to present to CD4 T cells and whether they might require signals via their BCR to become activated is not fully elucidated. In the setting of persistent LCMV infection, uptake of antigen by LCMV-unspecific B cells is independent of complement receptors (CRs) and FcγR, as knockout mice still display hypergammaglobulinemia (202). A recent study showed in the setting of an acute disseminated encephalomyelitis model with influenza infection, that uptake of the self-antigen myelin oligodendrocyte glycoprotein (MOG) via the BCR could occur concurrent with influenza hemagglutinin (HA). This led to the simultaneous presentation of MOG and HA on the MHC II surface molecules of MOG-specific B cells and subsequently their activation via HA-specific CD4 T cells (208). This scenario could serve as explanation for the activation of self-reactive B cells in the setting of persistent viral infections and would also indicate participation of BCR signaling pathways. However, this model does not account for virus-unspecific antibody responses toward non-self-antigens, such as for instance against the hapten nitrophenol (202). Another possible pathway that has been proposed to contribute to the uptake of viral antigen by virus-unspecific B cells in the setting of persistent viral infections is pinocytosis (202). Assumingly, due to the high viral burden, the concentration of viral particles and therefore viral antigen would be sufficient to induce sufficient uptake via this mechanism from the environment.
Regarding the contribution of TFH cells to dysregulated B cell responses, it has been shown before in settings of autoimmunity that prolonged maintenance of TFH cells, and therefore prolonged maintenance of GC B cells, is one cause for the emergence of autoreactive antibodies (58, 209–213). The selection threshold is lowered in GCs when TFH numbers are increased; thereby permitting the survival of low affinity and self-reactive B cells (214)—a situation that is met during persistent viral infections.
Analogous, in HIV and SIV infection, the expansion of TFH cells observed in LNs of infected individuals correlated with hypergammaglobulinemia and polyclonal B cell activation as well as the deletion of circulating memory B cells (42, 43, 215). Treatment of HIV-infected individuals with antiretroviral therapy reduced TFH cell numbers and at the same time B cell dysfunctions (42, 200), which indicates a connection between expansion of the TFH cell population in persistent HIV and SIV infections and dysregulated B cell responses. Similarly, in persistent HBV infection, the frequency of cTFH cells correlated with the emergence of autoantibodies (205).
Are there Organ-Specific Differences in TFH Cell Expansion and Function?
It also should be considered when discussing TFH accumulation and its impact on the antibody response that organ-specific differences might exist in specific persistent viral infections. This has been recently addressed in the context of SIV infection (107). Most studies upon persistent SIV and HIV infection have been conducted in blood or LN samples of infected animals/patients. Yet, recently, TFH responses have been analyzed in spleens of SIV-infected RMs (107). In contrast to results obtained from LNs of SIV-infected RMs, the TFH cell frequency in spleen was drastically reduced already in the acute phase of SIV infection as compared with healthy animals. This phenomenon was maintained in the persistent phase of SIV infection. In addition, TFH cells in spleen of SIV-infected RMs expressed less of the TFH-associated transcription factors Bcl-6 and c-Maf and instead upregulated transcription factors that counter-regulate TFH cell fate, i.e., Krüppel-like factor-2. This decrease in TFH cell frequency was further associated with reduced titers of SIV-specific IgG antibodies (107). However, TFH frequencies were similar or elevated in LNs of these infected RM as compared with healthy animals and in accordance with previous reports (43, 107, 215). Interestingly, the depletion of TFH cells in the spleen of SIV-infected RMs occurred in the context of severe destruction of the splenic architecture (107). Therefore, it might be possible that differences concerning the preservation of the lymphoid tissue could account for the observed organ-specific differences. Probably, due to the severe destruction of splenic architecture, SIV-infected TFH cells might have enhanced contact with cytotoxic CD8 T cells in the acute phase of infection, which might cause deletion of TFH cells in the spleen. Possibly, also differences in the recruitment of effector cells or different cytokine milieus in the LN and the spleen might influence the maintenance of TFH cells upon SIV infection.
Therefore, organ-specific differences in TFH cell frequency and function have to be taken into consideration as together they might account for the outcome of the virus-specific antibody response.
Concluding Remarks
Follicular T helper cell function and optimal interactions between TFH cells and cognate B cells often are hampered during persistent viral infections due to several factors. These include sustained increase of TFH cells, leading to non-specific B cell activation and hypergammaglobulinemia at the expense of virus-specific antibodies, destruction of the lymphoid tissue architecture, B cell exhaustion, skewed ratios of regulatory cells to TFH/GC B cells or in case of HIV/SIV infection TFH cells being directly infected. Due to these dysregulations, protective virus-specific antibody responses are delayed. Moreover, viruses use different mechanisms to evade recognition by antibodies using, e.g., variable loops or glycan shields to protect neutralizing epitopes. Furthermore, constant viral evolution leads to continued selection of escape variants upon exerted pressure by neutralizing antibodies, which fuels a molecular arms race between virus and the humoral immune response.
Nevertheless, it is clear that sustained activity of TFH cells is essential for the induction of neutralizing, protective antibody responses upon persistent viral infection and that the eventual emergence of these antibodies can afford control of the persistent infection in absence of overt immunopathology.
Therefore, targeting mechanisms that promote optimal TFH cell function and interactions with cognate B cells as well as understanding the underlying mechanisms of the arms race between virus and humoral immune response might serve to improve the induction of neutralizing antibody responses and reduce B cell dysfunctions, thereby improving control of persistent viral infections.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by ETH Zürich, the Swiss National Science Foundation (grant no. 310030_146140 and 310030_166078 to AO), and the Promedica Foundation.
References
2. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi:10.1038/nri3862
3. Vella LA, Herati RS, Wherry EJ. CD4(+) T cell differentiation in chronic viral infections: the Tfh perspective. Trends Mol Med (2017) 23(12):1072–87. doi:10.1016/j.molmed.2017.10.001
4. Hangartner L, Zellweger RM, Giobbi M, Weber J, Eschli B, McCoy KD, et al. Nonneutralizing antibodies binding to the surface glycoprotein of lymphocytic choriomeningitis virus reduce early virus spread. J Exp Med (2006) 203(8):2033–42. doi:10.1084/jem.20051557
5. Hangartner L, Zinkernagel RM, Hengartner H. Antiviral antibody responses: the two extremes of a wide spectrum. Nat Rev Immunol (2006) 6(3):231–43. doi:10.1038/nri1783
6. Bergthaler A, Flatz L, Hegazy AN, Johnson S, Horvath E, Löhning M, et al. Viral replicative capacity is the primary determinant of lymphocytic choriomeningitis virus persistence and immunosuppression. Proc Natl Acad Sci U S A (2010) 107(50):21641–6. doi:10.1073/pnas.1011998107
7. Greczmiel U, Krautler NJ, Pedrioli A, Bartsch I, Agnellini P, Bedenikovic G, et al. Sustained T follicular helper cell response is essential for control of chronic viral infection. Sci Immunol (2017) 2(18):eaam8686. doi:10.1126/sciimmunol.aam8686
8. Harker JA, Lewis GM, Mack L, Zuniga EI. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science (2011) 334(6057):825–9. doi:10.1126/science.1208421
9. Cook KD, Shpargel KB, Starmer J, Whitfield-Larry F, Conley B, Allard DE, et al. T follicular helper cell-dependent clearance of a persistent virus infection requires T cell expression of the histone demethylase UTX. Immunity (2015) 43(4):703–14. doi:10.1016/j.immuni.2015.09.002
10. Harker JA, Dolgoter A, Zuniga EI. Cell-intrinsic IL-27 and gp130 cytokine receptor signaling regulates virus-specific CD4(+) T cell responses and viral control during chronic infection. Immunity (2013) 39(3):548–59. doi:10.1016/j.immuni.2013.08.010
11. Eschli B, Zellweger RM, Wepf A, Lang KS, Quirin K, Weber J, et al. Early antibodies specific for the neutralizing epitope on the receptor binding subunit of the lymphocytic choriomeningitis virus glycoprotein fail to neutralize the virus. J Virol (2007) 81(21):11650–7. doi:10.1128/JVI.00955-07
12. Murira A, Lapierre P, Lamarre A. Chapter three – evolution of the humoral response during HCV infection: theories on the origin of broadly neutralizing antibodies and implications for vaccine design. Adv Immunol (2016) 129:55–107. doi:10.1016/bs.ai.2015.09.004
13. Recher M, Lang KS, Hunziker L, Freigang S, Eschli B, Harris NL, et al. Deliberate removal of T cell help improves virus-neutralizing antibody production. Nat Immunol (2004) 5(9):934–42. doi:10.1038/ni1102
14. Coutelier JP, Johnston SJ, El Idrissi MEA, Pfau CJ. Involvement of CD4+ cells in lymphocytic choriomeningitis virus-induced autoimmune anaemia and hypergammaglobulinaemia. J Autoimmun (1994) 7(5):589–99. doi:10.1006/jaut.1994.1043
15. Ciurea A, Hunziker L, Zinkernagel RM, Hengartner H. Viral escape from the neutralizing antibody response: the lymphocytic choriomeningitis virus model. Immunogenetics (2001) 53(3):185–9. doi:10.1007/s002510100314
16. Ciurea A, Klenerman P, Hunziker L, Horvath E, Senn BM, Ochsenbein AF, et al. Viral persistence in vivo through selection of neutralizing antibody-escape variants. Proc Natl Acad Sci U S A (2000) 97(6):2749–54. doi:10.1073/pnas.040558797
17. Pircher H, Moskophidis D, Rohrer U, Bürki K, Hengartner H, Zinkernagel RM. Viral escape by selection of cytotoxic T cell-resistant virus variants in vivo. Nature (1990) 346:629. doi:10.1038/346629a0
18. Geiss YD. Ursula: catch me if you can – the race between HIV and neutralizing antibodies. AIDS Rev (2015) 17(2):107–13.
19. Burton DR, Mascola JR. Antibody responses to envelope glycoproteins in HIV-1 infection. Nat Immunol (2015) 16(6):571–6. doi:10.1038/ni.3158
20. Burton DR, Stanfield RL, Wilson IA. Antibody vs. HIV in a clash of evolutionary titans. Proc Natl Acad Sci U S A (2005) 102(42):14943–8. doi:10.1073/pnas.0505126102
21. Bukh J. The history of hepatitis C virus (HCV): basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control. J Hepatol (2016) 65(1 Suppl):S2–21. doi:10.1016/j.jhep.2016.07.035
22. Richman DD, Wrin T, Little SJ, Petropoulos CJ. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc Natl Acad Sci U S A (2003) 100(7):4144–9. doi:10.1073/pnas.0630530100
23. Vinuesa CG, Linterman MA, Yu D, MacLennan ICM. Follicular helper T cells. Annu Rev Immunol (2016) 34(1):335–68. doi:10.1146/annurev-immunol-041015-055605
24. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325(5943):1006–10. doi:10.1126/science.1175870
25. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science (2009) 325(5943):1001–5. doi:10.1126/science.1176676
26. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor BCL-6 directs T follicular helper cell lineage commitment. Immunity (2009) 31(3):457–68. doi:10.1016/j.immuni.2009.07.002
27. Ansel KM, McHeyzer-Williams LJ, Ngo VN, McHeyzer-Williams MG, Cyster JG. In vivo–activated Cd4 T cells upregulate Cxc chemokine receptor 5 and reprogram their response to lymphoid chemokines. J Exp Med (1999) 190(8):1123–34. doi:10.1084/jem.190.8.1123
28. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express Cxc chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med (2000) 192(11):1545–52. doi:10.1084/jem.192.11.1545
29. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. Cxc chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med (2000) 192(11):1553–62. doi:10.1084/jem.192.11.1553
30. Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, et al. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J Exp Med (2011) 208(7):1377–88. doi:10.1084/jem.20102065
31. MacLennan ICM, Toellner K-M, Cunningham AF, Serre K, Sze DMY, Zúñiga E, et al. Extrafollicular antibody responses. Immunol Rev (2003) 194(1):8–18. doi:10.1034/j.1600-065X.2003.00058.x
32. O’Connor BP, Vogel LA, Zhang W, Loo W, Shnider D, Lind EF, et al. Imprinting the fate of antigen-reactive B cells through the affinity of the B cell receptor. J Immunol (2006) 177(11):7723–32. doi:10.4049/jimmunol.177.11.7723
33. Taylor JJ, Pape KA, Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med (2012) 209(3):597–606. doi:10.1084/jem.20111696
34. Barnett LG, Simkins HMA, Barnett BE, Korn LL, Johnson AL, Wherry EJ, et al. B cell antigen presentation in the initiation of follicular helper T cell and germinal center differentiation. J Immunol (2014) 192(8):3607–17. doi:10.4049/jimmunol.1301284
35. Deenick EK, Chan A, Ma CS, Gatto D, Schwartzberg PL, Brink R, et al. Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity (2010) 33(2):241–53. doi:10.1016/j.immuni.2010.07.015
36. Goenka R, Barnett LG, Silver JS, O’Neill PJ, Hunter CA, Cancro MP, et al. Cutting edge: dendritic cell-restricted antigen presentation initiates the follicular helper T cell program but cannot complete ultimate effector differentiation. J Immunol (2011) 187(3):1091–5. doi:10.4049/jimmunol.1100853
37. Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T, et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity (2011) 34(6):961–72. doi:10.1016/j.immuni.2011.03.025
38. Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, McGavern DB, Brooks DG. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med (2011) 208(5):987–99. doi:10.1084/jem.20101773
39. Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, et al. Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science (2014) 345(6200):1058–62. doi:10.1126/science.1257861
40. Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, et al. T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature (2015) 517(7533):214–8. doi:10.1038/nature13803
41. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2012) 30(1):429–57. doi:10.1146/annurev-immunol-020711-075032
42. Lindqvist M, van Lunzen J, Soghoian DZ, Kuhl BD, Ranasinghe S, Kranias G, et al. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J Clin Invest (2012) 122(9):3271–80. doi:10.1172/JCI64314
43. Petrovas C, Yamamoto T, Gerner MY, Boswell KL, Wloka K, Smith EC, et al. CD4 T follicular helper cell dynamics during SIV infection. J Clin Invest (2012) 122(9):3281–94. doi:10.1172/JCI63039
44. Osokine I, Snell LM, Cunningham CR, Yamada DH, Wilson EB, Elsaesser HJ, et al. Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc Natl Acad Sci U S A (2014) 111(20):7409–14. doi:10.1073/pnas.1401662111
45. Kuller LH, Tracy R, Belloso W, Wit SD, Drummond F, Lane HC, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med (2008) 5(10):e203. doi:10.1371/journal.pmed.0050203
46. Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity (2013) 38(3):596–605. doi:10.1016/j.immuni.2012.11.020
47. Butler NS, Kulu DI. The regulation of T follicular helper responses during infection. Curr Opin Immunol (2015) 34:68–74. doi:10.1016/j.coi.2015.02.007
48. Chakarov S, Fazilleau N. Monocyte-derived dendritic cells promote T follicular helper cell differentiation. EMBO Mol Med (2014) 6(5):590–603. doi:10.1002/emmm.201403841
49. Eto D, Lao C, DiToro D, Barnett B, Escobar TC, Kageyama R, et al. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS One (2011) 6(3):e17739. doi:10.1371/journal.pone.0017739
50. Karnowski A, Chevrier S, Belz GT, Mount A, Emslie D, D’Costa K, et al. B and T cells collaborate in antiviral responses via IL-6, IL-21, and transcriptional activator and coactivator, Oct2 and OBF-1. J Exp Med (2012) 209(11):2049–64. doi:10.1084/jem.20111504
51. Hatzi K, Nance JP, Kroenke MA, Bothwell M, Haddad EK, Melnick A, et al. BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J Exp Med (2015) 212(4):539–53. doi:10.1084/jem.20141380
52. Weber JP, Fuhrmann F, Feist RK, Lahmann A, Al Baz MS, Gentz L-J, et al. ICOS maintains the T follicular helper cell phenotype by down-regulating Krüppel-like factor 2. J Exp Med (2015) 212(2):217–33. doi:10.1084/jem.20141432
53. Lee JY, Skon CN, Lee YJ, Oh S, Taylor JJ, Malhotra D, et al. The transcription factor KLF2 restrains CD4+ T follicular helper cell differentiation. Immunity (2015) 42(2):252–64. doi:10.1016/j.immuni.2015.01.013
54. Liu X, Chen X, Zhong B, Wang A, Wang X, Chu F, et al. Transcription factor achaete-scute homologue 2 initiates follicular T-helper-cell development. Nature (2014) 507(7493):513–8. doi:10.1038/nature12910
55. Chtanova T, Tangye SG, Newton R, Frank N, Hodge MR, Rolph MS, et al. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J Immunol (2004) 173(1):68–78. doi:10.4049/jimmunol.173.1.68
56. Haynes NM, Allen CDC, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol (2007) 179(8):5099–108. doi:10.4049/jimmunol.179.8.5099
57. Reinhardt RL, Liang H-E, Locksley RM. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol (2009) 10(4):385–93. doi:10.1038/ni.1715
58. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity (2011) 34(6):932–46. doi:10.1016/j.immuni.2011.03.023
59. Kim CH, Rott LS, Clark-Lewis I, Campbell DJ, Wu L, Butcher EC. Subspecialization of Cxcr5+ T cells: B helper activity is focused in a germinal center–localized subset of Cxcr5+ T cells. J Exp Med (2001) 193(12):1373–82. doi:10.1084/jem.193.12.1373
60. Suan D, Nguyen A, Moran I, Bourne K, Hermes JR, Arshi M, et al. T follicular helper cells have distinct modes of migration and molecular signatures in naive and memory immune responses. Immunity (2015) 42(4):704–18. doi:10.1016/j.immuni.2015.03.002
61. Luthje K, Kallies A, Shimohakamada Y, Belz GT, Light A, Tarlinton DM, et al. The development and fate of follicular helper T cells defined by an IL-21 reporter mouse. Nat Immunol (2012) 13(5):491–8. doi:10.1038/ni.2261
62. Weber JP, Fuhrmann F, Hutloff A. T-follicular helper cells survive as long-term memory cells. Eur J Immunol (2012) 42(8):1981–8. doi:10.1002/eji.201242540
63. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med (2011) 17(8):983–8. doi:10.1038/nm.2426
64. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17(8):975–82. doi:10.1038/nm.2425
65. Wollenberg I, Agua-Doce A, Hernández A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol (2011) 187(9):4553–60. doi:10.4049/jimmunol.1101328
66. Coffey F, Alabyev B, Manser T. Initial clonal expansion of germinal center B cells takes place at the perimeter of follicles. Immunity (2009) 30(4):599–609. doi:10.1016/j.immuni.2009.01.011
67. Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity (2011) 34(6):947–60. doi:10.1016/j.immuni.2011.03.024
68. Okada T, Miller MJ, Parker I, Krummel MF, Neighbors M, Hartley SB, et al. Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PLoS Biol (2005) 3(6):e150. doi:10.1371/journal.pbio.0030150
69. Paus D, Phan TG, Chan TD, Gardam S, Basten A, Brink R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J Exp Med (2006) 203(4):1081–91. doi:10.1084/jem.20060087
70. Green JA, Suzuki K, Cho B, Willison LD, Palmer D, Allen CDC, et al. The sphingosine 1-phosphate receptor S1P2 maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat Immunol (2011) 12(7):672–80. doi:10.1038/ni.2047
71. Okada T, Cyster JG. B cell migration and interactions in the early phase of antibody responses. Curr Opin Immunol (2006) 18(3):278–85. doi:10.1016/j.coi.2006.02.005
72. Allen CDC, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol (2004) 5(9):943–52. doi:10.1038/ni1100
73. Allen CDC, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity (2007) 27(2):190–202. doi:10.1016/j.immuni.2007.07.009
74. Allen CDC, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science (2007) 315(5811):528–31. doi:10.1126/science.1136736
75. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell (2010) 143(4):592–605. doi:10.1016/j.cell.2010.10.032
76. Mesin L, Ersching J, Victora GD. Germinal center B cell dynamics. Immunity (2016) 45(3):471–82. doi:10.1016/j.immuni.2016.09.001
77. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell (2000) 102(5):553–63. doi:10.1016/S0092-8674(00)00078-7
78. Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem (1999) 274(26):18470–6. doi:10.1074/jbc.274.26.18470
79. Teng G, Papavasiliou FN. Immunoglobulin somatic hypermutation. Annu Rev Genet (2007) 41:107–20. doi:10.1146/annurev.genet.41.110306.130340
80. Batista FD, Neuberger MS. B cells extract and present immobilized antigen: implications for affinity discrimination. EMBO J (2000) 19(4):513–20. doi:10.1093/emboj/19.4.513
81. Yoshino T, Kondo E, Cao L, Takahashi K, Hayashi K, Nomura S, et al. Inverse expression of bcl-2 protein and Fas antigen in lymphoblasts in peripheral lymph nodes and activated peripheral blood T and B lymphocytes. Blood (1994) 83(7):1856–61.
82. Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, et al. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol (2016) 17(7):861–9. doi:10.1038/ni.3460
83. Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, et al. High affinity germinal center B cells are actively selected into the plasma cell compartment. J Exp Med (2006) 203(11):2419–24. doi:10.1084/jem.20061254
84. Smith KGC, Light A, Nossal GJV, Tarlinton DM. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J (1997) 16(11):2996–3006. doi:10.1093/emboj/16.11.2996
85. Seiler P, Kalinke U, Rülicke T, Bucher EM, Böse C, Zinkernagel RM, et al. Enhanced virus clearance by early inducible lymphocytic choriomeningitis virus-neutralizing antibodies in immunoglobulin-transgenic mice. J Virol (1998) 72(3):2253–8.
86. McIlwain DR, Grusdat M, Pozdeev VI, Xu HC, Shinde P, Reardon C, et al. T-cell STAT3 is required for the maintenance of humoral immunity to LCMV. Eur J Immunol (2015) 45(2):418–27. doi:10.1002/eji.201445060
87. Clouthier DL, Zhou AC, Wortzman ME, Luft O, Levy GA, Watts TH. GITR intrinsically sustains early type 1 and late follicular helper CD4 T cell accumulation to control a chronic viral infection. PLoS Pathog (2015) 11(1):e1004517. doi:10.1371/journal.ppat.1004517
88. Kang SG, Liu WH, Lu P, Jin HY, Lim HW, Shepherd J, et al. MicroRNAs of the miR-17 approximately 92 family are critical regulators of T(FH) differentiation. Nat Immunol (2013) 14(8):849–57. doi:10.1038/ni.2648
89. Cook KD, Kline HC, Whitmire JK. NK cells inhibit humoral immunity by reducing the abundance of CD4+ T follicular helper cells during a chronic virus infection. J Leukoc Biol (2015) 98(2):153–62. doi:10.1189/jlb.4HI1214-594R
90. Velu V, Mylvaganam GH, Gangadhara S, Hong JJ, Iyer SS, Gumber S, et al. Induction of Th1-biased T follicular helper (Tfh) cells in lymphoid tissues during chronic simian immunodeficiency virus infection defines functionally distinct germinal center Tfh cells. J Immunol (2016) 197(5):1832–42. doi:10.4049/jimmunol.1600143
91. Yamamoto T, Lynch RM, Gautam R, Matus-Nicodemos R, Schmidt SD, Boswell KL, et al. Quality and quantity of TFH cells are critical for broad antibody development in SHIVAD8 infection. Sci Transl Med (2015) 7(298):298ra120. doi:10.1126/scitranslmed.aab3964
92. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-1+CXCR3−CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity (2013) 39(4):758–69. doi:10.1016/j.immuni.2013.08.031
93. Buranapraditkun S, Pissani F, Teigler JE, Schultz BT, Alter G, Marovich M, et al. Preservation of peripheral T follicular helper cell function in HIV controllers. J Virol (2017) 91(14):e497–417. doi:10.1128/JVI.00497-17
94. Feng J, Lu L, Hua C, Qin L, Zhao P, Wang J, et al. High frequency of CD4+ CXCR5+ TFH cells in patients with immune-active chronic hepatitis B. PLoS One (2011) 6(7):e21698. doi:10.1371/journal.pone.0021698
95. Cubas R, van Grevenynghe J, Wills S, Kardava L, Santich BH, Buckner CM, et al. Reversible reprogramming of circulating memory T follicular helper cell function during chronic HIV infection. J Immunol (2015) 195(12):5625–36. doi:10.4049/jimmunol.1501524
96. Blackburn MJ, Zhong-Min M, Caccuri F, McKinnon K, Schifanella L, Guan Y, et al. Regulatory and helper follicular T cells and antibody avidity to simian immunodeficiency virus glycoprotein 120. J Immunol (2015) 195(7):3227–36. doi:10.4049/jimmunol.1402699
97. Hu TT, Song XF, Lei Y, Hu HD, Ren H, Hu P. Expansion of circulating TFH cells and their associated molecules: involvement in the immune landscape in patients with chronic HBV infection. Virol J (2014) 11:54. doi:10.1186/1743-422X-11-54
98. Li Y, Ma S, Tang L, Li Y, Wang W, Huang X, et al. Circulating chemokine (C-X-C motif) receptor 5(+) CD4(+) T cells benefit hepatitis B e antigen seroconversion through IL-21 in patients with chronic hepatitis B virus infection. Hepatology (2013) 58(4):1277–86. doi:10.1002/hep.26489
99. Boswell KL, Paris R, Boritz E, Ambrozak D, Yamamoto T, Darko S, et al. Loss of circulating CD4 T cells with B cell helper function during chronic HIV infection. PLoS Pathog (2014) 10(1):e1003853. doi:10.1371/journal.ppat.1003853
100. Xu H, Wang X, Malam N, Lackner AA, Veazey RS. Persistent simian immunodeficiency virus infection causes ultimate depletion of follicular Th cells in AIDS. J Immunol (2015) 195(9):4351–7. doi:10.4049/jimmunol.1501273
101. Haynes BF, Shaw GM, Korber B, Kelsoe G, Sodroski J, Hahn BH, et al. HIV-host interactions: implications for vaccine design. Cell Host Microbe (2016) 19(3):292–303. doi:10.1016/j.chom.2016.02.002
102. Klein F, Mouquet H, Dosenovic P, Scheid JF, Scharf L, Nussenzweig MC. Antibodies in HIV-1 vaccine development and therapy. Science (2013) 341(6151):1199–204. doi:10.1126/science.1241144
103. Scandella E, Bolinger B, Lattmann E, Miller S, Favre S, Littman DR, et al. Restoration of lymphoid organ integrity through the interaction of lymphoid tissue-inducer cells with stroma of the T cell zone. Nat Immunol (2008) 9(6):667–75. doi:10.1038/ni.1605
104. Odermatt B, Eppler M, Leist TP, Hengartner H, Zinkernagel RM. Virus-triggered acquired immunodeficiency by cytotoxic T-cell-dependent destruction of antigen-presenting cells and lymph follicle structure. Proc Natl Acad Sci U S A (1991) 88(18):8252–6. doi:10.1073/pnas.88.18.8252
105. Battegay M, Moskophidis D, Waldner H, Bründler MA, Fung-Leung WP, Mak TW, et al. Impairment and delay of neutralizing antiviral antibody responses by virus-specific cytotoxic T cells. J Immunol (1993) 151(10):5408–15.
106. Levesque MC, Moody MA, Hwang K-K, Marshall DJ, Whitesides JF, Amos JD, et al. Polyclonal B cell differentiation and loss of gastrointestinal tract germinal centers in the earliest stages of HIV-1 infection. PLoS Med (2009) 6(7):e1000107. doi:10.1371/journal.pmed.1000107
107. Moukambi F, Rabezanahary H, Rodrigues V, Racine G, Robitaille L, Krust B, et al. Early loss of splenic Tfh cells in SIV-infected rhesus macaques. PLoS Pathog (2015) 11(12):e1005287. doi:10.1371/journal.ppat.1005287
108. Wang L, Qiu J, Yu L, Hu X, Zhao P, Jiang Y. Increased numbers of CD5+CD19+CD1dhighIL-10+ Bregs, CD4+Foxp3+ Tregs, CD4+CXCR5+Foxp3+ follicular regulatory T (TFR) cells in CHB or CHC patients. J Transl Med (2014) 12(1):251. doi:10.1186/s12967-014-0251-9
109. Estes JD, Wietgrefe S, Schacker T, Southern P, Beilman G, Reilly C, et al. Simian immunodeficiency virus-induced lymphatic tissue fibrosis is mediated by transforming growth factor beta 1-positive regulatory T cells and begins in early infection. J Infect Dis (2007) 195(4):551–61. doi:10.1086/510852
110. Zeng M, Smith AJ, Wietgrefe SW, Southern PJ, Schacker TW, Reilly CS, et al. Cumulative mechanisms of lymphoid tissue fibrosis and T cell depletion in HIV-1 and SIV infections. J Clin Invest (2011) 121(3):998–1008. doi:10.1172/JCI45157
111. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science (2013) 340(6129):202–7. doi:10.1126/science.1235208
112. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KCF, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science (2013) 340(6129):207–11. doi:10.1126/science.1235214
113. Matter MS, Hilmenyuk T, Claus C, Marone R, Schürch C, Tinguely M, et al. Destruction of lymphoid organ architecture and hepatitis caused by CD4+ T cells. PLoS One (2011) 6(9):e24772. doi:10.1371/journal.pone.0024772
114. Song H, Cerny J. Functional heterogeneity of marginal zone B cells revealed by their ability to generate both early antibody-forming cells and germinal centers with hypermutation and memory in response to a T-dependent antigen. J Exp Med (2003) 198(12):1923–35. doi:10.1084/jem.20031498
115. Cinamon G, Matloubian M, Lesneski MJ, Xu Y, Low C, Lu T, et al. Sphingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat Immunol (2004) 5(7):713–20. doi:10.1038/ni1083
116. Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, et al. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature (2007) 450(7166):110–4. doi:10.1038/nature06287
117. Wilkins BS, Davis Z, Lucas SB, Delsol G, Jones DB. Splenic marginal zone atrophy and progressive CD8+ T-cell lymphocytosis in HIV infection: a study of adult post-mortem spleens from Côte d’Ivoire. Histopathology (2003) 42(2):173–85. doi:10.1046/j.1365-2559.2003.01569.x
118. Kaplan DE, Sugimoto K, Newton K, Valiga ME, Ikeda F, Aytaman A, et al. Discordant role of CD4 T-cell response relative to neutralizing antibody and CD8 T-cell responses in acute hepatitis C. Gastroenterology (2007) 132(2):654–66. doi:10.1053/j.gastro.2006.11.044
119. Frebel H, Richter K, Oxenius A. How chronic viral infections impact on antigen-specific T-cell responses. Eur J Immunol (2010) 40(3):654–63. doi:10.1002/eji.200940102
120. Oxenius A, Zinkernagel RM, Hengartner H. Comparison of activation versus induction of unresponsiveness of virus-specific CD4+ and CD8+ T cells upon acute versus persistent viral infection. Immunity (1998) 9(4):449–57. doi:10.1016/S1074-7613(00)80628-7
121. Fuller MJ, Zajac AJ. Ablation of CD8 and CD4 T cell responses by high viral loads. J Immunol (2003) 170(1):477–86. doi:10.4049/jimmunol.170.1.477
122. Moir S, Fauci AS. B-cell exhaustion in HIV infection: the role of immune activation. Curr Opin HIV AIDS (2014) 9(5):472–7. doi:10.1097/COH.0000000000000092
123. Cubas RA, Mudd JC, Savoye A-L, Perreau M, van Grevenynghe J, Metcalf T, et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med (2013) 19(4):494–9. doi:10.1038/nm.3109
124. Spaan M, Kreefft K, de Graav GN, Brouwer WP, de Knegt RJ, ten Kate FJW, et al. CD4+CXCR5+ T cells in chronic HCV infection produce less IL-21, yet are efficient at supporting B cell responses. J Hepatol (2015) 62(2):303–10. doi:10.1016/j.jhep.2014.09.024
125. Havenar-Daughton C, Carnathan DG, Torrents de la Peña A, Pauthner M, Briney B, Reiss SM, et al. Direct probing of germinal center responses reveals immunological features and bottlenecks for neutralizing antibody responses to HIV Env trimer. Cell Rep (2016) 17(9):2195–209. doi:10.1016/j.celrep.2016.10.085
126. Chowdhury A, Del Rio PME, Tharp GK, Trible RP, Amara RR, Chahroudi A, et al. Decreased T follicular regulatory cell/T follicular helper cell (TFH) in simian immunodeficiency virus–infected rhesus macaques may contribute to accumulation of TFH in chronic infection. J Immunol (2015) 195(7):3237–47. doi:10.4049/jimmunol.1502269
127. Miles B, Miller SM, Folkvord JM, Kimball A, Chamanian M, Meditz AL, et al. Follicular regulatory T cells impair follicular T helper cells in HIV and SIV infection. Nat Commun (2015) 6:8608. doi:10.1038/ncomms9608
128. Miles B, Miller SM, Connick E. CD4 T follicular helper and regulatory cell dynamics and function in HIV infection. Front Immunol (2016) 7:659. doi:10.3389/fimmu.2016.00659
129. Preite S, Baumjohann D, Foglierini M, Basso C, Ronchi F, Rodriguez BMF, et al. Somatic mutations and affinity maturation are impaired by excessive numbers of T follicular helper cells and restored by Treg cells or memory T cells. Eur J Immunol (2015) 45(11):3010–21. doi:10.1002/eji.201545920
130. Feng C, Cao L-J, Song H-F, Xu P, Chen H, Xu J-C, et al. Expression of PD-L1 on CD4+CD25+Foxp3+ regulatory T cells of patients with chronic HBV infection and its correlation with clinical parameters. Viral Immunol (2015) 28(8):418–24. doi:10.1089/vim.2015.0062
131. Gong Y, Zhao C, Zhao P, Wang M, Zhou G, Han F, et al. Role of IL-10-producing regulatory B cells in chronic hepatitis B virus infection. Dig Dis Sci (2015) 60(5):1308–14. doi:10.1007/s10620-014-3358-1
132. Liu Y, Cheng LS, Wu SD, Wang SQ, Li L, She WM, et al. IL-10-producing regulatory B-cells suppressed effector T-cells but enhanced regulatory T-cells in chronic HBV infection. Clin Sci (2016) 130(11):907–19. doi:10.1042/CS20160069
133. Oxenius A, Bachmann MF, Zinkernagel RM, Hengartner H. Virus-specific major MHC class II-restricted TCR-transgenic mice: effects on humoral and cellular immune responses after viral infection. Eur J Immunol (1998) 28(1):390–400. doi:10.1002/(SICI)1521-4141(199801)28:01<390::AID-IMMU390>3.0.CO;2-O
134. Yin L, Calvo-Calle JM, Cruz J, Newman FK, Frey SE, Ennis FA, et al. CD4+ T cells provide intermolecular help to generate robust antibody responses in vaccinia virus-vaccinated humans. J Immunol (2013) 190(12):6023–33. doi:10.4049/jimmunol.1202523
135. Kohler SL, Pham MN, Folkvord JM, Arends T, Miller SM, Miles B, et al. Germinal center T follicular helper cells are highly permissive to HIV-1 and alter their phenotype during virus replication. J Immunol (2016) 196(6):2711–22. doi:10.4049/jimmunol.1502174
136. Perreau M, Savoye A-L, De Crignis E, Corpataux J-M, Cubas R, Haddad EK, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med (2013) 210(1):143–56. doi:10.1084/jem.20121932
137. Xu H, Wang X, Malam N, Aye PP, Alvarez X, Lackner AA, et al. Persistent simian immunodeficiency virus infection drives differentiation, aberrant accumulation, and latent infection of germinal center follicular T helper cells. J Virol (2015) 90(3):1578–87. doi:10.1128/JVI.02471-15
138. Miller SM, Miles B, Guo K, Folkvord J, Meditz AL, McCarter MD, et al. Follicular regulatory T cells are highly permissive to r5-tropic HIV-1. J Virol (2017) 91(17):e430–417. doi:10.1128/JVI.00430-17
139. Fukazawa Y, Lum R, Okoye AA, Park H, Matsuda K, Bae JY, et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med (2015) 21(2):132–9. doi:10.1038/nm.3781
140. Connick E, Mattila T, Folkvord JM, Schlichtemeier R, Meditz AL, Ray MG, et al. CTL fail to accumulate at sites of HIV-1 replication in lymphoid tissue. J Immunol (2007) 178(11):6975–83. doi:10.4049/jimmunol.178.11.6975
141. Mylvaganam GH, Rios D, Abdelaal HM, Iyer S, Tharp G, Mavinger M, et al. Dynamics of SIV-specific CXCR5+ CD8 T cells during chronic SIV infection. Proc Natl Acad Sci U S A (2017) 114(8):1976–81. doi:10.1073/pnas.1621418114
142. Miles B, Miller SM, Folkvord JM, Levy DN, Rakasz EG, Skinner PJ, et al. Follicular regulatory CD8 T cells impair the germinal center response in SIV and ex vivo HIV infection. PLoS Pathog (2016) 12(10):e1005924. doi:10.1371/journal.ppat.1005924
143. Drake JW, Holland JJ. Mutation rates among RNA viruses. Proc Natl Acad Sci U S A (1999) 96(24):13910–3. doi:10.1073/pnas.96.24.13910
144. Simmonds P, Domingo E. Virus evolution. Curr Opin Virol (2011) 1(5):410–2. doi:10.1016/j.coviro.2011.10.021
145. Sevilla N, de la Torre JC. Arenavirus diversity and evolution: quasispecies in vivo. Curr Top Microbiol Immunol (2006) 299:315–35.
146. Hunziker L, Ciurea A, Recher M, Hengartner H, Zinkernagel RM. Public versus personal serotypes of a viral quasispecies. Proc Natl Acad Sci U S A (2003) 100(10):6015–20. doi:10.1073/pnas.1031671100
147. Grande-Perez A, Sierra S, Castro MG, Domingo E, Lowenstein PR. Molecular indetermination in the transition to error catastrophe: systematic elimination of lymphocytic choriomeningitis virus through mutagenesis does not correlate linearly with large increases in mutant spectrum complexity. Proc Natl Acad Sci U S A (2002) 99(20):12938–43. doi:10.1073/pnas.182426999
148. Sevilla N, Domingo E, de la Torre JC. Contribution of LCMV towards deciphering biology of quasispecies in vivo. Curr Top Microbiol Immunol (2002) 263:197–220.
149. von Hahn T, Yoon JC, Alter H, Rice CM, Rehermann B, Balfe P, et al. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology (2007) 132(2):667–78. doi:10.1053/j.gastro.2006.12.008
150. Logvinoff C, Major ME, Oldach D, Heyward S, Talal A, Balfe P, et al. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc Natl Acad Sci U S A (2004) 101(27):10149–54. doi:10.1073/pnas.0403519101
151. Gray ES, Moore PL, Choge IA, Decker JM, Bibollet-Ruche F, Li H, et al. Neutralizing antibody responses in acute human immunodeficiency virus type 1 subtype C infection. J Virol (2007) 81(12):6187–96. doi:10.1128/JVI.00239-07
152. Moore JP, Cao Y, Ho DD, Koup RA. Development of the anti-gp120 antibody response during seroconversion to human immunodeficiency virus type 1. J Virol (1994) 68(8):5142–55.
153. Zhao J, Nie J, Jiao Y, Li L, Zhang T, Liu Q, et al. Effect of the maturation of neutralizing antibodies on human immunodeficiency virus (HIV) envelope evolution in HIV-infected subjects. Infect Genet Evol (2016) 38:82–9. doi:10.1016/j.meegid.2015.12.012
154. Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, et al. Antibody neutralization and escape by HIV-1. Nature (2003) 422(6929):307–12. doi:10.1038/nature01470
155. Rong R, Li B, Lynch RM, Haaland RE, Murphy MK, Mulenga J, et al. Escape from autologous neutralizing antibodies in acute/early subtype C HIV-1 infection requires multiple pathways. PLoS Pathog (2009) 5(9):e1000594. doi:10.1371/journal.ppat.1000594
156. Ogata N, Alter HJ, Miller RH, Purcell RH. Nucleotide sequence and mutation rate of the H strain of hepatitis C virus. Proc Natl Acad Sci U S A (1991) 88(8):3392–6. doi:10.1073/pnas.88.8.3392
157. Farci P, Shimoda A, Wong D, Cabezon T, De Gioannis D, Strazzera A, et al. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc Natl Acad Sci U S A (1996) 93(26):15394–9. doi:10.1073/pnas.93.26.15394
158. Leonard CK, Spellman MW, Riddle L, Harris RJ, Thomas JN, Gregory TJ. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J Biol Chem (1990) 265(18):10373–82.
159. Helle F, Duverlie G, Dubuisson J. The hepatitis C virus glycan shield and evasion of the humoral immune response. Viruses (2011) 3(10):1909–32. doi:10.3390/v3101909
160. Helle F, Vieyres G, Elkrief L, Popescu C-I, Wychowski C, Descamps V, et al. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J Virol (2010) 84(22):11905–15. doi:10.1128/JVI.01548-10
161. Sommerstein R, Flatz L, Remy MM, Malinge P, Magistrelli G, Fischer N, et al. Arenavirus glycan shield promotes neutralizing antibody evasion and protracted infection. PLoS Pathog (2015) 11(11):e1005276. doi:10.1371/journal.ppat.1005276
162. Bonhomme CJ, Capul AA, Lauron EJ, Bederka LH, Knopp KA, Buchmeier MJ. Glycosylation modulates arenavirus glycoprotein expression and function. Virology (2011) 409(2):223–33. doi:10.1016/j.virol.2010.10.011
163. Binley JM, Wyatt R, Desjardins E, Kwong PD, Hendrickson W, Moore JP, et al. Analysis of the interaction of antibodies with a conserved, enzymatically deglycosylated core of the HIV type 1 envelope glycoprotein 120. AIDS Res Hum Retrovirus (1998) 14(3):191–8. doi:10.1089/aid.1998.14.191
164. Koch M, Pancera M, Kwong PD, Kolchinsky P, Grundner C, Wang L, et al. Structure-based, targeted deglycosylation of HIV-1 gp120 and effects on neutralization sensitivity and antibody recognition. Virology (2003) 313(2):387–400. doi:10.1016/S0042-6822(03)00294-0
165. Ma B-J, Alam SM, Go EP, Lu X, Desaire H, Tomaras GD, et al. Envelope deglycosylation enhances antigenicity of HIV-1 gp41 epitopes for both broad neutralizing antibodies and their unmutated ancestor antibodies. PLoS Pathog (2011) 7(9):e1002200. doi:10.1371/journal.ppat.1002200
166. Qi Y, Jo S, Im W. Roles of glycans in interactions between gp120 and HIV broadly neutralizing antibodies. Glycobiology (2016) 26(3):251–60. doi:10.1093/glycob/cwv101
167. McLellan JS, Pancera M, Carrico C, Gorman J, Julien J-P, Khayat R, et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature (2011) 480(7377):336–43. doi:10.1038/nature10696
168. Pejchal R, Doores KJ, Walker LM, Khayat R, Huang P-S, Wang S-K, et al. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science (2011) 334(6059):1097–103. doi:10.1126/science.1213256
169. Kong L, Lee JH, Doores KJ, Murin CD, Julien J-P, McBride R, et al. Supersite of immune vulnerability on the glycosylated face of HIV-1 envelope glycoprotein gp120. Nat Struct Mol Biol (2013) 20(7):796–803. doi:10.1038/nsmb.2594
170. Pancera M, Shahzad-ul-Hussan S, Doria-Rose NA, McLellan JS, Bailer RT, Dai K, et al. Structural basis for diverse N-glycan recognition by HIV-1–neutralizing V1–V2–directed antibody PG16. Nat Struct Mol Biol (2013) 20(7):804–13. doi:10.1038/nsmb.2600
171. Garces F, Sok D, Kong L, McBride R, Kim HJ, Saye-Francisco KF, et al. Structural evolution of glycan recognition by a family of potent HIV antibodies. Cell (2014) 159(1):69–79. doi:10.1016/j.cell.2014.09.009
172. Doria-Rose NA, Klein RM, Manion MM, O’Dell S, Phogat A, Chakrabarti B, et al. Frequency and phenotype of human immunodeficiency virus envelope-specific b cells from patients with broadly cross-neutralizing antibodies. J Virol (2009) 83(1):188–99. doi:10.1128/JVI.01583-08
173. Sather DN, Armann J, Ching LK, Mavrantoni A, Sellhorn G, Caldwell Z, et al. Factors associated with the development of cross-reactive neutralizing antibodies during human immunodeficiency virus type 1 infection. J Virol (2009) 83(2):757–69. doi:10.1128/JVI.02036-08
174. Simek MD, Rida W, Priddy FH, Pung P, Carrow E, Laufer DS, et al. Human immunodeficiency virus type 1 elite neutralizers: individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J Virol (2009) 83(14):7337–48. doi:10.1128/JVI.00110-09
175. West AP Jr, Scharf L, Scheid JF, Klein F, Bjorkman PJ, Nussenzweig MC. Nussenzweig: structural insights on the role of antibodies in HIV-1 vaccine and therapy. Cell (2014) 156(4):633–48. doi:10.1016/j.cell.2014.01.052
176. Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien J-P, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature (2011) 477(7365):466–70. doi:10.1038/nature10373
177. Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TYK, et al. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science (2011) 333(6049):1633–7. doi:10.1126/science.1207227
178. Verkoczy L, Kelsoe G, Moody MA, Haynes BF. Role of immune mechanisms in induction of HIV-1 broadly neutralizing antibodies. Curr Opin Immunol (2011) 23(3):383–90. doi:10.1016/j.coi.2011.04.003
179. Briney BS, Willis JR, Crowe JE Jr. Human peripheral blood antibodies with long HCDR3s are established primarily at original recombination using a limited subset of germline genes. PLoS One (2012) 7(5):e36750. doi:10.1371/journal.pone.0036750
180. Burton DR, Ahmed R, Barouch DH, Butera ST, Crotty S, Godzik A, et al. A blueprint for HIV vaccine discovery. Cell Host Microbe (2012) 12(4):396–407. doi:10.1016/j.chom.2012.09.008
181. Wu X, Zhang Z, Schramm CA, Joyce MG, Kwon YD, Zhou T, et al. Maturation and diversity of the VRC01-antibody lineage over 15 years of chronic HIV-1 infection. Cell (2015) 161(3):470–85. doi:10.1016/j.cell.2015.03.004
182. Fera D, Schmidt AG, Haynes BF, Gao F, Liao H-X, Kepler TB, et al. Affinity maturation in an HIV broadly neutralizing B-cell lineage through reorientation of variable domains. Proc Natl Acad Sci U S A (2014) 111(28):10275–80. doi:10.1073/pnas.1409954111
183. Doria-Rose NA, Schramm CA, Gorman J, Moore PL, Bhiman JN, DeKosky BJ, et al. Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature (2014) 509(7498):55–62. doi:10.1038/nature13036
184. Liao H-X, Lynch R, Zhou T, Gao F, Alam SM, Boyd SD, et al. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature (2013) 496(7446):469–76. doi:10.1038/nature12053
185. Wibmer CK, Bhiman JN, Gray ES, Tumba N, Abdool Karim SS, Williamson C, et al. Viral escape from HIV-1 neutralizing antibodies drives increased plasma neutralization breadth through sequential recognition of multiple epitopes and immunotypes. PLoS Pathog (2013) 9(10):e1003738. doi:10.1371/journal.ppat.1003738
186. Sather DN, Carbonetti S, Malherbe DC, Pissani F, Stuart AB, Hessell AJ, et al. Emergence of broadly neutralizing antibodies and viral coevolution in two subjects during the early stages of infection with human immunodeficiency virus type 1. J Virol (2014) 88(22):12968–81. doi:10.1128/JVI.01816-14
187. Gao F, Bonsignori M, Liao H-X, Kumar A, Xia S-M, Lu X, et al. Haynes: cooperation of B cell lineages in induction of HIV-1-broadly neutralizing antibodies. Cell (2014) 158(3):481–91. doi:10.1016/j.cell.2014.06.022
188. Rusert P, Kouyos RD, Kadelka C, Ebner H, Schanz M, Huber M, et al. Determinants of HIV-1 broadly neutralizing antibody induction. Nat Med (2016) 22(11):1260–7. doi:10.1038/nm.4187
189. Smith SA, Burton SL, Kilembe W, Lakhi S, Karita E, Price M, et al. Diversification in the HIV-1 envelope hyper-variable domains V2, V4, and V5 and higher probability of transmitted/founder envelope glycosylation favor the development of heterologous neutralization breadth. PLoS Pathog (2016) 12(11):e1005989. doi:10.1371/journal.ppat.1005989
190. Piantadosi A, Chohan B, Panteleeff D, Baeten JM, Mandaliya K, Ndinya-Achola JO, et al. HIV-1 evolution in gag and env is highly correlated but exhibits different relationships with viral load and the immune response. AIDS (2009) 23(5):579–87. doi:10.1097/QAD.0b013e328328f76e
191. Doria-Rose NA, Klein RM, Daniels MG, O’Dell S, Nason M, Lapedes A, et al. Breadth of human immunodeficiency virus-specific neutralizing activity in sera: clustering analysis and association with clinical variables. J Virol (2010) 84(3):1631–6. doi:10.1128/JVI.01482-09
192. Euler Z, van den Kerkhof TLGM, van Gils MJ, Burger JA, Edo-Matas D, Phung P, et al. Longitudinal analysis of early HIV-1-specific neutralizing activity in an elite neutralizer and in five patients who developed cross-reactive neutralizing activity. J Virol (2012) 86(4):2045–55. doi:10.1128/JVI.06091-11
193. Carter CC, Wagner GA, Hightower GK, Caballero G, Phung P, Richman DD, et al. HIV-1 neutralizing antibody response and viral genetic diversity characterized with next generation sequencing. Virology (2015) 474:34–40. doi:10.1016/j.virol.2014.10.019
194. Murugan R, Buchauer L, Triller G, Kreschel C, Costa G, Pidelaserra Marti G, et al. Clonal selection drives protective memory B cell responses in controlled human malaria infection. Sci Immunol (2018) 3(20):eaa8029. doi:10.1126/sciimmunol.aap8029
195. Heesters BA, Lindqvist M, Vagefi PA, Scully EP, Schildberg FA, Altfeld M, et al. Follicular dendritic cells retain infectious HIV in cycling endosomes. PLoS Pathog (2015) 11(12):e1005285. doi:10.1371/journal.ppat.1005285
196. Keele BF, Tazi L, Gartner S, Liu Y, Burgon TB, Estes JD, et al. Characterization of the follicular dendritic cell reservoir of human immunodeficiency virus type 1. J Virol (2008) 82(11):5548–61. doi:10.1128/JVI.00124-08
197. Alter G, Barouch DH. Natural evolution of broadly neutralizing antibodies. Cell (2015) 161(3):427–8. doi:10.1016/j.cell.2015.04.007
198. Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature (2014) 509(7502):637–40. doi:10.1038/nature13300
199. Lane HC, Masur H, Edgar LC, Whalen G, Rook AH, Fauci AS. Abnormalities of B-cell activation and immunoregulation in patients with the acquired immunodeficiency syndrome. N Engl J Med (1983) 309(8):453–8. doi:10.1056/NEJM198308253090803
200. Moir S, Fauci AS. B cells in HIV infection and disease. Nat Rev Immunol (2009) 9(4):235–45. doi:10.1038/nri2524
201. Meyer K, Ait-Goughoulte M, Keck Z-Y, Foung S, Ray R. Antibody-dependent enhancement of hepatitis C virus infection. J Virol (2008) 82(5):2140–9. doi:10.1128/JVI.01867-07
202. Hunziker L, Recher M, Macpherson AJ, Ciurea A, Freigang S, Hengartner H, et al. Hypergammaglobulinemia and autoantibody induction mechanisms in viral infections. Nat Immunol (2003) 4(4):343–9. doi:10.1038/ni911
203. Shirai A, Cosentino M, Leitman-Klinman SF, Klinman DM. Human immunodeficiency virus infection induces both polyclonal and virus-specific B cell activation. J Clin Invest (1992) 89(2):561–6. doi:10.1172/JCI115621
204. Rosa D, Saletti G, De Gregorio E, Zorat F, Comar C, D’Oro U, et al. Activation of naïve B lymphocytes via CD81, a pathogenetic mechanism for hepatitis C virus-associated B lymphocyte disorders. Proc Natl Acad Sci U S A (2005) 102(51):18544–9. doi:10.1073/pnas.0509402102
205. Lei Y, Hu T, Song X, Nie H, Chen M, Chen W, et al. Production of autoantibodies in chronic hepatitis B virus infection is associated with the augmented function of blood CXCR5+CD4+ T cells. PLoS One (2016) 11(9):e0162241. doi:10.1371/journal.pone.0162241
206. Di Niro R, Lee SJ, Vander Heiden JA, Elsner RA, Trivedi N, Bannock JM, et al. Shlomchik: Salmonella infection drives promiscuous B cell activation followed by extrafollicular affinity maturation. Immunity (2015) 43(1):120–31. doi:10.1016/j.immuni.2015.06.013
207. Jellison ER, Guay HM, Szomolanyi-Tsuda E, Welsh RM. Dynamics and magnitude of virus-induced polyclonal B cell activation mediated by BCR-independent presentation of viral antigen. Eur J Immunol (2007) 37(1):119–28. doi:10.1002/eji.200636516
208. Sanderson NSR, Zimmermann M, Eilinger L, Gubser C, Schaeren-Wiemers N, Lindberg RLP, et al. Cocapture of cognate and bystander antigens can activate autoreactive B cells. Proc Natl Acad Sci U S A (2017) 114(4):734–9. doi:10.1073/pnas.1614472114
209. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41(4):529–42. doi:10.1016/j.immuni.2014.10.004
210. Simpson N, Gatenby PA, Wilson A, Malik S, Fulcher DA, Tangye SG, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum (2010) 62(1):234–44. doi:10.1002/art.25032
211. Ma J, Zhu C, Ma B, Tian J, Baidoo SE, Mao C, et al. Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin Dev Immunol (2012) 2012:827480. doi:10.1155/2012/827480
212. Szabo K, Papp G, Barath S, Gyimesi E, Szanto A, Zeher M. Follicular helper T cells may play an important role in the severity of primary Sjögren’s syndrome. Clin Immunol (2013) 147(2):95–104. doi:10.1016/j.clim.2013.02.024
213. Szabó K, Papp G, Szántó A, Tarr T, Zeher M. A comprehensive investigation on the distribution of circulating follicular T helper cells and B cell subsets in primary Sjögren’s syndrome and systemic lupus erythematosus. Clin Exp Immunol (2016) 183(1):76–89. doi:10.1111/cei.12703
214. Pratama A, Vinuesa CG. Control of TFH cell numbers: why and how? Immunol Cell Biol (2014) 92(1):40–8. doi:10.1038/icb.2013.69
215. Hong JJ, Amancha PK, Rogers K, Ansari AA, Villinger F. Spatial alterations between CD4+ T follicular helper, B, and CD8+ T cells during simian immunodeficiency virus infection: T/B cell homeostasis, activation, and potential mechanism for viral escape. J Immunol (2012) 188(7):3247–56. doi:10.4049/jimmunol.1103138
Keywords: follicular helper cells, chronic viral infection, antibody responses, germinal center, viral evolution
Citation: Greczmiel U and Oxenius A (2018) The Janus Face of Follicular T Helper Cells in Chronic Viral Infections. Front. Immunol. 9:1162. doi: 10.3389/fimmu.2018.01162
Received: 06 February 2018; Accepted: 09 May 2018;
Published: 25 May 2018
Edited by:
Georgia Fousteri, San Raffaele Hospital (IRCCS), ItalyReviewed by:
Ramon Arens, Leiden University Medical Center, NetherlandsKarl Lang, University of Essen, Germany
Copyright: © 2018 Greczmiel and Oxenius. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annette Oxenius, b3hlbml1c0BtaWNyby5iaW9sLmV0aHouY2g=